
Abstract
Current catalysts face challenges with low formate selectivity at high current densities during the CO2 electroreduction. Here, we showcase a versatile strategy to enhance the formate production on p-block metal-based catalysts by incorporating noble metal atoms on their surface, refining oxygen affinity, and tuning adsorption of the critical oxygen-bound *OCHO intermediate. The formate yield is observed to afford a volcano-like dependence on the *OCHO binding strength across a series of modified catalysts. The rhodium-dispersed indium oxide (Rh/In2O3) catalyst exhibits impressive performances, achieving Faradaic efficiencies (FEs) of formate exceeding 90% across a broad current density range of 0.20 to 1.21 A cm−2. In situ Raman spectroscopy and theoretical calculations reveal that the oxophilic Rh site facilitates *OCHO formation by optimizing its adsorption energy, placing Rh/In2O3 near the volcano-shaped apex. A bipolar electrosynthesis system, coupling the CO2 reduction at the cathode with the formaldehyde oxidative dehydrogenation at the anode, further boosts the FE of formate to nearly 190% with pure hydrogen generation under an ampere-level current density and a low cell voltage of 2.5 V in a membrane electrode assembly cell.
Similar content being viewed by others

Introduction
The electrochemical carbon dioxide reduction reaction (CO2RR) to valuable chemicals and fuels opens avenues to upgrade CO2 utilization and recycling, especially when driven by renewable electricity1. Among various products, formate (HCOO−)/formic acid (HCOOH) is an appealing feedstock with considerable market potential in fields such as pharmaceuticals, agriculture, and energy storage1. To date, high selectivity for CO2RR toward HCOO− has been demonstrated using p-block elements1,2, including indium (In), tin (Sn), lead (Pb), and bismuth (Bi). However, maintaining catalytic performance, particularly with high selectivity at industrial-grade current densities exceeding 500 mA cm−2, remains a great challenge1. This difficulty is primarily due to the large overpotentials required to sustain the reaction at such high current densities, as well as the increased competition from the hydrogen evolution reaction (HER)3.
Diverse efforts have been devoted to the development of more active CO2RR catalysts, including nanostructure construction, valence state tuning, facet engineering, doping, and alloying, motivated by different mechanisms2. It would be a significant step forward if a more generalized principle could be provided to guide the rational design strategy in the CO2-to-HCOO− conversion. Numerous studies have elucidated that the adsorption strength of the oxygen-bound intermediate (*OCHO) plays a decisive role in determining the selectivity toward HCOO− (refs. 4,5). Thereby, it is reasonable to propose that modulating surface oxygen affinity could influence the stabilization and activation of *OCHO species5. Inspired by our recent findings on tuning the reactivity of copper (Cu) surface via noble metal single site modification6, we attempt to apply a similar strategy to the HCOO−-generating catalysts, such as indium oxide (In2O3) and tin oxide (SnO2), aiming to alter their oxygen affinities, and accordingly optimize the adsorption energy of the *OCHO intermediate toward a high reactivity of the CO2-to-HCOO− conversion.
To assess the profitability and rationality of CO2RR technology, both cathodic and anodic reactions should be considered for the entire electrolysis system. The oxygen evolution reaction (OER), typically employed as the counter anodic half-reaction, consumes over 90% of input energy on the basis of Gibbs free energy in an ideal CO2 electrolyzer7,8. Replacing OER with a thermodynamically more favorable organic oxidation reaction offers a promising approach to lower the electricity consumption and co-produce value-added chemicals9. For example, the electrooxidation of glycerol paired with the CO(2)RR was reported to dramatically reduce the full-cell voltage7,8. However, the wide distribution of C1 to C3 products from glycerol oxidation poses a major challenge for the downstream separation10. In addition, viscous glycerol likely impedes the mass transfer7, particularly under an industrial-relevant current density, limiting its practical implementation. Considering economic benefits, many popular anodic reactions, such as the electrooxidation of urea, ammonia, hydrazine, and alcohols, result in less expensive final products than the raw reagents, thereby showing inadequate viability9. Alternatively, the electrocatalytic oxidative dehydrogenation (EOD) of aldehydes produces both valuable carboxylates and H2 at ultra-low potentials (close to 0 V vs. RHE)11,12. We speculate that coupling CO2RR with the EOD of aldehyde would be an economically feasible combination to simultaneously optimize the input energy and output product value.
In this work, we highlight the importance of optimizing surface oxygen affinity in the stabilization and activation of the key oxygen-bound *OCHO intermediate for maximizing the CO2-to-HCOO− conversion by using a noble metal modification method. Combined experimental and mechanistic investigations on various modified samples demonstrate the presence of a volcano-type relationship between the HCOO− yield and the *OCHO adsorption energy. Specifically, the Rh-dispersed In2O3 (Rh/In2O3) catalyst emerges as the most active catalyst, achieving a Faradaic efficiency (FE) of HCOO− up to 94% at a partial current density of 808 mA cm−2, corresponding to a generation rate of 15.1 mmol h−1 cm−2. We further corroborate the compatibility and feasibility of dual HCOO− production in a membrane electrode assembly (MEA) cell by coupling CO2RR at the cathode with EOD of formaldehyde at the anode. This paired system not only drastically reduces electricity consumption but also boosts the bipolar HCOO− production rate to 52.0 mmol h−1 cm−2 with a FE of approximately 190%.
