
Abstract
While switch-like gene expression (“on” in some individuals and “off” in others) has been linked to biological variation and disease susceptibility, a systematic analysis across tissues is lacking. Here, we analyze genomes, transcriptomes, and methylomes from 943 individuals across 27 tissues, identifying 473 switch-like genes. The identified genes are enriched for associations with cancers and immune, metabolic, and skin diseases. Only 40 (8.5%) switch-like genes show genetically controlled switch-like expression in all tissues, i.e., universally switch-like expression. The rest show switch-like expression in specific tissues. Methylation analysis suggests that genetically driven epigenetic silencing explains the universally switch-like pattern, whereas hormone-driven epigenetic modification likely underlies the tissue-specific pattern. Notably, tissue-specific switch-like genes tend to be switched on or off in unison within individuals, driven by tissue-specific master regulators. In the vagina, we identified seven concordantly switched-off genes linked to vaginal atrophy in females. Experimental analysis of vaginal tissues shows that low estrogen levels lead to decreased epithelial thickness and ALOX12 expression. We propose that switched-off driver genes in basal and parabasal epithelia suppress cell proliferation, leading to epithelial thinning and vaginal atrophy. Our findings underscore the implications of switch-like genes for diagnostic and personalized therapeutic applications.
Similar content being viewed by others


Introduction
The study of gene expression began in earnest with the characterization of lactose-metabolizing on-versus-off switch-like genes in E. coli1. The presence of lactose triggered the production of enzymes needed to metabolize it, while the absence of lactose was accompanied by the absence of these enzymes. These genes acted like switches, toggling between “on” and “off” states based on the presence or absence of lactose, respectively. In subsequent decades, the discovery of enhancer elements2,3,4, epigenetic modifications5,6,7,8, and transcription factor dynamics9 revealed that gene expression in humans is more nuanced, resembling a dimmer more often than a simple on-and-off mechanism. Consequently, the study of switch-like genes in humans was largely relegated to the narrow realm of Mendelian diseases10,11,12.
The recent availability of population-level RNA-sequencing data from humans has made it possible to systematically identify switch-like versus dimmer-like genes. For dimmer-like genes in a given tissue, we expect expression levels across individuals to be continuously distributed with a single mode, i.e., a unimodal distribution. In contrast, expression levels of switch-like genes in a given tissue are expected to exhibit a bimodal distribution, with one mode representing the “off” state and the other representing the “on” state.
As we will detail, bimodal expression across individuals is a characteristic of a gene in a specific tissue, referred to as a gene-tissue pair. We define a gene as switch-like if it exhibits bimodal expression in at least one tissue. Most of the recent studies on bimodal gene expression are related to cancer biology, associating on and off states to different disease phenotypes and their prognoses13,14,15. These cancer studies have already produced promising results for personalized medicine16. However, to our knowledge, the only study focusing on switch-like genes in non-cancerous tissues across individuals restricted their analysis to muscle tissue17. As a result, the dynamics of switch-like expression across the multi-tissue landscape remain unknown. We hypothesize that switch-like expression is ubiquitous but often tissue-specific. We further hypothesize that these tissue-specific expression trends underlie common disease states. Therefore, the analysis of switch-like genes across tissues and individuals may provide a means for early diagnosis and prediction of human disease.
Here, we systematically identified switch-like genes across individuals in 27 tissues. We find that these switch-like genes are enriched for disease associations. Moreover, our results explain the genetic and epigenetic regulatory bases of switch-like expression in humans, highlighting genomic structural variation as a major source of switch-like expression of genes in multiple tissues. We also find that sex-bias is ubiquitous among breast-specific switch-like genes. Furthermore, we identified a group of switch-like genes in the vagina wherein the “off” state predisposes individuals to vaginal atrophy. Overall, these findings improve our understanding of the regulation of switch-like genes in humans. They also suggest promising future paths for preventative biomedical interventions.
Results
Phenotypic relevance of switch-like genes
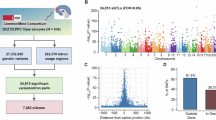
The misregulation of highly expressed genes often has consequences for health and fitness. To systematically identify biomedically relevant switch-like genes in humans, we focused on 19,121 genes that are highly expressed (mean TPM > 10) in at least one of the 27 tissues represented in the GTEx database Fig. 1A, B; and Supplementary Table 1). For each of the 516,267 gene-tissue pairs (19,121 genes x 27 tissues), we applied the dip test of unimodality18 to the expression level distribution across individuals (Fig. 1C). Controlling for confounders, using \(p\)-value recalibration for the dip test, and correcting for multiple hypotheses, we identified 473 switch-like genes (Fig. 1C; and Supplementary Data 1 and 2; Methods; Supplementary Notes 1–3; Supplementary Figs. 1–4). The expression of these genes is bimodally distributed in at least one tissue, such that it is switched “off” for one subset of individuals and switched “on” for the rest of the individuals.

A List of 27 tissues used in this study. B Distribution of 19,121 genes by the number of tissues in which they are highly expressed. C Bimodal expression is a property of a gene-tissue pair. We tested 516,267 gene-tissue pairs (19,121 genes x 27 tissues) for bimodal expression across individuals. When a gene-tissue pair exhibits switch-like (bimodal) expression, the individuals divide into two subpopulations: one with the gene switched off, and the other with the gene switched on. D Switch-like genes show an enrichment for biological processes related to skin, metabolism, and immunity. Only enrichments with \(q\)-value < 0.05 are reported. E Switch-like genes exhibit an enrichment for disease categories related to infections, tumors, etc. Only enrichments with \(q\)-value < 0.05 are reported. A and C were designed entirely in BioRender; all panels were assembled as Fig. 1 in BioRender. Aqil, A. (2025) https://BioRender.com/8jr185j. Source data are provided as a Source Data file.
To gauge the phenotypic effects of the 473 switch-like genes, we performed two enrichment analyses: (1) gene ontology (GO) enrichment for biological processes using GREAT19 and (2) disease ontology enrichment20. In the GO analysis, the switch-like genes were significantly enriched in biological processes related to skin, immunity, and metabolism, including keratinization (fold enrichment = 5.4; hypergeometric test \(q\)-value = 9.6 × 10−10), the B-cell receptor signaling pathway (fold enrichment = 4.6; hypergeometric test \(q\)-value = 1.2 × 10−2), and the D-ribose metabolic process (fold enrichment = 9.6; hypergeometric test \(q\)-value = 2.0 × 10−3) (Fig. 1D). Similarly, disease ontology analysis revealed enrichment in integumentary, immune-related, and metabolic diseases (Fig. 1E). Notably, switch-like genes were also enriched in diseases related to cell proliferation, including pre-malignant neoplasms (fold enrichment = 3.46; \(q\)-value < 10-4), benign neoplasms (fold enrichment = 1.49; \(q\)-value = 4.4 × 10−3), and cancers (fold enrichment = 1.26; \(q\)-value = 1.8 × 10−2). These findings align with previous studies that have linked switch-like genes to cancer13,14,15,16, further highlighting their potential role in oncogenesis.
