
Abstract
New York City (NYC) was a major hotspot during the 2022 multi-country Mpox virus (MPXV) outbreak in the U.S. To investigate the genomic characteristics of MPXV in NYC, we sequenced 1138 specimens from 758 individuals using PrimalSeq and Illumina technology and performed phylogenomic analyses alongside 2967 global MPXV sequences. Nextclade lineage assignment revealed a NYC-specific B.1.12 lineage, with phylogenetic analysis showing unique clusters in NYC and North America. MPXV lineage B-specific mutations were predominantly driven by APOBEC3 activity (53/58 mutations). APOBEC3-associated mutations were significantly higher in 2022 outbreak sequences compared to those before 2022. Intra-host genome diversity was investigated in individuals with multiple specimens (n = 360). This analysis showed that 6.4% of these individuals had distinct genomic profiles, with 4.2% likely due to co-infections with distinct MPXV strains. This study identified unique MPXV genomic profiles in NYC, estimated concurrent infections with multiple MPXV strains and emphasized the need for enhanced surveillance and public health strategies to address co-infections, especially through collaboration with labs, clinics, and community groups.
Similar content being viewed by others


Introduction
The causative agent of mpox in humans is the Mpox virus (MPXV), a zoonotic orthopox virus1,2. Mpox symptoms include fever, fatigue, headache, and a multi-stage rash that can spread between body sites. The genomic diversity of MPXV is represented in two clades, I and II. Clade I further subdivided into Clade Ia and newly identified Clade Ib3. Clade Ia variants include sequences from Central Africa and are typically more transmissible and pathogenic2. The first mpox outbreak outside the African continent was reported in the United States in 20034 and belonged to Clade II, which includes two subclades, IIa and IIb and has reduced pathogenicity5,6. Within Clade IIb, Lineages A and B emerged with subsequent sublineages7. In 2022, rapid transmission promoted the spread of MPXV, which resulted in a global outbreak6. This outbreak was driven by MPXV lineage B.1, which gave rise to B.1.1 and other sublineages7. The first diagnosis of mpox in New York City (NYC) was on May 19, 20228. Since then, a total of 3,821 and 204 confirmed cases were reported in NYC in 2022 and 2023, respectively9. MPXV outbreaks have primarily spread through sex and other intimate contact among social networks of gay, bisexual, and other men who have sex with men (MSM)8. Risk factors for mpox-related hospitalization included immunosuppression and HIV co-infection10.
Several genomic studies have been published on the 2022 outbreak in different countries around the world to understand the transmission and the evolution of the virus; however, most of these studies were limited by the number of genomes analyzed (n < 402)1,6,11,12,13,14,15,16,17,18,19,20,21,22,23,24. Studying the intra-host genomic diversity of MPXV can provide additional resolution6,16 to study virus transmission networks. The intra-host genomic diversity of MPXV can result from multiple infections, viral evolution, or a combination of both within the host. Often, multiple infections in the same host can be identified on phylogenetic trees based on polyphyletic relations. However, multiple infections resulting from a close-knit transmission cluster involving related strains can be monophyletic on a phylogenetic tree25. Such relationships are usually over-represented for viruses like HIV among MSM communities and in people who inject drugs25. Notably, 38-57% of mpox cases in the US are in people living with HIV26 and may show similar patterns to HIV on a phylogenetic tree due to potentially similar behavioral risk factors.
MPXV has a slow mutation rate, with 1-2 substitutions per year27. However, accelerated evolution (6 to 12-fold increase in mutation rate) has been documented for MPXV due to the genomic editing activities of host enzymes such as apolipoprotein B mRNA-editing catalytic polypeptide-like 3 (APOBEC3)6,18,24,28,29,30. Newly acquired mutations can alter viral transmissibility6 and reduce the efficacy of therapeutics and vaccines31. The probability of these mutations increases in cases of immunocompromised individuals, who may facilitate the emergence of new variants32.
An analysis of intra- and inter-host MPXV diversity is needed with a representative sample to enable a deeper understanding of MPXV evolution and transmission patterns. In this study, we used a large genomic dataset for MPXV infections in NYC (n = 1138 sequences from 758 total individuals) to analyze genomic diversity both within and between individuals aiming to estimate the occurrence of simultaneous infections with multiple MPXV strains. The sequencing data included 390 sequences collected from a single lesion from 390 individuals, and 748 sequences collected from two or more lesions from 368 individuals. Here we studied the genomic epidemiology of MPXV in NYC in comparison to MPXV genomes from around the world (n = 2967) using Nextclade and phylogenetic analysis. Mutational analysis was done to determine sublineage-defining mutations, APOBEC3 signatures, and the prevalence of clinically relevant mutations. Intra-host genomic diversity was assessed between different lesions within an individual, and inter-host genomic diversity was used to estimate co-infection rates in NYC. Finally, this study aimed to analyze sequences from epidemiologically linked mpox cases in NYC using phylogenetics.
Results
Overview of MPXV genomes dataset
This study included 1138 MPXV specimens collected between May 2022 and February 2023 and sequenced by the NYC Public Health Lab (PHL) (Supplementary Data 1). The spatiotemporal distribution (Supplementary Fig. 1A and 1B) shows a peak of specimen collection in July 2022 with the highest frequency of genomes sequenced from residents of Manhattan. Of 758 individuals in this study, 390 had single MPXV genomes collected from one lesion (34% of genomes), 361 had two genomes collected from two lesions (62% of genomes), and the remainder had three or more genomes collected from 3-4 lesions (4% of the genomes). Of the individuals with one genome, most of the sequenced specimens were from the genital area (62%, 241/390) (Supplementary Fig. 1C). Similarly, for individuals with multiple genomes, the majority of sequenced specimens (54%, 404/748) were from the genital area, 30% (228/748) were from upper body, and 10% (72/748) were from lower body sites. Specimens were collected from two lesions from the same anatomical site (e.g., genital area) for 133 sequences and from different anatomical sites (e.g., one lesion from the hand and one lesion from the genital area) for 468 sequences (Supplementary Fig. 1D).