Results
Synthesis and characterization of Rh/In2O3 catalyst
A precipitation approach was first employed to obtain the In2O3 matrix. Subsequently, Rh/In2O3 was synthesized via a modified wet impregnation method followed by calcination (Methods). The Rh content was determined by the inductively coupled plasma optical emission spectroscopy (ICP-OES; Supplementary Table 1). The optimal Rh content is 2.06 wt% as will be discussed below. The X-ray diffraction (XRD) pattern shows that cubic In2O3 is the only crystalline phase present in the Rh/In2O3 catalyst (Supplementary Fig. 1), without visible Rh diffraction peaks, suggesting that Rh species are well dispersed. High-magnification high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) reveals that the Rh/In2O3 catalyst has a small particle size of ~15 nm, with well-defined lattice fringes indexed to the (222) planes of In2O3 (Fig. 1a and Supplementary Fig. 2). Energy-dispersive X-ray spectroscopy (EDS) mapping shows a uniform distribution of Rh over the In2O3 matrix (Fig. 1b and Supplementary Fig. 3). The absence of obvious Rh particles implies the atomic dispersion of Rh sites. For comparison, the pristine In2O3 nanoparticles were also prepared as a control sample. The In2O3 catalyst exhibits similar crystallinity and morphology to Rh/In2O3 (Supplementary Figs. 1 and 2), indicating that the incorporation of Rh causes minimal effects on the structure of In2O3.

a, b HAADF-STEM image (a) and STEM-EDS mapping (b) of Rh/In2O3. c High-resolution In 3d XPS spectra of In2O3 and Rh/In2O3. d High-resolution Rh 3d XPS spectrum of Rh/In2O3. e, f XANES spectra at the In K-edge (e) and Rh K-edge (f) of Rh/In2O3. g EXAFS spectra at the Rh K-edge of Rh/In2O3. The corresponding spectra of In2O3, Rh2O3, and metal foils are used as references in e-g. Source data are provided as a Source Data file.
The high-resolution X-ray photoelectron spectroscopy (XPS) analysis of In 3d confirms that the valence state of In is +3 for both In2O3 and Rh/In2O3 (Fig. 1c)13. Both catalysts also display similar O 1s XPS spectra (Supplementary Fig. 4). In the high-resolution Rh 3d XPS spectrum (Fig. 1d), two peaks at 308.5 eV and 313.2 eV can be ascribed to the 3d5/2 and 3d3/2 of Rh3+, respectively14. X-ray absorption spectroscopy (XAS) analysis was performed to further explore the chemical state and coordination environment of the Rh site on the In2O3 matrix. In agreement with the XPS analysis, the In K-edge X-ray absorption near-edge structure (XANES) spectrum of Rh/In2O3 closely resembles that of In2O3 (Fig. 1e). The similar extended X-ray absorption fine-structure (EXAFS) spectra at the In K-edge confirm that the coordination structures of In2O3 matrix remain intact with and without Rh incorporation (Supplementary Fig. 5). The Rh K-edge XANES spectrum indicates that the Rh oxidation state in Rh/In2O3 is very close to +3 (Fig. 1f). The corresponding Rh K-edge EXAFS spectrum displays a prominent peak at ~1.5 Å assigned to the first shell Rh–O scattering path (Fig. 1g). The absence of a distinct Rh–Rh bond at a distance of ~2.4 Å in Rh/In2O3 indicates the lack of Rh particles. The high dispersion of Rh atoms is further corroborated by the EXAFS fitting results (Supplementary Fig. 6 and Supplementary Table 2).
Electrocatalytic evaluation of Rh/In2O3 catalyst for CO2RR
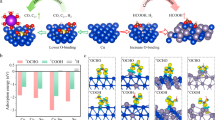
The electrocatalytic performance of Rh/In2O3 was assessed in a flow cell with 1.0 M KOH as the electrolyte. Compared to pristine In2O3, the reactivity of Rh/In2O3 for CO2RR to HCOO− improves with increasing Rh content up to 2.06 wt%, concurrently suppressing the formation of CO and H2 (Fig. 2a and Supplementary Fig. 7). However, a further increase in Rh content leads to a decrease in selectivity toward HCOO−, likely due to Rh aggregation favoring H2 evolution (Supplementary Figs. 8 and 9). Thus, the optimal catalytic performance is achieved with Rh/In2O3 containing 2.06 wt% of Rh, and all the subsequent CO2RR experiments were carried out using this catalyst unless otherwise noted. The FE of HCOO− on the Rh/In2O3 catalyst reaches 94% at −0.88 V vs. RHE, meanwhile the partial current density of HCOO− (jHCOO−) reaches 808 mA cm−2, which are about 1.17- and 3.76-fold higher than those on the pristine In2O3, respectively (Fig. 2a). Over a wide cathodic potential window, the Rh/In2O3 catalyst consistently demonstrates dramatically higher FE and jHCOO− than pristine In2O3. (Fig. 2b, c). Remarkably, steady FEs of HCOO− (> 90%) are kept across a broad current density range (0.20 <jtotal < 1.21 A cm−2). An impressive jHCOO− of 1.09 A cm−2 with a HCOO− FE of 90% is obtained at −0.94 V vs. RHE, corresponding to a formation rate of 20.4 mmol h−1 cm−2 (Fig. 2c and Supplementary Fig. 10). The Rh/In2O3 catalyst outperforms state-of-the-art electrocatalysts for CO2-to-HCOO− conversion, achieving superior performance in both productivity and selectivity (Fig. 2d and Supplementary Table 3).