Tissue-specificity of bimodal expression
Expression of different switch-like genes may be bimodally distributed in different numbers of tissues. We contend that genes that are bimodally expressed across all tissues are likely so due to a germline genetic polymorphism driving switch-like expression across tissues. If this is the case, the expression of these genes would be highly correlated in most pairs of tissues. Given this insight, discovering universally bimodal genes is more tractable using tissue-to-tissue co-expression of each gene. Therefore, for each gene, we calculated pairwise tissue-to-tissue correlation of expression levels for all tissue pairs (Methods; Supplementary Data 3; Supplementary Note 4; Supplementary Fig. 5). To visualize tissue-to-tissue co-expression patterns of genes, we performed principal component analysis (PCA) on the tissue-to-tissue gene co-expression data (Supplementary Data 4). We emphasize that we are referring to the co-expression of the same gene across pairs of tissues instead of the co-expression of pairs of genes in the same tissue. In the space spanned by the first two principal components (explaining 40.6% and 2.98% of the variance, respectively), switch-like genes form two major clusters (cluster 1 and cluster 2; Methods), dividing along PC1 (Fig. 2A). Applying PCA exclusively to switch-like genes reveals the further division of cluster 2 into two distinct subclusters – cluster 2 A and cluster 2B – in the space spanned by the first two principal components (explaining 74.5% and 5.31% of the variance, respectively) (Fig. 2B; and Supplementary Data 5).

A PCA analysis of tissue-pair correlations of gene expression. Each point represents a gene. When we perform PCA on the tissue-to-tissue co-expression vectors for 19,121 genes, the switch-like genes divide into two clusters. Cluster 1 primarily represents genes that are bimodally expressed in a tissue-specific manner, while cluster 2 represents genes that are bimodally expressed in at least all non-sex-specific tissues. B Performing PCA on the co-expression vectors of only switch-like genes further divides cluster 2 into two subclusters: cluster 2 A, which contains genes that are bimodally expressed across all 27 tissues, and cluster 2B, which contains genes that are bimodally expressed in all 22 tissues common to both sexes, but not in the five sex-specific tissues. C–E Violin plots display the expression levels in all 27 tissues for representative genes from cluster 1, cluster 2A, and cluster 2B, respectively. log(TPM + 1)C refers to the value of log(TPM + 1) corrected for the effects of technical confounders. All panels were assembled as Fig. 2 in BioRender. Aqil, A. (2025) https://BioRender.com/bird6wi. Source data are provided as a Source Data file.
Manual inspection reveals that cluster 1, which contains 432 genes, represents genes, such as KRT17, with bimodal expression in a small subset of tissues (Fig. 2C; Supplementary Note 5). Cluster 2 A consists of 32 genes, such as GPX1P1, with bimodal expression in all tissues (Fig. 2D; and Supplementary Fig. 6; Supplementary Note 6). Lastly, cluster 2B represents eight genes, such as EIF1AY, with bimodal expression in all non-sex-specific tissues but not in sex-specific tissues (Fig. 2E; and Supplementary Fig. 7). We will refer to genes in cluster 1 as “tissue-specific switch-like genes.” Although some of them are bimodally expressed in more than one tissue, these genes tend to exhibit high tissue specificity in their bimodal expression. Genes in cluster 2 will be referred to as “universally switch-like genes.”
Genetic variation underlies universally switch-like genes
We found that 8.5% of all switch-like genes (i.e., the proportion of switch-like genes that are in cluster 2) show clear bimodal expression, at least in all tissues common to both sexes. We contend that germline genetic variation across individuals likely drives this universally switch-like gene expression, specifically due to four major types of genetic variants. Firstly, we expect genes on the Y chromosome to show bimodal expression in all tissues common to both sexes since these genes are present in males and absent in females (Fig. 3A). Consistent with this reasoning, seven out of the eight genes in cluster 2B lie within the male-specific region of the Y-chromosome21; the remaining gene in cluster 2B is XIST, showing female-specific expression. Secondly, structural variation affecting a gene could lead to universally bimodal expression for that gene; for example, a homozygous gene deletion would result in the gene being switched off (Fig. 3B). We found eight such genes in cluster 2 A for which genomic structural variants likely underlie the observed universally switch-like expression; four genes are affected by gene deletions, two by polymorphic duplication of the gene, and two by polymorphic insertion into the gene. Thirdly, structural variation in a regulatory element can also switch off a gene in a part of the population (Fig. 3C). FAM118A is likely an instance of this scenario. Lastly, a loss-of-function single-nucleotide variant (SNV) or short indel, which disrupts gene function, can switch off the gene (Fig. 3D). We identified 13 genes in cluster 2 A where such SNVs cause universal bimodality.

A Genes located on the Y chromosome are expressed only in males, leading to bimodal expression in tissues shared by both sexes (i.e., tissues except the prostate, testis, uterus, ovary, and vagina). B Common structural variants, such as deletions, insertions, or duplications, may lead to increased, decreased, or no expression in all tissues relative to individuals who carry the alternative allele. C Common structural variants affecting a genomic region regulating a gene may lead to increased, decreased, or no expression in all tissues, relative to individuals who carry the alternative allele. D Common single-nucleotide variants or short indels affecting a gene or its regulatory region may lead to increased, decreased, or no expression in all tissues relative to individuals who carry the alternative allele. A–D were created in BioRender. Aqil, A. (https://BioRender.com/wk8gcin).
Collectively, we could genetically explain the expression of 22 out of 32 (69%) cases in cluster 2 A despite the small number of genes fitting our conservative definition for universally switch-like genes. SNVs underlie 13 of these cases (Fig. 3B), while structural variants underlie the remaining nine cases (Fig. 3D). Thus, out of the 22 cases where we can explain the genetic underpinnings of switch-like expression, 41% involve genomic structural variation, highlighting the importance of this type of genetic variation (see Supplementary Note 7 and Supplementary Figs. 8–10 for a discussion on why an expression distribution driven by three genotypes at a polymorphic site might still appear bimodal). Although we could not identify the genetic variation underlying the bimodal expression of the remaining 10 genes in cluster 2 A, their consistent and highly correlated switch-like expression across all tissues strongly suggests a genetic basis. We anticipate that better resolution assemblies and detailed regulatory sequence annotations will help identify the genetic variants responsible for the remaining universally switch-like genes.
