
Abstract
The platelet-derived growth factor receptor (PDGFR) family of receptor tyrosine kinases consists of two receptors, PDGFRα and PDGFRβ, that homodimerize and heterodimerize upon ligand binding. Here, we tested the hypothesis that differential internalization and trafficking dynamics of the various PDGFR dimers underlie differences in downstream intracellular signaling and cellular behavior. Using a bimolecular fluorescence complementation approach, we demonstrated that PDGFRα/β heterodimers are rapidly internalized into early endosomes. We showed that PDGFRα/β heterodimer activation does not induce downstream phosphorylation of ERK1/2 and significantly inhibits cell proliferation. Further, we identified MYO1D as a protein that preferentially binds PDGFRα/β heterodimers and demonstrated that knockdown of MYO1D leads to retention of PDGFRα/β heterodimers at the plasma membrane, increased phosphorylation of ERK1/2 and increased cell proliferation. Collectively, our findings impart valuable insight into the molecular mechanisms by which specificity is introduced downstream of PDGFR activation to differentially propagate signaling and generate distinct cellular responses.
Similar content being viewed by others
Introduction
The platelet-derived growth factor receptor (PDGFR) family of receptor tyrosine kinases (RTKs) allows cells to communicate with one another by binding to growth factors at the plasma membrane and activating intracellular signaling pathways to elicit responses such as migration, proliferation, survival and differentiation1,2. In humans, aberrant PDGF signaling has been shown to cause differences in craniofacial development, cancer, vascular disorders and fibrotic diseases3,4,5,6,7,8,9,10,11,12. The mammalian PDGF family is composed of four dimeric ligands, PDGF-AA, PDGF-BB, PDGF-CC and PDGF-DD, which variously signal through two receptors, PDGFRα and PDGFRβ. These receptors possess an extracellular ligand-binding domain harboring five immunoglobulin-like loops, a single transmembrane domain and an intracellular domain containing a split, catalytic tyrosine kinase13. PDGFRα and PDGFRβ share the highest amino acid homology in the kinase domains, with significantly reduced homology in the extracellular and interkinase domains14. Upon ligand binding, the PDGFRs dimerize to form PDGFRα homodimers, PDGFRα/β heterodimers or PDGFRβ homodimers14,15,16,17,18. Through an undetermined mechanism, dimerization promotes tyrosine kinase activity, resulting in the trans-autophosphorylation of intracellular tyrosine residues16,19. Signaling molecules and adapter proteins containing Src homology 2 phosphotyrosine recognition motifs subsequently bind to specific phosphorylated residues in the receptors to initiate downstream intracellular signaling cascades1,13.
The PDGFRs play roles in developmental and postnatal contexts in the mouse. Both PDGFRα and PDGFRβ signaling are active in the nervous system, neural crest, kidney and gonads. PDGFRα signaling additionally functions in the thymus, lung, gut, adrenal gland and somites, while PDGFRβ signaling is also active in vascular cells2. Based on in vivo mouse knockout phenotypes, the ligands PDGF-AA and PDGF-CC have been shown to solely activate PDGFRα signaling during development, while PDGF-BB activates PDGFRβ signaling20,21,22,23,24. While this data is often interpreted as representing the functional roles of PDGFR homodimers, compelling evidence has emerged indicating that PDGFRα/β heterodimers also form during development. Analysis of autophosphorylation mutant knockin alleles in mice suggested that PDGFRβ is able to compensate for the loss of PI3K signaling through PDGFRα upon heterodimer formation25. Embryos homozygous for an allele (PdgfraPI3K) in which PDGFRα is unable to bind PI3K26 die perinatally and exhibit palatal clefting, among other abnormalities18,25. However, these embryos do not display the full range of skeletal defects, such as overt facial clefting, observed in Pdgfra-null embryos21. In support of PDGFRα/β heterodimer formation, whereas PdgfrbPI3K/PI3K mice do not exhibit craniofacial defects27, PdgfraPI3K/PI3K;PdgfrbPI3K/PI3K double-homozygous mutant embryos in which PI3K signaling cannot be engaged through PDGFRα/β heterodimers phenocopy Pdgfra-null embryos25. More recently, we and others have demonstrated that PDGFRα and PDGFRβ genetically and physically interact in the murine embryonic mesenchyme, primarily in response to PDGF-BB ligand18,28,29,30. However, definitive proof of the formation of active PDGFRα/β heterodimers has remained elusive.
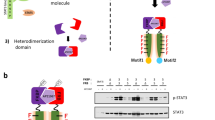
Technical limitations had previously prevented an unbiased analysis of PDGFR dimer-specific dynamics, as antibodies cannot differentiate between receptors present as monomers or engaged in homodimers versus heterodimers. Further, ligand identity alone cannot inform dimer-specific conclusions, as some PDGF ligands, especially PDGF-BB, are promiscuous and can result in the formation of multiple dimers18. To overcome these limitations, we recently adapted a bimolecular fluorescence complementation (BiFC)31,32 approach previously used with the ErbB family of RTKs33. We cloned plasmids expressing C-terminal fusions of each PDGFR with BiFC fragments corresponding to the N-terminal (V1) or C-terminal (V2) regions of the Venus fluorescent protein32. The individual N- and C-terminal fragments are non-fluorescent when bound to monomeric receptors. However, upon receptor dimerization, the N- and C-terminal fragments colocalize, resulting in a functional Venus protein that can be visualized by fluorescence microscopy or purified biochemically using a GFP-Trap nanobody that has an epitope spanning V1 and V233. Using this approach, we demonstrated differences in the timing and extent of PDGFRα homodimer and PDGFRβ homodimer dimerization, activation, internalization, trafficking and downstream signaling34. While these findings provided considerable insight into mechanisms by which specificity is introduced downstream of PDGFR activation, the protein interactions that mediate the internalization and trafficking of the various PDGFR dimers are incompletely understood.
Here, we employ the same BiFC approach to examine PDGFRα/β heterodimers, revealing significant differences from what we previously observed for the PDGFR homodimers34. Further, we generate the first PDGFR dimer-specific interactome, identifying proteins that differentially bind the three PDGFR dimers and contribute to receptor internalization following activation at the plasma membrane. Combined, these findings substantially expand our understanding of the mechanisms by which the various PDGFR dimers propagate downstream signaling to generate distinct cellular outputs.
Results
Generation and validation of a PDGFRα/β-BiFC cell line
To investigate PDGFRα/β-specific dynamics, we stably integrated PDGFRα-V1 and PDGFRβ-V2 sequences (Supplementary Fig. 1A) into the human-derived HCC15 cell line via lentiviral transduction. We chose this cell line due to its mesenchymal expression profile, lack of PDGFRA and PDGFRB expression, minimal expression of PDGF ligands, and a spread out morphology that allows for examination of trafficking events35,36. Quantitative real-time PCR (qRT-PCR) revealed that the transcripts encoding PDGFRA and PDGFRB were expressed at similar levels in the PDGFRαV1/βV2 heterodimer cell line (Supplementary Fig. 1B). Further, we demonstrated that both PDGFRα and PDGFRβ proteins were expressed and migrated at the expected masses via western blotting (Supplementary Fig. 1C).
To confirm the Venus complementation event upon exogenous ligand treatment, we starved the cells for 24 h in HITES medium lacking any growth factors, photobleached and stimulated the cells with PDGF-BB ligand for 5 min. We confirmed that 10 ng/ml PDGF-BB stimulation resulted in the greatest Venus fluorescence intensity in our PDGFRαV1/βV2 heterodimer cell line as compared to the other PDGF ligands and to other PDGF-BB ligand concentrations (Supplementary Fig. 2A, B). We observed little to no Venus expression in the absence of ligand treatment (Fig. 1A) and robust Venus expression surrounding the nucleus and at the periphery of the cell following ligand stimulation (Fig. 1B). Quantification of fluorescence intensity demonstrated a significant increase in Venus intensity between no ligand treatment and 5 min of PDGF-BB treatment (Fig. 1C). The extent of colocalization of the Venus signal with signals from anti-PDGFRα and anti-PDGFRβ antibodies was comparable in the PDGFRαV1/βV2 heterodimer cell line (Fig. 1D). The observed Pearson’s correlation coefficient values less than 1 indicated that the presence of the BiFC fragments alone was not driving heterodimerization (Fig. 1D). The regions of receptor expression that were not Venus positive (Fig. 1E, F) likely represent monomeric receptors and/or potential PDGFRαV1/αV1 and especially PDGFRβV2/βV2 homodimers, which would not be expected to fluoresce.

A, B Venus expression (white or green) as assessed by fluorescence analysis of HCC15 cells transduced with PDGFRα-V1 and PDGFRβ-V2 in the absence (A) or presence (B) of 10 ng/ml PDGF-BB ligand for 5 min. Nuclei were stained with DAPI (blue; A, B). Images representative of three experiments. Scale bars = 20 μm. C Scatter dot plot depicting fluorescence intensity for the PDGFRαV1/βV2 heterodimer cell line in the absence or presence of 10 ng/ml PDGF-BB ligand for 5 min. Data are mean ± s.e.m. P = 0.03 (two-tailed, paired t-test). Colored circles correspond to independent experiments. Summary statistics from biological replicates consisting of independent experiments (large circles) are superimposed on top of data from all cells; n = 38 technical replicates for biological replicate 1, n = 40 technical replicates for biological replicates 2 and 3. D Scatter dot plots depicting Pearson’s correlation coefficient of the PDGFRαV1/βV2 heterodimer cell line Venus signal with an anti-PDGFRα antibody and an anti-PDGFRβ antibody signal following 10 ng/ml PDGF-BB ligand stimulation for 5 min. Data are mean ± s.e.m. Colored circles correspond to independent experiments. Summary statistics from biological replicates consisting of independent experiments (large circles) are superimposed on top of data from all cells; n = 25 technical replicates across each of three biological replicates. E, F PDGFRα (E) and PDGFRβ (F) antibody signal (white or magenta; E, F) and/or Venus expression (white or green; E, F) as assessed by (immuno)fluorescence analysis of the PDGFRαV1/βV2 heterodimer cell line. Insets in E and F are regions where white arrows are pointing. Nuclei were stained with DAPI (blue; E, F). White arrows denote colocalization; white outlined arrows denote lack of colocalization. Images representative of three experiments. Scale bars = 20 μm (main images), 3 μm (insets). Source data are provided as a Source Data file.
PDGFRα/β heterodimers dimerize within several minutes
To confirm the Venus complementation event upon exogenous ligand treatment biochemically, we stimulated the cells with PDGF-BB ligand in a time course from 2 min to 4 h. Lysates were immunoprecipitated with the GFP-Trap nanobody, which should only pull down the dimerized receptors, and subjected to western blotting for PDGFRα and PDGFRβ. The immunoprecipitation signal was normalized to the whole-cell lysate signal for each receptor. We found that the receptors heterodimerize relatively quickly, with peak dimerization following 5 min of ligand treatment for both PDGFRα and PDGFRβ (Fig. 2A, B). As would be expected for heterodimers, the relative induction values for the two receptors were not significantly different at any timepoint of ligand stimulation (Fig. 2B). The presence of signal in the absence of ligand stimulation was in line with our previous findings for both PDGFR homodimers34 and a prior in silico study predicting an inactive dimerization state for PDGFRα37.

