
Abstract
The oxidative dehydrogenation of propane has the potential to reduce significantly the energy demand of propylene production. However, low propylene selectivity and the need for expensive air separation hamper its commercial implementation. In the herein presented chemical looping-based scheme for propylene production, gaseous oxygen is supplied in situ by a perovskite oxygen carrier (Sr1−xCaxFeO3−δ) while propane is dehydrogenated over a physically separated catalyst (VOx/SiO2), thus allowing for high systematic flexibility, as each component can be optimized individually. To prevent over-oxidation of propane to COx at the gas-oxygen carrier interface, a NaNO3-based surface modification is developed, which melts under operating conditions and wets the surface of the oxygen carrier, forming a non-porous diffusion barrier for gaseous hydrocarbons, thereby completely inhibiting over-oxidation. We demonstrate stable operation over 250 redox cycles at 500 °C (14.5% propane conversion at 68% propylene selectivity), matching the performance of the benchmark VOx/SiO2 with co-fed oxygen.
Similar content being viewed by others


Introduction
Propylene is one of the most important platform chemicals in industry and is used to produce bulk commodities such as polypropylene or further high-value chemical intermediates, including isopropanol, acetone, and acrolein1,2,3. The global production of polypropylene amounted to 130 Mt a−1 in 2019 and is estimated to rise to 190 Mt a−1 by 20301. Today, more than 80% of the propylene demand is met by the steam or fluid catalytic cracking of petroleum-derived sources, which have several disadvantages, such as high CO2 emissions due to energy-intensive reaction conditions and costly product separation1. Further, over the last decade, the production capacities of traditional cracking processes have not been capable to satisfy the propylene demand4. Thus, alternative, on-purpose propylene production routes such as the dehydrogenation of propane (PDH) have attracted increasing interest. Several PDH technologies have been commercialized, e.g., the Oleflex (SC3H6 = 84%, XC3H8 = 40%) and Catofin (SC3H6 = 88%, XC3H8 = 53%) processes, which account for up to 10% of the current propylene production1,2. Nevertheless, the endothermicity of PDH (Eq. 1) that translates into a significant energy demand, as well as catalyst deactivation through coking, present serious drawbacks of the PDH process. To resolve these issues, propylene production via the oxidative dehydrogenation of propane (ODHP) has been proposed.
In ODHP (Eq. 2), water instead of hydrogen is formed as a by-product, which renders the overall
reaction exothermic5. Moreover, achieving a high conversion of propane in ODHP is thermodynamically feasible, and catalyst deactivation due to coking generally does not occur1,6. Despite these advantages, ODHP has not yet reached commercial competitiveness with PDH. Co-feeding pure oxygen and propane necessitates costly air separation units, and the introduction of oxygen favors the over-oxidation of propane, i.e., its conversion to COx, thereby significantly reducing the propylene selectivity.
The realization of ODHP through a chemical looping (CL) scheme is a solution to avoid the need for air separation. In CL-ODHP, the oxidative dehydrogenation reaction is split into spatially or temporally separated half-reactions that form a closed redox loop through a redox catalyst7,8. The redox catalyst, commonly a mixed metal oxide, donates its lattice oxygen to the reaction while also catalyzing the ODHP in the reduction step. The redox catalyst is subsequently re-oxidized in air during the oxidation step of the redox loop. Furthermore, the two exothermic half-reactions allow for process optimization through tunable heat integration. A study evaluating Mn2O3/MnO as a redox catalyst for CL-ODHP found theoretical energy savings of up to 45% compared to commercial propylene production processes9. Similar results (i.e., energy savings of up to 45% and a reduction of CO2 emissions by ~40%) were obtained for a ceria-vanadia-based CL-ODHP system when compared to the industrial Oleflex process10.
In light of these benefits, several CL-ODHP concepts have been proposed, mostly involving vanadium oxide-based redox catalysts that catalyze the ODHP and donate lattice oxygen to the reaction5,11,12,13,14,15,16,17,18. VOx-based materials are also among the most studied catalysts for ODHP processes that involve the co-feeding of oxygen, and it has been established that isolated VO4 surface species are the most selective active sites for the ODHP. This observation restricts the vanadium loading on the catalyst supports, as less selective V2O5 nanoclusters form at higher vanadium loadings6,19,20,21. Chen et al.15 showed that in CL-ODHP, the propylene yield over VOx on TiO2 is maximized at vanadium loadings between 0.05 and 1 wt.%, i.e., the range in which isolated VO4 species remain the main catalytically active sites. With increasing VOx content and the formation of crystalline V2O5 nanoparticles, the propane conversion is enhanced at the cost of a decreasing propylene selectivity and increasing COx selectivity. However, to yield an efficient CL-ODHP process in terms of quantity of propylene produced per redox cycle and amount of redox catalyst, the redox catalyst is required to possess an oxygen storage capacity >1 wt.%, which would result in a theoretical propylene production > \(55.1\frac{{{mg}}_{{C}_{3}{H}_{6}}}{{g}_{{redox\; cat}}}\) per redox cycle. Yet, an oxygen storage capacity >1 wt.% requires VOx loadings >3.5–5 wt.%. Wu et al.13 investigated a VOx-based catalyst (supported on Al2O3) with a relatively high vanadium loading of 10 wt.%, reaching an oxygen storage capacity of up to 1.7 wt.%. However, this redox catalyst achieved a comparatively low propylene selectivity of ~54% at 6% propane conversion \((55.8\frac{{{mg}}_{{C}_{3}{H}_{6}}}{{g}_{{redox\; cat}}}{{{\rm{per\; cycle}}}})\), which can be attributed to the formation of V2O5 crystallites that show a low selectivity to propylene. It is therefore desirable to find a material engineering solution that allows for a high oxygen storage capacity without affecting the speciation of the catalytic sites and compromising propylene selectivity.
Herein, a reaction concept is proposed to circumvent the apparent trade-off between a high propylene selectivity and a high oxygen storage capacity. This CL scheme, which has been proposed initially for the oxidative dehydrogenation of ethane by Luongo et al.22,23 differs from previously reported CL-ODHP schemes, as the dual functionality of the redox catalyst, serving both as an oxygen donor and catalyst for the oxidative dehydrogenation reaction, is separated into two materials: An oxygen carrier that provides gaseous oxygen through CL with oxygen uncoupling (CLOU), and a physically separated ODHP catalyst placed further downstream. This concept is henceforth termed CLOU-ODHP, and it provides a high flexibility with regards to the structure of the oxygen carrier and the catalyst, allowing e.g., for the use of materials with specifically tailored oxygen carrier properties such as favorable oxygen release kinetics for the desired process without affecting the structure of the active sites of the catalyst. Specifically, in the CLOU-ODHP scheme presented here, a Ca-doped SrFeO3 perovskite provides gaseous oxygen, while the catalytic conversion of propane occurs further downstream over a VOx/SiO2 catalyst (Fig. 1). In such a configuration it is paramount that the oxygen carrier does not combust the propane feed during its reduction, as this would lower the propylene selectivity and hence the overall efficiency of the process. To inhibit the oxidation of propane to COx (over-oxidation) by the oxygen carrier, molten alkali halide and carbonate coatings have been explored as surface modifications in CL alkane dehydrogenation schemes. Yet, these surface modifications have often shown some reactivity with the hydrocarbon feed, leading to its partial over-oxidation to COx24,25. Here, we developed a NaNO3-based surface modification, which, unlike commonly used surface modifications such as carbonate coatings, melts under the applied operating temperature (500 °C), thereby wetting the surface of the oxygen carrier uniformly and in turn inhibiting completely any over-oxidation of the hydrocarbon feedstock, while allowing gaseous oxygen to permeate through. It is demonstrated that the proposed CL concept avoids the necessity of a separate air separation and yields propylene yields comparable to that of a conventional (i.e., oxygen co-fed) ODHP scheme. Since the catalyst and the oxygen carrier can be chosen independently, and the NaNO3-based surface modification is compatible with a wide range of oxygen carrier materials, the CL scheme introduced here is expected to be applicable to a variety of catalytic reactions that require gaseous oxygen as a reactant.

The CLOU-ODHP scheme consists of an oxygen carrier (Ca-doped SrFeO3 perovskite) providing gaseous oxygen for the ODHP reaction and a separate ODH (oxidative dehydrogenation) catalyst (VOx/SiO2).
