
Abstract
Obesity-related osteoarthritis (OA) and the molecular mechanisms governing multiple joint structural changes that occur with obesity are not well understood. This study investigated the progression of obesity in mice and validated the results using human joint samples post-arthroplasty. The results show that obesity is associated with the degeneration of the cartilage layer and abnormal remodeling of the subchondral bone layer, and this occurs alongside aging and DNA damage in chondrocytes, osteoclasts, and stem cells. Regulation of p53-FOXO3 gene loop expression in response to DNA damage effectively inhibits chondrocyte apoptosis, catabolism, and excessive osteoclast differentiation, while the intra-articular delivery of a lentivirus expressing FOXO3 to mouse joints alleviates the progression of OA. The excessive differentiation of subchondral bone marrow osteoclasts is ferroptosis-dependent and driven by the senescence-associated secretory phenotype. The results have identified multiple potential targets for future research into the progression of obesity-related OA.
Similar content being viewed by others
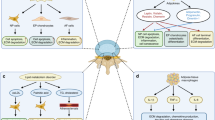


Introduction
Osteoarthritis (OA) is a degenerative joint disease characterized by the degeneration of articular cartilage, subchondral bone sclerosis, synovitis, and the formation of osteophytes1. The World Health Organization has found that approximately 10% of the global population is affected by OA to varying degrees2. In clinical settings, knee OA is primarily characterized by joint pain, crepitus, and restricted joint mobility. This condition significantly impairs patients’ ability to engage in physical activities and adversely affects their quality of life, resulting in substantial negative impacts on their mental well-being. The pathogenic factors and mechanisms underlying OA are highly complex. Common contributing factors include aging, joint injury, obesity, genetics, sex, and anatomical factors related to joint shape and alignment3,4,5,6; each of which possesses unique mechanisms that promote the development of OA. Among these factors, obesity, a metabolic abnormality, has attracted considerable attention.
The ongoing obesity epidemic is one of the greatest challenges in public health in modern societies7. In recent decades, the prevalence of obesity-related OA, associated with a high body weight, has been consistently increasing worldwide8,9. Weight loss has been documented as a highly effective strategy for individuals with OA, particularly those who are obese10,11. However, in metabolic obesity-related OA, the mechanisms of cartilage destruction and abnormal subchondral bone remodeling defy the conventional concept of “wear and tear” diseases, and the joint overload theory inadequately explains the robust correlation between obesity and OA12,13. This evidence collectively suggests that in individuals with obesity-related OA, the systemic metabolic changes induced by obesity have a greater impact on the progression of OA than merely increasing the joint load.
In this study, it was observed that obesity induced by a high-fat diet concurrently led to aging and DNA damage in chondrocytes in the articular cartilage, osteoclasts in the subchondral bone, and mesenchymal stem cells in the bone marrow. The p53-FOXO3 pathway, known for its swift response to DNA damage, orchestrates phenotypic changes in chondrocyte catabolism and the excessive differentiation of osteoclasts. Notably, the intra-articular delivery of a lentivirus expressing FOXO3 was found to effectively alleviate high-fat diet-induced cartilage degeneration and subchondral bone remodeling. This amelioration of subchondral bone remodeling is accomplished through the regulation of osteoclast differentiation, which is dependent on ferroptosis. Finally, and unexpectedly, the heightened adipogenic differentiation of BMSCs induced by aging, coupled with the subsequent secretion of a senescence-associated secretory phenotype (SASP) by aging adipocytes, was identified as a pivotal driving force promoting ferroptosis in osteoclasts.
Results
High fat diet accelerates the progression of osteoarthritis in mice
To understand whether obesity has an adverse effect on the degeneration of knee joint cartilage, that is, whether it leads to the progression of spontaneous OA, mice were fed a normal or high-fat diet (HFD) for 2 months. During this process, we regularly detected the weight, blood sugar, blood lipids, and other information of the mice to determine the success of establishing the obesity model (Supplementary Fig. 1). X-ray and micro-CT data revealed that obesity induced in mice by the HFD alone was adequate for the induction of joint-level wear and the narrowing of the joint space (Fig. 1a, b). The HFD also lead to a dramatic loss of articular cartilage, which is manifested by a significant increase in the OARSI score and a significant loss of cartilage (Fig. 1c, g). Synovitis scores also suggested the acceleration and worsening effects of a high-fat diet on synovial inflammation (Supplementary Fig. 2a). At the molecular level, compared with the normal diet group, the HFD group showed a decrease in Col2a1-positive cells and a sharp increase in MMP13-positive cells (Fig. 1c, j, k).

a X-ray imaging of the lower limbs (top) and knee joints (bottom) of mice subjected to normal diet as well as high-fat diet conditions. Further categorization was performed based on whether the mice underwent DMM surgery. White arrows indicate joint space narrowing and bone damage. b Micro-CT reveals the osteophyte volume in joints of mice subjected to various diets and surgical procedures. Osteophyte scores (h) was quantified. n = 6 mice per group. c SO&FG staining (top), H&E staining (middle), and immunohistochemistry (IHC) staining (bottom) of the knee joints of mice in (a, b). Scale bars, 100 µm and 40 µm. OARSI scoring was conducted through SO&FG staining (g). Col2a1+ (j) and Mmp13+ (k) chondrocytes were imaged and quantified. n = 6 mice per group. d Three-dimensional high-resolution µCT images of the tibial subchondral bone medial compartment (top), subchondral bone SO&FG staining (middle), and TRAP staining (bottom). Scale bar, 40 µm. Relative volume of trabecular bone (BV/TV) (i) and TRAP+ (l) cells were quantified. n = 6 mice per group. e Immunofluorescence staining of IL-6 in the synovial membrane and subchondral bone. Representative images are shown in (e). Scale bar, 40 µm. Quantification of IL-6+ cells in the synovial membrane and subchondral bone are presented in (m) and (n), respectively. n = 6 mice per group. f Immunofluorescence staining of Perilipin-1 in the subchondral bone. Representative images are shown in (f). Scale bar, 40 µm. Quantification of Perilipin-1+ cells are presented in (o). n = 6 mice per group. Data are represented as mean ± s.d. Two-way ANOVA was performed. All exact p-values have been indicated in the figures. Source data are provided as a Source Data file.
We further aimed to elucidate whether the HFD exacerbated pathological changes in OA. This investigation targeted individuals predisposed to, or afflicted with, OA. To achieve this, a destabilized medial meniscus (DMM) was performed. Imaging data indicated that when combined with the DMM surgical treatment, the HFD not only promoted but also exacerbated the formation of osteophytes around the joints. It contributes to joint space wear and stenosis and may even lead to joint fusion (Fig. 1a, b, h). Furthermore, the HFD enhanced the degeneration of the articular cartilage layer and exacerbated a reduction in type II collagen induced by DMM surgery. Conversely, a substantial and rapid upregulation of MMP13 in the cartilage layer was evident (Fig. 1c, j, k). These findings collectively indicate that, at the articular cartilage level, whether administered alone or in conjunction with DMM surgery, the HFD not only spontaneously induces the progression of OA but also exacerbates the ongoing pathological processes.
OA is a degenerative joint disease characterized by cartilage deterioration due to chondrocyte damage and loss, and also the abnormal remodeling of the subchondral bone14,15,16. The tibial subchondral bone volume in mice receiving an HFD underwent significant changes compared to that in the control mice before or after surgery, as revealed by 3D micro-CT analysis (Fig. 1d, i). TRAP staining revealed an elevated number of osteoclasts in the subchondral bone in the HFD group, leading to sustained osteoclastic bone resorption and formation of expansive bone marrow cavities, with or without concurrent DMM surgery (Fig. 1d, l).
Prior studies have demonstrated a significant upregulation of IL-6 levels in obese patients17. IL-6 is known to play a crucial role in promoting the progression of OA and osteoclast differentiation18. Our findings revealed a substantial upregulation of IL-6 levels in both the synovial tissue and subchondral bone, regardless of whether the mice were subjected to HFD alone or in conjunction with DMM surgery (Fig. 1e, m, n). Thus, obesity induced by an HFD autonomously accelerates the development of associated pathological changes in OA progression and intensifies the advancement of pre-existing OA. This regulatory process spans multiple levels, encompassing cartilage, subchondral bone marrow, and joint synovial membranes.
Chondrocytes exposed to fatty acids display characteristics of apoptosis, catabolism, and senescence
As a prevalent saturated fatty acid, the addition of palmitic acid to the culture medium effectively establishes a high-fat environment, mimicking the physiological and metabolic conditions associated with obesity6,19. The results showed a gradual decrease in chondrocyte viability as the palmitate concentration increased (Fig. 2a). Furthermore, in the presence of two well-known inflammatory cytokines, the treatment of chondrocytes with palmitate resulted in a further decrease in cell viability compared to the inflammatory cytokines alone (Fig. 2b). The results of annexin V-FITC staining also revealed that the exposure of chondrocytes to fatty acids resulted in an increase in apoptosis levels (Fig. 2c).