A Immunoprecipitation (IP) of dimerized PDGFRα/β receptors with GFP-Trap nanobody from cells that were unstimulated or treated with 10 ng/ml PDGF-BB ligand for 2 min to 4 h followed by western blotting (WB) with an anti-PDGFRα or an anti-PDGFRβ antibody. WCL, whole-cell lysates. Images representative of three experiments. Uncropped blots in Source Data. B Line graph depicting quantification of band intensities from n = 3 biological replicates as in A. Data are mean ± s.e.m. P = 0.05 for PDGFRβ 2 min, P = 0.02 for PDGFRβ 5 min (two-tailed, ratio paired t-test). C Immunoprecipitation of dimerized PDGFRα/β receptors with GFP-Trap nanobody from cells that were unstimulated or treated with 10 ng/ml PDGF-BB ligand for 2 min to 4 h followed by western blotting with an anti-phospho (p)-PDGFR antibody. Images representative of three experiments. Uncropped blots in Source Data. D Line graph depicting quantification of band intensities from n = 3 biological replicates as in C. Data are mean ± s.e.m. P = 0.02 for 5 min, P = 0.04 for 15 min, P = 0.01 for 30 min, P = 0.02 for 60 min (two-tailed, ratio paired t-test). Source data are provided as a Source Data file.
We next examined activation of the receptors by assessing receptor autophosphorylation. Cells were stimulated with PDGF-BB ligand in a time course from 2 min to 4 h. Whole-cell lysates were immunoprecipitated with the GFP-Trap nanobody and subjected to western blotting with an anti-phospho-PDGFR antibody, which recognizes a phosphorylated tyrosine residue within the tyrosine kinase domains of both PDGFRα and PDGFRβ. Consistent with previous findings which demonstrated that both ligand binding and dimerization are required for receptor activation19,34,38, we observed minimal phospho-PDGFR signal in the absence of ligand stimulation (Fig. 2C). However, the receptors were significantly autophosphorylated in response to ligand treatment, with peak activation following 5 min of PDGF-BB stimulation (Fig. 2C, D). The anti-phospho-PDGFR blots possessed a single band representing the glycosylated, phosphorylated versions of the receptors upon ligand treatment34.
PDGFRα/β heterodimers are rapidly internalized
Following dimerization and activation at the cell membrane in response to ligand binding, PDGFRs are internalized and trafficked through the early endosome before being trafficked to the lysosome via late endosomes for degradation or recycled to the membrane via recycling endosomes for continued signaling34,39,40,41,42,43. To investigate the trafficking dynamics of PDGFRα/β heterodimers in response to ligand treatment, we examined colocalization of the Venus signal in our PDGFRαV1/βV2 cell line with markers of various subcellular compartments. We found that PDGFRα/β heterodimers were rapidly internalized, as indicated by a significant decrease in colocalization with the plasma membrane marker Na+/K+-ATPase44 from the 1 min to 5 min ligand treatment timepoints (Fig. 3A–C). Further, analysis of Venus colocalization with the early endosome marker RAB545 demonstrated that PDGFRα/β heterodimers were internalized into early endosomes, where they dwelled at relatively stable levels from 2 min to 30 min following ligand treatment (Fig. 3D–F). PDGFRα/β heterodimers were primarily found in signaling endosomes during this time based on colocalization values for the signaling endosome marker APPL146 at 2 min and 30 min of ligand treatment (Fig. 3G–I) compared to those for the non-signaling endosome marker EEA147 at these same timepoints (Supplementary Fig. 3A–C). Following trafficking to the early endosomes, the PDGFRα/β heterodimers were subsequently trafficked to RAB4-positive rapid recycling endosomes45 (Supplementary Fig. 3D, E), RAB11-positive slow recycling endosomes45 (Supplementary Fig. 3F, G) and more so to RAB7-positive late endosomes45 (Supplementary Fig. 3H, I). Colocalization of Venus signal with Na+/K+-ATPase at 60 min and 90 min of ligand treatment (Supplementary Fig. 3J, K) was comparable to that following 1 min of ligand treatment described above (Fig. 3A, B), demonstrating that PDGFRα/β heterodimers can repopulate the plasma membrane following ligand stimulation.

A, D, G Scatter dot plots depicting Pearson’s correlation coefficient of the PDGFRαV1/βV2 heterodimer cell line Venus signal with an anti-Na+/K+-ATPase antibody (A), an anti-RAB5 antibody (D) or an anti-APPL1 antibody (G) signal following 10 ng/ml PDGF-BB ligand stimulation from 1–5 min (A) or 2–30 min (D, G). Data are mean ± s.e.m. P = 0.001 for Na+/K+-ATPase 1 vs 5 min, P = 0.02 for RAB5 2 vs 10 min, P = 0.04 for RAB5 10 vs 30 min (two-tailed, unpaired t-test with Welch’s correction). Colored circles correspond to independent experiments. Summary statistics from biological replicates consisting of independent experiments (large circles) are superimposed on top of data from all cells; exact number (n) of technical replicates across each of three biological replicates can be found in Source Data. B, C, E, F, H, I Na+/K+-ATPase antibody signal (white or magenta; B, C), RAB5 antibody signal (white or magenta; E, F) or APPL1 antibody signal (white or magenta; H, I) and/or Venus expression (white or green; B, C, E, F, H, I) as assessed by (immuno)fluorescence analysis of the PDGFRαV1/βV2 heterodimer cell line. Insets in B, C, E, F, H and I are regions where white arrows are pointing. Nuclei were stained with DAPI (blue; B, C, E, F, H, I). White arrows denote colocalization; white outlined arrows denote lack of colocalization. Images representative of three experiments. Scale bars = 20 μm (main images), 3 μm (insets). Source data are provided as a Source Data file.
To test the possibility of PDGFRα/β heterodimer recycling, we performed flow cytometry, assessing the percentage of cells with expression of Venus and surface expression of PDGFRα at multiple timepoints of PDGF-BB ligand stimulation in our PDGFRαV1/βV2 heterodimer cell line. As PDGF-BB ligand does not induce formation of PDGFRα homodimers18, these double-positive cells are a read-out of PDGFRα/β heterodimer formation at the plasma membrane. These experiments were performed in the presence of cycloheximide to inhibit translation of new receptors. We observed a significant increase in the percentage of double-positive cells between 0 min and 1 min of ligand treatment (Supplementary Fig. 4A, B and E), which decreased to baseline levels by 5 min (Supplementary Fig. 4C, E). The percentage of double-positive cells was further decreased, though not significantly so, at 90 min of ligand treatment (Supplementary Fig. 4D, E). Collectively, these data indicate that PDGFRα/β heterodimers are rapidly internalized into signaling endosomes, where they dwell for extended lengths of time, before being trafficked for recycling and, more likely, degradation.
Phospho-ERK1/2 is not induced by PDGFRα/β heterodimers
To examine the effects of the above PDGFRα/β heterodimer-specific trafficking dynamics on downstream intracellular signaling, we performed a time course of ligand stimulation from 2 min to 4 h followed by western blotting of whole-cell lysates for phosphorylation of two effector molecules downstream of PDGFR activation, ERK1/2 and AKT18,34,48. Parental HCC15 cells have a heterozygous mutation in NRAS that results in constitutive activation of the protein49 and increased phosphorylation of ERK1/2 at baseline in the absence of exogenous PDGF ligand stimulation34. Baseline ERK1/2 phosphorylation was not different between parental HCC15 and PDGFRαV1/βV2 cell lines (Supplementary Fig. 5A, B). PDGFRα/β heterodimer activation did not induce downstream phosphorylation of ERK1/2 (Fig. 4A, B). Alternatively, PDGFRα/β heterodimer activation led to a transient phospho-AKT response, with a peak at 5 min of ligand stimulation and a return to near baseline levels by 15 min of PDGF-BB treatment that remained stable throughout the rest of the time course (Fig. 4C, D).

A, C Western blot (WB) analysis of whole-cell lysates (WCL) from the PDGFRαV1/βV2 heterodimer cell line following a time course of 10 ng/ml PDGF-BB ligand stimulation from 2 min to 4 h with anti-phospho (p)-ERK1/2 (A) or anti-phospho (p)-AKT (C) antibodies. Images representative of three experiments. Uncropped blots in Source Data. B, D Line graphs depicting quantification of band intensities from n = 3 biological replicates as in A and C. Data are mean ± s.e.m. P = 0.005 for p-AKT 5 min (two-tailed, ratio paired t-test). E, F Colony growth in soft agar anchorage-independent growth assays for the HCC15 cell line (E) or the PDGFRαV1/βV2 heterodimer cell line (F) after 10 days in RPMI growth medium in the absence or presence of 10 ng/ml PDGF-BB ligand in six-well plate wells. Images representative of six experiments. G, H Scatter dot plots depicting quantification of colony count (G) or colony area (H) from n = 6 biological replicates as in E and F. Data are mean ± s.e.m. P = 0.0002 for colony count HCC15 + PDGF-BB vs PDGFRα/β + PDGF-BB, P = 0.0004 for colony count PDGFRα/β vs PDGFRα/β + PDGF-BB, P = 0.009 for colony area HCC15 vs PDGFRα/β, P < 0.0001 for colony area HCC15 + PDGF-BB vs PDGFRα/β + PDGF-BB (two-tailed, paired t-test within each cell line and a two-tailed, unpaired t-test with Welch’s correction between each cell line). Colored circles correspond to independent experiments. Source data are provided as a Source Data file.
We then assessed phosphorylation of ERK1/2 and AKT in parallel across all three of our PDGFR-BiFC cell lines, including over a shorter time course on the same blots (Supplementary Fig. 6A–D) and over the full 4 h time course in separate blots for each dimer (Supplementary Fig. 6E–L, Supplementary Fig. 7). We demonstrated that PDGFRα homodimer signaling did not induce downstream phosphorylation of ERK1/2 nor AKT (Supplementary Fig. 6E, H, I and L). Alternatively, PDGFRβ homodimer signaling led to significant phospho-ERK1/2 and phospho-AKT responses (Supplementary Fig. 6G, H, K and L). Combined, these results demonstrate that PDGFRβ homodimers are the only PDGFR dimers that generate a phospho-ERK1/2 response in this setting, while phospho-AKT is induced downstream of PDGFRβ homodimers and, to a lesser extent, PDGFRα/β heterodimers.
