
Abstract
Aqueous organic redox flow batteries offer promising prospects for large-scale, high-safety, and cost-effective energy storage systems with no reliance on scarce mineral resources. However, challenges such as limited water solubility and poor stability hinder the practical application of organic redox molecules in aqueous organic redox flow batteries. Herein, we report the design and synthesis of an artificial redox-active α-amino acid molecule by functionalizing 1,5-dihydroxyanthraquinone with natural cysteine side group, which exhibits enhanced aqueous solubility and redox reversibility in alkaline aqueous organic redox flow batteries. Owing to its unique zwitterionic structure and abundant hydrogen bonds, the negolyte based on artificial α-amino acid molecule exhibits a very low capacity decay rate of 0.00025% per cycle (equivalent to 0.011% per day) under 1 M electron transfer. Theoretical simulations and spectroscopic analyses underscore the importance of the symmetric distribution and abundant hydrogen-bonding interactions of amphipathic amino acid side chains in enhancing the stability of the anthraquinone redox core and reducing its dimerization, as well as enhancing its water solubility and redox reversibility. This study presents the promising potential of nature-inspired principles in designing electrochemically stable, redox-active organic molecules, contributing to the advancement of large-scale, biocompatible, and sustainable aqueous organic redox flow batteries.
Similar content being viewed by others


Introduction
The urgent task of replacing fossil fuels with renewable energy sources, demands the development of economical and safe energy storage technologies1,2,3. Aqueous redox flow batteries (ARFBs) offer numerous advantages such as high scalability, moldable structure, intrinsic safety and low cost, making them highly competitive4,5,6,7,8. However, the resource limits and cross-contamination of metal ions are obstacles to the widespread application of ARFBs9,10. Aqueous organic redox flow batteries (AORFBs) have garnered increasing interests due to resource abundance and environmental friendliness11,12,13. So far, a series of AORFBs based on various classes of organic molecules, including quinone, phenazine, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), and viologen derivatives, have been demonstrated1,7,13,14,15,16. Among them, quinone derivatives with multiple carbonyl groups have emerged as promising negolyte materials, but their aqueous solubility and cycling stability need to be further improved via rational molecular engineering17,18,19. Currently, various functional groups, such as sulfonic acid17,20,21, carboxyl22,23,24,25, phenolic hydroxyl26,27,28, phosphate29, polyethylene glycol30, and so on31,32,33,34,35 have been proposed for anthraquinone derivatives. Nonetheless, biomimetic α-amino acid groups, which possess the combination of high hydrophilicity, biocompatibility, and environmental friendliness, have not been hitherto investigated in AORFBs.
Herein, we harness the bionics principle, employing natural α-amino acid group (cysteine, Cys), to functionalize poorly water-soluble 1,5-dihydroxyanthraquinone (1,5-DHAQ, 0.2 M). This molecular engineering strategy yields a unique artificial α-amino acid grafted anthraquinone derivative (namely Cys-DHAQ), characterized by enhanced water solubility (0.63 M) and redox reversibility, rendering it suitable for use as an advanced negolyte in AORFBs. As an artificial bionic α-amino acid, Cys-DHAQ features abundant amphoteric hydrophilic groups (−NH2 and −COOH), which enhance aqueous solubility and also facilitate the formation of numerous hydrogen bonds with water molecules. This interaction effectively suppresses the activity of water molecules, resulting in inhibited hydrogen evolution and high Coulombic efficiency. By introducing two symmetric α-amino acid side-groups, potential side reactions at the α-position of the phenolic hydroxyl group are minimized. Simultaneously, abundant hydrogen-bond interactions anchor more water molecules within the solvation shell of Cys-DHAQ, reducing its dimerization and thereby improving molecular stability and electrochemical reversibility. In consequence, the 0.1 M Cys-DHAQ||K4[Fe(CN)6] AORFB exhibits a stable capacity with a very low fading rate (0.00038% cycle−1 or 0.086% day−1) over 1100 cycles. This performance represents an obvious improvement compared to that of 1,5-DHAQ (0.0039% cycle−1 or 0.821% day−1). The 0.5 M Cys-DHAQ||K4[Fe(CN)6] AORFB also showed a very low attenuation rate of 0.011% day−1. Furthermore, the structural variations of Cys-DHAQ during cycling, along with its interaction with the solvent through hydrogen bonding, were investigated using detailed spectroscopic analyses, including 1H nuclear magnetic resonance (NMR), ultraviolet–visible (UV–Vis) spectroscopy, electron paramagnetic resonance (EPR), and in-suit Fourier transform infrared spectroscopy (in-situ FTIR). It reveals that the capacity decay of Cys-DHAQ||K4[Fe(CN)6] AORFB primarily stems from the detachment of α-amino acid side-groups and the rare formation of anthrone. This work presents the high potential of biomimetic molecular engineering strategy for creating redox-reversible and electrochemically stable organic molecules, which may contribute to the development of scalable and environmentally friendly energy storage systems.
Results
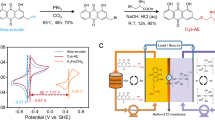
The redox-active biomimetic α-amino acid, Cys-DHAQ, was synthesized through a convenient three-step reaction process, as depicted in Fig. 1a. This process involved the hydroxymethylation of 1,5-DHAQ36, followed by the bromination of hydroxymethyl anthraquinone and finally the grafting of L-cysteine37. The reactants and final product were characterized using FTIR spectroscopy (Fig. 1b and Supplementary Fig. 1), with the characteristic FTIR bands thoroughly identified in Supplementary Table 1. Furthermore, 1H NMR spectroscopy validated the molecular structure and high chemical purity of the product after each synthesis step (Supplementary Figs. 2–8). Regarding the optimization of the reaction pathway, we analyzed the raw material costs in the synthesis processes to assess the economic feasibility of Cys-DHAQ and compared it with 3,3’-(9,10-anthraquinone-diyl)bis(3-methylbutanoic acid) (DPivOHAQ) (Supplementary Tables 2–3)19. The results show that the synthesis route for Cys-DHAQ reduces the stringent requirements for reaction conditions and the need for high-value auxiliary materials, thereby enhancing the overall sustainability of AORFBs. In the current estimated pricing, the synthesis costs of Cys-DHAQ and DPivOHAQ are primarily determined by the costs of 1,5-DHAQ and 9,10-di-hydroanthracene, respectively. Considering the industrial synthesis conditions and costs of both molecules, Cys-DHAQ demonstrates strong potential for practical applications. By utilizing UV–Vis absorption spectroscopy, a comparative analysis of the solubilities of 1,5-DHAQ under alkaline conditions (pH 14) and Cys-DHAQ at different pH values from 1 to 14 were conducted (Supplementary Figs. 9–10 and Supplementary Table 4). The solubility of Cys-DHAQ was determined to be 0.63 M under pH 14, much higher than that of 1,5-DHAQ (merely 0.2 M). These results verified that the two amphiphilic α-amino acid side chains contributed to the enhanced dissociation of Cys-DHAQ and clearly improved its water solubility through extensive hydrogen bonding with water molecules. This finding demonstrated the high efficacy of biomimetic design in regulating the physicochemical properties of organic redox-active molecules.

a Synthesis route of Cys-DHAQ conducted at room temperature (~20 °C). b FTIR spectra comparison of Cys, 1,5-DHAQ and Cys-DHAQ. c CV curves of 5 mM 1,5-DHAQ and Cys-DHAQ dissolved in 1.0 M KOH solutions at a scan rate of 100 mV s−1. d Structural configuration of the AORFB based on Cys-DHAQ negolyte and potassium ferrocyanide posolyte.
The electrochemical characteristics of 1,5-DHAQ and Cys-DHAQ were investigated by cyclic voltammetry (CV) in a pH 14 aqueous solution using a 3 mm glassy carbon electrode (Fig. 1c). The redox potential of Cys-DHAQ, referenced to the Ag/AgCl electrode, was determined to be −0.692 V. This value was observed to be 13 mV higher than that of 1,5-DHAQ, which was attributed to the electron-withdrawing effect of the Cys- side chains. Moreover, Cys-DHAQ exhibited different redox reversibility compared to 1,5-DHAQ, owing to the symmetrical grafting of Cys groups that strongly altered the solvation structure of a molecule.
Figure 1d visually depicted the structural configuration of Cys-DHAQ||K4[Fe(CN)6] AORFB. When pairing the as-prepared Cys-DHAQ negolyte with potassium ferrocyanide posolyte, the theoretical equilibrium cell voltage reached 1.03 V at pH 14. A low-cost custom-made sulfonated PEEK (SPEEK) membrane separator was employed as a substitute for the expensive conventional Nafion membranes. The custom-made SPEEK membrane was characterized by scanning electron microscopy, revealing a thickness of 35 μm (Supplementary Fig. 11). Electrochemical impedance spectroscopy (EIS) analysis indicated that the resistance of SPEEK and Nafion-212 membranes in a 1 M KOH solution amounted to 0.32 and 0.39 Ω cm², respectively (Supplementary Fig. 12). The reduced electrochemical impedance observed from the SPEEK membrane signified its faster ion transfer rate. From a cost perspective, the SPEEK membrane was priced at less than $100 m2 and projected to experience further reduction via mass production, making it a more cost-effective alternative to the Nafion-212 membrane retailing at $500–1000 m2 38,39.