a Comparison of the HCOO– FEs and jHCOO– at −0.88 V vs. RHE for Rh/In2O3 catalysts with various Rh contents. b, c Comparison of the HCOO– FEs as a function of jtotal (b), and the jHCOO– as a function of potential (c) for In2O3 and Rh/In2O3. Potentials have been 100% iR corrected. Data are presented as mean ± s.d. Error bars represent s.d. from measurements of three independent electrodes. d Performance comparison of Rh/In2O3 with reported electrocatalysts for CO2RR to HCOO–. Details are given in Supplementary Table 3. Source data are provided as a Source Data file.
Elucidating the role of surface oxygen affinity in CO2RR toward HCOO−
The Rh/In2O3 catalyst exhibits a lower onset potential and significantly higher electrochemically active surface area (ECSA)-normalized jHCOO− than pristine In2O3 (Supplementary Figs. 11, 12 and Supplementary Table 4). This signifies that the surface roughness is not the main factor contributing to the enhanced catalytic performance. Systematic characterizations of the post-catalytic Rh/In2O3 confirm well-preserved morphology and uniformly distributed Rh, despite slight reduction in the oxidation states of both In and Rh species (Supplementary Fig. 13). The time-dependent in situ Raman measurements were conducted to monitor the structural changes of Rh/In2O3 at −0.88 V vs. RHE, where the maximum catalytic performance is achieved. The characteristic Raman bands associated with oxide species are present, supporting the robustness of oxidized indium under CO2RR conditions (Supplementary Fig. 14). In line with prior studies, the surface oxide site may partially remain and actively participate in the adsorption and activation of intermediates during CO2RR (refs. 15,16,17,18,19,20). We propose that the metal oxide serves as both the support matrix for the atomic noble metal and an active species collaborating with the noble metal to direct the CO2RR to HCOO− pathway.
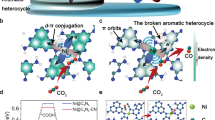
In situ Raman spectra were measured as a function of potential to identify possible intermediate adsorption behaviors. Pronounced peaks at ~1350 and ~1540 cm−1 are attributed to the symmetric stretching of O−C−O from oxygen-bound *OCHO intermediate and the asymmetric stretching of O−C−O from HCOO−, respectively (Fig. 3a)21,22,23. Compared to bare In2O3, the Rh/In2O3 catalyst always displays much stronger band intensities at various potentials, signifying a more favorable formation of oxygen-bound intermediates and accelerated HCOO− production rate. The stabilization of the *OCHO intermediate dictates the effective CO2-to-HCOO− conversion.

a Potential-dependent in situ Raman spectra for In2O3 and Rh/In2O3. Potentials are referenced to the RHE. b Reaction free energy diagrams of CO2RR-to-HCOOH for In2O3 and Rh/In2O3 at −0.88 V vs. RHE. Inset: adsorption configurations of the *OCHO intermediate on In2O3 and Rh/In2O3. The pink, blue, red, brown, and white spheres represent In, Rh, O, C, and H atoms, respectively. c Comparison of HCOO– FEs and jHCOO– for different catalysts at −0.88 V vs. RHE. Potentials have been 100% iR corrected. The molar content of noble metal species is kept at ~5 at%. d Relationship between ECSA-normalized experimental HCOO– activity at −0.88 V vs. RHE and DFT-calculated adsorption free energy of the *OCHO intermediate. The dashed line shows a volcano-shaped relationship to guide the eye. The corresponding potential-limiting step is listed next to each leg of the plot. Data are presented as mean ± s.d. Error bars represent s.d. from measurements of three independent electrodes. Source data are provided as a Source Data file.
We used density functional theory (DFT) calculations to evaluate the favorability of *OCHO formation on In2O3 and Rh/In2O3 surface sites (see Supplementary Note 1 for details). The reduction pathway of CO2 to HCOOH requires two electron-proton transfer steps: (1) * + CO2 + H+ + e− → *OCHO and (2) *OCHO + H+ + e− → HCOOH + *. The endothermic reaction free energy of the first step indicates that the *OCHO formation is the potential-limiting step on the pristine In2O3 surface (Fig. 3b and Supplementary data 1). The high oxophilicity of Rh stabilizes the *OCHO intermediate substantially via the Rh–O coordination, making HCOOH production much more energetically favorable on Rh/In2O3 (Fig. 3b). Therefore, manipulating the oxygen affinity of the catalyst surface via heterometal incorporation proves to be a promising strategy for modulating the binding strength of oxygen-bound intermediates, particularly *OCHO, in the HCOOH formation pathway.
To further validate our hypothesis, we also tested if Ir, which has a similar oxygen affinity to Rh (refs. 24,25), shows a similar effect when incorporated into the In2O3 matrix using the same approach (Supplementary Fig. 15). The resulting Ir-dispersed In2O3 (Ir/In2O3) catalyst also boosts the HCOO− FE to 92% and the jHCOO− to 604 mA cm−2 at −0.88 V vs. RHE (Fig. 3c and Supplementary Fig. 16). DFT analysis also shows that the Ir site can promote the stabilization of the *OCHO intermediate and the favorability of HCOOH generation (Supplementary Figs. 17–20).