PDGFRα/β heterodimer activation inhibits cell proliferation
Proliferation is increased downstream of both PDGFRα and PDGFRβ signaling in settings where both receptors are active30,50,51. To determine the effect of PDGFRα/β heterodimer activation on cell activity, we assessed cell proliferation via a soft agar anchorage-independent growth assay in RPMI growth medium with 10% fetal bovine serum (FBS) over the course of 10 days (Fig. 4E–H, and Supplementary Fig. 8, Supplementary Fig. 9A, B). Given that HCC15 cells are hyperproliferative at baseline, these anchorage-independent growth assays represent a rigorous assay to assess differential cell growth in response to ligand treatment and between cell lines. As expected, HCC15 cells, which do not express PDGFRs, did not have significantly increased colony count nor colony area upon PDGF-BB ligand stimulation (Fig. 4E, G and H). Parental HCC15 cells grew visible colonies by the end of the assay (Fig. 4E). PDGFRαV1/βV2 cells, however, proliferated less than parental HCC15 cells, and significantly so in the presence of PDGF-BB ligand (Fig. 4F–H). TUNEL staining revealed no significant differences in the percentage of apoptotic cells between HCC15 cells and PDGFRαV1/βV2 cells upon PDGF-BB ligand stimulation (Supplementary Fig. 10A–E), establishing that the disparities between the two cell lines in the anchorage-independent growth assays are due to inhibition of proliferation upon PDGFRα/β activation.
Given that PDGFRβ homodimers and PDGFRα/β heterodimers bind the same ligand, PDGF-BB, together, these findings indicate that PDGFRα/β heterodimers may serve as sinks that bind PDGF-BB and prevent the ligand from activating PDGFRβ homodimers. To test this hypothesis, we stimulated PDGFRαV1/βV2 cells with PDGF-BB ligand, resulting in the potential formation of both PDGFRα/β heterodimers and PDGFRβ homodimers. Following immunoprecipitation with the GFP-Trap nanobody to pull down PDGFRα/β heterodimers, the supernatant was immunoprecipitated with an anti-PDGFRβ antibody to isolate PDGFRβ monomers and PDGFRβ homodimers. Each sample was then subjected to western blotting with the anti-phospho-PDGFR antibody to identify activated dimers (Supplementary Fig. 11A). This experiment revealed that both active PDGFRα/β heterodimers and PDGFRβ homodimers formed in the PDGFRαV1/βV2 cells (Supplementary Fig. 11B). However, normalization of ligand-stimulated phospho-PDGFR levels to unstimulated phospho-PDGFR levels for each sample indicated lower relative induction approaching significance for PDGFRβ homodimers following 5 min of ligand stimulation in the PDGFRαV1/βV2 cell line compared to this same timepoint in the PDGFRβV1/βV2 cell line (Supplementary Fig. 11C), indicating that PDGFRα/β heterodimers may indeed serve as ligand sinks.
MYO1D preferentially binds PDGFRα/β heterodimers
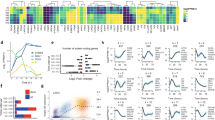
In light of our findings that the three PDGFR dimers have distinct internalization and trafficking dynamics34, we next combined BiFC with stable isotope labeling by amino acids in cell culture (SILAC) and quantitative mass spectrometry to identify proteins that differentially interact with the various PDGFR dimers at the time of internalization. We used a control cell line with myristoylated, membrane-targeted Venus (Supplementary Fig. 12A, B), as well as our PDGFRαV1/αV234, PDGFRαV1/βV2 and PDGFRβV1/βV234 cell lines representing each of the three PDGFR dimers. Cells were either grown in the presence of medium with light isotopes and left unstimulated or grown in the presence of medium with heavy isotopes and stimulated with the relevant PDGF ligand for 5 min. Following cell lysis and extraction, we mixed light and heavy samples 1:1 for each cell line, immunoprecipitated the receptor dimers and interacting proteins with the GFP-Trap nanobody, and generated tryptic peptides for mass spectrometry analysis (Fig. 5A). Following total signal normalization and removal of common contaminants52 that were not differentially detected across samples, we filtered the proteins based on three criteria to identify those with increased binding to one or more PDGFR dimer in response to PDGF ligand treatment (Fig. 5B). These parameters generated a list of 350 total proteins (Supplementary Data 1). Of these, 71 proteins were shared amongst all three PDGFR dimers, 51 were unique to PDGFRα homodimers, 67 were unique to PDGFRα/β heterodimers, and 60 were unique to PDGFRβ homodimers (Fig. 5B). Gene ontology (GO) analysis of the identified proteins with the Enrichr tool53 demonstrated common and distinct biological processes for the individual dimers (Supplementary Fig. 13).

A Schematic depicting experimental workflow for identification of PDGFR dimer-specific interacting proteins. B Filters applied to the mass spectrometry dataset and Venn diagram displaying shared protein identifications between samples within the filtered data. C Heat map of heavy (H)/light (L) SILAC ratios for identified proteins involved in trafficking. D Immunoprecipitation (IP) of myristoylated Venus and dimerized PDGFR receptors with GFP-Trap nanobody from cells that were unstimulated or treated with 10 ng/ml PDGF ligand for 5 min followed by western blotting (WB) with an anti-MYO1D antibody. WCL, whole-cell lysates. Images representative of three experiments. Uncropped blots in Source Data. E Scatter dot plot depicting relative interaction from n = 3 biological replicates as in D. Data are mean ± s.e.m. Colored circles correspond to independent experiments. F Scatter dot plot depicting Pearson’s correlation coefficient of the PDGFRαV1/αV2 homodimer, PDGFRαV1/βV2 heterodimer or PDGFRβV1/βV2 homodimer cell line Venus signal with an anti-MYO1D antibody signal following 10 ng/ml PDGF ligand stimulation for 5 min as in G. Data are mean ± s.e.m. P = 0.0001 for PDGFRαV1/αV2 vs PDGFRαV1/βV2, P = 0.0004 for PDGFRαV1/βV2 vs PDGFRβV1/βV2 (two-tailed, unpaired t-test with Welch’s correction). Colored circles correspond to independent experiments. Summary statistics from biological replicates consisting of independent experiments (large circles) are superimposed on top of data from all cells; n = 10 technical replicates across each of three biological replicates. G MYO1D antibody signal (white or magenta) and/or Venus expression (white or green) as assessed by (immuno)fluorescence analysis of the PDGFRαV1/βV2 heterodimer cell line following 10 ng/ml PDGF-BB ligand stimulation for 5 min. Inset in G is a region where white arrows are pointing. Nuclei were stained with DAPI (blue). White arrows denote colocalization; white outlined arrows denote lack of colocalization. Images representative of three experiments. Scale bars = 20 μm (main images), 3 μm (insets). Source data are provided as a Source Data file.
Given our demonstration that the various PDGFR dimers have different trafficking dynamics34, we next performed GO analysis of our filtered proteins to identify those involved in trafficking that differentially interact with the various PDGFR dimers (Fig. 5C). While some of these 20 proteins had been implicated in RTK binding, none had previously been reported to interact with the PDGFRs. One potential issue with our approach of purification after sample mixing is a replacement of PDGFR dimer-interacting proteins labeled with heavy isotopes with proteins labeled with light isotopes, especially in the case of transient interactions54. To validate our mass spectrometry findings, we performed immunoprecipitation of whole-cell lysates from each cell line with the GFP-Trap nanobody followed by western blotting with antibodies against several of these trafficking proteins (Supplementary Fig. 14). We chose to focus our initial studies on unconventional myosin-Id (MYO1D), as this protein was not detected in the control myristoylated Venus sample and had differential interaction with the various PDGFR dimers upon ligand stimulation, with the greatest binding to PDGFRα/β heterodimers (Fig. 5C). Further, MYO1D had previously been shown to bind and affect the subcellular localization of the RTKs EGFR, ERBB2 and ERBB455. We confirmed that MYO1D preferentially binds PDGFRα/β heterodimers via immunoprecipitation of whole-cell lysates from each cell line with the GFP-Trap nanobody followed by western blotting with an anti-MYO1D antibody (Fig. 5D, E).
Of note, we observed increased MYO1D expression in whole-cell lysates from the PDGFRαV1/βV2 cell line compared to both the PDGFRαV1/αV2 homodimer and PDGFRβV1/βV2 homodimer cell lines (Fig. 5D). Quantitative RT-PCR indicated that this upregulation is observed on the transcript level as well (Supplementary Fig. 15). This upregulation could stem from a mutation in the clone chosen for the PDGFRαV1/βV2 cell line, disruption of MYO1D regulatory elements upon insertion of the PDGFR-BiFC sequences, and/or increased MYO1D transcriptional activity or transcript stability upon PDGFRα/β heterodimer activation. In favor of the latter hypotheses, MYO1D expression increased with passage number for the PDGFRαV1/βV2 cell line (Supplementary Fig. 15). Finally, we examined colocalization of MYO1D with the various PDGFR dimers following 5 min of PDGF ligand stimulation via immunofluorescence analysis, demonstrating significantly increased colocalization with PDGFRα/β heterodimers compared to either PDGFRα homodimers or PDGFRβ homodimers (Fig. 5F, G).
MYO1D knockdown alters PDGFRα/β heterodimer dynamics
To examine the effects of MYO1D knockdown on PDGFRα/β heterodimer trafficking, we transfected PDGFRαV1/βV2 cells with a Silencer Select negative control siRNA or a combination of two Silencer Select siRNAs targeting MYO1D for 48 h. We confirmed efficient knockdown of MYO1D with the siRNAs targeting MYO1D via western blotting of whole-cell lysates at this timepoint (Supplementary Fig. 16). We found a significant increase in colocalization of the Venus signal with Na+/K+-ATPase upon treatment with siRNAs targeting MYO1D compared to the negative control following 5 min of ligand stimulation (Fig. 6A–C). Accordingly, we observed a significant decrease in colocalization with RAB5 upon treatment with siRNAs targeting MYO1D compared to the negative control at 2 min of ligand stimulation (Fig. 6D–F). We did not detect differences in colocalization with RAB7 between the two treatments at 1 h of ligand stimulation (Fig. 6G–I), indicating that knockdown of MYO1D primarily affects early internalization and trafficking dynamics of the PDGFRα/β heterodimers.

A, D, G Scatter dot plots depicting Pearson’s correlation coefficient of Venus signal from the PDGFRαV1/βV2 heterodimer cell line treated with Silencer Select negative control or Silencer Select siRNAs against MYO1D with an anti-Na+/K+-ATPase antibody (A), an anti-RAB5 antibody (D) or an anti-RAB7 antibody (G) signal following 10 ng/ml PDGF-BB ligand stimulation for 5 min (A), 2 min (D) or 60 min (G). Data are mean ± s.e.m. P = 0.05 for Na+/K+-ATPase, P = 0.008 for RAB5 (two-tailed, unpaired t-test with Welch’s correction). Colored circles correspond to independent experiments. Summary statistics from biological replicates consisting of independent experiments (large circles) are superimposed on top of data from all cells; n = 10 technical replicates across each of three biological replicates. B, C, E, F, H, I Na+/K+-ATPase antibody signal (white or magenta; B, C), RAB5 antibody signal (white or magenta; E, F) or APPL1 antibody signal (white or magenta; H, I) and/or Venus expression (white or green; B, C, E, F, H, I) as assessed by (immuno)fluorescence analysis of the PDGFRαV1/βV2 heterodimer cell line. Insets in B, C, E, F, H and I are regions where white arrows are pointing. Nuclei were stained with DAPI (blue; B, C, E, F, H, I). White arrows denote colocalization; white outlined arrows denote lack of colocalization. Images representative of three experiments. Scale bars = 20 μm (main images), 3 μm (insets). Source data are provided as a Source Data file.