MPXV global genome sequences (n = 2967, excluding NYC sequences) included in this study were collected from 1985 through 2023 with representative sequences from five continents except Oceania. In this dataset, 2877 genomes belong to lineage B of clade IIb, 63 genomes belong to lineage A of clade IIb, and 27 genomes from clade Ia serve as the outgroup (Supplementary Data 2).
Lineage assignment and phylogenetic analysis of MPXV genomes
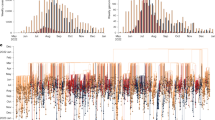
In the combined NYC and global dataset, Nextclade designated 4,015 of the 4105 sequences as lineage B.1 of MPXV clade IIb. In NYC, the most frequently observed B.1 sublineages were B.1.2, B.1.12, B.1.3 and B.1.7 ( > 50 sequences per sublineage) (Fig. 1A). Nextclade lineage assignment had 99% and 98% agreement with the observed phylogenetic clades for sublineages observed in the global and NYC phylogenies, respectively (Figs. 1B, 2A). The phylogenetic clade that belonged to sublineage B.1.12 included predominantly sequences from NYC (94%) (Figs. 1B and 2A, red asterisk). For genomes designated as B.1 lineage by Nextclade, at least nine distinct clusters were observed in the MPXV phylogeny with sequences in each cluster predominantly from NYC and/or North America (Fig. 2B, Supplementary Fig. 2, Table 1).

A) B.1 and sublineages assignment of the 2022 MPXV genome sequences from NYC, North America (Excluding NYC) and Europe. Sequences from the Africa, Asia and South America were excluded from this figure due to small sample size (Africa: n = 2; Asia: n = 4; South America: n = 75). Lineage and sequence count per source were represented with x-axis and y-axis, respectively. Apart from the parent lineage B.1, the most frequently observed B.1 sublineages in Europe were B.1.1, B.1.2, B.1.7, B.1.3 and B.1.5 ( > 50 sequences per sublineage). In North America (excluding NYC), the most frequently observed B.1 sublineages were B.1.2, B.1.11, B.1.4, B.1.3, and B.1.1 ( > 50 sequences per sublineage). The most frequently observed NYC B.1 sublineages ( > 50 sequences per sublineages) were B.1.2, B.1.12, B.1.3 and B.1.7. B) Global phylogeny of MPXV genome sequences (including NYC sequences). The tree visualized with 4078 sequences where 63 and 4,015 sequences were from lineage A and B, respectively. The collection dates ranged from 10/09/2017 to 01/09/2023. In the phylogeny, the branches were colored by Nextclade lineage assignment. The outer ring was colored by geographical region. Sublineages associated with specific geographical regions were shown using colored asterisks. All the B.1 sublineages were placed as distinct clades and had a 98.9% agreement with the Nextclade lineage assignment. Most sequences (93.6%) from sublineage B.1.12 were from NYC (red asterisk). Most sequences from sublineages B.1.11, B.1.13, B.1.2 and, B.1.4 were primarily from North America (excluding NYC) (orange asterisks); and sublineages B.1.1, B.1.7 and B.1.9 were primarily from Europe (blue asterisks). B.1.6 sublineage was predominantly from South America (purple asterisk). The source data files for Fig. 1 are available in the following GitHub repository: https://github.com/sakther-NYCDOHMH/nyc_mpox_genomic_epidemiology/.

A) A subtree of the global MPXV tree from Fig. 1 containing 1138 NYC sequences. The branches were colored by Nextclade lineage assignment. The B.1 sublineages in the phylogeny were in 98.0% agreement with the Nextclade lineage assignment. The B.1.12 sublineage was predominantly NYC specific (red asterisk). B) Clusters emerged within B.1 assigned sequences. Lineage assignment was based on Nextclade. Six distinct clusters were observed in the NYC genome phylogeny (see also Supplementary Fig. 2) where sequences in each cluster were predominantly NYC specific and had at least one cluster-specific nonsynonymous mutation except for Cluster #6 (Table 1, Supplementary Fig. 3). The source data files for Fig. 2 are available in the following GitHub repository: https://github.com/sakther-NYCDOHMH/nyc_mpox_genomic_epidemiology/.
MPVX lineage defining mutations, deletions, and the rate of evolution
A total of 58 MPXV lineage-defining mutations were observed in lineage B, in which eight mutations were intergenic, 22 were synonymous, and 28 were nonsynonymous mutations (Supplementary Table 1). Most of these B lineage-specific mutations (91%) had APOBEC3 signatures (Supplementary Table 1). Genomic analyses showed that some B.1 sublineages harbored additional high frequency mutations that were not considered for defining lineage calls by Nextclade. For example, the B.1.13 sublineage observed in the MPXV global phylogeny had one high frequency intergenic mutation in position 132,520 (allele frequency=0.88) along with the lineage defining-mutation in position 175,093 (Fig. 1B, Supplementary Fig. 3, Table 1). Additionally, the NYC-specific sublineage B.1.12 had two high frequencies nonsynonymous mutations in positions 98,233 (allele frequency=0.93) and 98,455 (allele frequency=0.93) along with the lineage-defining mutation in position 182950 (Supplementary Fig. 3, Table 1). Genome sequences from six of the nine phylogenetic clusters that were predominantly from NYC and/or North America had at least one cluster-specific nonsynonymous mutation (Table 1). The presence of these cluster-specific mutations may qualify the sequences in those clusters to be designated as MPXV B.1 sublineages.
No deletions were found in surface glycoprotein B21R13,19 and the TNF receptor crmB in the genomes sequenced by the NYC PHL. Other large-scale deletions were also rare in NYC sequences. Five occurrences of deletions were observed in 11 sequences collected from 10 individuals (Table 2). Sequences with a deletion from position 11,326:12,237/8 in the OPG023 gene were grouped into two distinct clades in the NYC phylogeny, likely two independent mutational events due to convergent evolution (Supplementary Fig. 4).
The MPXV genome evolution rate was estimated to be 4.28e-5 subs/site/year using MPXV sequences collected from 2021 to 2023 (Supplementary Fig. 5). Analysis of the amino acid changes in the phylogeny indicated that certain genes (OPG109, OPG110, OPG048) were more likely to have mutations (Supplementary Fig. 6).
