
Abstract
Despite the STING-type-I interferon pathway playing a key role in effective anti-tumor immunity, the therapeutic benefit of direct STING agonists appears limited. In this study, we use several artificial intelligence techniques and patient-based multi-omics data to show that Ectonucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1), which hydrolyzes STING-activating cyclic GMP-AMP (cGAMP), is a safer and more effective STING-modulating target than direct STING agonism in multiple solid tumors. We then leverage our generative chemistry artificial intelligence-based drug design platform to facilitate the design of ISM5939, an orally bioavailable ENPP1-selective inhibitor capable of stabilizing extracellular cGAMP and activating bystander antigen-presenting cells without inducing either toxic inflammatory cytokine release or tumor-infiltrating T-cell death. In murine syngeneic models across cancer types, ISM5939 synergizes with targeting the PD-1/PD-L1 axis and chemotherapy in suppressing tumor growth with good tolerance. Our findings provide evidence supporting ENPP1 as an innate immune checkpoint across solid tumors and reports an AI design-aided ENPP1 inhibitor, ISM5939, as a cutting-edge STING modulator for cancer therapy, paving a path for immunotherapy advancements.
Similar content being viewed by others


Introduction
Activation of the STING pathway promotes the production of type I interferons (IFNs), thereby enhancing antitumor immunity via cell-autonomous and non-autonomous mechanisms1. STING activation in antigen-presenting cells (APCs), such as dendritic cells (DCs) and macrophages, licenses the cells for enhanced antigen presentation and upregulates co-stimulatory molecules, thereby enhancing the ability to prime and activate cytotoxic T cells2,3,4. Additionally, STING signaling in the tumor microenvironment (TME) has been demonstrated to dampen tumorigenic and pro-metastatic processes, such as myeloid-derived suppressor cell activation, T-cell suppression, and the tumor-intrinsic epithelial-mesenchymal transition5. The pathway’s central role in regulating immune responses makes it a promising target for cancer immunotherapy.
Despite its potential as an activator of antitumor immunity, the development of direct STING agonists has so far encountered substantial difficulty. Cyclic dinucleotide (CDN) STING agonists, such as ADU-S100 and MK-1454, suffer from poor bioavailability and can only be administrated intratumorally, with a marginal therapeutic benefit6,7,8. Non-CDN STING agonists, such as MSA-2 and SR-717, designed with improved bioavailability, lead to systemic inflammatory responses and toxic cytokine release when administered systemically in preclinical models9,10, thus presenting substantial safety concerns. Given the significant challenges associated with direct STING agonists, there is a pressing need for developing alternative strategies to safely and effectively modulate the STING pathway.
Detection of cytoplasmic DNA by cyclic GMP–AMP synthase (cGAS) catalyzes formation of second-messenger cyclic GMP-AMP (cGAMP), which then activates STING11. The cellular export and import of cGAMP among the various cell types within the TME facilitates endogenous STING agonism and effective antitumor immunity5,12,13,14,15,16. However, the intracellular accumulation of imported cGAMP is significantly impeded by cell surface-bound ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), which is the main degrader of cGAMP12,17,18,19. High expression of ENPP1 within the tumor bed in various solid tumors suggests a link between ENPP1’s role in antagonizing cGAMP-STING-mediated antitumoral immunity and cancer onset, progression, and metastasis13,15,20,21 Inhibition of ENPP1 results in increased extracellular cGAMP, allowing for sustained, controlled activation of the STING pathway by promoting consistent and localized STING activation without the systemic toxicity associated with direct STING agonists. Accordingly, ENPP1 inhibitors show promise as next-generation STING modulators effective in overcoming resistance to immunotherapy characteristic of “immune-cold” cancers22.
However, the development of ENPP1 inhibitors has so far proved challenging, with only a few candidates such as TXN10128, RBS2418, and SR-8541A entering phase 1 studies. Common shortcomings include low potency and specificity at physiological pH, as many inhibitors lose efficacy outside optimal assay conditions (e.g., pH 9)18, as well as unfavorable pharmacokinetic properties23. Moreover, any such inhibitor must also sufficiently avoid the challenges common among small-molecule inhibitors, including poor oral absorption, low overall efficacy in physiological settings, and non-specific interactions, such as blocking cGAMP export. Accordingly, there is an urgent need to develop a highly potent and selective ENPP1 inhibitor that can be administered orally to enhance cancer immunotherapy.
Artificial intelligence (AI)-driven molecular design has recently emerged as an efficient strategy to discover hit compounds24,25,26 by facilitating and integrating the discovery of disease-associated molecular targets27,28,29, designing compounds specific to those targets30,31,32,33,34,35,36, and identifying disease indications and patient populations most likely to respond to targeted therapy37,38,39,40,41,42,43,44. Several generative chemistry approaches have designed experimentally validated molecules30, with Chemistry4245 emerging as one of the most frequently validated multi-model platforms30.
In this work, our generative AI-integrated workflow extended ENPP1 as an immune checkpoint among multiple solid tumors and assisted in developing a highly specific oral ENPP1 inhibitor, ISM5939. Mechanistically, ISM5939 specifically suppresses cGAMP degradation and augments non-cell autonomous STING signaling in the tumor microenvironment, which synergizes with current standard-of-care treatments, including anti-PD(L)1 therapy and DNA-destabilizing agents. The identification and development of ISM5939 as a specific oral ENPP1 inhibitor marks a significant advance in cancer immunotherapy, offering a promising strategy to harness the cGAS-STING pathway for enhanced antitumor immunity with minimized systemic toxicity.
Results
Integrative analysis validates ENPP1 as a potential innate immune checkpoint in multiple solid tumor types
ENPP1 supports immune evasion in triple-negative breast cancer (TNBC)46, but its role as an innate immune checkpoint across a broad range of solid tumor types remains to be fully elucidated. To address this, we used PandaOmics, a commercially available target-discovery platform which integrates large-scale patient omics data sets with multiple omics- and text-based algorithms to explore additional oncology indications34. Briefly, we utilized the indication prioritization tools embedded in PandaOmics to rank indications by composite score for potential response to ENPP1 inhibition. This score incorporates biological relationships (e.g., gene networks, expression, pathways, causal directionality) and text-based information (e.g., publication text-mining, research trends, strength of evidence) to nominate diseases in which ENPP1 may play a central, pathogenic role. Several cancer types, such as TNBC, hepatocellular carcinoma, acute myeloid leukemia, ovarian carcinoma, colorectal adenocarcinoma, colorectal cancer, breast cancer, head and neck cancer and ER-negative breast cancer, were identified as top candidates for ENPP1 inhibitor treatment (Fig. 1A). Complementing the PandaOmics prediction, we also analyzed the expression of ENPP1 and its correlation with prognostically beneficial STING-type I interferon signaling across all solid tumor types in the TCGA database. We predicted that cancer indications consistently fulfilling the following criteria would emerge as prime candidates for ENPP1-targeted therapies: 1) high expression of cGAS, which catalyzes the production of cGAMP; 2) presence of ENPP1 in tumor tissue (either on malignant cells or mesenchymal/immune cells within tumors); 3) a negative correlation between ENPP1 expression and STING-type I IFN signaling; and 4) association of STING-type I IFN pathway expression with favorable prognosis. Based on our analysis, eight solid tumor types stand out as priority indications: breast carcinoma, gastric carcinoma, colorectal carcinoma, head and neck carcinoma, esophageal carcinoma, ovarian carcinoma, lung squamous carcinoma, and cervical squamous carcinoma (Supp. Figs. S1A, S1B). Together, the robust insights provided by PandaOmics coupled with the TCGA analysis underscore the potential of ENPP1 as a therapeutic target across a diverse array of solid tumors.

A Indication prioritization tools embedded in PandaOmics to rank indications for ENPP1. B The correlation analysis of type-I IFN related immune parameters across cancer types: Correlation between ENPP1 expression in tumor cells and ISG score in conventional dendritic cells (cDC) in gastric cancer (OMIX001073, n = 10) and colorectal cancer (CRC, GSE132465, n = 23), as well as correlation between ENPP1 expression in tumor cells and CXCL10 expression in dendritic cells (DC) and cDC percentage in triple-negative breast cancer (TNBC, GSE176078, n = 10). Each dot represents a single patient sample. The x-axis indicates the ENPP1 expression in tumor cells and the y-axis displays the corresponding immune parameters: ISG score, CXCL10 expression or cDC percentage. The P value and correlation were performed using Pearson’s correlation coefficient, with the resulting trend line depicted in blue. The gray shaded area surrounding the trend line represents the 95% confidence intervals. Source data are provided as a Source Data file. C Spatial distribution of ENPP1, ISG score and predicted proportion of CD8 T and pro-inflammatory macrophage cells per spot in histology slide SN84_A120838_Rep2 from CRC patient (Zenodo record: 7760264). The color represents the value of ENPP1 expression, ISG score and predicted proportion of DC and NK cells per spot. Source data are provided as a Source Data file. D Spatial distribution of ENPP1, ISG score and predicted proportion of CD4 T and DC cells per spots in slide GSM6433591_094A from a TNBC patient (GEO: GSE210616). The color represents the value of ENPP1 expression, ISG score and predicted proportion of DC and NK cells per spot. Source data are provided as a Source Data file. E Spatial distribution of ENPP1, ISG score and predicted proportion of DC and NK cells per spot in slide 21_00734_LI_SING from a GC patient (GEO: GSE251950). The color represents the value of ENPP1 expression, ISG score and predicted proportion of DC and NK cells per spot. Source data are provided as a Source Data file.
A study recently reported the role of ENPP3 in cGAMP degradation and immune modulation47, raising the questions of relative contribution and sufficiency of targeting ENPP1. We found that ENPP1 is likely the predominant enzyme responsible for cGAMP degradation in various solid tumors, as evidenced by its higher and more widespread expression across cancer cell lines (Supp. Fig. S2A) and various tumor tissues (Supp. Fig. S2B). Single-cell gene expression data showed ENPP1 was widely expressed across diverse cell types within the tumor microenvironment, whereas ENPP3 was mainly restricted to fibroblasts and mast cells (Supp. Fig. S2C). We also found that across various cancer subtypes and treatment regimens, ENPP1 exhibited a significant association with resistance to standard of care (SOC) treatments, but ENPP3 expression was not significantly correlated with the response to either chemotherapy or immune checkpoint therapy (Supp. Fig. S2D). Given its broad expression and clinical relevance, targeting ENPP1 may be more effective in overcoming treatment resistance and improving therapeutic outcomes.
Given the critical role of type-I IFN signaling in tumor antigen presentation mediated by conventional dendritic cells (cDCs), we utilized single-cell sequencing data from tumor tissues of patients with TNBC, gastric cancer (GC), and colorectal cancer (CRC) to investigate the correlation between the tumor cell-intrinsic expression of ENPP1 and cDC-intrinsic STING-type I IFN signaling. A significant negative correlation was found between ENPP1 expression in tumor cells and the expression of CXCL10, a typical chemokine downstream of type I IFN48, or the interferon-stimulated gene (ISG) score49,50,51 in DCs in all three cancer types (Fig. 1B), consistent with the immunosuppressive role of tumor-derived ENPP1 and decreased cGAMP in the TME.
Further, due to intratumoral heterogeneity common across solid tumors52,53,54, we posited that within a given tumor tissue, regions with high ENPP1 expression would exhibit more “immune desert”-like characteristics, such as low immune cell infiltration and activation. To test this hypothesis, we integrated single-cell RNA sequencing (scRNA-seq) data and spatial transcriptome data from tumor samples55. In CRC, TNBC, and GC samples, regions abundant in ENPP1 were found to be mutually exclusive with those with high ISG scores and those enriched with CD8+ T or pro-inflammatory macrophage cells (Fig. 1C–E)56,57,58, further supporting the role of ENPP1 in modulating the immune composition within the TME. Collectively, our integrative analysis reveals ENPP1 as a negative regulator of the STING-type-I IFN pathway and as a therapeutic target in multiple solid tumors.
Discovery of potent, selective, and orally bioavailable ENPP1 inhibitor ISM5939
We implemented structure-based drug design of ENPP1 inhibitors via the multi-model, commercially available Chemistry42 generative AI platform45, which has so far yielded among the most experimentally validated molecules across generative chemistry approaches30. We started with the known binding poses of previously reported ENPP1 inhibitors QS1 and QPS259,60,61 and designated key ligand-interacting amino acids of ENPP1 as mandatory binding points. Diverse candidate structures were selected for validation based on medicinal chemistry filters, self-organizing maps (SOM) rewards, pharmacophore hypothesis rewards, synthetic accessibility, and novelty, leading to the discovery of initial hit compound ISM7516 (Fig. 2A). ISM7516 is a sulfonamide with a 7-phenyl-1H-imidazo[4,5-c]pyridine core scaffold, which inhibited the hydrolysis of cGAMP with an IC50 of 2.55 nM, whereas QS1 in the same assay exhibited an IC50 of 142 nM. Furthermore, ISM7516 stabilized extracellular cGAMP secreted by MDA-MB-231 breast cancer cells transfected with double-stranded DNA (dsDNA), with an EC50 of 0.50 µM (Fig. 2D). To optimize this promising hit compound, we conducted molecular docking of ISM7516 with ENPP1 (Fig. 2B), aiming to explore critical binding interactions and elucidate the structure-activity relationship (SAR). The docking results suggested that the sulfonamide and 7-phenyl-1H-imidazo[4,5-c]pyridine core of ISM7516 form major contacts with ENPP1, while the benzyl group provides additional hydrophobic interactions (Fig. 2B, C), which were iteratively modified to obtain potentially improved structures. Alchemistry62, an efficient physics-based module within Chemstry42 to estimate binding free energy, was employed to prioritize the compounds with lower calculated binding energy (△Gcal) than ISM7516 (△Gcal = −13.29 kcal/mol) (Supp. Fig. S3), ultimately leading to the discovery of ISM3576 (△Gcal = −14.59 kcal/mol) (Fig. 2D).

A Flow scheme for AI-facilitated ENPP1 inhibitor discovery. ReRSA, retrosynthesis related synthetic accessibility; SA, synthetic accessibility; PXR, pregnane xenobiotic receptor. Novelty calculated based on the dataset compiled from ChEMBL. Alchemistry, accurately estimates the relative binding free energy to prioritize molecules with efficient physics-based methods. ADMET prediction, predict physicochemical and ADMET molecular properties. B Docking pose of ISM7516 with human ENPP1. The source data are provided as a Source Data file. ENPP1 (white) is shown in ribbon and ISM7516 (cyan) is drawn in sphere, while key residues (white) and ISM7516(cyan) are shown in stick. C Docking pose of ISM5939 with human ENPP1. The source data are provided as a Source Data file. ISM5939 (yellow) was superimposed with AMP (purple, PDB ID: 6wfj) and QPS2 (green, PDB ID: 6wev). Zn atoms are shown as gray sphere and dashed lines indicate hydrogen bonds or coordination bonds. D Biochemical/cellular activity, △Gcal, CYP3A4 induction, hERG inhibition and predicted hERG inhibition value of candidate inhibitors. The data are colored by profiling results. Green represents ideal profiling, orange indicates potential issues, black represents acceptable results, and red represents unacceptable outcomes. CYP3A4 induction calculated as ratio of CYP3A4 mRNA following compound treatment at 10 µM compared to rifampicin. △Gcal was calculated by Alchemistry tool. hERG, human ether-a-go-go-related gene. Predicted hERG inhibition values were achieved by ADMET prediction module.