Further, we assessed phosphorylation of ERK1/2 and AKT in cells treated with negative control versus siRNAs targeting MYO1D (Fig. 7A–F, Supplementary Fig. 17). PDGFRα/β heterodimer activation did not induce downstream phosphorylation of ERK1/2 upon treatment with the negative control siRNA (Fig. 7A, C). However, treatment with siRNAs targeting MYO1D led to significant increases in phosphorylation of ERK1/2 at 2, 5 and 15 min of PDGF-BB ligand treatment compared to baseline levels at 0 min of ligand stimulation (Fig. 7B, C). Alternatively, there were less significant increases in phospho-AKT levels upon treatment with siRNAs targeting MYO1D compared to treatment with negative control siRNA at 5, 15 and 60 min of PDGF-BB ligand stimulation (Fig. 7D–F). Finally, we assessed cell proliferation downstream of PDGFRα/β heterodimer activation upon knockdown of MYO1D (Fig. 7G–J, Supplementary Fig. 18, Supplementary Fig. 19A, B). Soft agar anchorage-independent growth assays revealed that PDGFRαV1/βV2 cells treated with negative control (Fig. 7G) proliferated less than the same cells treated with siRNAs targeting MYO1D (Fig. 7H) in the absence and presence of PDGF-BB ligand (Fig. 7I, J), and significantly so as assessed by colony count (Fig. 7I). Taken together, these data demonstrate that knockdown of MYO1D leads to retention of PDGFRα/β heterodimers at the plasma membrane, further resulting in increased downstream phosphorylation of ERK1/2 and increased cell proliferation.

A, B, D, E Western blot (WB) analysis of whole-cell lysates (WCL) from the PDGFRαV1/βV2 heterodimer cell line treated with Silencer Select negative control (A, D) or Silencer Select siRNAs against MYO1D (B, E) following a time course of 10 ng/ml PDGF-BB ligand stimulation from 2 min to 4 h with anti-phospho (p)-ERK1/2 (A, B) or anti-phospho (p)-AKT (D, E) antibodies. Images representative of six experiments. Uncropped blots in Source Data. C, F Line graphs depicting quantification of band intensities from n = 6 biological replicates as in A, B, D and E. Data are mean ± s.e.m. P = 0.05 for p-ERK1/2 siRNA MYO1D 2 min, P = 0.02 for p-ERK1/2 siRNA MYO1D 5 min, P = 0.01 for p-ERK1/2 siRNA MYO1D 15 min, P < 0.0001 for p-AKT negative control 5 min, P = 0.0006 for p-AKT negative control 15 min, P = 0.01 for p-AKT negative control 60 min, P = 0.04 for p-AKT siRNA MYO1D 5 min, P = 0.03 for p-AKT siRNA MYO1D 15 min (two-tailed, ratio paired t-test). G, H Colony growth in soft agar anchorage-independent growth assays for the PDGFRαV1/βV2 heterodimer cell line treated with Silencer Select negative control (G) or Silencer Select siRNAs against MYO1D (H) after 10 days in RPMI growth medium in the absence or presence of 10 ng/ml PDGF-BB ligand in six-well plate wells. Images representative of six experiments. I, J Scatter dot plots depicting quantification of colony count (I) or colony area (J) from n = 6 biological replicates as in G and H. Data are mean ± s.e.m. P = 0.03 for colony count negative control vs siRNA MYO1D, P = 0.01 for colony count negative control + PDGF-BB vs siRNA MYO1D + PDGF-BB (two-tailed, unpaired t-test with Welch’s correction between each siRNA treatment). Colored circles correspond to independent experiments. Source data are provided as a Source Data file.
Discussion
In this study, we used the BiFC technique to visualize and purify PDGFRα/β heterodimers, demonstrating that this continues to be an invaluable approach for studying RTK dimer-specific properties33,34,56. Comparing our findings across the three PDGFR-BiFC cell lines, all three dimers exhibited peak dimerization following 5 min of ligand stimulation, with PDGFRα/β heterodimers exhibiting an intermediate speed of dimerization compared to the two homodimers34. Further, the PDGFRα/β heterodimers had considerably reduced levels of autophosphorylation compared to either homodimer, particularly at early ligand stimulation timepoints34. Next, PDGFRα/β heterodimers were internalized into early endosomes more quickly and dwelled at that location for longer periods of time than either homodimer34. The peak extent of colocalization of the Venus signal from PDGFRα/β heterodimers with markers of rapid (15 min) and slow (60 min) recycling endosomes was higher than those for both PDGFR homodimers at these same timepoints, though closer to the values from PDGFRβ homodimers34. Alternatively, Venus signal from PDGFRα/β heterodimers had an intermediate level of colocalization with a marker of late endosomes at 90 min of ligand stimulation compared to either homodimer34. Taken together, these findings indicate that PDGFRα/β heterodimers and PDGFRβ homodimers undergo recycling with similar dynamics and that both dimers are less likely to be degraded than PDGFRα homodimers34. However, multiple lines of evidence indicate that degradation is the predominant outcome of intracellular trafficking for all three PDGFR dimers34. Surprisingly, phosphorylation of ERK1/2 was not induced in response to PDGFRα/β heterodimer activation, and the phospho-AKT response, while reaching a higher relative induction than upon PDGFRα homodimer activation, was transient in the PDGFRα/β heterodimer cell line upon PDGF-BB ligand treatment. Accordingly, PDGFRα/β heterodimer activation did not generate a proliferative response.
While this study represents a first step in the analysis of PDGFRα/β heterodimer dynamics, we acknowledge the possibility that tagged PDGFRs may not function the same as untagged receptors. One limitation of this study is that we cannot rule out that differences in expression levels of the PDGFRs across our three PDGFR-BiFC cell lines contribute to the disparities in results discussed above. Though we were able to identify a PDGFRα/β heterodimer clone with relatively equal expression of PDGFRA and PDGFRB among the hundreds of clones screened, the clone had reduced overall levels of these transcripts compared to our homodimer clones34. In the future, it will be critical to compare the dynamics of the various PDGFRs at endogenous expression levels, ideally in a cell type in which there is evidence that all three dimers form, such as the craniofacial mesenchyme18. In that setting, BiFC should continue to serve as a robust approach to both visualize and purify the individual PDGFR dimers. An additional limitation of our work is that we cannot directly monitor the kinetics of receptor internalization and trafficking in response to sustained PDGF ligand stimulation. Live-cell imaging over such long timepoints is not possible in our system due to photobleaching of Venus. However, the combination of more photostable fluorescent proteins with the BiFC technique should prove a powerful approach to overcome this limitation in the future.
Our colocalization studies demonstrated that PDGFRα/β heterodimers are rapidly internalized into the signaling endosome subpopulation of early endosomes, where they dwell for extended lengths of time. The fact that PDGFRα/β heterodimers are not long-lived at the plasma membrane indicates that they are unlikely to generate a robust signaling response at this subcellular platform. Given that the signaling endosome marker APPL146 has previously been shown to associate with the RTKs EGFR and TrkA and is required for Akt and Erk1/2 signaling downstream of these receptors57,58,59,60, it was unexpected that PDGFRα/β heterodimers did not induce robust and sustained phospho-ERK1/2 and phospho-AKT signals from this site either. It is possible, however, that PDGFRα/β heterodimers may sequester APPL1, thereby preventing it from propagating signaling downstream of other RTKs. In fact, our results point to a similar role for PDGFRα/β heterodimers in serving as sinks that prevent PDGF-BB ligand from binding and activating PDGFRβ homodimers. While the biological significance of this potential function of PDGFRα/β heterodimers is unknown, it is tempting to speculate that heterodimer formation may serve to attenuate PDGFR signaling in instances of cell crowding and/or increased PDGF-BB ligand availability. Though assays comparing the binding affinity of PDGFRα/β heterodimers and PDGFRβ homodimers to PDGF-BB ligand are beyond the scope of this work, such studies would likely shed further light on the role of PDGFRα/β heterodimers.
Our mass spectrometry findings represent the first PDGFR dimer-specific interactome and will be a rich resource for the field. Known PDGFR-binding proteins were detected in our mass spectrometry screen. For example, the catalytic subunit of PI3K, p110β, was bound to both PDGFRα/β heterodimers and PDGFRβ homodimers, however only in a single biological replicate for each dimer. Further, of the unfiltered proteins that we identified, 49% overlapped with proteins containing phospho-tyrosine residues from a previous SILAC-based mass spectrometry screen in which human fibroblasts were stimulated with PDGF-BB ligand, including MYO1D61. Of note, our screen revealed an enrichment for proteins that function in mRNA processing in binding all three PDGFR dimers. Interestingly, we had previously demonstrated in the context of the facial mesenchyme that phosphorylation targets of Akt downstream of PI3K-mediated PDGFRα homodimer signaling are similarly enriched for proteins involved in RNA processing62 and further, that alternative RNA splicing is the predominant mechanism of gene expression regulation downstream of this signaling pathway63,64. These results indicate that proteins that function in mRNA processing engage with PDGFR signaling on multiple levels: through binding to the receptors and via phosphorylation by downstream effector(s). Moreover, our finding that PDGFRα homodimers interact with proteins that function in organization of the actomyosin cytoskeleton and in the regulation of signal transduction by p53 is consistent with prior work62,65. Though the full scope of PDGFRα/β heterodimer function remains to be determined, our mass spectrometry results suggest that activation of this dimer may contribute to nucleosome assembly and organization, and regulation of autophagy, immunoglobulin production and gene expression. Given our findings that PDGFR dimers are dynamically trafficked throughout the cell for hours following ligand treatment34, similar approaches coupling biochemical purification of the dimers followed by mass spectrometry at multiple timepoints following ligand stimulation should provide a more complete understanding of the PDGFR dimer-specific interactome in various subcellular compartments.