APOBEC3 mutations in MPXV genomes
We assessed the prevalence of putative APOBEC3 signatures comparing the 2022 outbreak with previous years. The MPXV dataset was divided into three groups: (i) pre-2022 outbreak sequences retrieved from NCBI (1985-2021, n = 81); (ii) 2022 global outbreak sequences, excluding NYC, retrieved from NCBI (n = 2,877); (iii) 2022 NYC outbreak sequences sequenced by the NYC PHL (n = 1138). Compared to pre-outbreak sequences, both global and NYC MPXV sequences had significantly higher numbers of APOBEC3 signatures (p < 0.0001) (Fig. 3). MPXV sequences in NYC also had slightly higher numbers of APOBEC signatures when compared to the global sequences during the 2022 outbreak (p < 0.0001).

In the box plots, data points represent the total number of mutations identified with (red) and without (blue) APOBEC-3 signatures in individual MPXV genome sequences. The values are shown on y-axis for: (i) Sequences retrieved from NCBI for MPXV genomes prior to the 2022 outbreak (n = 81); (ii) 2022 outbreak global sequences excluding NYC (n = 2877); and (iii) 2022 NYC outbreak MPXV genomes sequenced by NYC PHL (n = 1138). The center line of each box denotes the median number of mutations per MPXV genome sequences. The whiskers extend to the minimum and maximum number of mutations per sequence within 1.5 times the interquartile range from the first and third quartiles. Mutation counts outside this range are shown as individual outlier points, representing genomes with unusually high or low numbers of mutations. The number of mutations with APOBEC3 signatures was significantly higher in the 2022 outbreak sequences (both global and NYC MPXV sequences) compared to the pre-outbreak sequences, as determined by a two sample t-test (Pre-Outbreak Global vs Outbreak Global: p.adj = 9.18e−24; Pre-Outbreak Global vs Outbreak NYC: p.adj = 1.99e−24; Outbreak Global vs Outbreak NYC: p.adj = 1.61e−114). In the figure, “****“ indicates a p-value < 0.0001. The source data file for Fig. 3 is available in the following GitHub repository: https://github.com/sakther-NYCDOHMH/nyc_mpox_genomic_epidemiology/.
We screened 1138 NYC MPXV genomes and found 455 (42%), out of the total of 1076 mutations, had the “GA > AA” APOBEC3 signature. Four hundred twenty-nine mutations (40%) had the “TC > TT” APOBEC3 signature, and 192 (18%) did not have any APOBEC signatures (Supplementary Data 3). Amongst the 884 APOBEC3 mutations, 6% APOBEC3 signatures were MPXV B lineage-defining, 44% occurred in two or more genomes and the remaining APOBEC3 signatures (51%) occurred only once in a genome (Supplementary Table 1 & Supplementary Data 3).
F13L mutations in NYC MPXV genome sequences
Mutations in the MPXV F13L gene (encodes the VP37 protein that form membrane to surrounds the mature virus) homolog have been previously reported to be associated with tecovirimat (TPOXX) resistance. The NYC MPXV genomes showed a low frequency of F13L mutations (n = seven different mutations), four mutations of which were previously confirmed to be associated with TPOXX resistance33 (Table 3). MPXV sequences with a TPOXX-associated mutation were from four individuals who had advanced HIV disease and were immunocompromised. These patients had been previously treated with TPOXX and were regarded as severe mpox cases. However, the risk of TPOXX resistance after treatment in the general population could not be evaluated due to a lack of information available on TPOXX treatment and outcomes in NYC.
Infections with genetically distinct MPXV strains
Nearly half of individuals in this study (n = 360) had two or more MPXV genomes collected from two or more lesions (66% of genomes). When analyzing individuals for intra-host variation, we observed individuals that had multiple sequences with a high degree of variation between sequences (>10 SNPs). Amongst the individuals that had more than one lesion sequenced, 94% of individuals (337/360) had the same lineage assignment, 6% of individuals (20/360) had lineage assignments that were sublineages of the other sequence(s), and only <1% of individuals (3/360) had multiple genomes assigned as different lineages. To ensure the robustness of our multiple infection analysis, we chose only high-quality sequences in our NYC dataset and further masked genomic regions where many sequences had ambiguous bases. The final dataset used to infer a phylogenetic tree specific to this analysis consisted of 1114 sequences.
Of the 360 individuals with MPXV genomes from multiple lesions, we identified 15 individuals whose genomes were polyphyletic in the phylogeny (i.e., at least two distinct viral genomes mapped to disparate clades diverged at the root of the phylogeny; see Methods for details) (Fig. 4). Eight additional sets of genomes were found to be distantly related but did not pass through the root (Supplementary Fig. 7). The remaining 337 individuals had sequences that were monophyletic, direct ancestors (i.e., closely related, and consistent with potential intra-host variation), or closely related (i.e., genomes did not have enough variation to be considered due to multiple infections). When considering only individual genomes where the branches separating the sequences pass through the root of the tree, we estimated 4.2% (15/360) of mpox cases had multiple infections with distinct MPXV strains in NYC. Expanding the definition to include the eight individuals with a node distance of at least four, the estimate for infection with multiple MPXV strains was 6.4% of mpox cases. These cases occurred in July 2022, the height of the outbreak in NYC (Supplementary Fig. 8).

This NYC MPXV tree was inferred from 1114 NYC sequences by masking the regions with low depth of coverage (Supplementary Table 2). In the figure, PID stands for an individual. Links in the figure were used to connect the sequences from the same individual that diverged at root using their phylogenetic placement. For Individual PID 187, one out of four sequences collected was found in a separate clade. The remaining three were found within the same clade, but one was distantly related by at least 4 nodes. The source data files for Fig. 4 are available in the following GitHub repository: https://github.com/sakther-NYCDOHMH/nyc_mpox_genomic_epidemiology/.
Intra-host variation in NYC mpox outbreak specimens
The intra-host variation of NYC MPXV sequences was evaluated in 344 individuals with sequenced MPXV genomes from multiple specimens (i.e., more than one lesion, majority sampled on the same day) and that were not due to multiple infections with distinct MPXV strains. We found that MPXV sequences from individuals with multiple lesions were identical in 172 individuals. Of the remaining 172 individuals, 119 individuals had multi-lesion sequences differing by only 1–2 mutations and 53 individuals had multi-lesion sequences differing by more than three mutations. Most intra-host variation had APOBEC3 signatures (Fig. 5A). All the intra-host variation in 66% (114/172) of the individuals had APOBEC3 signatures, and at least 50% of the intra-host variation showed APOBEC3 signatures in 22% (38/172) of the individuals. Only 9% (16/172) of the individuals had intra-host variation that did not have APOBEC3 signatures (Fig. 5B).

