
Abstract
Gossypium barbadense is renowned for its superior fiber quality, particularly its extra-long fibers, although its fiber yield is lower compared to G. hirsutum. Here, to further reveal fiber-related genomic variants of G. barbadense, we de novo assemble 12 genomes of G. barbadense that span the wild-to-domesticated continuum, and construct a graph-based pangenome by integrating these assemblies and 17 publicly available tetraploid cotton genome assemblies. We uncover the divergent evolutionary trajectories and subsequent exchanges between G. barbadense and G. hirsutum through investigation of structural variants (SVs). We perform the SV-based GWAS analysis in G. barbadense and identify four, three, and seven candidate SVs for fiber length, fiber strength, and lint percentage, respectively. Furthermore, we detect the underlying candidate genes and uncover the origin and distribution of favorable alleles, and reveal the tradeoff between lint percentage and fiber quality. These pangenome and trait-associated SVs provide insights into and resources for improving cotton fiber.
Similar content being viewed by others


Introduction
Cotton (Gossypium L.) is a globally important fiber crop whose production accounts for the majority of natural textile fibers worldwide1. While four cotton species with spinnable fiber were independently domesticated in diverse geographic regions, two cultivated allotetraploid cotton species (i.e., G. hirsutum and G. barbadense) currently dominate the worldwide cotton market. Although the high-yielding cotton species G. hirsutum accounts for more than 90% of cultivated cotton worldwide, the lower-yielding G. barbadense is utilized in specialty applications for its superior extra-long fiber2,3. Detecting the causal variants of fiber quality in G. barbadense is a primary goal for improving cotton fiber through plant breeding.
More than 41 genome assemblies from 19 cotton species have been published during the past ten years, which have significantly stimulated cotton functional genomics4. Several cultivated G. barbadense draft and reference genomes have also been assembled2,3,5,6,7,8,9,10,11, which provide insights into the genomic foundation of superior extra-long fiber. Additional genome resequencing projects have yielded insight into the origin, population structure, and domestication history of G. barbadense12,13. Based on insight from resequencing data, interspecific introgression from G. hirsutum into G. barbadense has been characterized, which accompanied the improved fiber yield and quality during domestication and improvement of G. barbadense2,9,10,12,13,14,15. Furthermore, haploblocks associated with agronomic trait improvement and their distribution have revealed population differentiation in G. barbadense10. Traditional whole-genome association studies based on single-nucleotide polymorphism (SNP-GWAS) have also revealed several variants underlying fiber quality and yield in G. barbadense12,14,16,17,18,19. However, unlike G. hirsutum20,21, few causal variants related to fiber length have been identified in G. barbadense.
Functional genomic studies based on limited genetic resources may miss some key variants, especially large structural variants (SVs)22. SVs play important roles in conferring agronomically important characteristics. For example, SVs may influence the expression of nearby genes by altering the sequence, composition, or position of cis-regulatory sequences23,24, which may affect domestication traits such as non-shattering25,26, yield27, and plant architecture28. At the population genetic level, SVs have also been used to infer introgression23,29. A pangenome aims to discover the genomic variants that exist in a collection of diverse individuals; these variants include single-nucleotide polymorphisms (SNPs), short insertions and deletions (InDels), and larger structural variants (SVs). Constructing the pangenome of a species can stimulate insight into the origin, evolution, selection, and functionality of genomes22,30,31.
Recent advances in plant pan-genomics include the development of graph-based pangenomes32, which aim to capture all genomic variants, especially SVs, within or among species and without any reference bias. In parallel, advanced breeding populations, such as those used in GWAS analysis, have been used in multiple crop species to identify causal variants of interest24,25,33,34,35. Recently, cotton interspecific graph pangenomes were constructed, and SV-based GWAS analysis was used to identify candidate variants affecting fiber quality and yield in G. barbadense9,10. However, these recent studies did not incorporate the broader range of G. barbadense genomes represented by the spectrum of natural diversity, including wild or primitively domesticated forms, and thus, they likely overlooked some SVs that could be associated with phenotypic traits, particularly those involved in domestication. This realization motivated the present study, where genetic variation among G. barbadense accessions spanning the wild-to-domesticated continuum was studied with the aim of discovering genomic diversity and its relationship to the unique fiber properties of domesticated G. barbadense.
Previously, we resolved relationships among tetraploid cotton species and gene pools and elucidated multiple aspects of the parallel domestication of G. hirsutum and G. barbadense13. To further document genetic diversity and mine favorable variants affecting fiber quality in G. barbadense, we de novo assembled 12 G. barbadense genomes using HiFi sequencing reads. These accessions include 7 primitive lines, 2 landraces, and 3 cultivars. We clarify the phylogenetic relationship among G. barbadense accessions. Signals of selection and interspecies introgression are identified using the assembled pangenome in conjunction with resequencing of 671 G. barbadense genomes. Furthermore, we use the graph pangenome to identify G. barbadense SVs and associate them with cotton phenotypes through SV-GWAS analysis for three key agronomic fiber traits, i.e., fiber length, fiber strength, and lint percentage. Additionally, we infer that multiple favorable alleles have historically been combined, which significantly improved fiber quality or yield. Taken together, these analyses (Supplementary Fig. 1) demonstrate the utility of graph-based pangenome analysis to reveal agronomically relevant SVs and introgression within plant breeding improvement efforts in G. barbadense.
Results
De novo assembly of 12 G. barbadense genomes and pangenome analysis
We selected 12 representative G. barbadense accessions based on previous phylogenomic work13 (Supplementary Fig. 2), including 7 primitive accessions from South America, 2 Sea Island landrace accessions from the Caribbean region, and 3 cultivated accessions. An average of 63.1 GB (approximately 25-fold) high-fidelity (HiFi) reads were generated for the 12 accessions (Supplementary Data 1). Genome assessments showed low heterozygosity rates among the accessions, with accession GB0262 exhibiting the highest heterozygosity at 0.5% (Supplementary Fig. 3). HiFi reads were initially assembled via hifiasm36 into individual genomes ranging from 2.21 to 2.25 GB in size (Table 1) and with contig N50 values ranging from 55.0 Mb to 77.9 Mb (average = 65.0 Mb), indicating greater contiguity than the previously published genomes of G. barbadense and its wild sister-species G. darwinii (Table 1 and Fig. 1a). These assembled contigs from individual genomes were further anchored and ordered into 26 pseudo-chromosomes based on the 3–79 reference genome3,37, covering an average of 98.40% (range 97.48%–98.93%) of the 3–79 assembly (Fig. 1b, Supplementary Fig. 4 and Fig. 1c). Multiple metrics were used to validate the assembly quality of the 12 G. barbadense genomes (Supplementary Data 2). The assembly consensus quality value (QV) was estimated with Merqury38, and the high QVs (average = 69.64) indicated high accuracy of 12 G. barbadense genomes. Completeness estimates were similarly high; more than 99.5% of the Benchmarking Universal Single-Copy Orthologue (BUSCO) genes were mapped to the twelve assemblies39, over 98.1% of the Illumina reads mapped to their assemblies, and Merqury estimated completeness >98.7%. Purge_Dups40 revealed low false duplication rates in the 12 G. barbadense genomes, and LTR Assembly Index (LAI) scores were indicative of reference-quality genomes (LAI = 13.78–15.18 per genome)41 (Table 1). Additionally, centromeric regions were evaluated using public ChIP-seq data2. The results of centromeric region identification showed that most of the assemblies had centromeric regions for all 26 chromosomes, except GB0262, which missed only 1 putative centromeric region, which represents significantly greater centromeric recovery than in previously published assemblies (Supplementary Data 3).

a Contig NG(x) of 17 G. barbadense accessions and its wild sister-species G. darwinii. NG(x) values were the length of the contig at the top x percent of the estimated genome size, consisting of the longest contigs. b Sequence alignment between the K101 assembly and the 3–79 reference genome. c Whole-genome collinearity plot between the 12 de novo assembled genomes and the 3–79 reference genome. d Genomic components and phylogenetic relationships of 17 G. barbadense and its wild sister-species G. darwinii. The order of species corresponds to the phylogeny shown. The phylogenetic tree was constructed using high-quality SNP sets and G. darwinii (AD5) as the outgroup. Source data are provided as a Source Data file.
Transposable element annotation yielded repeat sequence contents ranging from 68.92% to 72.89%, with long terminal repeat retrotransposons (LTR-RTs) representing the most abundant class (Fig. 1d and Supplementary Data 4), as expected2,3,6. Gene annotation also varied among these genomes, ranging from 70,810 to 75,729 protein-coding genes, with BUSCO scores ranging from 98.6% to 99.3% (Table 1).
Pan-genome analyses were performed with the 12 de novo assemblies along with 5 previously released G. barbadense genomes (Table 1). A total of 1,224,794 genes from 17 accessions clustered into 54,321 gene families. Furthermore, there were 21,230 singleton genes that were found exclusively in individual G. barbadense accessions, ranging from 275 to 3440 singletons per accession (Supplementary Data 5). Pangenome size permutation data showed that 14 accessions included more than 99% of gene families, and the curve of the number of gene families nearly reached a plateau, indicating that these 17 G. barbadense accessions were both representative and collectively complete (Fig. 2a). These 54,321 gene families were classified into four categories based on their frequency of occurrence: 27,337 (50.32%) gene families were present in all 17 accessions and were defined as core; 6603 (12.16%) gene families were present in 15–16 accessions and were defined as near-core; 19,260 (35.46%) gene families were present in 2–15 accessions and were defined as dispensable; and only 1121 (2.16%) gene families were present in one accession alone and were defined as private (Fig. 2b). Although the dispensable and private gene families collectively accounted for one third (35.46%) of the total number of gene families in the 17 accessions, they only accounted for an average of 12.6% of the genes in individual accessions (Supplementary Data 5). We found that 86.5% of the core genes and 73.2% of the near-core genes contained InterPro domains, much higher than the percentages in the dispensable and private genes (62.7% and 41.8%) (Fig. 2c). The CDS length of core genes and near-core genes is longer than that of dispensable and private genes (Fig. 2d). Moreover, nucleotide diversity (π) and Ka/Ks were higher in dispensable and/or private genes than in core and near-core genes (Fig. 2e, f), indicating that core and near-core genes were more functionally conserved than dispensable and private genes. Gene Ontology (GO) and Pfam enrichment analyses showed that core and near-core genes were enriched in biological processes related to plant growth and development, whereas dispensable and private genes were enriched for abiotic and biotic response genes, consistent with findings in other plants (Fig. 2g, h)42.