a, b, Cell viability in mouse chondrocytes treated with palmitate alone (a) or in combination with IL-1β or TNF-α (b). n (Control) = 3, n (0.5) = 3, n (1) = 3, n (1.5) = 3, n (2) = 3, n (Control, -) = 3, n (IL-1β, -) = 3, n (IL-1β, +) = 3, n (TNF-α, -) = 3, n (TNF-α, +) = 3. c Detection of cell apoptosis using recombinant Annexin V labeled with FITC (red). Scale bar, 25 µm. d Micromass culture of mouse chondrocytes treated with vehicle or palmitate. e Immunoblot detection of Sox9, Collagen II, Collagen X, Adamts5, Mmp13, Cleaved PARP, and Bcl-2 in mouse chondrocytes. Palmitate, IL-1β, or TNF-α was added to the mouse chondrocytes 24 h before harvesting. f The mRNA expression levels of Collagen II, Aggrecan, Sox9, Mmp13, Adamts5, and Mmp3 in mouse chondrocytes. The cell treatment method was the same as in (e). n (Control) = 3, n (Palmitate) = 3, n (IL-1β) = 3, n (TNF-α) = 3, n (Palmitate + IL-1β) = 3, n (Palmitate + TNF-α) = 3. g, h Immunofluorescence staining of Collagen II (green) (g) and MMP13 (red) (h) in mouse chondrocytes. Scale bars, 100 µm and 25 µm. i–k The primary mouse chondrocytes were isolated and treated with palmitate, IL-1β, TNF-α, or a combination of palmitate with the inflammatory cytokines. After treatment, cells were fixed and subjected to immunostaining for γH2AX (red) (i) and Lamin-B1 (red) (j), as well as SA-β-Gal (green) staining (k), respectively. The palmitate concentration used in (b–k) is 1 mM. Scale bars, 20 µm and 100 µm. The above experiments were repeated three times. Data are represented as mean ± s.d. Two-tailed Student’s t tests and one-way ANOVA were performed. All exact p-values have been indicated in the figures. For western blot analyses in (e) and RT-qPCR in (f), GAPDH was utilized as the control. Source data are provided as a Source Data file.
The relevant characteristics of chondrocyte ECM metabolism were then assessed. Micromass cultures of chondrocytes with Alcian blue staining confirmed that exposure to palmitate affected their collagen synthesis (Fig. 2d). Western blot analysis using protein extracts from chondrocytes stimulated with palmitate, inflammatory cytokines, or a combination of both indicated that the palmitate treatment downregulated the chondrocyte collagen synthesis markers Sox9 and Col2a1. Furthermore, it simultaneously upregulated the chondrocyte hypertrophy marker Col10a1, as well as the matrix metalloproteinases Mmp13 and Adamts5. Importantly, when palmitate was combined with inflammatory cytokines, the changes in these markers were more pronounced than when inflammatory cytokines alone were used (Fig. 2e, f). Two indicators of cell apoptosis are Cleaved PARP and Bcl-2, and they showed upregulated and downregulated trends, respectively, indicating that palmitate promotes chondrocyte apoptosis (Fig. 2e). Immunofluorescence (IF) showed a reduction in Col2a1 synthesis and an upregulation in Mmp13 expression in chondrocytes following the palmitate treatment (Fig. 2g, h). Chondrocytes treated with palmitate combined with inflammatory cytokines showed a completely senescent subtype, including high expression levels for γ-H2AX and a loss of Lamin-B1 (Fig. 2i, j). In addition, the high expression of β-gal further suggested that the level of cellular senescence was sharply upregulated (Fig. 2k). After palmitate treatment, chondrocytes showed decreased cell viability, increased apoptosis, and metabolic disorders of the extracellular matrix, causing them to exhibit a completely senescent phenotype.
p53-AKT-FOXO3 orchestrates fatty acid-induced apoptosis and catabolism in response to chondrocyte ageing and damage
Notably, γH2AX is not only a marker of cellular senescence but also of DNA damage20. The results showed that palmitate significantly increased the expression and co-localization of γH2AX and p53 in the nucleus of chondrocytes (Fig. 3a, b). It has been suggested that with fatty acid exposure, chondrocytes activate p53 and promote its expression and functional responses. At the same time, as a potential p53-binding protein21, the results showed that p53 could also activate and phosphorylate downstream AKT (Fig. 3c, d). These results suggest that p53 activation in response to aging and DNA damage may play an important role in the regulation of HFD-induced obesity-related OA (Fig. 3e). The next step in the investigation was to study the impact of alterations in p53-AKT on chondrocyte apoptosis and ECM metabolism in response to palmitate exposure. To achieve this, the specific inhibitor of p53, Pifithrin-β (PFT-β), was used while treating chondrocytes with palmitate. The results demonstrated that the combined treatment of chondrocytes with palmitate significantly enhanced the downregulation of markers such as Sox9, Col2a1, and Bcl-2, compared to the use of IL-1β or TNF-α alone. Additionally, it led to a notable upregulation of markers, including Col10a1, Mmp13, Adamts5, Cleaved PARP, and Cleaved Caspase-3 (Fig. 3f–h). Moreover, with the combined use of p53 inhibitors, this downregulation or upregulation was further promoted (Fig. 3f–h). Although the inhibition of p53 promoted the upregulation of autophagy in chondrocytes, it still manifested as catabolism and the upregulation of apoptosis when exposed to fatty acids (Fig. 3f). Furthermore, using the classic AKT-selective inhibitor MK-2206, similar experimental conclusions consistent with the inhibition of p53 were obtained (Fig. 3f–h). This result made us excited and curious, because in the presence of non-fatty acids, inhibiting p53 or AKT in chondrocytes alone promotes the anabolism of chondrocytes rather than inhibits it (Supplementary Fig. 3). IF experiments were also used to confirm whether it was the inhibition of p53 or AKT, the downregulated Col2a1 showed a further reduction in expression (Fig. 3i). The results of the micromass analysis also showed that the inhibition of p53 or AKT significantly inhibited collagen synthesis (Fig. 3j).

a, b Confocal images of IF staining against γH2AX (red) and p53 (green) in mouse chondrocytes treated with vehicle or palmitate (1 mM) (a). Scale bar, 25 µm. The nuclear levels of γH2AX and p53 were quantified (b) (n = 15). c, d Confocal images of immunofluorescence staining against p53 (green) and pAKT (red) in mouse chondrocytes treated with vehicle or palmitate (1 mM) (c). Scale bar, 25 µm. The nuclear levels of p53 and pAKT were quantified as well (d) (n = 15). e A pattern diagram illustrating nuclear DNA damage and p53 response in chondrocytes induced by a high-fat diet. Created in BioRender. Zhao, C. (2024) https://BioRender.com/s11w531. f Immunoblot detection of Sox9, Collagen II, Collagen X, Adamts5, Mmp13, Cleaved Caspase-3, Cleaved PARP, Bcl-2, and LC3 in mouse chondrocytes. g, h The mRNA expression levels of Collagen II, Aggrecan, Sox9, Mmp13, Adamts5, and Mmp3 in mouse chondrocytes (n = 3). i IF staining of Collagen II (green). Scale bar, 100 µm. j Micromass culture of mouse chondrocytes. k Confocal images of IF staining against γH2AX (red) and p21 (green) in mouse chondrocytes. Infection with FOXO3-expressing lentivirus was combined using palmitate (1 mM). Scale bar, 5 µm. l Immunoblot detection of Sox9, Adamts5, Mmp13, Cleaved PARP, and Bcl-2 in mouse chondrocytes. m The mRNA expression levels of Collagen II, Aggrecan, Sox9, Mmp13, Adamts5, and Mmp3 in mouse chondrocytes (n = 3). n Micromass culture of mouse chondrocytes. o SA-β-Gal (green) staining for mouse chondrocytes transfected with or without FOXO3. Scale bar, 100 µm. p IF staining of Collagen II (red). Scale bar, 50 µm. q The mRNA levels of Collagen II, Aggrecan, Sox9, Mmp13, Adamts5, and Mmp3 in mouse chondrocytes (n = 3). r Micromass culture of mouse chondrocytes. s The mRNA levels of Collagen II, Aggrecan, Sox9, Mmp13, Adamts5, and Mmp3 in mouse chondrocytes (n = 3). t Micromass culture of mouse chondrocytes. The above experiments were repeated at least three times. Data are represented as mean ± s.d. Two-tailed Student’s t tests and one-way ANOVA were performed. All exact p-values have been indicated in the figures. For western blot analyses in (f, l) and RT-qPCR in (g, h, m, q, s), GAPDH was utilized as the control. Source data are provided as a Source Data file.
AKT and the FOXO protein family have a crucial interplay, primarily manifested through AKT phosphorylating FOXO proteins to regulate their functions in cellular physiological processes22. At the same time, a recent cutting-edge study showed that p53 can also directly bind FOXO3 protein21. In view of the important regulatory role of the FOXO3 protein in OA, we conducted further exploration and excavation around this preliminary research basis. FOXO3 exhibits reduced expression upon inactivation, accompanied by its translocation from the nucleus to the cytoplasm (Supplementary Fig. 4). Next, we transduced chondrocytes with lentiviruses expressing FOXO3 and verified the success of the FOXO3 overexpression using western blotting and RT-qPCR (Supplementary Fig. 5). Through IF experiments, it was observed that chondrocytes transduced with lentivirus no longer exhibited an upregulation senescence response and DNA damage markers, γH2AX and p21, in response to the palmitate stimulation (Fig. 3k). Western blotting further confirmed the protective effect of FOXO3 overexpression in chondrocytes (Fig. 3l). The data obtained from the RT-qPCR and IF experiments substantiated the conclusions drawn from the protein-level experiments (Fig. 3m, p). Alcian blue staining showed significant antagonism to palmitate in cells transduced with FOXO3-expressing lentivirus (Fig. 3n). The results from the β-gal staining also demonstrated an amelioration of the cellular senescence phenotype (Fig. 3o).