In addition to In2O3-based materials, we investigated the impact of the Rh modification on SnO2, another typical HCOO−-selective catalyst (Supplementary Fig. 21). Interestingly, the Rh-dispersed SnO2 (Rh/SnO2) catalyst shows enhanced reactivity for CO2RR to HCOO− than pristine SnO2 (Supplementary Fig. 22), despite SnO2 being a highly oxophilic species26,27. In particular, the Rh/SnO2 catalyst yields a HCOO− FE of 88% with a jHCOO− of 383 mA cm−2 at −0.88 V vs. RHE, whereas bare SnO2 only shows a HCOO− FE of 78% and a jHCOO− of 184 mA cm−2 at a similar potential (Fig. 3c). The DFT computation reveals that SnO2 has the most favorable reaction free energy of the initial electron-proton transfer step (CO2 → *OCHO) among the investigated samples, including In2O3, Rh/In2O3, Ir/In2O3, SnO2, and Rh/SnO2 (Supplementary Fig. 20). However, such strong *OCHO binding on SnO2 hinders the following *OCHO reduction and desorption step (*OCHO → HCOOH), resulting in a sluggish HCOOH production rate (Supplementary Fig. 20). By contrast, the Rh site, with weaker oxophilicity than SnO2, has a weaker *OCHO adsorption strength alleviating the thermodynamic desorption barrier for converting *OCHO to HCOOH on the Rh/SnO2 surface (Supplementary Fig. 20).
Collectively, the In2O3- and SnO2-based materials in our study exhibit varying affinities to the key oxygen-bound *OCHO intermediate, leading to different potential-limiting steps in converting CO2 to HCOO−. Through changes in surface oxygen affinity, noble metal modification influences the stabilization and activation of the *OCHO intermediate, particularly taking the binding affinity of the *OCHO intermediate to a moderate level for both cases of In2O3 and SnO2. A volcano-shaped relationship emerges when correlating the experimentally measured HCOO− activities with the *OCHO adsorption free energies across different samples containing similar molar contents of the noble metal (Fig. 3d and Supplementary Fig. 23). Note that CO2 transport is not the limiting factor within the investigated potential range in our flow cell according to the Tafel plots (Supplementary Fig. 24). Thus, an optimal *OCHO binding is crucial for maximizing the CO2-to-HCOO− conversion, as it balances the energetic barriers of the two protonation steps in the reaction pathway. In our work, Rh/In2O3 with the *OCHO binding strength of −0.07 eV tends to be the most active catalyst for HCOO− production.
Coupling CO2RR and FOR in MEA cell
The cathodic CO2RR process is conventionally paired with an anodic OER. However, OER is kinetic-sluggish and generates a low-value O2 product. Recently, we discovered that the formaldehyde (HCHO) oxidation reaction (FOR) in alkaline media can undergo an EOD pathway on Cu-based materials at low potenitals28. Distinguishing from the conventional oxidation (HCHO + 3OH− → HCOO− + 2H2O + 2e−), our Cu-foam-supported Cu-Ag catalyst (CuAg/CF) enables the oxidative dehydrogenation of HCHO to co-produce H2 and HCOO− at a drastically lower anodic potential (2HCHO + 4OH− → 2HCOO−+ H2 + 2H2O + 2e−)12,28. The highly porous foam substrate benefits mass transport and gas desorption. The Ag modification facilitates C−H bond dissociation of the diol intermediate on CuAg/CF (ref. 28). Taking these advantages, the CuAg/CF electrode reaches anodic current density of 500 mA cm−2 at merely 0.36 V vs. RHE for the FOR (Supplementary Fig. 25), which is a negative shift of ~1.4 V compared to water oxidation on the Ni-foam-supported Ni nanoparticles (Ni/NF). More importantly, the reaction rate of CO2RR to HCOO− on the Rh/In2O3 catalyst is compatible with that of FOR on CuAg/CF, benefiting the high-productivity co-electrosynthesis of HCOO− within a single system under practical conditions.
We are thus motivated to design a reaction system by pairing CO2RR with FOR that driven by the EOD mechanism (CO2RR//FOR), anticipating a dual production of HCOO− at both poles of the electrolyzer with high current densities under low cell voltages (Fig. 4a). CO2RR//FOR was performed in an MEA cell by using CuAg/CF as the anode and the Rh/In2O3 catalyst as the cathode. Paraformaldehyde (10.0 g l−1) as a representative feedstock was fed to the anode containing Ar-saturated KOH (1.0 M) analyte (see Methods for details)28,29. The linear sweep voltammetry (LSV) curve shows that a current density of 500 mA cm−2 requires a cell voltage of ~2.1 V for the CO2RR//FOR system (Fig. 4b), much smaller than that of ~3.5 V for the conventional CO2RR//OER system. We then assessed the HCOO− production using a chronoamperometry model. The conventional CO2RR//OER system delivers a HCOO− FE of ~90% with a jHCOO− of 847 mA cm−2 and a production rate of 15.8 mmol h−1 cm−2 at a cell voltage of 3.9 V (Supplementary Fig. 26). The majority of HCOO− products are collected at the anode owing to the strong electromigration through the anion-exchange membrane (AEM)30,31. On the other hand, the CO2RR//FOR system can achieve HCOO− FEs of ~190% at cell voltages ranging from 2.1 to 2.5 V (Fig. 4c and Supplementary Fig. 27). A peak HCOO− formation rate is ramped up to 52.0 mmol h−1 cm−2 (Fig. 4d), along with pure H2 simultaneously produced at the anode with a rate of 17.9 mmol h−1 cm−2 (Supplementary Fig. 28). Compared to other systems coupling alternative anodic feedstocks for a bipolar HCOO− production, our integrated system achieves a superior current density with a comparable HCOO− selectivity at a relatively lower cell voltage (Fig. 4e and Supplementary Table 5). Moreover, the long-term stability of CO2RR//FOR retains an average HCOO− formation rate of 27.7 mmol h−1 cm−2 at a cell voltage of 2.1 V, producing ~51.3 g of potassium formate and ~5.3 l of pure H2 after 22 h of electrolysis (Supplementary Fig. 29).