ISM3576 showed in vitro biochemical and cellular potency superior to ISM7516, likely attributable to the substitution of the methoxyl group with a methyl group and the addition of two fluorine atoms at the 3,5-positions of the benzyl group in ISM7516. However, ISM3576 was suboptimal in its strong CYP3A4 enzyme induction in human hepatocytes, a phenomenon that can indicate severe drug-drug interactions or self-induction in clinical use predominately mediated through the activation of pregnane xenobiotic receptor (PXR)63. Our strategy to reduce PXR binding involved fine-tuning the geometry of the difluorophenyl group to disrupt hydrogen bonds, as well as modifying the 7-phenyl-1H-imidazo[4,5-c]pyridine core to decrease the hydrophobic interactions engaged in PXR binding, while maintaining affinity for ENPP1, as these components also contribute to ENPP1 binding (Supp Figs. S4 andS5). ISM3576 also demonstrated increased potential for cardiotoxic inhibition of hERG channels (hERG IC50 = 11.68 µM) compared to ISM7516 (hERG IC50 > 30 µM) (Fig. 2D), so the newly designed compounds were further screened for predicted hERG inhibition in the Chemistry42 ADMET prediction module along with △Gcal. This filtering aimed to balance CYP3A4 induction, hERG liability, and ENPP1 inhibition (Fig. 2A). Through systematic SAR exploration and AI-enabled filtering (Fig. 2A, Supplemental Figs. S4 and S5), we eventually identified ISM5939 (△Gcal = −14.06 kcal/mol, predicted hERG inhibition value = 23.44 µM), which exhibited promising drug-like features, such as low CYP3A4 induction, high ENPP1 inhibitory potency, high metabolic stability, minimal hERG liability, and favorable animal pharmacokinetic (PK) profiles (Fig. 2D, Table 1). ISM5939 was thus nominated for further development, highlighting the ability for integrated AI-driven platforms like Chemistry42 to accelerate the entire drug discovery hit-to-lead process.
According to the docking results, ISM5939 has a modified imidazole[4,5-c]pyridine core pack between Phe257 and Tyr340 with a hydrogen bond forming with Lys295 in the adenine pocket (Fig. 2C). The 7-phenyl substitution on the core of ISM5939 projects deep into the pocket surrounded by multiple aromatic amino acids. The additional π-π interactions with Phe257, Phe321 and Tyr371 may contribute to the increased affinity of ISM5939, considering that a methoxy group in QPS2 was extended into this pocket instead (Fig. 2C). We superimposed ISM5939 with AMP (Kd = 103 nM), a tightly bound hydrolysis nucleotide product of ATP and cGAMP that acts as a natural ENPP1 ligand. The fused ring of ISM5939 occupies the same region as the adenine of AMP but extends further into the pocket. Additionally, the 5-fluoropyridine extends out of the adenine pocket and occupies a similar space as the ribose ring of AMP, but the sulfonamide group does not project to the zinc site like the phosphate of AMP but instead forms hydrogen bonds with Glu373 and Asp376. Despite the different interactions of ISM5939 and AMP with the zinc site, the virtual analysis suggests that ISM5939 binds to the ATP/cGAMP active site and competes with natural substrates for ENPP1 binding.
ISM5939 is a potent and ENPP1-selective Inhibitor
To evaluate the inhibitory effect of ISM5939, we used recombinant ENPP1 protein and both 2,3-cGAMP and ATP as substrates. Under physiological pH conditions (pH 7.4), ISM5939 demonstrated a remarkable inhibition of ENPP1 (Fig. 3A), with IC50 values of 0.63 nM against the 2,3-cGAMP degradation and 9.28 nM against ATP hydrolysis (Fig. 3B). This potency far surpasses that of ENPP-IN-164, the positive control in the assay, which exhibited significantly higher IC50 values of 259.0 nM for 2,3-cGAMP and 1328.0 nM for ATP, highlighting the superior inhibitory efficacy of ISM5939. Under tumor microenvironment-mimicking pH conditions (pH 6.5), ISM5939 retained significant inhibitory activity against ENPP1, with IC50 values of 11.41 nM for 2,3-cGAMP degradation and 170.30 nM for ATP degradation (Fig. 3B). In alignment with the docking analysis (Supplemental Fig. S4), the ENPP1 inhibition assay revealed that increasing concentrations of either ATP or cGAMP led to a decrease in the inhibitory potency of ISM5939 (Fig. 3C). This suggests that the inhibitory action of ISM5939 is competitive with the natural substrates of ENPP1. Additionally, enzymatic assays were performed using recombinant ENPP1 proteins from cynomolgus monkeys, dogs, and mice, which yielded comparable inhibition effects of ISM5939 on ENPP1 (Supp. Figs. S6A–C). Furthermore, to assess the selectivity of ISM5939 for ENPP1, we simultaneously evaluated its inhibitory effects on ENPP2 and ENPP3, the most closely related members within the ecto-nucleotide pyrophosphatase/phosphodiesterase (ENPP) family, themselves often inhibited by ENPP1-targeting inhibitors65,66,67. For the ENPP2 assay (Fig. 3D), we included the reported ENPP2 inhibitor HA13068 as a positive control. To verify the specificity of the ENPP3 assay (Fig. 3E), we used STF-108417, a recognized potent ENPP1 inhibitor, as the positive control, and also included a range of unrelated compounds such as HA130 (an ENPP2 inhibitor)68, SN-38 (a topoisomerase I inhibitor)69, and paclitaxel (a microtubule stabilizer)70 as negative controls. The IC50 value for ENPP1 was at the sub-nanomolar level, while the IC50 values for ENPP2 and ENPP3 were over 15,000 and 3400 times greater, respectively (Fig. 3F), suggesting that ISM5939 exhibits high selectivity for ENPP1. In addition, we also evaluated the effect of ISM5939 on the ICESTP™ SafetyOne44 panel, which encompasses a comprehensive set of 44 clinically relevant off-targets, including GPCRs, ion channels, enzymes, nuclear receptors, and key transporters (Supp. Fig. S6C). The results demonstrated that ISM5939 does not exhibit any significant agonistic or antagonistic effects on any of these targets, with the maximum observed induction or inhibition being less than 20% (Fig. 3G, Supplementary Data. 1). This comprehensive evaluation underscores the specificity and safety profile of ISM5939 as a potential therapeutic agent targeting ENPP1.

A Concentration response curves of ISM5939 or positive control (ENPP-1-IN-1) in ENPP1 enzymatic assay (pH 7.4, n = 2 biological replicates, 2 independent experiments). Left, 2,3- cGAMP as substrate; right, ATP as substrate. B IC50 values for ISM5939 against human ENPP1 catalytic activity using 2’,3’ cGAMP or ATP as substrates at pH 7.4 and pH 6.5. C Concentration response curves of ISM5939 in the ENPP1 enzymatic assay at different concentrations of ATP and cGAMP (n = 2 biological replicates). D–F The inhibitory activity of ISM5939 against the catalytic activity of (D) human ENPP2 (E) human ENPP3 and (F) table summary for IC50 values (n = 2 biological replicates). G The Rador map illustrating the effect of ISM5939 (10 μM) on SafetyOne44 Panel targets (n = 2 biological replicates). H Inhibitory effects of ISM5939 on cGAMP degradation mediated by soluble ENPP1 in human plasma detected by ELISA. I Stabilization of cGAMP in vivo in murine plasma by ISM5939 (n = 3 biological replicates). Data were analyzed by two‐tailed, unpaired Student’s t‐test. P-value < 0.05 were shown. J Potency of ISM5939 in maintaining extracellular cGAMP levels when applied to cancer cells in vitro (n = 2 biological replicates). Data were represented as mean ± SEM. Source data are provided as a Source Data file.
Subsequently, we evaluated the inhibitory effects of ISM5939 on cGAMP degradation mediated by both soluble ENPP1 in plasma and membrane-bound ENPP1 on tumor cells. We found that cGAMP was degraded in plasma samples within 3 h (Fig. 3H) or when intravenously injected in vivo into mice within 1 h (Fig. 3I); however, degradation was inhibited with the addition of ISM5939 to the assay system (Fig. 3H, I). We then assessed the potency of ISM5939 in maintaining extracellular cGAMP levels in the presence of ENPP1-expressing cancer cells, including MDA-MB-231 breast cancer cells and H1792 lung adenocarcinoma cells, finding an IC50 of approximately 40 nM (Fig. 3J). Notably, this potency was absent in MV4-11 cells, which naturally lack expression of ENPP1 (Fig. 3H, Supp. Fig. S6E).
ISM5939 stabilizes tumor-secreted cGAMP and orchestrates non-cell autonomous STING activation in antigen-presenting cells
In the tumor microenvironment, tumor cells serve as the predominant source of active cGAMP secretion17, which can be taken up by neighboring APCs to activate the STING pathway and enhance antigen presentation71,72. To determine whether ISM5939 could enhance the accumulation of tumor cell-secreted cGAMP, a panel of human (MDA-MB-231, ZR-75-30, 786-O, H1792) and murine (4T1, MC38, EMT6, B16F10) cancer cell lines that naturally express high levels of ENPP1 were transfected with dsDNA to induce cGAS activation. The cells were treated with a range of concentrations of ISM5939 followed by extracellular cGAMP measurement. Our results confirmed that ISM5939 potently promotes the accumulation of extracellular cGAMP, in a dose-dependent manner (Fig. 4A, Supp. Fig. S7A). In contrast, this effect is much weaker in ENPP1-deficient CT26 tumor cells (Supp. Fig. S7B, C). Consistent with cGAMP accumulation, co-culturing dsDNA-transfected cancer cells with THP1-Dual cells, a monocytic line expressing an IFN reporter, revealed that ISM5939 significantly enhanced dsDNA-induced IFN expression (Fig. 4B, C, Supp. Fig. S7D, E). However, the reporter signal response to ISM5939 was completely inhibited by the addition of the STING inhibitor H-151 (Fig. 4B, C, Supp. Fig. S7D, E). Ablation of IRF3, a key downstream transcriptional factor of STING signaling, reversed the IFN reporter signal gain in response to ISM5939 (Fig. 4D, E, Supp. Fig. S7E). These results together demonstrate that ISM5939 effectively activates non-cell autonomous STING signaling in APCs via stabilizing cancer cell-secreted cGAMP.

A Dose-dependent accumulation of extracellular cGAMP by ISM5939. Cancer cell lines as indicated were treated with a gradient of ISM5939 concentrations followed by detection of supernatant cGAMP levels via ELISA (n = 2 biological replicates, two independent experiments). The color represents cGAMP concentrations. B, C Type I interferon (IFN) pathway activation in THP1-Dual cells co-cultured with (B) MDA-MB-231 (B) or (C) ZR-75-30 tumor cells transfected with dsDNA (n = 2 biological replicates, two independent experiments). D, E Type I IFN pathway activation in IRF3-deficient THP1-dual cells co-cultured with (D) MDA-MB-231 or (E) ZR-75-30 tumor cells transfected with dsDNA (n = 2 biological replicates, two independent experiments). F, G 4T1 syngeneic tumor-bearing mice treated with a single dose of ISM5939 (10 mg/kg, p.o.), and plasma and tumor samples assayed for (F) drug concentrations of ISM5939 and (G) cGAMP (orange) and IFNβ (green) in tumor tissues (n = 3 biological replicates). H Quantification of tumor infiltrating immune cells by flow cytometry in 4T1 syngeneic tumor bearing mice treated with ISM5939 (n = 6 biological replicates). Data were analyzed by two‐tailed Student’s t‐test. P-value < 0.05 were shown. All data were represented as mean ± SEM. Source data are provided as a Source Data file.
To investigate whether this effect indeed occurs in vivo, 4T1 syngeneic tumor-bearing mice were orally administered a single dose of ISM5939 at 10 mg/kg followed by monitoring both the drug exposure and STING signaling in tumor tissue. Along with the rapid absorption of ISM5939 into plasma and distribution within tumor tissues (Fig. 4F), both cGAMP and IFN-β levels increased in tumors and were retained for up to 48 h (Fig. 4G), indicating sustained activation of STING signaling. Additionally, long-term treatment with ISM5939 remodeled the tumor immune microenvironment into a more immunologically active state, characterized by enhanced polarization of macrophages into M1 status and augmented production of granzyme B and IFN-γ from CD8+ T cells (Fig. 4H). These results show that ISM5939 inhibits ENPP1 in vivo, thereby suppressing cGAMP degradation and subsequently augmenting STING signaling and antitumor immune programs in TME-resident immune cells.
To directly compare ISM5939 with other ENPP1 inhibitors that have progressed to phase 1 clinical testing, we synthesized analogs based on available patents73,74,75 and conducted a series of comparative analyses (Supp. Fig. S8A). ISM5939 demonstrated superior potency in enzymatic inhibition (Supplementary Fig. 8B, C), in preventing cancer cell-mediated cGAMP degradation (Supplementary Fig. 8D), and in binding affinity for ENPP1 compared to the synthesized analogs (Supplementary Fig. 8E). ISM5939 also demonstrated potential PK advantage over the other inhibitors with higher systemic exposure and significantly lower clearance in mice compared to analogs of RBS2418 and SR-8541A (Supp. Fig. S8F). Using in vitro-in vivo extrapolation (IVIVE) and allometric scaling analyses (Supplementary Data. 9), we predicted a projected human dose range of 70 to 358 mg/day, which is significantly lower than the RBS2418 dosage of 100 to 800 mg twice daily (ClinicalTrials.gov ID: NCT05270213). Although STF-108417, a potential analog of the ENPP1 inhibitor ANG-3132 (Angarus Therpeutics), which is nearing phase 1 clinical trials, exhibits similar potency to ISM5939, its administration is restricted to intravenous or subcutaneous routes. This limitation may impact patient convenience and quality of life76,77.