a Pan and Core size in terms of the number of gene families. b Composition of the pangenome and individual genomes. The histogram shows the number of gene families in the 17 genomes with different frequencies. Pie chart shows the proportion of the gene family by composition. c Proportion of genes with InterPro domains in Core, Near-core, Dispensable, and Private subsets. Yellow barplot indicates the genes with InterPro domain annotation; the purple barplot indicates the gene without InterPro domain annotation. d The CDS Length of Core, Near-core, Dispensable, and Private genes. Number of gene pairs used in Core, Near-core, Dispensable, and Private genes are 896,642, 167,949, 157,003, and 3180, respectively. e Nucleotide diversity (π) in Core, Near-core, Dispensable, and Private genes. The nucleotide diversity was calculated with SNPs of 671 G. barbadense accessions. Number of gene pairs used in Core, Near-core, Dispensable, and Private genes are 32,671, 7677, 7110, and 90, respectively. f Non-synonymous/synonymous substitution ratios (Ka/Ks) within Core, Near-core, and Dispensable genes. Kruskal–Wallis test was used to determine significance. Number of gene pairs used in Core, Near-core, and Dispensable genes are 15,122, 3153, and 9127, respectively. Pairwise comparisons were performed, using Fisher’s least significant difference. The level of significance used in the post hoc test was 0.001. The upper and lower edges of the boxes represent the 75% and 25% quartiles, the central line denotes the median, and the whiskers extend to 1.5 × IQR in (d–f). g Gene ontology analysis in Core and Near-core (upper panel) and Dispensable and Private (lower panel) genes. h Pfam enrichment analyses in Core and Near-core (upper panel) and Dispensable and Private (lower panel) genes. Source data are provided as a Source Data file.
Structural variation identification in G. barbadense and other tetraploid cotton genome assemblies
SVs (larger than 50 bp) relative to the 3–79 assembly were identified in 16 G. barbadense assemblies based on whole-genome alignment. A total of 357,450 SVs were found with an average of 22,341 per genome. SVs were classified into four main types: presence/absence variation (PAV), duplication (DUP), inversion (INV), and translocation (TRANS). Among these, PAVs were the most frequent (average: 18,452; range: 9962 [Yuma] to 34,513 [GB0262]). The other SV categories were far less frequent, encompassing an average of 3062 DUPs (973 [Yuma] to 5757 [GB0262]); 635 TRANSs (135 [Yuma] to 1596 [GB0262]); and 192 INVs (138 [Yuma] to 379 [PimaS6]) (Supplementary Data 6). To check the presence of these SVs, 30 PAVs were randomly selected and all of them were positively validated by PCR amplification (Supplementary Fig. 5 and Supplementary Data 7). Furthermore, to confirm large inversions, we mapped the Hi-C data of four accessions (3–79, Hai7124, Pima90, and Tanguis) to the corresponding genome assemblies, 30 inversions were randomly selected, and all of them were validated by Hi-C data (Supplementary Fig. 6 and Supplementary Data 8).
We also detected SVs using only the HiFi reads mapped to the reference genome (see “Methods” section). In all cases, the number of identified SVs was substantially lower (21.9%) in the read-mapping dataset than in the genome alignments (Fig. 3a). The undetected rates for SVs identified in the aligned HiFi reads were positively correlated with SV size, indicating bias towards overlooking large SVs using read-mapping data alone. These results are congruent with earlier results in rice genomes that suggest the vast majority of large SVs can only be identified using high-quality genome assemblies33. Accordingly, these high-quality reference genomes allowed us to identify large SVs that were missed by a read-mapping approach, such as the large translocation between chromosomes A01 and A13 identified in GB0414, which was further confirmed by PCR amplification and Sanger sequencing (Supplementary Figs. 7, 8).

a Comparisons between PAVs identified by SyRI pipelines and HiFi read-mapping pipelines among 13 G. barbadense accessions. The number of “Detected” and “Undetected” PAVs, respectively, indicated the number of SVs that are identified and not identified using HiFi reads. b The number of SV terms of four types in each G. barbadense accession and the number of non-redundant SV sets. c The frequency of SVs in G. barbadense accessions indicated that most SVs were present in one or only a few accessions. d Percentage of SVs sharing overlap with different genomic regions in 16 G. barbadense accessions. The upper and lower edges of the boxes represent the 75% and 25% quartiles, the central line denotes the median, and the whiskers extend to 1.5 × IQR. n = 16 in each group. e Distribution of PAV density between repeats and non-repeated regions. f The number of the repeat sequence overlaps with the PAVs. g Distribution of the SVs when an SV was present in half of the 16 G. barbadense accessions. Source data are provided as a Source Data file.
To construct a set of non-redundant SVs, we merged all overlapping SVs across accessions. In total, 129,673 non-redundant SVs affecting 3.05 GB genomic sequence were cataloged, including 99,082 PAVs; 24,192 duplications, 5451 translocations, and 947 inversions (Fig. 3b). We found that the length distribution of different SV types in G. barbadense was similar to that in soybeans24. Most PAVs were smaller than 1 kb, whereas the size ranges for duplications and translocations were both concentrated between 10 and 20 kb; inversions were generally the largest, ranging from 100 to 200 kb (Supplementary Fig. 9). Most SVs were present only in one or a few accessions (Fig. 3c), consistent with reports of SV frequency in 100 tomato accessions23. Most non-redundant SVs were found in intergenic regions. The 7.6% of SVs that overlapped genic regions (i.e., coding regions, introns, and ±2 kb of coding regions) affected more than 27,569 nearby genes among the 16 G. barbadense accessions, with an average of 7924 genes affected per genome (4817 [Yuma] to 12,622 [GB0262]) (Fig. 3d and Supplementary Data 9). Congruent with previous research implicating variation in repetitive sequences in structural differentiation24,25, we found that the PAVs detected in these 16 genomes frequently occur in repetitive regions, primarily those occupied by LTR-retrotransposons (Fig. 3e, f). Among the 16 G. barbadense genomes studied here, the 8 primitive accessions were considered unimproved, while the 2 Sea Island landrace accessions, and 6 cultivated accessions were considered improved (Fig. 1c). Many SVs differentiated these two groups of accessions; for example, 2041 SVs were detected in 8 accessions and more than half of these (1029/2041) of the SVs were only present in the one group, being absent from the other group. These SVs represented fixed differences between unimproved and improved G. barbadense accessions (Fig. 3g and Supplementary Data 10).
To elucidate further the differentiation between unimproved and improved G. barbadense accessions, we extended our analysis of SVs to include 12 previously published genomes of other tetraploid cotton species5,6,43,44,45, including seven G. hirsutum accessions and five other wild tetraploid cotton species. Among all 28 allotetraploid cotton genomes, we identified 721,532 SVs relative to the 3–79 genomes based on whole-genome alignment (Supplementary Data 6; see “Methods” section). These were merged into a set of 350,995 non-redundant SVs representing structural diversity among the seven species of tetraploid cotton. Based on the allele frequency of these SVs, we classified the SVs into four categories: core (present in all 28 samples), near-core (present in 26–27 samples), dispensable (present in 2–25 samples), or private (present in only one sample). We found that two G. barbadense accessions from previous studies, PimaS6 and Hai7124, had the highest ratios of private SVs (Supplementary Fig. 10). These two assemblies were likely outliers because they were highly fragmented and created with an earlier generation of sequencing technology24.
Genetic diversity, population structure, and selection in G. barbadense
To identify genetic diversity within a broader sampling of G. barbadense, 671 representative G. barbadense accessions2,13,14,16,17,18,20 (with an average depth of 17.2×) were collected and genotyped (Supplementary Data 11; see “Methods” section). A total of 5,985,215 high-quality SNPs (maf >0.05 and missing <0.1) were identified. Additionally, 6 wild G. mustelinum and 33 primitive G. hirsutum accessions were collected and genotyped. Neighbor-Joining (NJ) analysis of fourfold degenerate sites was used to evaluate phylogenetic relationships within 710 cotton accessions. In addition to wild G. mustelinum (G0) and G. hirsutum (G1), the 671 G. barbadense accessions were classified into seven groups, a result also supported by population structure and principal component analyses (Supplementary Fig. 11a, b). Group G3 represents primitive G. barbadense (n = 156) in the west of the Andes, which is the native range, and from where G. barbadense later spread under domestication46 to northeastern South America and the Caribbean. The Sea Island landrace (G2; n = 28) includes accessions collected from the Caribbean region, the southeast coast of the United States, and Egypt; these accessions have a complex geographic origin and history46. The remaining G. barbadense groups are the cultivar group representing the majority of G. barbadense grown around the world, including G4 (n = 80) representing Egyptian cotton; G5 (n = 191), mainly grown in Central Asia; G6 (n = 38), representing American Pima cotton; G7 (n = 103), representing the earlier Xinjiang extra-long-staple cotton; and G8 (n = 75), representing the recently developed Xinhai cotton in Xinjiang (Supplementary Fig. 11c). Genetic diversity and pairwise fixation (FST) values in G. barbadense were also calculated. Among the cultivated groups, the FST values of G6 (Pima) versus G8 (Xinhai) were the greatest (0.128), but both of these cultivated groups had the smallest amount of genetic diversity (Supplementary Fig. 11d).
To reveal SV diversity in G. barbadense, a cotton graph-based pangenome was constructed through integrating the SVs from 28 tetraploid cotton genome assemblies. The graph pangenome was used to genotype SVs for 671 G. barbadense and 39 other tetraploid cotton accessions. A total of 43,855 SVs (maf >0.05) were identified (size ranges 51–296,239 bp, mean = 3112 bp, median = 485 bp). In a recent study, short SVs (>20 bp) were reported to contribute to phenotypic variation in maize34, but this possibility has not been explored in G. barbadense. Toward this end, we identified 75,324 short SVs (20–50 bp) based on the graph pangenome and merged the 43,855 large SVs and 75,324 short SVs into a Pan-SV set. Phylogenetic analysis using Pan-SVs differentiated the G. barbadense accessions, separating them into seven distinct groups that are consistent with SNP-based phylogenomic relationships (Supplementary Fig. 12).