A The frequency distribution of the maximum total SNP distance with (red) and without (blue) APOBEC3 signatures between sequences from the same individual. Out of 172 individuals, sequences from 119 (69%) individuals were closely related with 1-2 total SNPs between sequences, and 53 Individuals had sequences that were divergent with >=3 SNPs. Majority of these mutational differences were due to APOBEC3. B The proportion of intra-host SNP variation due to APOBEC. For 66% (114/172) of individuals, the observed mutational difference between sequences was completely due to APOBEC3. For 22% (38/172) of individuals, APOBEC3 contributed to 50% or more (but not 100%) of the mutational differences. Only 9% (16/172) of individuals had observed mutational differences that could not be attributed to APOBEC3. C Intra-host phylogeny. Sequences from 17 individuals that had the greatest phylogenetic distance between them were annotated with colored tips and plotted on the NYC-only phylogeny. Two individuals’ sequences (J2665014/light blue and NG930408/light green) resulted in non-monophyletic placements of sequences. The source data files for Fig. 5 are available in the following GitHub repository: https://github.com/sakther-NYCDOHMH/nyc_mpox_genomic_epidemiology/.
When these samples were evaluated on the NYC phylogeny, 21 individuals had samples with a genetic distance greater than 0.000021 substitutions/site. Sequences from the 17 individuals with the greatest within-host genetic distance are shown in Fig. 5C, and the remaining four are shown in Supplementary Fig. 9. Sequences from three individuals (two in Fig. 5C and one in Supplementary Fig. 9) were also found in different clades on the NYC phylogeny, illustrating that MPXV evolution within the same individual can be divergent enough to obscure the phylogenetic relationships between sequences. Although a rare occurrence (0.9% of individuals), this observation contrasts with Rueca et al.16 wherein longitudinally sampled individual sequences clustered together on a phylogenetic tree.
Epidemiologically linked mpox cases in NYC
Contact tracing identified 43 mpox cases who were part of epidemiologically linked pairs where both patients were tested at the NYC PHL; these case-pairs were then further divided into 17 groups. (Table 4, Supplementary Data 4). MPXV sequences from groups #1, #3, #5, #8, and #15 were genetically related (Fig. 6, Table 4). Two MPXV sequences were genetically related in group #2, and five MPXV sequences were genetically related in group #6. The genetic relationship for the rest of the sequences from these two groups could not be resolved due to their placement in the basal part of the phylogenetic tree (Fig. 6, Table 4). MPXV sequences from groups #7, #9, #11, #16, and #17 were not monophyletic but shared a common ancestor in the same clade in the phylogeny and were therefore considered potentially genetically related (Fig. 6, Table 4). MPXV sequences from groups #10, #12, #13 and #14 were placed in different clades in the phylogeny and were not genetically related. Intra-host sequence pairs from groups #10, #12, and #13 were categorized as “Distantly Related” based on the multiple infection analysis performed in the previous section (Fig. 6, Table 4). For example, group #12 included sequences from two lesions belonging to one individual. One of these intra-host sequences was assigned to the B.1 lineage (Accession #OQ469282) while the other sequence was assigned as B.1.7 (Accession #OQ469283) (Table 4, Supplementary Data 4). This observation suggests that sequencing only one lesion can result in inaccurate reconstruction of genomic transmission networks. Overall, 13% of sequences from patients with epidemiological links were not genetically linked based on the phylogeny.

The symbols on the branches were colored by putative groups of epidemiologically linked cases. The outer ring (Sequence Type) in the figure was colored by the genome sequences that belonged to the same or different individual of epidemiologically linked cases in a particular group. The middle ring (Phylogenomic Category) is colored by the phylogenetic placement of the genome sequences of epidemiologically linked cases within a group. Epidemiologically linked cases in groups with sequences placed on the same or sister branches were designated as “monophyletic”. Epidemiologically linked cases in groups for which genome sequences were in the same clade in the phylogeny and shared a common ancestor with other NYC sequences in this clade were designated as “Shared Ancestor”. Epidemiologically linked cases in groups for which most of the genome sequences were placed in the basal part of the phylogenetic tree were designated as “inconclusive”. Epidemiologically linked cases in groups for which genome sequences were in different clades in the phylogeny were designated as “Not-linked”. The inner ring (Intra-host category) was colored by the categories that were assigned using the phylogenetic placement of the genome sequences from the same individual (See multiple infection result section: Fig. 4 and Supplementary Fig. 7). The source data files for Fig. 6 are available in the following GitHub repository: https://github.com/sakther-NYCDOHMH/nyc_mpox_genomic_epidemiology/.
Discussion
This study used a large collection of MPXV sequences (1,138 sequences from NYC and 2967 sequences for the global dataset) to understand the genomic epidemiology of MPXV in NYC. This study also included a large dataset of sequences sampled from two or more lesions in the same individual (748 sequences). We found that some highly divergent sequences sampled from the same individual were non-monophyletic on the phylogenetic tree (Fig. 5C). In addition, infection with multiple MPXV strains in the same individual occurred when case counts were high (Supplementary Fig. 8). The estimation of infections with multiple MPXV strains in the same individuals in NYC was 4.2%, which is likely an under-estimation of the true prevalence. The phylogenetic analysis was based on consensus sequences derived from major alleles, and accounting for the minor alleles could likely have identified additional cases of infections with multiple MPXV strains. Incorporating within-host viral diversity in phylogenetic inference can improve the robustness of detecting clusters34. Therefore, future MPXV surveillance efforts can benefit from considering genomic variation outside of consensus sequences when attempting to identify potential transmission chains and clusters.