ISM5939 exhibits a wider therapeutic index compared with direct systemic STING agonists
The development of direct STING agonists has been hampered substantially by the risk of eliciting systemic cytokine release and low antitumor efficacy in clinical trials78. This has led to a series of studies focused on STING-modulation strategies that aim to retain efficacy while curbing potential adverse effects, such as the encapsulation of direct STING agonists in nanoparticles79. Given ENPP1’s selective modulation of the STING pathway in the tumor microenvironment, we anticipated that ISM5939 exhibits a much broader therapeutic index than traditional STING agonists– that is, exhibiting less toxicity at similar, effective dosages. To this end, we compared the impact of ISM5939 and ADU-S100, a CDN STING agonist, on the function of human peripheral blood mononuclear cells (PBMCs). In contrast to ADU-S100, which induced a significant release of inflammatory cytokines such as IFN-α, IL-6, and TNF-α, no such effect was observed with ISM5939 treatment, even at high concentrations (Fig. 5A). In vivo studies yielded similar results, with systemic administration of the STING agonists diABZI and MSA-2 at dosages known to induce modest to optimal antitumor effects80,81,82,83 leading to a marked increase in inflammatory cytokine levels in mouse plasma. In contrast, no significant cytokine induction was observed in the ISM5939-treated group (Fig. 5B). To further establish the safety profile of our compound, we conducted in vitro evaluations of cytokine induction and 28-day Good Laboratory Practice (GLP) toxicity studies in Sprague Dawley (SD) rats and Beagle dogs. Consistent with the absence of cytokine induction in rat and dog PBMCs (Supp. Fig. S9A), long-term treatment with ISM5939 at varying doses was well-tolerated, with no obvious alterations in the ratios of T cell subtypes (Supp. Fig. S9B, C).

A Drug-induced cytokine release from human PBMCs treated with ISM5939 or ADU-S100 for 24 h, followed by cytokine detection in the supernatant using ELISA. B Cytokine concentrations in plasma obtained from mice administered with STING agonists (diABZI, MSA-2) or ISM5939 for 7 days via ELISA (n = 5 biological replicates). C Viability of T cells isolated from cultured human PBMCs treated with ISM5939 or STING agonists by flow cytometry (n = 2 biological replicates). The color represents cell numbers. D–F Proportions and immunophenotypes of immune cells collected from plasma and tumor samples of MC38 tumor-bearing mice treated with STING agonists (orange) or ISM5939 (green) by flow cytometry 24 h after treatment (n = 6 biological replicates). D Live CD8 + T cells and CD4 + T cells within all live immune cells from blood. E Live total immune cells (left), CD8+ (middle) and CD4+ T cells (right) within all live immune cells in the tumor. F IFN-γ + (left) and TNF-α + (right) CD8 + T cells within all tumor-infiltrating CD8 + T cells. For B and D–F, data were analyzed by two‐tailed Student’s t‐test. P-value < 0.05 were shown. Data were represented as mean ± SEM. Source data are provided as a Source Data file.
In addition, oral ISM5939 did not cause any severe adverse change in clinical signs, body weight, clinical pathology, or microscopic examination in 28-day repeat dose toxicity studies in rats (Supplementary Data 2 and 5) and dogs (Supplementary Data 6 and 8) at doses up to 150 mg/kg/day and 30 mg/kg/day, respectively. The main adverse findings were limited to clinical pathology and histopathological findings of kidneys in rats, which had completely or partially recovered by the conclusion of the recovery period (Supplementary Data 2 and 5). No adverse findings were noted in dogs (Supplementary Data 6 and 8). These data support a strong safety profile of ISM5939, further emphasizing its distinct advantage over STING agonists in mitigating systemic inflammation and enhancing patient safety.
Another challenge for direct STING agonists is that STING activation can induce apoptosis within various cell subtypes, such as lymphocytes, thus diminishing therapeutic efficacy84. Considering that extracellular cGAMP cannot freely diffuse into cells and relies on the limited expression of cGAMP importers85,86, we predicted that modulating cGAMP levels through ISM5939 would be less likely to cause massive lymphocyte cell death than direct STING agonists. To test this hypothesis, we compared the impact of ISM5939 and three STING agonists on T-cell viability. In stark contrast to the STING agonists, which demonstrated a dose-dependent reduction in CD4+ and CD8+ T cell viability, both ISM5939 and cGAMP itself did not exhibit such an effect, even at very high concentrations (Fig. 5C). To further validate these findings in vivo, we administered ISM5939 or a single dose of a STING agonist (MSA-2 or diABZI) at therapeutic levels to mice bearing MC38 tumors. While treatment with MSA-2 and diABZI resulted in decreased T cell proportions in circulation (Fig. 5D) and in harvested tumor tissues (Fig. 5E), no such reduction was observed in ISM5939-treated mice (Fig. 5D, E). In contrast, both the IFN-γ and TNF-α producing T cells significantly increased in the tumor bed upon ISM5939 treatment (Fig. 5F, Supp. Fig. S9D), indicating enhanced antitumor immunity. These data confirmed that ISM5939 exhibits a wider therapeutic index than conventional STING agonists, supporting its development as a safer and potentially more effective option for antitumor therapy.
ISM5939 synergizes with anti-PD(L)1 therapy
We next asked whether ISM5939 could improve the efficacy of mechanistically orthogonal immune-stimulatory therapy, such as anti-PD(L)1 checkpoint inhibition. To this end, we assessed the expression of ENPP1 in tumor tissues and its correlation to the efficacy of anti-PD-1 therapy in breast cancer patients87. Interestingly, we found that non-responders had significantly higher baseline tumoral expression of ENPP1 compared to responders (Fig. 6A). Additionally, in a proportion of esophageal adenocarcinoma patients with high level of LRRC8A88 —a transporter necessary for both the efflux and influx of cGAMP89—non-responders to atezolizumab (anti-PD-L1) had higher expression of ENPP1 (Fig. 6B). These findings suggest ENPP1 may act as an intrinsic driver or biomarker of immune checkpoint blockade resistance, particularly in tumor microenvironments with the potential to utilize cGAMP.

A Baseline ENPP1 expression levels in Paclitaxel+anti-PD1 responsive or non-responsive breast cancer patients (GEO: GSE194040). n = 38 for responders and n = 31 for non-responders. B Baseline ENPP1 expression levels in anti-PD-L1 responsive or non-responsive esophageal adenocarcinoma patients with high LRRC8A expression. Data were extracted from GSE165252. n = 6 for responders and n = 3 for non-responders. For boxplots, boxes indicate 25th and 75th percentiles, the lines within boxes mark medians, whiskers extend over 1.5 times the interquartile range (IQR, the distance from 25th to 75th percentile); dots represent patient samples. C Combinational effect of ISM5939 and anti-PD-L1 therapy. MC38 tumor‐bearing C57BL/6 J mice were treated with anti‐PD‐L1 antibody (3 mg kg−1, twice per week) alone or in combination with ISM5939 (30 mg/kg, twice a day) for indicated days (n = 6 biological replicates per group). D Combinational effect of ISM5939 and anti-PD1 therapy. MC38 tumor‐bearing C57BL/6 J mice were treated with anti‐PD1 antibody (5 mg kg−1, twice per week) alone or in combination with ISM5939 at the indicated doses (n = 8 biological replicates per group). E–H Tumor infiltrating immune cells analyzed by flow cytometry. MC38 tumor‐bearing C57BL/6 J mice were treated with anti‐PD1 antibody (5 mg kg−1, twice per week) alone or in combination with ISM5939 (20 mg kg−1, twice a day) for 7 consecutive days (n = 6 biological replicates). E, F The proportion of the indicated immune cells in tumor infiltrating CD45+ cells; G The ratio of CD8+ T cells (left) and CD4+ T cells (right) to Tregs. H CD69 positive ratios among the indicated immune subsets. I Tumor growth of CT26 syngeneic tumors. Tumor bearing mice (n = 6 biological replicates per group) were treated with anti-PD1 antibody alone or in combination with ISM5939 (20 mg kg−1, twice a day). Data are represented as mean ± SEM. For (A, B) and (E–G), data were analyzed by two‐tailed Student’s t‐test. P value < 0.05 were shown. Data were represented as mean ± SEM. Source data are provided as a Source Data file.
To translate these clinical findings into therapeutic potential, the combination of ISM5939 with a suboptimal dose of either anti-PD-1 (5 mg kg-1) or anti-PD-L1 (3 mg kg-1) was evaluated in the MC38 tumor model, which has been shown to recapitulate the microsatellite instable (MSI) tumor subset, characterized by spontaneous cGAS activation90,91. Compared to each monotherapy group, ISM5939 combined with either anti-PD-L1 (Fig. 6C) or anti-PD-1 antibodies (Fig. 6D) significantly suppressed tumor growth, with negligible body weight change (Supp. Figs. S10A-B). The coefficient drug interaction (CDI) confirmed synergy between ISM5939 with either anti-PD-L1 (CDIday20 = 0.40) or anti-PD-1(For ISM5939 at 5 mg kg-1, CDIday16 = 0.80; For ISM5939 at 20 mg kg−1, CDIday16 = 0.34). Tumor-infiltrating lymphocytes were then quantified to determine how the tumor immune microenvironment was remodeled with treatment. ISM5939 alone and in combination with anti-PD-1 significantly increased the ratio of M1 to M2 macrophages (Fig. 6E). Additionally, the ratios of CD8+ T cells to regulatory CD4+ T cells (Tregs) and effector CD4+ T cells to Tregs were also enhanced upon ISM5939 treatment (Fig. 6F). Of note, both the infiltration of T cells, especially of CD8+ T cells (Fig. 6G, Supp. Fig. S10C), and the expression level of CD69, an activation marker of lymphocytes (Fig. 6H), were highest in mice receiving the combination regimen, consistent with a reduction in immunosuppressive cell types and an increase in cytotoxic cell types in the TME. We also conducted efficacy study in LLC1, a murine lung cancer model that is highly resistant to anti-PD1 therapy42. Despite a relatively lower ranking in the indication prioritization list for lung cancer (Supp. Fig. 1B), this model also demonstrated a significant antitumor effect for the combination of ISM5939 with an anti-PD1 antibody (Supp. Fig. S10D).
To identify mechanisms driving response and patient populations potentially most likely to benefit from ENPP1 inhibition, we studied the efficacy of ISM5939 against CT26 tumors, a validated microsatellite stable (MSS) CRC model with low ENPP1 and cGAS expression (Supp. Fig. S10E). We speculated that low basal cGAS activation or the lack of ENPP1 in the tumor would make this model relatively insensitive to further cGAMP-mediated immune activation and therefore ISM5939 combinational synergism. As expected, while anti-PD-1 alone achieved some tumor inhibition, the addition of ISM5939 failed to show a significant therapeutic advantage (Fig. 6I, Supp. Fig S10F). Altogether, these results demonstrated ISM5939 as a potentially effective and safe combinatorial strategy to sensitize tumors to anti-PD(L)1 therapy, especially in tumors with high expression of the ENPP1-cGAS axis.
ISM5939 synergizes with genome-destabilizing chemotherapeutic drugs
Chemotherapy remains the mainstay of frontline cancer therapy, whose induction of DNA damage and chromosome instability not only kills tumor cells but also triggers immune-stimulating signal cascades, including activation of cGAS due to increased cytosolic dsDNA92. We hypothesized that ISM5939 has the potential to synergize with chemotherapeutics by boosting activation of the cGAS-STING pathway in the TME mediated by tumor-secreted cGAMP. To explore the relationship between ENPP1 levels and chemotherapy responses, we analyzed a diverse collection of clinical cohorts covering multiple solid tumor types87,88,93,94,95,96. These cohorts included patient responses to chemotherapy along with gene expression profiles in tumor tissues (Fig. 7A). In breast cancer cohorts, ENPP1 expression was consistently higher in chemotherapy non-responsive patients, regardless of the presence of estrogen receptor (ER) and human epidermal growth factor receptor 2 (HER2). Similarly, chemotherapy non-responsive CRC patients exhibited significantly higher ENPP1 level compared to responders. Notably, ovarian cancer patients showed significantly increased levels of ENPP1 following chemotherapy compared to their levels prior to treatment (Supp. Fig. S11A), reinforcing the role of ENPP1 as an underlying driver of resistance to chemotherapy. Given these results, we next evaluated the effect of ISM5939 in combination with multiple chemotherapeutic agents on cGAMP accumulation. As expected, in two human (MDA-MB-231 and HCC1395) and one murine (EMT6) breast cancer cell lines, the concurrent use of ISM5939 with either cisplatin or paclitaxel resulted in significant cGAMP accumulation in culture medium (Fig. 7B, Supp. Fig. S11B). Similar results were obtained for the combination of ISM5939 with either oxaliplatin or SN-38, the active metabolite of irinotecan, in murine colorectal cancer cells (Supp. Fig. S11C).

A Baseline ENPP1 expression levels in chemotherapy responsive or non-responsive TNBC, HER2 positive breast cancer, ER positive breast cancer and colorectal cancer patients. Data were extracted from GSE22513, GSE50948, GSE22093 and GSE28702. For TNBC, n = 8 for responders and n = 20 for non-responders; for HER2 positive breast cancer, n = 22 for responders and n = 71 for non-responders; for ER positive breast cancer, n = 10 for responders and n = 32 for non-responders; for CRC, n = 42 for responders and n = 41 for non-responders. For boxplots, boxes indicate 25th and 75th percentiles, the lines within boxes mark medians, whiskers extend over 1.5 times the interquartile range (IQR, the distance from 25th to 75th percentile); dots represent patient samples. B Maximal cGAMP release induced by combined use of cisplatin or paclitaxel with ISM5939 (n = 2 biological replicates, two independent experiments). C–E EMT6 syngeneic tumor bearing mice treated with cisplatin and ISM5939 alone or in combination for 2 weeks to profile (C) differential tumor growth and (D, E) endpoint tumor sample bulk RNA sequencing showing (D) pathway enrichment (The color represents adjust P-value) and (E) deconvoluted immune cell populations in the tumor microenvironment post-treatment (n = 6 biological replicates for tumor growth detection, n = 3 biological replicates for RNA sequencing). Color represents z-scores of cell population, values range from red (low population) to green (high population). F Tumor growth of 4T1 orthotopic tumor-bearing mice treated with docetaxel and ISM5939 alone or in combination. G In vitro release of cGAMP by BRCA-proficient (MDA-MB-231, ID8) or BRCA-deficient (MDA-MB-436, UWB1.289) cells treated with Olaparib or ISM5939 alone or in combination via ELISA (n = 2 biological replicates). H Tumor growth of 4T1 orthotopic tumor-bearing mice treated with Olaparib and ISM5939 alone or in combination (n = 7 biological replicates). Data were represented as mean ± SEM. For (A), (C), (F) and (H), data were analyzed by two‐tailed Student’s t‐test. P value < 0.05 were shown. Source data are provided as a Source Data file.