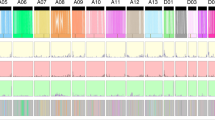
To identify possible regions under selection during breeding and improvement in G. barbadense, putative selective sweep regions were identified using SNPs. Because the modern gene pools of cultivated G. barbadense were derived from landrace (G2) accessions, putative selection signals in cultivated G. barbadense were identified by comparing the diversity in the landrace group to the diversity in the cultivated group. The top 5% regions of πlandrace/πcultivar (=4.09 greater in landrace than in cultivar) and the top 5% regions of FST value (0.41) were recognized as putative regions of selection. Using these criteria, we identified 427 regions (253 regions in AT and 174 regions in DT) that contained 3649 putatively selected genes (2044 genes in AT and 1605 genes in DT; Fig. 4a, Supplementary Fig. 13, 14, Supplementary Data 12, 13). Some of these regions contained genes encoding transcription factors that have been previously associated with fiber development, such as MYB2547, HOX348, and HD149. Furthermore, we identified 5590 SVs among the 16 G. barbadense accessions in the selective sweep regions, including 4670 PAVs, 740 DUPs, 148 TRANS, and 32 INVs. For example, a 887-kb inversion was found on the D11 chromosome from 57.72 to 58.61 Mb of the 3–79 genome, and this inversion was only identified in four unimproved accessions and its wild sister-species G. darwinii (Supplementary Fig. 15a). This inversion was further confirmed by Hi-C (Supplementary Fig. 15b). Three major haplotypes were identified in this inversion among 671 G. barbadense accessions. Hap1 and Hap2 were found in primitive and landrace groups, while Hap3 was found in the cultivars (Supplementary Fig. 15c, d). Notably, six genes at the boundaries of this inversion are involved in various abiotic and biotic stress responses. At one boundary, two genes Gbar_D11G002900 and Gbar_D11G002901 are homologous to ATL650 in Arabidopsis thaliana, which encodes a putative RING-H2 zinc finger protein responsive to environmental stresses; at the other boundary, four genes from Gbar_D11G002862 to Gbar_D11G002865, contain the NB-ARC domain, which mediates plant resistance. These putatively selected SV regions thus provide insights for past and potentially future crop improvement.

a Diversity and FST plots detecting putative regions of selection in G. barbadense, with purple lines indicating the 5% threshold. The approximate position of functional genes known to be associated with fiber development is indicated by their respective gene names. b Scatter plots showing Pan-SV frequencies in landrace and cultivar (adj p-value computed using two-sided Fisher’s exact test). c Frequency pattern of improvement-related Pan-SVs. Lines in red and green indicate impSVs during improvement. d Venn plot of improvement genes that were identified by the Pan-SVs and SNPs. e Pan-SV frequency captures some genes that were not identified by SNPs. The distribution of FST values (purple curve) and πlandrace/πcultivar (yellow curve), the dotted line (purple and yellow) represents the top 5% value of FST and πlandrace/πcultivar, respectively (Upper). The red blocks represent the SVs that were potentially selected during in G. barbadense improvement (Middle). SV frequency for each group is shown in the bar charts (Lower).
To further identify possible targets of selection, Pan-SVs frequencies between landrace and cultivated G. barbadense were compared, and a total of 10,015 SVs exhibited significantly different frequencies (Fig. 4b). Among these, 3142 SVs that have consistently elevated or reduced frequencies in both landrace and cultivated accessions were identified as improvement SVs (impSVs)25 (Fig. 4c and Supplementary Data 14). A total of 1097 genes were affected by these impSVs, but only 211 genes were identified by both Pan-SV frequency and selective sweeps based on SNPs (Fig. 4d). These genes, which included the drought stress response gene WRKY3351 and the cellulose synthase gene CESA152, were detected by both methods (Supplementary Data 15). Additionally, the Pan-SV frequency data captured some genes that were not identified by SNPs, such as the fiber development genes FLA153 and PDF254, which are perhaps relevant to fiber quality improvement in G. barbadense (Fig. 4e). Furthermore, a total of 2149 impSVs were detected among 336 G. barbadense accessions18. These impSVs were then associated with 14 agronomic traits, with 1260 impSVs directly related to at least one of 14 traits (Supplementary Data 16). For example, the reference haplotype of SV_10_95043356 was positively correlated with fiber yield (LP and SI) and negatively with fiber quality (FL, FS, and FE) improvement, early maturity (GP), and plant height (PH), while the alternative haplotype has opposite relationships with these traits (Supplementary Fig. 16). We found that the fiber yield (reference) haplotype was selected during domestication and early improvement, while the fiber quality (alternative) haplotype was more recently under selection during later crop improvement, especially Xinhai serial cultivars in Xinjiang region (Supplementary Fig. 17). These impSVs provide insight into the genetic basis of fiber yield and quality improvement in G. barbadense.
Interspecific introgression and genomic divergence in G. barbadense
Natural and artificial selection have driven genomic divergence accompanying geographic differentiation in multiple crops20,55. The G. barbadense genomic divergence located on chromosome A01 has been reported previously2,10,12,15, but its origin and evolution have not been clear. Our extensive sampling of G. barbadense accessions created a robust phylogenomic framework and allowed us to identify and characterize the evolution of this region. We clustered the SNP genotypes of the G. hirsutum and G. barbadense accessions on chromosome A01, and four general haplotypes (Hap1 to Hap4) were identified in G. barbadense, with only one haplotype identified in G. hirsutum (Hap0) (Supplementary Data 11). Pairwise FST comparisons among G. barbadense accessions showed that the peak of the divergent regions ranged from ~41 to ~110 Mb on chromosome A01 (Fig. 5a); this region is larger than previously reported based on nine G. barbadense accessions2. In addition, eight inversions were identified in this introgression region that exist in all eight unimproved G. barbadense, but are not present in improved G. barbadense and G. hirsutum (Supplementary Fig. 18). Because these inversions are low-frequency variants among multiple species56, and multiple independent occurrences of the same naturally occurring SV are improbable, the eight inversions provide robust evidence for the introgression interpretation. For the G. barbadense haplotype, nearly all primitive G. barbadense accessions carry Hap1, which represents the primitive G. barbadense haplotype (Fig. 5b). Hap4, discovered in four Sea Island landrace accessions from Caribbean regions, is similar to Hap0 and supports the idea that G. barbadense spread throughout the Caribbean and hybridized with G. hirsutum13,57,58. The Sea Island landrace represents the gene pool of cultivated G. barbadense, and it was subsequently introduced into America and Egypt57. Two recombinant haplotypes (Hap2 and Hap3) were identified in Egypt, perhaps derived from the hybridization between Hap1 and Hap4. We found the introgression regions of Hap2 at 52–75 Mb and 93–102 Mb and the introgression region of Hap3 only at 93–102 Mb, which corroborate previous reports using different populations and different variation identification technologies2,12. Three haplotypes (Hap2, Hap3, and Hap4) further spread as G. barbadense became a global crop, with genomic divergence among different planting areas accompanying this geographic diversification. For example, Hap2 is highly polymorphic in Xinjiang cotton, Hap3 reaches comparably high frequency in Central Asia, and Hap4 attains high frequency in Pima cottons (Fig. 5c). In summary, the occurrence and evolution of introgression on the A01 chromosome provide insights for understanding the genetic history of G. barbadense.

a Genomic divergence and haplotype classification of chromosome A01. Colored lines represent FST values for pairwise groups (top). Haplotype classification of the A01 chromosome in 710 accessions (bottom). Diagrams left of the y-axis represent the major haplotypes. The colored bar represents the samples of 671 G. barbadense and 33 primitive G. hirsutum accessions (left). b Haplotype category of each group. The colors of the pie charts of (b) also correspond to the bars in (a). c Geographic distribution of the 671 G. barbadense accessions. The number of accessions collected from each region is indicated by the size of the pie, and the ratio of haplotypes for each region is shown in the pies.
To further explore interspecific introgression from G. hirsutum into G. barbadense, we identified introgressed SVs (iSVs) throughout the genome of G. barbadense. An SV that was shared with improved G. barbadense accessions and at least three G. hirsutum accessions (including race punctatum TX1000) but was absent in all 8 unimproved G. barbadense accessions and its wild sister-species G. darwinii59, was inferred to be an iSV. A total of 23,411 non-redundant iSVs (including 19,613 PAVs, 2837 duplications, 838 translocations, and 123 inversions) were identified, which overlapped with 6824 genes (including 2 kb flanking both the transcriptional start and end sites) (Supplementary Data 17). The introgressed regions are typically large (an average length of 87.7 kb; see “Methods” section). Phylogenetic analysis of flanking sequences (50 kb surrounding the iSVs) was used to validate the quality of iSV inference (Supplementary Fig. 19). A total of 100 iSVs were randomly selected, and phylogenetic analysis of flanking sequences (50 kb surrounding the iSVs) was conducted. More than 96% of the iSVs were confirmed using this approach (Supplementary Data 18). We found that 22,735 iSVs were detected in Caribbean landrace accessions, especially in accession GB0262 (21,083 iSVs), and that only 2889 iSVs were detected in cultivated accessions, implying that most introgressed regions did not make it into the cultivated G. barbadense gene pool (Supplementary Fig. 20a and Supplementary Data 19). D statistics also showed more introgressed regions were detected in Caribbean landrace accessions, especially in GB0262 (Supplementary Data 19). The distribution of iSVs in GB0262 is widespread across the genome (Supplementary Fig. 21), whereas in the other 7 improved accessions, the 3947 iSVs were primarily detected in chromosomes A01, A08, and D11 (Supplementary Fig. 22). For the 2889 iSVs detected in the cultivated G. barbadense gene pool, half of the SVs (577/1029) differentiated between unimproved and improved G. barbadense accessions (Fig. 3g), documenting the extent to which cotton improvement involved interspecies introgression. Furthermore, comparing the introgression regions detected by SNPs and iSVs, we found half (1591) of the iSVs were not detected in previous work17. Noting that interspecific introgression may possibly be relevant to selection, such as that which accompanied the origin of photoperiod neutrality of G. barbadense (which possibly was acquired from G. hirsutum in the Caribbean57), we found a 54-bp iSV in the Caribbean landrace accessions (GB0262) that is fixed in cultivated G. barbadense (Supplementary Fig. 20b, c). This iSV is positioned 392 bp downstream of Gbar_D11G002753, a gene that is homologous to the flowering time regulator FLK60 in Arabidopsis thaliana.