The optimized structures, free energy of solvation, and electrostatic potentials of 1,5-DHAQ and Cys-DHAQ were investigated through density functional theory (DFT) simulations (Fig. 2)40,41. The symmetrical amphiphilic Cys groups in Cys-DHAQ resulted in a more negative electrostatic potential distribution and a larger solvation free energy compared to 1,5-DHAQ (Fig. 2a, b). The atomic coordinates of the optimized computational models are provided in Supplementary Table 5. Additionally, theoretical simulations of the hydrogen bonding interactions between 1,5-DHAQ and Cys-DHAQ with water molecules revealed that the amphiphilic Cys groups in Cys-DHAQ facilitated the formation of more hydrogen bonds with water molecules than 1,5-DHAQ (Supplementary Fig. 13). The atomic coordinates of the optimized computational models are provided in Supplementary Table 6. These hydrogen bonds enhanced the intermolecular interactions between Cys-DHAQ and water molecules, thereby improving the aqueous solubility of Cys-DHAQ.

Optimized structures and electrostatic potentials of (a) 1,5-DHAQ and (b) Cys-DHAQ. The solvation free energy of the corresponding molecule is presented below. c FTIR spectra of Cys-DHAQ solutions with different concentrations. UV–Vis absorption spectra of (d) 1,5-DHAQ and (e) Cys-DHAQ solutions at different pH values. f No-D 1H NMR spectra showing the water peaks in Cys-DHAQ negolytes with different concentrations between 0.03 and 0.5 M. g 3D snapshot obtained by MD simulations and representative Cys-DHAQ4− solvation structure in the 0.5 M Cys-DHAQ negolyte. The results of MD simulations include (h) the optimized number of hydrogen bonds around 1,5-DHAQ2− and Cys-DHAQ4− molecules, (i) the schematic diagrams of their solvation structures, and (j) the distribution of hydrogen bond distances.
As an artificial α-amino acid, the Cys-DHAQ molecule exhibited amphiphilic characteristics akin to natural amino acids. Changes in the dissociation equilibrium of the Cys side chains under varying pH levels altered the intramolecular charge distribution of Cys-DHAQ. This charge redistribution was manifested as a color change of the aqueous solution at different pH values (Supplementary Fig. 14). The impact of the dissociation equilibrium of Cys groups on intramolecular charge distribution was assessed using UV–Vis absorption spectroscopy across a pH range of 1–14. The 1,5-DHAQ molecule ungrafted with Cys groups was practically insoluble in aqueous solutions at pH <11. At pH >11, the 1,5-DHAQ solution exhibited three main peaks and one shoulder peak, with the color transitioning from brown-red to red (Fig. 2d). In contrast, the UV–Vis absorption spectrum of Cys-DHAQ from 190 to 700 nm exhibited multiple main peaks and shoulder peaks (Fig. 2e). With the increase of pH, the absorbance peak of Cys-DHAQ shifted from 454 nm (at pH 1) to 515 nm (at pH 14). To simulate the UV–Vis absorption spectra of these two molecules, time-dependent density functional theory (TDDFT) calculations were performed to compute the contributions of various excited states (Supplementary Fig. 15). Compared to 1,5-DHAQ, the simulated spectrum of Cys-DHAQ showed a slight red-shift with more contributions from excited states in the UV–Vis region, attributed to the introduction of two Cys side chains.
A more detailed comparison on the dissociation behaviors of 1,5-DHAQ and Cys-DHAQ was performed based on their UV–Vis absorption spectra at different pH values. As shown in Fig. 2d, 1,5-DHAQ exhibited a significant increase in absorbance at 482 nm between pH 11–12, corresponding to the dissociation of phenolic hydroxyl group on anthraquinone (pKa = 11.5). Notably, the phenolic hydroxyl group on the 1,5-DHAQ molecule was close to the carbonyl group, which could easily form hydrogen bonds with water molecules, thereby hindering the complete dissociation of the phenolic hydroxyl group. This resulted in a lower charge density distribution on the 1,5-DHAQ molecule, leading to its relatively low solubility in alkaline solutions (0.20 M). Unlike 1,5-DHAQ, the synthetic α-amino acid, Cys-DHAQ, exhibited typical zwitterionic behavior, undergoing multiple dissociation equilibria in aqueous solutions. The shifts in the absorption peaks of Cys-DHAQ between 454 and 514 nm were used to determine three pKa values (Fig. 2e and Supplementary Fig. 16). The pKa1, pKa2, and pKa3 values of Cys-DHAQ were 3.0, 8.5, and 11.5, corresponding to the dissociation of carboxyl groups, amino groups, and phenolic hydroxyl groups (Supplementary Fig. 17), respectively. Interestingly, by analyzing the saturated aqueous solubility curve of Cys-DHAQ as a function of pH value, we observe that the isoelectric point of Cys-DHAQ is around pH 7 (Supplementary Table 4 and Supplementary Fig. 18).
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of 1,5-DHAQ and Cys-DHAQ in their oxidized/reduced states were compared through DFT calculations (Supplementary Fig. 19)40,41. The atomic coordinates of the optimized computational models are provided in Supplementary Table 7. The results indicate that the grafting of Cys side groups modulated the HOMO/LUMO energy levels. Cys-DHAQ has a lower LUMO energy level compared to 1,5-DHAQ, indicating that Cys-DHAQ is more easily reduced. On the other hand, re-Cys-DHAQ exhibits a higher HOMO energy level than re-1,5-DHAQ, suggesting that Cys-DHAQ is more easily oxidized in its reduced state. These results also indicate that Cys-DHAQ possesses better electrochemical kinetics.
Using the rotating disk electrode (RDE), the diffusion coefficients (D) and kinetic reaction rate constants (k0) of 1,5-DHAQ and Cys-DHAQ were measured (Supplementary Fig. 20–21)24. Linear sweep voltammogram curves of 2 mM 1,5-DHAQ and Cys-DHAQ in 1 M KOH on a glassy carbon electrode were recorded at different rotation rates between 200 and 2000 rpm. Through the Levich equation, the diffusion coefficients of 1,5-DHAQ and Cys-DHAQ were calculated to be 3.77 × 10−6 cm2 s−1 and 1.03 × 10−6 cm2 s−1, respectively, similar to most anthraquinone derivatives. Correspondingly, by using the Koutecky-Levich equation, the k0 values of 1,5-DHAQ and Cys-DHAQ were determined to be 2.70 × 10−3 cm s−1 and 0.88 × 10−3 cm s−1, respectively. These differences arise from the fact that Cys-DHAQ possesses a larger molecular volume, more negative charges, and stronger hydrogen bonding interactions compared to DHAQ. Accordingly, it exhibits a higher solvation reorganization energy, as estimated by the Marcus-Hush theory42. Consequently, the diffusion and reaction kinetics of Cys-DHAQ are slightly slower; however, they still remain very competitive among anthraquinone-based compounds.
Through CV analyses of 1,5-DHAQ and Cys-DHAQ at different pH values, the corresponding Pourbaix diagrams were obtained to further study the proton/electron transfer kinetics during the redox process (Supplementary Fig. 22). The potential-pH diagram of 1,5-DHAQ could be fitted into a continuous curve, displaying a slope of −53.6 mV pH−1. In contrast, the potential-pH diagram of Cys-DHAQ could be fitted into two distinctly different curves, with a slope of −32 mV pH−1 in the pH range of 9–13.5, indicating a two-electron, one-proton process. When the pH value exceeded 13.5, the slope approaches 0 mV pH−1, indicating a two-electron, zero-proton process. The variations in the potential-pH diagram of Cys-DHAQ are originated from the intricate hydrogen bonding interactions inherent to the Cys side chain. As a result, the rearrangement and redistribution of intramolecular and intermolecular hydrogen bonds significantly alter the thermodynamic equilibrium state of the system (Supplementary Fig. 23). Notably, under conditions where the pH falls below 12 and further to 9, the redox peaks of both 1,5-DHAQ and Cys-DHAQ converge into indistinguishable features, accompanied by a notable increase in irreversibility. The primary reasons for this phenomenon are the reduced solubility as the pH decreases and the formation of intramolecular hydrogen bonds during the reduction process under mildly alkaline conditions, which further suppresses its solubility (Supplementary Table 4 and Supplementary Fig. 23). The atomic coordinates of the optimized computational models are provided in Supplementary Table 8.