Building on these promising in vitro results, we then explored the therapeutic benefits of ISM5939 with chemotherapeutic agents in vivo, using the EMT6 TNBC model, which naturally bears high ENPP1 levels (Supp. Fig. S7C). While either ISM5939 or cisplatin alone achieved modest tumor inhibition, their combination significantly synergized (CDI = 0.70), rendering a superior tumor inhibition (Fig. 7C) with good tolerability (Supp. Fig. S11D). Tumors harvested from mice treated with the combination regimen showed the highest levels of cGAMP compared to those from untreated or monotherapy-only groups, as measured by LC-MS (Supp. Fig. S11E). To further investigate how the tumor immune microenvironment was remodeled, tumor tissues from all groups were subjected to bulk RNA sequencing followed by pathway enrichment analysis. The combination of ISM5939 and cisplatin demonstrated a pronounced enhancement of both innate and adaptive immune pathways, consistent with the increased tumoral cGAMP accumulation. Key features included the interferon gamma response, inflammation, and interferon-alpha response (Fig. 7D). Cell type deconvolution pointed to a concomitant increase of CD8+ T cells, M1 macrophages, and dendritic cells, along with a reduction in M2 macrophages, effects uniquely observed in the combination group (Fig. 7E). Extending these findings, we observed similar results with the combined use of ISM5939 and docetaxel, which showed a superior synergistic effect in retarding tumor growth in a 4T1 orthotopic breast cancer model (CDI = 0.86, Fig. 7F). Interestingly, despite the lack of ENPP1 expression in CT26 tumor cells, the combination of ISM5939 with oxaliplatin, the standard-of-care treatment for colorectal cancers, resulted in a moderate reduction in tumor burden (Supp. Fig. S11F), suggesting that inhibition of non-tumor–derived ENPP1 contributes to the efficacy of chemotherapeutics. Taken together, these data suggest that administration of ISM5939 is a promising strategy to enhance the efficacy of chemotherapeutic agents.
Finally, considering the capability of Poly (ADP-ribose) polymerase (PARP) inhibitors to induce cGAS activation97, we explored the potential of combining ISM5939 with olaparib. Using both BRCA1/2-proficient and -deficient breast and ovarian cancer cell lines, we found that the concurrent use of ISM5939 with olaparib resulted in maximal cGAMP accumulation in the extracellular space, regardless of the mutation status of BRCA1/2 (Fig. 7G). In accordance with these findings, ISM5939 and olaparib exhibited striking synergy in a 4T1 orthotopic tumor model in vivo (CDI = 0.93, Fig. 7H).
Discussion
Modulating the STING pathway effectively and safely to enhance antitumor immunity remains a challenge for targeted drug development. In this study, we employed PandaOmics, the commercially available target-discovery platform, and integrated patient-based multi-omic data to highlight ENPP1 as a STING-modulating target among broad hard-to-treat solid tumors. Leveraging the Chemistry42 drug design platform, we also created ISM5939, an AI-aided oral ENPP1 inhibitor, which demonstrates favorable bioavailability and high potency in enhancing non-cell autonomous STING signaling in the TME (Fig. 8). In multiple murine tumor models, ISM5939 synergized well with anti-PD(L)1 antibodies and genome-destabilizing chemotherapy regimens in inhibiting tumor growth with good tolerance. Our findings thus provide ISM5939 as a cutting-edge STING modulator for cancer therapy with the potential of combining with standard-of-care treatments, paving a path for immunotherapy advancements.

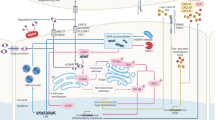
ISM5939 facilitates the accumulation of extracellular cGAMP by blocking ENPP1-dependent degradation, thereby triggering the cGAS-STING signaling pathway in antigen-presenting cells (APCs). This activation effectively enhances the activity of CD8 + T cells. Additionally, ISM5939 diminishes adenosine production by inhibiting ENPP1-mediated ATP hydrolysis, potentially mitigating the suppression of tumor-infiltrating T cells.
Despite the growing interest in ENPP1 as a tumor target, it remains unclear which patients, beyond those with breast cancer, would benefit from ENPP1 inhibition. Leveraging the PandaOmics platform, we have expanded the potential application of ENPP1 targeting strategies to a wide range of hard-to-treat solid tumors, including gastric, colorectal, esophageal, ovarian, and lung carcinomas. While the combination of anti-PD(L)1 therapy or chemotherapy with ISM5939 has demonstrated significant potency in syngeneic tumor models, the optimal combination for a given patient can vary based on cancer type and certain key features. Because neither ISM5939 nor anti-PD(L)1 stimulates the cGAS pathway on their own, we speculate their combination is more suitable for cancers with microsatellite instability-high (MSI-H), chromosomal instability, or ATM mutations98,99. These cancers exhibit cytosolic dsDNA accumulation and endogenous cGAS activation, facilitating abundant cGAMP release. Additionally, our analysis of esophageal adenocarcinoma patients revealed that higher ENPP1 levels are enriched in anti-PD-1 non-responders, but only in those who highly express the cGAMP transporter, LRRC8A. This suggests that the expression level of LRRC8A can serve as a predictive biomarker for selecting patients for this treatment, potentially paving the way for the development of a companion diagnostic tool in this therapeutic context. Chemotherapy, on the other hand, elicits broad cytotoxic effects and offers the advantage of fewer biomarker requirements and less stringent patient selection criteria compared to immunotherapy. Complementing chemotherapy’s mechanism of action, ISM5939 enhances overall efficacy regardless of specific genetic or molecular markers. This flexibility makes the ISM5939-chemotherapy combination a potentially more universally applicable treatment option, increasing its relevance for a wider patient population with diverse tumor profiles.
Our ENPP1 inhibitor offers a wider safety margin compared to direct STING agonists. Systemic administration of non-CDN STING agonists often leads to systemic inflammatory responses, such as cytokine storms, which restrict their clinical application. However, in the context of cancer patients, malignant cells serve as the predominant source of extracellular cGAMP due to dsDNA accumulation in the cytosol caused by endogenous DNA damage and genome instability17,100. The effects of tumor-derived cGAMP have been shown to be spatially restricted to the TME, which presents an opportunity for exploiting this mechanism therapeutically. It has been shown that depletion of CD8+ T cells in tumor-bearing mice ablates the abscopal antitumor activity of intratumoral cGAMP injection but, importantly, not its proximal activity101, suggesting that non-CD8+ T cell-mediated antitumor effects of cGAMP are restricted to the local cGAMP-high microenvironment. Other studies have found intercellular transfer of cGAMP mediated by direct cell-cell connexin channels dependent on cell density102. Together with our safety profiling experiments finding no systemic cytokine induction or changes in circulating T cell phenotypes, this supports our hypothesis that ENPP1 inhibition allows for the localized activation of STING in tumors, thereby avoiding the risk of systemic inflammation.
Moreover, STING agonists often face limited clinical efficacy, partly due to the pleiotropic functions of STING across various immune cell types. While activation of STING in APCs is beneficial for licensing antitumor T cells, intrinsic STING activation in T cells can induce their apoptosis, hindering effective antitumor responses84. In contrast to most synthesized STING agonists, the diffusion of endogenous cGAMP into immune cells of the TME is gatekept by limited expression of cGAMP transporters, thus avoiding unrestricted STING activation within the TME. Particularly, our in vitro head-to-head assessment demonstrated that cGAMP had a negligible impact on the viability of CD8+ and CD4+ T cells, even at high concentrations, in stark contrast to other synthesized STING agonists that adversely affect T cell viability. In vivo treatment in diverse animal models also supports the safety of ISM5939 for endogenous levels of T cells and circulating cytokines. These findings confirm that enhancing extracellular cGAMP levels via ENPP1 inhibition is a feasible strategy to induce non-cell autonomous STING activation in APCs while preserving the viability of antitumor T cells.
Although our work describes an ENPP1 inhibitor that acts broadly and safely across solid tumors, challenges remain to fully characterize its application for use in the clinic. While the number of indications supported by our PandaOmics analysis and experimental testing is encouraging, understanding why some cancer types express ENPP1 highly but have no association between ENPP1 expression and STING-type I IFN response, and even have negative prognostic scoring for STING-IFN pathway expression, such as sarcoma and uterine carcinosarcoma, is important to identify determinants of response to STING agonism and immunotherapy more broadly. Because we identified the cGAMP transporter LRRC8A as a potential biomarker for ISM5939 use in patients non-responsive to anti-PD-L1 monotherapy, it can be reasoned that related mechanisms can render patients resistant to ISM5939. Mutations in LRRC8A have been associated with tumorigenesis and resistance to chemotherapeutics in various cancers, including cervical103, ovarian, alveolar104, mammary105, and gastric cancers106, so elucidating how mutations in or expression of the various components of the cGAS-STING pathway, including LRRC8A and ENPP1 itself, affect response to ENPP1 inhibition is crucial for predicting response to treatment and identifying patients who are most likely to benefit. Our work began to investigate potential drivers of resistance by showing lower efficacy against ENPP1-low CT26 cells in combination with anti-PD-L1 in vivo, but future work will need to carefully profile potential mechanisms of resistance and incorporate the significant heterogeneity of TME cell types to pinpoint the most informative cell types and biomarkers for response.
So far, no ENPP1 inhibitors have been approved for clinical use, but three have progressed to clinical trials, including RBS2418 (Riboscience), TXN10128 (Txinno Bioscience), and SR-8541A (Stingray Therapeutics). We therefore conducted head-to-head comparison with best-guess molecules based on patent literature, as their structures have not been disclosed yet. ISM5939 demonstrates significant advantage over other current ENPP1 inhibitors in binding affinity, potency, and PK profile, making ISM5939 a compelling candidate for further clinical development likely to outperform existing ENPP1 inhibitors. As such, the FDA has approved our dose design (IND: 172399) for a Phase 1 clinical trial of ISM5939.
In conclusion, the development and application of ENPP1 inhibitors, particularly ISM5939, offers a promising direction in cancer immunotherapy. By expanding therapeutic indications, ensuring localized immune activation with minimal systemic toxicity, and providing a robust, orally bioavailable therapeutic option, our ENPP1 inhibitor ISM5939 has the potential to revolutionize cancer treatment, particularly when combined with existing therapies such as anti-PD(L)1 and chemotherapy.
Methods
Ethical statement
All animal experiments followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals. For in vivo efficacy studies in mice, the protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Wuxi AppTec (Code: AUF# 105-7), Pharmaron Beijing Co., Ltd. (Code: ON-SYN-06012023), Biometas (Code: BMM-23001061) and HD Biosciences (Code: AUF# 105-7). For all tumor-bearing mice studied in this article, the maximal allowable tumor size/burden was defined as an individual tumor volume of ≤2000 mm³. Throughout all in vivo studies, these limits were strictly adhered to.
For in vivo PK studies, the protocols and procedures were approved by the IACUC of Shanghai Medicilon Inc. (For Mice PK, Code: 09013-21275/21277/21299/22372/22446/22451/ 22457/22504/24792; For Rat PK, Code: 09013-22545; For Dog PK, Code: 09013-22638).
For 28-day GLP, an animal care and use application for studies was reviewed and approved by Pharmaron TSP IACUC before the initiation of study (For dog GLP, Code: No.: 23-333; For Rat GLP. Code: No.: 23-332).
PandaOmics and TCGA indication expansion analysis
Indication prioritization tools embedded in PandaOmics (Insilico Medicine) was applied to rank indications for ENPP1 inhibition within the realm of oncology therapy. The target, ENPP1, was used as the input gene in the Indication Prioritization tool. The composite score encompassed models for network neighbors, causal inference, pathways, relevance, expression and text-based information (evidence, attention score, and trend), resulting in the top 10 ranking indications regarded as the priority indications.
The expression of ENPP1, the STING-type I IFN score, and patient clinical information were downloaded from TCGA Pan-Cancer (PANCAN) in UCSC Xena. The correlation between the ENPP1 and STING-type I IFN score was calculated using Pearson correlation for each cancer type. Prognostic analyses were conducted using the Cox proportional hazards regression model. The analysis was performed using the ‘survival’ package in R software version 4.1.2. The model was fitted to assess the association between STING-type I IFN score and overall survival (OS)/progression-free survival (PFS). Hazard ratios (HR) with 95% confidence intervals (CI) were calculated to estimate the risk of events. Statistical significance was considered at a p-value < 0.05. The proportional hazards assumption was tested using the Schoenfeld residuals method.
Correlation analysis between ENPP1 expression and inflammatory signatures
Previously published single-cell RNA-seq data from patients with TNBC (GEO: GSE176078)107, GC (Omix: OMIX001073)108, and CRC (GEO: GSE132465)109 were downloaded from their respective repositories. Samples from each dataset were sequenced using the 10x Genomics Chromium platform. Downstream analysis was performed using the Seurat package (version 4.3.0) in R. The data underwent quality control, scaling, and normalization according to protocols described by the original authors. Cell annotations were determined by the original authors.
TNBC: ENPP1 expression in tumor cells was calculated as the mean expression of ENPP1 in the ‘CAFs’, ‘Cancer Epithelial’, ‘PVL’, and ‘Normal Epithelial’ clusters within each sample. CXCL10 expression in DCs was calculated as the mean expression of CXCL10 in the ‘Myeloid_c0_DC_LAMP3’ cluster within each sample. The cell percentage of cDC2 was calculated as the percentage of ‘Myeloid_c11_cDC2_CD1C’ cells among all quality-controlled cells within each sample.
GC: ENPP1 expression in tumor cells was calculated as the mean expression of ENPP1 in the ‘Endocrine cells’, ‘Epithelial cells’, and ‘Fibroblasts’ clusters within each sample. The ISG signature in cDC2 cells was calculated as the mean expression of ISG signature genes50,51 in the ‘cDC2_CD1C’ cluster within each sample.
CRC: ENPP1 expression in tumor cells was calculated as the mean expression of ENPP1 in the ‘Epithelial cells’ and ‘Stromal cells’ clusters within each sample. The ISG signature in cDC cells was calculated as the mean expression of ISG signature genes in the ‘cDC’ cluster within each sample.
Pearson correlation analysis was performed using the ‘cor.test’ function in R.
Spatial Transcriptomics Analysis
Spatial transcriptomics data for colorectal cancer (CRC, https://zenodo.org/records/7760264, slide SN84_A120838_Rep2;)56, gastric cancer (GC, GSE251950)58, and triple-negative breast cancer (TNBC, GSE210616)57 were obtained from publicly available repositories, and single-cell RNA-seq data was obtained for TNBC (GSE176078)107 and GC (GSE183904)110.
The scRNA-seq data sets were processed using Seurat. Quality control steps included filtering out cells with less than 500 nFeature_RNA and more than 3000 nFeature_RNA. Mitochondrial gene expression was used to identify and remove low-quality or dying cells, with a cutoff set at 15% mitochondrial gene content. Gene expression was normalized using a global-scaling normalization method, where the counts were log-transformed and scaled to 10,000 transcripts per cell. To address batch effects arising from different samples, we employed Harmony111 for batch correction. The integrated analysis was performed utilizing the Harmony function in Seurat. Briefly, the principal component analysis (PCA) space was computed using the 3,000 most variable genes, and Harmony was applied to the PCA embeddings to align datasets and reduce batch-associated variances. Following batch correction, the Harmony-corrected embeddings were used for clustering analysis. The Uniform Manifold Approximation and Projection (UMAP) method was utilized for dimensionality reduction and visualization of the single-cell data. Clustering was performed using the Louvain algorithm, implemented in the FindNeighbors and FindClusters functions in Seurat, with a significance resolution parameter tuned to ensure optimal clustering granularity.