SVs are related to the differentiation of fiber quality and yield differences between G. hirsutum and G. barbadense
Structural variation has potentially contributed to fiber development in cotton, and has been identified among cultivated G. barbadense and G. hirsutum2,3,6. This earlier work likely overlooked some SVs due to reciprocal introgression and a more limited sampling of genomes. To further define interspecific divergence between G. hirsutum and G. barbadense, we identified 34,939 species-specific SVs, defined here as those that occurred in most accessions (more than 80%) of one species (G. barbadense or G. hirsutum) and were absent in the other. These species-specific SVs were further manually checked using the Integrative Genomics Viewer (IGV)61 (Supplementary Fig. 23). These 7035 species-specific SVs overlapped with 7938 nearby genes (2 kb region flanking the transcriptional start and end sites) (Supplementary Data 20). Among these genes, we found multiple genes associated with fiber development (Supplementary Fig. 24 and Supplementary Data 21). For example, JAZ2[ 62, bHLH18 63, and MYB transcription factors such as MYB25-like64, MYB2547, MYB106, and MYB365, play key roles in fiber initiation. Transcriptome data66 show that fiber initiation is inhibited in G. barbadense relative to G. hirsutum during the earliest stage of fiber initiation (−3 to 1 DPA) (Supplementary Fig. 25). Increasing the fiber initial number per seed has the potential to improve fiber yield47. In addition, MYB10967, VIN168, and APX169 are more highly expressed in G. barbadense than in G. hirsutum during the fiber elongation stage (3–25 DPA) (Supplementary Fig. 26), correlated with the longer and stronger fibers in G. barbadense. These species-specific SVs and transcriptome data provided clues into the differentiation of fiber quality and yield between G. barbadense and G. hirsutum.
GWAS analysis in G. barbadense
To further identify genetic contributions to fiber quality and yield, 336 G. barbadense accessions18 were subjected to both SNP-based and SV-based GWAS analysis with three key quality and yield traits, i.e., fiber length, fiber strength, and lint percentage. For SNP-based GWAS analysis, our results are consistent with previous studies12,18 (Supplementary Data 22). SV-based GWAS analysis revealed 14 SVs loci, including four fiber length-related loci (FL1, FL2, FL3, and FL4), three fiber strength-related loci (FS1, FS2, and FS3), and seven lint percentage-related loci (LP1, LP2, LP3, LP4, LP5, LP6, and LP7) (Fig. 6a, Supplementary Figs. 27–29). Among these, three lint percentage-related loci (LP1, LP4, and LP6) were first discovered here, and the 11 remaining SV loci overlapped with those previously reported9,10,12,17,18. We further explored the underlying candidate genes and the origin and distribution of favorable alleles (Supplementary Data 23). The linkage between individual SVs and surrounding SNPs (50 kb flanking the SVs) was evaluated, revealing that more than 76.5% of the SVs had loose linkage to a nearby SNP (r2 < 0.5), suggesting that some genomic regions were overlooked in SNP-GWAS studies (Supplementary Fig. 30).

a Manhattan plots of GWAS for fiber length (Upper), fiber strength (Middle), and Lint Percentage (Lower). Pie charts represent allelic frequencies of each locus in the GWAS population. Gray and blue dotted lines indicate the significant thresholds of GWAS at α = 0.05 and α = 1, respectively. b Dot plots show the effects of allelic combinations for fiber length (Upper), fiber strength (Middle), and Lint Percentage (Lower) in the population. Green dots represent the accessions categorized according to different allelic combinations. NA indicates accessions carrying no favorable allele. The upper and lower edges of the boxes represent the 75% and 25% quartiles, the central line denotes the median, and the whiskers extend to 1.5 × IQR. c Schematic illustrates the origin and recombination of favorable alleles related to lint percentage and fiber quality during improvement of G. barbadense.
G. barbadense is well known for its superior fiber quality. Among the fiber length and strength-related loci, the FL1 locus and FS3 locus, on chromosomes A05 and D09, respectively, as inferred to improve fiber length and fiber strength, with the favorable allele of FL1 was inferred to have been introgressed from G. hirsutum and the favorable allele of FS3 was inferred to have originated from primitive G. barbadense (Supplementary Fig. 31 and Supplementary Fig. 32). FL3 was localized here to chromosome A07 and we detected the candidate variation as a 47-bp insertion positioned 9419 bp downstream of a gene encoding a Pectin methylesterase inhibitor (Gbar_A07G000183), which was associated with a 6.3% improvement of fiber length, and the presence of this insertion decreased Gbar_A07G000183 expression during fiber elongation (10 DPA and 15 DPA) relative to samples containing the ordinary allele. This favorable allele originated from G. hirsutum and was introgressed into G. barbadense (Supplementary Fig. 33).
As with G. hirsutum20, there is significant positive correlation between fiber length and fiber strength, with two major fiber strength loci FS1 and FS2 co-located with two fiber length loci FL2 and FL4, respectively (Supplementary Fig. 34a). FL2/FS1 and FL4/FS2 were detected on chromosomes A06 and D03, respectively. We further detected candidate variants consisting of a 29-bp deletion positioned 6593 bp upstream of a gene encoding an early nodulin-like protein 22 (Gbar_A06G000806) and a 20-bp insertion positioned in the sixth intron of a gene encoding a Sec14p-like phosphatidylinositol transfer protein (Gbar_D03G000163) (Supplementary Fig. 34b, c). The former was associated with an increase of fiber length (7.7%) and fiber strength (10.8%) and increased the expression of Gbar_A06G000806 during the fiber elongation stage (10 DPA) relative to ordinary (Supplementary Fig. 34d–f). The latter was associated with an increase in fiber length (12.1%) and fiber strength (22.1%), and decreased Gbar_D03G000163 expression during the fiber elongation stages at 10 DPA and 25 DPA, relative to ordinary (Supplementary Fig. 34g–i). These favorable allele combinations, FL2/FS1, were inferred to have originated from primitive G. barbadense, and the FL4/FS2 were introgressed from G. hirsutum (Supplementary Fig. 34j, k).
Among seven lint percentage-related loci, LP1, on chromosome A01, exhibited candidate variation as a 36-bp deletion positioned in the sixth intron of a gene encoding the microtubule-associated protein WDL4 (Gbar_A01G002501). This deletion was associated with a markedly improved lint percentage (12.0%), and its presence increased the expression of Gbar_A01G002501 in the accession TM1 (G. hirsutum (AD1)) in developing lint-bearing ovules at 3 and 5 DPA relative to Hai7124 (G. barbadense (AD2)) and 3–79 (G. barbadense (AD2)) (Supplementary Fig. 35a–d). The favorable allele of LP1 was inferred to have been introgressed from G. hirsutum (Supplementary Fig. 35h). The candidate variation of LP2 was reported in a previous study11, and the favorable allele is inferred to have originated from primitive G. barbadense (Supplementary Fig. 35i). We detected candidate variation in LP3 on chromosome A10, which is a 24-bp insertion positioned in the 3’ UTR region of SDH3-1 (Gbar_A10G001515) that encodes one of the membrane anchor subunits of mitochondrial respiratory complex II. The insertion is associated with a 12.4% improved lint percentage. Gbar_A10G001515 was highly expressed in developing lint-bearing ovules at 3 and 5 DPA, and G. hirsutum TM1 had higher expression at 5 DPA compared to accessions with the Hai7124 and 3–79 (Supplementary Fig. 35e–g). The favorable allele of LP3 was inferred to have been introgressed from G. hirsutum (Supplementary Fig. 35j). LP4 was localized here to chromosome A10, exhibited candidate variation in the form of a 47-bp deletion positioned 791 bp upstream of a gene (Gbar_A10G002525) encoding a RNA-binding family protein, and was associated with a reduced lint percentage of more than 7.1%. Its presence decreased expression of Gbar_A10G002525 in developing fibers at 3 and 5 DPA compared to Hai7124 and 3–79 (Supplementary Fig. 36). The LP5 locus on A13 chromosome and has a favorable allele that significantly improves lint percentage (Supplementary Fig. 37). LP6 on chromosome D04 exhibits candidate variation as a 24-bp deletion positioned 791 bp upstream of a gene encoding a RNA helicase family protein (Gbar_D04G001079); here, the favorable allele of LP6 was associated with an increase of 6.8% lint percentage (Supplementary Fig. 38). The LP7 locus on D13 chromosome and we detected the candidate variation as a 47-bp insertion positioned 1284 bp upstream of a gene (Gbar_D13G002432) encoding an alpha/beta-Hydrolases superfamily protein, which originated from G. hirsutum and was associated with an increase of 6.7% lint percentage. Its presence impacted the expression of Gbar_D13G002432. Expression of the shared Hai7124 and 3–79 allele was lower in developing ovules at 3 and 5 DPA relative to the G. hirsutum reference TM1 (Supplementary Fig. 39).
The genetic architecture of fiber quality and yield in G. barbadense
To evaluate the pyramiding effects of favorable alleles for each candidate SV in 336 G. barbadense accessions, we compared fiber yield and quality among accessions carrying multiple favorable allele combinations. As expected, accessions carrying more favorable alleles exhibited increased fiber length, fiber strength, and lint percentage (Fig. 6b). Because lint percentage was negatively correlated with both fiber length (Pearson r = -0.41, p-value = 2.62e-15) and fiber strength (Pearson r = -0.19, p-value = 0.0003), most accessions with higher fiber quality likely carried few favorable alleles for lint percentage (Supplementary Fig. 40, Supplementary Data 24). To further explore the relationship between fiber quality and lint percentage, we performed association analysis for each candidate SV among fiber length, fiber strength, and lint percentage. The favorable alleles of all lint percentage loci are significantly negatively correlated with fiber length and/or fiber strength, making it difficult to improve lint percentage without compromising fiber quality (Supplementary Fig. 41). The fiber quality-related loci could affect both fiber length and fiber strength except FL3, and three candidate SVs (FL1, FL3, and FL4/FS2) could improve fiber quality without compromising lint percentage (Supplementary Fig. 42). We found that most accessions carried the favorable allele for FL1, but few accessions carried the favorable allele for FL3. The favorable allele of FL4/FS2 exhibited more significant enhancement effects on both fiber length (reaching a maximum of 12.1%) and fiber strength (reaching a maximum of 22.4%), and this allele combination is enriched in the Xinhai (G8) (Fig. 6a and Supplementary Fig. 34k). For fiber strength, half of the accessions (n = 185) didn’t carry favorable alleles, except Xinhai accessions (G8) which combined three favorable alleles (Supplementary Figs. 32c and 34j, k).