Results
Characterization of the oxygen carrier and ODHP catalyst
Ca-doped strontium ferrite (Sr1−xCaxFeO3−δ) perovskites were synthesized via a sol-gel Pechini route, and the X-ray diffraction (XRD) patterns of the Sr1−xCaxFeO3−δ series matched the characteristic diffraction pattern of a strontium ferrite perovskite (Fig. 2a). Upon increasing the calcium concentration, the diffraction peaks shifted to higher angles (Fig. 2b), indicating a decrease in the lattice parameters due to the successful substitution of A-site Sr2+ cations (rSr2+ = 1.27 Å) with smaller Ca2+ cations (rCa2+ = 1.06 Å). A Rietveld refinement of Sr0.8Ca0.2FeO3−δ was carried out to determine the crystal structure of the perovskites synthesized (Supplementary Fig. 1). Fitting a cubic space group (Pm-3m) to the diffraction pattern yielded a lattice parameter of a = 3.85 Å (goodness of fit X2 = 2.43); fitting an orthorhombic space group (Cmmm), the lattice parameters were calculated as a = 10.91 Å, b = 7.67 Å, and c = 5.45 Å, and the goodness of fit decreased to X2 = 1.39. Due to the improved fit and the fact that minor reflections in the diffraction pattern at 2Θ values of e.g., 36.11° and 43.2° could be modeled only by the space group Cmmm, it was concluded that the synthesized materials crystallized as orthorhombic perovskites. Further, inductively coupled plasma optical emission spectroscopy (ICP-OES) measurements show that the desired cation ratios were achieved within a standard deviation <1 mol% (Supplementary Fig. 2), and a homogeneous distribution of the elements within the material was verified by transmission electron microscopy energy dispersive X-ray spectroscopy (TEM-EDS) (Supplementary Fig. 3).

a, b XRD patterns of the series of Sr1−xCaxFeO3−δ-based oxygen carriers. The dashed line serves as a visual aid to illustrate the peak shift upon doping. c SEM image of SCFO (Sr0.8Ca0.2FeO3−δ). d Thermogravimetric analysis of Sr1−xCaxFeO3−δ in N2 at 500 °C. Shadings in blue and orange indicate the gas atmospheres air and N2, respectively. e Oxygen storage capacity of Sr1−xCaxFeO3−δ over ten redox cycles (reduction in N2 for 40 min, oxidation in air for 20 min) at 500 °C. f XRD patterns of fresh, reduced, and re-oxidized SCFO (reference patterns are shown for Sr2Fe2O5 and SrFeO3).
To assess whether the doping of the A-site of a SrFeO3 with Ca leads to a significant decrease in the onset temperature upon which the oxygen carrier releases gaseous oxygen in a reducing environment (e.g., N2), the doping concentration of Ca in the A-site of the perovskite was varied from 0 to 25 mol%, and its effect on the rate of oxygen release and oxygen storage capacity during a 40 min reduction in N2 at 500 °C was investigated by thermogravimetric analysis (TGA). The rate of oxygen release increased significantly upon the incorporation of Ca in the A-site of the perovskite (Fig. 2d) and the fastest oxygen release was observed for the perovskite with the highest doping concentration, i.e., Sr0.75Ca0.25FeO3−δ. Indeed, undoped SrFeO3, as well as Sr0.9Ca0.1FeO3−δ, did not reach their equilibrium oxygen stoichiometry after 40 min of reduction in N2 at 500 °C. Next, the oxygen storage capacity of all materials was evaluated over ten redox cycles in a TGA experiment to determine their cycling stability (Fig. 2e). Generally, the oxygen storage capacity of all tested materials did not decrease over ten redox cycles, indicating the absence of irreversible structural changes over repeated redox cycles. During its reduction, the perovskite transitions to a brownmillerite phase, yet the perovskite phase is completely restored in the subsequent re-oxidation step in air (Fig. 2f for Sr0.8Ca0.2FeO3−δ, Supplementary Fig. 4 for other Ca dopant concentrations). While Sr0.8Ca0.2FeO3−δ and Sr0.75Ca0.25FeO3−δ possess a very similar oxygen storage capacity of 2.05 and 2.11 wt.%, respectively, undoped SrFeO3 released only 1.15 % of its weight as gaseous oxygen during the 40 min reduction step due to its rather slow oxygen release properties. Hence, the extent of oxygen release under the chosen conditions was nearly doubled by incorporating Ca into the A-site of SrFeO3. Overall, the TGA results demonstrate the tunability of the oxygen release rates and oxygen storage capacity of the oxygen carrier, which can be tailored to the desired process conditions by adjusting the type and concentration of the dopant (e.g., La, Sm, K, Co, Ni, Ti)26,27,28.
In the following, 500 °C was chosen as the reaction temperature for the coupled CL-ODHP process to inhibit gas-phase cracking reactions. Under such reaction conditions, Sr0.8Ca0.2FeO3−δ (henceforth abbreviated as SCFO) was used as the oxygen carrier due to its high oxygen storage capacity when compared to Sr0.9Ca0.1FeO3−δ and its more gradual release of oxygen when compared to Sr0.75Ca0.25FeO3−δ, resulting in a relatively constant oxygen supply during the reduction step in the coupled CL-ODHP process. The actual oxygen concentration generated by the SCFO during a 30 min reduction step at 500 °C was determined in a separate oxygen release experiment, demonstrating an average oxygen concentration of ~1.4% over 50 redox cycles (Supplementary Fig. 5).
To catalyze the ODHP reaction, a vanadium oxide-based catalyst was used. The weight loading of vanadium on SiO2 was ~2.5% as determined by ICP-OES, and the XRD pattern of the prepared ODHP catalyst showed no characteristic peaks due to vanadium oxides (Supplementary Fig. 5), indicating that vanadium was largely present as VO4 surface species and small V2O5 nanoclusters that were below the detection limit of the XRD instrument. The presence of active VO4 surface species and V2O5 nanoclusters was confirmed by Raman spectroscopy (Supplementary Fig. 6), with their characteristic bands at 1039 cm−1 and 995 cm−1, respectively29,30.
Surface modification and over-oxidation of the oxygen carrier
The sole purpose of the oxygen carrier in the CLOU-ODHP scheme introduced here is to supply gaseous oxygen for the ODHP that proceeds over a physically separate catalyst. Hence, the oxygen carrier must not react with propane. However, when exposing unmodified SCFO to propane at 500 °C, a substantial quantity of propane was combusted to CO2 during the initial 15 min of reduction (Fig. 3a). It is therefore critical to modify the surface of the oxygen carrier, to inhibit (or at least mitigate) its interaction with the hydrocarbon feed.

a Propane oxidation over NaNO3 modified and unmodified SCFO using 50 mL min−1 of 3 vol.% propane diluted in N2 at 500 °C (1 g of the oxygen carrier). b Oxidation of propane over SCFOs modified via different approaches over five redox cycles at 500 °C using 0.5 g of the oxygen carrier. c, d XRD patterns of as-synthesized, modified SCFOs, and reference materials. e Raman spectra of as-synthesized, modified SCFOs, and reference materials (Supplementary Fig. 10 shows enlarged plots of c–e). The bars colored in orange, brown, and gray indicate peak positions for NaNO3, Na2CO3, and SrCO3, respectively.
Surface modifications of oxygen carriers relying on the addition of an alkali metal carbonate have been reported to reduce the over-oxidation of ethane in the CL oxidative dehydrogenation of ethane, albeit at relatively high operating temperatures of 700 °C, at which the carbonate surface modifications (e.g., Li2CO3) are in a liquid state25. At operating temperatures in which the contribution of gas-phase pyrolysis can be excluded to accurately assess the catalytic performance of the system, i.e., ~500 °C, such alkali metal carbonate-based surface modifications are in a solid state and inefficient in preventing over-oxidation. Generally, surface modifications inhibiting the over-oxidation of propane in CL-ODHP systems have received only little attention, as most studies either focus on the more facile ODH of ethane or aim to achieve high propylene selectivities through doped redox catalysts12,31,32,33,34,35. In the following, a series of surface modification approaches that rely on different preparation methods, coating materials, and concentrations, were explored as to whether they can prevent the direct interaction of a hydrocarbon feed with the oxygen carrier surface. All modified oxygen carrier materials and their method of preparation are summarized in Table 1. The effectiveness of the different surface modifications was evaluated by assessing the extent of propane oxidation over up to five redox cycles at 500 °C (Fig. 3b). Unmodified SCFO displayed the highest extent of propane over-oxidation (i.e., 0.57 mmol of CO2 per redox cycle) at a propane conversion of ~10.2% and oxygen storage capacity consumption of 89%. A surface modification of 20 wt.% Na2CO3 by dry (20Na2CO3-d) or wet impregnation (20Na2CO3-w) lowered the propane conversion to 4.5 and 4.2%, respectively, and decreased the extent of over-oxidation by 55–60%. Reducing the quantity of Na2CO3 to 10 wt.% increased the propane conversion and propane over-oxidation to 6.2% and 0.34 mmol of CO2 per cycle, respectively (Supplementary Fig. 7), indicating an inhomogeneous and/or incomplete surface coverage, or an insufficient thickness of the surface layer for small weight fractions of Na2CO3. In another set of experiments, the oxygen carrier was impregnated with NaNO3 (dry impregnation) and subsequently heated to 750 °C in a tube furnace under air flow for 12 h (20NaNO3c-d) to melt and carbonate the NaNO3. 20NaNO3c-d produced ~0.27 mmol of CO2 per redox cycle, corresponding to an average propane conversion of 5.1%, which was similar to that of 20Na2CO3-w and 20Na2CO3-d. Rather surprisingly, SCFO modified with 20 wt.% NaNO3 by dry impregnation, yet without the additional calcination treatment (20NaNO3-d), completely inhibited any over-oxidation of propane, and no propane conversion was observed over five redox cycles (Fig. 3b).