For spatial transcriptomics, data preprocessing was conducted using the scanpy package. For each individual sample, we filtered out spots for which the number of UMI counts detected were below 1000 or above 40000. In addition, spots containing a fraction of more than 0.2 mitochondrial genes were not considered in the analysis. We normalized the UMI counts from the remaining spots using global-scaling normalization. Spatial transcriptomics deconvolution was performed using the Cell2Location method or RCTD112. Spatial distribution of Interferon-Stimulated Genes (ISGs), ENPP1, and immune cellular populations were visualized using scanpy.
In vitro ENPP1 inhibition assay
The inhibitory effect of ISM5939 on ENPP1 enzyme activity at two different pH conditions (6.5 and 7.4) was evaluated using the AMP-Glo luminescence-based assay (Promega). Recombinant human ENPP1 protein (R&D) or purified mouse/dog/monkey ENPP1 protein (Biometas, see below) was incubated with a final concentration of 2.5 µM of either ATP (Promega) or 2’3’-cGAMP (InvivoGen) in assay buffer adjusted to the respective pH. ISM5939 was added at varying concentrations diluted in DMSO, and the reaction was initiated by the addition of AMP-Glo Reagent (Promega) followed by Kinase Detection Reagent (Promega), according to the kit instructions. ENPP-1-IN-1 (WO2019046778A1), the reported ENPP1 inhibitor was used as the positive control. Luminescence signal was measured using a microplate reader (BMG LABTECH), and percentage of inhibition was calculated relative to a control group without the test compound. IC50 values were determined using nonlinear regression analysis in GraphPad Prism 8 software.
Expression and purification of mouse, dog or monkey ENPP1 protein
For protein expression, Expi293F cells (Invitrogen, A14257) were cultured at 1.5 × 10^6 cells/ml and passaged 24 h before transfection in a shaking incubator set to 120 rpm, 37 °C, and 8% CO2. On the day of transfection, the cell density was increased to 3×10^6 cells/ml with fresh OPM-293 CD05 medium (OPM, 81075-001). The respective DNA plasmids for mouse (pcDNA3.4-kappa-mENPP1(80-906)-His), dog (pcDNA3.4-kappa-8His-dog ENPP2(144-239)-dog ENPP1(182-582)-ENPP2(627-958)), and monkey (pcDNA3.4-kappa-8His-cyno ENPP2(149-237)-cyno ENPP1(189-589)-ENPP2(678-1034)) were diluted in Opti-MEM medium (Invitrogen, 31985088), filtered through a 0.22μm filter, and used at a final concentration of 1ug/ml. PEIMAX (Polyscience, 24765-1) was diluted in Opti-MEM and mixed with the DNA at a 1:4 DNA/PEI (w:w) ratio, incubated for 20 minutes at room temperature before addition to the cell culture.
Twenty-four hours post-transfection, 1/20th of the culture volume of 10% peptone (Sigma, P0521-1KG) was added to the flask, and glucose (Sigma, G7528-5KG) was supplemented at 4 g/L when the supernatant glucose concentration fell below 2 g/L. Cell growth and viability were monitored with Countstar six days after transfection. The cells were then harvested by centrifugation at 4000 rpm for 40 minutes in shaking flasks.
The purification process included affinity chromatography, size exclusion chromatography, and concentration steps. In affinity chromatography, a pre-packed 5 mL Ni-Excel column (Cytiva, 17371203) was pre-equilibrated with 4 column volumes of PBS at pH 7.4, loaded with the culture supernatant, and washed with PBS at pH 7.4 containing 20 mM imidazole. The protein was eluted with the same buffer containing 500 mM imidazole, and purity was assessed by SDS-PAGE. For size exclusion chromatography, a 120 ml Superdex 200 column (Cytiva, 17-1043-01) was pre-equilibrated with two column volumes of PBS at pH 7.4. The elution fractions from the Ni column were concentrated using a 30 kDa cut-off concentrator at 4000RPM, and 2 mL of the sample was injected into the column. Purity was again evaluated by SDS-PAGE. Finally, selected fractions were combined and concentrated to the desired concentration using centrifugation at 4000 rpm with Amicon Ultra centrifugal filters (Millipore, MWCO 30 kDa, UFC9030).
Novelty calculation
The compound ISM7516 was represented as a Morgan fingerprint and compared against the fingerprints of molecules in ChEMBL dataset. For each molecule in ChEMBL, the Tanimoto Coefficient was calculated to determine the similarity between the fingerprint of ISM7516 and the molecule from the dataset. The Tanimoto Coefficient is defined as:
\(\left|{\mbox{A}}\cap {\mbox{B}}\right|\) is the number of bits that are shared between the two sets being compared; A is the number of bits in fingerprint of ISM7516. B is the number of bits in fingerprint of the molecule from ChEMBL. The novelty score is determined as:
\({{{\rm{Tanimoto}}}(A,B)}_{\max }\) represents the highest level of similarity found between ISM7516 and compound in ChEMBL.
Molecular docking
Molecular docking studies were carried out using the Molecular Operating Environment (MOE) (2022.02; Chemical Computing Group ULC,1010Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A2R7, 2022). The active site of the protein was defined as a sphere with a 5 Å radius around the co-crystalized inhibitor. The template-based docking was employed to position the compounds within the protein’s active site. The optimal pose was selected manually considering both the similarity to the reference ligand binding mode and the scoring provided by the software’s default scoring function.
Medicinal chemistry analysis of ISM5939
CYP3A4 induction
55,000 hepatocytes were plated per well in a collagen I-coated 96-well plate for 24 h. Fresh dosing medium with the test compounds at 10 µM or the appropriate control was added to the cells. The positive control inducer for CYP3A4 was rifampicin (10 µM), flumazenil (30 µM) was used as the negative control compound, and DMSO (0.1%) was used as vehicle control. The dosing medium was renewed every 24 h, and all incubations were performed in triplicate. After 72-h of treatment, cell viability was determined using a Cell Titer-Fluor™ cell viability assay kit. Quantitative PCR was performed with the Cells-to-Ct kit (Thermo Fisher) to detect mRNA expression of CYP3A4. Gene expression was calculated as 2Ct(ACTB)-Ct(induced).
The percentage of positive control was calculated as following:
hERG channel inhibition
CHO-hERG cells were cultured in a humidified incubator at 37 °C and 5% CO2. 24 to 48 h prior to electrophysiological recordings, the cells were plated on glass cover slips placed in culture dishes and maintained under the same incubation and media conditions. The HEKA EPC 10 USB patch clamp amplifier (HEKA Elektronik, Germany) was used in the whole-cell recording. A cover slip with single CHO hERG cells on the surface was removed and placed into a continuously perfused (approximately 1 ml/minute) recording chamber mounted on an inverted microscope. hERG channel currents were recorded from single cells using standard whole-cell recording techniques. The cells were voltage clamped at a holding potential of −80 mV. The hERG current was activated by depolarizing at +20 mV for 5 sec, after which the current was taken back to −50 mV for 5 sec to remove the inactivation and observe the deactivating tail current. The K+ tail current through hERG channels observed during this step was allowed to stabilize under continuous bath perfusion. Cells were then superfused with drug until steady state block was achieved. Steady state was considered reached when three consecutive super-imposable current records were collected. At this point, cells were once again superfused with extracellular solution until the current amplitude returned to values close to those measured before application of drug. One or more compounds or concentrations of drugs were tested on each cell with washout in between each drug application. Cisapride was used in the experiments to ensure the normal response and good quality of the hERG cells. Data were analyzed using Assay Software provided by Patchmaster and Graphpad Prism.
Kinetic solubility
10 μL of 10 mM solution of test and control compounds in DMSO was added into lower chambers of Whatman mini-uniprep vials. 490 μL of 50 mM PB (pH 7.4) was added into lower chambers of the Whatman mini-uniprep vials. Solubility samples were vortexed for at least 2 minutes. Mini-uniprep vials were placed on a shaker for 24 h at room temperature at the speed of 800 rpm. Vials were then centrifuged for 20 minutes at 4000 rpm. Mini-unipreps were compressed to prepare the filtrates for injection into HPLC-UV & UPLC-UV systems to calculate the concentrations.
Caco-2 permeability assay
Caco-2 cells (ATCC) were seeded onto polyethylene membranes (PET) in 96-well Corning Insert plates at 1 ×105 cells/ cm2. Media was refreshed every 4-5 days until a confluent cell monolayer was reached at 21 to 28 days. Compounds were tested at 2.00 μM bi-directionally in duplicate. Digoxin was tested at 10.0 μM bi-directionally in duplicate, and nadolol and metoprolol were tested at 2.00 μM in A to B direction in duplicate. The transport buffer was HBSS with 10.0 mM HEPES at pH 7.40 ± 0.05, and final DMSO concentration was adjusted to less than 1%. Concentrations of test and control compounds in starting, donor, and receiver solutions were quantified by LC-MS/MS using peak area ratio of analyte/internal standard. Following the transport assay, lucifer yellow rejection assay was applied to determine the Caco-2 cell monolayer integrity.
Liver microsome metabolic stability assay
Microsomes (final concentration 0.5 mg/mL), 100 mM phosphate buffer pH 7.4, and compound (final concentration 1 μM) were added to the assay plate and allowed to preincubate for 10 min at 37 °C. The reaction was initiated by the addition of NADPH (final concentration 1 mM), and the plate was constantly shaken at 37 °C. After 0, 5, 15, 30, 45, and 60 minutes, aliquots were taken, and the reaction was quenched using cold acetonitrile. The samples were shaken for 10 min, then centrifuged at 4000 rpm for 20 min at 4 °C and analyzed by LC-MS/MS. The in vitro intrinsic clearance was calculated from the rate of compound disappearance.
Hepatocyte metabolic stability assay
All incubations were carried out in duplicate in a 95% humidified, 5% CO2 incubator at 37 °C, and the final volume of the mixture was 200 μL containing test compound (1 μM) and human/dog/rat/mouse hepatocytes (0.5 × 106 cells/mL) in Williams’ Medium E. After incubations, 25 μL of each sample at each time point (15, 30, 60 and 90 min) were transferred to wells containing 125 μL of ice-cold stop solution (ACN containing 200 ng/mL tolbutamide and 200 ng/mL labetalol as internal standards). For T0, 125 μL/well of ACN stop solution were added prior to the addition of cell suspensions. Medium Control (MC) sample plates (labeled as T0-MC and T90-MC) are prepared at T0 and T90 by adding the same components to each well except cell suspensions. The plates were then sealed and shaken for 10 minutes prior to centrifugation at 4000 rpm and 4 °C for 20 minutes. The resulting supernatant was diluted 1:3 with pure water and sealed and shaken for 10 minutes prior to LC-MS/MS analysis.
Plasma protein binding assay
The extent of protein binding was determined by equilibrium dialysis with a 96-well device. The test compound was spiked into CD-1 mouse, SD rat, Beagle dog and human plasma and the final concentration was 2 μM. 100 μL of plasma samples were dialyzed against an equal volume of dialysis buffer (Phosphate Buffered Saline, 100 mM, pH 7.4 ± 0.1) for 4 h with 5% CO2 at 37 °C. Triplicate incubations were performed. An aliquot of plasma sample was harvested before the incubation and used as T0 samples for recovery calculation. After incubation, 50 μL of samples were transferred from the plasma side as well as the buffer side into new 96-well plates. Each sample was added with equal volume of opposite blank matrix (buffer or plasma) to reach a final volume of 100 μL with 1:1 ratio of plasma: Dialysis Buffer volume in each well. 300 μL stop solution (Acetonitrile/Methanol (50:50, v:v)) containing internal standards (tolbutamide at 200 ng/mL) was then added. The resulting supernatant was diluted 1:1 with pure water and sealed and shaken for 10 minutes prior to LC-MS/MS (API 4000, AB Sciex, MA, USA) analysis.
CYP Inhibition
Pooled human liver microsomes supplied by Corning (Cat No. 452117) were used. CYP isoform and substrate-specific probe reactions were employed as previously reported: Phenacetin O-deethylation for CYP1A2, diclofenac 4-hydroxylation for CYP2C9, S-mephenytoin 4-hydroxylation for CYP2C19, dextromethorphan O-demethylation for CYP2D6, midazolam 1′-hydroxylation for CYP3A4. The incubation systems consisted of 100 mM potassium phosphate buffer (pH 7.4), 33 mM MgCl2, 10 mM NADPH and HLM (0.253 mg/mL). Substrates, compounds/positive controls, HLM, and NADPH solutions were added to corresponding wells. The plate was mixed and incubated for 10 minutes for all CYPs at 37 °C. The reaction was terminated by adding 400 uL of cold stop solution (200 ng/ml Tolbutamide and Labetalol in CAN). Samples were centrifuged at 4000 rpm for 20 minutes to precipitate protein. 200 uL supernatant was transferred to 100 uL of HPLC water and shaken for 10 minutes. Samples were analyzed by LC/MS/MS. XL fit was used to plot the percent of vehicle control versus the test compound concentrations, and for non-linear regression analysis of the data.
In vivo mouse and rat PK studies
Male CD-1 mice (at the age of 6 to 8 weeks, Shanghai Medicilon Inc., IACUC No.: 09013-21275/21277/21299/22372/22446/22451/22457/22504/24792) (n = 3) and male SD rats (at the age of 6 to 8 weeks, Shanghai Medicilon Inc., IACUC No.: 09013-22545) (n = 3) received either a single intravenous (bolus) injection or single oral administration (by gavage) of the compound in a cocktail preparation. Doses of 1 mg/kg (per compound, intravenous administration) and 5 mg/kg (per compound, perorally) were given as solutions in different formulations as needed, e.g. 5% DMSO + 95% (10% HP-β-CD in saline). Consecutive blood samples were taken via submandibular vein or other suitable vein from n = 3 animals per route of administration after 0.083 (iv only), 0.25, 0.5, 1, 2, 4, 6 (po only), 8 and 24 h and were further processed to obtain plasma.
In vivo dog PK studies
Male Beagle dogs (at the age of 1 to 3 years, Shanghai Medicilon Inc., IACUC No.: 09013-22638) received either a single intravenous (bolus) injection or single oral administration (by gavage) of the compound in a cocktail preparation. Doses of 0.5 mg/kg (intravenous administration) and 1 mg/kg (perorally) were given as solutions in different formulations as needed, e.g. 5% DMSO + 95% (10% HP-β-CD in saline). Consecutive blood samples were taken via jugular vein or other suitable vein from n = 3 animals per route of administration after 0.083 (iv only), 0.25, 0.5, 1, 2, 4, 6 (po only), 8, 24 and 48 h and were further processed to obtain plasma.