a Schematic illustrations of the CO2RR//FOR and CO2RR//OER systems with half-cell equilibrium potentials (E0)28,38. b Comparison of LSV curves for the CO2RR//FOR and CO2RR//OER systems at a scan rate of 20 mV s–1. Voltages are non-iR corrected. c, d HCOO– FEs (c) and HCOO– formation rates (d) for the CO2RR//FOR system at various cell voltages without iR compensation. Data are presented as mean ± s.d. Error bars represent s.d. from three independent measurements. e Performance comparison of paired CO2RR and representative anodic reaction systems for the bipolar HCOO– formation based on performance metrics as jtotal, HCOO– FE, and cell voltage in a flow-type or MEA electrolyzer. Details are given in Supplementary Table 5. f Plant-gate levelized cost per tonne of HCOOH and the corresponding quantity of H2 produced on the CO2RR//FOR and CO2RR//OER systems at a jtotal of 500 mA cm–2. The total reference price of HCOOH and H2 is marked by the dashed line. Source data are provided as a Source Data file.
To assess the economic viability of the HCOO− electrosynthesis powered by renewable energy, a preliminary techno-economic assessment (TEA) was performed considering the costs of capital, separation, electricity, input chemicals, installation, balance of plant, other operations, and carbonate regeneration (Supplementary Fig. 30 and Supplementary Note 2)32,33,34,35. The HCOOH and H2 products are considered as the final products for sale in the analysis. The TEA calculation, based on experimental data at a jtotal of 500 mA cm−2 in the MEA cell, reveals that the plant-gate levelized cost for 1 tonne of HCOOH and the corresponding quantity of H2 is projected to be less than the sum of their reference prices in CO2RR//FOR (Fig. 4f). In specific, its plant-gate levelized cost mostly depends on the cost of input chemicals since formaldehyde is supplied as the anodic feedstock. However, CO2RR//FOR markedly reduces the costs of electricity usage and carbonate regeneration by 5.37 and 3.22 times, respectively, compared to those in the CO2RR//OER system (Fig. 4f). These findings manifest that CO2RR//FOR using the Rh/In2O3 catalyst and a CuAg/CF electrode exhibits better compatibility and economic feasibility than traditional CO2RR//OER (Supplementary Fig. 31).
Discussion
In summary, we report an efficient Rh/In2O3 catalyst that demonstrates ampere-level performance in converting CO2 to HCOO−. The jHCOO− can be ramped up to 1.09 A cm−2, where the HCOO− FE still presents 90%. The mechanistic insight reveals that the affinity for the pivotal oxygen-bound *OCHO intermediate governs the CO2RR reactivity toward HCOO−. This assertion is supported by the observed volcano-type relationship between the ECSA-normalized HCOO− yield and the *OCHO binding energy. We emphasize that different optimizing directions should be aimed when designing HCOO−-favorable catalysts, as their oxygen affinities may vary the potential-limiting step in the CO2RR. For catalysts with a relatively weak binding of *OCHO, such as In2O3, incorporating stronger oxophilic species like Rh and Ir onto the surface favors to stabilize the *OCHO intermediate. This alteration effectively transforms the uphill step of CO2 protonation to *OCHO into a downhill process. Conversely, in the case of SnO2 that exhibits very strong binding strength to *OCHO, the modification with Rh reduces its surface oxygen affinity. This mitigation facilitates the protonation of *OCHO to HCOOH. Thus, we demonstrate the applicability and universality of regulating surface oxygen affinity through noble metal modification to influence the adsorption of the oxygen-coordinated intermediate in CO2RR. We also deduce that the modifier can extend to other elements with an appropriate oxophilicity, structure, and construction method. Moreover, we discover that pairing the CO2RR on Rh/In2O3 with the FOR on CuAg/CF can boost the HCOO− FE to nearly 190% along with pure H2 generation at the anode, merely requiring a low cell voltage of 2.5 V to achieve an ampere-level current density. Integrating the EOD of aldehydes, an alternative anodic reaction, with the high-performance CO2RR holds the potential for considerable productivity and economic advantages in the simultaneous production of valuable chemicals.
Methods
Chemicals and materials
Indium(III) chloride (InCl3, 98%), tin(IV) chloride (SnCl4, 98%), sodium hexachlororhodate(III) (Na3RhCl6, 99%), sodium hexachloroiridate(III) (Na3IrCl6, 99%), and sodium carbonate (Na2CO3, 99.5%) were purchased from Sigma Aldrich and used as received.