Four of 17 groups with epidemiologically linked pairs of individuals infected with mpox had MPXV sequences that were not genetically related. The failure to link these individuals using phylogenetic analysis may be due to the inability to generate high-quality MPXV genomes from available specimens or due to the lack of a representative sample of specimens that are available for sequencing. For example, individuals might have had multiple partners, some of whom might not have been tested at the NYC PHL so would not have been included in our analysis. Additionally, the epidemiologically linked cases did not distinguish between direct or indirect exposures (e.g., attending common events). Nevertheless, it has been shown previously in HIV investigations among MSM in NYC that epidemiological linkage does not necessarily imply a genetic linkage consistent with viral transmission, and the percentage of named partners that had similar genomes varies depending on the number of partners and the perceived stigma of their behaviors35. Many of the groups with epidemiologically linked cases had unresolved genomic relationships, appearing highly related due to the slow-evolving nature of MPXV genomes, which had minimal diversity during the early weeks of the outbreak. Thus, phylogenetics as a way to infer direct transmission needs to be approached with extreme caution because genomic similarity could be a result of slow evolution rather than transmission, and genomic divergence could be a result of intra-host evolution mediated by APOBEC3 or infection with multiple MPXV strains within an epidemiological cluster.
The MPXV genome evolution rate was estimated to be 4.28e-5 substitutions/site/year using MPXV sequences collected from 2021 to 2023 (Supplementary Fig. 5), which was consistent with a previous rate (5e-5 substitutions/site/year) estimated by analyzing 1,900 global MPXV genomes collected from 1958 to 202236. The MPXV genome evolutionary rate in this study was 4.8-fold higher than a previously reported rate (9e-6 substitutions/site/year) estimated using 87 MPXV genomes collected from 1978 until the early months of the 2022 outbreak. This discrepancy could be due to differences in collection date ranges and sample size. MPXV genomes had a higher evolutionary rate in NYC comparison to MPXV genomes from around the world during the 2022 outbreak. Additionally, nine clusters were identified with mutational profiles that were unique to NYC and North America (Table 1). Based on the phylogenetic placement and mutations, these clusters were likely to be MPXV sublineages that had emerged and spread within NYC or North America during the 2022 outbreak. Consistent with these findings, several unique lineages had emerged in the Midwest of the United States during the 2022 outbreak20. However, at the time of writing, these lineages were still classified as B.1 by Nextclade. The emergence of clusters for subgroups of genomic sequences may present a challenge when using only Nextclade lineage assignment to determine genomic similarity or divergence.
Most of the genomic variations detected had APOBEC3 signatures (Fig. 3, Supplementary Table 1), which is consistent with recent reports4,23,28. This APOBEC3-driven deamination has been observed with many DNA viruses and retroviruses and has been shown to be a driver for MPXV evolution during human-to-human transmission29,30. Despite this increased mutation rate, we found that the frequency of TPOXX-associated mutations was low (7/1138) in our MPXV sequences. As a result, it is likely that additional factors, such as weakened immunity, contributed to these mutations in the four severe mpox cases. Therefore, information on the duration of infection, co-morbidities (e.g., advanced HIV), or immunocompromised status can help better understand the virus’s genomic features19. There were several limitations in this study. On a global level, sequences from Africa and Asia were excluded from the global phylogeny due to the small sample size. On a local level, the NYC PHL was the sole testing facility for MPXV within the first seven weeks of the mpox outbreak in NYC. Specimen testing volume at PHL dropped once commercial lab testing was available. Despite these challenges, the number of individuals sequenced was reflective of the city’s epidemic curve37; and while randomly selected, sequences from global regions reflect the genomic diversity reported across the world38. Additionally, clinical data and time of infection (e.g., acute, shedding, or recovery) were not available to study any associations between MPXV genomic characteristics and clinical outcomes. A potential bias in sample selection could be attributed to the lower real-time PCR Ct values required for sequencing (<30), which might have introduced bias towards a certain tissue, collection site, or phase of the infection (e.g., macular, papular, vesicular, and pustular stages) where viral concentrations are higher. Most genomic sequences were collected from the genital region. As a result, it is unclear how well genomic variations of MPXV can be captured in other specimen types where viral concentration is lower (e.g., blood, saliva, oral/rectal swabs)39. Longitudinal sampling from the same lesions and hosts could have also provided valuable information on the intra-host evolution of the virus. However, intra-host variations that were observed in this study were primarily from samples from the same individual collected during the same day. The multiple infection analysis was therefore focused on the genomic diversity within an individual at the time of diagnosis. Last, but not least, epidemiological contact tracing data may be incomplete due to perceived stigma related to the number of named or unnamed partners and sexual behaviors35.
In conclusion, this study included a genomic dataset collected during the 2022 mpox outbreak, featuring a large archive of specimens from the same individuals. The integration of genomic and epidemiological data revealed transmission relationships involving individuals infected with multiple MPXV strains. Analyzing infections with multiple MPXV strains showed that individual lesions often do not represent the complete diversity of within-host variation and may have distinct genomic profiles for MPXV. Some of the exposures from contacts documented through traditional epidemiological methods were not supported by the viral genomic diversity between individuals, which may have been underrepresented due to a variety of biological as well as technical, and sampling reasons. Improving concordance between genomic and epidemiological clusters may require improving the reconstruction of high-quality genomes from different lesion sites as well as improving the representativeness of collected samples that are available for sequencing by expanding partnerships with testing sites and collaborations with community groups, which may be a challenge during resource-constrained times. Results from this study highlight the importance of gathering thorough epidemiological, clinical, and case data for outbreak investigations followed by thorough and careful interpretation of the data to perform genomic epidemiology for MPXV.
Methods
This work was determined to be human subjects exempt by the New York City Department of Health and Mental Hygiene IRB, as the study involved the secondary use of biospecimens and data. All necessary patient or participant consents were obtained, and the required institutional forms were archived. Any identifiers used were not known outside the organization and cannot be used to identify individuals.
Specimen selection
Lesion swabs from one or more body sites from suspected mpox cases were submitted to the New York City Public Health Laboratory (NYC PHL) for testing using Non-Variola Orthopoxvirus (NVO) Real-Time PCR. DNA was extracted from lesion swabs or swabs in viral transport media using manual or automated DNA extraction platforms and the QIAGEN QIAamp® DSP DNA Blood Mini Kit or the QIAGEN EZ1&2 DNA Tissue Kit, respectively. NVO detection was performed using the CDC Laboratory Response Network (LRN) Non-Variola Orthopoxvirus real-time PCR Assay targeting the E9L gene target found in MPXV. Specimens with a Ct value < 37 were considered positive for Non-Variola Orthopoxvirus DNA. Specimen with Ct values <= 30 were sequenced after DNA extraction.