Chrondocytes were then investigated to determine if FOXO3 overexpression conferred a protective effect against p53 and AKT knockdown in combination with palmitate stimulation. The PCR results were particularly noteworthy; whether in combination with a p53 inhibitor or an AKT inhibitor, and regardless of whether palmitate combined with IL-1β or TNF-α, the overexpression of FOXO3 in the cells not only failed to exert a balancing effect on the ECM metabolism but further exacerbated and deteriorated catabolic metabolism (Fig. 3q, s). The results of the Alcian blue staining further confirmed that the promotive effect of FOXO3 overexpression on collagen synthesis was no longer present when p53 or AKT was inhibited (Fig. 3r, t). In summary, under conditions where p53-AKT regulation is artificially inhibited, the overexpression of FOXO3 fails to protect chondrocytes and may potentially exacerbate phenotypic expression associated with further degradation.
Intra-articular injection of lentiviruses expressing FOXO3 attenuates OA lesions caused by a HFD in mice
Through IF experiments, we first determined the successful regulation of FOXO3 expression levels in the cartilage layer and subchondral bone marrow layer by injection of lentivirus expressing FOXO3 into the joint cavity (Supplementary Fig. 6). Micro-CT scans and histomorphometric analyses of knee joint sections stained with SO&FG and H&E were conducted to evaluate the extent of articular repair. As anticipated, mice treated with lentiviruses expressing FOXO3 exhibited improvements in OA lesions compared to obese mice fed HFD. Specifically, there was a significant increase in cartilage thickness, reduction in osteophyte formation, and decrease in OARSI scores in mice overexpressing FOXO3 (Fig. 4a–e). The effective overexpression of FOXO3 in the joint cavity also alleviates the synovial inflammation promoted by abnormal metabolic factors (Supplementary Fig. 2b). At the molecular level, intra-articular delivery of FOXO3 effectively enhanced the collagen marker, Collagen II, in the cartilage. Simultaneously, it significantly downregulated the crucial cartilage-degrading enzyme Mmp13 (Fig. 4h–k).

a, b SO&FG staining (a) as well as H&E staining (b) was performed to depict the cartilage area of joints in mice fed with a high-fat diet after intra-articular injection of lentiviruses expressing FOXO3. Black boxes and dashed lines indicate representative areas of joint cartilage, which were further magnified for detailed examination. n = 6 mice per group. c The extent of cartilage damage was assessed using the OARSI scoring system based on the staining in (a). d, e Micro-CT (e) was conducted to assess the osteophyte volume (top) in joints from mice fed with a high-fat diet after intra-articular injection of lentiviruses expressing FOXO3. Osteophyte scores were quantified (d). n = 6 mice per group. f, g Quantitative micro-CT analysis was performed on the tibial subchondral bone (e), measuring trabecular bone volume per total volume (BV/TV) (f) and trabecular thickness (Tb.Th) (g) in joints from mice fed with an HFD after intra-articular injection of lentiviruses expressing FOXO3. h–k, IHC staining of Col2a1 (h) and Mmp13 (j) was performed on the articular cartilage of joints from mice fed with an HFD after intra-articular injection of lentiviruses expressing FOXO3. The percentage of positive cells in the cartilage was quantified. n = 6 mice per group. l, m TRAP staining on the tibial subchondral bone in mice fed with an HFD after intra-articular injection of lentiviruses expressing FOXO3. The percentage of positive cells in the subchondral bone of the cartilage was quantified. n = 6 mice per group. n–s IF staining of TRAP (red) (n), γH2AX (red) (p), and Perilipin-1 (red) (r) was performed on the subchondral bone of joints from mice fed with an HFD after intra-articular injection of lentiviruses expressing FOXO3. The percentage of positive cells in the subchondral bone of the cartilage was quantified. n = 6 mice per group. The sham group received an injection of a lentiviral blank vector, while the FOXO3 group received an injection of a lentivirus expressing FOXO3. Data are represented as mean ± s.d. One-way ANOVA was performed. All exact p-values have been indicated in the figures. Source data are provided as a Source Data file.
A previous study found that mammalian FOXO3 also plays a role at the G2 to M checkpoint of the cell cycle and triggers the repair of damaged DNA23. The growth arrest and DNA damage response gene Gadd45a appear to be direct targets of FOXO3a, mediating part of FOXO3’s role in DNA repair. Through in vitro cell experiments, we initially confirmed that overexpression of FOXO3 in chondrocytes promotes an increase in Gadd45a expression levels in chondrocytes treated with or without fatty acids (Supplementary Fig. 7a, b). Furthermore, through animal experiments, we further reinforced this conclusion at the cellular and molecular level in the cartilage layer, suggesting that FOXO3’s protective effect on chondrocytes may involve various regulatory mechanisms, including catabolism and DNA damage (Supplementary Fig. 7c).
In the preceding sections of this study, mice fed an HFD exhibited not only articular cartilage degeneration but also significant remodeling of the subchondral bone, accompanied by a marked enhancement in osteoclast differentiation (Fig. 1d, l). Intra-articular injection of FOXO3-expressing lentivirus effectively mitigated the diminished bone volume (Fig. 4e–g). TRAP staining indicated a significant reduction in the number of osteoclasts in mice treated with lentivirus compared to the HFD group (Fig. 4l, m). IF further demonstrated that after viral treatment, the number of TRAP-positive cells exhibited a trend consistent with the staining results (Fig. 4n, o). These results indicate that intra-articular overexpression of FOXO3 not only effectively restrains the degeneration of the articular cartilage in obese mice but also mitigates the loss and remodeling of subchondral bone marrow volume.
Excessive adipogenesis in subchondral bone of human osteoarthritic cartilage
The replaced knee joints from patients who underwent total knee arthroplasty were collected (Supplementary Fig. 8a). As anticipated, there was a reduction in aggrecan-containing cartilage and a notable elevation in the OARSI score with OA when compared with healthy cartilage (Supplementary Fig. 8b, d). Moreover, an apparent enhancement in adipogenic differentiation was observed within the subchondral bone marrow of the arthritic joint bed (Supplementary Fig. 8c).
RANKL-induced osteoclastogenesis is regulated by p53-AKT-FOXO3
Compared to the control group, mice fed an HFD had enriched levels of p53-pFOXO3 in the subchondral bone marrow (Fig. 5a). The role of the p53-AKT-FOXO3 axis in osteoclast differentiation was then determined in vitro. TRAP staining revealed that treatment with PFT β, MK-2206, or lentivirus expressing FOXO3 significantly inhibited the formation of osteoclasts (Fig. 5b, c). The expression levels of various molecular markers that regulate osteoclast differentiation was validated. Inhibition of either p53 or AKT using their respective inhibitors was found to result in marker suppression, indicating the inhibition of osteoclast differentiation (Fig. 5d, e). Conversely, FOXO3 overexpression within the cells led to the inhibition of these markers (Fig. 5f). In summary, these findings suggest that the regulatory effects on RANKL-stimulated osteoclast differentiation are achieved through the targeted modulation of the p53-AKT-FOXO3 pathway.

a IF staining of p53 (green) and pFOXO3 (red) was performed on the subchondral bone of joints from mice fed with an HFD. n = 6 mice per group. Scale bar, 100 µm. b, c Representative images of TRAP staining (b) and quantification of mature TRAP+ osteoclasts (c) were performed after culturing with PFT β (10 μM), MK-2206 (10 μM), or infection with lentivirus expressing FOXO3, followed by treatment with RANKL for 6 days (n = 4-6). Scale bar, 100 µm. d The mRNA expression levels of Acp5, Atp6v1h, Ctsk, Dcstamp, Fos, and Nfatc1 in mouse osteoclasts. During the differentiation of BMDMs into osteoclasts, a combination of the p53 inhibitor PFT β (10 μM) and treatment with RANKL for 6 days was applied. n (Control) = 3, n (PFT β) = 3. e The mRNA expression levels of Acp5, Atp6v1h, Ctsk, Dcstamp, Fos, and Nfatc1 in mouse osteoclasts. During the differentiation of BMDMs into osteoclasts, a combination of the AKT inhibitor MK-2206 and treatment with RANKL (100 ng/ml) for 6 days was applied. n (Control) = 3, n (MK-2206) = 3. f The mRNA expression levels of Acp5, Atp6v1h, Ctsk, Dcstamp, Fos, and Nfatc1 in mouse osteoclasts. n (Lv-NC) = 3, n (Lv-FoxO3a) = 3. Mouse BMDMs were transfected with lentivirus expressing FOXO3 or an empty vector, followed by treatment with RANKL (100 ng/ml) for 6 days. Data are represented as mean ± s.d. Two-tailed Student’s t tests was performed. All exact p-values have been indicated in the figures. For RT-qPCR in (d–f), GAPDH was utilized as the control. Source data are provided as a Source Data file.
FOXO3 regulates the vicious cycle of chondrocyte-osteoclast differentiation
In addition to the reasons for lentiviral penetration into the underlying osteoclast layer, we aimed to further elucidate whether a direct impact on chondrocytes can similarly regulate the differentiation of the underlying osteoclasts. To mimic the vicious cycle within the joint microenvironment, chondrocytes were co-cultured with BMDM following the RANKL treatment (Fig. 6a). The upper chondrocytes were pre-transfected with either a FOXO3-expressing lentivirus or an empty vector. Mature osteoclasts were detected using TRAP staining. The results showed that when co-cultured with FOXO3-overexpressing chondrocytes in the upper layer, while the control group showed obvious osteoclast differentiation and maturity on the fourth day of cell culture, this group was still in the process of pre-osteoclast differentiation (Fig. 6b, c). The data revealed that upon exposure of the upper layer of chondrocytes to palmitate, the lower layer of osteoclasts exhibited significantly heightened osteoclast fusion and expression compared with the control group. When subjected to the inflammatory cytokines IL-1β or TNF-α, the upper layer of chondrocytes also notably promoted the differentiation and fusion of the underlying osteoclasts. Furthermore, the combination of palmitate with the two inflammatory cytokines for the treatment of upper chondrocytes resulted in peak differentiation of the lower osteoclasts. However, when the upper chondrocytes were transfected with FOXO3-expressing lentivirus, the significant promotion of osteoclast differentiation was reversed (Fig. 6d, e).