In vitro ENPP2 and ENPP3 inhibition assay
The Amplex® Red-based fluorescence assay was used to evaluate the inhibitory effects of test compounds on human ENPP2 (or Autotaxin, ATX) and ENPP3. Compounds were dissolved in DMSO and prepared into 10 mM stock solutions. Working solutions with 10 concentration gradients were prepared by diluting the stock solutions with DMSO. The assay buffer for ATX contained choline oxidase, horseradish peroxidase (HRP), and Amplex® Red reagent (Invitrogen), while the buffer for ENPP3 contained Tris-HCl, MgCl2, and ZnCl2. For the ENPP2 assay, compounds were transferred to a 384-well plate using an Echo liquid handler, followed by the addition of ENPP2 protein (Echelon Biosciences, Cat No. E-4000), HRP, choline oxidase, LPC, and Amplex® Red reagent, according to the manufacturer’s instructions. HA130, the reported ENPP2 inhibitor68 was used as the positive control. The plate was incubated at room temperature for 30 minutes and fluorescence was measured at 530 nm excitation and 590 nm emission using an Envision plate reader (PerkinElmer). For the ENPP3 assay, compounds were transferred to a 384-well plate using an Echo liquid handler, followed by the addition of recombinant human ENPP3 protein (ACRO, Cat No. EN3-H52H4), TMP-pNP, and the assay buffer, according to the manufacturer’s instructions. The reported ENPP3 inhibitor was used as a positive control, while the ENPP2 inhibitor HA130, the topoisomerase I inhibitor SN-38, and the microtubule stabilizer paclitaxel were selected as the negative control. The plate was incubated at room temperature and fluorescence was measured at 405 nm excitation and 492 nm emission using an Envision plate reader (PerkinElmer). Data analysis was performed using GraphPad Prism software to determine IC50 values and inhibition percentages.
In vitro safety pharmacology profiling
The SafetyOne44 Panel (https://en.ice-biosci.com/index/show.html?catname=safety4490&id=173) which assessed the in vitro safety pharmacology of ISM5939 was performed by ICE Bioscience. For ion channel modulation, both manual and automated patch clamp techniques were utilized. The manual patch clamp method involved the formation of whole-cell configurations, followed by the application of defined voltage protocols to measure the resulting ionic currents for Cav1.2, IKs, and NR1/NR2B channels. The automated patch clamp system, QPatch 48 X, was employed for the assessment of Nav1.5 and hERG channels, providing high-throughput data with consistent quality control. In the realm of G-protein coupled receptors (GPCRs), the FLIPR assay was used to investigate the effects of test compounds on GABAA (α1β2γ2), 5-HT3A, and nAChRα4β2 receptors. This assay utilized fluorescent dyes to detect changes in membrane potential and calcium flux, offering a rapid and sensitive method for screening potential modulators. For enzymatic targets, a series of assays were conducted to measure the inhibitory effects of test compounds on COX1, COX2, PDE3A, PDE4D2, LCK, AchE, and MAO A. These assays utilized a combination of fluorescence and luminescence-based detection methods, providing quantitative data on the potency of the test compounds against these enzymes. For the study of transporter targets, a reuptake assay for DAT, NET, and 5-HTT was performed. This assay measured the ability of test compounds to inhibit the reuptake of neurotransmitters into cells, using a fluorescent detection method that provided a quantitative assessment of the compounds’ potency.
Surface plasmon resonance (SPR) for ENPP1
SPR assay was performed by Pharmaron Inc. The BIAcoreTM S200 instrument was used for data collection in surface plasmon resonance experiments, and the data collection temperature was set to 25°. In this study, the ENPP1 (MCE) protein was immobilized on the surface of a CM5 sensor chip (Cytiva) via amine-coupling method, and the operation was performed in strict accordance with the standard S-series CM5 sensor chip instructions. The ENPP1 protein was diluted to a concentration of 20 ug/mL using running buffer HBS-P (Cytiva), and the injection time was set to 1200 seconds, and the final chip surface density reached ~15000 response units. A certain number of diluted compounds were set to the surface of the CM5 sensor chip at a flow rate of 30 uL/min, the association time was 120 s, and the dissociation time was 3000 s in a single-cycle mode. The running buffer is HBS-P (Cytiva), 2% DMSO. The data were solvent corrected for different proportions of diluted DMSO concentrations. All data are double subtracted to exclude the effects of buffers and reference channels. The data were processed and analyzed using BIAcoreTM S200 evaluation software version 1.1. The binding constant Ka, the dissociation constant Kd and the affinity constant KD were confirmed by 1:1 binding model fitting. The protein immobilization amounts on the surface of the CM5 sensor chip is calculated according to the following formula:
Affinity constant KD was confirmed according to the following formula:
Cell lines
Cancer cell lines, including MDA-MB-231 (ATCC Cat# HTB-26), MDA-MB-436 (ATCC Cat# HTB-130), ZR-75-30 (ATCC Cat# CRL-1504), MV411 (ATCC Cat# CRL-9591), NCI-H1792 (ATCC Cat# CRL-5895), 786-O (ATCC Cat# CRL-1932), 4T1 (ATCC Cat# CRL-2539), EMT6 (ATCC Cat# CRL-2755), B16F10 (ATCC Cat# CRL-6475), CT26 (ATCC Cat# CRL-2638), HCC1395 (ATCC Cat# CRL-5868), and UWB1.289 (ATCC Cat# CRL-2945) were purchased from ATCC. Additionally, the MC38 cell line (Cobioer Cat# CBP60825) was acquired from Cobioer. the ID8 cell line (SUNNCELL Cat# SNL-498) was obtained from SUNNCELL. the THP1-DUAL cell line (InvivoGen Cat# thpd-nfis) was obtained from InvivoGen.
Cell culture
The THP1-DUAL, ZR-75-30, NCI-H1792, 786-O, 4T1, EMT6, CT26, HCC1395, and UWB1.289 cell lines were cultured in RPMI1640 medium (Invitrogen), supplemented with 10% FBS (Gibco) and 1% P/S, except UWB1.289, which also included 50% MEGM and 3% FBS. The MDA-MB-231 and MDA-MB-436 cell lines were cultured in L-15 medium (Invitrogen), supplemented with 10% FBS, 1% P/S, and additional factors for MDA-MB-436: 10ug/ml insulin and 16ug/ml glutathione. The MC38, MV411, and B16F10 cell lines were cultured in DMEM (Invitrogen), supplemented with 10% FBS and 1% P/S. The ID8 cell line was also cultured in DMEM with the same supplements. All cell lines were maintained in a humidified incubator at 37 °C with 5% CO2, except MDA-MB-231 and MDA-MB-436, which were cultured without CO2. All cell lines were authenticated by STR DNA profiling and tested for Mycoplasma contamination. All cell-based experiments were conducted within 15 passages.
Coculture assay
THP1-Dual™ cell is a human monocyte cell line engineered to express the Lucia luciferase reporter gene under the control of an ISG54 promoter and IFN-stimulated response elements. Lucia luciferase activity serves as a measure of IRF pathway activation. MDA-MB-231, ZR-75-30, or MC38 cells were pretreated with ISM5939, H-151, or their combination at the indicated concentrations for 24 h, followed by transfection with dsDNA using Lipofectamine 3000 (Thermo Fisher). Briefly, for dsDNA transfection, dsDNA at the final concentration of 800 or 2000 ng/ml was incubated with P3000 reagent for 5 min at 37 °C, then Lipofectamine 3000 reagent was added, incubated at 37 °C for 5 min, and then 10 µL of transfection mix was added to each 96-well plate well containing cells and incubated for 24 h, according to the manufacturer’s protocol. ISM5939 was dissolved in DMSO and diluted to 10,000 nM, 1,000 nM, and 100 nM in cell culture medium, while H-151 STING inhibitor (InvivoGen) was diluted to 10 μM. After transfection, dsDNA-containing medium was removed, and THP1-Dual™ cells were seeded on top of tumor cells at 1:1 (ZR-75-31, MC38 and 4T1) or 3:1 (MDA-MD-231) tumor:immune cells ratios. Co-cultures were incubated with indicated compounds for an additional 24 or 48 h. Supernatants were collected at 24 and 48 h post-tumor cell seeding, and Lucia luciferase activity was measured using the QUANTI-Luc™ Lucia kit and a luminometer.
Detection of cancer cell secreted cGAMP
For detection of dsDNA-induced cGAMP release, the compounds were diluted as indicated and incubated with MV4-11, MDA-MB-231, H1792, ZR-75-30, 786-O, 4T1, MC38, EMT6, or B16F10 cells, followed by transfection with dsDNA, as described above. At time points indicated, the supernatant was analyzed for cGAMP levels using an ELISA kit (Cayman Chemical), while cell viability was assessed using the CellTiter-Glo® 2.0 assay (Promega). The data were analyzed using standard curve fitting to determine cGAMP concentrations (XLFIT software) and GraphPad Prism software to generate column charts representing cell viability.
For detection of compound-induced cGAMP release, MDA-MB-231, MDA-MB-436, ID8, UWB1.289, HCC1395, or EMT6 cancer cell lines were seeded at a confluence of 30% and treated with ISM5939 (1 µM) alone or in combination with either Cisplatin (5 µM, 1 µM, or 0 µM; MedChemExpress), Paclitaxel (10 nM, 5 nM, 1 nM, or 0 nM; MedChemExpress), Olaparib (5 µM, 2 µM, 0.5 µM, 0.2 µM, or 0 µM; MedChemExpress), or SN-38 (0.1 µM; MedChemExpress, HY-13704) for 48-72 h. Cell confluence was monitored using an Incucyte live-cell imaging system. At the end of the treatment period, cell supernatants were collected and analyzed for cGAMP levels using a 2’3’-cGAMP ELISA kit (Cayman). Data analysis involved calculating cGAMP concentrations from a standard curve and generating bar charts to visualize the impact of treatments on cGAMP secretion and cell confluence.
Plasma mediated cGAMP degradation
To assess the effect of test compounds on cGAMP degradation in human and mouse plasma, plasma was isolated from 8-10 week-old C57BL/6 J female mice (Shainghai Lingchang, Ltd) and human heparinized whole blood (Shanghai Liquan hospital, approved by hospital ethics committee, Code: [2023]−0) by centrifugation at 4 °C at 2000 × g for 15 minutes. Aliquots of 50 μL of plasma were mixed with 0.5 μM or 3 μM cGAMP (MedChemExpress) and 10 μM or 1 μM ISM033-154 or vehicle control in 1.5 mL EP tubes and incubated for 0, 3, or 6 h. The samples were then stored at −80 °C until 2’3’-cGAMP levels were quantified using the Cayman 2’3’-cGAMP ELISA Kit (Cat.No. 501700). The absorbance was read at the specified wavelength to determine the remaining cGAMP concentrations.
RT-qPCR
Total RNA was extracted from cell lysates using the MagMAX™ mir Vana™ Total RNA Isolation Kit (Applied Biosystems). Reverse transcription was conducted with the High-Capacity RNA-to-cDNA Kit (Invitrogen), followed by real-time PCR with the PowerUp™ SYBR™ Green Master Mix (Invitrogen), according to manufacturers’ protocols. Relative gene expression was calculated using the 2-ΔΔCt method, with ACTB as the internal reference gene. The primers used for amplification were as follows: human ENPP1 forward: CAAAGGTCGCTGTTTCGAGAG,
human ENPP1 reverse: TGCACGTCTCCTGGTAATCTAAA;
human cGAS forward: ACATGGCGGCTATCCTTCTCT,
human cGAS reverse: GGGTTCTGGGTACATACGTGAAA;
human ACTB forward: CATGTACGTTGCTATCCAGGC,
human ACTB reverse: CTCCTTAATGTCACGCACGAT;
mouse ENPP1 forward: CTGGTTTTGTCAGTATGTGTGCT,
mouse ENPP1 reverse: CTCACCGCACCTGAATTTGTT;
mouse cGAS forward: GAGGCGCGGAAAGTCGTAA,
mouse cGAS reverse: TTGTCCGGTTCCTTCCTGGA;
mouse ACTB forward: GGCTGTATTCCCCTCCATCG,
mouse ACTB reverse: CCAGTTGGTAACAATGCCATGT;
In vivo pharmacokinetics and efficacy
Female Balb/cAnN mice at the age of 6 to 8 weeks (n = 3 per timepoint) were implanted with 5 × 105 4T1 cells into the mammary fat pad and allowed to grow until tumor volume reached 60-80 mm3 before single oral dosing with ISM5939 at 10 mg/kg or vehicle control. Blood and tumors were collected from mice at 0 hr, 0.25 hr, 0.5 hr, 1 hr, 2 hr, 4 hr, 8 hr, 24 hr, 48 hr post-dosing, and ISM5939 was detected with liquid chromatography with tandem mass spectrometry (LC-MS/MS), cGAMP was detected with LS-MS/MS, and IFN-β was detected by ELISA (PBL Assay Science, Cat No. 41410-1) according to manufacture instructions.
HD Biosciences Co. (Shanghai, China) analyzed anti-tumor efficacy of ISM5939 as a monotherapy or in combination against MC38 tumors. Briefly, C57BL/6 J mice at the age of 6 to 8 weeks (n = 6 per condition; Beijing Vital River Laboratory Animal Technology Co.) were inoculated subcutaneously with 2×105 MC38 cells in the right flank and treated with orally administered ISM5939 (30 mg/kg, twice a day) and/or anti-PD-L1 antibody (5 mg kg−1, twice a week]] (Bio X Cell, Cat No. BE0101) injected intravenously when tumors reached 51 mm3. Data analysis included reporting mean and SEM, with statistical differences calculated with one-way ANOVA or Student’s t-test. Synergy was evaluated with the coefficient of drug interaction (CDI), calculated by the formula CDI = AB/(A×B), AB is the ratio of the combination group to the control group in mean TV; A or B is the ratio of the corresponding single drug group to the control group in mean TV. CDI < 1 indicates synergism, CDI < 0.7 indicates a significant synergistic effect, CDI = 1 indicates additive and CDI > 1 indicates antagonism.
The efficacy of ISM5939 as a single or combination therapy against 4T1 orthotopic tumors was carried out in 8-10 week-old female Balb/cAnN mice (n = 6 per condition) implanted with 5 × 105 4T1 cells into the mammary fat pad and allowed to grow until tumor volume reached 60-80 mm3 before single oral dosing with ISM5939 (10 mg/kg, BID), docetaxel (10 mg/kg, i.v., QW), olarparib (50 mg/kg, oral, QD), and/or cisplatin (5 mg/kg, i.p., QW), or control non-treatment.
The efficacy study in the CT26 syngeneic model was conducted by Pharmaron (Beijing). Briefly, 6 to 8-week female BALB/cAnN mice (Vital River Laboratory Animal Technology Co., Ltd.) were subcutaneously inoculated with 3 × 105 CT26 cells and treated with orally administered ISM5939 and/or anti-PD1 (Bio X Cell, BE0146) when tumors reached 70 mm3 (n = 8 per condition). The measurement of tumor size was conducted three times per week with calipers and the tumor volume (mm3) was estimated using the formula: TV = a × b 2/2 throughout the study, where a and b are the long and short diameters of a tumor, respectively.