In addition to inhibiting the over-oxidation of propane to COx, the surface modification also affected the reduction kinetics of the oxygen carrier. When the unmodified SCFO was subjected to propane at 500 °C, no further oxygen was released after the initial period of propane oxidation (15 min). On the other hand, 20NaNO3-d displayed slower reduction kinetics, releasing oxygen throughout the entire reduction period (30 min) without combusting propane (Fig. 3a). To confirm the faster reduction of the unmodified SCFO, reference experiments were conducted in different reducing environments (N2 and propane diluted in N2, Supplementary Fig. 8). Indeed, unmodified SCFO released gaseous oxygen throughout the entire 40 min of reduction in N2, whereas in diluted propane, the conversion of propane occurred largely within the first 25 min and a switch to N2 (after 30 min) did not lead to a noticeable further release of oxygen, indicating that SCFO was reduced almost completely to its equilibrium oxygen non-stoichiometry after 25 min. In contrast, 20NaNO3-d did not show any significant change in its oxygen release profile when reduced in diluted propane or N2 (Supplementary Fig. 8). These results show that in propane unmodified SCFO is reduced faster than the modified SCFO, possibly due to the presence of highly reactive oxygen species at its surface, which oxidize propane very rapidly (and with a high selectivity) to CO2.
Carbonation behavior of the oxygen carrier
To understand how a surface modification can inhibit propane over-oxidation, the bulk and surface structures of the modified oxygen carrier materials were characterized by XRD and Raman spectroscopy, respectively (Fig. 3c, d). As opposed to oxygen carriers modified by dry impregnation, the XRD pattern of all of the oxygen carriers modified by wet impregnation exhibited peaks due to SrCO3 (in addition to peaks due to the perovskite phase). It has been reported that Sr contained in perovskite structures can readily carbonate when exposed to water36,37: Sr segregation is believed to occur due to the relatively large size of Sr2+ cations, assisted by a distortion of the perovskite lattice due to water absorption38. Subsequently, the segregated Sr cations react with water, forming strontium hydroxide hydrate complexes that readily carbonate with atmospheric CO2 at ambient conditions38. To validate the carbonation of SCFO when in contact with water and air at ambient conditions, an in situ Raman spectroscopy experiment was carried out, in which the surface of a SCFO pellet was wetted by a few droplets of deionized water, while exposing the system to a constant stream of air (Supplementary Fig. 9). Indeed, 35 min after the initial exposure to water, a Raman band assigned to the ν1 symmetric stretching mode of the carbonate moiety in SrCO3 (at 1073 cm−1) emerged, thus verifying the carbonation of SCFO when in contact with water at ambient conditions.
A partial carbonation of the oxygen carrier can also occur through annealing in a CO2-enriched atmosphere. For example, when SCFO was modified by wet impregnation with 5 wt.% NaNO3, followed by a treatment in a packed reactor at 550 °C in 6 vol.% CO2 for 3 h (5NaNO3-CO2-w), XRD of the resulting material exhibited a much higher intensity of the SrCO3 peak relative to the reflections assigned to the perovskite phase when compared to 20Na2CO3-w (Supplementary Fig. 11), indicating that a considerable fraction of the bulk perovskite structure was carbonated to SrCO3. Small reflections due SrCO3 were also observed for spent (redox cycled), 20Na2CO3-d, which did not contain substantial amounts of SrCO3 prior to the redox experiments (Supplementary Fig. 11). However, 20NaNO3-d, which, unlike 20Na2CO3-d, completely inhibited the over-oxidation of propane, did not show any peaks of SrCO3 in the XRD analysis of the spent sample. Consequently, as the surface modification of SCFO with Na2CO3 (20Na2CO3-d) did not completely avoid propane over-oxidation, it can be concluded that propane oxidation (to CO2) resulted in a partial carbonation of the SCFO. Furthermore, modified SCFO that did not completely inhibit propane over-oxidation, e.g., 20NaNO3c-d, produced CO2 not only during their reduction in a diluted propane feed, but also during their re-oxidation in air (Supplementary Fig. 12), indicating the decomposition of surface carbonates or the oxidation of coke during re-oxidation in air. The deposition of coke on the surface of unmodified SCFO and 20NaNO3c-d after reduction was evidenced by Raman spectroscopy (Supplementary Fig. 12). In contrast, 20NaNO3-d did not produce any over-oxidation products, neither during its reduction in propane, nor during its subsequent re-oxidation in air (Supplementary Fig. 12), emphasizing that the interaction of the surface of the oxygen carrier and propane was completely inhibited by the NaNO3-based surface modification.
Characterization of Na2CO3-based surface modifications
20NaNO3c-d, 20Na2CO3-w, and 20Na2CO3-d, all of which only partially inhibited the over-oxidation of propane, displayed reflections due to crystalline Na2CO3 in their diffraction patterns, albeit at very low intensity (Fig. 3c, d). Particularly in 20Na2CO3-d, which was calcined at 900 °C to melt Na2CO3 and to obtain, in turn, a homogeneous surface layer, the diffraction peak assigned to Na2CO3 was of very low intensity. Previous studies on alkali metal carbonate-based surface modifications on perovskites have reported an amorphization of the carbonate shell after impregnation and calcination above the carbonate´s melting point, which might explain their rather low intensity in the diffraction patterns25. Raman spectroscopy of 20Na2CO3-w and 20Na2CO3-d revealed Na2CO3 bands at the surface of the modified oxygen carriers (Fig. 3e). However, the Na2CO3 bands were present in only some of the recorded spectra, indicating an inhomogeneous surface coverage by Na2CO3 in both samples. On the other hand, a Raman map of 20NaNO3c-d suggested a homogeneous surface coverage, as the Na2CO3 band at 1078 cm−1 was present in all of the 144 individual scans (Supplementary Fig. 13), coinciding also with a prominent reflection due to Na2CO3 in its XRD pattern. Despite its homogeneous surface coverage, 20NaNO3c-d was not able to completely inhibit the over-oxidation of propane (5.2 % propane conversion in oxidation experiments), indicating that a solid Na2CO3 surface layer may reduce, but does not completely inhibit, the interaction of a hydrocarbon feed with the surface of an oxygen carrier. This may be attributed to either the diffusion of gaseous propane through the potentially porous surface layer of Na2CO3 or the presence of exposed SCFO surface sites that were not detected by Raman spectroscopy. The distribution of Na in the surface-modified SCFOs was further investigated by TEM-EDS (Supplementary Fig. 14). For all three samples (20Na2CO3-w, 20Na2CO3-d, and 20Na2CO3c-d), Na2CO3 was present at the surface of SCFO, yet TEM-EDS revealed a rather inhomogeneous distribution of Na for all three surface-modified oxygen carriers, which might indicate the formation of Na2CO3 clusters at the surface of SCFO. Particularly in the case of 20Na2CO3-w, EDS also revealed that Sr and Ca cations had migrated from SCFO in/onto the Na-based surface modification.