We highlight a clear example of a common structural variant leading to universally switch-like expression (Fig. 3B). USP32P2 and FAM106A – both universally switch-like genes – are bimodally expressed in all 27 tissues. Both genes show high levels of tissue-to-tissue co-expression. A common 46 kb deletion (esv3640153), with a global allele frequency of ~25%, completely deletes both genes (Fig. 4A, B). We propose that this deletion accounts for the universal switch-like expression of both USP32P2 and FAM106A in all tissues. For illustration, we show the expression level distributions of USP32P2 and FAM106A in the cerebellum (Fig. 4C, D). Indeed, the haplotype harboring this deletion is strongly associated with the downregulation of both genes in all 27 tissues (\(p\) < 10-5 for every single gene-tissue pair, Methods). We note that the under-expression of USP32P2 in sperm is associated with male infertility22, and plausibly, homozygous males for the deletion may be prone to infertility. Additionally, FAM106A interacts with SARS-CoV-2 and is downregulated after infection, at least in lung-epithelial cells23,24,25. Individuals with FAM106A already switched off may develop more severe COVID-19 symptoms upon infection, though further investigation is needed. The case of FAM106A and USP32P2 exemplifies the link between disease and bimodal gene expression.

A FAM106A and USP32P2 (not drawn to scale) are overlapping genes on chromosome 17. Two alternative haplotype classes exist for these genes: one in which both genes are present (haplotype 1) and the other with both genes deleted (haplotype 2). B Frequency distribution of the deletion across diverse populations. Each pi chart represents one of the 26 populations from the 1000 Genomes Project. Purple indicates the frequency of the deletion, while gray indicates the frequency of the alternative haplotype. C, D Expression level distribution in the cerebellum (as an example) across individuals for FAM106A and USP32P2, respectively. The gene deletion presumably leads to the switched-off expression state in both genes. log(TPM + 1)C refers to the value of log(TPM + 1) corrected for technical confounders. A was designed entirely in BioRender; the map in (B) was designed in BioRender; all panels were assembled as Fig. 4 in BioRender. Aqil, A. (https://BioRender.com/dsfjyaa). Source data are provided as a Source Data file.
Sex is a frequent modulator of tissue-specific switch-like gene expression
Tissue-specific expression patterns are crucial for tissue function. Thus, we now turn our attention to tissue-specific switch-like genes. We found that the breast, colon, intestine, kidney, and vagina show a higher number of tissue-specific switch-like genes than other tissues (Fig. 5A). Moreover, within these tissues, the expression of switch-like genes is not independent; instead, at least a subset of switch-like genes exhibit high pairwise gene-to-gene co-expression (Fig. 5B, C; and Supplementary Data 6). This pattern is most pronounced in the breast and colon, where tissue-specific switch-like genes tend to be either all switched off or switched on within an individual. A distinct co-expression pattern emerges in the vagina, where switch-like genes form two communities. Within each community, genes exhibit positively correlated expression levels, meaning they tend to be switched on or off in unison in a given individual. In contrast, genes from different communities show negatively correlated expression. The presence of these communities is consistent with synchronized hormonal regulation, likely driven by estrogen, as discussed later in the manuscript. Specifically, genes in one community are switched on when systemic hormone levels exceed a certain threshold, while genes in the other community are simultaneously switched off under the same conditions. Overall, these results suggest a shared regulatory mechanism for the expression of these genes in each tissue. Given that hormonal regulation plays a substantial role in shaping tissue-specific expression patterns26,27, we hypothesize that hormones may regulate genes that are bimodally expressed in specific tissues (cluster 1; Fig. 2B).

A Number of tissue-specific switch-like genes showing bimodal expression in each of the 27 tissues. The breast, colon, intestine, kidney, and vagina show a greater number of tissue-specific switch-like genes than other tissues. B An illustration of how Pearson’s correlation coefficients were calculated for each pair of bimodally expressed tissue-specific switch-like genes within the breast, colon, intestine, kidney, and vagina. We show the scatterplots for two arbitrarily chosen gene pairs for each of the five tissues. The axes in each dot plot represent the log(TPM + 1)C for the labeled gene in the relevant tissue. Panel C was generated using the pairwise correlation coefficients thus obtained. C Tissue-specific switch-like genes within the five tissues shown are highly co-expressed, at least for a subset of genes. Tissue-specific master regulators, such as endocrinological signals, likely drive their concordant on and off states. The icons in (B) are created with BioRender; all panels were assembled as Fig. 5 in BioRender. Aqil, A. (https://BioRender.com/5inwfv7). Source data are provided as a Source Data file.
Sexual differences in hormonal activity are well documented28,29. To explore this further, we investigated whether hormone-mediated sex-biased expression underlies the co-expression of tissue-specific switch-like genes within tissues. Under this scenario, a gene would be largely switched on in one sex and off in the other in a given tissue. Among tissue-specific switch-like genes, we identified 163 gene-tissue pairs with sex-biased bimodal expression (Fig. 6A; and Supplementary Data 7). These instances are biologically relevant; for example, we found switch-like immunoglobulin genes with female-biased expression in the muscle (IGKJ5) and adrenal gland (IGHJ5), and male-biased expression in the minor salivary gland (IGHV5-10-1). This observation may relate to previous findings30,31 of higher antibody responses to diverse antigens in females than in males. We note that the X-chromosome gene FAM9C shows female-biased, bimodal expression in the spleen. This female bias results from its escape from X-chromosome inactivation32,33, leading to a higher dosage in females. Additionally, the rs3037872 (C/CTG) variant switches the gene off specifically in haploid C (males) or diploid C/C (females), but not in other genotypes, contributing to bimodality. Despite this double dosage being present across tissues, FAM9C clusters with tissue-specific switch-like genes (Cluster 1) rather than universally sex-biased switch-like genes (Cluster 2B). This is because it is switched off in all individuals across most tissues, precluding high tissue-to-tissue co-expression.