We identified MYO1D as a protein interacting with the PDGFRs and demonstrated that it preferentially binds to dimerized, phosphorylated PDGFRα/β heterodimers. We do not know at this time whether MYO1D binds PDGFRα and/or PDGFRβ in the heterodimer complex, nor at what site(s) in the receptor(s) this binding occurs. These questions will be the subject of future research. However, given that the heavy/light ratio in our mass spectrometry screen was also greater than 1 for the PDGFRα homodimers, we speculate that MYO1D binds PDGFRα within the heterodimer complex upon ligand treatment. We found that MYO1D contributes to the rapid internalization of PDGFRα/β heterodimers from the plasma membrane into early endosomes in HCC15 lung cancer cells. Interestingly, these results are in direct contrast to what has previously been demonstrated for EGFR, ERBB2 and ERBB4 in Caco2 colorectal cancer cells. For these members of the ERBB family, MYO1D was shown to bind the unphosphorylated receptors and anchor them in the plasma membrane55. As such, siRNA-mediated knockdown of MYO1D resulted in decreased EGFR, ERBB2 and ERBB4 levels at the plasma membrane55. It is possible that these discrepancies arise due to differing cellular contexts and/or expression levels of the various RTKs in their respective settings. Alternatively, these results may reflect opposing roles for MYO1D in regulating RTK localization and represent a means of divergence across RTK families.
Here, we demonstrated that PDGFRα/β heterodimers are rapidly internalized from the plasma membrane into early endosomes, resulting in an absence of phospho-ERK1/2 induction and a transient phospho-AKT response. Alternatively, retention of PDGFRα/β heterodimers at the plasma membrane via knockdown of MYO1D led to increased phospho-ERK1/2 levels and decreased phospho-AKT levels at early timepoints of ligand treatment. These findings indicate that ERK1/2 and AKT phosphorylation downstream of PDGFRα/β activation may primarily occur at the plasma membrane and the early endosome, respectively, immediately following ligand stimulation. Our previous results upon inhibition of clathrin-mediated endocytosis of PDGFRβ homodimers via Dyngo-4a treatment suggest that engagement of these signaling molecules occurs at the same subcellular platforms for PDGFRα/β heterodimers and PDGFRβ homodimers34. Alternatively, we previously demonstrated that rapid internalization of PDGFRα homodimers plays an important role in the propagation of downstream signaling, indicating that this dimer primarily signals from early endosomes34. Future work will explore whether binding of the PDGFR homodimers to one or more of the proteins identified in our mass spectrometry screen mediates receptor trafficking to affect intracellular signaling. Taken together, our findings support a model in which protein interactions and subcellular localization represent mechanisms by which specificity is introduced downstream of PDGFR activation to differentially propagate signaling and generate distinct cellular responses.
Methods
Generation of the PDGFRα/β-BiFC and myr-Venus cell lines
The PDGFRαV1/αV2 homodimer and PDGFRβV1/βV2 homodimer cell lines were generated as previously described34. For the PDGFRαV1/βV2 heterodimer cell line, PDGFRα-V1/pLVX-Puro34 and PDGFRβ-V2/pLVX-Puro34 lentiviral constructs (10 mg each) and packaging vectors pCMV-VSV-G66 and pCMV-dR8.9167 (5 mg each) were transfected into HEK 293T/17 cells (American Type Culture Collection) using Lipofectamine LTX (Thermo Fisher Scientific, Waltham, MA, USA). For the myristoylated-Venus cell line, the pCAG:myr-Venus sequence (#32602, Addgene, Watertown, MA, USA) was amplified by PCR using the following primers (Integrated DNA Technologies, Inc., Coralville, IA, USA): 5’-GATAGGATCCGCCACCATG-3’ and 5’-CGTTTCTAGATTACTTGTACAGCTCG-3’. The sequence was cloned into the pLVX-Puro vector using BamHI and XbaI sites. The myr-Venus/pLVX-Puro lentiviral construct (10 mg) and packaging vectors pCMV-VSV-G and pCMV-dR8.91 (5 mg each) were transfected into HEK 293T/17 cells using Lipofectamine LTX. In each case, medium containing lentivirus was collected 48 h and 72 h following transfection and filtered using a 13 mm syringe filter with a 0.45 μm PVDF membrane (Thermo Fisher Scientific) following addition of 4 mg/ml polybrene (Sigma-Aldrich, St Louis, MO, USA). Lentiviral-containing medium was added to HCC15 lung cancer cells (see below) every 24 h for 2 days, and cells were subsequently grown in the presence of 2 μg/ml puromycin for 10 days. Individual Venus-positive cells were isolated on a Moflo XDP 100 cell sorter (Beckman Colter Inc., Brea, CA, USA), following 5 min of stimulation with 10 ng/ml PDGF-BB (R&D Systems, Minneapolis, MN, USA) diluted from a 1.5 μg/ml working solution in 40 nM HCl containing 0.1% BSA in the case of the PDGFRαV1/βV2 heterodimer cell line, and expanded to generate clonal cell lines. The final clone chosen for each cell line was confirmed by PCR amplification of the inserted sequence(s) from genomic DNA using primers found in Supplementary Data 2. These PCR products were subcloned into the Zero Blunt TOPO vector (Thermo Fisher Scientific) and Sanger sequenced.
PDGFR-BiFC and myr-Venus cell culture
The HCC15 lung cancer cell line was obtained from the laboratory of Dr. Lynn Heasley (University of Colorado Anschutz Medical Campus). The cell line was authenticated through short tandem repeat analysis and tested for mycoplasma contamination every 10 passages using the MycoAlert Mycoplasma Detection Kit (Lonza Group Ltd, Basel, Switzerland). PDGFR-BiFC and myr-Venus stable cells were cultured in RPMI growth medium [RPMI 1640 (Gibco, Thermo Fisher Scientific) supplemented with 100 U/ml penicillin (Gibco), 100 μg/ml streptomycin (Gibco) containing 10% FBS (Hyclone Laboratories Inc., Logan, UT, USA)] at 37 °C in 5% CO2. Once the stable cell lines were established, they were split at a ratio of 1:5 for maintenance. PDGFRα/β heterodimer cells were used for experiments at passages 10–17, PDGFRα homodimer cells were used for experiments at passages 39–44, PDGFRβ homodimer cells were used for experiments at passages 48–53, and myr-Venus cells were used for experiments at passages 10–13. When serum starved, cells were grown in HITES medium [DMEM/F12 (Corning, Corning, NY, USA) supplemented with 0.1% bovine serum albumin (Fisher Scientific, Thermo Fisher Scientific), 10 mM β-estradiol (Sigma-Aldrich), 10 mM hydrocortisone (Sigma-Aldrich), 5 μg/ml insulin (Sigma-Aldrich), 100 U/ml penicillin (Gibco), 100 μg/ml streptomycin (Gibco), 1.2 mg/ml NaHCO3 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA), 30 nM Na3SeO3 (Sigma-Aldrich) and 10 μg/ml apo-transferrin (Sigma-Aldrich)].
qRT-PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions. First-strand cDNA was synthesized using a ratio of 2:1 random primers:oligo (dT) primer and SuperScript II RT (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions. qRT-PCR was performed on a CFX Connect Real-Time PCR Detection System and analyzed with CFX Manager software (version 3.1; Bio-Rad Laboratories, Inc., Hercules, CA, USA). All reactions were performed with SYBR Select Master Mix (Applied Biosystems, Thermo Fisher Scientific), 300 nM primers (Integrated DNA Technologies, Inc.) and cDNA in a 20 μl reaction volume. PCR primers for qRT-PCR analyses can be found in Supplementary Data 2. The following PCR protocol was used: step 1, 2 min at 50 °C; step 2, 2 min at 95 °C; step 3, 15 s at 95 °C; step 4, 1 min at 60 °C; repeat steps 3 and 4 for 39 cycles; step 5 (melting curve), 5 s per 0.5 °C increment from 65 °C to 95 °C. All samples were run in triplicate and normalized against an endogenous internal control, B2M. qRT-PCR experiments were performed across three independent experiments, each using a separate passage of cells. When applicable, statistical analyses were performed with Prism 9 (GraphPad Software Inc., San Diego, CA, USA) using a two-tailed, unpaired t-test with Welch’s correction between each cell line.
Immunoprecipitation and western blotting
PDGFR-BiFC cells and myr-Venus cells were cultured as described above. To induce PDGFRα homodimer, PDGFRα/β heterodimer or PDGFRβ homodimer signaling, PDGFRα homodimer cells and PDGFRβ homodimer cells at ~60–70% confluence or PDGFRα/β heterodimer cells at ~70–80% confluence were serum starved for 24 h in HITES medium and stimulated with 10 ng/ml PDGF-AA (PDGFRα homodimer cells) or PDGF-BB (PDGFRα/β heterodimer and PDGFRβ homodimer cells) ligand (R&D Systems) for the indicated length of time. When applicable, cells were transfected with siRNA (see below) for 48 h before ligand stimulation. Protein lysates for immunoprecipitation were generated by resuspending cells in ice-cold GFP-Trap buffer [M-PER mammalian protein extraction reagent (Thermo Fisher Scientific), 1x complete Mini protease inhibitor cocktail (Roche, MilliporeSigma, Burlington, MA, USA), 1 mM PMSF, 10 mM NaF, 1 mM Na3VO4, 25 mM β-glycerophosphate] and collecting cleared lysates by centrifugation at 13,400 g at 4 °C for 20 min. For immunoprecipitations, cell lysates (500 μg for PDGFRα homodimers and PDGFRβ homodimers, 1 mg for PDGFRα/β heterodimers) were incubated with GFP-Trap agarose beads (GTA020; Bulldog Bio, Inc., Portsmouth, NH, USA) for 1 h or overnight (only for ligand sink experiments) at 4 °C. Beads were washed three times with ice-cold GFP-Trap buffer and the precipitated proteins were eluted with Laemmli buffer containing 10% β-mercaptoethanol, heated for 10 min at 100 °C, and separated by SDS-PAGE. When applicable, supernatant from immunoprecipitation experiments was incubated with primary antibody overnight at 4 °C followed by incubation with 20 μL of Protein A/G Plus-agarose beads (Santa Cruz Biotechnology, Inc.) for 2 h at 4 °C the following day. Beads were washed five times with ice-cold GFP-Trap buffer and the precipitated proteins were eluted with Laemmli buffer containing 10% β-mercaptoethanol, heated for 5 min at 95 °C, and separated by SDS-PAGE. For western blotting analysis of whole-cell lysates, protein lysates were generated by resuspending cells in ice-cold GFP-Trap buffer and collecting cleared lysates by centrifugation at 13,400 g at 4 °C for 20 min. Laemmli buffer containing 10% β-mercaptoethanol was added to the lysates, which were heated for 5 min at 100 °C. Proteins were subsequently separated by SDS-PAGE. Western blot analysis was performed according to standard protocols using horseradish peroxidase-conjugated secondary antibodies. Blots were imaged using a ChemiDoc XRS+ (Bio-Rad Laboratories, Inc.) or a ChemiDoc (Bio-Rad Laboratories, Inc.). The following antibodies were used for western blotting: PDGFRα (1:1,000; D13C6; 5241; Cell Signaling Technology, Inc., Danvers, MA, USA); PDGFRβ (1:1,000; 28E1; 3169; Cell Signaling Technology, Inc.); β-tubulin (1:1,000; E7; E7; Developmental Studies Hybridoma Bank, Iowa City, IA, USA); GAPDH (1:50,000; 1E6D9; 60004-1-Ig; Proteintech Group Inc); phospho-PDGFRα (Tyr 849)/PDGFRβ (Tyr857) (1:1,000; C43E9; 3170; Cell Signaling Technology, Inc.); phospho-ERK1/2 (Thr202/Tyr204) (1:1,000; 9101; Cell Signaling Technology, Inc.); ERK1/2 (1:1,000; 9102; Cell Signaling Technology, Inc.); phospho-AKT (Ser473) (1:1,000; 9271; Cell Signaling Technology Inc.); AKT (1:1,000; 9272; Cell Signaling Technology Inc.); AAKG1 (1:1,000; PA5-104369; Invitrogen); RHG01 (1:500; PA5-82157; Invitrogen); SCFD2 (1:1,000; BS-17281R; Bioss, Thermo Fisher Scientific); VPS11 (1:1,000; 19140-1-AP; Proteintech Group Inc, Rosemont, IL, USA); Myosin ID (1:500; H-1; sc-515292; Santa Cruz Biotechnology, Inc); horseradish peroxidase-conjugated goat anti-rabbit IgG (1:20,000; 111035003; Jackson ImmunoResearch Inc., West Grove, PA, USA); and horseradish peroxidase-conjugated goat anti-mouse IgG (1:20,000; 115035003; Jackson ImmunoResearch Inc.). Quantifications of signal intensity were performed with ImageJ software (version 1.53t, National Institutes of Health, Bethesda, MD, USA). Immunoprecipitation and western blotting experiments were performed across at least three and as many as six independent experiments. Relative dimerized PDGFR levels were determined by normalizing GFP-Trap immunoprecipitated PDGFR levels to total PDGFR levels. Relative phospho-PDGFR levels were determined by normalizing to total PDGFR levels, except for ligand sink experiments in which relative phospho-PDGFR levels were determined by normalizing to unstimulated phospho-PDGFR signals. Relative phospho-ERK1/2 levels were determined by normalizing to total ERK1/2 levels. Relative phospho-AKT levels were determined by normalizing to total AKT levels. Relative MYO1D levels were determined by normalizing immunoprecipitated MYO1D levels to total MYO1D levels. When applicable, statistical analyses were performed with Prism 9 (GraphPad Software Inc.) using a two-tailed, ratio paired t-test within each cell line (comparing individual ligand treatment timepoint values to the no ligand 0 min timepoint value) or a two-tailed, unpaired t-test with Welch’s correction between each cell line or siRNA treatment.