a, A schematic diagram of an in vitro cell co-culture model. Chondrocytes are seeded in the upper insert, while osteoclasts derived from BMDMs are cultured in the lower layer. Created in BioRender. Zhao, C. (2024) https://BioRender.com/s11w531. b, c, Representative images of TRAP staining (b) and quantification of mature TRAP+ osteoclasts (c) were performed after infection of upper layer chondrocytes with lentivirus expressing FOXO3 (n = 5). Scale bar, 100 µm. d, e, Representative images of TRAP staining and quantification of mature TRAP+ osteoclasts were conducted after culturing upper layer chondrocytes with palmitate (1 mM), IL-1β (10 ng/ml), TNF-α (10 ng/ml), or infection with lentivirus expressing FOXO3 (n = 6). Scale bar, 100 µm. Data are represented as mean ± s.d. Two-tailed Student’s t tests and one-way ANOVA were performed. All exact p-values have been indicated in the figures. Source data are provided as a Source Data file.
Concomitant cellular ferroptosis during osteoclast differentiation
A recent study indicates that p53 transcriptionally represses VKORC1L1 expression to activate ferroptosis24. Our previous study also revealed that FOXO3 could modulate the progression of OA by regulating ferroptosis in chondrocytes25. Considering the pivotal roles of the upstream p53 and downstream FOXO3 in ferroptosis regulation, we aimed to explore whether the regulation of p53-FOXO3 on subchondral bone remodeling and osteoclast differentiation in HFD-induced obese mice is mediated through osteoclast ferroptosis regulation. Ferroptosis suppressor protein 1 (FSP1), glutathione peroxidase 4 (GPX4), and solute carrier family 7 member 11 (SLC7A11) are integral components of the regulatory framework of ferroptosis and serve as crucial suppressors of this iron-dependent form of regulated cell death26,27,28. The expression levels of these three proteins were significantly downregulated in mature osteoclasts (Fig. 7a). TRAP staining showed that the number of mature osteoclasts was significantly reduced when combined with the ferroptosis inhibitor Ferrostatin-1 (Fer-1) during the differentiation of BMDM into osteoclasts (Fig. 7b, c). The fluorescence intensity observed using FerroOrange, an Fe2+-specific probe, was markedly enhanced in cells treated with RANKL (Fig. 7d). Simultaneously, intracellular levels of reactive oxygen species (ROS) and lipid peroxidation were significantly upregulated during osteoclast differentiation (Fig, 7e, f). Collectively, these three indicators serve as crucial markers for the activation of cellular ferroptosis. Furthermore, the fluorescence intensity of the three ferroptosis suppressor proteins was markedly diminished in mature osteoclasts (Fig. 7g–i). In summary, results suggest that the differentiation of BMDM into osteoclasts is accompanied by the activation of intracellular ferroptosis, which indicates that the regulation of osteoclast ferroptosis may have important interventional value.

a Immunoblot detection of Fsp1, Slc7a11, and Gpx4 in mouse osteoclasts. b, c Representative images of TRAP staining (b) and quantification of mature TRAP + osteoclasts (c) (n = 6). In comparison to the control group stimulated solely with RANKL (100 ng/ml), the experimental group was co-treated with the ferroptosis inhibitor Fer-1 (10 μM). Scale bar, 100 µm. d–f Representative images of FerroOrange staining (red) (d), lipid peroxidation staining (green) (e), and ROS images (green) (f) are presented. The cell grouping and treatment methods were the same as in (a). g–i Immunofluorescence staining of Gpx4 (red), Slc7a11 (red), and Fsp1 (red). The cell grouping and treatment were the same as in (a). Scale bar, 25 µm. The above experiments were repeated at least three times. Data are represented as mean ± s.d. Two-tailed Student’s t tests was performed. All exact p-values have been indicated in the figures. For western blot analyses in (a), GAPDH was utilized as the control. Source data are provided as a Source Data file.
Intra-articular injection of lentiviruses expressing FOXO3 attenuates OA lesions and caused upregulation of ferroptosis in the subchondral bones of obese mice
Given the observed activation of intracellular ferroptosis during osteoclast differentiation, we aimed to elucidate whether remodeling of the subchondral bone in diet-induced obesity-related OA is associated with the regulation of ferroptosis. With the HFD, the key proteins involved in the regulation of ferroptosis were downregulated in subchondral bone (Fig. 8a–f). Moreover, intra-articular injection of lentiviruses expressing FOXO3, as previously mentioned, improved subchondral bone remodeling and inhibited osteoclast differentiation (Fig. 4l–o). This intervention partially upregulated the expression of Gpx4 and significantly elevated the expression of Slc7a11 and Fsp1 (Fig. 8a–f). The findings highlight the ability of FOXO3 to regulate the progression of OA by modulating ferroptosis in both chondrocytes and subchondral osteoclasts.

a–f IHC staining of Gpx4 (a), Slc7a11 (c), and Fsp1 (e) was performed on the subchondral bone of joints from mice fed with an HFD after intra-articular injection of lentiviruses expressing FOXO3. The percentage of positive cells in the subchondral bone was quantified. n = 6 mice per group. Scale bar, 100 µm. The sham group received an injection of lentiviral blank vector, while the FOXO3 group received an injection of lentivirus expressing FOXO3. Data are represented as mean ± s.d. One-way ANOVA was performed. All exact p-values have been indicated in the figures. Source data are provided as a Source Data file.
HFD-induced adipogenic differentiation of mesenchymal stem cells promote subchondral bone marrow osteoclast differentiation in a ferroptosis-dependent manner
During the initial exploratory phase of the study, a significant increase in the expression of adipogenic marker Perilipin-1 was noticed in the subchondral bones of mice fed an HFD (Fig. 1f, o). Through further analysis of the subchondral bone from joint replacement patients, it was tentatively concluded that as OA progressed, adipocyte differentiation in the subchondral bone marrow was significantly enhanced (Fig. 9a). It appears that we have tentatively resolved the question raised by Xiaonan et al. in their Discussion section, which was, with an increase in obesity level, the bone marrow adipose tissue mass increases significantly29. However, we have generated a new question: whether SASP factors from aging adipocytes have a similar effect on osteoclasts. To simulate the microenvironment of the interaction between osteoclasts and BMSCs undergoing adipogenic differentiation, preosteoclasts and BMSCs were co-cultured in an adipogenic differentiation medium (Fig. 9b). After co-culturing preosteoclasts and BMSCs undergoing adipogenic differentiation, the preosteoclasts in the lower layer differentiated into multinucleated osteoclasts as round giant cells with three or more nuclei earlier than those in the control group without adipogenic differentiation. Furthermore, the promotional effect of the BMSCs undergoing adipogenic differentiation on the differentiation of osteoclasts in the lower layer could be further reversed by a combination with the ferroptosis inhibitor Fer-1 (Fig. 9c, e). To further determine whether certain substances from differentiated adipocytes could promote early differentiation and enhance osteoclast differentiation in the subchondral bone marrow, the JAK inhibitor ruxolitinib (JAKi) was used to treat BMSCs in the upper layer, which is known to inhibit the release of SASP29,30. The results indicate that inhibiting SASP secretion from BMSCs undergoing adipogenic differentiation can also reverse the differentiation of osteoclasts (Fig. 9d, f).

a IF staining of Perilipin-1 (red) on the subchondral bone of joints from OA patients (n = 6). Scale bar, 100 µm. b A schematic diagram of an in vitro cell co-culture model involving murine BMSC and osteoclasts. Created in BioRender. Zhao, C. (2024) https://BioRender.com/s11w531. c TRAP staining. The upper layer of stem cells was subjected to two different culture media: normal medium and adipogenesis medium. Scale bar, 100 µm. d TRAP staining. In the last column, ruxolitinib (10 μM) was included in the adipogenesis medium. Scale bar, 100 µm. e, f Quantification of mature TRAP+ osteoclasts in (c, d) (n = 6). g, h TRAP staining (h) and quantification of mature TRAP+ osteoclasts (g) (n = 6). Scale bar, 100 µm. In the third column, the upper layer of stem cells was supplemented with PFT β (10 μM), and in the last two columns, the upper layer of stem cells was pre-infected with lentivirus expressing empty vector or FOXO3. i, j FerroOrange staining (red) (i) and lipid peroxidation staining (green) (j). Scale bar, 50 µm. k–m IF staining of Gpx4 (red), Slc7a11 (red) and Fsp1 (red). Scale bar, 50 µm. n, o Exchange of cells in the co-culture system (o). Created in BioRender. Zhao, C. (2024) https://BioRender.com/s11w531. The upper layer of pre-osteoclasts was subjected to normal medium, RANKL-containing medium, and RANKL + Fer-1-containing medium. Oil Red (red) staining was used to assess the extent of adipogenic differentiation in the lower layer of stem cells (n). Scale bar, 100 µm. p Oil Red (red) staining. The upper layer of pre-osteoclasts was treated with normal medium, RANKL-containing medium, RANKL plus PFT β-containing medium, or cells pre-infected with lentivirus. Scale bar, 100 µm. q–s IF staining of Perilipin (red) (q), γH2AX (red) (r), and Lamin-B1 (red) (s). The cell grouping and treatment methods were the same as in (n). Scale bar, 100 µm. t–v IF staining of Perilipin (red) (t), γH2AX (red) (u) and Lamin-B1 (red) (v). The cell grouping and treatment methods were the same as in (p). Scale bar, 100 µm. The above experiments were repeated at least three times. Data are represented as mean ± s.d. One-way ANOVA was performed. All exact p-values have been indicated in the figures. Source data are provided as a Source Data file.