Through further investigation, we analyzed the impact of complex hydrogen bonding interactions introduced by the natural α-amino acid moieties on the solvent environment of Cys-DHAQ negolyte. The FTIR spectra of Cys-DHAQ negolytes at varying concentrations revealed that the H − O bending vibration peak of water molecules shifted to lower wavenumbers as the concentration increased (Fig. 2c). Additionally, no-D 1H NMR of Cys-DHAQ negolytes at different concentrations (ranging from 0.03 to 0.5 M) were initially analyzed (Fig. 2f). When the concentration of Cys-DHAQ increased from 0.03 to 0.5 M, the water peak shifted significantly lower chemical shift, from 4.89 ppm to 4.80 ppm. The chemical shifts of the water peak at relatively high concentrations are ascribed to the aggregation of Cys-DHAQ molecules and the hydrogen bonds formed between Cys-DHAQ and water molecules43. With the increase of concentration, Cys-DHAQ molecules are prone to aggregation due to hydrogen bond interactions. Meanwhile, water molecules become situated within the shielding region amid multiple aromatic rings, resulting in heightened electron cloud density. The −O− anion in the Cys-DHAQ molecule acts as a proton acceptor, while the hydrogen atom in the water molecule acts as a proton donor, fostering the formation of strong hydrogen bond between them and further increasing the electron cloud density around the hydrogen atom in the water molecule, thus shifting its chemical shift up-field. Moreover, with increasing concentration of Cys-DHAQ, there is a heightened hydrogen bonding interaction between the −NH2 groups in Cys-DHAQ molecules with water molecules. This interaction diminishes the shielding effect on active hydrogen, resulting in a shift of the resonance absorption of active hydrogen to a lower field. Hence, the evolving trend of the chemical shift of hydrogen atoms in water molecules reveals that, as influenced by various factors, the hydrogen bond interaction in Cys-DHAQ negolyte intensifies with increasing concentration, leading to a rising electron cloud density around the water molecules. These findings indicate that the Cys side chains in Cys-DHAQ have formed strong hydrogen bonds with water molecules, thereby reducing the activity of water molecules in the electrolyte solution.
Snapshots from molecular dynamics (MD) simulations and representative solvation structures of 0.5 M 1,5-DHAQ and Cys-DHAQ electrolytes were presented in Fig. 2g and Supplementary Fig. 24. Additionally, based on the optimized MD results, we analyzed the number of hydrogen bonds formed between each 1,5-DHAQ2− or Cys-DHAQ4− species and surrounding water molecules, as well as the hydrogen bonds between water molecules in the respective electrolyte solutions (Fig. 2h and Supplementary Fig. 25)44. The atomic coordinates of the optimized computational models are provided in Supplementary Data 1. The results indicate that, due to the introduction of the Cys side chain, the number of hydrogen bonds between Cys-DHAQ4− and water molecules is five times greater than that between 1,5-DHAQ2− and water molecules. Correspondingly, the number of hydrogen bonds between water molecules in the Cys-DHAQ electrolyte is lower than in the 1,5-DHAQ electrolyte, confirming that water molecules preferentially form hydrogen bonds with Cys-DHAQ. Moreover, statistical analysis from the MD simulations demonstrates stronger hydrogen bonding interactions between Cys-DHAQ4− and water molecules (Fig. 2j). Based on both experimental and computational results, schematic solvation structures for 1,5-DHAQ2− and Cys-DHAQ4− were proposed (Fig. 2i). Compared to 1,5-DHAQ2−, the solvation shell of Cys-DHAQ4− contains more water molecules. According to the degradation mechanism of anthraquinone dimerization, the presence of a water molecule buffer layer between solvated Cys-DHAQ4− species likely reduces direct contact between redox molecules, thereby mitigating the formation of electrochemically inactive anthrones.
The power densities and rate performances of AORFBs based on low-concentration 1,5-DHAQ or Cys-DHAQ negolyte, potassium ferrocyanide posolyte, and SPEEK membrane separator were investigated (Fig. 3). The negolyte is 5 mL of 0.1 M 1,5-DHAQ or Cys-DHAQ in a 1.0 M KOH solution, and the posolyte is 40 mL of 0.1 M K4[Fe(CN)6] in a 1.0 M KOH solution. The polarization curves of the AORFBs at different state of charges (SOCs) of 20%, 50%, and 100% were measured, and the peak power densities were calculated accordingly (Fig. 3a, b). At 100% SOC, the Cys-DHAQ||K4[Fe(CN)6] AORFB demonstrated a power density of 117 mW cm−2, marginally lower than the 128 mW cm−2 observed in its 1,5-DHAQ counterpart. However, Cys-DHAQ demonstrates higher power densities at elevated concentrations, which is a critical advantage stemming from its obviously enhanced aqueous solubility (≈3.2 times higher than 1,5-DHAQ). This solubility-driven performance enhancement establishes Cys-DHAQ as a decisively more promising candidate for large-scale energy storage systems. Rate performance tests were also conducted on the AORFBs within a current density range of 20–150 mA cm−2 (Fig. 3c). At 20 mA cm−2, the capacity utilization ratios for 1,5-DHAQ||K4[Fe(CN)6] and (b) Cys-DHAQ||K4[Fe(CN)6] AORFBs were 90.6% and 90.4%, respectively, confirming that both underwent a thorough two-electron reaction process. These tow AORFBs exhibited similar rate performances, as shown in Fig. 3d and Supplementary Fig. 26, where the energy density and capacity utilization ratio decreased linearly with increasing current density, demonstrating the fixed voltammetric characteristics of the SPEEK membrane. When the current density reached 100 mA cm−2, the capacity utilization ratio of 0.1 M Cys-DHAQ||K4[Fe(CN)6] AORFB was 78.8%, with a corresponding energy efficiency value of 72.3%.

Polarization and power density curves of (a) 0.1 M 1,5-DHAQ||K4[Fe(CN)6] and (b) 0.1 M Cys-DHAQ||K4[Fe(CN)6] AORFBs collected at SOCs of 20%, 50%, and 100%. c Coulombic efficiencies, energy efficiencies, and specific capacities of 1,5-DHAQ||K4[Fe(CN)6] and Cys-DHAQ||K4[Fe(CN)6] AORFBs evaluated by galvanostatic charge/discharge at various current densities of 20, 40, 80, 100, and 150 mA cm−2. d Coulombic efficiency, energy efficiency, and capacity utilization ratio of the Cys-DHAQ||K4[Fe(CN)6] AORFB at various current densities. Charge/discharge profiles of (e) 1,5-DHAQ||K4[Fe(CN)6] and (f) Cys-DHAQ||K4[Fe(CN)6] AORFBs at the 10th, 500th, and 1000th cycles, respectively. g Cycling performances of 1,5-DHAQ||K4[Fe(CN)6] and Cys-DHAQ||K4[Fe(CN)6] AORFBs upon galvanostatic-potentiostatic cycling at a current density of 100 mA cm−2 ranging from 1.5 to 0.5 V.
The impact of different charge/discharge methods on the cycling performance of the AORFBs was also explored. Firstly, a galvanostatic charge/discharge process was employed for 1000 cycles. In Supplementary Fig. 27, the discharge capacity of 1,5-DHAQ||K4[Fe(CN)6] AORFB decreased from 4.96 Ah L−1 to 4.86 Ah L−1 after 1000 cycles (97.7 h), equivalent to a capacity decay rate of 0.002% per cycle or 0.42% per day. In contrast, the discharge capacity of Cys-DHAQ||K4[Fe(CN)6] AORFB decreased from 4.88 Ah L−1 to 4.84 Ah L−1 (97.6 h), corresponding to a capacity decay rate of 0.001% per cycle or 0.20% per day, exhibiting a better capacity retention performance. Subsequently, a galvanostatic-potentiostatic charge/discharge process was applied on the AORFBs for 1100 cycles. During charging, the potential was maintained at 1.5 V until the current density decreased to 8 mA cm−2. Similarly, during discharging, the potential was held at 0.5 V until the current density dropped to 8 mA cm−2. These conditions could prompt the active molecules to approach a 100% redox degree, thereby conducive to investigating their electrochemical stability during the thorough charge/discharge process. As shown in Fig. 3e–g, after 1100 cycles (116.0 h), the Cys-DHAQ||K4[Fe(CN)6] AORFB exhibited a capacity retention of 99.6%, reflecting a very low decay rate of 0.00038% per cycle or 0.086% per day. In contrast, the 1,5-DHAQ||K4[Fe(CN)6] AORFB showed a higher capacity decay rate of 0.0039% per cycle or 0.821% per day (93 h, after 300 cycles). These results suggested that the introduction of Cys side groups effectively improved the cyclic stability of Cys-DHAQ and reduced the impact of different SOCs on it.