Catalyst synthesis
Firstly, the In2O3 nanoparticles were prepared using a precipitation method. Na2CO3 (0.5 M) was added into InCl3 (0.5 M) under strong stirring until pH 10. The mixture was aged for 16 h and then collected by washing and centrifugation. After drying at 60 °C overnight, the precipitate was annealed at 300 °C for 5 h. The as-prepared In2O3 was then pretreated in 10 vol% H2/Ar at 130 °C for 2 h in order to create oxygen vacancies. The Rh/In2O3 catalyst was prepared by a wet impregnation method. The Na3RhCl6 aqueous solution (1.0 mg ml−1) was injected into the partially reduced In2O3 aqueous suspension (10.0 mg ml−1) using a syringe pump at a rate of 1.0 µl min−1 under N2 protection. The oxygen vacancy served as the anchoring site to trap RhCl63− (ref. 36). After vigorous stirring of 12 h under N2, the sample was washed, dried, and calcined in air at 300 °C for 3 h.
A similar procedure was applied to synthesize In2O3, Ir/In2O3, SnO2, and Rh/SnO2 by using corresponding salt precursors. ICP-OES was used to determine the mass contents of Rh and Ir in different samples (Supplementary Table 1).
Material characterizations
XPS data were collected from the PHI Quantera XPS instrument equipped with an Al Kα radiation source. XRD was carried out on the PANalytical X’Pert Pro MPD using Cu Kα radiation. HAADF-STEM and EDS elemental mapping were conducted using the JEOL ARM 200 F microscope, which is equipped with a cold field emission gun and operated at 200 kV voltage. XAS measurements were performed on the Beamline 8-ID of the National Synchrotron Light Source II (NSLS II) at Brookhaven National Laboratory. The Athena and Artemis software in the Demeter package was employed for data processing and analysis. The theoretical EXAFS signal was fitted to the experimental EXAFS data in R-space by Fourier transforming both the theoretical and experimental data.
Electrocatalytic measurements of CO2RR in the flow cell
The CO2RR performance was evaluated in a flow cell with 1.0 M KOH electrolyte (pH 14.0 ± 0.1) at 25 °C. The Rh/In2O3 catalyst ink, containing 5 wt% of Nafion, was spray-coated onto the gas diffusion layer (GDL, Sigracet 39BB) to form the gas diffusion electrode (GDE) as the cathode. The mass loadings of all samples were controlled at ~0.75 mg cm−2 by weighing the mass difference before and after the coating. A piece of Ni foam was used as the anode. A Nafion 117 membrane (Fuel Cell Store, 180 μm, 3 × 3 cm2) was used to separate cathodic and anodic compartments. The fresh electrolyte (50 ml) was prepared before each testing and fed by syringe pumps at 1 ml min−1 and 2 ml min−1 to the cathode and anode, respectively. CO2 gas was supplied to the cathode at 50 sccm via a mass flow controller (Alicat Scientific). A potentiostat (Gamry Interface 1010E) controlled a constant voltage to the flow cell and recorded the corresponding current. A programmable d.c. power supply (B&K Precision XLN3640) was employed when the current density exceeded 1 A cm−2. The cathode potential was measured relative to the Ag/AgCl (3 M KCl) reference electrode, and converted to the RHE scale using: ERHE = EAg/AgCl + 0.209 V + 0.0591 × pH. The reference electrode was calibrated using a standard hydrogen electrode before measurements. The resistance was 3.0 ± 0.4 Ω for an electrode area of 1 cm2, determined by potentiostatic electrochemical impedance spectroscopy (EIS). A 100% iR compensation was manually applied to each potential.
During the electrolysis, an in-line gas chromatograph (GC, SRI Instruments MultipleGas#5) was used to monitor the gas products. Argon, as an internal standard, was mixed with the effluent gas to calibrate its flow rate. The liquid products were collected and quantified via 1H NMR (Bruker NEO 400 MHz spectrometer). 500 μl electrolyte was mixed with 100 μl internal standard of 5 mM 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt in D2O. The standard deviations were calculated based on the measurements of three independent electrodes. The FE for liquid products is calculated as:
where z is the number of electrons transferred for producing a target product; F is the Faraday constant; c is the molarity of a target product determined by 1H NMR; V is the volume of electrolyte collected; Q is the total charge.
The formation rate for liquid products is calculated as:
where t is the electrolysis time; S is the geometric area of the electrode.
Electrocatalytic measurements in the MEA cell
In the CO2RR//OER system, the Rh/In2O3 GDE as the cathode and Ni/NF as the anode28 were separated by an AEM (Fumasep FAA-3-50, 50 μm, 3 × 3 cm2). Humidified CO2 at a rate of 50 sccm was supplied to the cathode. Fresh KOH (1.0 M) was pumped into the anode chamber at 20 ml min−1. A potentiostat (Gamry Interface 5000E) was applied to monitor current densities in a two-electrode system at different cell voltages without iR correction. A cold trap was placed downstream of the effluent gas at the cathode to separate gas and liquid products. Due to the liquid product crossover, the FEs of liquid products were determined based on the sum from both the anode and cathode sides during the same period. The rest procedure was identical to that in a flow cell.
For the CO2RR//FOR system, the CuAg/CF prepared by an electrodeposition method28 was applied to replace Ni/NF as the anode for the FOR. Because the commercial formaldehyde aqueous solution normally contains methanol as the stabilizer and its concentration is always limited (for example, 37 wt%), we adopted paraformaldehyde powder as the feedstock to investigate the FOR process in this case. The formaldehyde molecule would be released from paraformaldehyde in the aqueous solution28,29. As guided by our previous study28, an optimum composition of 10.0 g l−1 of paraformaldehyde in 1.0 M KOH was supplied to the anode. The following measurements were identical to those in the CO2RR//OER system.