Our attention was then turned back to p53-FOXO3 and aimed to clarify whether osteoclast differentiation could be regulated by controlling the expression of p53-FOXO3 in adipogenic differentiated BMSCs. The results of TRAP staining suggest that adding the p53 inhibitor Pifithrin-β to the adipogenic differentiation medium of BMSCs, or directly using BMSCs transfected with FOXO3-overexpressing lentivirus, could reverse the promotion effect of BMSCs undergoing adipogenic differentiation on the differentiation of osteoclasts (Fig. 9g, h). Notably, the inhibition of subchondral osteoclast differentiation in the co-culture system concurrently suppressed the activating effect of upper-layer adipogenically differentiated BMSCs on osteoclast ferroptosis (Fig. 9i–m).
Furthermore, the mature osteoclasts also affected BMSC adipogenic differentiation and aging, thereby establishing a vicious cycle. By exchanging cells between the upper and lower layers within the co-culture system, mature osteoclasts in the upper layer were observed to promote the adipogenic differentiation of BMSCs more rapidly in the lower layer. In contrast, inhibiting the activation of ferroptosis within the upper-layer osteoclasts was found to impede the rapid progression of this process (Fig. 9n, o). Simultaneously, by intervening in the p53-FOXO3 pathway within the upper-layer osteoclasts, either by inhibiting p53 or by overexpressing FOXO3, significant inhibition of the rapid progression of adipogenic differentiation in the lower-layer BMSCs was observed (Fig. 9p). Subsequently, cellular senescence was investigated in the later stages of adipogenesis when the majority of cells transitioned into mature adipocytes. When the upper layer was comprised of mature osteoclasts rather than BMDMs alone, the differentiation of lower-layer BMSCs into adipocytes was significantly accelerated (Fig. 9q). Concurrently, this phenomenon was accompanied by pronounced cellular senescence phenotypes, as evidenced by the upregulation of γH2AX expression and significant downregulation of Lamin-B1 expression (Fig. 9r, s). Interference with ferroptosis activation in upper osteoclasts also inhibited aging and adipogenic differentiation of the underlying cells (Fig. 9q–s). Intervention in the p53-FOXO3 pathway within upper-layer osteoclasts also modulated the differentiation of lower-layer BMSCs into mature adipocytes and impeded the rapid progression of the senescent phenotype (Fig. 9t–v). Finally, the results of in vivo animal experiments showed that the injection of a FOXO3-expressing virus into the joint cavity effectively alleviated HFD-induced subchondral bone-aging-related phenotypes and hindered the progression of rapid adipocyte differentiation (Fig. 4p–s). In summary, this study has validated the vicious cycle of mutual promotion between adipogenically differentiated BMSCs and mature osteoclasts. Interventions targeting osteoclast ferroptosis and the p53-FOXO3 axis in both cell types effectively regulated this process.
Cartilage degeneration and abnormal osteoclast differentiation can be alleviated by suppressing the release of SASP
Aging mice, as the most intuitive model, were used to study the intervention and therapeutic potential of inhibiting the release of SASP during the progression of aging, and to clarify the role of ferroptosis in this process. The loss of proteoglycans and significant upregulation of OARSI scores were identified in the articular cartilage of aging mice, as well as a protective effect of JAKi (Supplementary Fig. 9a, b). Correspondingly, the downregulation of Col2a1 and upregulation of Mmp13 in the articular cartilage layer of aging mice also showed significant improvements after the JAKi treatment (Supplementary Fig. 9c–f). Representative micro-CT images also showed that the continuous use of JAKi improved knee joint narrowing and significant osteophyte proliferation around the joints (Supplementary Fig. 9g, h). For the subchondral bone, continuous administration of the JAKi significantly alleviated the heightened osteoclast differentiation induced by aging and concurrently markedly suppressed the rapid progression of ferroptosis in the subchondral bone (Supplementary Fig. 9i–p). This further underscores that targeting obesity, as a distinctive form of aging or in direct aging scenarios, inhibits SASP originating from senescent cells and can markedly impede abnormal remodeling in the subchondral bone and degradation of the cartilage layer.
The combined use of two genetic tools further clarified that the regulatory effect of FOXO3 on the progression of obesity-related OA involves multiple cells, such as chondrocytes, subchondral bone stem cells, and osteoclasts, as well as multiple mechanisms, such as ferroptosis, aging, and adipogenesis
At the end of this article, we aim to further ascertain whether the protective and reversal effects against the progression of obesity-related OA induced by high-fat diet feeding are regulated by FOXO3. To achieve this experimental objective, we subjected mice to continuous HFD for one month and introduced another common gene intervention tool, small interfering RNA, alongside lentivirus. Firstly, the results of safranin O-fast green staining and HE staining suggest that the cartilage degeneration induced by continuous HFD, which was reversed by intra-articular injection of lentivirus expressing FOXO3, reappeared with a deteriorating trend after injection of small interfering RNA targeting FOXO3 (Supplementary Fig. 10a, b, j, k). Furthermore, through IF staining targeting FOXO3 in the articular cartilage layer, we can confirm that this effect is due to changes in FOXO3 levels (Supplementary Fig. 10c, l). Subsequently, the altered expression trends of Col2a1 and Mmp13 further indicate that changes in FOXO3 levels within articular chondrocytes can affect the ECM metabolism of obesity-related osteoarthritic chondrocytes (Supplementary Fig. 10d, e, m, n). Turning our attention to the subchondral bone, we confirmed that the intra-articular injection of lentivirus expressing FOXO3 and small interfering RNA targeting FOXO3 successfully achieved regulation of FOXO3 levels in the subchondral bone marrow (Supplementary Fig. 10f, o). Moreover, this regulatory effect is closely associated with changes in ferroptosis within the subchondral bone marrow, suggesting that FOXO3’s inhibition of subchondral bone remodeling and osteoclast activity relies on its intracellular ferroptotic activity (Supplementary Fig. 10g, h, p, q). It is evident that changes in the expression levels of the adipogenic marker align with alterations in FOXO3 and ferroptosis levels (Supplementary Fig. 10i, r). In summary, these findings allow us to outline the research framework of this article once again, illustrating that the regulatory role of FOXO3 in obesity-related osteoarthritis is comprehensive and multi-layered.
Discussion
OA is a complex, multifactorial disease with several distinct clinical subtypes; of these, obesity-related OA differs because of its association with obesity or metabolic syndrome, low-grade systemic inflammation, early onset, and rapid progression. Furthermore, those with metabolic syndrome and obesity exhibit an increased risk of developing knee osteoarthritis when compared with those without12. However, the cartilage degradation and subchondral bone remodeling with obesity-related OA cannot be explained by the conventional concept of “wear and tear“13.
The increasing prevalence of obesity-related OA necessitates a better understanding of the intricate mechanisms through which abnormalities in lipid metabolism led to joint damage. Oscar et al. identified the aberrant activation of synovial inflammation in affected joints of obesity-related OA patients19. Sung et al. revealed that during this process, the abnormal accumulation of excessive lipid droplets within chondrocytes can lead to their aberrant apoptosis6. These studies examined the pathogenic mechanisms of obesity-related OA from different angles and have attempted to propose potential solutions. However, to date, research specifically investigating the role of subchondral bone marrow cells in the development of obesity-related OA has been limited.
In this study, the joint microenvironment and degenerative changes associated with HFD-induced obesity were investigated from three perspectives: chondrocytes within the cartilage, osteoclasts in the subchondral bone, and mesenchymal stem cells within the bone marrow. Numerous alterations related to the obesity process were delineated in detail. Moreover, the significant therapeutic potential of gene interventions targeting the p53- FOXO3 regulation of cartilage degeneration at the cartilage level and abnormal remodeling in the subchondral bone during obesity-induced degeneration were elucidated. The exciting discovery that HFD-induced obesity is associated with the aging of bone marrow mesenchymal stem cells and their rapid differentiation into adipocytes was also reported. Remarkably, the SASP produced and released by aging adipocytes was able to promote the abnormal remodeling of subchondral bone through the activation of osteoclast ferroptosis. These results contribute to our understanding of the complex interplay between obesity, bone marrow mesenchymal stem cells, and subchondral bone reshaping in OA.
When cells are exposed to DNA damage, p53 is rapidly activated and translocated from the cytoplasm to the nucleus31,32. Inspired by a recent study, we subsequently developed a deeper interest in this discovery21. Research suggests that p53, enriched in the nucleus due to DNA damage, can further phosphorylate AKT, forming a p53-AKT complex in the cell nucleus. Phosphorylated AKT (p-AKT) subsequently phosphorylates multiple downstream effectors, including the third member of the O subfamily of the forkhead transcription factor (FOX) family, FOXO3. FOXO3 has been the subject of significant research within our group, and notable progress has been made21,22,33. Research by White et al. demonstrated that FOXO3a reduces genomic instability in mouse embryonic fibroblasts (MEFs) treated with agents inducing DNA double-strand breaks (DSBs), known as clastogens34. Moreover, FOXO3 can interact with the ATM-Chk2-p53 complex, enhancing complex phosphorylation and inducing the formation of nuclear foci in cells upon DNA damage35. Building upon the foundation of these studies, further investigations were conducted using both cell experiments and animal models to explore the role of the p53- FOXO3 complex in obesity-related OA. The preliminary results suggest that the inhibition of either p53 or AKT could further enhance ECM degradation and chondrocyte apoptosis. This implies that, under the premise of metabolic abnormalities induced by obesity, the protective effects of traditional AKT inhibition in chondrocytes with conventional arthritis may be reversed36,37. Subsequently, lentiviruses were constructed for the overexpression of the FOXO3 gene to further validate that in chondrocytes, this can effectively inhibit apoptosis and elevated expression of MMPs induced by a fatty acid treatment. Animal experiments further confirmed that the overexpression of FOXO3 within the joint cavity of mice could effectively alleviate the degeneration of the articular cartilage promoted by HFD.