To assess the advantages of custom-made SPEEK membrane, Nafion-212 was used as a comparison in the 0.1 M Cys-DHAQ||K4[Fe(CN)6] AORFB for extended cycling tests under both galvanostatic and galvanostatic-potentiostatic conditions (Supplementary Fig. 28). During 170 cycles of a galvanostatic test, the initial capacity of Nafion-212 based AORFB decreased from 4.22 Ah L−1 to 3.84 Ah L−1, corresponding to a capacity retention of 99.95% per cycle or 84.6% per day. In the galvanostatic-potentiostatic test, the capacity of Nafion-212 based AORFB decreased from 4.24 Ah L−1 to 3.84 Ah L−1 after 530 cycles, exhibiting a capacity retention of 99.98% per cycle or 95.7% per day. This result was much inferior to the SPEEK membrane based AORFB (99.9996% per cycle or 99.91% per day). To understand the reasons behind this difference, the cycled negolyte and posolyte were analyzed using CV method (Supplementary Fig. 29). The results showed that the SPEEK membrane based AORFB did not exhibit any signs of electrolyte crossover. In contrast, an obvious CV peak of potassium ferrocyanide was observed in the negolyte of Nafion-212 based AORFB. These results highlight the effectiveness of custom SPEEK membrane in preventing the crossover of potassium ferrocyanide and thus enhancing the cycling stability of the AORFB.
Considering practical applications, we studied the electrochemical performances of Cys-DHAQ||K4[Fe(CN)6] AORFBs at a high negolyte concentration of 0.5 M (Fig. 4). In Fig. 4a, the open-circuit voltage (OCV) of high-concentration Cys-DHAQ||K4[Fe(CN)6] AORFBs was recorded as it transitioned from 10% state of charge (SOC) to 100% SOC. At 50% SOC, the OCV was measured to be 1.03 V, aligning with the CV results in Fig. 1c. The polarization curves at 20%, 50%, and 100% SOC were obtained using linear sweep voltammetry, and the power densities were subsequently calculated (Fig. 4b). Notably, at 100% SOC, the observed peak power density stands at 290 mW cm−2, a value that is highly competitive among the state-of-the-art quinone-based AORFBs (Supplementary Table 9). The improvement in power density under high-concentration conditions is attributed to the increase in ionic conductivity, which reduces ohmic polarization, as well as the enhanced availability of active materials. The rate performance of high-concentration Cys-DHAQ||K4[Fe(CN)6] AORFBs was measured at current densities of 20, 40, 80, 100, and 150 mA cm−2 (Fig. 4c, d). As the current density escalated from 20 mA cm−2 (21.5 Ah L−1) to 150 mA cm−2 (17.8 Ah L−1), the discharge capacitance remained at 83%. This observation demonstrates the promising rate performance of Cys-DHAQ||K4[Fe(CN)6] AORFBs, supporting their potential use in high-power energy storage scenarios.

a The measured open circuit voltages of Cys-DHAQ negolyte under different charging states. b Polarization and power density curves of 0.5 M Cys-DHAQ||K4[Fe(CN)6] AORFBs collected at SOCs of 20%, 50%, and 100%. c Coulombic efficiencies, energy efficiencies, and discharge capacities of 0.5 M Cys-DHAQ||K4[Fe(CN)6] AORFBs at various current densities. d Charge/discharge profiles of 0.5 M Cys-DHAQ||K4[Fe(CN)6] AORFBs at 20, 40, 80, 100, and 150 mA cm−2. Long-term (e) galvanostatic and (f) galvanostatic-potentiostatic cycling performances of 0.5 M Cys-DHAQ||K4[Fe(CN)6] AORFBs tested at 100 mA cm−2. The inset shows the charge/discharge profiles at different cycles.
The galvanostatic cycling stability of high-concentration Cys-DHAQ||K4[Fe(CN)6] AORFBs, employing the SPEEK membrane separator, was assessed at a current density of 100 mA cm−2 (Fig. 4e). During the duration of 600 cycles (277 h), the capacity decreased from 24.52 Ah L−1 to 24.38 Ah L−1, corresponding to a capacity decay rate of 0.00096% per cycle or 0.05% per day. Furthermore, galvanostatic- potentiostatic charge/discharge tests were carried out for the 0.5 M Cys-DHAQ||K4[Fe(CN)6] AORFBs (Fig. 4f). During a rigorous testing regimen of 890 cycles (equivalent to 506 h) at 100 mA cm−2, the capacity exhibited a reduction from its initial value of 26.64 Ah L−1 to 26.59 Ah L−1, representing a capacity fading rate of 0.00025% per cycle or 0.011% per day. Once again, these findings exhibit the fascinating influence of amphiphilic Cys side groups in augmenting the electrochemical stability of anthraquinone derivatives (Supplementary Table 9).
To further elucidate the electrochemical redox alteration and decomposition mechanisms of Cys-DHAQ molecules during the circulation process, comprehensive characterizations were conducted on Cys-DHAQ negolytes employing EPR, in-situ FTIR, UV–Vis, and NMR spectroscopies (Fig. 5a–e). The oxidation mechanism of Cys-DHAQ in pH 14 aqueous solution was further investigated employing EPR spectroscopy. As depicted in Fig. 5a, Cys-DHAQ exhibited a signal peak at the magnetic field is 3511.8 G (corresponding to a g factor of 2.004), which is assigned to Cys-DHAQ5−• radical anion. With the progression of charging process, the intensity and broadness of the EPR signal increase, reaching a peak at 50% SOC, after which the signal intensity begins to decline and split. The signal broadening is primarily caused by dipolar interactions, which originate from magnetic interactions between adjacent unpaired electron spins and become more pronounced at higher concentrations due to reduced intermolecular distances. Additionally, the hyperfine interaction between the unpaired electron spin magnetic moment and the nuclear spin magnetic moment results in the split of EPR signal from single line to multiple lines.

a EPR spectra of Cys-DHAQ negolytes under different SOCs. b UV–Vis spectra of Cys-DHAQ negolytes under different SOCs. c Contour plot and (d) original curves of in-situ FTIR spectra of Cys-DHAQ negolytes measured during charge-discharge cycles. e Redox cycle and hydrolysis of Cys-DHAQ during charge/discharge processes. 1H NMR spectra (500 MHz, D2O) of (f) 0.1 M DHAQ and (g) 0.1 M Cys-DHAQ negolytes at pristine state and fully discharged state after different cycles. h CFF calculations of 1,5-DHAQ and Cys-DHAQ performed by using Gaussian 16 and the MultiWFN 3.8 programs at the DFT B3LYP/6-311 G(d,p) level.
The structural evolution of Cys-DHAQ becomes more evident through alterations in absorption peaks observed in UV–Vis spectra at various charging states (Fig. 5b). As the charging progresses, the absorption peak at λ514 nm diminishes gradually, while a new absorption peak emerges at λ426 nm and gains intensity, coinciding with the color transition of Cys-DHAQ negolyte from red to tan. The modulation of the absorption peak during charging aligns with the conversion of −C=O double bonds into −C−O single bonds. In-situ FTIR analysis was conducted to investigate the structural changes of 1,5-DHAQ and Cys-DHAQ during charge-discharge cycles (Fig. 5c, d and Supplementary Fig. 30). For 1,5-DHAQ, the C=O stretching vibrations were observed at 1618 cm−1 and 1580 cm−1, corresponding to the free carbonyl groups and the carbonyls involved in intramolecular hydrogen bonding with adjacent hydroxyl groups, respectively. These peaks gradually disappeared during reduction and reappeared during oxidation. Regarding C–O bonds, the C–O stretching vibration at 1357 cm−1 in the pristine state shifted to 1377 cm−1 during reduction, with a new vibration peak emerging at 1409 cm−1, attributed to the reduction of C=O groups. Notably, a reversible O–H bending vibration was observed at 1284 cm−1, indicating that some phenolic hydroxyl groups formed during reduction remain non-dissociated (Supplementary Fig. 31). These findings are consistent with our computational results. The reversible changes in these peaks confirm the high reversibility of 1,5-DHAQ. For Cys-DHAQ, similar changes were also observed during the charge-discharge processes: C=O vibration peaks (1614 cm−1 and 1579 cm−1), C–O vibration peaks (1334 cm−1 and 1407 cm−1), and O–H vibration peaks (1319 cm−1) all exhibited reversible behaviors akin to 1,5-DHAQ. Additionally, upon full oxidation of Cys-DHAQ, a vibration peak at 1584 cm−1 was observed, corresponding to the C=O groups in the Cys side chains (Supplementary Fig. 32). These results further demonstrate the favorable chemical stability of Cys-DHAQ.