Because we detected multiple loci for fiber quality and yield and placed these in a phylogenomic context, we were able to reconstruct aspects of fiber quality and yield enhancement in G. barbadense. For fiber quality, the favorable allele combination FL2/FS1 + FL4/FS2 + FS3 was found in Xinhai (G8), suggesting that this allele combination was a target of selection (Fig. 6c and Supplementary Figs. 32c, 34j, k). For fiber yield, we found three favorable alleles for lint percentage (LP1, LP2, and LP3) had stronger linkage and were combined in Pima cotton, with the favorable allele of LP2 originating from the Tanguis accession (primitive G. barbadense), and the favorable alleles of LP1 and LP3 having been introgressed from G. hirsutum (Supplementary Fig. 35h–j). The Pima S1 gene pool is the one of most important contributors of the modern Pima gene pool, which was created by intercrossing three diverse G. barbadense germplasm sources (Pima, Sea Island and Tanguis) and one G. hirsutum accession (Stoneville)57,70 (Supplementary Fig. 43). We speculate that three favorable alleles affecting lint percentage were unknowingly but intentionally combined into Pima S1 through this breeding scheme (that includes interspecific hybridization) and were inherited in the G6 group (Fig. 6c).
Discussion
Allotetraploid cotton is an attractive model for studying the origin and domestication of agronomically important crop species, having originated from a hybridization and chromosome doubling event approximately 1–2 million years ago (mya), and subsequently diverging into at least seven species, two of which (G. hirsutum and G. barbadense) were independently domesticated71,72,73,74. G. hirsutum has high yield and wide adaptability, while G. barbadense has longer and stronger fibers2,3. Of special interest here was the detection of favorable variants affecting fiber quality in G. barbadense and which may have facilitated fiber improvement, as well as phylogenetically determining their origin and diffusion pathways under domestication.
Pangenome analysis in plants has provided insights into evolution and domestication, revealing genetic diversity among wild relatives and offering insights into crop improvement25,33,75. In this study, we de novo assembled 12 high-quality genomes from diverse accessions of G. barbadense spanning the wild-to-domesticated continuum, and performed pangenome analysis on these genomes and 5 previously released G. barbadense genomes. Pangenome size permutation data showed that the curve of the number of gene families nearly reached a plateau, which indicated that these 17 G. barbadense accessions were both representative and collectively complete. In addition, we identified SVs relative to the reference genome 3–79 among 28 tetraploid cotton genomes (i.e., seven G. hirsutum genomes, sixteen G. barbadense genomes, and one genome for each of the other tetraploid cotton species), which enabled us to identify species-specific SVs between G. hirsutum and G. barbadense, introgressed SVs from G. hirsutum into G. barbadense, and SVs that putatively have experienced selective sweeps in G. barbadense. Analysis of these SVs sheds light on past selection, the history of interspecific introgression between the two cultivated tetraploid species G. hirsutum and G. barbadense, and divergence among the species. Furthermore, the Pan-SVs were genotyped based on the cotton graph pangenome, and SV-GWAS analysis was used to detect more candidate SVs than was possible using only SNP-based GWAS analysis. In summary, we constructed the pangenome of tetraploid cotton and illustrated its utility for revealing phylogenomic history and crop improvement.
Interspecific introgression has long been suspected to have been important in facilitating the improvement of yield or quality in G. barbadense9,10,12,14,17,20,76. Deep sequencing data, including nine G. barbadense and ten G. hirsutum accessions, had identified an introgressed region from 43.10 Mb to 92.00 Mb on chromosome A01 of G. barbadense and revealed the differentiation of introgressed regions2. In a recent study, haplotype block distinctions have further revealed the genomic basis of geographic differentiation in cultivated G. hirsutum10. Here, we performed haplotype block analysis with 704 tetraploid cotton accessions and confirmed that the introgressed region on chromosome A01 ranged in size from ~41 to ~110 Mb. We further identified four general haplotypes in this region, and determined that the introgression occurred in the Caribbean region, with two recombinant haplotypes possibly originating in Egypt (Fig. 5c). These analyses complement the results of deep sequencing data, showing a larger introgressed region and revealing more haplotypes and the details of their origin in cultivated G. barbadense. In addition, our synthesis of SVs among these species and genomes permitted the identification of 23,411 introgressed SVs (iSVs) that are phylogenetically inferred to have been transferred from G. hirsutum into G. barbadense during crop improvement as the two species became sympatric, first in the American tropics and subtropics, and in the last several hundred years as cotton spread globally. We also show that most iSVs are not in the cultivated G. barbadense gene pool. Also, half of the remaining iSVs were not detected in the previous study based on SNPs, which revealed that iSVs offer additional levels of insights not apparent from SNP data alone.
G. hirsutum has a high yield relative to G. barbadense, and this has led to fiber yield improvement in cultivated G. barbadense from interspecific introgression. Two yield-related QTLs (q-D03-SCW and q-D03-LW) are located in the Gb_INT13 introgression region, which significantly improved fiber yield in G. barbadense14. Multiple introgression regions are associated with yield traits, such as five boll weight (BW)- and six LP-associated IS regions, which improved G. barbadense yield, and two interspecific Gh-i regions (Gh-i3 and Gh-i5) that increase LP12,17. As shown earlier, the accumulation of higher LP haplotypes from G. hirsutum gradually increased LP in G. barbadense10. In addition, SV-GWAS analysis identified a large-effect SV to improve lint percentage, the favorable allele having originated in primitive G. barbadense and introduced into cultivated Pima cotton11. Here we identified seven candidate SVs related to lint percentage, and further found that favorable alleles of three loci (LP1, LP2, and LP3) had stronger linkage and were combined into Pima S1 through interspecies hybridization and transmission into modern Pima cotton.
For fiber quality, SV-GWAS revealed that the favorable alleles on chromosomes A01, A06, and D09 were introduced into the Xinjiang type G. barbadense (GbCHN) from the Egyptian type in Egypt (GbEGY) and the Central Asian type (GbCA), consistent with Chinese ELS cotton breeding history9. Genome-wide association study and haplotyping analyses have shown that interspecies introgression also increased the fiber quality of G. barbadense. For example, two fiber micronaire-related haplotype blocks were detected, and a G. hirsutum-introgressed haplotype block (FM2) that might enhance FM in a small subset of G. barbadense cultivars was described17. In a different study, a total of 25 G. hirsutum ISs were related to fiber quality of G. barbadense, and 13 ISs showed a positive role in fiber quality improvement19. In this study, we detected 4 fiber length and 3 fiber strength-related candidate SVs and found that a favorable allele combination, FL2/FS1 + FL4/FS2 + FS3, became recombined in the Xinhai group (G8), revealing the introgressive and historical origin of the genomic basis of improved fiber quality in this cultivar.
For fiber yield and quality improvement, interspecific introgression has been known to have yield-increasing potential, but often with negative tradeoffs, such as that between yield and fiber quality12. Here we also found that the favorable alleles of lint percentage loci are significantly negatively correlated with fiber quality (fiber length and/or fiber strength), and it is difficult to improve both lint percentage and fiber quality simultaneously. Different strategies appear to have been used to surmount these challenges during the G. barbadense improvement processes. For example, the favorable allele combination of fiber quality (FL2/FS1 + FL4/FS2 + FS3) originated through recombination in Xinhai (G8) through selection and introgression, and the favorable alleles of three lint percentage loci (LP1, LP2, and LP3) were combined in Pima cotton (G6) through interspecies hybridization. Correspondingly, the Xinhai (G8) and Pima cotton (G6) had the greatest genetic divergence among these cultivated G. barbadense accessions. Both of these exemplify the history of artificial selection and interspecies introgression that influence population divergence and the improvement of agronomic traits in G. barbadense.
Methods
Data collection
Genome sequences of the tetraploid Gossypium species (Supplementary Data 25), including TM1 (AD1)77, ZMS24 (AD1)77, NDM8 (AD1)6, B713 (AD1)43, Bar32 (AD1)43, TX1000 (AD1)45, Hai7124 (AD2)2, Pima90 (AD2)6, G. tomentosum (AD3)5, G. mustelinum (AD4)5, G. darwinii (AD5)5, G. ekmanianum (AD6)45, and G. stephensii (AD7)45 were downloaded from CottonGen78. The 3–79 (AD2)7, PimaS6 (AD2)8, Gb_M210936 (AD2)10, and CRI12 (AD1)44 genomes were also downloaded from the National Center for Biotechnology Information database.
Library construction and sequencing
Twelve accessions of G. barbadense were cultivated in the greenhouse in Wuhan, China, in 2021. Fresh young leaves were collected from one single plant for each accession and immediately frozen in liquid nitrogen. High-quality genomic DNA was extracted from leaves using a modified CTAB method. High-molecular-weight nuclear DNA isolated from young leaf tissue was used to construct both Illumina sequencing libraries (including short-insert libraries) and PacBio SMRT libraries for each sample, following standard protocols. Subsequent libraries were sequenced on the MGI2000 platform for Illumina libraries, and two or three PacBio cells were sequenced on the PacBio Sequel II platform to generate HiFi reads by Frasergen in Wuhan or Biohuaxing in Beijing.
RNA sequencing and data analysis
Mixed tissues (at least two tissues of leaf, stem, fiber, ovule) from 11 accessions (GB0262 missing) were collected for RNA sequencing. RNA purity and integrity were monitored by a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and a Bioanalyzer 2100 system (Agilent Technologies, CA, USA). RNA contamination was assessed by a 1.5% agarose gel. In total, 1 μg RNA per sample was used as the input material, and mRNA was purified from the total RNA using poly-T oligo-attached magnetic beads. Sequencing libraries were generated using the VAHTS Universal V6 RNA-seq Library Kit for MGI (Vazyme, Nanjing, China) following the manufacturer’s recommendations and adding index codes to each sample. Library quantification and size were measured using a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and a Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Subsequently, sequencing was performed on the MGI-SEQ 2000 platform.