Research indicates that there is a complex interrelationship between obesity and iron metabolism38,39. The results of this study have revealed that, compared to the control group, the levels of anti-ferroptosis proteins in the subchondral bones of obesity-induced mice were significantly downregulated. This strongly suggests that ferroptosis may also be involved in regulating the abnormal remodeling process of the subchondral bone in obesity-related OA. In addition, the in vitro cell experiments indicated that during the differentiation of BMDMs into osteoclasts, there was a significant downregulation of anti-ferroptosis proteins accompanied by the accumulation of lipid peroxides, ferrous ions, and ROS. This indicates that an iron overload occurs during OC differentiation. It was thus speculated that ferroptosis may play a role in regulating this process.
Prior to addressing this issue, we first answered a question raised in the Discussion section of a previous study, regarding whether there is indeed an accumulation of bone marrow adipocytes with HFD-induced obesity in mice29. During the process of skeletal ageing, senescent cells can promote the aging of neighboring cells within the bone by secreting SASP40,41. In these aging skeletal systems, a prominent feature is the senescence of BMSCs and the consequent augmentation of adipogenic differentiation. Wei et al. indicated that mesenchymal stem cells can promote the differentiation and activity of osteoclasts by secreting factors such as RANKL42. In addition, Leilei et al. indicated that adipogenic differentiation of BMSCs impedes bone formation43. Our further data also indicate that by co-culturing adipogenically induced mesenchymal stem cells with pre-osteoclasts in vitro, it is possible to effectively induce premature differentiation and maturation of osteoclasts. During this process, there is an elevation in intracellular iron overload and ferroptosis levels within osteoclasts. Intervention in the activation of ferroptosis within osteoclasts or inhibition of the release of SASP from the adipocytes can alleviate the enhanced premature differentiation of osteoclasts. Moreover, after exchanging the upper and lower layers of cells, mature osteoclasts can accelerate the differentiation of BMSCs into fully senescent adipocytes, thereby forming a vicious cycle. During this vicious cycle, interference with p53-FOXO3 may play a regulatory role. Finally, it was confirmed that inhibiting the release of SASP in the skeletal system could rescue the degeneration of the cartilage layer caused by aging. Therefore, whether it is because of the special forms of aging caused by obesity or the direct aging conditions, multidimensional and intercellular communication between joints should be the focus of future studies, as disease progression should be viewed from multiple perspectives.
Methods
Primary cell cultures
According to a previously reported method, primary chondrocytes were extracted from newborn mice44. The extracted primary chondrocytes were cultured in high-glucose DMEM (GIBCO) medium containing 10% FBS and 1% penicillin/streptomycin for subsequent use. After cell adherence, the relevant viruses were introduced, and the success rate of infection was assessed 48 h post-transfection.
For bone marrow-derived macrophages (BMDMs), 6–8-week-old mice were selected for experiments. After euthanasia and dissection of the hind limbs, femurs and tibias were harvested. The bone epiphyses at both ends were removed, and the bone marrow was flushed out using a 1 mL insulin syringe. The harvested cells were cultured either in a 10 cm dish or transferred to a T75 flask. The culture medium comprised complete medium containing 30 ng/mL M-CSF, 10% fetal bovine serum (FBS), and penicillin-streptomycin. After two days, the cells were observed with a microscope, and the medium changed for fresh medium (α-MEM was chosen as the culture medium). After an additional two days, numerous mononuclear macrophages derived from the bone marrow were visible under the microscope, indicating the successful extraction of BMDMs. For further induction of their differentiation into osteoclasts, BMDMs were trypsin-digested, plated, and after cell adhesion, cultured for 5–6 consecutive days in α-MEM complete medium containing 30 ng/mL M-CSF and 100 ng/mL RANKL. The observation of a significant number of ginkgo leaf-like fused cells indicates the initiation of osteoclast fusion. If fusion did not occur by day 8 or 9, the experiment was considered unsuccessful.
The isolation method for bone marrow mesenchymal stem cells (BMSCs) is similar to that for BMDMs; however, the cells obtained through isolation do not require the use of medium containing M-CSF.
Animals
All mice were housed under specific pathogen-free (SPF) conditions and maintained in a controlled environment with a temperature of 23 ± 2 °C, humidity between 40% and 50%, and a consistent 12 h light/dark cycle throughout the experiments. All C57BL/6 mice used were male. Animals were randomly divided into different groups. For mice subjected to a high-fat diet, male C57BL/6 J mice were fed a 60% HFD (D12492, Research Diets, Inc.) to induce obesity. For mice undergoing gene therapy, intra-articular injections were administered before the commencement of the high-fat diet feeding. In the experiments involving aged mice, the mice were 24 months old. For the ruxolitinib (JAKi) treatment, JAKi was dissolved in 10% DMSO and administered via oral gavage at a dose of 60 mg/kg daily for 2 months29. The body conditions and overall appearance of the mice were monitored daily.
All animal experiments were approved by the Institutional Research Ethics Committee of the Shanghai Ninth People’s Hospital. For mice that required a high-fat diet, adult C57BL/6 mice aged 8–12 weeks were selected and fed a high-fat diet containing 60 kcal% adipose for a continuous period of two months. To investigate whether a high-fat diet would accelerate the progression of pre-existing arthritis, medial meniscus destabilization (DMM) surgery was performed as previously described45,46. Briefly, mice were anesthetized, and their medial joint capsules were incised to expose the medial meniscotibial ligament (MMTL). The MMTL was severed using microsurgical scissors to release the ligaments attached to the tibial plateau, thereby destabilizing the medial meniscus. After the surgery, the incision was closed using sutures and disinfected. A sham operation was performed in the control animals in parallel by incising the medial knee joint capsule. For mice requiring gene therapy, lentiviruses expressing FOXO3 were intra-articularly injected.
The aged mice were randomly assigned to experimental groups for the relevant therapeutic experiments. Ruxolitinib (JAKi) was dissolved in 10% DMSO and administered via oral gavage at a dose of 60 mg/kg daily for two months until euthanasia. Young male C57BL/6 J mice aged 6–8 weeks were used as the controls.
Human pathology
Human knee joint samples were obtained from the discarded joints of patients who underwent knee replacement therapy for severe arthritis. All patients included in the article had a BMI index exceeding 28, indicating obesity. Histological staining was used to assess the conditions of both relatively intact and severely damaged areas of the articular cartilage layer. Immunofluorescence experiments were conducted to determine p53 expression levels in the cartilage and subchondral bones. Each participant gave written informed consent. Ethics approved by The Institutional Research Ethics Committee of the Shanghai Ninth People’s Hospital (SH9H-2024-TK349-1).
Histology, immunohistochemistry, and immunofluorescence
The knee joints of mice were fixed in 4% paraformaldehyde, decalcified, dehydrated, and embedded in paraffin. Serial sections were cut and stained with Safranin O and Fast Green (SO&FG), and Hematoxylin and Eosin (H&E) for morphological analysis according to the manufacturer’s instructions. TRAP staining was used to analyze the degree of osteoclast differentiation in subchondral bone. For IF staining, the sections were permeabilized with 0.5% Triton X-100, blocked with 5% BSA for 1 h, and then incubated with primary antibodies overnight at 4 °C. On the second day, after washing three times, the sections were incubated with goat anti-rabbit IgG H&L (Alexa Fluor® 555; Abcam) or goat anti-mouse IgG H&L (Alexa Fluor® 488; Abcam) secondary antibodies (diluted 1:200) for 1 h. For immunohistochemistry, sections were deparaffinized in xylene, followed by hydration with graded ethanol. Subsequently, the sections were treated with 3% H2O2 and 5% bovine serum albumin (BSA) at and incubated overnight with specific antibodies. Afterward, a secondary antibody conjugated with horseradish peroxidase (HRP) was applied to the sections, followed by staining with 3,3’-diaminobenzidine (DAB; Sigma-Aldrich)47. Histological scoring and quantitative IF staining analyses were performed in a double-blinded manner. The Osteoarthritis Research Society International (OARSI) score was based on the SO&FG staining of each sample48. The antibodies used in this study are listed in the Supplementary Table. 1.
Radiographic analysis
Digital X-ray imaging of the right lower limb was conducted following the manufacturer’s guidelines49, employing a 21 lp/mm detector in the anteroposterior axis. The detector provided up to 5 × geometric magnification (Faxitron VersaVision; Faxitron Bioptics LLC, Tucson, AZ, USA).
Micro-computed tomography (micro-CT)
The knee joints of the mice were dissected so that they were free from soft tissue, then fixed them overnight in 70% ethanol and analyzed using by high-resolution μCT (Skyscan1275, Bruker micro-CT, Kontich, Belgium). The scanner was set to a voltage of 50 kVp and a current of 200 µA, with a resolution of 5.7 µm per pixel. Sagittal images of the tibial subchondral bone were used for three-dimensional morphometric analysis. We defined a region of interest covering the entire subchondral bone compartment and used a total of five consecutive images from the medial tibial plateau for three-dimensional reconstruction and analysis.