A Number of tissue-specific switch-like genes that show female- and male-biased expression. We only display the tissues that have at least one tissue-specific switch-like gene showing sex bias. The number in the central grid underneath each tissue image represents the number of tissue-specific switch-like genes in that tissue. The orange numbers to the left indicate the count of female-biased genes in each of the five tissues shown. The blue numbers to the right indicate the count of male-biased genes. B Heatmap showing the expression of the 157 sex-biased switch-like genes in the breast. Each column represents a gene, and each row represents an individual. These genes tend to be switched on in females. They also tend to be switched on in approximately one half of the males and switched off in the other half. C Violin plots showing the expression level distribution in the breast for five female-biased tissue-specific switch-like genes discussed in the main text. D CDYL2 and TP53INP2 exhibit age-driven bimodality in the uterus. Each point in these graphs represents an individual. In addition to the black best-fit lines, D also shows a brown horizontal line representing a hypothetical switching threshold. The age bias in the expression of these genes can be understood as the proportion of females below the brown line, which differs between those younger than 50 and those older. All panels were assembled as Fig. 6 in BioRender. Aqil, A. (https://BioRender.com/ebd39qz). Source data are provided as a Source Data file.
More dramatically, we found that 157 out of 158 tissue-specific switch-like genes (cluster 1) in the breast tissue are female-biased (Fig. 6A). This bias may be a result of differential hormonal milieu in males versus females. However, the sex-based disparity in the on-versus-off states of these genes is not absolute, but rather a statistical tendency. In other words, the gene is not switched off in all males and switched on in all females. Instead, the proportion of individuals with the gene switched on significantly differs between sexes; almost all females have it switched on, while about half of the males have it switched off (Fig. 6B). Notably, multiple sex-biased switch-like genes—including SPINT1 and SPINT234, multiple keratin genes35, and the oxytocin receptor gene36,37 (OXTR; Fig. 6C)—in the breast tissue are differentially expressed in breast cancers relative to matched non-cancerous tissues. Future investigations could reveal whether the toggling of these genetic switches affects breast cancer risk in females. We caution that sex-biased switch-like expression in the breast may result from differences in cell-type abundance between females and males. Nevertheless, the differential expression of some genes between sexes might developmentally drive such differences in cell-type abundance. In summary, our results indicate that sex is a major contributor to bimodal gene expression, with breast tissue standing out as particularly sex-biased in this context.
In addition to sex, other factors may influence the coordinated expression of switch-like genes within tissues. To explore this, we tested for correlations between the expression levels of tissue-specific switch-like genes and two factors: body mass index (BMI) and age. Using an FDR threshold of 5% and |\(r\)| > 0.3, we assessed whether gene expression levels were associated with either variable. (Supplementary Data 7). We observed no significant correlation between body mass index (BMI) and the expression of these genes. However, the expression levels of two genes, CDYL2 and TP53INP2, both switch-like in the uterus, were significantly correlated with age (Fig. 6D). Notably, TP53INP2 is differentially expressed in endometrial glandular cells, residing in the inner lining of the uterus, in women with recurrent implantation failure38. This differential expression suggests TP53INP2’s involvement in implantation. Since endometrial aging is linked to decreased fertility39, these findings indicate that age-driven shifts in TP53INP2 expression may contribute to the uterine environment becoming less receptive to implantation with increasing age.
Regardless of whether it is associated with sex or age, the aforementioned intra-tissue co-expression of switch-like genes implies an upstream regulatory mechanism shared among switch-like genes in a given tissue. We hypothesize that these genes are regulated by a master regulator, likely a hormone or an external environmental factor. If regulated hormonally, multiple genes may be switched off simultaneously when systemic hormone levels drop below a critical threshold. Such master regulators could coordinate gene expression within a given tissue through two potential mechanisms: (1) by simultaneously inducing epigenetic modifications, such as DNA methylation, that synchronously affect gene activity, or (2) by modulating the expression of a transcription factor that subsequently regulates multiple genes. Next, we investigate whether upstream master regulators result in switch-like gene expression by inducing differential epigenetic modification across individuals.
Epigenetic regulation of bimodal gene expression by DNA methylation
DNA methylation at CpG sites, particularly in promoter regions, is a key epigenetic mechanism regulating gene expression. Hypermethylation in a promoter is typically associated with transcriptional repression, while hypomethylation is linked to gene activation40,41,42 (Fig. 7A). To test whether differential methylation across individuals contributes to switch-like expression, we analyzed methylation at CpG sites in the vicinity of switch-like genes using GTEx data43 (Methods). Our analysis focused on six tissues (prostate, lung, muscle, ovary, colon, and breast) with at least 30 individuals having both gene expression and methylation data. Among 247 tissue-specific switch-like genes (cluster 1) in this analysis, 73 (30%) exhibited a significant negative correlation between expression and methylation (FDR < 5%; |\(r\)| > 0.5) in the tissue where they were bimodally expressed (Supplementary Data 8). Two out of the 73 genes exhibited a significant correlation between expression and methylation in the prostate (e.g., TBX4; Fig. 7B). Strikingly, the remaining 71 genes were all bimodally expressed in the breast (e.g., KRT6B; Fig. 7C), suggesting that differential methylation across individuals plays a key role in regulating switch-like expression in this tissue.

A A schematic showing that methylation at CpG sites in gene promoters typically represses transcription. B Correlation in the prostate between expression of tissue-specific switch-like gene TBX4 and methylation at a CpG site close to the transcription start site for the gene. C Correlation in the breast between expression of tissue-specific switch-like gene KRT6B and methylation at a CpG site close to the transcription start site for the gene. D A schematic showing how the polymorphic deletion of a gene and a nearby unmethylated CpG site can lead to a negative correlation between the expression level of the gene and the methylation level at the CpG site across individuals. This mechanism is not an example of epigenetic silencing. E The expression of GSTM1 is negatively correlated with methylation levels at a nearby CpG site across individuals in multiple tissues because of a polymorphic deletion affecting both the gene and the CpG site. F The genetic variant rs393329 (T- > C) represses NPIPB2 expression across tissues via cg16495772 (a CpG site in the vicinity of NPIPB2) methylation. The x-axis represents genotypes of the variant. The figure on the left shows methylation level distributions of cg16495772 for each genotype in six tissues. The figure on the right shows expression level distributions of NPIPB2 for each genotype in six tissues. In each violin, data points below the 1st percentile and above the 99th percentile were excluded to minimize the impact of outliers on the distribution. In B, C, E, and F, log(TPM + 1)C refers to the values of log(TPM + 1) corrected for the effects of technical confounders. A and D were designed entirely with BioRender. All panels were assembled as Fig. 7 in BioRender. Aqil, A. (2025) https://BioRender.com/eet7v3w. Source data are provided as a Source Data file.