Detection of immune cells and cytokines in vitro and in vivo
2.5 × 105 Human PBMCs (Hemarea, CNHBC100C) were incubated in 96-well plates and treated with ISM5939 or STING agonist ADU-S100 (InvivoGen) at the indicated concentrations for 6 h. Supernatants were collected for analysis with Cytometric Bead Array (CBA; BD Biosciences) for TNF-α (Cat No. 558273), IFN-α (Cat No. 560379), and IL-6 (Cat No. 558276).
For rat or dog PBMCs, 0.1 × 105 PBMCs ([SOURCE]) were treated with ISM5939, ADU-S100 or diABZI at the indicated concentrations for 6 h. Rat TNF-α was detected by CBA (BioLegend, Cat No.741391), while dog TNF-α was detected by ELISA (AssayGenie, CNFI00020).
The proportions of circulating TAM and T cell subtypes were quantified in 4T1 orthotopic Balb/cAnN mice receiving ISM5939 or vehicle control (see above), C57BL/6 J mice bearing MC38 subcutaneous tumors receiving oral ISM5939 (10 mg/kg or 30 mg/kg BID) or STING agonists diABZI (1.5 mg/kg, i.v., single dose) and MSA-2 (60 mg/kg, oral, single dose), and in rat and dog models without tumors. For cell typing and quantification, tumors and blood were collected and single-cell suspensions were analyzed by flow cytometry for immune subtypes using markers for live/dead (Zombie NIR dye), CD45, CD3, CD4, CD8, CD25, Foxp3, CD44, CD62L, CD69, NK1.1, CD19, IFN-γ, TNF-α. For cytokine detection, plasma from the MC38 tumor-bearing C57BL/6 J mice treated with ISM5939, diABZI, or MSA-2 at the indicated concentrations was collected and analyzed for IFN-β by ELISA (R&D, VAL612), or TNF-α and IL-6 using CBA (BD Biosciences, Cat No. 558301 and 558299) according to manufacturer instructions.
28-day repeat dose GLP-compliant toxicity studies with 28-day recovery period were conducted in Sprague–Dawley (SD) rats (both the male and female; 7 weeks; n = 10 per condiion) and Beagle dogs (both the male and female, 7.2-8 months, n = 5 per condition) by Pharmaron (Beijing) TSP Services Limited. The SD rats and Beagle dogs were obtained from Vital River Laboratory Animal Technology, Co. Ltd and Beijing Marshall Biotechnology, Co. Ltd., respectively. The animals were dosed by oral gavage once daily for 28 consecutive days. The SD rats were administrated at 15, 50 or 150 mg/kg/day in a 20 ml/kg dose volume and the beagle dogs were at 3, 10 or 30 mg/kg/day in a 7.5 ml/kg dose volume. Approximately 1.0 mL of the whole blood was collected via puncture of peripheral vessels from dogs and via puncture of abdominal aorta from rats at the end of dosing period and recovery period.
Blood samples from SD rats were used to assess the absolute numbers and percentages of Total T cells (CD45 + CD3 + ), Helper T cells (CD45 + CD3 + CD4 + CD8a-) and Cytotoxic T cells (CD45 + CD3 + CD8a + CD4-) using flow cytometry (FCM).
The percentages and absolute numbers of lymphocyte subpopulations of Total T cells (CD3e + ), Helper T cells (CD3e + CD4 + CD8a-) and Cytotoxic T cells (CD3e + CD4-CD8a + ) in the peripheral blood from Beagle dogs were assessed by flow cytometry (FCM).
RNA-seq for post-treatment TME analysis
Tumors from subcutaneous syngeneic EMT6 tumor-bearing mice (n = 6 per condition) were treated with ISM5939 and/or cisplatin, as described above. Tumors were collected when tumors reached 36-79 mm3 and processed for bulk RNA-seq. Total RNA was isolated from cell pellets using the Magnetic Tissue/Cell/Blood Total RNA Kit (TIANGEN, DP761) on the KingFisher Flex. RNA samples were quantitated with the Quant-IT RNA kit (Invitrogen, R11490) and evaluated for integrity by using Agilent 5400 Bioanalyzer. RNA-seq libraries were constructed using KAPA mRNA HyperPrep Kit (Roche, KR1352) in the Insilico Robotics Lab (Suzhou, China) on the Biomek i7 workstation with custom scripts. The libraries were sequenced on the Illumina NovaSeq6000 platform with 150 nt paired-end reads. Read quality control was conducted with FastQC, and raw reads were filtered and mapped to the mouse mm10 reference genome using the DRAGEN RNA Pipeline (Illumina). The quantitation of gene expression was based on raw read counts using transcripts per million normalization.
Differential expression analysis was performed using DESeq2. A ranked gene list was generated based on log2-fold change. Pathway enrichment was done with R package “clusterProfiler” according to the pre-ranked gene lists. The enrichment results were evaluated based on Normalized Enrichment Score (NES) and False Discovery Rate (FDR). The FDR < 0.05 is regarded as significant. Immune cell proportions were estimated by CIBERSORT113 according to the gene TPM matrix of the bulk RNA-seq samples. All analyses were performed using R (v. 4.1.2).
Analysis of tumor ENPP1 and ENPP3 gene expression for association with response to treatments
Previously published pre-treatment gene expression data from breast cancer tissue from patients treated with combination paclitaxel+anti-PD1 therapies were accessed from GEO: GSE19404087; from esophageal adenocarcinoma patients treated with anti-PDL1 therapy, subset to patients with high LRRC8A expression (top 25%), was accessed from GEO: GSE16525288; from TNBC patients treated with paclitaxel + radiation were accessed from GEO: GSE2251393; from HER2 + BC patients treated with doxorubicin/paclitaxel followed by cyclophosphamide/methotrexate/fluorouracil plus trastuzumab were accessed from GEO: GSE5094894; from ER + BC patients treated with various chemotherapy regimens were accessed from GEO: GSE2209395; and from CRC patients treated with FOLFOX therapy were accessed from GEO: GSE2870296 and were stratified by response. Statistical significance was calculated by Student’s t-test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data of EMT6 tumor tissue generated in this study have been deposited in the GEO database under the accession code GSE277788.The publicly available single-cell or bulk RNA-seq and spatial transcriptomics used in this study are available in public database. The spatial Transcriptomics of human colorectal cancer are in the Zenodo repository under accession code 7760264, the single-cell RNA-seq of human colorectal cancer are available in the GEO database under accession code GSE132465,the single-cell RNA-seq of human breast cancers are available in the GEO database under accession code GSE176078, the single cell RNA-seq of gastric cancer form single-cell atlas are available in the GEO database under accession code GSE183904, the Spatial transcriptomics of triple negative breast cancer are available in the GEO database under accession code GSE210616, the Spatial transcriptomics of gastric cancers are available in the GEO database under accession code GSE251950, the expression profiling by array of breast cancer are available in the GEO database under accession code GSE194040, the RNA-seq of esophageal adenocarcinoma patients are available in the GEO database under accession code GSE165252, the expression profiling by array of breast cancer are available in the GEO database in I-SPY2-990 under accession code GSE22513, the expression profiling by array of HER2-positive breast cancer are available in the GEO database under accession code GSE50948, the expression profiling by array from breast cancer FNA biopsies from patients are available in the GEO database under accession code GSE22093, the expression profiling by array of colorectal cancer under are available in the GEO database accession code GSE28702, and the single-cell RNA of human gastric tumor are available in the OMIX database under accession code OMIX001073.The structures used as template for molecular docking and comparison in this study have been deposited in the PDB database under accession code: 6wfj; 6wev; 6hty.The molecules for novelty comparison and calculation in this study are available in the public ChEMBL dataset: https://www.ebi.ac.uk/chembl/. The experimental data generated in this study are provided in the Supplementary Information/Source Data file. The raw numbers for charts and graphs are provided in the Source Data file, where applicable. Source data are provided with this paper.
Code availability
PandaOmics and Chemistry42 are industry-grade commercial software platforms used by the industry since 2020. Both platforms are available at https://pandaomics.com and http://chemistry42.com, respectively. Demonstrations and trial access to the platforms are available from the corresponding author upon request. The other codes and data are deposited on GitHub at https://github.com/HUICUI1992/Other-codes-and-data-for-ENPP1 and available under Creative Commons Attribution 4.0 International License on Zenodo at https://zenodo.org/records/15194102114.
References
Zhu, Y. et al. STING: a master regulator in the cancer-immunity cycle. Mol. Cancer 18, 152 (2019).
Caiazza, C. et al. The Lack of STING Impairs the MHC-I Dependent Antigen Presentation and JAK/STAT Signaling in Murine Macrophages. Int. J. Mol. Sci. 23, 14232 (2022).
McKee, A. S. et al. Host DNA released in response to aluminum adjuvant enhances MHC class II-mediated antigen presentation and prolongs CD4 T-cell interactions with dendritic cells. Proc. Natl. Acad. Sci. 110, E1122–E1131 (2013).
Li, G. et al. cGAS-STING pathway mediates activation of dendritic cell sensing of immunogenic tumors. Cell. Mol. Life Sci. 81, 149 (2024).
Cheng, H. et al. Activation of STING by cGAMP Regulates MDSCs to Suppress Tumor Metastasis via Reversing Epithelial-Mesenchymal Transition. Front. Oncol. 10, 896 (2020).
Meric-Bernstam, F. et al. Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas. Clin. Cancer Res. 28, 677–688 (2022).
Meric-Bernstam, F. et al. Combination of the STING Agonist MIW815 (ADU-S100) and PD-1 Inhibitor Spartalizumab in Advanced/Metastatic Solid Tumors or Lymphomas: An Open-Label, Multicenter, Phase Ib Study. Clin. Cancer Res. 29, 110–121 (2023).
Chang, W. et al. Discovery of MK-1454: A Potent Cyclic Dinucleotide Stimulator of Interferon Genes Agonist for the Treatment of Cancer. J. Med. Chem. 65, 5675–5689 (2022).
Chen, X. et al. Chemically programmed STING-activating nano-liposomal vesicles improve anticancer immunity. Nat. Commun. 14, 4584 (2023).
Chin, E. N. et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 369, 993–999 (2020).
Decout, A., Katz, J. D., Venkatraman, S. & Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21, 548–569 (2021).
Li, J. et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov 11, 1212–1227 (2021).
Lau, W. M. et al. Enpp1: A Potential Facilitator of Breast Cancer Bone Metastasis. PLOS ONE 8, e66752 (2013).
Goswami, A. et al. AVA-NP-695 Selectively Inhibits ENPP1 to Activate STING Pathway and Abrogate Tumor Metastasis in 4T1 Breast Cancer Syngeneic Mouse Model. Molecules 27, 6721 (2022).
Hu, M. et al. Dysregulated ENPP1 increases the malignancy of human lung cancer by inducing epithelial-mesenchymal transition phenotypes and stem cell features. Am. J. Cancer Res. 9, 134–144 (2019).
Li, T. et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci. Rep. 6, 19049 (2016).
Carozza, J. A. et al. Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nat. Cancer 1, 184–196 (2020).
Carozza, J. A. et al. Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem. Biol. 27, 1347–1358.e5 (2020).
Carozza, J. A. et al. ENPP1’s regulation of extracellular cGAMP is a ubiquitous mechanism of attenuating STING signaling. Proc. Natl. Acad. Sci. 119, e2119189119 (2022).
Takahashi, R. et al. Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat. Commun. 6, 7318 (2015).
Wang, H., Ye, F., Zhou, C., Cheng, Q. & Chen, H. High expression of ENPP1 in high-grade serous ovarian carcinoma predicts poor prognosis and as a molecular therapy target. PLOS ONE 16, e0245733 (2021).
Duan, Q., Zhang, H., Zheng, J. & Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 6, 605–618 (2020).
Guan, D. et al. Ecto-nucleotide pyrophosphatase/phosphodiesterase 1 inhibitors: Research progress and prospects. Eur. J. Med. Chem. 267, 116211 (2024).
Zhavoronkov, A. Artificial Intelligence for Drug Discovery, Biomarker Development, and Generation of Novel Chemistry. Mol. Pharm. 15, 4311–4313 (2018).
Jayatunga, M. K. P., Xie, W., Ruder, L., Schulze, U. & Meier, C. AI in small-molecule drug discovery: a coming wave? Nat. Rev. Drug Discov. 21, 175–176 (2022).
Mak, K.-K., Wong, Y.-H. & Pichika, M. R. Artificial Intelligence in Drug Discovery and Development. in Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays (eds. Hock, F. J. & Pugsley, M. K.) 1–38 (Springer International Publishing, Cham, 2022). https://doi.org/10.1007/978-3-030-73317-9_92-1.
Pun, F. W., Ozerov, I. V. & Zhavoronkov, A. AI-powered therapeutic target discovery. Trends Pharmacol. Sci. 44, 561–572 (2023).
Schulte-Sasse, R., Budach, S., Hnisz, D. & Marsico, A. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat. Mach. Intell. 3, 513–526 (2021).
Pun, F. W. et al. Hallmarks of aging-based dual-purpose disease and age-associated targets predicted using PandaOmics AI-powered discovery engine. Aging 14, 2475–2506 (2022).
Du, Y. et al. Machine learning-aided generative molecular design. Nat. Mach. Intell. 6, 589–604 (2024).
Cheng, Y., Gong, Y., Liu, Y., Song, B. & Zou, Q. Molecular design in drug discovery: a comprehensive review of deep generative models. Brief. Bioinform. 22, bbab344 (2021).
Zeng, X. et al. Deep generative molecular design reshapes drug discovery. Cell Rep. Med. 3, 100794 (2022).
Sousa, T., Correia, J., Pereira, V. & Rocha, M. Generative Deep Learning for Targeted Compound Design. J. Chem. Inf. Model. 61, 5343–5361 (2021).
Ren, F. et al. A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models. Nat. Biotechnol. 1–13 https://doi.org/10.1038/s41587-024-02143-0 (2024).
Xu, J. et al. Discovery of a Novel and Potent Cyclin-Dependent Kinase 8/19 (CDK8/19) Inhibitor for the Treatment of Cancer. J. Med. Chem. 67, 8161–8171 (2024).
Fu, Y. et al. Intestinal mucosal barrier repair and immune regulation with an AI-developed gut-restricted PHD inhibitor. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02503-w (2024).
Koromina, M., Pandi, M.-T. & Patrinos, G. P. Rethinking Drug Repositioning and Development with Artificial Intelligence, Machine Learning, and Omics. OMICS J. Integr. Biol. 23, 539–548 (2019).
Long, X. et al. AI-enabled cancer target prioritization with optimal profiles balancing novelty, confidence and commercial tractability. Future Med. AI 2, https://doi.org/10.2217/fmai-2023-0019 (2024).
Vanhaelen, Q. et al. Design of efficient computational workflows for in silico drug repurposing. Drug Discov. Today 22, 210–222 (2017).
Tanoli, Z., Vähä-Koskela, M. & Aittokallio, T. Artificial intelligence, machine learning, and drug repurposing in cancer. Expert Opin. Drug Discov 16, 977–989 (2021).