In situ characterization of SCFO surface modified by NaNO3
20NaNO3-d, which was found to completely inhibit propane over-oxidation over five redox cycles, showed signatures due to NaNO3 in its XRD pattern and its Raman spectra, both in its fresh and spent state (Supplementary Fig. 11). Neither SrCO3, nor Na2CO3 was detected by XRD or Raman spectroscopy in spent 20NaNO3-d. Particularly, the absence of SrCO3 infers that any interaction between gaseous hydrocarbons and the surface of the oxygen carrier was inhibited for 20NaNO3-d, as it was shown that the oxidation of propane on the surface of SCFO will lead to its carbonation and consequently the formation of SrCO3. Interestingly, despite the successful inhibition of propane over-oxidation, Raman spectroscopy of spent 20NaNO3-d at room temperature revealed an inhomogeneous distribution of NaNO3 (Supplementary Fig. 15). Hence, to probe the coverage of the surface of SCFO by NaNO3 under operating conditions, in situ Raman measurements were carried out (Fig. 4). When heating a NaNO3 impregnated SCFO pellet in N2, the ν1 symmetric stretching vibration band of NaNO3 at ~1069 cm−1 shifted slightly towards lower wavenumbers upon increasing the temperature. Once the melting point was reached at ~320 °C, the ν1 band disappeared and a new, less intense band appeared at ~1055 cm−1. The disappearance of the ν1 Raman band and transition into a new low intensity band at 1055 cm−1 has been reported for liquid NaNO3 or highly saturated NaNO3 solutions39, and can therefore be attributed to molten NaNO3 covering the (entire) surface of the impregnated SCFO. When cooling down, the band assigned to molten NaNO3 completely disappeared, and the Raman band due to crystalline NaNO3 did not reemerge. However, once the in situ cell was reheated to 500 °C, the Raman band due to molten NaNO3 reappeared once the temperature exceeded 320 °C.

a Series of Raman spectra when heating to 500 °C, cooling down to room temperature, and reheating to 500 °C under a constant flow of N2, and the temperature profile inside the in situ Raman cell during the experiment. b Graphic illustration of the textural changes of the NaNO3 surface modification during the in situ Raman experiment. c In situ Raman maps of 20NaNO3-d at a selected wavenumber of 1069 cm−1 (when recorded at 30 °C) and 1055 cm−1 (when recorded at 350 °C). Before mapping at 350 °C, the in situ cell was first heated to 500 °C and subsequently cooled down to 350 °C, to keep the surface modification in a molten state, while increasing the quality of the Raman signal, which is compromised at elevated temperatures. Each Raman map consists of 529 individual scans, corresponding to an investigated grid of 69 × 69 μm. d Selected Raman spectra of the in situ maps at 30 and 350 °C. The bars colored in green and blue indicate the Raman bands for crystalline and molten NaNO3, respectively.
During the in situ experiment, the Raman laser remained in the same spot. Hence, the disappearance of the Raman band due to the formation of crystalline NaNO3 after cooling down implies the formation of relatively heterogeneously distributed crystallites of NaNO3 below 300 °C, which did not homogeneously cover the entire surface of SCFO. Consequently, the complete inhibition of propane over-oxidation on 20NaNO3-d was due to the melting of NaNO3 under operating conditions, leading in turn to a fully wetted surface, thereby preventing the direct contact between propane and the surface of SCFO. The melting of the surface modification of 20NaNO3-d was validated further by monitoring the heat flow during a TGA experiment, indicating a melting event at ~305 °C, which agrees with the results of the in situ Raman spectroscopy experiment and the melting point of NaNO3 reported in the literature (Supplementary Fig. 16)40.
In a separate set of in situ Raman experiments, 20NaNO3-d was mapped using a grid of 529 individual scans (covering an area of 69 × 69 μm) at different temperatures (30 °C and 350 °C) to validate the reproducibility of a complete NaNO3 surface coverage under operating conditions. The Raman map of the initial 20NaNO3-d at ambient conditions showed a NaNO3 ν1 band (1069 cm−1) at very high intensity at some spots, while the band was absent in others; this observation agrees with the inhomogeneous surface coverage evidenced by EDS (Supplementary Fig. 17). When heating the in situ Raman cell to 350 °C, the Raman band due to molten NaNO3 (1055 cm−1) was detected in each of the 529 recorded spectra, thus demonstrating a homogeneous surface coverage under operating conditions. After cooling down to ambient conditions, the ν1 band of NaNO3 was not observed in all 529 spots, indicating an inhomogeneous surface coverage with (solid) NaNO3 after cooling down. When 20NaNO3-d was subsequently reheated to 350 °C, the band of molten NaNO3 at 1055 cm−1 was again detected in every sampling spot, confirming the repeatability of NaNO3 wetting the entire surface of SCFO.
The thermal stability of NaNO3 was investigated by studying its decomposition under a flow of Ar in a fixed-bed quartz reactor coupled with an off-gas analysis using a mass spectrometer (MS). The decomposition products O2 and NO were detected in the off-gas only when the temperature exceeded 600 °C, i.e., the reported temperature of NaNO3 decomposition (Supplementary Fig. 18)41. In addition, the Na content of 20NaNO3-d after 0, 5, 20, and 100 redox cycles at 500 °C was determined by ICP-OES, and no significant loss of Na was observed (Supplementary Fig. 19). However, when 20NaNO3-d was calcined in air at 550 °C for 18 h, the resulting material exhibited Raman bands due to both NaNO3 and Na2CO3, indicating a slow carbonation of NaNO3 at 550 °C in air (Supplementary Fig. 20). Thus, NaNO3-based surface modifications showed a high stability and continued inhibition of propane over-oxidation at 500 °C. However, for operating temperatures >550 °C, NaNO3 decomposes slowly and carbonates in a CO2-containing environment, thereby deteriorating the performance of the surface modification in preventing propane over-oxidation, due to the formation and growth of solid Na2CO3 clusters and/or increasing the porosity of the coating.
To summarize, in situ Raman spectroscopy of the NaNO3-based surface modification revealed an inhomogeneous NaNO3 surface coverage at ambient conditions, while at 500 °C, NaNO3 melts and wets the surface of SCFO completely, thereby providing a diffusion barrier for propane and successfully inhibiting its oxidation. The melting point of Na2CO3 (Tm,Na2CO3 = 851 °C) significantly exceeds the temperature of the CLOU-ODHP process (T = 500 °C). Consequently, Na2CO3-based surface modifications remain solid under operating conditions, and their potentially porous structure and/or inhomogeneous surface distribution does not prevent the interaction between propane and the SCFO surface, leading to a partial over-oxidation of propane.
Interaction between released oxygen and the NaNO3 modification
To probe the interaction between the oxygen released by the oxygen carrier and the surface modification during redox cycling, 18O-labeled SCFO modified with 20 wt.% NaNO3 (SCF18O 20NaNO3-d) and SCFO modified with 20 wt.% 18O-labeled NaNO3 (SCFO 20NaN18O3-d) were oxidized and reduced at 500 °C in air and N2, respectively, while analyzing the off-gas by mass spectrometry (Fig. 5a). For SCF18O 20NaNO3-d and SCFO 20NaN18O3-d, labeled oxygen species were detected at the reactor outlet in reducing and oxidizing environments, and the amount of labeled oxygen species (36O2 and 34O2) released decreased over cycling as the 18O content in the samples was continuously depleted. The concentration of 36O2 reached its maximum in the first reduction segment (Fig. 5a). In both SCF18O 20NaNO3-d and SCFO 20NaN18O3-d, the concentration of 34O2 in the off-gas was significantly higher than that of 36O2, which may be attributed to the fact that not all of the redox-active lattice oxygen of the oxygen carrier was replaced by 18O and also to an exchange of oxygen between the NaNO3 surface modification and the SCFO during reduction and re-oxidation.

a Redox cycling of SCF18O 20NaNO3-d (top) and SCFO 20NaN18O3-d (middle) in air and N2 at 500 °C. The spikes in the MS signal were caused by spikes in pressure when switching between different gas atmospheres. The ratio of the intensity of the MS signal of 36O2 to all of the released oxygen species, i.e., I36O2/IO2,tot (bottom). Shadings in gray and white mark the oxidizing and reducing half-cycles, respectively. b Raman spectroscopy of SCF18O 20NaNO3-d before and after redox cycling. c Raman spectroscopy of SCFO 20NaN18O3-d before and after redox cycling. The reference NaN18O3 shows the Raman spectrum of as-prepared 18O-labeled NaNO3 that was used to impregnate the oxygen carrier.