To minimize the influence of the Cannizzaro reaction of aldehydes in the alkaline condition, the analyte was acidified by 2 M HCl after the electrolysis and conducted 1H NMR measurements immediately28. Due to the crossover of HCOO− product, both CO2RR and FOR would account for the observed formation rates of HCOO− at the anode in the MEA cell. To determine the FEs of HCOO−, we utilized a water displacement method to quantify the H2 production at the anode, which could transfer to the formation rates and FEs of HCOO− in FOR according to the stoichiometry. Then, the contributions of HCOO− formation rates in CO2RR can be obtained through subtracting those in FOR from the total formation rates of HCOO− at the anode. Meanwhile, the in-line GC was employed to check if CO2 and O2 were generated at the anode, which are possibly originated from HCOO− oxidation and OER, respectively.
In situ Raman measurements
In situ Raman experiment was carried out using a modified flow cell developed by our group37 (Supplementary Fig. 14a). The Raman spectra were recorded on a Renishaw inVia Raman microscope with a 785 nm laser. For each in situ Raman measurement, the acquisition time was 15 s, and the accumulation of scans was 3. Cathode potentials were applied in the potentiostatic mode and converted to the RHE scale accordingly.
Data availability
All the data that support the findings of this study are available in the main text and the Supplementary Information, or from the corresponding authors upon reasonable request. Source data are provided in this paper.
References
Fernández-Caso, K., Díaz-Sainz, G., Alvarez-Guerra, M. & Irabien, A. Electroreduction of CO2: Advances in the Continuous Production of Formic Acid and Formate. ACS Energy Lett. 8, 1992–2024 (2023).
Nitopi, S. et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 119, 7610–7672 (2019).
Jouny, M., Luc, W. & Jiao, F. General Techno-Economic Analysis of CO2 Electrolysis Systems. Ind. Eng. Chem. Res. 57, 2165–2177 (2018).
Feaster, J. T. et al. Understanding Selectivity for the Electrochemical Reduction of Carbon Dioxide to Formic Acid and Carbon Monoxide on Metal Electrodes. ACS Catal. 7, 4822–4827 (2017).
Zhi, X., Vasileff, A., Zheng, Y., Jiao, Y. & Qiao, S.-Z. Role of oxygen-bound reaction intermediates in selective electrochemical CO2 reduction. Energy Environ. Sci. 14, 3912–3930 (2021).
Li, Z. et al. Directing CO2 electroreduction pathways for selective C2 product formation using single-site doped copper catalysts. Nat. Chem. Eng. 1, 159–169 (2024).
Verma, S., Lu, S. & Kenis, P. J. A. Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption. Nat. Energy 4, 466–474 (2019).
Yadegari, H. et al. Glycerol Oxidation Pairs with Carbon Monoxide Reduction for Low-Voltage Generation of C2 and C3 Product Streams. ACS Energy Lett. 6, 3538–3544 (2021).
Na, J. et al. General technoeconomic analysis for electrochemical coproduction coupling carbon dioxide reduction with organic oxidation. Nat. Commun. 10, 5193 (2019).
Wu, J., Yang, X. & Gong, M. Recent advances in glycerol valorization via electrooxidation: Catalyst, mechanism and device. Chin. J. Catal. 43, 2966–2986 (2022).
Wang, T. et al. Combined anodic and cathodic hydrogen production from aldehyde oxidation and hydrogen evolution reaction. Nat. Catal. 5, 66–73 (2022).
Liu, H. et al. Ultra-low voltage bipolar hydrogen production from biomass-derived aldehydes and water in membrane-less electrolyzers. Energy Environ. Sci. 15, 4175–4189 (2022).
Detweiler, Z. M., Wulfsberg, S. M., Frith, M. G., Bocarsly, A. B. & Bernasek, S. L. The oxidation and surface speciation of indium and indium oxides exposed to atmospheric oxidants. Surf. Sci. 648, 188–195 (2016).
Marot, L., Mathys, D., Temmerman, G. D. & Oelhafen, P. Characterization of sub-stoichiometric rhodium oxide deposited by magnetron sputtering. Surf. Sci. 602, 3375–3380 (2008).
Li, J. et al. Probing the role of surface hydroxyls for Bi, Sn and In catalysts during CO2 Reduction. Appl. Catal. B Environ. 298, 120581 (2021).
Wang, W. et al. Carburized In2O3 Nanorods Endow CO2 Electroreduction to Formate at 1 A cm–2. ACS Catal. 13, 796–802 (2023).
Qiu, C. et al. MOF-Transformed In2O3-x@C nanocorn electrocatalyst for efficient CO2 reduction to HCOOH. Nano Micro Lett. 14, 167 (2022).
Li, J. et al. Two-dimensional SnO2 nanosheets for efficient carbon dioxide electroreduction to formate. ACS Sustain. Chem. Eng. 8, 4975–4982 (2020).
Deng, W. et al. Crucial role of surface hydroxyls on the activity andatability in electrochemical CO2 reduction. J. Am. Chem. Soc. 141, 2911–2915 (2019).