The mixed RNA-seq reads were respectively mapped to each genome for protein-coding prediction with Hisat2 v2.1.079. The publicly available RNA-seq data18,66 was mapped to the 3–797 genome with Hisat2 v2.1.079 for both gene expression levels estimation and for identifying key candidate genes via GWAS. TPM (Transcripts Per Million) values for each transcript were calculated using StringTie2 v2.1.480. RNA-seq data for 6 fiber development stages from 5 G. barbadense accessions were used to identify candidate genes for the fiber length and strength18, and RNA-seq data of 10 fiber development stages from 3 cotton samples were used to identify candidate genes for lint percentage66. RNA-seq data of Hai7124 were used to annotate genes2.
De novo assembly of 12 G. barbadense genomes
For each accession, HiFi reads longer than 10 kb were kept via seqkit v0.11.081 and subsequently assembled by hifiasm v0.16.1-r37536 with default parameters. The primary assemblies from the hifiasm output were aligned to the 3–79 genome3,37 based on our previous study to construct pseudo-chromosomes using Mummer v4.082, and the contigs were anchored and ordered based on the sequence synteny using an in-house Perl script (https://github.com/qymeng1996/Pangenome). NGenomeSyn83 was used to display the whole-genome collinearity plot between the 12 de novo assembled genomes and the 3–79 reference.
Repeat sequence annotation and protein-coding prediction
Transposable elements were annotated in the 12 de novo and 5 previously released assemblies using the Extensive De-Novo TE Annotator (EDTA v1.9984), and redundant TE libraries for each accession were removed using the cleanup_nested.pl script from the EDTA package with the parameters “-cov 0.95 -minlen 80 -miniden 80”. The resulting pangenome TE library was then used to annotate whole-genome TEs in the 17 cotton genomes using RepeatMasker v4.1.0 (Smit, AFA, Hubley, R & Green, P. RepeatMasker Open-4.0. 2013-2015 RepeatMasker Home Page) with parameters “-q -no_is -norna -nolow -div 40 -cutoff 225”.
Protein-coding genes were annotated through a combination of ab initio-based prediction, homology-based prediction, and transcript-based prediction. For the homology-based approach, GeMoMa v1.885 was applied using protein sequences from the G. barbadense3 and G. arboreum86 genome sequences downloaded from Cottongen78, as well as G. raimondii87, Theobroma cacao88, Oryza sativa89, Glycine max90, and Arabidopsis thaliana91 downloaded from Phytozome92. For transcript-based prediction, the RNA-seq data from the mixed tissues and public data2 were mapped to repeat-masked genomes using Hisat2 v2.1.079 to identify putative exonic regions and splicing junctions, and then Cufflinks v2.2.193 and StingTie2 v2.1.480 were used to assemble the mapped reads into gene models. For ab initio-based prediction, Augustus v3.3.394, GeneID v1.495, GlimmerHMM v3.0.496, and SNAP (2006-07-28)97 were used to predict the coding regions in the repeat-masked genomes. Of these, three software (Augustus, SNAP, and GlimmerHMM) were trained based on the PASA v2.3.398 gene models, while GeneID used existing A. thaliana models. Gene models generated from all methods were integrated by EvidenceModeler98. A weighted and non-redundant gene set was further revised by PASA to generate untranslated regions and alternative splicing variation information. The longest transcript of each predicted gene model was used to represent that gene in subsequent analyses. Gene models were further filtered on the basis of their Cscore, protein coverage, and coding sequence overlapping TEs2. Genes were functionally annotated using InterProScan v5.55-88.099 to predict potential protein domains based on sequence signatures, with parameters “-appl PANTHER -appl Pfam -iprlookup -goterms”.
Identification of the centromeric regions by CenH3 ChIP-seq data
Public G. barbadense ChIP-seq data2 were downloaded from NCBI (PRJNA488416). After filtering low-quality reads using Trimmomatic v0.39100 with the parameters “LEADING:10 SLIDINGWINDOW:4:15 MINLEN:50”, FastUniq101 was used to remove duplicate read pairs under default parameters. High-quality Illumina reads were mapped to all 17 G. barbadense genome assemblies with Bowtie2 v2.3.2102 and the parameter ‘-N 1‘, and the output was converted to a sorted Binary Alignment Map (BAM) using SAMtools v1.9103. Only high-quality mapping reads (-F 4 -q 30) were kept for further analysis. The number of mapped reads was counted with SAMtools v1.9103 for each 10 kb non-overlapping window, and read density was then calculated by dividing the total number of mapped reads by the total number of mapped nucleotides in each genomic window. To remove the impact of non-specific binding by rabbit serum, the read density was adjusted for background signal by using mock control data. CenH3 domains were identified via SICER2104 with ‘ -g 400 --significant_reads‘, and defined as regions where the fold change (versus control) was ≥5 and the false-discovery rate (FDR) was <0.01, using 200-bp windows and allowing gaps of 400 bp.
Genome assembly quality assessment
Standard descriptive information for genome assemblies (sequence length, Contig N50, etc.) was calculated using in-house Perl scripts (https://github.com/qymeng1996/Pangenome). Assembly completeness, including the genome and protein level, was evaluated using BUSCO v5.2.239 with the “embryophyte_odb10” dataset under default parameters. The false duplication rate was estimated by Purge_Dups40. Base-level accuracy and completeness were estimated by Merquey38. LTR-RT candidates were obtained using LTRharvest v1.6.1105 and LTR_FINDER_parallel v1.1106. After identifying LTR-RTs and generating high-quality LTR libraries with LTR_retriever v2.9.0107, the LTR Assembly Index (LAI) for each assembly was calculated using default parameters41.
Pangenome analysis in G. barbadense
The OrthoFinder (v.2.3.8)108 software was used to construct gene families among G. barbadense genomes. The pangenome size was calculated using all permutations of different combinations and numbers of genome sequences (C(17, N) = 17!/(N!*(17-N)!)). Non-synonymous/synonymous substitution ratios (Ka/Ks) within core, near-core, and dispensable gene families were computed using ParaAT (v2.0)109, the sequence alignment was calculated with muscle v3.8.1551110, and the Ka/Ks values were calculated with KaKs_Calculator3.0111. Functional enrichment was performed using Fisher’s exact tests in R v4.2.1, and the resulting raw P-values were subjected to Benjamini & Hochberg (BH) correction112. Functional classes with p-adj < 0.05 were regarded as significantly enriched.
Structural Variation identification and comparison
All 28 tetraploid cotton genome assemblies (including 12 de novo G. barbadense assemblies and 16 publicly released assemblies2,5,6,7,8,10,44,45,77) were aligned to the 3–79 reference7 using Mummer (v4.0)82 with the parameters: --maxmatch -c 500 -l 100. Raw alignment results were further filtered using the delta-filter with parameters: -m -i 90 -l 100. The resulting filtered delta files were used to detect structural variation using the SyRI pipeline with default parameters113. Using the definitions of sequence variation in SyRI, we converted these variations into four types of SVs: (1) PAVs (Presence/Absence Variation), (2) duplications, (3) inversions, and (4) translocations. SVs carrying ‘N’ sequences were removed. SVs from different accessions were merged into a non-redundant SV set. For the PAVs and duplications, which were merged and normalized with BCFtools114, “bcftools merge -0 *vcf.gz | bcftools norm -m -any -N | bcftools norm -d none”. For the inversions and translocations, if the length of the overlapping sequence of two SVs exceeded 90% of the length of the non-redundant genome segment, they were merged into a non-redundant SV.
We separately detected PAVs using HiFi reads from the 13 (including Gb_M21093610) G. barbadense accessions. We mapped HiFi reads to 3–797 using NGLMR v0.2.7115 with the default parameters, and three callers: Sniffles v2.0.7116, SVIM v2.0.0117, and CuteSV v2.0.2118 with the default parameters used for variant calling in each accession. In addition, HiFi reads were mapped to 3–797 using Minimap2 v2.1.7119 and PBSV v2.9.0 (https://github.com/PacificBiosciences/pbsv) with the default parameters used for variant calling in each accession. The PAVs from four callers were merged using Survivor v1.0.6120 with parameters ‘1000 2 1 1 0 30’.
We next compared PAVs from the two SV callsets, the PAVs obtained based on the HiFi reads pipeline, and those obtained from whole-genome comparison of high-quality G. barbadense assemblies. For those SVs absent relative to 3–79 (Absent SV), if the overlapping ratio of two Absent SVs (the length of the overlapping sequence/the length of non-redundant genome segment covered by the two SVs) exceeded 90%, then we determined that these two Absence SVs from two call sets were the same. Similarly, for Presence SVs relative to 3–79, if the overlapping ratio of two Presence SVs (the length of shorter SV/the length of longer SV) exceed 90%, and their positions in the 3–79 genome was less than 5 bp apart, then we determined that the two Presence SVs from the two call sets were the same.
SV validation
For PAVs, a total of 30 PAVs were randomly selected, and the primers were designed at the upstream and downstream of target PAVs, which were further validated by PCR amplification. For translocation, the primers were designed at the translocation boundary, and the translocations were further validated by PCR amplification and Sanger sequencing. Four Hi-C libraries were downloaded from NCBI (BioProject: PRJNA396502 for 3–79, PRJNA505106 for Hai7124, PRJNA680449 for Pima90, PRJNA810935 for Tanguis). The Hi-C-Pro121 pipeline (v2.11.14) was used to generate chromatin interaction matrices, which were visualized with juicebox122. For large-sized SVs, a total of 30 inversions were randomly selected and were further validated with Hi-C matrices.
Variant calling, population structure of G. barbadense
G. barbadense resequencing data2,12,17,18,20 were trimmed and filtered using Trimmomatic v0.39100. The clean data were mapped to the 3–79 genome, and single-nucleotide polymorphisms (SNPs) were called using the Sentieon (v202112) pipeline123. High-quality SNPs with a sequencing depth greater than 5, minor allele frequency (MAF) ≥ 0.05, and missing rate ≤0.1 were retained for further analysis. A phylogenetic tree was constructed with VCF2Dis (https://github.com/BGI-shenzhen/VCF2Dis) and visualized with the online tool iTOL v6.8.1124. PCA was carried out using GCTA v1.93125. Population structure was constructed by FastStructure v1.0126. Phylogenetic and principal component analysis (PCA) of 1308 G. barbadense accessions led to the exclusion of 637 samples due to germplasm misidentification or low resequencing depth, leaving 671 accessions for further analysis. For these, population fixation (FST) and the nucleotide diversity (π) of each subgroup were calculated using VCFtools127 with 200 kb sliding windows and a step size of 20 kb. The candidate selective sweep regions were identified as the overlap between the top 5% of genomic regions exhibiting the greatest reduction in diversity (πlandrace/πcultivar) values and the top 5% of regions with the greatest FST between groups.