Since the experiments reported above confirmed that NaNO3 does not decompose at 500 °C in inert gases (Ar and N2), no labeled oxygen species would be expected during the reduction of SCFO 20NaN18O3-d in the absence of a direct interaction between the NaNO3 surface modification and the oxygen carrier. Indeed, in a reference experiment using unsupported 18O-labeled NaNO3, no labeled oxygen species were detected in the gas phase under redox cycling at 500 °C. The absence of any oxygen exchange between 18O-labeled NaNO3 and air (in the absence of SCFO) was validated further by Raman spectroscopy (Supplementary Fig. 21). Consequently, it can be inferred that the 34O2 observed during the reduction of SCFO 20NaN18O3-d in N2 originated from an oxygen exchange between the 18O-labeled surface modification and the 16O released by the SCFO. The oxygen exchange between SCFO and NaNO3 was confirmed by Raman spectroscopy of cycled SCF18O 20NaNO3-d (three redox cycles at 500 °C), as Raman bands due to 18O-labeled NaNO3 were observed (Fig. 5b). On the other hand, the Raman spectrum of cycled SCFO 20NaN18O3-d revealed that the majority of 18O had been released and exchanged for 16O, which, due to the absence of oxygen exchange in unsupported 18O-labeled NaNO3 under redox conditions, is expected to have originated from the SCFO (Fig. 5c). The depletion of 18O was in agreement with the continuously decreasing release of labeled oxygen species over multiple redox cycles. Nevertheless, it is worth noting that the release of 36O2 into the gas phase during the reduction of SCF18O 20NaNO3-d is a strong indicator that gaseous oxygen may also be released without interacting with the NaNO3 surface modification. Summarizing, the 18O-labeling experiments confirm an oxygen exchange between SCFO and NaNO3 during oxidation and reduction, as labeled oxygen species were detected in the NaNO3 surface modification of SCF18O 20NaNO3-d after redox cycling and reference experiments showed that unsupported NaN18O3 did not exchange any 18O with air under redox conditions. This oxygen exchange may be attributed to the presence of surface defects on SCFO (e.g., oxygen vacancies) and contribute to a fast permeation of oxygen through the NaNO3 melt.
Catalytic performance of the coupled CLOU-ODHP system
First, a series of reference measurements was performed to assess under what operating conditions (e.g., residence time, heated (post-catalytic) reactor volume, temperature gradients), ODHP occurs non-catalytically through gas-phase pyrolysis42. To this end, the temperature of an empty reactor was increased from 500 °C to 550 °C in 10 °C increments while co-feeding diluted propane (9.5 vol.%) and oxygen (3 vol.%). For T < 550 °C, no propane conversion was observed (Supplementary Fig. 22). Hence, to exclude a non-catalytic propane conversion, the performance of the CLOU-ODHP processes was assessed at 500 °C.
Due to its stable catalytic performance and prominent role in ODHP research, VOx/SiO2 was chosen as the catalyst. The ODHP reaction over VOx-based catalysts proceeds via a Mars-van Krevelen mechanism43. Hence, conceptually, VOx/SiO2 could also be used as a redox catalyst, donating its lattice oxygen for the ODHP reaction during the reduction step and replenishing the oxygen-depleted material in the subsequent oxidation step. For reference, VOx/SiO2 (without the addition of an oxygen carrier) achieved an average propylene selectivity of 87% under CL-ODHP conditions, albeit at a relatively low propane conversion of ~2% due to its low oxygen storage capacity of 0.05 wt.% (Supplementary Figs. 23 and 24).
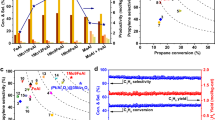
The performance of the coupled CLOU-ODHP scheme proposed here was investigated in a fixed-bed reactor in which the ODHP catalyst VOx/SiO2 was placed downstream of the oxygen carrier (20NaNO3-d). In the first CLOU-ODHP half cycle, the average propane conversion was 7.9% and the propylene selectivity was 68.5%, whereas the average selectivities towards the main side products CO and CO2 were 20.6% and 10.8%, respectively (Fig. 6a). Over 50 redox cycles, the average propane conversion decreased slightly to 7.5%, while the propylene selectivity marginally increased to 68.8%, demonstrating a very stable performance. During one ODHP half cycle, the propane conversion increased rapidly during the first 3 min, reaching 10.5%, and decreased subsequently more gradually to 6.4% by the end of the cycle (Supplementary Fig. 25). The propylene selectivity reached ~65% within the first 5 min and subsequently increased continuously to 73%. This behavior can be attributed to a decreasing oxygen concentration in the gas feed with time on stream (Supplementary Fig. 5), resulting in a high propylene selectivity, yet at the cost of a lower propane conversion as the concentration of gaseous oxygen in the reactor decreases.

a 50 redox cycles performed in a packed bed reactor containing 2 g 20NaNO3-d and 100 mg VOx/SiO2 (GHSV of 16,200 mL(gcath)−1) with 30 min of ODHP half cycle at 500 °C. b Comparison of different ODHP catalysts when co-feeding oxygen and CL(OU)-ODHP at 500 °C. The examined ODHP catalysts are hexagonal boron nitride (h-BN), MgO-supported CrOx (CrOx/MgO), MoVNbTeOx (M1), and SiO2-supported VOx (VOx/SiO2). A detailed discussion on the choice of ODHP catalyst is provided in the supplementary discussion. CLOU-ODHP was carried out utilizing 2 g SCFO/20NaNO3-d and 100 mg VOx/SiO2 (GHSV of 16,200 mL(gcath)−1), with 30 min of ODHP half cycle. CL-ODHP of VOx/SiO2 was carried out under identical conditions as CLOU-ODHP but in the absence of an oxygen carrier. c Performance of h-BN and VOx-based ODHP catalysts when co-feeding oxygen and VOx-based CL(OU)-ODHP under comparable operating conditions at 500 °C. The performance referring to the herein presented CLOU-ODHP scheme was achieved using 2 g SCFO 20NaNO3-d and 300 mg VOx/SiO2 (GHSV of 5400 mL(gcath)−1) at 500 °C, with 15 min of ODHP half cycle (a list of references is provided in the Supplementary Table 1).
Further, the selectivities of the CLOU-ODHP process towards CO and CO2 were very similar to co-feeding gaseous oxygen and propane over VOx/SiO2, which indicates that the observed extent of propane over-oxidation in the CLOU-ODHP process was due to the VOx/SiO2 catalyst, without any over-oxidation originating from the modified oxygen carrier. In fact, the propylene selectivity of the CLOU-ODHP process (68.7% over 50 cycles) slightly exceeded the propylene selectivity when co-feeding oxygen and propane over VOx/SiO2 (Fig. 6b). Without the NaNO3-based surface modification of the oxygen carrier, the propylene selectivity of the coupled CLOU-ODHP process dropped significantly to 21.6% (Fig. 6b).
The oxygen concentration measured at the outlet of the reactor decreased quickly to zero during the oxygen release/reduction step, indicating that the released oxygen was fully consumed in the ODHP reaction (Supplementary Fig. 25) and that the propane conversion was limited by the supply of gaseous oxygen by the oxygen carrier. Hence, a further improvement of the process can, e.g., be achieved by using an oxygen carrier that can generate a higher oxygen partial pressure and/or adjusting the process parameters. As such, by decreasing the reduction time in the reactor from 30 to 15 min, and thus increasing the average oxygen concentration in the reactor during propane dehydrogenation, the average propane conversion was increased by 30%. In fact, lowering the duration of the reduction step to 15 min and increasing the catalyst amount from 100 to 300 mg of VOx/SiO2, led to an overall increase in propylene yield by 90% (from YC3H6 = 5.2% (Fig. 6b) to YC3H6 = 9.9% (Fig. 6c)). This performance is maintained over 250 redox cycles, corresponding to 6 days of continuous operation, proving long-term stability of the CLOU-ODHP process (Supplementary Fig. 26). Overall, the presented CLOU-ODHP scheme compares favorably to reported VOx-based redox catalysts in CL reaction schemes, as well as VOx-based catalysts when operated in a classic ODHP in which oxygen and propane are co-fed (Fig. 6c). It is noteworthy, that propylene yields >20% have been reported for CL-ODHP under different conditions (e.g., higher temperature)5,10,12,31. However, while high propylene yields were achieved, this generally seems to come at a cost of very short redox cycles, making the CL process difficult to operate. Moreover, important process parameters to estimate the potential productivity per redox cycle, such as the oxygen storage capacity of the redox catalyst, are often not reported. As demonstrated, the herein presented CLOU-ODHP scheme matches the catalytic performance of the conventionally operated ODHP catalyst. Consequently, when paired with recently developed, better-performing ODHP catalysts, such as modified h-BN catalysts44,45,46, the CLOU-ODHP scheme has the potential to exceed propylene yields of 20%, while simultaneously providing an in situ oxygen supply and high oxygen storage capacity.