We also tested whether the expression of universally switch-like genes (cluster 2 A) correlated with methylation at CpG sites in their vicinity using the same thresholds (FDR < 5%; |\(r\)| > 0.5; Supplementary Data 9). If methylation contributes to their bimodal expression across multiple tissues, we expect to see significant correlations between expression and methylation in multiple tissues. We observed this pattern for 9 out of 32 cluster-2A genes. Notably, five (GSTM1, USP32P1, USP32P2, FAM106A, KANSL1-AS1) of these nine genes are linked to copy number variation, likely driving both expression and methylation differences across individuals. This result is expected given how methylation levels (β-values) are measured44. β is defined as the hybridization intensity of the probe for the unmethylated CpG site divided by the total intensity of both probes (methylated and unmethylated). In individuals homozygous for a deletion affecting both a gene and a nearby unmethylated CpG site, the hybridization intensities of both probes are near background levels. Consequently, the β-value is intermediate, and the expression is low. In individuals without the deletion, the CpG site is intact, and the hybridization signal for the unmethylated probe is much stronger than the background signal of the methylated probe, yielding a low β-value coupled with high expression. As a result, expression levels and methylation show a negative correlation (Fig. 7D). Indeed, the cluster-2A gene GSTM1 and the nearby CpG site cg16180556, both affected by a polymorphic deletion, show this pattern in all six tissues analyzed (Fig. 7E). Importantly, we observe these patterns despite that the methylation data had already been normalized using the normal-exponential-out-of-band (Noob) method to correct for background fluorescence. We emphasize that such cases are not instances of epigenetic silencing by methylation; instead, they are examples of copy number variation affecting both expression and methylation measurements.
In contrast, the remaining four genes unlinked to copy number variation represent cases of genuine epigenetic regulation of transcription, specifically through genetic variants that influence methylation45,46. For example, we identify an intronic variant (rs393329) in NPIPB2 that functions as both an expression quantitative trait locus (eQTL) and a methylation quantitative trait locus (mQTL; Fig. 7F). Across all tissues studied, this variant consistently represses NPIPB2 expression while simultaneously increasing methylation at multiple CpG sites in the vicinity of NPIPB2. One of these CpG sites is cg16495772, located 593 bases upstream of the variant. The increased methylation at this site likely facilitates gene silencing by recruiting transcriptional repressors or other regulatory machinery. Such epigenetic modifications are genetically codified and thus transmitted faithfully from one generation to another. These findings illustrate how common genetic variation can drive universally bimodal gene expression by orchestrating methylation differences between individuals.
Concordantly switched-off genes in females result in vaginal atrophy
We found that among switch-like genes in the vagina, genes linked to vaginal atrophy in postmenopausal women are significantly overrepresented (86-fold enrichment; \(p\) < 10−4; “see Methods”). Vaginal atrophy, affecting nearly half of postmenopausal women, is triggered by sustained low levels of systemic estrogen and is marked by increased microbial diversity, higher pH, and thinning of the epithelial layer in the vagina47,48. It is also known as atrophic vaginitis, vulvovaginal atrophy, estrogen-deficient vaginitis, urogenital atrophy, or genitourinary syndrome of menopause, depending on the specialty of the researchers. Symptoms experienced by women include dryness, soreness, burning, decreased arousal, pain during intercourse, and incontinence49. Our analysis of switch-like genes in the vagina provides insights into the development of vaginal atrophy.
Specifically, we overlapped a previously published list50 of the top 40 differentially expressed genes in vaginal atrophy with our list of bimodally expressed genes in the vagina. We found that the genes ALOX12, DSG1, KRT1, SPINK7, CRISP3, SBSN, and CRCT1 are both bimodally expressed in the vagina (Fig. 8A) and among the top transcriptionally downregulated genes in women with vaginal atrophy. We refer to these genes as “atrophy-linked switch-like genes.” Indeed, these seven genes are either all switched on, or all switched off concordantly in 73% (113/156) of the vaginal samples we studied. The pairwise concordance rates (percentage of individuals with both genes switched on or both genes switched off) for these genes are shown in Fig. 8B. Among postmenopausal women with this concordant gene expression, 50% are in the “off” state – a fraction that closely matches the prevalence of vaginal atrophy in postmenopausal women47,51. Therefore, our data suggest that estrogen-dependent transcription underlies concordant expression of atrophy-linked switch-like genes, with the “off” state of these genes associated with vaginal atrophy.

A The distribution of expression levels in the vagina of the seven switch-like genes implicated in vaginal atrophy. log(TPM + 1)C represents values of log(TPM + 1) corrected for technical confounders. B Pairwise concordance rates (percentage of individuals in which the two genes are either both switched on or both switched off). C A schematic model for the etiology of vaginal atrophy. High levels of estrogen keep the driver genes switched on in basal and parabasal epithelium, impelling basal and parabasal cells to proliferate and mature, resulting in healthy vaginal mucosa. Conversely, low estrogen levels switch off the driver genes. The lack of basal and parabasal cell proliferation leads to a thin vaginal epithelium, resulting in vaginal atrophy. D Representative immunohistochemical staining of Arachidonate 12-Lipoxygenase (ALOX12) from healthy (\(n\) = 35) and atrophic (\(n\) = 28) vaginal tissues. Scale bars = 50 µm. We show healthy vaginal tissue from a woman with higher systemic estrogen levels and a thicker vaginal epithelial layer, along with atrophic vaginal tissue from a woman with low systemic estrogen levels and a thinner vaginal epithelial layer. There is no difference in ALOX12 expression in the basal or parabasal cells between healthy and atrophic epithelium groups, implicating it as a passenger gene. Images were taken with Axio Observer Z1 (Carl Zeiss AG) with a 20X objective. Panel C was designed entirely with BioRender. All panels were assembled as Fig. 8 in BioRender. Aqil, A. (2025) https://BioRender.com/l6mphqf.