Fluorescence analysis
For fluorescence intensity and marker colocalization experiments, cells were seeded onto glass coverslips coated with 5 μg/ml human plasma fibronectin purified protein (MilliporeSigma) at a density of 80,000 cells, 100,000 cells and 40,000 cells per 24-well plate well for the PDGFRα homodimer cell line, the PDGFRα/β heterodimer cell line and the PDGFRβ homodimer cell line, respectively, in RPMI growth medium. After 24 h, cells were washed with 1x phosphate buffered saline (PBS) and serum starved in HITES medium. HITES medium was replaced 23 h later. After 54 min, coverslips were photobleached for 1 min with an Axio Observer 7 fluorescence microscope (Carl Zeiss Microscopy LLC, White Plains, NY, USA) using the 2.5x objective and 488 nm laser. Cells were allowed to recover for 5 min and were treated with 10 ng/ml PDGF-AA, PDGF-CC or PDGF-DD ligand (R&D Systems) or 1–100 ng/ml PDGF-BB ligand (R&D Systems) for the indicated amount of time. When applicable, cells were transfected with siRNA (see below) for 48 h before ligand stimulation. Cells were fixed in 4% paraformaldehyde (PFA) in PBS with 0.1% Triton X-100 for 10 min and washed in PBS. Cells were blocked for 1 h in 5% normal donkey serum (Jackson ImmunoResearch Inc.) in PBS and incubated overnight at 4 °C in primary antibody diluted in 1% normal donkey serum in PBS. After washing in PBS, cells were incubated in Alexa Fluor 546-conjugated donkey anti-rabbit secondary antibody (1:1,000; A21206; Invitrogen) or Alexa Fluor 546-conjugated donkey anti-mouse secondary antibody (1:1,000; A10036; Invitrogen) diluted in 1% normal donkey serum in PBS with 2 μg/ml DAPI (Sigma-Aldrich) for 1 h. Cells were mounted in VECTASHIELD HardSet Antifade Mounting Medium (Vector Laboratories, Inc., Burlingame, CA, USA) and photographed using an Axiocam 506 mono digital camera (Carl Zeiss Microscopy LLC) fitted onto an Axio Observer 7 fluorescence microscope (Carl Zeiss Microscopy LLC) with the 63x oil objective with a numerical aperture of 1.4 at room temperature. The following antibodies were used for immunofluorescence analysis: PDGFRα (1:1000; D13C6; 5241; Cell Signaling Technology, Inc.); PDGFRβ (1:1000; 28E1; 3169; Cell Signaling Technology, Inc.); Na+/K+-ATPase (1:500; EP1845Y; ab76020; Abcam); RAB5 (1:200; C8B1; 3547; Cell Signaling Technology Inc.); APPL1 (1:200; D83H4; 3858; Cell Signaling Technology Inc.); EEA1 (1:200; C45B10; 3288; Cell Signaling Technology Inc.); RAB7 (1:100; D95F2; 9367; Cell Signaling Technology Inc.); RAB4 (1:200; ab13252; Abcam); RAB11 (1:100; D4F5; 5589; Cell Signaling Technology Inc.) and Myosin ID (1:500; H-1; sc-515292; Santa Cruz Biotechnology, Inc). For fluorescence intensity measurements, three independent trials, or biological replicates, were performed. For each biological replicate, at least 38 technical replicates consisting of individual cells were imaged with Z-stacks (0.24 μm between Z-stacks with a range of 2-8 Z-stacks). For marker colocalization experiments, three independent trials, or biological replicates, were performed. For each biological replicate, at least 10 technical replicates consisting of individual cells were imaged with Z-stacks (0.24 μm between Z-stacks with a range of 2-8 Z-stacks) per timepoint. For Fig. 3 and Supplementary Fig. 3, in cases where technical replicates were less than 20, all BiFC-positive cells were imaged. Images were deconvoluted using ZEN Blue software (Carl Zeiss Microscopy LLC) using the ‘Better, fast (Regularized Inverse Filter)’ setting. For all images, extended depth of focus was applied to Z-stacks using ZEN Blue software (Carl Zeiss Microscopy LLC) to generate images with the maximum depth of field. For fluorescence intensity measurements, background was subtracted using rolling background subtraction with a radius of 30 pixels using Fiji software (version 2.1.0/1.53c). A region of interest (ROI) was drawn around each Venus-positive cell, and integrated density was measured and recorded as the fluorescence intensity. For marker colocalization measurements, an ROI was drawn around each Venus-positive cell in the corresponding Cy3 (marker) channel using Fiji software (version 2.1.0/1.53c). For each image with a given ROI, the Cy3 channel and the EGFP channel were converted to 8-bit images. Colocalization was measured using the Colocalization Threshold function, where the rcoloc value [Pearson’s correlation coefficient (PCC)] was used in statistical analysis. When applicable, statistical analyses were performed on the average values from each biological replicate with Prism 9 (GraphPad Software Inc.) using a two-tailed, paired t-test within each cell line (comparing individual ligand treatment timepoint values to the no ligand 0 min timepoint value) or a two-tailed, unpaired t-test with Welch’s correction between each cell line, siRNA treatment or non-0 min ligand treatment timepoints.
Flow cytometry
PDGFRα/β heterodimer cells at ~70-80% confluence were serum starved for 24 h in HITES medium, pretreated with 10 μg/ml cycloheximide (Sigma-Aldrich) in DMSO for 30 min and stimulated with 10 ng/ml PDGF-BB ligand (R&D Systems) for the indicated length of time. Cells were washed twice in ice-cold PBS and suspended by adding ice-cold 5 mM EDTA-PBS (pH 8.0) spiked with 0.0025% trypsin/EDTA followed by centrifugation at 181 g at 4 °C for 5 min. Cells were washed twice with ice-cold FACS buffer [2% normal donkey serum (Jackson ImmunoResearch Inc.) in PBS] and co-stained with PDGFRα XP Rabbit mAb (Alexa Fluor 647 conjugate) antibody (1:50; D13C6; 5876; Cell Signaling Technology, Inc.) and a cell viability dye (LIVE/DEAD Fixable Violet Dead Cell Stain Kit, for 405 nm excitation; Thermo Fisher Scientific) diluted in FACS buffer at 4 °C for 30 min. Cells were washed three times with ice-cold FACS buffer and resuspended in FACS buffer for analysis. Sample collection was performed on a NovoCyte Penteon flow cytometer (Agilent Technologies, Inc., Santa Clara, CA, USA) and data were analyzed using NovoExpress software (version 1.5.6; Agilent Technologies, Inc.). Each biological replicate consisted of at least 50,000 cells per treatment. Singlets made up at least 93% of cells, at least 50% of which were alive. Only singlet, live cells were analyzed. Flow cytometry experiments were performed across three independent experiments, each using a separate passage of cells. Statistical analyses were performed with Prism 9 (GraphPad Software Inc.) using a two-tailed, paired t-test.
Anchorage-independent growth assay
For measurement of anchorage-independent cell growth, 25,000 cells were suspended in 1.5 ml RPMI growth medium with 0.35% Difco Agar Noble (Becton, Dickinson and Company, Franklin Lakes, NJ) and overlaid on a base layer containing 1.5 ml RPMI growth medium with 0.5% Difco Agar Noble (Becton, Dickinson and Company) in six-well plate wells. A feeding layer of 2 ml RPMI growth medium supplemented with 10 ng/ml PDGF-BB ligand (R&D Systems) and/or 0.01 μM siRNA (see below) was added on top of the agar and replaced every day. Plates were incubated at 37 °C in 5% CO2 for 10 days, and viable colonies were stained overnight with 1 mg/ml Nitrotetrazoleum Blue (Sigma-Aldrich) in PBS at 37 °C. Following a second overnight incubation at 4 °C, wells from each independent trial were photographed using a COOLPIX S600 digital camera (Nikon Inc., Melville, NY, USA). Images were made binary using Adobe Photoshop (version 25.0.1; Adobe Inc., San Jose, CA, USA), and colony number and area were quantified using MetaMorph imaging software (Molecular Devices, LLC, San Jose, CA, USA) and separately using ImageJ software (version 1.53t, National Institutes of Health, Bethesda). Samples in which agar was broken were excluded from statistical analyses. These included Trials 1 and 3 of HCC15 in the absence of ligand stimulation in Fig. 4. Statistical analyses were performed on values from each of six independent trials, or biological replicates, with Prism 9 (GraphPad Software Inc.) using a two-tailed, paired t-test within each cell line or siRNA treatment and a two-tailed, unpaired t-test with Welch’s correction between each cell line or siRNA treatment.