Discussion
A CL scheme for ODHP is introduced in which a surface-modified oxygen carrier provides gaseous oxygen to oxidatively dehydrogenate propane over a physically separate catalyst within one reactor. The oxygen carrier, Ca-doped SrFeO3, releases oxygen at 500 °C in low oxygen partial pressure environments, and the speed of the oxygen release can be tailored to the desired reaction by adjusting the type and quantity of the dopant. To inhibit the over-oxidation of propane by the oxygen carrier, a NaNO3-based surface modification was developed. NaNO3 melts under operating conditions, thereby wetting the surface of the oxygen carrier uniformly and avoiding direct contact between propane and the surface of the oxygen carrier. The proposed NaNO3-based surface modification is easily applicable to any oxygen carrier material and completely avoids the combustion of hydrocarbons by the oxygen carrier over multiple redox cycles in the temperature range of 350–500 °C. On the other hand, Na2CO3-based surface modifications remain in a solid state under CLOU-ODHP operating conditions and were found to be unsuccessful in completely inhibiting the over-oxidation of propane. However, it is noteworthy that at temperatures exceeding 550 °C, the NaNO3-based surface modification will slowly deteriorate by NaNO3 decomposition and, depending on the reaction environment, by its partial conversion into Na2CO3, thereby exposing the surface of the oxygen carrier to interact with the hydrocarbon feed.
The coupled CLOU-ODHP process that combines the NaNO3-modified oxygen carrier (20NaNO3-d) with VOx/SiO2 in a sequential fashion in a packed bed reactor shows a stable propane conversion of 7.5–7.9% at ~70% propylene selectivity, thereby outperforming the identical VOx/SiO2 catalyst under conventional ODHP conditions in which propane and oxygen are co-fed. Moreover, the addition of an oxygen carrier significantly increases the productivity of the process compared to using VOx/SiO2 as a redox catalyst by substantially increasing the oxygen storage capacity of the system. By tuning the operating parameters of CLOU-ODHP, its performance could be improved to 14.5% propane conversion at 68% propylene selectivity at high stability over 250 redox cycles. The presented CL concept demonstrates how oxygen is provided in situ for the ODHP reaction, thereby avoiding the costly air separation necessary in conventional ODHP. Through the in situ generation of oxygen and overall advantages of the oxidative dehydrogenation, such as reduced energy consumption due to the exothermic reaction and facilitated downstream processing9,10,23, the CLOU-ODHP process can offer significant economic and ecological benefits over state-of-the-art propylene production processes. Overall, the separation of the oxygen-providing and catalytic functionalities enables great flexibility, as each component can be tailored individually. This concept may therefore be applied to further reactions and catalytic processes involving the necessity of gaseous oxygen.
Methods
Oxygen carrier synthesis
Calcium-doped strontium ferrite perovskites (Sr1-xCaxFeO3−δ, 0 ≤ x ≤ 0.25) were prepared by the Pechini method. A-site doping concentrations x ≥ 30 mol% were not considered, as these exceed the incorporation limit of Ca in the A-site of the perovskite and lead to the formation of brownmillerite-type Ca1.5Sr0.5Fe2O5 and Ca2Fe2O5 phases47. In a typical synthesis for ~3.5 g of oxygen carrier, a total 0.1 mol of metal cations in the form of calcium nitrate tetrahydrate (Acros Organics, 99 %), strontium nitrate (Sigma-Aldrich, ACS Reagent, ≥99%), and iron(III) nitrate nonahydrate nitrate (Sigma-Aldrich, ACS Reagent, ≥98%), was dissolved in the desired stoichiometric ratio in 100 mL deionized water under continuous stirring at room temperature. Citric acid (Thermo Scientific, 99%) was slowly added to the metal nitrate solution in a molar ratio of 3:1 (citric acid to metal ions) and subsequently transferred to an oil bath heated to 80 °C. After 20 min, ethylene glycol (Sigma-Aldrich, ReagentPlus, ≥99%) was added to the metal citrate solution in a molar ratio of 1.2:1 (ethylene glycol to citric acid), and the solution was kept at 80 °C under continuous stirring until a gel formed. The gel was dried at 130 °C for 20 h, crushed, and calcined in air at 500 °C for 7 h (heating ramp 3 °C min−1) to combust the nitrates. The powder (<100 μm) was then recalcined in air at 1200 °C for 10 h to form the perovskite phase. Surface modifications of Sr0.8Ca0.2FeO3−δ with 5, 10, or 20 wt.% of NaNO3 (Sigma-Aldrich, ACS Reagent, ≥99%) or Na2CO3 (Acros Organics, 99.5 %) were prepared by wet or dry impregnation. For the modification by wet impregnation, the respective amounts of NaNO3 or Na2CO3 were dissolved in 5 mL deionized water. 2 g of the Sr0.8Ca0.2FeO3−δ powder was added to the aqueous solution, and the suspension was stirred for 5 min at room temperature. After drying in an oven at 80 °C overnight, the impregnated oxygen carrier was calcined in air at 500 °C for 3 h (heating ramp 5 °C min−1). Analogously, for dry impregnation, the respective amount of NaNO3 or Na2CO3 was ground together with 2 g of Sr0.8Ca0.2FeO3−δ in a mortar for 20 min to obtain a homogeneous powder. The powder was calcined in air for 3 h at 900 °C for an impregnation with Na2CO3 (heating ramp 5 °C min−1). To improve handling, the materials were pressed into pellets (diameter 14 mm) under 5 tons, crushed, and then sieved to the size fraction of 425–850 μm.
18O-labeled NaNO3 was synthesized by first mixing 200 μL of HNO3 (ACS Reagent, 70%) with 280 μL of H218O (CMR—Center of Molecular Research, isotope enrichment ≥ 98%) and aged in a closed vial at 100 °C for 3 days to equilibrate the diluted nitric acid. Subsequently, 164.8 mg of Na2CO3 (Acros Organics, 99.5%) was added to neutralize the nitric acid. After the dissolution of Na2CO3, the solution was dried in a vacuum oven at 100 °C and 50 mbar for 24 h to yield the final product of 18O enriched NaNO3 with a 18O content of ~45%, as determined using Raman spectroscopy by calculating the ratio of the peak intensities attributed to labeled and unlabeled NaNO3 (Supplementary Fig. 27).
18O-labeled Sr0.8Ca0.2FeO3−δ was produced by reducing 200 mg of Sr0.8Ca0.2FeO3−δ powder in a thermogravimetric analyzer (Mettler Toledo TGA/DSC 3+) at 1000 °C in 7 % H2/N2 for 12 h, followed by re-oxidation in a N2 stream saturated with H218O at 900 °C for 6 h. Thermodynamically, this procedure limited the re-oxidation to the brownmillerite phase Sr0.8Ca0.2FeO2.5, which contained ~15 wt.% of 18O (Sr0.8Ca0.2Fe16O18O1.5). Prior to the experiments involving 18O-labeled oxygen carrier, the oxygen carrier was oxidized completely in air at 500 °C, such that its redox-active lattice oxygen also contained a small fraction (<20%) of 16O.
ODHP catalyst synthesis
The ODHP catalyst VOx/SiO2 was prepared by first dispersing Aerosil 300 SiO2 (Evonik Industries) in deionized water until a homogeneous slurry was obtained. The slurry was dried in an oven at 120 °C for 16 h, and the resulting SiO2 aggregates were crushed gently, sieved to a size fraction of 212–300 μm, and calcined in air at 600 °C for 4 h (heating ramp 4 °C min−1). Subsequently, 3.2 mL of a 0.297 M aqueous ammonium metavanadate (Acros Organics, ACS Reagent) solution was prepared and mixed with 3.2 mL of a 0.55 M aqueous oxalic acid solution (Sigma-Aldrich, ≥99%) until the ammonium metavanadate was completely dissolved. Subsequently, 2 g of the prepared SiO2 support was impregnated with the NH4VO3 solution by incipient wetness impregnation. The impregnated catalyst was calcined in air at 600 °C for 4 h (heating ramp 4 °C min−1).
Structural and compositional characterization
The bulk crystal structure of the materials was analyzed with a PANalytical Empyrean X-ray powder diffractometer equipped with an X’Celerator Scientific ultrafast line detector and Bragg-Brentano high-density incident beam optics. The diffraction patterns were collected for 1 h over the 2Θ range of 10–90 °C with a step size of 0.016° using Cu Kα radiation (45 kV, 40 mA). All materials were prepared as finely ground powders on silicon wafers, zero-background sample holders.
The elemental composition of the perovskite oxygen carriers, as well as the catalyst loading, was determined by ICP-OES using an Agilent 5100 VDV, calibrated with the multi-element standard 5 (Sigma-Aldrich). Generally, ~3 mg of sample was dissolved in 5 mL of aqua regia facilitated by a microwave digestion system (Anton Paar, Multiwave GO), and subsequently diluted with deionized water to a total volume of 25 mL.