The three-dimensional model visualization software CTVol was utilized for the analysis of trabecular bone parameters in the epiphysis as well as for the overall reconstruction of the knee joint.
Lentivirus production
HEK293T cells were seeded in 10 cm culture dishes to achieve 85-90% confluency at the time of transfection. Lipofectamine 3000 was used for co-transfection of the transfer plasmid psPAX2 and pDM2.G packaging plasmids. In brief, 9 μg of transfer plasmid, 6 μg of psPAX2, 3 μg of pDM2.G, and 36 μl of p3000 reagent were added to 1.5 ml of serum-free MEM medium. 23 μl of Lipofectamine 3000 reagent was diluted in 1.5 ml of serum-free MEM medium. Subsequently, the diluted plasmids were mixed with Lipofectamine 3000 and incubated at 24 °C for 15 min. After the incubation, 3 ml of culture medium was removed, and 3 ml of transfection mixture was added. The transfection medium was refreshed 6 hours later. The viral supernatant was harvested 24 and 48 h post-transfection. After ultracentrifugation at low temperature (12,000 rpm), the virus was dissolved in 100 ml of PBS, aliquoted, and stored at − 80 °C50.
RNA interference (RNAi)
Negative control and FOXO3 siRNAs were purchased from Genomeditech (Shanghai, China). Plasmid and siRNA transfections were performed using Lipofectamine 3000 (Thermo Fisher Scientific), according to the manufacturer’s instructions.
Cell counting kit-8 (CCK-8) cell proliferation assay
For the cell proliferation assay, cells were infected with lentivirus, seeded onto 96-well plates at a density of 1 × 104 cells/well, and incubated for 24 h. Subsequently, the cells were treated with different chemical reagents. At the designated endpoint, cell proliferation was assessed using the CCK-8 assay (C0037; Beyotime). Absorbance at 450 nm (mean optical density) was measured using an Infinite M200 Pro multimode microplate reader (Tecan Group Ltd., Männedorf, Switzerland).
Annexin V-FITC apoptosis detection
Apoptosis was detected according to the manufacturer’s instructions (C1062S; Beyotime). After inducing apoptosis, the cell culture medium was aspirated, and the cells were washed once with PBS. Then, 195 μL of Annexin V-FITC binding solution was added, along with 5 μL of Annexin V-FITC, and gently mixed. Subsequently, 10 μL of propidium iodide staining solution was added and gently mixed. The samples were then incubated in the dark at room temperature (20–25 °C) for 10–20 min, followed by placement on ice. Light avoidance can be achieved using aluminum foil. Finally, cells were observed under a fluorescence microscope (Leica DMi8).
Cell high-density culture and Alcian blue staining
To evaluate chondrogenic differentiation, 1.5 × 105 chondrocytes were suspended in 10 μL of incomplete DMEM (GIBCO) and seeded as micromasses at the bottom of a 24-well plate. The cells were allowed to adhere for 1 h at 37 °C, following which 0.5 mL of DMEM containing 10 ng/mL ITS and 2% FBS was added. Media were refreshed every other day, and after 14 d, the micromasses were stained with Alcian blue for 24 h at room temperature (RT).
Western blot
Cultured cells were lysed using RIPA lysis buffer supplemented with phosphatase and protease inhibitors. Total protein was quantified using the bicinchoninic acid assay (Thermo Fisher Scientific). Equal amounts of extracted protein (20–30 μg) were then separated using 12% or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto 0.22-μm PVDF membranes (MilliporeSigma, Burlington, MA, USA). The membranes were blocked with 5% BSA-PBS (Beyotime Biotechnology) at room temperature (RT = 25 °C) for 1 h, followed by overnight incubation with primary antibodies at 4 °C. The following day, the membranes were washed with Tris-buffered saline (TBS)−0.1% Tween 20 (TBST) and subsequently incubated with an anti-rabbit IgG (H + L) secondary antibody (cat. no. 5151; DyLight™ 800 4X PEG Conjugate; Cell Signaling Technology; 1:10000) for 1 h at RT in the dark. After washing with TBST, the protein immunoreactivity was detected using an Odyssey Fluorescence Imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Reverse-transcriptase quantitative PCR (RT–qPCR)
Total RNA was isolated from cultured cells using the TRIzol® reagent (Thermo Fisher Scientific, USA) following the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized by reverse transcription using TaKaRa reverse transcription reagents (TaKaRa Bio Inc., Kusatsu, Shiga, Japan), and RT-qPCR was conducted using Real-Time PCR Mix (TaKaRa Bio Inc.). The expression levels of target genes were determined utilizing the 2-ΔΔCq method51, with GAPDH serving as the internal reference control. The primers used were listed in Supplementary Table. 2.
Senescence β-Galactosidase Staining
For cells cultured in 6-well plates, the culture medium was aspirated, the cells rinsed once with PBS, and then 1 mL of β-galactosidase staining fixative added (C0602, Beyotime). The mixture was then incubated at room temperature for 15 min. The volume of the fixative and subsequent solutions was adjusted proportionally to the other types of culture plates. The fixative was removed, and the cells were washed three times with PBS for 3 min each. After the final PBS wash, 1 mL of the staining working solution was added to each well. Working solutions were prepared according to the formula provided in the reagent kit. Cells where then incubate at 37 °C overnight and observed under a standard optical microscope.
Tartrate-resistant acid phosphatase (TRAP) staining assay
BMDMs were plated in 96-well plates at a density of 1 × 104 cells per well and cultured with M-CSF (30 ng/mL) and RANKL (100 ng/mL), either in the presence of external chemical stimuli or after pre-infection with a FOXO3-expressing lentivirus. The culture medium was renewed every 2 days until osteoclast (OCs) formation was observed on day 5. Following a 20 min fixation with 4% paraformaldehyde (PFA), cells were stained with TRAP staining solution at 37 °C for 1 h. OCs were identified as TRAP-positive cells with more than three nuclei. Images were captured using an optical microscope (Olympus). The OC number and area were then quantified using ImageJ software (NIH, Bethesda, MD, USA).
Cell co-culture and TRAP staining
A dual co-culture assay and TRAP staining were conducted to study the osteoclasts. Preosteoclasts derived from murine bone marrow macrophages (1 × 105 cells/well) were directly seeded into the wells of 12-well co-culture plates, while chondrocytes (5 × 104 cells/well) were seeded into Transwell® cell culture inserts (Costar) within the co-culture plates. The following day, chondrocytes adhered to the membrane surface of the inserts, and various stimuli, such as chemical agents, inflammatory cytokines, or viruses, were added to the top of the chondrocyte layer. The upper co-culture assays were conducted in DMEM/high-glucose medium supplemented with 10% FBS, with media changes occurring every 2 days. The lower chambers were maintained using α-MEM complete medium, supplemented with M-CSF (30 ng/mL) and RANKL (100 ng/mL). On day 4, TRAP staining of osteoclasts was performed, and TRAP-positive multinucleated cells were identified as mature osteoclasts and quantified.
FerroOrange staining
Pre-cultured osteoclasts were seeded in a fluorescent culture dish and incubated overnight at 37 °C with 5% CO2. The following day, the cells were washed three times with serum-free medium and treated with a solution containing drugs, recombinant proteins, or lentiviruses. The cells were then incubated at 37 °C with 5% CO2. FerroOrange working solution (1 μmol/L; DOJINDO) was added to the cells and incubated at 37 °C with 5% CO2. The cells were then observed under a fluorescence microscope without washing.
Lipid peroxidation detection
Osteoclasts were seeded into specific culture dishes or well plates. After removing the supernatant, cells were washed once with a serum-free medium. Then, an appropriate amount of Liperfluo (1 mM; DOJINDO) working solution was added and the cells were incubated at 37 °C for 30 min. After removing the supernatant, the cells were washed twice with a serum-free medium. Finally, cells were observed under a fluorescence microscope.
Intracellular reactive oxygen species (ROS) detection
The intracellular ROS content was measured using DCFH-DA and confocal scanning microscopy. DCFH-DA was diluted 1:1000 with serum-free medium to a final concentration of 10 μM/L. After removing the culture medium, an appropriate volume of the diluted DCFH-DA was added to each wells. The volume added was sufficient to cover the cells. The cells were then incubated at 37 °C for 20 min in a cell culture incubator. After incubation, the cells were washed thrice with serum-free medium to remove any unincorporated DCFH-DA. For samples with in situ-loaded probes, a laser confocal microscope can be used for direct observation.
Adipogenic induction of BMSCs and co-culture with osteoclasts
BMSCs were isolated using the same method as was used for the BMDMs. Freshly isolated single-cell suspensions were plated at a density of 5 × 105 cells in 10 cm plates using BMSCs growth media (α-MEM supplemented with 10% FBS, 100 μ·mL−1 penicillin, 100 μg·mL−1 streptomycin). Cells were allowed to proliferate for 3 days prior to aspirating the supernatant, rinsing with PBS three times, and changing the medium three times per week for 2 weeks. Upon reaching 80% confluence as observed under a microscope, the cells were trypsinized and seeded either on the upper or lower layer of a cell co-culture plate, as required, for the osteoclast co-cultures.
For adipogenic differentiation, BMSCs were cultured in adipogenic media (mesenchymal stem cell adipogenic differentiation and staining kit, Meilunbio) for a specified number of days, with medium changes three times a week.