For background, the vaginal epithelial layers are differentiated from the inside out. The basal and parabasal layers of the epithelium consist of mitotic progenitor cells with differentiation potential, while the outermost layer comprises the most differentiated cells52,53. When basal and parabasal cells stop proliferating, the death of mature cells leads to a thin epithelium, causing vaginal atrophy. Given this background, atrophy-linked switch-like genes may either be a cause or a consequence of vaginal atrophy. In particular, if an atrophy-linked switch-like gene encodes a protein necessary for the continued proliferation and differentiation of basal and parabasal cells, we call it a “driver” gene. In the absence of the driver gene’s protein, cell differentiation ceases, and the outer layer gradually disappears, resulting in vaginal atrophy (Fig. 8C). On the other hand, if the product of an atrophy-linked switch-like gene is not required for basal and parabasal cell proliferation, we refer to it as a “passenger” gene, borrowing the terminology from cancer literature54. In healthy vaginas with a thick epithelium, there are more cells in which passenger genes would be expressed. By contrast, in atrophic vaginas, the epithelium thins, resulting in fewer cells where these genes can be expressed. This contrast would lead to the bimodal expression of passenger genes across vagina samples in whole-tissue RNA-sequencing datasets. We hypothesize that at least some of the atrophy-linked switch-like genes are driver genes.
Two key findings allowed us to construct this hypothesis. Firstly, switch-like genes in the vagina show an 8-fold ontological enrichment for the skin development (FDR = 0.016) and a 13-fold enrichment for keratinocyte differentiation (FDR = 0.013), both related to epithelial thickness and differentiation. Notably, two atrophy-linked switch-like genes in the vagina that we identified, DSG1 and KRT1, are involved in the differentiation of epithelial cells in the vagina55,56. Protein stainings available through Human Protein Atlas57 show that all seven atrophy-linked switch-like genes are expressed at the protein level, predominantly in the vaginal epithelium. Secondly, administering 17β-estradiol (a type of estrogen) to postmenopausal women with vaginal atrophy leads to the upregulation of five (ALOX12, DSG1, KRT1, SPINK7, and CRISP3) of these seven genes, causing symptoms to subside58. According to our hypothesis, administering estrogen activates the expression of the driver switch-like genes in the vagina, resuming the proliferation of basal and parabasal cells in the epithelium. This process leads to the reformation of a thick and healthy vaginal mucosa, thereby alleviating the symptoms of vaginal atrophy.
Thus, it is essential to distinguish driver genes from passenger genes to understand the etiology of vaginal atrophy. However, we expect driver and passenger genes to show the same expression patterns in healthy versus atrophic vaginas using bulk RNA-sequencing data. In order to make this distinction, we need comparative expression data, specifically from the basal and parabasal epithelium from healthy versus atrophic vaginas. We expect driver genes to be differentially expressed in the basal and parabasal layers of the epithelium. By contrast, we expect passenger genes to show no differential expression in the basal and parabasal layers between healthy and atrophic vaginas.
To look at the expression levels in the basal and parabasal layers of the epithelium, we arbitrarily chose ALOX12 from the seven atrophy-linked switch-like genes for immunohistochemical staining of its protein product in the vaginal mucosa (which includes the epithelium and the underlying connective tissue). We found that the ALOX12 protein is present in the epithelial cells, and its abundance directly correlates with epithelial thickness, as expected from our transcriptomic analysis. However, we found no significant difference in the staining of the ALOX12 protein in the basal or parabasal epithelial layers between healthy and atrophic samples (Fig. 8D). This suggests that the gene is not differentially expressed in the basal or parabasal layers of the vaginal epithelium between healthy and atrophic vaginas. Therefore, ALOX12 is a passenger gene for vaginal atrophy. Comparative immunohistochemical staining of the protein product of the other switch-like genes in the vagina, especially the remaining six atrophy-linked switch-like genes, may identify the driver gene in the future. Indeed, the KRT1 protein is recognized as a marker of basal cell differentiation in mouse vaginas59, a finding that may also be true for humans. Overall, our results open up several paths for potential pre-menopausal risk assessment and intervention frameworks targeting cell differentiation pathways in the clinical setting.
Discussion
In this study, we investigated factors underlying switch-like gene expression and its functional consequences by integrating genomic, methylomic, and transcriptomic data from 943 individuals. Our systematic analysis revealed 473 switch-like genes, with 6.8% of them bimodally expressed in all tissues, suggesting a genetic basis, while the majority display tissue-specific bimodality. Notably, tissue-specific switch-like genes tend to be concordantly switched on or off within individuals, implicating tissue-specific master regulators such as hormonal signals. Our methylation analysis suggests that hormonally driven epigenetic modification is a key source for achieving breast-specific bimodal expression. In contrast, genetically driven epigenetic silencing leads to universally bimodal expression for genes unlinked to structural variation. We also found that a drop in systemic estrogen levels in the vagina triggers the coordinated silencing of seven vagina-specific switch-like genes, resulting in vaginal atrophy. More broadly, our findings connect switch-like genes to cancers, infections, endocrine disorders, and skin-related conditions. Furthermore, this study has paved two major paths forward toward early medical interventions, as we discuss below.
First, we emphasize that bimodal expression that is correlated across all tissues is driven by genetic polymorphisms. However, the genetic bases for 13/23 universally switch-like genes remain elusive. We propose that the underlying genetic bases for these universally switch-like genes are structural variants, which are not easily captured by short-read DNA sequencing. These structural variants may be discovered in the future as population-level long-read sequencing becomes more common. The first biomedical path forward is to use long-read DNA sequencing to pinpoint the genetic polymorphisms responsible for the bimodal expression of disease-related genes. Of particular interest are the genes CYP4F24P and GPX1P1, both long non-coding RNAs, which are implicated in nasopharyngeal cancer. While we have identified the genetic basis for the bimodal expression of CYP4F24P, that of GPX1P1 remains unknown. CYP4F24P is significantly downregulated in nasopharyngeal cancer tissues60, while GPX1P1 is significantly upregulated in nasopharyngeal carcinomas treated with the potential anticancer drug THZ161. Investigating whether individuals with naturally switched-off GPX1P1 and CYP4F24P are at a higher risk of nasopharyngeal cancer will enable genotyping to identify individuals at elevated risk for nasopharyngeal cancer, facilitating early interventions and improving patient outcomes.
Secondly, switch-like genes present a promising avenue for exploring gene-environment interactions, an area of growing interest. Recent studies indicate that environmental factors can significantly modulate genetic associations62,63. Polymorphisms that result in switch-like gene expression have already been linked to several diseases within specific environmental contexts64. For instance, the deletion of GSTM1 has been associated with an increased risk of childhood asthma, but only in cases where the mother smoked during pregnancy65. Even more critically, switch-like genes can create different cellular environments depending on whether they are switched on or off. These environments may, in turn, influence how genetic variants affect disease risk. Thus, much like current gene-environment association studies that control for factors such as birthplace, geography, and behaviors like smoking, it is conceivable that controlling for switch-like gene expression states could enhance the power of such studies. By cataloging these switch-like genes and developing a framework to classify them as “on” or “off” in various samples, our work lays the groundwork for more robust association studies in future research.