A FEI Talos F200X TEM was operated at an operating voltage of 200 kV. The instrument was equipped with a high-brightness field-emission gun, a high-angle annular dark-field detector, and a large collection-angle EDS detector. The samples were prepared by dispersion in ethanol in an ultrasonication bath for 15–20 min, and their subsequent deposition onto a carbon-coated Cu grid.
Structural surface analysis was carried out with a Thermo Fisher DXR 2 Raman spectrometer equipped with a 532 nm excitation laser. Two different objectives with 10- and 50-fold magnification were used, resulting in estimated spot sizes of 2.1 and 0.7 μm in diameter, respectively. The Raman spectra were recorded in a range of 120–3500 cm−1 with a resolution of 900 lines mm−1. In situ experiments were performed using a Linkam CCR1000 stage, by heating the sample to 500 °C at a ramp of 5 °C min−1. The gas atmosphere inside the in situ cell was controlled by a steady flow of 50 mL min−1 of the respective gas used. For sample preparation and to obtain a smooth surface for characterization, the materials were pressed into 3 mm pellets using a hand pelletizer.
Thermogravimetric analysis of the oxygen carrier
The oxygen storage capacity and the rates of oxygen release of the oxygen carriers were assessed by TGA in a Mettler Toledo TGA/DSC 3+. In a typical TGA experiment, 30 mg of sample was filled into a 70 μL alumina crucible. The total gas flow rate into the furnace chamber was kept constant at 150 mL min−1, including a 25 mL min−1 nitrogen purge to protect the balance. The sample was first heated to 800 °C in air for 1 h to decompose surface carbonates that influence the rate of oxygen release47. Isothermal redox cycling was carried out at 500 °C by alternating the gas flow between 125 mL min−1 of nitrogen during the reduction (oxygen release) step (40 min) and 125 mL min−1 of air during the oxidation step (20 min). The oxygen storage capacity was determined by dividing the difference of the sample mass in its oxidized (mox) and reduced (mred) state by its oxidized mass:
Redox cycling and evaluation of catalytic performance
The redox behavior of the oxygen carrier, as well as the catalytic activity of the ODHP catalyst and the coupled system of Sr0.8Ca0.2FeO3−δ and VOx/SiO2, was assessed in a U-shaped quartz reactor with an inner diameter of 5 mm and a total volume of ~9 mL. In a typical redox cycling experiment of the coupled system, the packed bed reactor contained 2 g of the modified oxygen carrier and 0.1 g of the VOx/SiO2 catalyst that was placed further downstream and separated from the oxygen carrier by a small plug of quartz wool with a length of ~5–10 mm. A redox cycle consisted of an oxidation step in air for 15–20 min, and a reduction step using 9.5 % propane diluted in nitrogen for 15–30 min, with a 2 min purge in nitrogen between each redox half step. The total gas flow rate was 27 mL min−1, as measured and controlled using Bronkhorst mass flow controllers at normal temperature and pressure, leading to a superficial velocity of ~0.025 m s−1. The reaction was carried out at ambient pressure and 500 °C, resulting in a gas density of 450–500 g m−3 when assuming a mixture of ideal gases. The composition of the outlet gas was evaluated by a paramagnetic analyzer for oxygen (ABB EL3020, Magnos 206), a nondispersive infrared analyzer for CO and CO2 (ABB EL3020, Uras 26), a thermal conductivity analyzer for H2 (ABB EL3020, Caldos 27), and an FTIR analyzer (MKS, Precisive 5) for the hydrocarbons. It is noteworthy that the gas analyzers were set up in series, which affected the measured gas concentrations in the initial few seconds after a gas switch due to gas mixing effects in the analyzer cells and the transfer lines. For this reason, the first 60 s after the gas switch were not considered when calculating the average conversion and selectivities for each cycle. To reduce back-mixing effects and latency within the sequence of gas analyzers, the outlet gas of the reactor was diluted with nitrogen (typically equaling the total gas flow rate through the reactor). Over-oxidation experiments and catalytic reference measurements were carried out in the same reactor setup. The quartz reactor was loaded with 0.5 g of oxygen carrier, and the total flow rate was kept constant at 50 mL min−1 throughout the redox cycling. The sample was reduced in 3 % propane in nitrogen for 30 min and oxidized in air for 20 min with a 1-min nitrogen purge between each redox half step. To determine the partial pressure of oxygen that is achieved in the coupled process under reaction conditions, 2 g of the modified oxygen carrier was cycled under CLOU-ODHP conditions, except for the reduction step, which was carried out in pure nitrogen instead of diluted propane. Subsequently, the oxygen carrier inside the reactor was replaced by inert quartz chips of the same volume and size fraction as the oxygen carrier, to record a blank experiment of the oxygen profile during redox cycling. The blank oxygen profile of the redox cycling experiment was subtracted from the oxygen release of the oxygen carrier to obtain the oxygen concentration produced during the CLOU-ODHP process. The operating conditions for the catalytic reference experiments were chosen to mimic the coupled CLOU-ODHP conditions. Thus, the average oxygen concentration produced by the oxygen carrier during a reduction step (~1.4%) was co-fed to 9.5% of propane in nitrogen at a constant total gas flow of 27 mL min−1. Subsequently, the temperature was increased incrementally from 450 to 550 °C in 10 °C intervals. Note that due to the small isothermal reactor volume and therefore limited loading capacity, typical O2:C3H8 ratios in ODHP (0.5–1 for V-based catalysts or 1–2 for BN-based catalysts)1 could not be achieved with the oxygen carrier at reasonable flow rates (>20 mL min−1) in our setup.
Due to the relatively high dilution of the reactive gas in inert nitrogen (1:9), the total molar gas flow rate ṅtot was approximated to be constant and ideal gas properties were assumed to calculate the molar gas flow rates for a specific component i from the measured gas concentration \({x}_{i}\):
In Eq. 4, \(R\) denotes the ideal gas constant, \({\dot{V}}_{{{{\rm{tot}}}}}\) the total volumetric flow rate into the reactor, and \({p}_{{{{\rm{cal}}}}}\) and \({T}_{{{{\rm{cal}}}}}\) are the pressure and temperature for which the mass flow controllers were calibrated and operated. The propane conversion XC3H8, propylene selectivity SC3H6, propylene yield YC3H6, and overall carbon balance Cbal were calculated as follows:
Data availability
The data generated in this study have been deposited in the Zenodo database and are freely available under the Digital Object Identifier https://doi.org/10.5281/zenodo.15167212, as well as from the corresponding author upon request.
References
Carter, J. H. et al. Direct and oxidative dehydrogenation of propane: From catalyst design to industrial application. Green Chem. 23, 9747–9799 (2021).
Yan, W., Sun, Q. & Yu, J. Dehydrogenation of propane marches on. Matter 4, 2642–2644 (2021).
Gambo, Y. et al. Catalyst design and tuning for oxidative dehydrogenation of propane—a review. Appl. Catal. A Gen. 609, 117914 (2021).
Marsh, M. & Wery, J. Filling the propylene gap—shaping the future with on-purpose technologies. (UOP LLC (a Honeywell company), 2019). https://pages2.honeywell.com/rs/828-DHL-685/images/Filling%20the%20Propylene%20Gap%20On%20purpose%20technologies.pdf.
Rostom, S. & De Lasa, H. I. Propane oxidative dehydrogenation using consecutive feed injections and fluidizable VOx/γAl2O3 and VOx/ZrO2-γAl2O3 catalysts. Ind. Eng. Chem. Res. 56, 13109–13124 (2017).
Carrero, C. A., Schloegl, R., Wachs, I. E. & Schomaecker, R. Critical literature review of the kinetics for the oxidative dehydrogenation of propane over well-defined supported vanadium oxide catalysts. ACS Catal. 4, 3357–3380 (2014).
Liu, J. & Li, F. Mixed oxides as multi-functional reaction media for chemical looping catalysis. Chem. Commun. 59, 10–28 (2022).
Zhu, X., Imtiaz, Q., Donat, F., Müller, C. R. & Li, F. Chemical looping beyond combustion—a perspective. Energy Environ. Sci. 13, 772–804 (2020).
Wu, T., Yu, Q. & Qin, Q. Energy analysis of chemical looping oxidative dehydrogenation of propane. Petrol. Sci. Technol. 36, 266–272 (2018).
Chen, S. et al. Concerted oxygen diffusion across heterogeneous oxide interfaces for intensified propane dehydrogenation. Nat. Commun. 14, 2620 (2023).