Zhang, J. et al. Grain boundary-derived Cu+/Cu0 interfaces in CuO nanosheets for low overpotential carbon dioxide electroreduction to ethylene. Adv. Sci. 9, 2200454 (2022).
Shen, H. et al. In situ constructuring of copper-doped bismuth catalyst for highly efficient CO2 electrolysis to formate in ampere-level. Adv. Energy Mater. 13, 2202818 (2023).
Hayden, B. E., Prince, K., Woodruff, D. P. & Bradshaw, A. M. An iras study of formic acid and surface formate adsorbed on Cu(110). Surf. Sci. 133, 589–604 (1983).
Ichinohe, Y., Wadayama, T. & Hatta, A. Electrochemical reduction of CO2 on silver as probed by surface-enhanced Raman scattering. J. Raman Spectrosc. 26, 335–340 (1995).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
Wu, F. et al. Modulating the oxophilic properties of inorganic nanomaterials for electrocatalysis of small carbonaceous molecules. Nano Today 29, 100802 (2019).
Ye, K. et al. In Situ Reconstruction of a Hierarchical Sn-Cu/SnOx Core/Shell Catalyst for High-Performance CO2 Electroreduction. Angew. Chem. Int. Ed. 59, 4814–4821 (2020).
Sopiha, K. V., Malyi, O. I., Persson, C. & Wu, P. Chemistry of Oxygen Ionosorption on SnO2 Surfaces. ACS Appl. Mater. Interfaces 13, 33664–33676 (2021).
Li, G. et al. Dual hydrogen production from electrocatalytic water reduction coupled with formaldehyde oxidation via a copper-silver electrocatalyst. Nat. Commun. 14, 525 (2023).
Suenobu, T., Isaka, Y., Shibata, S. & Fukuzumi, S. Catalytic hydrogen production from paraformaldehyde and water using an organoiridium complex. Chem. Commun. 51, 1670–1672 (2015).
Li, Y. C. et al. Bipolar Membranes Inhibit Product Crossover in CO2 Electrolysis Cells. Adv. Sustain. Syst. 2, 1700187 (2018).
Zhang, J., Luo, W. & Züttel, A. Crossover of liquid products from electrochemical CO2 reduction through gas diffusion electrode and anion exchange membrane. J. Catal. 385, 140–145 (2020).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, eaav3506 (2019).
Lum, Y. et al. Tuning OH binding energy enables selective electrochemical oxidation of ethylene to ethylene glycol. Nat. Catal. 3, 14–22 (2020).
Leow, W. R. et al. Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density. Science 368, 1228–1233 (2020).
Wang, X. et al. Efficient electrosynthesis of n-propanol from carbon monoxide using a Ag–Ru–Cu catalyst. Nat. Energy 7, 170–176 (2022).
Ye, X. et al. Highly selective hydrogenation of CO2 to ethanol via designed bifunctional Ir1–In2O3 single-atom catalyst. J. Am. Chem. Soc. 142, 19001–19005 (2020).
Li, Z. et al. Planar defect-driven electrocatalysis of CO2-to-C2H4 conversion. J. Mater. Chem. A 9, 19932–19939 (2021).
Kuhl, K. P. et al. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 136, 14107–14113 (2014).
Acknowledgements
This work is supported by the U.S. Department of Energy (DOE) Office of Energy Efficiency and Renewable Energy under IEDO contract DE-EE0010836. Y.S. acknowledges the support of the National Science Foundation (NSF) grant CHE 2328176. T.P.S. and P.W. acknowledge support by the NSF grant CBET 2143941. S.Y. acknowledges the use of facilities within the Eyring Materials Center at Arizona State University, supported in part by NNCI-ECCS-1542160. Z.L. acknowledges the URC Graduate Student Stipend awarded by the Office of Research at the University of Cincinnati. A.I.F. and S.X. acknowledge support by the NSF grant CHE 2102299. S.D.S. is supported by a DOE Early Career Award. J.D.J. is supported by the Brookhaven National Laboratory Goldhaber Distinguished Fellowship. The work carried out at Brookhaven National Laboratory was supported by the DOE, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences (GSGB) Division, Catalysis Science Program under contract DE-SC0012704. The XAS measurements used resource 8-ID of the National Synchrotron Light Source II, a DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under contract DE-SC0012704. We would like to thank E. Stavitski for helping with XAFS data collection. X.L. acknowledges that the research was sponsored by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory, managed by UT-Battelle, LLC, for the DOE. Z.L. and G.H. appreciate experimental support by G. Li.
Author information
Authors and Affiliations
Contributions
Z.L., P.W., and G.H. conceptualized the project under the supervision of Y.S., T.P.S., and J.W.; Z.L. and G.H. synthesized catalysts, performed electrochemical tests, and analyzed experimental data. P.W. performed a DFT simulation. S.Y., S.R., X.L., R.L., P.M.A, J.L., and A.S. conducted catalyst characterizations. Z.L., V.K.R.K., and V.S. carried out in situ Raman measurements. S.X., J.D.J., S.D.S., and A.I.F. carried out XAS measurements and analyses. Z.L., P.W., T.P.S., and J.W. wrote the paper. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Guoxiong Wang, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Wang, P., Han, G. et al. Ampere-level co-electrosynthesis of formate from CO2 reduction paired with formaldehyde dehydrogenation reactions. Nat Commun 16, 4850 (2025). https://doi.org/10.1038/s41467-025-60008-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60008-9