Graph-based pangenome construction and population-scale SV genotyping
The reference sequence of 3–79 and the genomic variation comparing PAVs, duplications, and InDels (>20 bp) from 28 tetraploid cotton genomes were built into a variant graph using the ‘construct’ subcommand of vg without removing any alternate alleles128. The indices of the giraffe algorithms were created with ‘autoindex’. The snarls (i.e., superbubble vertices) were generated using ‘snarls’. Illumina paired-end reads were subsequently mapped against the graph genome by ‘giraffe’, and alignments in the GAM format were generated. Alignments with mapping quality <5 or base quality <5 were excluded. Finally, a compressed coverage index was calculated using ‘pack’. In total, 710 resequencing datasets (for 671 G. barbadense accessions, 33 primitive G. hirsutum accessions, and 6 G. mustelinum accessions) and 336 G. barbadense accessions18 were genotyped for the Pan-SVs using ‘call’, respectively. The resulting Pan-SVs from each resequencing accession were merged using the BCFtools (v1.8) and filtered for lower frequency Pan-SVs (maf <0.05) using the VCFtools (v0.1.16)114,127.
The phylogenetic tree based on the Pan-SVs was constructed with VCF2Dis (https://github.com/BGI-shenzhen/VCF2Dis) and visualized with the online tool iTOL v6.8.1124.
Introgression analysis
A larger introgression haplotype on A01 chromosome in cultivated G. barbadense2,9,10,12, and the genotype of G. barbadense and G. hirsutum accessions on the A01 chromosome were calculated using the Perl script (https://github.com/qymeng1996/Pangenome/tree/main/04_HaplotypeIdentify) reported in a previous study20. Genotypes were plotted and clustered with Pheatmap (CRAN - Package pheatmap (r-project.org)) using R v4.2.1.
D statistics were calculated for each and all of the eight improved G. barbadense accessions throughout the genome using Dsuite129. The putative introgressed regions were detected as windows with the highest X% of fdM values, where X was determined by the proportion of introgression estimated based on the f4 ratio statistic. A total of 80.1 Mb putative introgressed regions were identified in the 8 improved G. barbadense accessions with an average length of 87.7 kb. Therefore, the flanking sequence (50 kb surrounding the iSV) was used to validate the quality of iSVs.
Selection signal identification
To evaluate the possible significance of SVs during G. barbadense improvement, the occurrence frequencies of each SV were calculated in landrace and cultivar groups. The significance of the difference in the frequencies for each SV was determined using Fisher’s exact test, and the resulting raw p-values of all SVs were subjected to Benjamini & Hochberg (BH) correction112. SVs with significantly different frequencies (p-adj < 0.05 and fold change > 2) were identified as those potentially under selection.
Genome-wide association analysis
The extensive phenotypic data of 336 G. barbadense accessions were collected, including the phenotypic data across four environments (Korla-1 (E1), Korla-2 (E2), Baotou Lake (E3), Awat (E4)) in 2019 year and the BLUP values. Three key agronomic traits, i.e., lint percentage, fiber length, and fiber strength, were subjected to association analysis18.
The 3,900,000 SNPs and 79,988 SVs were utilized for three key agronomic traits, i.e., lint percentage, fiber length, and fiber strength, with GEMMA130, respectively. The top three principal components (PCs) were used to build the S matrix for population structure correction, and the matrix of simple matching coefficients was used to build the K matrix. The effective number of genomic variants was calculated with GEMMA. The modified Bonferroni correction was used to determine the genome-wide significance thresholds of the SV-based GWAS, based on nominal levels of α = 0.05 and α = 1, corresponding to -log10(P) values of 6.24 and 4.96, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
PacBio HiFi reads, RNA-seq reads, and re-sequence data have been deposited in the NCBI BioProject database under accession PRJNA1049502 and PRJNA1061756. Genome assemblies and annotation of 12 de novo G. barbadense accessions, structural variation, and Pan-SVs have been deposited in Figshare [https://figshare.com/projects/Pangenome_of_Gossypium_barbadense/189915] and Genome Sequence Archive (GSA) under accession PRJCA039983. Source data are provided with this paper.
Code availability
Code used for the pangenome analysis is available at Zenodo [https://doi.org/10.5281/zenodo.15366284].
References
Wen, X. et al. A comprehensive overview of cotton genomics, biotechnology and molecular biological studies. Sci. China Life Sci. 66, 2214–2256 (2023).
Hu, Y. et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 51, 739–748 (2019).
Wang, M. et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 51, 224–229 (2019).
Yang, Z. et al. Recent progression and future perspectives in cotton genomic breeding. J. Integr. Plant Biol. 65, 548–569 (2023).
Chen, Z. J. et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 52, 525–533 (2020).
Ma, Z. et al. High-quality genome assembly and resequencing of modern cotton cultivars provide resources for crop improvement. Nat. Genet. 53, 1385–1391 (2021).
Chang. X. et al. High-quality Gossypium hirsutum and Gossypium barbadense genome assemblies reveal the centromeric landscape and evolution. Plant Commun. 5, 100722 (2023).
Chávez Montes, R. A. et al. Assembly and annotation of the Gossypium barbadense L. ‘Pima-S6’ genome raise questions about the chromosome structure and gene content of Gossypium barbadense genomes. BMC Genomics 24, 11 (2023).
Jin, S. et al. Structural variation (SV)-based pan-genome and GWAS reveal the impacts of SVs on the speciation and diversification of allotetraploid cottons. Mol. Plant 16, 678–693 (2023).
Wang, N. et al. Genome and haplotype provide insights into the population differentiation and breeding improvement of Gossypium barbadense. J. Adv. Res. 54, 15–27 (2023).
Yuan, D. et al. The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 5, 17662 (2015).
Fang, L. et al. Divergent improvement of two cultivated allotetraploid cotton species. Plant Biotechnol. J. 19, 1325–1336 (2021).
Yuan, D. et al. Parallel and intertwining threads of domestication in allopolyploid cotton. Adv. Sci. 8, 2003634 (2021).
Nie, X. et al. High-density genetic variation maps reveal the correlation between asymmetric interspecific introgressions and improvement of agronomic traits in Upland and Pima cotton varieties developed in Xinjiang, China. Plant J. 103, 677–689 (2020).
Fang, L. et al. Genomic insights into divergence and dual domestication of cultivated allotetraploid cottons. Genome Biol. 18, 33 (2017).
Yu, J. et al. Whole-genome resequencing of 240 Gossypium barbadense accessions reveals genetic variation and genes associated with fiber strength and lint percentage. Theor. Appl. Genet. 134, 3249–3261 (2021).
Wang, P. et al. Introgression from Gossypium hirsutum is a driver for population divergence and genetic diversity in Gossypium barbadense. Plant J. 110, 764–780 (2022).
Zhao, N. et al. Genomic and GWAS analyses demonstrate phylogenomic relationships of Gossypium barbadense in China and selection for fibre length, lint percentage and Fusarium wilt resistance. Plant Biotechnol. J. 20, 691–710 (2022).
Song, X. et al. Combined genome and transcriptome analysis of elite fiber quality in Gossypium barbadense. Plant Physiol. 195, 2158–2175 (2024).
He, S. et al. The genomic basis of geographic differentiation and fiber improvement in cultivated cotton. Nat. Genet. 53, 916–924 (2021).
Ma, Z. et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 50, 803–813 (2018).
Liu, Y. & Tian, Z. From one linear genome to a graph-based pan-genome: a new era for genomics. Sci. China Life Sci. 63, 1938–1941 (2020).
Alonge, M. et al. Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182, 145–161.e123 (2020).
Liu, Y. et al. Pan-genome of wild and cultivated soybeans. Cell 182, 162–176.e113 (2020).
He, Q. et al. A graph-based genome and pan-genome variation of the model plant Setaria. Nat. Genet. 55, 1232–1242 (2023).
Liu, H. et al. Transposon insertion drove the loss of natural seed shattering during foxtail millet domestication. Mol. Biol. Evol.39, msac078 (2022).
Chen, W. et al. Convergent selection of a WD40 protein that enhances grain yield in maize and rice. Science 375, eabg7985 (2022).
Xin, W. et al. BSA-seq and fine linkage mapping for the identification of a novel locus (qPH9) for mature plant height in rice (Oryza sativa). Rice 15, 26 (2022).
Xia, X. et al. Structural variation and introgression from wild populations in East Asian cattle genomes confer adaptation to local environment. Genome Biol. 24, 211 (2023).
Jayakodi, M. et al. Building pan-genome infrastructures for crop plants and their use in association genetics. DNA Res. 28, dsaa030 (2021).
Della Coletta, R. et al. How the pan-genome is changing crop genomics and improvement. Genome Biol. 22, 3 (2021).
Paten, B. et al. Genome graphs and the evolution of genome inference. Genome Res. 27, 665–676 (2017).
Qin, P. et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 184, 3542–3558.e3516 (2021).
Tian, T. et al. Genome assembly and genetic dissection of a prominent drought-resistant maize germplasm. Nat. Genet. 55, 496–506 (2023).
Zhou, Y. et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 606, 527–534 (2022).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Meng, Q. et al. Comparative analysis of genome sequences of the two cultivated tetraploid cottons, Gossypium hirsutum (L.) and G. barbadense (L.). Ind. Crops Prod.196, 116471 (2023).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Manni, M. et al. BUSCO: assessing genomic data quality and beyond. Curr. Protoc. 1, e323 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Ou, S. et al. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126–e126 (2018).
Tang, D. et al. Genome evolution and diversity of wild and cultivated potatoes. Nature 606, 535–541 (2022).
Perkin, L. C. et al. Genome assembly of two nematode-resistant cotton lines (Gossypium hirsutum L.). G311, jkab276 (2021).