TUNEL assay
For assessment of apoptosis, cells were seeded onto glass coverslips coated with 5 μg/ml human plasma fibronectin purified protein (MilliporeSigma) at a density of 100,000 cells per 24-well plate well in RPMI growth medium. After 24 h, cells were washed with 1x PBS and serum starved in HITES medium. HITES medium was replaced 23 h later and 1 h later cells were left untreated or treated with 10 ng/ml PDGF-BB. After 24 h, apoptotic cells were identified using the In Situ Cell Death Detection Kit, Fluorescein (Sigma-Aldrich) according to the manufacturer’s instructions for adherent cells. Cells were mounted in VECTASHIELD HardSet Antifade Mounting Medium (Vector Laboratories, Inc.) and photographed using an Axiocam 506 mono digital camera (Carl Zeiss Microscopy LLC) fitted onto an Axio Observer 7 fluorescence microscope (Carl Zeiss Microscopy LLC) with the 40x air objective with a numerical aperture of 0.75 at room temperature. Three independent trials, or biological replicates, were performed for each cell line. For each biological replicate, 5 technical replicates consisting of individual fields of cells were imaged. All TUNEL-positive signals were confirmed by DAPI staining. Statistical analyses were performed on the average values from each biological replicate with Prism 9 (GraphPad Software Inc.) using a two-tailed, paired t-test within each cell line and a two-tailed, unpaired t-test with Welch’s correction between each cell line.
Mass spectrometry
The control myr-Venus and three PDGFR-BiFC cell lines were processed and analyzed across two biological replicates each. The following amino acids were used for stable isotope labeling: unlabeled (light) L-LYSINE:2HCL, unlabeled (light) L-ARGININE:HCL, stable isotope-labeled (heavy) L-LYSINE:2HCL (13C6, 99%; 15N2, 99%) and stable isotope-labeled (heavy) L-ARGININE:HCL (13C6, 99%; 15N4, 99%) (Cambridge Isotope Labs, Inc. Andover, MA). Light SILAC medium was generated by adding unlabeled L-LYSINE:2HCL (final concentration 146.0 mg/ml) and unlabeled L-ARGININE:HCL (final concentration 42.0 mg/ml) to RPMI SILAC growth medium [RPMI 1640 SILAC (Gibco, Thermo Fisher Scientific) supplemented with 100 U/ml penicillin (Gibco), 100 μg/ml streptomycin (Gibco) containing 10% FBS (Hyclone Laboratories Inc.)]. Heavy isotope SILAC medium was generated by adding heavy labeled L-LYSINE:2HCL (13C6, 99%; 15N2, 99%) (final concentration 151.3 mg/ml) and heavy labeled L-ARGININE:HCL (13C6, 99%; 15N4, 99%) (final concentration 44.0 mg/ml) in RPMI SILAC growth medium. Each SILAC medium was filtered through a 0.22 μm syringe filter. Each PDGFR-BiFC cell line was cultured in light SILAC medium and separately in heavy SILAC medium for at least 6 cell passages to achieve > 95% labeling. PDGFRα homodimer cells at ~75% confluence, PDGFRα/β heterodimer cells at ~70–80% confluence and PDGFRβ homodimer cells at ~65% confluence were serum starved for 24 h in HITES medium. Cells grown in light SILAC medium were left unstimulated and cells grown in heavy SILAC medium were stimulated with 10 ng/ml PDGF-AA (PDGFRα homodimer cells) or 10 ng/ml PDGF-BB (PDGFRα/β heterodimer and PDGFRβ homodimer cells) ligand for 5 min. Myr-Venus cells were cultured in light SILAC medium, serum starved at ~70% confluence for 24 h in HITES medium and left unstimulated. Protein lysates for immunoprecipitation were generated by resuspending cells in ice-cold GFP-Trap buffer and collecting cleared lysates by centrifugation at 13,400 g at 4 °C for 20 min. Before immunoprecipitation, light SILAC samples were combined with heavy SILAC samples in a 1:1 ratio per cell line. For immunoprecipitations, cell lysates (500 μg for myr-Venus, PDGFRα homodimers and PDGFRβ homodimers, 1 mg for PDGFRα/β heterodimers) were incubated with GFP-Trap agarose beads (GTA020; Bulldog Bio, Inc.) for 1 h at 4 °C. Beads were washed three times with ice-cold GFP-Trap buffer.
To prepare for trypsin digestion, beads were resuspended with 8 M urea, 50 mM ammonium bicarbonate. Tris (2-carboxyethyl)phosphine (TCEP) was added to a final concentration of 10 mM and samples were incubated at 30 °C for 1 h. Alkylation was performed by adding chloroacetamide (CAA) to 25 mM and incubating in the dark at room temperature for 30 min. Following alkylation, urea was diluted to 1 M and proteins digested overnight with modified sequencing grade trypsin (Promega, Madison, WI, USA) at a 1:50 enzyme:protein ratio. Beads were then centrifuged for 15 min at 18,000 g and supernatant digest solution was collected. Digested peptides were then desalted using Pierce C18 spin tips (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Digested peptides were loaded onto individual Evotips (Evosep Biosystems, Odense, Denmark) following the manufacturer’s instructions and separated on an Evosep One chromatography system (Evosep Biosystems) using a Pepsep column (150 mm inner diameter, 15 cm) packed with ReproSil C18 1.9 mm, 120 Å resin. Samples were analyzed using the instrument default “30 samples per day” LC gradient. The system was coupled to the timsTOF Pro mass spectrometer (Bruker Daltonics, Bremen, Germany) via the nano-electrospray ion source (Captive Spray, Bruker Daltonics). The mass spectrometer was operated in PASEF mode. The ramp time was set to 100 ms and 10 PASEF MS/MS scans per topN acquisition cycle were acquired. MS and MS/MS spectra were recorded from m/z 100 to 1700. The ion mobility was scanned from 0.7 to 1.50 Vs/cm2. Precursors for data-dependent acquisition were isolated within ±1 Th and fragmented with an ion mobility-dependent collision energy, which was linearly increased from 20 to 59 eV in positive mode. Low-abundance precursor ions with an intensity above a threshold of 500 counts but below a target value of 20,000 counts were repeatedly scheduled and otherwise dynamically excluded for 0.4 min.
Data was searched using PEAKS X Pro (version 10.6; Bioinformatics Solutions, Inc., Ontario, Canada). Precursor tolerance was set to ± 25 ppm and fragment tolerance was set to ± 0.2 Da. Data was searched against SwissProt (20,379 sequences) restricted to Homo sapiens. Each fraction was searched independently using a fully specific trypsin cleavage definition allowing for 2 missed cleavages and a maximum of 5 modifications per peptide. Fixed modifications were set as carbamidomethyl (C). Variable modifications were set as oxidation (M), acetylation (K, protein N-term), and phosphorylation (STY). SILAC isotope modifications 13C(6) 15N(4) (R) and 13C(6) 15N(2) (K) were also set as variable modifications. The minimum peptide length was set at 7 amino acids and protein identifications required a minimum of 1 unique peptide identification. Results were filtered to 1% FDR at the peptide and protein levels. SILAC quantification was performed using a “Quantification” task in PEAKS X Pro set for SILAC 2-plex (R10, K8) analysis. Quantification retention time range was set to 1.0 min, with an ion mobility range of 0.05 1/k0. Heavy conditions were set to include 100% R(+10.01) and K(+8.01) modifications. All results were filtered to 1% FDR. Auto-normalization was applied to the quantification results within PEAKS before further processing. Statistical testing was performed on filtered, normalized data using a one-way ANOVA with multiple comparison correction within the SILAC “Quantification” task in PEAKS X Pro. Gene ontology (GO) analysis of the identified proteins was performed with the Enrichr tool53.
siRNA-mediated knockdown of Myosin ID
PDGFRα/β heterodimer cells at ~60% confluence were transfected with Silencer Select Negative Control No. 1 siRNA (4390843; Invitrogen, Thermo Fisher Scientific) and separately two Silencer Select siRNAs targeting human Myosin ID (4392420; ID s9203 and s9204; Invitrogen, Thermo Fisher Scientific) at a final concentration of 0.01 μM using Lipofectamine RNAiMAX (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions for the indicated amount of time.
Statistics
All experiments in the main text were performed across at least three independent, biological replicates with similar results, with the exception of mass spectrometry, which was performed across two independent, biological replicates with similar results. The number of independent experiments, number of technical replicates and statistical tests applied are indicated in the relevant “Methods” section and figure legend. Representative images of western blot, (immuno)fluorescence, flow cytometry density dot plots and soft agar anchorage-independent growth assays are shown.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data generated in this study have been deposited in the ProteomeXchange Consortium via the PRIDE68 partner repository under dataset identifier PXD048421. The mass spectrometry proteomics data generated in this study are provided in Supplementary Data 1 file. The remaining data generated in this study are provided within the article and its Supplementary Information. Source data are provided with this paper.
References
Heldin, C. H. & Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 79, 1283–1316 (1999).
Hoch, R. V. & Soriano, P. Roles of PDGF in animal development. Development 130, 4769–4784 (2003).
Bolger, G. B. et al. Chromosome translocation t(14;22) and oncogene (c-sis) variant in a pedigree with familial meningioma. N. Engl. J. Med. 312, 564–567 (1985).
Golub, T. R., Barker, G. F., Lovett, M. & Gilliland, D. G. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell 77, 307–316 (1994).
Cools, J. et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N. Engl. J. Med. 348, 1201–1214 (2003).
Heinrich, M. C. et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299, 708–710 (2003).
Nakanishi, G. et al. A novel fusion gene of collagen type I alpha 1 (exon 31) and platelet-derived growth factor B-chain (exon 2) in dermatofibrosarcoma protuberans. Eur. J. Dermatol 17, 217–219 (2007).
Cheung, Y. H. et al. A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am. J. Hum. Genet 92, 996–1000 (2013).
Keller, A. et al. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat. Genet 45, 1077–1082 (2013).
Nicolas, G. et al. Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80, 181–187 (2013).
Johnston, J. J. et al. A point mutation in PDGFRB causes autosomal-dominant Penttinen Syndrome. Am. J. Hum. Genet 97, 465–474 (2015).
Takenouchi, T. et al. Novel overgrowth syndrome phenotype due to recurrent de novo PDGFRB mutation. J. Pediatr. 166, 483–486 (2015).
Williams, L. T. Signal transduction by the platelet-derived growth factor receptor. Science 243, 1564–1570 (1989).
Matsui, T. et al. Isolation of a novel receptor cDNA establishes the existence of two PDGF receptor genes. Science 243, 800–804 (1989).