Whole genome sequencing
NYC PHL utilized PrimalSeq40 for MPXV whole genome sequencing. PrimalSeq for MPXV is a tiling PCR approach which consists of 163 MPXV-specific primer pairs divided into two primer pools for the initial PCR step. These primers spanned nearly the entire length of the genome (from base 356 to 196424) and produced overlapping amplicons with an average length of approximately 2000 bp. These amplicon pools were cleaned of primers using Beckman Coulter Ampure XP beads (Cat. # A63880). The bead-cleaned amplicons were subjected to a tagmentation-based library preparation for small PCR amplicon input using the Illumina DNA Prep Kit (Cat. # 20018705) with IDT for Illumina Unique Dual Index Sets (Cat. #s: 20027213(UD-A), 20027214(UD-B), 20042666(UD-C), 20042667(UD-D)). The libraries were run on an Illumina MiSeq at 151 bp paired-end reads with an Illumina MiSeq Reagent Kit 600-cycle v3 kit (Cat. # MS-102-3003) or a NextSeq2000 with a NextSeq 1000/2000 P2 Reagent 300-cycle kit v3 kit (Cat. # 20046813).
Assembly and variant calling of NYC PHL MPXV outbreak genomes
Index sequences were removed as part of FASTQ generation. Reads were quality trimmed using Trimmomatic 0.3641 before mapping to the MPXV reference genome NC_063383 using minimap2 v2.17-r94142. Samtools v1.1343 was used to sort and index mapped reads before trimming primer sequences from alignment files with iVar44. Variant Call Files (VCF) were created by BCFtools using a minimum quality score of 20, depth of coverage (DP) of 20, and frequency threshold of 0.65 from the primer-trimmed alignment files. The primer-trimmed alignment files were also used to generate consensus sequences with iVar consensus, with the same minimum quality score, DP, and frequency thresholds that were used to generate the consensus sequences. For phylogenetic analyses, Some Quick Rearranging to Resolve Evolutionary Links (squirrel)45 was used to mask the inverted terminal repeat (ITR) regions as well as other problematic regions. Sequences with a genome coverage <90% were excluded from downstream analyses, resulting in a total of 1138 MPXV genomes from NYC PHL. The complete list of 1138 NYC PHL MPXV sequences is available in Supplementary Data 1.
Curation of the global MPXV genome sequences dataset
Global MPXV genome sequences (n = 2967, excluding any sequences from NYC) and associated metadata were obtained from the National Center for Biotechnology Information (NCBI) virus database (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/) on February 1st, 2023. Partial sequences and/or sequences with missing collection dates and geographic origins were excluded from downstream analyses. MPXV global genome sequences included in this study were collected from 1985 through 2023 with representative sequences from five continents except Oceania. The complete list of 2967 global MPXV sequences is available in Supplementary Data 2.
Lineage assignment
The lineages of global and NYC MPXV genome sequences were assigned using Nextclade (https://clades.nextstrain.org/) with MPXV reference genome NC_063383 (collected from Nigeria, August 2018) and Dataset name hMPXV. The combined dataset, which contains 4105 MPXV sequences was aligned to the reference using MAFFT v7.49046 with the --6merpair option. A maximum-likelihood tree was inferred by IQTree247 with the GTR + F + I model of nucleotide evolution and NNI search option. Trees were visualized using iTOL48. MPXV genome divergence rate was calculated using Nextstrain49 with the reference genome and the previously generated IQTree2 maximum-likelihood tree. A time-resolved tree was obtained, but many samples were filtered out by the --clock-filter-iqd 4 option. This led to only sequences from 2021 onward being included for a total of 3936 sequences. The rate of divergence and the number of changes at each nucleotide position in the MPXV phylogeny were estimated using Nextstrain49 and visualized with https://auspice.us/ (Supplementary Figs. 5 and 6).
Characterization of MPXV mutations, deletions, and TPOXX resistance
Genome variants in MPXV sequences were called using NucDiff50 against the reference genome, and the resulting VCFs were annotated with SnpEff51. Ambiguous bases (N) were excluded from further analyses. Variant analyses were performed using a custom-written R script and visualized in R with ggplot2. Potential APOBEC3 signatures in the MPXV genomes were detected using the Mutation profile (https://github.com/insapathogenomics/mutation_profile)6. To identify cluster-specific mutations and other clinically relevant mutations, we restricted our analysis to genomes sequenced at the NYC PHL due to limitations in the availability of alignment and VCF data from the global submissions. To detect deletions, Samtools depth was used to flag regions of the genome with a DP of 0. To reduce false positives, deletion regions were determined to be amplicon dropouts and discarded if the surrounding regions had a DP less than 20. Furthermore, the remaining deletion regions were manually reviewed using IGV52. TPOXX (tecovirimat) resistance-associated mutations were identified using SnpEff and searching for F13L mutations listed in the FDA Microbiology Review on TPOXX33. Functional annotations of the mpox proteome were accessed from UniProtKB (https://www.uniprot.org/uniprotkb: embl-MT903340).
Intra-host variations in NYC mpox outbreak specimens
snp-dists (https://github.com/tseemann/snp-dists) was used to determine the nucleotide differences between sequences from the same individual (i.e., differences in ambiguous bases are ignored). Sequences that were divergent enough to be considered potential multiple infections were excluded from this analysis. Genomic variation between sequences from the same individual was then annotated as having an APOBEC signature or not. The phylogenetic distance between these sequences was determined using the R package adephylo and manually assessed by plotting on the NYC-specific phylogenetic tree.
Inferring Infection with multiple MPXV strains
Selecting sequences to identify multiple infections and genome masking
Sequences with more than 5% N in the squirrel masked45 sequences were excluded, resulting in 1114 sequences from 742 individuals. Most individuals (n = 382) had only a single individual sequenced MPXV genome; however, 360 individuals had MPXV genomes sequenced from at least 2 lesions (n = 732 genomes).