Our study has shown that 1,5-DHAQ and Cys-DHAQ exhibit different electrode equilibrium reaction pathways in pH 14 solutions. However, in-situ FTIR analyses reveal similar O–H vibration peak changes. To explain this, we conducted a more detailed analysis of the entire redox process, dividing it into two parts: the electrode reduction process and the dissociation process in solution. By using DFT calculations, we performed geometric optimizations and energy calculations to investigate the reaction of the reduction product, 1,5-DHAQ2H2−, with OH− at pH 14. The detailed computational procedures are provided in the Supporting Information. The results show that the reaction between 1,5-DHAQ2H2− and OH− has a ΔG of −0.47 eV, indicating that it proceeds spontaneously (Supplementary Fig. 23). In contrast, the reaction between 1,5-DHAQH3− and OH− has a ΔG of 0.04 eV, suggesting it is non-spontaneous. Although Cys-DHAQ has a redox core similar to 1,5-DHAQ at positions 1 and 5, it exhibits different electrode equilibrium behavior. This discrepancy arises from the Cys side chains that alter the solvation layer and hydrogen-bonding network around the anthraquinone redox core, thereby affecting the electrode equilibrium process. Similarly, we also computed the reaction of the reduction product Cys-DHAQ6− with OH− at pH 14. The results indicate a ΔG of −0.05 eV, suggesting a spontaneous reaction. However, the reaction between 1,5-DHAQH3− and OH− at pH 14 has a ΔG of 0.25 eV, confirming it is non-spontaneous. These findings demonstrate that Cys-DHAQ6− can undergo partial protonation in aqueous solution. It suggests that despite their different electrode reaction processes, the reduced forms of 1,5-DHAQ and Cys-DHAQ, driven by thermodynamics, tend to form unilateral intramolecular hydrogen bonds. Previous studies have shown that the dissociation behavior of reduced hydroxy-anthraquinone derivatives is influenced by the distribution of intramolecular phenolic hydroxyl groups45. As a comparison, we also calculated the ΔG for the reaction of 2,6-DHAQ2H2− with OH−. The ΔG values for the reactions of 2,6-DHAQ2H2− and 1,5-DHAQH3− with OH− were calculated to be −1.58 eV and −1.03 eV, respectively, indicating that the dissociation processes are spontaneous. It confirms that the dissociation of phenolic hydroxyl groups in reduced hydroxy-anthraquinone derivatives is significantly influenced by the positional distribution of the hydroxyl groups on the anthraquinone core. Based on the above findings, the redox behavior of Cys-DHAQ during the charging process can be elucidated as follows (Fig. 5e)46:
To elucidate the degradation mechanism of Cys-DHAQ, we conducted 1H NMR spectroscopy analyses on the negolytes after different cycle numbers for both 0.1 M 1,5-DHAQ||K4[Fe(CN)6] and 0.1 M Cys-DHAQ||K4[Fe(CN)6] AORFBs (Fig. 5f, g and Supplementary Fig. 33). Although 1,5-DHAQ exhibits enhanced cycling stability compared to previously reported hydroxyanthraquinone derivatives, the capacity decay rate of 1,5-DHAQ||K4[Fe(CN)6] AORFB is still higher than that observed from Cys-DHAQ||K4[Fe(CN)6] AORFB. During prolonged cycling, the NMR spectra of 1,5-DHAQ negolyte exhibit the emergence of new peaks in the aromatic region (marked with “*”), which can be attributed to the reported degradation mechanism of conventional hydroxyanthraquinone derivatives involving the formation of anthrone and anthrone dimers (Supplementary Fig. 34a)46. In contrast, the NMR signals of Cys-DHAQ remain nearly identical to their initial state during the early stages of cycling testing. As cycling progresses, minimal new peaks emerge in the aliphatic region of NMR spectra, whereas the aromatic region shows no significant changes. As illustrated in Fig. 5g and Supplementary Fig. 34b, these newly emerged aliphatic peaks are attributed to the fact that a trace amount of Cys-DHAQ underwent side-group loss. The retention of soluble hydroxyl groups on the anthraquinone core maintains molecular solubility and prevents precipitation, thereby ensuring that even a minimal amount of side-chain loss has a negligible impact on battery capacity stability. The slight capacity decay observed during long-term cycling primarily results from minor electrolyte exfiltration and an increase in the internal resistance of the full cell. In conjunction with theoretical simulations, the reduced formation of anthrone dimers in Cys-DHAQ is attributed to its hydrogen-bond-rich structure, which facilitates strong interactions with surrounding water molecules, leading to the formation of a hydration buffer layer. This buffer layer effectively mitigates dimerization reactions, thereby contributing to the enhanced electrochemical stability of Cys-DHAQ negolyte.
Existing studies indicate that hydroxyanthraquinone derivatives exhibit high activity at the α-sites of phenolic hydroxyl groups under alkaline conditions46,47. By introducing Cys groups at these reactive α-sites, potential side reactions can be minimized, thereby significantly enhancing the stability of hydroxyanthraquinone derivatives. To evaluate the molecular stability before and after Cys group grafting, we employed Kenichi Fukui’s frontier orbital theory to predict electrophilic reaction sites based on the contributions of HOMO or LUMO orbitals. This involves examining the total electron density (ρi = |φi|2) of a given orbital (i) around each atom. Consequently, sites with larger Fukui function values exhibit greater reactivity. We calculated and visualized the condensed Fukui function (CFF) models for electrophilic/nucleophilic reactions using Gaussian 16 software at the DFT B3LYP/6-311 G(d, p) level, combined with the MultiWFN 3.8 program48,49. The atomic coordinates of the optimized computational models are provided in Supplementary Table 7. The calculations reveal that, compared to 1,5-DHAQ, the electrophilic reactivity at both the ortho and para positions of the phenolic hydroxyl groups in Cys-DHAQ is significantly reduced, indicating that the grafting of the Cys group greatly enhances molecular stability (Fig. 5h). Additionally, the results show potential electrophilic reactivity in the C−S−C bond of the Cys-DHAQ molecule, which aligns with post-cycle observations from the 1H NMR spectra. However, the peak intensity in the 1H NMR suggests minimal detachment of the Cys group due to hydrolysis, and this trace amount of side-chain loss does not compromise the redox stability. This underscores the critical role of the Cys side chain in improving the redox stability of the molecule.
Discussion
In summary, we report the design and synthesis of a biomimetic anthraquinone derivative symmetrically functionalized by two natural α-amino acid cysteine groups, Cys-DHAQ, which exhibits high water solubility, rapid redox kinetics, and enhanced electrochemical stability. Upon testing under highly alkaline conditions (pH 14), the Cys-DHAQ||K4[Fe(CN)6] AORFBs exhibited higher operational voltage and improved cycling stability across diverse concentration ranges, exhibiting a very low capacity decay rate of 0.00038% per cycle or 0.086% per day over 1100 cycles. This represents a notable reduction in capacity decay rate compared to 1,5-DHAQ (0.0039% per cycle or 0.821% per day). Furthermore, detailed analyses were conducted to investigate the structural changes and degradation mechanism of Cys-DHAQ. These studies highlight the critical role of the amphiphilic amino acid side chain and abundant hydrogen bonding interactions in Cys-DHAQ. These features enhance the stability of the redox core while minimizing dimerization, thereby improving both its water solubility and redox reversibility. This study reveals the desirable promise of biogenic amino acid functionalization strategy to modify redox-active organic molecules, which may contribute to the development of large-scale, safe, and environmentally sustainable electrochemical energy storage systems.
Methods
Chemicals and materials
The 1,5-dihydroxyanthraquinone (1,5-DHAQ, 85%) was purchased from Shanghai Yuanye Biotechnology Co., Ltd. Formaldehyde (HCHO, 37 wt.% in H2O) was purchased from Nanjing Wanqing Chemical Glass Instrument Co., Ltd. Cyanuric chloride (TCT, 99%) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd., L-cysteine was purchased from Shanghai D&B Biotechnology Technology Co., Ltd. Sodium Hydrosulfite (Na2S2O4, 90%) was purchased from Shanghai Acmec Biochemical Technology Co., Ltd. Methanol (CH3OH, 99.7%), Hydrogen peroxide (H2O2, 30%), Sodium bromide (NaBr, 99.7%) and Concentrated sulfuric acid (H2SO4, 98.3%) were purchased from Nanjing Chemical Reagent Co., Ltd. Potassium ferrocyanide (K4Fe(CN)6, 99%), potassium ferricyanide (K3Fe(CN)6, 99%), Sodium Carbonate (Na2CO3, 99%), HCl (36%), and KOH (99%) were purchased from Sinopharm Chemical Reagent Co., Ltd. All the reagents were used without further purification. Graphite felts and graphite plates were purchased from SGL Corp., Germany. Nafion-212 membranes were purchased from DuPont Corp., USA. The copper plates (99.98%, 1 mm thickness) were purchased from Jiangxi Jinpin Copper Technology Co., Ltd.