Seifert, R. A. et al. Two different subunits associate to create isoform-specific platelet-derived growth factor receptors. J. Biol. Chem. 264, 8771–8778 (1989).
Herren, B. et al. Dimerization of extracellular domains of platelet-derived growth factor receptors. A revised model of receptor-ligand interaction. J. Biol. Chem. 268, 15088–15095 (1993).
Shim, A. H. et al. Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc. Natl Acad. Sci. USA 107, 11307–11312 (2010).
Fantauzzo, K. A. & Soriano, P. PDGFRbeta regulates craniofacial development through homodimers and functional heterodimers with PDGFRalpha. Genes Dev. 30, 2443–2458 (2016).
Kelly, J. D. et al. Platelet-derived growth factor (PDGF) stimulates PDGF receptor subunit dimerization and intersubunit trans-phosphorylation. J. Biol. Chem. 266, 8987–8992 (1991).
Bostrom, H. et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85, 863–873 (1996).
Soriano, P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 124, 2691–2700 (1997).
Ding, H. et al. A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat. Genet 36, 1111–1116 (2004).
Leveen, P. et al. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 8, 1875–1887 (1994).
Soriano, P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 8, 1888–1896 (1994).
Klinghoffer, R. A., Hamilton, T. G., Hoch, R. & Soriano, P. An allelic series at the PDGFalphaR locus indicates unequal contributions of distinct signaling pathways during development. Dev. Cell 2, 103–113 (2002).
Yu, J. C. et al. Tyrosine mutations within the alpha platelet-derived growth factor receptor kinase insert domain abrogate receptor-associated phosphatidylinositol-3 kinase activity without affecting mitogenic or chemotactic signal transduction. Mol. Cell Biol. 11, 3780–3785 (1991).
Heuchel, R. et al. Platelet-derived growth factor beta receptor regulates interstitial fluid homeostasis through phosphatidylinositol-3’ kinase signaling. Proc. Natl Acad. Sci. USA 96, 11410–11415 (1999).
McCarthy, N. et al. Pdgfra and Pdgfrb genetically interact during craniofacial development. Dev. Dyn. 245, 641–652 (2016).
Fantauzzo, K. A. & Soriano, P. Generation of an immortalized mouse embryonic palatal mesenchyme cell line. PLoS One 12, e0179078 (2017).
Mo, J., Long, R. & Fantauzzo, K. A. Pdgfra and Pdgfrb genetically interact in the murine neural crest cell lineage to regulate migration and proliferation. Front Physiol. 11, 588901 (2020).
Hu, C. D., Chinenov, Y. & Kerppola, T. K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798 (2002).
Shyu, Y. J., Liu, H., Deng, X. & Hu, C. D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques 40, 61–66 (2006).
Croucher, D. R. et al. Bimolecular complementation affinity purification (BiCAP) reveals dimer-specific protein interactions for ERBB2 dimers. Sci. Signal 9, ra69 (2016).
Rogers, M. A., Campana, M. B., Long, R. & Fantauzzo, K. A. PDGFR dimer-specific activation, trafficking and downstream signaling dynamics. J. Cell Sci. 135, jcs259686 (2022).
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Cancer Cell Line Encyclopedia, C. & Genomics of Drug Sensitivity in Cancer, C. Pharmacogenomic agreement between two cancer cell line data sets. Nature 528, 84–87 (2015).
Polyansky, A. A. et al. Atomistic mechanism of the constitutive activation of PDGFRA via its transmembrane domain. Biochim Biophys. Acta Gen. Subj. 1863, 82–95 (2019).
Yang, Y., Xie, P., Opatowsky, Y. & Schlessinger, J. Direct contacts between extracellular membrane-proximal domains are required for VEGF receptor activation and cell signaling. Proc. Natl Acad. Sci. USA 107, 1906–1911 (2010).
Mori, S., Tanaka, K., Omura, S. & Saito, Y. Degradation process of ligand-stimulated platelet-derived growth factor beta-receptor involves ubiquitin-proteasome proteolytic pathway. J. Biol. Chem. 270, 29447–29452 (1995).
Miyake, S., Lupher, M. L. Jr., Druker, B. & Band, H. The tyrosine kinase regulator Cbl enhances the ubiquitination and degradation of the platelet-derived growth factor receptor alpha. Proc. Natl Acad. Sci. USA 95, 7927–7932 (1998).
Miyake, S., Mullane-Robinson, K. P., Lill, N. L., Douillard, P. & Band, H. Cbl-mediated negative regulation of platelet-derived growth factor receptor-dependent cell proliferation. A Crit. role Cbl tyrosine kinase-binding domain J. Biol. Chem. 274, 16619–16628 (1999).
Karlsson, S. et al. Loss of T-cell protein tyrosine phosphatase induces recycling of the platelet-derived growth factor (PDGF) beta-receptor but not the PDGF alpha-receptor. Mol. Biol. Cell 17, 4846–4855 (2006).
Hellberg, C., Schmees, C., Karlsson, S., Ahgren, A. & Heldin, C. H. Activation of protein kinase C alpha is necessary for sorting the PDGF beta-receptor to Rab4a-dependent recycling. Mol. Biol. Cell 20, 2856–2863 (2009).
Skou, J. C. The identification of the sodium-pump as the membrane-bound Na+/K+-ATPase: a commentary on ‘The Influence of Some Cations on an Adenosine Triphosphatase from Peripheral Nerves’. Biochim Biophys. Acta 1000, 435–438 (1989).
Zerial, M. & McBride, H. Rab proteins as membrane organisers. Nat. Rev. Mol. Cell Biol. 2, 107–117 (2001).
Miaczynska, M. et al. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell 116, 445–456 (2004).
Mu, F. T. et al. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J. Biol. Chem. 270, 13503–13511 (1995).
Vasudevan, H. N., Mazot, P., He, F. & Soriano, P. Receptor tyrosine kinases modulate distinct transcriptional programs by differential usage of intracellular pathways. Elife 4, e07186 (2015).
Der, C. J., Finkel, T. & Cooper, G. M. Biological and biochemical properties of human rasH genes mutated at codon 61. Cell 44, 167–176 (1986).
He, F. & Soriano, P. A critical role for PDGFRalpha signaling in medial nasal process development. PLoS Genet 9, e1003851 (2013).
He, F. & Soriano, P. Sox10ER(T2) CreER(T2) mice enable tracing of distinct neural crest cell populations. Dev. Dyn. 244, 1394–1403 (2015).
Mellacheruvu, D. et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 (2013).
Xie, Z. et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 1, e90 (2021).
Wang, X. & Huang, L. Defining dynamic protein interactions using SILAC-based quantitative mass spectrometry. Methods Mol. Biol. 1188, 191–205 (2014).
Ko, Y. S. et al. MYO1D binds with kinase domain of the EGFR family to anchor them to plasma membrane before their activation and contributes carcinogenesis. Oncogene 38, 7416–7432 (2019).
Kennedy, S. P. et al. Targeting promiscuous heterodimerization overcomes innate resistance to ERBB2 dimerization inhibitors in breast cancer. Breast Cancer Res. 21, 43 (2019).
Lin, D. C. et al. APPL1 associates with TrkA and GIPC1 and is required for nerve growth factor-mediated signal transduction. Mol. Cell Biol. 26, 8928–8941 (2006).
Varsano, T. et al. GIPC is recruited by APPL to peripheral TrkA endosomes and regulates TrkA trafficking and signaling. Mol. Cell Biol. 26, 8942–8952 (2006).
Zoncu, R. et al. A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell 136, 1110–1121 (2009).
Lee, J. R. et al. Adaptor protein containing PH domain, PTB domain and leucine zipper (APPL1) regulates the protein level of EGFR by modulating its trafficking. Biochem Biophys. Res Commun. 415, 206–211 (2011).
Martinez-Martinez, E. et al. Increased abundance of Cbl E3 ligases alters PDGFR signaling in recessive dystrophic epidermolysis bullosa. Matrix Biol. 103-104, 58–73 (2021).
Fantauzzo, K. A. & Soriano, P. PI3K-mediated PDGFRalpha signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes Dev. 28, 1005–1017 (2014).
Dennison, B. J. C., Larson, E. D., Fu, R., Mo, J. & Fantauzzo, K. A. Srsf3 mediates alternative RNA splicing downstream of PDGFRalpha signaling in the facial mesenchyme. Development 148, dev199448 (2021).
Forman, T. E., Sajek, M. P., Larson, E. D., Mukherjee, N. & Fantauzzo, K. A. PDGFRalpha signaling regulates Srsf3 transcript binding to affect PI3K signaling and endosomal trafficking. Elife 13, RP98531 (2024).
Vasudevan, H. N. & Soriano, P. SRF regulates craniofacial development through selective recruitment of MRTF cofactors by PDGF signaling. Dev. Cell 31, 332–344 (2014).
Stewart, S. A. et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501 (2003).
Zufferey, R., Nagy, D., Mandel, R. J., Naldini, L. & Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15, 871–875 (1997).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
We thank Damian Garno, Jessica Johnston, Daniel Fuhr and Erin Binne for technical assistance. Cell sorting and flow cytometry were performed at the University of Colorado Cancer Center Flow Cytometry Shared Resource with assistance from Dr. Dmitry Baturin and Kristina Terrell. Mass spectrometry was performed at the University of Colorado School of Medicine Proteomics Core Facility with assistance from Sean Maroney. We are grateful to the laboratory of Dr. Rytis Prekeris for pCAG:myr-Venus, lentiviral and packaging plasmids, the laboratory of Dr. Lynn Heasley for HCC15 cells and advice on anchorage-independent growth assays, and Dr. Eric Clambey and Christine Childs for advice on flow cytometry experiments. We thank members of the Fantauzzo laboratory and Drs. Rytis Prekeris, Mary Reyland and David Clouthier for their critical comments on the manuscript. This work was supported by National Institutes of Health grants R01DE027689 (to K.A.F.), K02DE028572 (to K.A.F.) and F32DE032554 (to M.B.C.). The Flow Cytometry Shared Resource and Proteomics Core Facility are supported by National Institutes of Health grant P30CA046934.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.B.C., M.A.R., K.A.F.; Methodology: M.B.C., M.A.R., K.A.F.; Formal analysis: M.B.C., M.C.M., K.A.F.; Investigation: M.B.C., M.A.R., M.C.M., A.N., K.A.F.; Writing—Original Draft: M.B.C., K.A.F.; Writing – Review & Editing: M.A.R., M.C.M., A.N., E.D.L.; Visualization: M.B.C., E.D.L., K.A.F.; Supervision: K.A.F.; Funding acquisition: M.B.C., K.A.F.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Campaña, M.B., Perkins, M.R., McCabe, M.C. et al. PDGFRα/β heterodimer activation negatively affects downstream ERK1/2 signaling and cellular proliferation. Nat Commun 16, 4754 (2025). https://doi.org/10.1038/s41467-025-59938-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59938-1