Lu, X. et al. A high-quality assembled genome and its comparative analysis decode the adaptive molecular mechanism of the number one Chinese cotton variety CRI-12. GigaScience 11, giac019 (2022).
Peng, R. et al. Evolutionary divergence of duplicated genomes in newly described allotetraploid cottons. Proc. Natl Acad. Sci. USA 119, e2208496119 (2022).
Viot, C. R. & Wendel, J. F. Evolution of the cotton genus, Gossypium, and its domestication in the Americas. Crit. Rev. Plant Sci. 42, 1–33 (2023).
Machado, A. et al. The MYB transcription factor GhMYB25 regulates early fibre and trichome development. Plant J. 59, 52–62 (2009).
Shan, C.-M. et al. Control of cotton fibre elongation by a homeodomain transcription factor GhHOX3. Nat. Commun. 5, 5519 (2014).
Ding, M. et al. The Hairless Stem Phenotype of Cotton (Gossypium barbadense) Is Linked to a Copia-Like Retrotransposon Insertion in a Homeodomain-Leucine Zipper Gene (HD1). Genetics 201, 143–154 (2015).
Xu, G. et al. FERONIA phosphorylates E3 ubiquitin ligase ATL6 to modulate the stability of 14-3-3 proteins in response to the carbon/nitrogen ratio. J. Exp. Bot. 70, 6375–6388 (2019).
Wang, N.-N. et al. The cotton WRKY transcription factor (GhWRKY33) reduces transgenic Arabidopsis resistance to drought stress. Sci. Rep. 9, 724 (2019).
Khan, G. A. et al. Phosphate starvation regulates cellulose synthesis to modify root growth. Plant Physiol. 194, 1204–1217 (2024).
Huang, G.-Q. et al. A fasciclin-like arabinogalactan protein, GhFLA1, is involved in fiber initiation and elongation of cotton Plant. Physiology 161, 1278–1290 (2013).
Qin, Y. et al. Single-cell RNA-seq reveals fate determination control of an individual fibre cell initiation in cotton (Gossypium hirsutum). Plant Biotechnol. J. 20, 2372–2388 (2022).
Liu, Y. et al. Genomic basis of geographical adaptation to soil nitrogen in rice. Nature 590, 600–605 (2021).
Zhang, X. et al. Pan-genome of Raphanus highlights genetic variation and introgression among domesticated, wild, and weedy radishes. Mol. Plant 14, 2032–2055 (2021).
Percy R. The Worldwide Gene Pool of Gossypium barbadense L. and Its Improvement. In: Genetics and Genomics of Cotton, (ed. Paterson, A. H.) pp. 53–68 (Springer US. New York, NY, 2009).
Percy, R. G. & Wendel, J. F. Allozyme evidence for the origin and diversification of Gossypium barbadense L. Theor. Appl. Genet. 79, 529–542 (1990).
Wendel, J. F. & Percy, R. G. Allozyme diversity and introgression in the Galapagos Islands endemic Gossypium darwinii and its relationship to continental G. barbadense. Biochem. Syst. Ecol. 18, 517–528 (1990).
Mockler, T. C. et al. Regulation of flowering time in Arabidopsis by K homology domain proteins. Proc. Natl Acad. Sci. USA 101, 12759–12764 (2004).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Hu, H. et al. GhJAZ2 negatively regulates cotton fiber initiation by interacting with the R2R3-MYB transcription factor GhMYB25-like. Plant J. 88, 921–935 (2016).
Gao, Z. et al. GhbHLH18 negatively regulates fiber strength and length by enhancing lignin biosynthesis in cotton fibers. Plant Sci. 286, 7–16 (2019).
Walford, S.-A. et al. GhMYB25-like: a key factor in early cotton fibre development. Plant J. 65, 785–797 (2011).
Shangguan, X. et al. Function analysis of a cotton R2R3 MYB transcription factor GhMYB3 in regulating plant trichome development. Plant Biol. 23, 1118–1127 (2021).
Zhang. J. et al. Transcriptome time-course analysis in the whole period of cotton fiber development. Front. Plant Sci. 13, 864529 (2022).
Pu, L. et al. The R2R3 MYB transcription factor GhMYB109 is required for cotton fiber development. Genetics 180, 811–820 (2008).
Wang, L. et al. Evidence that high activity of vacuolar invertase is required for cotton fiber and arabidopsis root elongation through osmotic dependent and independent pathways, respectively. Plant Physiol. 154, 744–756 (2010).
Li, H.-B. et al. A cotton ascorbate peroxidase is involved in hydrogen peroxide homeostasis during fibre cell development. N. Phytol. 175, 462–471 (2007).
The Story of Pima S-1. Progressive Agriculture in Arizona Vol. 12, No. 3 (1960), (1960).
Endrizzi, J. E. et al. Genetics, cytology, and evolution of Gossypium. In Advances in Genetics (eds Caspari, E. W. & Scandalios, J. G.) (Academic Press, 1985).
Grover, C. E. et al. Re-evaluating the phylogeny of allopolyploid Gossypium L. Mol. Phylogenet. Evol. 92, 45–52 (2015).
Wendel, J. & Grover, C. Taxonomy and Evolution of the Cotton Genus, Gossypium (2015).
Wendel, J. F. New World tetraploid cottons contain Old World cytoplasm. Proc. Natl Acad. Sci. USA 86, 4132–4136 (1989).
Li, N. et al. Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species. Nat. Genet. 55, 852–860 (2023).
Fang, L. et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 49, 1089–1098 (2017).
Yang, Z. et al. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars. Nat. Commun. 10, 2989 (2019).
Yu, J. et al. CottonGen: The Community Database for Cotton Genomics, Genetics, and Breeding Research (2021).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Shen, W. et al. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 11, e0163962 (2016).
Marçais, G. et al. MUMmer4: a fast and versatile genome alignment system. PLoS Computat. Biol. 14, e1005944 (2018).
He, W. et al. NGenomeSyn: an easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics 39, btad121 (2023).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Keilwagen, J. et al. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinform. 19, 189 (2018).
Huang, G. et al. Genome sequence of Gossypium herbaceum and genome updates of Gossypium arboreum and Gossypium hirsutum provide insights into cotton A-genome evolution. Nat. Genet. 52, 516–524 (2020).
Paterson, A. H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427 (2012).
Motamayor, J. C. et al. The genome sequence of the most widely cultivated cacao type and its use to identify candidate genes regulating pod color. Genome Biol. 14, r53 (2013).
Kawahara, Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 4 (2013).
Valliyodan, B. et al. Construction and comparison of three reference-quality genome assemblies for soybean. Plant J. 100, 1066–1082 (2019).
Cheng, C.-Y. et al. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 89, 789–804 (2017).
Goodstein, D. M. et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 (2012).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Keller, O. et al. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763 (2011).
Blanco, E. et al. Using geneid to identify genes. Curr. Protoc. Bioinform. 18, 4.3.1-4.3.28 (2007).
Delcher, A. L. et al. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27, 4636–4641 (1999).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120 (2005).
Bolger, A. M. et al. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Xu, H. et al. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS ONE 7, e52249 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Zang, C. et al. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958 (2009).
Ellinghaus, D. et al. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 18 (2008).
Ou, S. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mob. DNA 10, 48 (2019).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Zhang, Z. et al. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 419, 779–781 (2012).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Zhang, Z. KaKs_Calculator 3.0: calculating selective pressure on coding and non-coding sequences. Genom. Proteom. Bioinform. 20, 536–540 (2022).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol. 57, 289–300 (1995).
Goel, M. et al. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 277 (2019).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Sedlazeck, F. J. et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018).
Smolka M, et al. Detection of mosaic and population-level structural variants with Sniffles2. Nat. Biotechnol. 42, 1571–1580 (2024).
Heller, D. & Vingron, M. SVIM: structural variant identification using mapped long reads. Bioinformatics 35, 2907–2915 (2019).
Jiang, T. et al. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 21, 189 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Jeffares, D. C. et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 8, 14061 (2017).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Donald, F. et al. The sentieon genomics tools - a fast and accurate solution to variant calling from next-generation sequence data. bioRxiv https://doi.org/10.1101/115717 (2017).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Yang, J. et al. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Raj, A. et al. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573–589 (2014).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Garrison, E. et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 36, 875–879 (2018).
Malinsky, M. et al. Dsuite - fast D-statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. 21, 584–595 (2021).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824 (2012).
Acknowledgements
This work was financially supported by the National Key Research and Development Program (NO. 2021YFF1000102), the Natural Science Foundation of Xinjiang Uygur Autonomous Region (NO. 2023D01E02), the National Science and Technology Innovation 2030 Major Projects (NO. 2023ZD04038), the Fundamental Research Funds for the Central Universities (NO. 2021ZKPY013) and the development fund for Xinjiang talents XL. We thank the high-performance computing platform at the National Key Laboratory of Crop Genetic Improvement in Huazhong Agricultural University.
Author information
Authors and Affiliations
Contributions
D.Y. conceived and managed the entire project. Q.M., P.X., J.T., J.G. and S.J. collected re-sequence data and conducted PacBio HiFi and Illumina sequencing. Q.M., P.X., Z.X. and J.Z. performed de novo genome assembly, pangenome construction and structural variation identification. J.K. provided the phenotype data. Q.M., L.H., X.G. and Y.R. performed panel GWAS analysis. S.H. and X.G. performed the PCR amplification validation. Q.M. prepared the figures and wrote the manuscript draft. J.A.U., C.E.G., J.F.W., K.Z., Q.C. and J.K. contributed to the project discussion. J.A.U., C.E.G., J.F.W., M.W., X.N., Z.L., S.J., X.Z. and D.J revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Murukarthick Jayakodi, Marc-André Lemay, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Meng, Q., Xie, P., Xu, Z. et al. Pangenome analysis reveals yield- and fiber-related diversity and interspecific gene flow in Gossypium barbadense L.. Nat Commun 16, 4995 (2025). https://doi.org/10.1038/s41467-025-60254-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60254-x
This article is cited by
-
Genetic basis and role of exotic accessions in cultivated cotton fiber quality improvement
Theoretical and Applied Genetics (2025)
-
Integrative genomic structural variation analysis unveils genetic architecture underlying important traits in Gossypium barbadense
Theoretical and Applied Genetics (2025)