Synthesis 1,5-dihydroxy-2,6-bis(hydroxymethyl)anthracene-9,10-dione
Briefly, 2.4 g of 1,5-DHAQ was dissolved in 40 mL of MeOH, following by adding 20 mL of 1 M KOH. Then, 20 mL of 0.5 M NaS2O4 was slowly added into the mixture. After the color change, 3.0 g of 40% formaldehyde solution was added and reacted for 12 h. Subsequently, 11 g of 3% H2O2 was dropped into the mixture, and then the mixture was acidified with HCl. The precipitate was collected, and washed with water several times, and dried at 60 °C for 24 h, with a yield of 90%. 1H NMR (500 MHz, DMSO-d6) δ 12.83 (s, 2H), 7.89 (d, J = 7.5 Hz, 2H), 7.76 (d, J = 7.7 Hz, 2H), 5.47 (t, J = 5.5 Hz, 2H), 4.64 (d, J = 5.1 Hz, 4H), 13C NMR (101 MHz, DMSO) δ 187.64, 158.53, 139.11, 133.30, 130.75, 118.74, 114.51, 57.40.
Synthesis of 2,6-bis(bromomethyl)-1,5-dihydroxyanthracene-9,10-dione
Briefly, 2,4,6-trichlorotriazine (1.83 g, 10.0 mmol) was added to 2 mL of DMF, stirred at room temperature to form white solid. Then, 25 mL of dichloromethane and 1.95 g of NaBr were added in to the mixture, stirred for 8 h. Subsequently, 1.5 g of 1,5-dihydroxy-2,6-bis(hydroxymethyl)anthracene-9,10-dione was added and stirred for 12 h. Then, 20 mL of water was added, then the product was washed with 15 mL of saturated Na2CO3 solution, and then the mixture was washed with 1 M HCl solution several times, filtered and dried at 60 °C for 24 h, with a yield of 95%. 1H NMR (500 MHz, DMSO-d6) δ 12.43 (s, 2H), 7.85 (d, J = 8.1, 2H), 7.43 (d, J = 8.3, 2H), 4.87 (s, 4H), 13C NMR (126 MHz, DMSO) δ 187.71, 160.22, 138.55, 130.71, 129.65, 117.70, 115.57, 28.87.
Synthesis of (2 R,2’R)-3,3’-(((1,5-dihydroxy-9,10-dioxo-9,10-dihydroanthracene-2,6-diyl)bis(methylene))bis(sulfanediyl))bis(2-aminopropanoic acid)
Briefly, 2,6-bis(bromomethyl)-1,5-dihydroxyanthracene-9,10-dione (1.28 g, 3 mmol) and Cys (0.97 g, 8 mmol) were added to 40 mL of 1 M KOH solution, and stirred overnight at 60 °C. After acidification and washing, the target product Cys-DHAQ was obtained, with a yield of 82%. 1H NMR (500 MHz, D2O) δ 7.50 (d, J = 7.3 Hz, 2H), 7.22 (dd, J = 7.4, 3.3 Hz, 2H), 3.77 (s, 4H), 3.47 (ddd, J = 7.7, 5.2, 3.0 Hz, 2H), 2.88 (dd, J = 13.4, 5.4 Hz, 2H), 2.78 (dd, J = 13.6, 6.8 Hz, 2H).
Preparation of membranes
The preparation of the SPEEK membrane was carried out following the previously reported method, with necessary modifications24: Polyetheretherketone (PEEK) powder (8 g) was mixed with 100 mL of 98 wt.% sulfuric acid and vigorously stirred at 60 °C for 2 h. After the reaction, the mixture was poured into an ice-water bath and thoroughly washed with potassium hydroxide solution and deionized water until the pH reached neutral, and then vacuum-dried for 24 h. The dried SPEEK was dissolved in dimethylacetamide (DMAc) to prepare a 16–17 wt.% solution, which was uniformly cast onto a clean glass plate and dried at 60 °C for 12 h. Finally, the SPEEK membrane was carefully peeled off from the glass plate.
The treatment of the Nafion-212 membrane was carried out following the previously reported method50: The Nafion-212 ion-exchange membrane underwent a stepwise pre-treatment process involving sequential immersion and heating in three different solutions: an aqueous mixture of 5% H2O2 and 1.0 M KOH, deionized water, and another 1.0 M KOH solution. Each heating step was maintained for 30 min. Subsequently, the membrane was cooled down and then stored in deionized water, ensuring its readiness for subsequent applications.
Material characterizations
Attenuated total-reflectance Fourier transform infrared spectroscopy (ATR-FTIR, Nicolet iS50 spectrometer) were employed to analyze the chemical bond features of samples. The nuclear magnetic resonance (NMR) spectra of 1,5-DHAQ and Cys-DHAQ were recorded on a Bruker AVANCE III HD (400 MHz and 500 MHz) in dimethylsulfoxide-d6 (DMSO-d6) or D2O. The chemical shifts (δ) were reported in ppm with respect to tetramethylsilane (TMS) or 3-(Trimethylsilyl)-1-propane sulfonic acid sodium salt (DSS). The 2D 1H-detected heteronuclear multiple bond correlation (HMBC) spectra of Cys and Cys-DHAQ were recorded on a Bruker AVANCE III HD 500 MHz, in D2O with 1.0 M NaOH, the chemical shifts (δ) were reported in ppm with respect to DSS. EPR spectra were obtained using a Bruker EMX plus-6/1 variable-temperature apparatus. The EPR samples were sealed in a quartz capillary and detected at room temperature immediately without adding any free radical catching agent. The used standard sample was Bruker’s commercial product α,γ-bisdiphenylene-β-phenylallyl (BDPA), with a g value of 2.00268. The parameters for EPR analysis are listed as follows: center magnetic field of 3519.70 G, sweep width of 200.0 G, sweep time of 30 s, modulating magnetic field of 9.851938 GHz, modulation amplitude of 1 G, modulation frequency of 100 kHz, microwave power of 20.00 mW, and a conversion time of 15 ms. The ultraviolet-visible (UV–Vis) absorption spectra of the samples at different concentrations and pH values were measured using a UV–Vis spectrometer (UV-2600, Shimadzu Scientific Instrument) and quartz spectrophotometer cells (Aldrich, 10 mm optical path length) in a wavelength range of 200–700 nm, with a 0.2 nm interval. The scanning electron microscope (SEM) images were taken on a Japan Hitachi S-3400N.
Measurements of ionization constants (pKa)
For the measurement of ionization constants by UV–Vis absorption analysis, 1,5-DHAQ or Cys-DHAQ was dissolved in aqueous solution with a concentration of ~0.1 mM. The pH value was adjusted by HCl and KOH. A pH meter (pHS-3C, INESA, China) was calibrated by pH 4.00 and PH 6.86 buffer solutions, and then used to measure the pH value.
Electrochemical characterizations
Cyclic voltammetry (CV) analysis was conducted on a CS-350H electrochemical workstation (Wuhan Corrtest Instruments, China). A three-electrode configuration was employed, including a glassy carbon working electrode (GC 130, 3 mm in diameter), which was polished with ~50 nm alumina particles before use. A platinum (Pt) plate (0.75 cm × 1.5 cm) served as the counter electrode, while an Ag/AgCl reference electrode (0.199 V vs. SHE) was pre-soaked in a saturated KCl solution. A series of 2 mM 1,5-DHAQ or Cys-DHAQ solutions with pH values ranging from 13.98 to 0.25 were prepared, and their CV curves were recorded over a potential range from –1.3 V to 0.0 V at a scan rate of 100 mV s−1. The pH values were measured using a pH meter (pHS-3C, INESA, China) calibrated with standard buffer solutions at pH 4.00 and pH 6.86.
Rotating disk electrode (RDE) experiments were performed using an ALS RRDE-3A system. A 5 mm glassy carbon electrode was used as the working electrode, a Pt plate (0.75 cm × 1.5 cm) served as the counter electrode, and an Ag/AgCl reference electrode (pre-soaked in saturated KCl solution) was employed. The working electrode was rotated at controlled speeds ranging from 200 to 2000 rpm. Linear sweep voltammetry (LSV) was conducted using a CHI-760E electrochemical workstation (ChenHua Instruments, Shanghai) at a scan rate of 25 mV s–1 within the potential range of 0 to –1.30 V vs. Ag/AgCl.
Electrochemical impedance spectroscopy (EIS) measurements of the AORFBs were conducted before the cycling test. Sinusoidal voltage oscillations with a 5 mV amplitude was applied over a frequency range of 100 kHz to 0.1 Hz, with data collected at 10 points per decade.
All electrochemical tests were conducted at room temperature (~25 °C) under ambient atmospheric conditions, with no additional environmental control measures applied.