Oil Red O
After induction of adipogenic differentiation, the supernatant was discarded, each well was washed with PBS 2–3 times, and 2 mL of 4% paraformaldehyde solution was added to each well for 30 min to allow for fixation. Next, the Oil Red O staining working solution was prepared by mixing saturated Oil Red O dye with distilled water in a 3:2 ratio, stirring well, and filtering through neutral filter paper. The 4% paraformaldehyde was removed, washed with PBS 2 times, and 1 mL of Oil Red O staining working solution was added per well for staining, with a staining time of 30 min. The Oil Red O staining working solution was removed, and the cells were washed with PBS 2–3 times. Then 1 mL of PBS was added per well, and the lipid-staining effects observed under an inverted microscope. Cells undergoing adipogenic differentiation exhibited orange-to-red lipid droplets, demonstrating the typical characteristics of adipogenic differentiation.
Statistical analysis
Statistical analysis of all data was conducted using GraphPad Prism software (version 8.0). The results are presented as the mean ± s.d. Comparisons between two groups were performed using an unpaired two-sided Student’s t test (p < 0.05 was deemed significant). One- or two-way analysis of variance was used for comparisons involving multiple conditions. Specific values of n are provided in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in the article and Supplementary Information files. Source data are provided in this paper.
References
Felson, D. T. Clinical practice. Osteoarthritis of the knee. N. Engl. J. Med. 354, 841–848 (2006).
Katz, J. N., Arant, K. R. & Loeser, R. F. Diagnosis and treatment of hip and knee osteoarthritis: A review. Jama 325, 568–578 (2021).
Conaghan, P. G., Kloppenburg, M., Schett, G. & Bijlsma, J. W. Osteoarthritis research priorities: a report from a EULAR ad hoc expert committee. Ann. Rheum. Dis. 73, 1442–1445 (2014).
Lotz, M. Cytokines in cartilage injury and repair. Clin Orthop Relat Res, S108–S115, https://doi.org/10.1097/00003086-200110001-00011 (2001).
Choi, W. S. et al. The CH25H-CYP7B1-RORalpha axis of cholesterol metabolism regulates osteoarthritis. Nature 566, 254–258 (2019).
Lee, S. W. et al. Dietary fat-associated osteoarthritic chondrocytes gain resistance to lipotoxicity through PKCK2/STAMP2/FSP27. Bone Res. 6, 20 (2018).
Alrubaian, F. & Mulla, Z. Governments policy measures to address obesity among adults: a scoping review of the global evidence. Lancet 402, S20 (2023).
Liu, M., Jin, F., Yao, X. & Zhu, Z. Disease burden of osteoarthritis of the knee and hip due to a high body mass index in China and the USA: 1990-2019 findings from the global burden of disease study 2019. BMC Musculoskelet. Disord. 23, 63 (2022).
Zhao, G. et al. Global Burden of osteoarthritis associated with high body mass index in 204 countries and territories, 1990-2019: findings from the Global Burden of Disease Study 2019. Endocrine 79, 60–71 (2023).
Arden, N. K. et al. Non-surgical management of knee osteoarthritis: comparison of ESCEO and OARSI 2019 guidelines. Nat. Rev. Rheumatol. 17, 59–66 (2021).
Fernandes, L. et al. EULAR recommendations for the non-pharmacological core management of hip and knee osteoarthritis. Ann. Rheum. Dis. 72, 1125–1135 (2013).
Sowers, M. et al. Knee osteoarthritis in obese women with cardiometabolic clustering. Arthritis Rheum. 61, 1328–1336 (2009).
Zhuo, Q., Yang, W., Chen, J. & Wang, Y. Metabolic syndrome meets osteoarthritis. Nat. Rev. Rheumatol. 8, 729–737 (2012).
Hu, W., Chen, Y., Dou, C. & Dong, S. Microenvironment in subchondral bone: predominant regulator for the treatment of osteoarthritis. Ann. Rheum. Dis. 80, 413–422 (2021).
Zhen, G. et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 19, 704–712 (2013).
Zhu, S. et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J. Clin. Invest. 129, 1076–1093 (2019).
Wunderlich, C. M. et al. Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat. Commun. 9, 1646 (2018).
Liao, Y. et al. Interleukin-6 signaling mediates cartilage degradation and pain in posttraumatic osteoarthritis in a sex-specific manner. Sci. Signal 15, eabn7082 (2022).
Alvarez-Garcia, O., Rogers, N. H., Smith, R. G. & Lotz, M. K. Palmitate has proapoptotic and proinflammatory effects on articular cartilage and synergizes with interleukin-1. Arthritis Rheumatol. 66, 1779–1788 (2014).
Seo, J. et al. Weight-bearing activity impairs nuclear membrane and genome integrity via YAP activation in plantar melanoma. Nat. Commun. 13, 2214 (2022).
Chen, M. et al. A p53-phosphoinositide signalosome regulates nuclear AKT activation. Nat. Cell Biol. 24, 1099–1113 (2022).
Hoxhaj, G. & Manning, B. D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20, 74–88 (2020).
Tran, H. et al. DNA Repair pathway stimulated by the forkhead transcription factor FOXO3a through the gadd45 protein. Science 296, 530–534 (2002).
Yang, X. et al. Regulation of VKORC1L1 is critical for p53-mediated tumor suppression through vitamin K metabolism. Cell Metab 35, 1474–1490 (2023).
Zhao, C. et al. Forkhead box O3 attenuates osteoarthritis by suppressing ferroptosis through inactivation of NF-κB/MAPK signaling. J. Orthop. Transl. 39, 147–162 (2023).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Liu, X. et al. Oxylipin-PPARgamma-initiated adipocyte senescence propagates secondary senescence in the bone marrow. Cell Metab. 35, 667–684 (2023).
van Deursen, J. M. The role of senescent cells in ageing. Nature 509, 439–446 (2014).
O’Brate, A. & Giannakakou, P. The importance of p53 location: nuclear or cytoplasmic zip code? Drug Resist. Updat. 6, 313–322 (2003).
Chen, S. H., Forrester, W. & Lahav, G. Schedule-dependent interaction between anticancer treatments. Science 351, 1204–1208 (2016).
Manning, B. D. & Toker, A. AKT/PKB Signaling: Navigating the network. Cell 169, 381–405 (2017).
White, R. R. et al. FOXO3a acts to suppress DNA double-strand break-induced mutations. Aging Cell 19, e13184 (2020).
Chung, Y. M. et al. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nat. Commun. 3, 1000 (2012).
Xie, J. et al. Sustained Akt signaling in articular chondrocytes causes osteoarthritis via oxidative stress-induced senescence in mice. Bone Res. 7, 23 (2019).
Xue, J. F., Shi, Z. M., Zou, J. & Li, X. L. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed. Pharmacother. 89, 1252–1261 (2017).
Tan, S. K. et al. Obesity-dependent adipokine chemerin suppresses fatty acid oxidation to confer ferroptosis resistance. Cancer Discov. 11, 2072–2093 (2021).
Zhang, S. et al. Ferroptosis increases obesity: Crosstalk between adipocytes and the neuroimmune system. Front. Immunol. 13, 1049936 (2022).
Admasu, T. D., Rae, M. & Stolzing, A. Dissecting primary and secondary senescence to enable new senotherapeutic strategies. Ageing Res. Rev. 70, 101412 (2021).
Teo, Y. V. et al. Notch signaling mediates secondary senescence. Cell Rep. 27, 997–1007 (2019).
Yu, W. et al. Bone marrow adipogenic lineage precursors promote osteoclastogenesis in bone remodeling and pathologic bone loss. J. Clin. Invest. 131, https://doi.org/10.1172/jci140214 (2021).
Zhong, L. et al. Single cell transcriptomics identifies a unique adipose lineage cell population that regulates bone marrow environment. Elife 9, https://doi.org/10.7554/eLife.54695 (2020).
Gosset, M., Berenbaum, F., Thirion, S. & Jacques, C. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 3, 1253–1260 (2008).
Glasson, S. S., Blanchet, T. J. & Morris, E. A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 15, 1061–1069 (2007).
Chen, D. et al. Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res. 5, 16044 (2017).
Wu, Y. et al. Metabolite asymmetric dimethylarginine (ADMA) functions as a destabilization enhancer of SOX9 mediated by DDAH1 in osteoarthritis. Sci. Adv. 9, eade5584 (2023).
Laverty, S., Girard, C. A., Williams, J. M., Hunziker, E. B. & Pritzker, K. P. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the rabbit. Osteoarthr. Cartil. 18, S53–S65 (2010).
Sohara, Y. et al. Lytic bone lesions in human neuroblastoma xenograft involve osteoclast recruitment and are inhibited by bisphosphonate. Cancer Res. 63, 3026–3031 (2003).
Jing, Y. et al. Genome-wide CRISPR activation screening in senescent cells reveals SOX5 as a driver and therapeutic target of rejuvenation. Cell Stem Cell 30, 1452–1471 (2023).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Acknowledgements
This research was supported by the National Key Research and Development Program of China (2023YFB3813000 to X.W.), National Natural Science Foundation of China (81871791 to X.W.), National Natural Science Foundation of China (81902194 to L.W.), and the Cross-disciplinary Research Fund of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (JYJC202229 to X.W.).
Author information
Authors and Affiliations
Contributions
C.Z. designed and performed most of the experiments, analyzed and interpreted the data, and wrote the manuscript. K.K. contributed critical reagents and collected clinical specimens. P.L., X.C., K.R., and P.Z. helped with some experiments. L.W. and X.W. contributed to funding, oversaw protocol logistics, and supervised the entire study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Amit Sharma, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, C., Kong, K., Liu, P. et al. Regulating obesity-induced osteoarthritis by targeting p53-FOXO3, osteoclast ferroptosis, and mesenchymal stem cell adipogenesis. Nat Commun 16, 4532 (2025). https://doi.org/10.1038/s41467-025-59883-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59883-z