While our findings regarding switch-like genes provide exciting topics for exploration, we caution that we base our results on expression at the RNA level. The bimodal expression of genes across individuals at the RNA level may not necessarily lead to bimodal expression at the protein level. For example, the universally switch-like expression of RPS26 at the RNA level can be explained by a single-nucleotide variant (rs1131017) in the gene’s 5’-untranslated region (UTR). In particular, RPS26 has three transcription states based on the SNV genotypes. The ancestral homozygote C/C corresponds to a high transcription state, the heterozygote C/G to a medium state, and the derived homozygote G/G to a low state. Remarkably, this pattern is reversed at the translation level66: Messenger RNA carrying the derived G allele produces significantly more protein. This reversal may be due to a SNV in the 5’-UTR that can abolish a translation-initiation codon67. This finding demonstrates how the same SNV can regulate a gene’s expression level in opposite directions during transcription and translation. This multi-level regulation in opposite directions likely serves to dampen protein expression variability. It has been shown previously that RNA variability is greater than protein variability in primates68,69; the presence of dampening variants discussed here may be one reason behind these findings. Such compensatory mechanisms for gene expression remain fascinating areas for future research.
In summary, our study has significant implications for understanding the fundamental biology of gene expression regulation and the biomedical impact of switch-like genes. Specifically, it contributes to the growing repertoire of methods for determining individual susceptibility to diseases, facilitating early therapeutic interventions. By providing an approach to studying gene expression states, our study will enhance the predictive accuracy of disease susceptibility and improve patient outcomes.
Methods
Ethics statement
This study complies with all relevant ethical regulations. We obtained the RNA-seq data from GTEx, which obtained institutional review board approval for all collected samples. All GTEx samples from the original study were collected with informed consent (from next-of-kin) under protocols approved by the GTEx consortium, as detailed in the original project documentation. Vaginal tissue sections reused in this work were collected and stained in a previous study70 under approval from the Regional Ethical Review Board, Uppsala, Sweden (record-/diary-number 2008/076). No new human samples were obtained for the present study; only previously approved, de-identified material was reanalyzed. The research conformed to the criteria set by the Declaration of Helsinki.
Data
The Genotype-Tissue Expression (GTEx) project is an ongoing effort to build a comprehensive public resource to study tissue-specific gene expression and regulation. The data we use are transcripts per million (TPM) obtained from human samples across 54 tissues and 56,200 genes (as of December 1st, 2023). We excluded laboratory-grown cell lines from our analysis. Since we need a reasonable number of individuals from each tissue, we excluded tissues with fewer than 50 individuals for our calculations. Of the remaining tissues, there were instances of multiple tissues from the same organ. In such cases, we arbitrarily chose one tissue per organ. We thus focus our analysis on 27 tissues (Fig. 1). Additionally, we retained only those genes for which the mean TPM across individuals was greater than 10 in at least one of the 27 focal tissues. This filter was applied because the analysis of lowly expressed genes may lead to false-positive calls for bimodal expression and, as a result, to assigning biological significance to cases where there is none. After these filtering steps, we are left with TPM data from 19,121 genes in each of the 27 tissues. We will refer to this set of 19,121 genes as G in our equations and the rest of the methods. We note that each tissue contains data from a different number of samples (individuals), totaling 943 across tissues: 312 females and 631 males.
We accessed GTEx protected data from dbGaP (phs000424.v10.p2) and the anvil platform (https://anvil.terra.bio/#workspaces/anvil-datastorage/AnVIL_GTEx_V9_hg38). For DNA methylation (DNAm) data, we used the file GTEx_v9_EPIC_data_noob_final_BMIQ_all_tissues_987.txt. This data, containing CpG site IDs and beta values for methylation, had undergone normal-exponential out-of-band normalization to correct for background fluorescence noise. Additionally, it had also undergone beta mixture quantile normalization to correct for the bias between Type I and Type II probes on Illumina methylation arrays.
Notations
We denote by \({S}_{i}\) the number of samples (individuals) available for tissue \(i\). We also denote by \({x}_{g,i,s}\) the TPM value for gene g in tissue i, for sample \(s\) ∈ {1, …, \({S}_{i}\)} and \(g\) \(\in\) \(G\). According to convention, we log-transform the TPM, specifically by log(\({x}_{g,i,s}\) + 1)71 to suppress the effect of outliers; TPM is extremely large for some samples. Note that log(\({x}_{g,i,s}\) + 1) conveniently maps \({x}_{g,i,s}\,\)= 0 to 0.
Correcting expression levels for confounders
To account for confounding variables, we adopted an approach wherein we use confounders to predict expression values via regression and then treat the residuals as the corrected expression. Regression models may perform poorly when predictors are highly correlated (see Supplementary Note 1 and Supplementary Fig. 1), a phenomenon known as multicollinearity. Using principal component analysis (PCA) to transform a set of intercorrelated confounders into an orthogonal set of principal components (PCs) ensures that the predictors used in the regression model are uncorrelated72. This orthogonality leads to more stable coefficient estimates, which in turn supports a more reliable calculation of the fitted values and thus the residuals. We combined up to six confounder vectors (corresponding to DTHHRDY, SMRIN, SMTSISCH, SMTSPAX, SMNABTCHT, and COHORT; the number varying by tissue) to form a confounder matrix for each tissue. COHORT and DTHHRDY are characteristics of individuals as opposed to that of tissue-individual pairs. Thus, for a given individual s, the values of COHORT and DTHHRDY were identical for every tissue-individual pair (\(i,s\)). Since only 14 individuals (12 males and 2 females) belonged to the surgical cohort, we removed them from all downstream analysis to prevent complications in confounder-correction. Then, we performed PCA on these matrices. The obtained PC score PCk,i,s represents the value of the kth PC in tissue \(i\) for individual \(s\). We then fitted the following generalized additive model (GAM) to regress out the effects of the confounders from the expression data:
where \({\alpha }_{g,i}\) is the intercept term for gene g in tissue i representing the expected log-expression when the effects of all confounders are zero; \({f}_{k,g,i}\)(⋅) is a smooth spline function for gene g in tissue \(i\) applied to kth principal component; Ki is the number of confounders considered for tissue i. \({\varepsilon }_{g,i,s}\) represents the random error term for gene \(g\) in tissue \(i\) for sample \(s\). The fitted values provided by the model are
We define the residual as
Here, \({\hat{\varepsilon }}_{g,i,s}\) represents the confounder-corrected expression value for gene \(g\) in tissue \(i\) for individual \(s\); that is, it reflects the component of the log-transformed expression that remains after removing the systematic effects of the technical confounders.