Redox flow battery tests
Unless stated otherwise, all AORFB tests were carried out in a nitrogen-filled glove box at room temperature (~25 °C). The pre-cut graphite felt electrodes (SGL, 5 mm thick, 2 cm × 2.5 cm) were first ultrasonically cleaned with ethanol and deionized water, followed by thermal treatment at 400 °C for 10 h. The treated graphite felt electrodes were then pressed onto graphite flow fields of the same size (2 cm × 2.5 cm) and sealed with PTFE gaskets. A custom-made SPEEK membrane (~35 μm, 6 cm × 6 cm) or a Nafion-212 membrane (50 μm, 6 cm × 6 cm) was used to separate the two electrodes. Electrolyte circulation was maintained using a dual-channel peristaltic pump (Zibo Newking Electromechanical Equipment, China) at a flow rate of 60 mL min−1.
The polarization curves at 20%, 50%, and 100% states of charge (SOC) were recorded using LSV at a scan rate of 100 mV s−1 on a CS-350H workstation equipped with a current booster (Wuhan Corrtest Instruments, China). To obtain the OCV profile at different SOCs, the cell was rest for 30 s after charging to each selected SOC before recording the corresponding voltage. The fully charged state at 1.5 V was defined as 100% SOC.
For the cycling tests of 0.1 M 1,5-DHAQ or Cys-DHAQ, the negolyte was 5 mL of 0.1 M 1,5-DHAQ or Cys-DHAQ solution at pH 14, and the posolyte was 40 mL of 0.1 M K4[Fe(CN)6] solution at pH 14. Galvanostatic cycling was conducted using a 5 V/1 A battery test station (Wuhan Land Instruments, China), at a current of 100 mA cm−2 between 1.5 V and 0.4 V. For the extended galvanostatic-potentiostatic charge–discharge tests of 0.1 M 1,5-DHAQ or Cys-DHAQ in 1 M KOH, the AORFBs were cycled between 0.5 and 1.5 V. At the end of each charge/discharge step, the voltage was held constant until the current decreased below 8 mA cm−2.
For the galvanostatic cycling tests of 0.5 M Cys-DHAQ, the negolyte was 5 mL of 0.5 M Cys-DHAQ solution at pH 14, and the posolyte was 40 mL of 0.3 M K4[Fe(CN)6] solution at pH 14. Galvanostatic cycling was conducted at a current of 100 mA cm−2 between 1.5 V and 0.5 V. The galvanostatic-potentiostatic cycling mode was applied in the same manner as for the 0.1 M Cys-DHAQ tests. We reported all battery test results in the main text, with only the battery based on 0.5 M Cys-DHAQ undergoing duplicate testing.
Computational details
The DFT calculations were performed using the Gaussian 16 software package at the m062x theoretical level with the def2tzvp basis set. The structural optimization and frequency calculations were performed for the reduced and oxidized forms of each molecule. The implicit solvent model used for calculating solvation free energy was SMD, while the default IEFPCM model was applied for other calculations to account for solvation effects in aqueous solutions. The optimized structures were confirmed to be true local minima with no imaginary frequencies. Following structural optimization, time-dependent DFT (TDDFT) calculations were performed at the B3LYP/6-311 G(d,p) level, considering the lowest 40 excited states.
All molecular dynamics (MD) simulations were performed using the Forcite module in the Materials Studio 2023 software package, with modifications adapted from previously reported work44. The COMPASS force field was employed to describe interatomic interactions. Long-range electrostatic interactions were calculated using the Ewald summation method, while van der Waals forces were evaluated with the atom-based cutoff approach. To obtain a stable molecular configuration, geometry optimization was conducted using the smart algorithm, which combines steepest descent, adapted basis Newton-Raphson (ABNR), and quasi-Newton methods. The convergence criteria were set to 2.0 × 10−5 kcal mol−1 for energy and 1.0 × 10−3 kcal mol−1 Å−1 for force. The system was then equilibrated under an NPT ensemble (constant pressure and temperature) at 298.15 K and 1 atm for 1 ns using the Nose thermostat and Berendsen barostat. Subsequently, a production run was performed under the NVT ensemble for 100 ns to collect simulation data.
The diffusion coefficient (D) was determined by the slope of fitted Levich equation:
where n = 2, the Faraday’s constant F = 96485 C mol−1, the electrode area A = 0.196 cm2, the 1,5-DHAQ and Cys-DHAQ concentrations c = 2 × 10−6 mol cm−3, the kinetic viscosity ν = 0.01 cm2 s−1.
The rate constant (k0) of 1,5-DHAQ and Cys-DHAQ were calculated from the Koutecky-Levich equation as follows:
where n = 2, the universal gas constant R = 8.314 J K−1, the temperature T = 293.15 K.
Protonation analysis of reduced 1,5-DHAQ, 2,6-DHAQ and Cys-DHAQ
The possibility of protonation of reduced 1,5-DHAQ, 2,6-DHAQ, and Cys-DHAQ were analyzed by DFT calculations (Supplementary Fig. 19).
Firstly, based on the Pourbaix diagram equation, it can be determined that the electrochemical reduction process of 1,5-DHAQ under pH 14 conditions involves the transfer of two electrons and two protons. Consequently, the reduction reaction equation for 1,5-DHAQ under these conditions can be expressed as follows:
The reaction process of 1,5-DHAQ2H2− product with OH− at pH 14:
Then the ΔG was calculated with pH corrections45:
Where kB is the Boltzmann constant and T is the temperature. The results indicate that the reaction between 1,5-DHAQ2H2− and OH− has a ΔG of −0.47 eV, suggesting that this reaction is thermodynamically spontaneous. In contrast, the reaction between 1,5-DHAQH3− and OH− shows a ΔG of 0.04 eV, indicating that this reaction is not spontaneous under the given conditions. The reduction products of 2,6-DHAQ were calculated using the same method.
Secondly, based on the Pourbaix diagram equation, it can be determined that the electrochemical reduction process of Cys-DHAQ under pH 14 conditions involves the transfer of two electrons and zero protons. Consequently, the reduction reaction equation for Cys-DHAQ under these conditions can be expressed as follows:
The reaction process of Cys-DHAQ6− product with H2O at pH 14:
The results show that the ΔG of the reaction between Cys-DHAQ6− and H2O was −0.05 eV, indicating that this reaction proceeds spontaneously. However, the ΔG of the reaction between Cys-DHAQH5− and H2O was 0.25 eV, indicating that this reaction does not proceed spontaneously, which shown that Cys-DHAQ6− can be partially protonated in aqueous solution.
Data availability
The authors declare that all data supporting the finding of this study are available within the paper and its Supplementary Information. Source data are provided with this paper.
References
Park, M., Ryu, J., Wang, W. & Cho, J. Material design and engineering of next-generation flow-battery technologies. Nat. Rev. Mater. 2, 16080 (2017).
Narayan, S. R. et al. Next-generation aqueous flow battery chemistries. Curr. Opin. Electrochem. 18, 72–80 (2019).
Jing, Y. et al. Anthraquinone flow battery reactants with nonhydrolyzable water-solubilizing chains introduced via a generic cross-coupling method. ACS Energy Lett. 7, 226–235 (2022).
Argyrou, M. C., Christodoulides, P. & Kalogirou, S. A. Energy storage for electricity generation and related processes: Technologies appraisal and grid scale applications. Renew. Sustain. Energy Rev. 94, 804–821 (2018).
Koohi-Kamali, S., Tyagi, V. V., Rahim, N. A., Panwar, N. L. & Mokhlis, H. Emergence of energy storage technologies as the solution for reliable operation of smart power systems: a review. Renew. Sustain. Energy Rev. 25, 135–165 (2013).
Guney, M. S. & Tepe, Y. Classification and assessment of energy storage systems. Renew. Sustain. Energy Rev. 75, 1187–1197 (2017).
Luo, J., Hu, B., Hu, M., Zhao, Y. & Liu, T. L. Status and prospects of organic redox flow batteries toward sustainable energy storage. ACS Energy Lett. 4, 2220–2240 (2019).
Zhang, H. & Sun, C. Cost-effective iron-based aqueous redox flow batteries for large-scale energy storage application: a review. J. Power Sources 493, 229445 (2021).
Lourenssen, K., Williams, J., Ahmadpour, F., Clemmer, R. & Tasnim, S. Vanadium redox flow batteries: a comprehensive review. J. Energy Storage 25, 100844 (2019).
Skyllas-Kazacos, M., Cao, L., Kazacos, M., Kausar, N. & Mousa, A. Vanadium electrolyte studies for the vanadium redox battery—a review. ChemSusChem 9, 1521–1543 (2016).
Gentil, S., Reynard, D. & Girault, H. H. Aqueous organic and redox-mediated redox flow batteries: a review. Curr. Opin. Electrochem. 21, 7–13 (2020).
Huang, J., Dong, X., Guo, Z. & Wang, Y. Progress of organic electrodes in aqueous electrolyte for energy storage and conversion. Angew. Chem. Int. Ed. 59, 18322–18333 (2020).