To minimize sequencing artefacts related to batch effects, amplicon dropout, sequencing error, or the 3’ terminal repeat region53, the proportion of sequences with genome positions with low DP (<20) at each position was calculated to identify potential regions to mask. Using k-means in R with three centers starting with 25 random centers, positions were categorized into three groups of proportions: low (0.1–9.16%), medium (9.25–51.9%), and high (52.1–100%). Positions classified as medium or high were used to identify larger regions to mask.
A BED file for masking the 1114 genomes was created by including all regions of medium or high proportions of low DP except the k-means group of regions with the smallest stretch of low DP. Additionally, the 3’ inverted terminal repeat (ITR) was masked due to poor read mapping quality (MAPQ). Genomic regions that were masked due to potential low-quality sequences and the 3’ ITR are listed in the Supplemental Table 2. In total, 12 regions were masked, resulting in high-quality genome coverage between 89.91% to 93.67% in the NYC dataset for the multiple infection analysis.
Phylogenetics
After masking, a phylogenetic tree comprising only MPXV genomes sequenced by NYC PHL (n = 1114 genomes) was inferred using IQ-Tree2 version 2.1.3 under a GTR + F + I substitution model, with a minimum branch length of 1e-10 substitutions/site and collapsing polytomies. For the 360 people with more than one sequenced viral genome, we determined the phylogenetic relationship and distance among these viruses using the ete3 Python package54. Genetic distance was assessed using HIV-TRACE55. Individuals were potential multiple infection with different MPXV strains if at two or more of their sequenced viruses were (i) separated by at least 3 nucleotide substitutions, (ii) not each other’s direct ancestor or descendant, (iii) were separated by at least four internal nodes in the phylogeny, (iv) were at least one nucleotide substitution from the basal polytomy at the root, and (v) shared a most recent common ancestor (MRCA) at the root of the phylogeny. As a sensitivity analysis, we also considered multiple infections if all these criteria were met except for sharing an MRCA at the root of the phylogeny.
NYC epidemiologically linked mpox outbreak specimens
Whole genome sequencing, lineage assignment, and phylogenetic analyses were performed to characterize specimens from epidemiologically linked mpox outbreak cases in which individuals had contact with one or more persons who had mpox that were tested at the NYC PHL, and transmission by the usual modes of transmission was deemed likely. Lineage assignment, phylogenetic inference, and visualization were performed using NextClade, IQTree2, and iTOL, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The NYC MPXV sequence data generated in this study have been summarized in Supplementary Data 1 and deposited in the NCBI database under BioProject: PRJNA949682. Sequencing data that were retrieved from NCBI for the Global Phylogeny are summarized in Supplementary Data 2.
Code availability
The source data files and code are available in the following GitHub repository: https://github.com/sakther-NYCDOHMH/nyc_mpox_genomic_epidemiology/.
References
Patino, L. H. et al. Phylogenetic landscape of Monkeypox Virus (MPV) during the early outbreak in New York City, 2022. Emerg. Microbes Infect. 12, e2192830 (2023).
Jezek, Z., Gromyko, A. I. & Szczeniowski, M. V. Human monkeypox. J. Hyg. Epidemiol. Microbiol Immunol. 27, 13–28 (1983).
Vakaniaki, E. H. et al. Sustained human outbreak of a new MPXV clade I lineage in eastern Democratic Republic of the Congo. Nat. Med. 30, 2791–2795 (2024).
Likos, A. M. et al. A tale of two clades: monkeypox viruses. J. Gen. Virol. 86, 2661–2672 (2005).
Alcami, A. Pathogenesis of the circulating mpox virus and its adaptation to humans. Proc. Natl Acad. Sci. USA 120, e2301662120 (2023).
Isidro, J. et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat. Med. 28, 1569–1572 (2022).
Happi, C. et al. Urgent need for a non-discriminatory and non-stigmatizing nomenclature for monkeypox virus. PLoS Biol. 20, e3001769 (2022).
Kyaw, N. T. T. et al. Notes from the field: Clinical and epidemiologic characteristics of Mpox cases from the initial phase of the outbreak - New York City, May 19-July 15, 2022. MMWR Morb. Mortal. Wkly Rep. 71, 1631–1633 (2022).
Health, N. Mpox. Accessed on 8/22/2023 from https://www.nyc.gov/site/doh/health/health-topics/monkeypox.page (2023).
Laurenson-Schafer, H. et al. Description of the first global outbreak of mpox: an analysis of global surveillance data. Lancet Glob. Health 11, e1012–e1023 (2023).
Baliere, C. et al. Complete genome sequences of Monkeypox Virus from a French clinical sample and the corresponding isolated strain, obtained using nanopore sequencing. Microbiol Resour. Announc 12, e0000923 (2023).
Vauhkonen, H. et al. Intrahost Monkeypox Virus Genome Variation in Patient with Early Infection, Finland, 2022. Emerg. Infect. Dis. 29, 649–652 (2023).
Brinkmann, A. et al. Possible adaption of the 2022 Monkeypox virus to the human host through gene duplication and loss. bioRxiv 10.1101 2022.10.21.512875 (2022).
Loconsole, D. et al. Monkeypox Virus Infections in Southern Italy: Is There a Risk for Community Spread? Int. J. Environ. Res. Public Health 19, 11719 (2022).
Forni, D. et al. Whole-genome sequencing of hMPXV1 in five Italian cases confirms the occurrence of the predominant epidemic lineage. J. Med. Virol. 95, e28493 (2023).
Rueca, M. et al. Temporal intra-host variability of mpox virus genomes in multiple body tissues. J. Med. Virol. 95, e28791 (2023).
Claro, I. M. et al. Shotgun metagenomic sequencing of the first case of monkeypox virus in Brazil, 2022. Rev. Inst. Med. Trop. Sao Paulo 64, e48 (2022).
Gigante, C. M. et al. Multiple lineages of monkeypox virus detected in the United States, 2021-2022. Science 378, 560–565 (2022).
Sereewit, J. et al. ORF-interrupting mutations in monkeypox virus genomes from Washington and Ohio, 2022. Viruses 14, 2393 (2022).