Al-Ghamdi, S. A. & De Lasa, H. I. Propylene production via propane oxidative dehydrogenation over VOx/γ-Al2O3 catalyst. Fuel 128, 120–140 (2014).
Chen, S. et al. Modulating lattice oxygen in dual-functional Mo-V-O mixed oxides for chemical looping oxidative dehydrogenation. J. Am. Chem. Soc. 141, 18653–18657 (2019).
Wu, T., Yu, Q., Roghair, I., Wang, K. & van Sint Annaland, M. Chemical looping oxidative dehydrogenation of propane: a comparative study of Ga-based, Mo-based, V-based oxygen carriers. Chem. Eng. Process. 157, 108137 (2020).
Wu, T. et al. Selecting suitable oxygen carriers for chemical looping oxidative dehydrogenation of propane by thermodynamic method. J. Therm. Anal. Calorim. 140, 1837–1843 (2020).
Chen, S. et al. Coverage-dependent behaviors of vanadium oxides for chemical looping oxidative dehydrogenation. Angew. Chem. Int. Ed. 59, 22072–22079 (2020).
Wu, T., Yu, Q., Wang, K. & van Sint Annaland, M. Development of V-based oxygen carriers for chemical looping oxidative dehydrogenation of propane. Catalysts 11, 1–18 (2021).
Song, H. et al. Chemical looping oxidative propane dehydrogenation controlled by oxygen bulk diffusion over FeVO4 oxygen carrier pellets. Chin. J. Chem. Eng. 53, 409–420 (2023).
Wang, W. et al. Tandem propane dehydrogenation and surface oxidation catalysts for selective propylene synthesis. Science 381, 886–890 (2023).
Love, A. M. et al. Understanding the synthesis of supported vanadium oxide catalysts using chemical grafting. Chemistry 26, 1052–1063 (2020).
Love, A. M. et al. Elucidation of anchoring and restructuring steps during synthesis of silica-supported vanadium oxide catalysts. J. Mater. Chem. 28, 5495–5504 (2016).
Wu, X. P. & Gong, X. Q. Unique electronic and structural effects in Vanadia/Ceria-catalyzed reactions. J. Am. Chem. Soc. 137, 13228–13231 (2015).
Luongo, G. et al. Highly selective oxidative dehydrogenation of ethane to ethylene via chemical looping with oxygen uncoupling through structural engineering of the oxygen carrier. Adv. Energy Mater. 12, 2200405 (2022).
Luongo, G., Donat, F., Krödel, M., Cormos, C. C. & Müller, C. R. Experimental data supported techno-economic assessment of the oxidative dehydrogenation of ethane through chemical looping with oxygen uncoupling. Renew. Sustain. Energy Rev. 149, 111403 (2021).
Gao, Y. et al. Alkali metal halide-coated perovskite redox catalysts for anaerobic oxidative dehydrogenation of n-butane. Sci. Adv. 8, 7343 (2022).
Gao, Y. et al. A molten carbonate shell modified perovskite redox catalyst for anaerobic oxidative dehydrogenation of ethane. Sci. Adv. 6, 1–10 (2020).
Luongo, G., Donat, F. & Müller, C. R. Structural and thermodynamic study of Ca A- or Co B-site substituted SrFeO3−δ perovskites for low temperature chemical looping applications. Phys. Chem. Chem. Phys. 22, 9272–9282 (2020).
Wang, X. et al. High-throughput oxygen chemical potential engineering of perovskite oxides for chemical looping applications. Energy Environ. Sci. 15, 1512–1528 (2022).
Fan, Q. et al. An investigation of the structural and electronic origins of enhanced chemical looping air separation performance of B-site substituted SrFe1−xCoxO3−δ perovskites. Phys. Chem. Chem. Phys. 26, 20511–20521 (2024).
Nadjafi, M. et al. Oxidative dehydrogenation of propane on silica-supported vanadyl sites promoted with sodium metavanadate. Catal. Sci. Technol. 10, 7186–7193 (2020).
Tadeo, I. J., Bhardwaj, D., Sheela, D., Krupanidhi, S. B. & Umarji, A. M. Highly photoresponsive VO2(M1) thin films synthesized by DC reactive sputtering. J. Mater. Sci. Mater. Electron. 31, 4687–4695 (2020).
Wang, X. et al. Coupling acid catalysis and selective oxidation over MoO3-Fe2O3 for chemical looping oxidative dehydrogenation of propane. Nat. Commun. 14, 2039 (2023).
Yusuf, S., Neal, L., Bao, Z., Wu, Z. & Li, F. Effects of sodium and tungsten promoters on Mg6MnO8-based core-shell redox catalysts for chemical looping—oxidative dehydrogenation of ethane. ACS Catal. 9, 3174–3186 (2019).
Wang, T. et al. Core-shell Na2WO4/CuMn2O4 oxygen carrier with high oxygen capacity for chemical looping oxidative dehydrogenation of ethane. Fuel 303, 121286 (2021).
Gao, Y., Haeri, F., He, F. & Li, F. Alkali metal-promoted LaxSr2-xFeO4-δ redox catalysts for chemical looping oxidative dehydrogenation of ethane. ACS Catal. 8, 1757–1766 (2018).
Oing, A., von Müller, E., Donat, F. & Müller, C. R. Material engineering solutions toward selective redox catalysts for chemical-looping-based olefin production schemes: a review. Energy Fuels 38, 17326–17342 (2024).
Østergaard, M. B., Strunck, A. B., Boffa, V. & Jørgensen, M. K. Kinetics of strontium carbonate formation on a Ce-doped SrFeO3 perovskite. Catalysts 12, 265 (2022).
Koo, B. et al. Sr segregation in perovskite oxides: Why it happens and how it exists. Joule 2, 1476–1499 (2018).
Hong, J., Heo, S. J. & Singh, P. Water mediated growth of oriented single crystalline SrCO3 nanorod arrays on strontium compounds. Sci. Rep. 11, 3368 (2021).
Rusli, I. T., Schrader, G. L. & Larson, M. A. Raman spectroscopic study of NaNO3 solution system - solute clustering in supersaturated solutions. J. Cryst. Growth 97, 345–351 (1989).
Lomonaco, A., Haillot, D., Pernot, E., Franquet, E. & Bédécarrats, J. P. Sodium nitrate thermal behavior in latent heat thermal energy storage: a study of the impact of sodium nitrite on melting temperature and enthalpy. Sol. Energy Mater. Sol. Cells 149, 81–87 (2016).
Freeman, E. S. The kinetics of the thermal decomposition of sodium nitrate and of the reaction between sodium nitrate and oxygen. J. Phys. Chem. 60, 1487–1493 (1956).
Nadjafi, M. et al. On the importance of benchmarking the gas-phase pyrolysis reaction in the oxidative dehydrogenation of propane. ChemCatChem 15, e202200694 (2023).
Jiang, X. et al. Multiple promotional effects of vanadium oxide on boron nitride for oxidative dehydrogenation of propane. JACS Au 2, 1096–1104 (2022).
Zhou, H. et al. Isolated boron in zeolite for oxidative dehydrogenation of propane. Science 372, 76–80 (2021).
Cao, L. et al. Antiexfoliating h-BN⊃In2O3 catalyst for oxidative dehydrogenation of propane in a high-temperature and water-rich environment. J. Am. Chem. Soc. 145, 6184–6193 (2023).
Liu, Z. et al. Plasma tuning local environment of hexagonal boron nitride for oxidative dehydrogenation of propane. Angew. Chem. Int. Ed. 60, 19691–19695 (2021).
Luongo, G. et al. Activation in the rate of oxygen release of Sr0.8Ca0.2FeO3−δ through removal of secondary surface species with thermal treatment in a CO2-free atmosphere. J. Mater. Chem. A 11, 6530–6542 (2023).
Acknowledgements
This publication was created as part of NCCR Catalysis (Grant Numbers 180544 and 225147, C.R.M.), a National Centre of Competence in Research funded by the Swiss National Science Foundation. The authors thank Dr Agnieszka Kierzkowska for contributing to this work by carrying out electron microscopy. The authors acknowledge the help of ScopeM at ETH Zurich for TEM and EDS analysis.
Author information
Authors and Affiliations
Contributions
A.O. wrote the original draft of the manuscript, performed the material synthesis, performance assessment and structural characterization. F.D. and C.R.M. supervised the project, wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oing, A., Donat, F. & Müller, C.R. Controlling selectivity in the chemical looping oxidative dehydrogenation of propane through interface engineering. Nat Commun 16, 5247 (2025). https://doi.org/10.1038/s41467-025-60428-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60428-7