Chen, Q. et al. Organic electrolytes for pH‐neutral aqueous organic redox flow batteries. Adv. Funct. Mater. 32, 2108777 (2022).
Lin, K. et al. Alkaline quinone flow battery. Science 349, 1529–1532 (2015).
Huskinson, B. et al. A metal-free organic-inorganic aqueous flow battery. Nature 505, 195–198 (2014).
Wang, C. et al. Molecular design of fused-ring phenazine derivatives for long-cycling alkaline redox flow batteries. ACS Energy Lett. 5, 411–417 (2020).
Gerhardt, M. R. et al. Anthraquinone derivatives in aqueous flow batteries. Adv. Energy Mater. 7, 1601488 (2017).
Bachman, J. E., Curtiss, L. A. & Assary, R. S. Investigation of the redox chemistry of anthraquinone derivatives using density functional theory. J. Phys. Chem. A 118, 8852–8860 (2014).
Wu, M. et al. Extremely stable anthraquinone negolytes synthesized from common precursors. Chem 6, 1432–1442 (2020).
Wu, M. et al. Highly stable, low redox potential quinone for aqueous flow batteries. Batter. Supercaps 5, e202200009 (2022).
Ruan, W., Mao, J. & Chen, Q. Redox flow batteries toward more soluble anthraquinone derivatives. Curr. Opin. Electrochem. 29, 100748 (2021).
Kwabi, D. G. et al. Alkaline quinone flow battery with long lifetime at pH 12. Joule 2, 1894–1906 (2018).
Wu, M., Bahari, M., Fell, E. M., Gordon, R. G. & Aziz, M. J. High-performance anthraquinone with potentially low cost for aqueous redox flow batteries. J. Mater. Chem. A. 9, 26709–26716 (2021).
Wang, C. et al. N-alkyl-carboxylate-functionalized anthraquinone for long-cycling aqueous redox flow batteries. Energy Storage Mater. 36, 417–426 (2021).
Amini, K. et al. An extremely stable, highly soluble monosubstituted anthraquinone for aqueous redox flow batteries. Adv. Funct. Mater. 33, 2211338 (2023).
Cao, J., Tao, M., Chen, H., Xu, J. & Chen, Z. A highly reversible anthraquinone-based negolyte for alkaline aqueous redox flow batteries. J. Power Sources 386, 40–46 (2018).
Wang, C. et al. Alkaline soluble 1,3,5,7-tetrahydroxyanthraquinone with high reversibility as negolyte for aqueous redox flow battery. J. Power Sources 524, 231001 (2022).
Tong, L., Jing, Y., Gordon, R. G. & Aziz, M. J. Symmetric all-quinone aqueous battery. ACS Appl. Energy Mater. 2, 4016–4021 (2019).
Ji, Y. et al. A phosphonate‐functionalized quinone redox flow battery at near‐neutral pH with record capacity retention rate. Adv. Energy Mater. 9, 1900039 (2019).
Jin, S. et al. A water-miscible quinone flow battery with high volumetric capacity and energy density. ACS Energy Lett. 4, 1342–1348 (2019).
Muench, S. et al. pNTQS: easily accessible high-capacity redox-active polymer for organic battery electrodes. ACS Appl. Energy Mater. 1, 3554–3559 (2018).
Cao, J. et al. A new redox-active conjugated polymer containing anthraquinone pendants as anode material for aqueous all-organic hybrid-flow battery. J. Power Sources 423, 316–322 (2019).
Hatakeyama-Sato, K. et al. Hydrophilic organic redox-active polymer nanoparticles for higher energy density flow batteries. ACS Appl. Polym. Mater. 1, 188–196 (2019).
Liu, Y. et al. Organic electrolytes for aqueous organic flow batteries. Mater. Today Energy 20, 100634 (2021).
Kwabi, D. G., Ji, Y. & Aziz, M. J. Electrolyte lifetime in aqueous organic redox flow batteries: a critical review. Chem. Rev. 120, 6467–6489 (2020).
Bredereck, K., Metwally, S. A., Koch, E. & Weckmann, R. Anthrachinone, VI. Hydroxymethylierung und Aminomethylierung von Anthrachinonen. Liebigs Ann. Chem. 1975, 972–987. https://doi.org/10.1002/jlac.197519750515 (1975).
De Luca, L., Giacomelli, G. & Porcheddu, A. An efficient route to alkyl chlorides from alcohols using the complex TCT/DMF. Org. Lett. 4, 553–555 (2002).
Chang, S. et al. A low-cost SPEEK-K type membrane for neutral aqueous zinc-iron redox flow battery. Surf. Coat. Technol. 358, 190–194 (2019).
Winardi, S. et al. Sulfonated poly (ether ether ketone)-based proton exchange membranes for vanadium redox battery applications. J. Membr. Sci. 450, 313–322 (2014).
Casida, M. E., Jamorski, C., Casida, K. C. & Salahub, D. R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 108, 4439–4449 (1998).
Stratmann, R. E., Scuseria, G. E. & Frisch, M. J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 109, 8218–8224 (1998).
Sharma, K., Sankarasubramanian, S., Parrondo, J. & Ramani, V. Electrochemical implications of modulating the solvation shell around redox active organic species in aqueous organic redox flow batteries. Proc. Natl. Acad. Sci. USA 118, e2105889118 (2021).
Wei, J. et al. Supramolecule-based excluded-volume electrolytes and conjugated sulfonamide cathodes for high-voltage and long-cycling aqueous zinc-ion batteries. ACS Energy Lett. 8, 762–771 (2023).
Xu, C. et al. Unravelling rechargeable zinc-copper batteries by a chloride shuttle in a biphasic electrolyte. Nat. Commun. 14, 2349 (2023).
Huang, S. et al. Molecular engineering of dihydroxyanthraquinone-based electrolytes for high-capacity aqueous organic redox flow batteries. Nat. Commun. 13, 4746 (2022).
Zhao, E. W. et al. Coupled in situ NMR and EPR studies reveal the electron transfer rate and electrolyte decomposition in redox flow batteries. J. Am. Chem. Soc. 143, 1885–1895 (2021).
Zhao, E. W. et al. In situ NMR metrology reveals reaction mechanisms in redox flow batteries. Nature 579, 224–228 (2020).
Fu, R., Lu, T. & Chen, F.-W. Comparing methods for predicting the reactive site of electrophilic substitution. Acta Phys. Chim. Sin. 30, 628–639 (2014).
Deng, Y. et al. Structure, aromaticity and reactivity of corannulene and its analogues: a conceptual density functional theory and density functional reactivity theory study. Mol. Phys. 116, 956–968 (2018).
Liu, Y. et al. Artificial α-amino acid based on cysteine grafted natural aloe-emodin for aqueous organic redox flow batteries. Nat. Commun. 16, 2965 (2025).
Acknowledgements
The authors appreciate the financial support from the National Natural Science Foundation of China (22479074 and 22475096, Z.J.), the General Project of the Joint Fund of Equipment Pre-research and the Ministry of Education (8091B02052407, Z.J.), the Natural Science Foundation of Jiangsu Province (BK20240400 and BK20241236, Z.J.), the Science and Technology Major Project of Jiangsu Province (BG2024013, Z.J.), the Scientific and Technological Achievements Transformation Special Fund of Jiangsu Province (BA2023037, Z.J.), the Academic Degree and Postgraduate Education Reform Project of Jiangsu Province (JGKT24_C001, Z.J.), the Key Core Technology Open Competition Project of Suzhou City (SYG2024122, Z.J.), the open research fund of Suzhou Laboratory (SZLAB-1308-2024-TS005, Z.J.), the Gusu Leading Talent Program of Scientific and Technological Innovation and Entrepreneurship of Wujiang District in Suzhou City (ZXL2021273, Z.J.), and the Chenzhou National Sustainable Development Agenda Innovation Demonstration Zone Provincial Special Project (2023sfq11, Z.J.).
Author information
Authors and Affiliations
Contributions
Z.J. conceived the idea of this study. Z.J. and P.B.Z. designed the experiments. P.B.Z., Y.Z.L., and J.W. performed the sample synthesis, electrochemical measurements, cell tests, data analysis. P.B.Z., Y.Z.L, did the theoretical calculations and analysis. P.B.Z., Y.Z.L., J.W., Z.A.W., X.M.S., G.C.D., H.Z.W., J.C.L., Z.X.T., and Z.J. analyzed the data and discussed the results. P.B.Z. and Z.J. co-wrote and revised the manuscript. Z.J. supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Evan Wenbo Zhao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, P., Liu, Y., Wei, J. et al. An amphoteric and hydrogen-bond-rich artificial α-amino acid for highly durable aqueous redox flow batteries. Nat Commun 16, 4727 (2025). https://doi.org/10.1038/s41467-025-59962-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59962-1