Bosmeny, M. S. et al. Global mpox lineage discovery and rapid outbreak tracking with nanopore sequencing. Virol. J. 20, 90 (2023).
Harrison, L. B. et al. Monkeypox in Montreal: Epidemiology, phylogenomics, and public health response to a large North American outbreak. Ann. Intern Med 176, 67–76 (2023).
Luna, N. et al. Phylogenomic analysis of the monkeypox virus (MPXV) 2022 outbreak: Emergence of a novel viral lineage?. Travel Med. Infect. Dis. 49, 102402 (2022).
Benko, M., Harrach, B. & Kajan, G. L. Phylogenetic analysis of all available monkeypox virus strains shows the close relatedness of contemporary ones. Heliyon 9, e12895 (2023).
Wang, L. et al. Genomic annotation and molecular evolution of monkeypox virus outbreak in 2022. J. Med. Virol. 95, e28036 (2023).
Wertheim, J. O. et al. Maintenance and reappearance of extremely divergent intra-host HIV-1 variants. Virus Evol. 4, vey030 (2018).
Saldana, C. S. et al. Mpox and HIV: a narrative review. Curr. HIV/AIDS Rep. 20, 261–269 (2023).
Firth, C. et al. Using time-structured data to estimate evolutionary rates of double-stranded DNA viruses. Mol. Biol. Evol. 27, 2038–2051 (2010).
Chen, Y., Li, M. & Fan, H. The monkeypox outbreak in 2022: adaptive evolution associated with APOBEC3 may account for. Signal Transduct. Target Ther. 7, 323 (2022).
Forni, D. et al. An APOBEC3 mutational signature in the genomes of human-infecting Orthopoxviruses. mSphere 8, e0006223 (2023).
Colson, P. et al. Sequencing of monkeypox virus from infected patients reveals viral genomes with APOBEC3-like editing, gene inactivation, and bacterial agents of skin superinfection. J. Med. Virol. 95, e28799 (2023).
Roper, R. L. et al. Monkeypox (Mpox) requires continued surveillance, vaccines, therapeutics and mitigating strategies. Vaccine 41, 3171–3177 (2023).
Ahmed, S. K. et al. Monkeypox (mpox) in immunosuppressed patients. F1000Res 12, 127 (2023).
FDA, FDA Mpox Response. Accessed on 8/23/2023 from https://www.fda.gov/emergency-preparedness-and-response/mcm-issues/fda-mpox-response#therapeutics. (2023).
Guang, A. et al. Incorporating within-host diversity in phylogenetic analyses for detecting clusters of new HIV diagnoses. Front. Microbiol. 12, 803190 (2021).
Wertheim, J. O. et al. Social and genetic networks of HIV-1 transmission in New York City. PLoS Pathog. 13, e1006000 (2017).
Luna, N. et al. Monkeypox virus (MPXV) genomics: A mutational and phylogenomic analyses of B.1 lineages. Travel Med. Infect. Dis. 52, 102551 (2023).
New York City Department of Health and Mental Hygiene, Mpox. Accessed on 8/23/2023 from https://www.nyc.gov/site/doh/health/health-topics/monkeypox.page.
CDC 2022 Mpox Outbreak Global Map. Accessed on 8/23/2023 from https://www.cdc.gov/poxvirus/mpox/response/2022/world-map.html. (2023).
Thomas, R. J. et al. Expansion of a low-cost, saliva-based PCR test for the detection of mpox virus. medRxiv, p. 2023.06.12.23291295. (2023)
Quick, J. et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Grubaugh, N. D. et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 20, 8 (2019).
O’Toole, Á. Some QUIck Rearranging to Resolve Evolutionary Links. https://github.com/aineniamh/squirrel. (2023).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Khelik, K. et al. NucDiff: in-depth characterization and annotation of differences between two sets of DNA sequences. BMC Bioinforma. 18, 338 (2017).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Zhao, K., Wohlhueter, R. M. & Li, Y. Finishing monkeypox genomes from short reads: assembly analysis and a neural network method. BMC Genom.17, 497 (2016).
Huerta-Cepas, J., Serra, F. & Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638 (2016).
Kosakovsky Pond, S. L. et al. HIV-TRACE (TRAnsmission Cluster Engine): a tool for large scale molecular epidemiology of HIV-1 and other rapidly evolving pathogens. Mol. Biol. Evol. 35, 1812–1819 (2018).
Acknowledgements
The authors acknowledge the Grubaugh Lab at Yale University for their help in providing the primers for the initial proof of concept. SA was supported by Cooperative Agreement Number NU60OE000104 (CFDA #93.322), funded by the Centers for Disease Control and Prevention (CDC) of the US Department of Health and Human Services (HHS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Association of Public Health Laboratories (APHL), CDC, HHS, or the US Government. This project was 100% funded with federal funds from a federal program of $120,402,978.
Author information
Authors and Affiliations
Contributions
S.A. designed the project, performed genome assembly, curated the global dataset, characterized genomic variants, performed evolutionary analyses, prepared the first draft and revised the manuscript. M.S. curated the metadata, characterized the deletion/TPOXX resistance, created the Nextstrain tree, identified intra-host variations and prepared the first draft of the manuscript. J.C.W. performed data curation, genome masking and prepared the first draft of the manuscript. HA performed genome assembly and APOBEC3 analysis. F.T. supervised the project, prepared the first draft and revised the manuscript. N.C. and M.C. prepared genomic DNA libraries and performed the genome sequencing. A.O. supervised the genome sequencing. TC curated the metadata and deposited the sequences into the NCBI databases. J.L., K.J., J.B. and M.W. curated epidemiological data and revised the manuscript. E.K., V.R. and M.L. performed testing, prepared genomic DNA's and revised the manuscript. R.F. supervised testing and revised the manuscript. J.O.W. performed evolutionary and multiple infection analyses and revised the manuscript. J.P., J.H. and T.V. performed evolutionary and multiple infection analyses. S.H. obtained funding and supervised the project. EO obtained funding, supervised the project and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Delia Djuicy and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Akther, S., Su, M., Wang, J.C. et al. Genomic epidemiology of mpox virus during the 2022 outbreak in New York City. Nat Commun 16, 8354 (2025). https://doi.org/10.1038/s41467-025-60486-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60486-x