Tari, L. et al. Identifying Novel Drug Indications through Automated Reasoning. PLOS ONE 7, e40946 (2012).
Han, Y., Klinger, K., Rajpal, D. K., Zhu, C. & Teeple, E. Empowering the discovery of novel target-disease associations via machine learning approaches in the open targets platform. BMC Bioinformatics 23, 232 (2022).
Aliper, A. et al. Deep Learning Applications for Predicting Pharmacological Properties of Drugs and Drug Repurposing Using Transcriptomic Data. Mol. Pharm. 13, 2524–2530 (2016).
Kamya, P. et al. PandaOmics: An AI-Driven Platform for Therapeutic Target and Biomarker Discovery. J. Chem. Inf. Model. 64, 3961–3969 (2024).
Ivanenkov, Y. A. et al. Chemistry42: An AI-Driven Platform for Molecular Design and Optimization. J. Chem. Inf. Model. 63, 695–701 (2023).
Wang, S. et al. ENPP1 is an innate immune checkpoint of the anticancer cGAMP–STING pathway in breast cancer. Proc. Natl. Acad. Sci. 120, e2313693120 (2023).
Mardjuki, R. et al. Identification of the extracellular membrane protein ENPP3 as a major cGAMP hydrolase and innate immune checkpoint. Cell Rep 43, 114209 (2024).
Tokunaga, R. et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation – A target for novel cancer therapy. Cancer Treat. Rev. 63, 40–47 (2018).
El-Sherbiny, Y. M. et al. A novel two-score system for interferon status segregates autoimmune diseases and correlates with clinical features. Sci. Rep. 8, 5793 (2018).
Buang, N. et al. Type I interferons affect the metabolic fitness of CD8 + T cells from patients with systemic lupus erythematosus. Nat. Commun. 12, 1980 (2021).
Liu, H. et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med. 25, 95–102 (2019).
Jia, Q., Wang, A., Yuan, Y., Zhu, B. & Long, H. Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp. Hematol. Oncol. 11, 24 (2022).
Huang, Y.-K. et al. Macrophage spatial heterogeneity in gastric cancer defined by multiplex immunohistochemistry. Nat. Commun. 10, 3928 (2019).
Szpor, J. et al. Dendritic Cells Are Associated with Prognosis and Survival in Breast Cancer. Diagnostics 11, 702 (2021).
Ahmed, R. et al. Single-Cell RNA Sequencing with Spatial Transcriptomics of Cancer Tissues. Int. J. Mol. Sci. 23, 3042 (2022).
Valdeolivas, A. et al. Profiling the heterogeneity of colorectal cancer consensus molecular subtypes using spatial transcriptomics. Npj Precis. Oncol. 8, 1–16 (2024).
Bassiouni, R. et al. Spatial Transcriptomic Analysis of a Diverse Patient Cohort Reveals a Conserved Architecture in Triple-Negative Breast Cancer. Cancer Res. 83, 34–48 (2023).
Lee, S. H. et al. Spatial dissection of tumour microenvironments in gastric cancers reveals the immunosuppressive crosstalk between CCL2+ fibroblasts and STAT3 -activated macrophages. Gut 74, 714–727 (2025).
Patel, S. D. et al. Quinazolin-4-piperidin-4-methyl sulfamide PC-1 inhibitors: Alleviating hERG interactions through structure based design. Bioorg. Med. Chem. Lett. 19, 3339–3343 (2009).
Shayhidin, E. E. et al. Quinazoline-4-piperidine sulfamides are specific inhibitors of human NPP1 and prevent pathological mineralization of valve interstitial cells. Br. J. Pharmacol. 172, 4189–4199 (2015).
Dennis, M. L. et al. Crystal structures of human ENPP1 in apo and bound forms. Acta Crystallogr. Sect. Struct. Biol. 76, 889–898 (2020).
Muegge, I. & Hu, Y. Recent Advances in Alchemical Binding Free Energy Calculations for Drug Discovery. ACS Med. Chem. Lett. 14, 244–250 (2023).
Hewitt, N. J., Lecluyse, E. L. & Ferguson, S. S. Induction of hepatic cytochrome P450 enzymes: methods, mechanisms, recommendations, and in vitro-in vivo correlations. Xenobiotica Fate Foreign Compd. Biol. Syst 37, 1196–1224 (2007).
William, M., Joshua, Gregory N. FLORIA, V., Chandregowda & Athisayamani, J. ECTONUCLEOTIDE PYROPHOSPHATASE-PHOSPHODIESTERASE 1 (ENPP-1) INHIBITORS AND USES THEREOF. WO Pat. Appl. 2019046778 (2019).
Gangar, M. et al. Design, synthesis and biological evaluation studies of novel small molecule ENPP1 inhibitors for cancer immunotherapy. Bioorganic Chem 119, 105549 (2022).
Ullah, S., Pelletier, J., Sévigny, J. & Iqbal, J. Synthesis and Biological Evaluation of Arylamide Sulphonate Derivatives as Ectonucleotide Pyrophosphatase/Phosphodiesterase-1 and −3 Inhibitors. ACS Omega 7, 26905–26918 (2022).
Semreen, M. H. et al. Synthesis, biological evaluation, and molecular docking study of sulfonate derivatives as nucleotide pyrophosphatase/phosphodiesterase (NPP) inhibitors. Bioorg. Med. Chem. 27, 2741–2752 (2019).
Albers, H. M. H. G. et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. USA. 107, 7257–7262 (2010).
Ramesh, M., Ahlawat, P. & Srinivas, N. R. Irinotecan and its active metabolite, SN-38: review of bioanalytical methods and recent update from clinical pharmacology perspectives. Biomed. Chromatogr. BMC 24, 104–123 (2010).
Barbuti, A. M. & Chen, Z.-S. Paclitaxel Through the Ages of Anticancer Therapy: Exploring Its Role in Chemoresistance and Radiation Therapy. Cancers 7, 2360–2371 (2015).
Woo, S.-R. et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 41, 830–842 (2014).
Li, T. & Chen, Z. J. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 (2018).
Cho, Y. et al. Discovery of Orally Bioavailable Phthalazinone Analogues as an ENPP1 Inhibitor for STING-Mediated Cancer Immunotherapy. J. Med. Chem. 66, 15141–15170 (2023).
Ronald, H. Bicyclic heteroaryl boronate derivatives as ectonucleotide pyrophosphatase phosphodiesterase 1 inhibitors. WO Pat. Appl. 2022197734 (2023).
Rao, S. et al. Inhibitors of ectonucleotide pyrophosphatase/phosphodiesterase 1 (enpp1) and methods of use thereof. WO Pat. Appl. 2021158829 (2021).
Borner, M., Scheithauer, W., Twelves, C., Maroun, J. & Wilke, H. Answering patients’ needs: oral alternatives to intravenous therapy. The Oncologist 6, 12–16 (2001).
Quante, M., Thate-Waschke, I. & Schofer, M. [What are the reasons for patient preference? A comparison between oral and subcutaneous administration]. Z. Orthopadie Unfallchirurgie 150, 397–403 (2012).
Le Naour, J., Zitvogel, L., Galluzzi, L., Vacchelli, E. & Kroemer, G. Trial watch: STING agonists in cancer therapy. OncoImmunology 9, 1777624 (2020).
Zhang, P. et al. STING agonist-loaded, CD47/PD-L1-targeting nanoparticles potentiate antitumor immunity and radiotherapy for glioblastoma. Nat. Commun. 14, 1610 (2023).
Xie, Z. et al. Structural insights into a shared mechanism of human STING activation by a potent agonist and an autoimmune disease-associated mutation. Cell Discov 8, 133 (2022).
Ramanjulu, J. M. et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564, 439–443 (2018).
Li, Z. et al. Branched glycopolymer prodrug-derived nanoassembly combined with a STING agonist activates an immuno-supportive status to boost anti-PD-L1 antibody therapy. Acta Pharm. Sin. B 14, 2194–2209 (2024).
Pan, B.-S. et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 369, eaba6098 (2020).
Zheng, W. et al. How the Innate Immune DNA Sensing cGAS-STING Pathway Is Involved in Apoptosis. Int. J. Mol. Sci. 24, 3029 (2023).
Cordova, A. F., Ritchie, C., Böhnert, V. & Li, L. Human SLC46A2 Is the Dominant cGAMP Importer in Extracellular cGAMP-Sensing Macrophages and Monocytes. ACS Cent. Sci. 7, 1073–1088 (2021).
Luteijn, R. D. et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438 (2019).
Wolf, D. M. et al. Redefining breast cancer subtypes to guide treatment prioritization and maximize response: Predictive biomarkers across 10 cancer therapies. Cancer Cell 40, 609–623.e6 (2022).
van den Ende, T. et al. Neoadjuvant Chemoradiotherapy Combined with Atezolizumab for Resectable Esophageal Adenocarcinoma: A Single-arm Phase II Feasibility Trial (PERFECT). Clin. Cancer Res. 27, 3351–3359 (2021).
Lahey, L. J. et al. LRRC8A:C/E Heteromeric Channels Are Ubiquitous Transporters of cGAMP. Mol. Cell 80, 578–591.e5 (2020).
Efremova, M. et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat. Commun. 9, 32 (2018).
Kim, J. C. et al. Brief report: STING expressed in tumor and non-tumor compartments has distinct roles in regulating anti-tumor immunity. Cancer Immunol. Immunother. 72, 1327–1335 (2023).
Zheng, J. et al. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol. Cancer 19, 133 (2020).
Bauer, J. A. et al. Identification of Markers of Taxane Sensitivity Using Proteomic and Genomic Analyses of Breast Tumors from Patients Receiving Neoadjuvant Paclitaxel and Radiation. Clin. Cancer Res. 16, 681–690 (2010).
Prat, A. et al. Research-Based PAM50 Subtype Predictor Identifies Higher Responses and Improved Survival Outcomes in HER2-Positive Breast Cancer in the NOAH Study. Clin. Cancer Res. 20, 511–521 (2014).
Iwamoto, T. et al. Gene Pathways Associated With Prognosis and Chemotherapy Sensitivity in Molecular Subtypes of Breast Cancer. JNCI J. Natl. Cancer Inst. 103, 264–272 (2011).
Tsuji, S. et al. Potential responders to FOLFOX therapy for colorectal cancer by Random Forests analysis. Br. J. Cancer 106, 126–132 (2012).
Shen, J. et al. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 79, 311–319 (2019).
Kakoti, S., Sato, H., Laskar, S., Yasuhara, T. & Shibata, A. DNA Repair and Signaling in Immune-Related Cancer Therapy. Front. Mol. Biosci. 7, 205 (2020).
Wang, S.-W. et al. Mechanism of immune activation mediated by genomic instability and its implication in radiotherapy combined with immune checkpoint inhibitors. Radiother. Oncol. 199, 110424 (2024).
Marcus, A. et al. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763.e4 (2018).
Demaria, O. et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. 112, 15408–15413 (2015).
Pépin, G. & Gantier, M. P. cGAS-STING Activation in the Tumor Microenvironment and Its Role in Cancer Immunity. in Regulation of Inflammatory Signaling in Health and Disease (ed. Xu, D.) 175–194 (Springer, Singapore, 2017). https://doi.org/10.1007/978-981-10-5987-2_8.
Chen, Y. et al. Upregulation of LRRC8A by m5C modification-mediated mRNA stability suppresses apoptosis and facilitates tumorigenesis in cervical cancer. Int. J. Biol. Sci. 19, 691–704 (2023).
Sørensen, B. H., Nielsen, D., Thorsteinsdottir, U. A., Hoffmann, E. K. & Lambert, I. H. Downregulation of LRRC8A protects human ovarian and alveolar carcinoma cells against Cisplatin-induced expression of p53, MDM2, p21Waf1/Cip1, and Caspase-9/−3 activation. Am. J. Physiol.-Cell Physiol. 310, C857–C873 (2016).
Widmer, C. A. et al. Loss of the Volume-regulated Anion Channel Components LRRC8A and LRRC8D Limits Platinum Drug Efficacy. Cancer Res. Commun. 2, 1266–1281 (2022).
Kurashima, K. et al. LRRC8A influences the growth of gastric cancer cells via the p53 signaling pathway. Gastric Cancer 24, 1063–1075 (2021).
Wu, S. Z. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 53, 1334–1347 (2021).
Sun, K. et al. scRNA-seq of gastric tumor shows complex intercellular interaction with an alternative T cell exhaustion trajectory. Nat. Commun. 13, 4943 (2022).
Lee, H.-O. et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 52, 594–603 (2020).
Kumar, V. et al. Single-Cell Atlas of Lineage States, Tumor Microenvironment, and Subtype-Specific Expression Programs in Gastric Cancer. Cancer Discov 12, 670–691 (2022).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517–526 (2022).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Hui, C. Other-codes-and-data-for: Oral ENPP1 inhibitor designed using generative AI as next generation STING modulator for solid tumors. Zenodo https://doi.org/10.5281/ZENODO.15194101 (2025).
Acknowledgements
We thank the following contract research organizations for performing in vitro and in vivo experiments and clinical PK sample analysis in this study: Wuxi AppTec (China), Pharmaron Beijing Co., Ltd. (China), Biometas (China), ICE Bioscience (China), HD Biosciences (China), and ChemPartner (China). We thank Nvidia for providing access to its latest GPUs throughout Insilico Medicine’s history and the lifetime of this project.
Author information
Authors and Affiliations
Contributions
A.Z. and F.R. conceived the project and supervised the study; M.Z. and L.Q. guided the study design and data analysis. C.P. and X.C designed the experiments, performed the research, analyzed the data and prepared the figures; H.C., H.Y., W.Z., Z.N. and S.C. provided AI platform or bioinformatics-based analysis, molecular docking, SAR exploration strategy and prepared the related figures; Y.Q., F.W., X.L. and L.W. performed part of experiments or provided technical assistance; C.P., H.C., X.C., D.G. and F.W.P wrote or refined the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The Authors declare the following competing interests: All listed authors are affiliated with Insilico Medicine, a global clinical-stage commercial generative AI company with several hundred patents and patent applications and commercially available software. Insilico Medicine is a company developing an AI-based end-to-end integrated pipeline for drug discovery and development that is engaged in drug-discovery programs for aging, fibrosis and oncology. C.P., H.C., H.Y., X.C., M.Z., L.Q., Z.N., W.Z., S.C., L.W., X.L., D.G., F.W.P., F.R., and A.Z. are affiliated with Insilico Medicine or were affiliated with Insilico Medicine when the studies were performed. The remaining authors declare no competing interest. No other conflicts are reported.
Peer review
Peer review information
Nature Communications thanks Geneviève Pépin, Tiago Rodrigues, Scott Weir and Guangchuang Yu for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pu, C., Cui, H., Yu, H. et al. Oral ENPP1 inhibitor designed using generative AI as next generation STING modulator for solid tumors. Nat Commun 16, 4793 (2025). https://doi.org/10.1038/s41467-025-59874-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59874-0