Dip test
We sought to determine whether a given gene–tissue pair exhibits bimodal expression across individuals. Although several tests of bimodality exist16,73, we employed the dip test via the R function dip.test() from the “diptest” package. Under the null hypothesis of unimodality, a well-calibrated dip test should yield \(p\)-values that are uniformly distributed over the interval [0,1].
To assess whether the test is well-calibrated, we examined a set of housekeeping genes, which are expected to exhibit unimodal expression, in each tissue. For housekeeping gene \(g\) in tissue \(i\), we applied the dip test to the distribution of confounder-corrected expression values \({\hat{\varepsilon }}_{g,i,s}\) across the \({S}_{i}\) samples. We observed that the resulting \(p\)-values were heavily skewed toward 1, indicating that the dip test is more conservative than implied by the raw \(p\)-values—a finding also confirmed using simulated unimodal distributions (Supplementary Note 2; Supplementary Fig. 2). To address this bias, we constructed an empirical null mapping from raw to calibrated \(p\)-values (calibration curve) by rank ordering the raw housekeeping-gene \(p\)-values (Supplementary Fig. 2C). We denote by \(n\) the number of housekeeping-gene \(p\)-values. We sorted the \(n\) \(p\)-values in ascending order and assigned the \(k\)th smallest \(p\)-value a recalibrated value of \(k\)/\(n\). We applied this calibration curve to recalibrate the raw \(p\)-values for all dip tests performed in this study before multiple-hypothesis correction, ensuring that the reported false discovery rates are accurate.
Specifically, we applied a two-round dip test procedure to identify gene-tissue pairs exhibiting bimodal expression across individuals: one round on raw expression data and another on confounder-corrected expression data. This two-round approach was carried out to explicitly gauge the impact of technical confounders on bimodality detection (Supplementary Note 3; and Supplementary Fig. 3), rather than simply relying on corrected data for analysis.
In the first round, for each pair of gene g and tissue \(i\), we carried out a dip test on the distribution of log(\({x}_{g,i,s}\) + 1) across the \({S}_{i}\) samples with 10,000 bootstrap samples. The resulting \(p\)-values were recalibrated using the empirical mapping derived from the housekeeping genes. Following recalibration, we applied the Benjamini–Hochberg procedure at a 5% false discovery rate and imposed an effect-size threshold on the dip statistic \(D\) to reduce false positives. We set this threshold based on the housekeeping null: specifically, we chose the threshold as the mean \(D\) plus three standard deviations from the dip test on housekeeping gene-tissue pairs, which yielded 0.033. This threshold ensures that the effect size is greater than that expected under unimodality. In addition, to penalize gene–tissue pairs with low mean expression, we defined our threshold as
where
We applied this penalization to eliminate cases of noise-driven bimodality since, in cases with low mean expression levels, noise tends to drown out the true biological signal.
In the second round, we reassessed these candidate genes using the confounder-corrected expression values \({\hat{\varepsilon }}_{g,i,s}\) in place of the raw log(\({x}_{g,i,s}\,\)+ 1) data. In this round, we applied the same dip-test procedure with 10,000 bootstrap iterations, followed by empirical \(p\)-value recalibration and Benjamini–Hochberg correction (5% FDR). However, because the residuals \({\hat{\varepsilon }}_{g,i,s}\) are centered around zero, the additional penalization for effect size applied in the first round of dip test was not appropriate. Therefore, we used a simplified criterion of \(D\) > 0.033. The genes that were thus identified as having bimodal distribution in at least one tissue are referred to as “switch-like genes”. We note that, to display gene expression levels in the figures, instead of using \({\hat{\varepsilon }}_{g,i,s}\), we use:
The quantity on the left-hand side of this equation is denoted as log(TPM + 1)C in the figures. Here the subscript \({{\rm{C}}}\) stands for “corrected.” We perform this transformation to ensure that 1) the average corrected levels roughly match the average raw expression levels, and 2) the corrected expression levels are not negative. The values of log(TPM + 1)C for all 473 switch-like genes in all tissue-individual pairs used in this study are available in Supplementary Data 10.
Functional enrichment among switch-like genes
We performed two enrichment analyses to investigate the biological roles and disease associations of switch-like genes. For Gene Ontology (GO) enrichment, we used the Genomic Regions Enrichment of Annotations Tool (GREAT)19 to evaluate overrepresentation of various biological processes among switch-like genes. GREAT provides both effect sizes and \(q\)-values for enriched GO terms.
For disease enrichment, we leveraged the Disease Ontology resource20 using the R package “ontologyIndex” to parse the disease ontology file. This file organizes diseases into a hierarchical structure with top-level (primary) categories and their corresponding daughter (secondary) categories. Our analysis focused on diseases in the seven top-level categories: (1) Disease by infectious agent; (2) Disease of anatomical entity; (3) Disease of cellular proliferation; (4) Disease of mental health; (5) Disease of metabolism; (6) Genetic disorder; and (7) Physical disorder. Each disease was further assigned to a secondary category based on its hierarchical structure (i.e., the daughter terms of the top-level categories). We then determined, for each secondary category, the number of genes in the complete set G and the number of switch-like genes associated with that category.
Fold enrichment for each secondary disease category among switch-like genes was calculated as the ratio of the proportion of switch-like genes (319 of the 473 switch-like genes were represented in this dataset) linked to that category to the corresponding proportion among all genes in \(G\) (18,342 out of the 19,121 genes in \(G\) were present in this dataset). To assess statistical significance, we generated 10,000 random gene sets of size 319 (which is the number of switch-like genes represented in the disease dataset) from 18,342 and computed the fold enrichment for each set. The \(p\)-value for a given secondary category was defined as the fraction of these random sets for which the fold enrichment was equal to or exceeded the value observed among the switch-like genes. Finally, we applied Benjamini–Hochberg correction to control the false discovery rate at 5%, and retained only those secondary categories for which at least 10 switch-like genes were linked.
Tissue-to-tissue co-expression of genes
We sought to identify switch-like genes exhibiting bimodal expression in all tissues. One seemingly straightforward approach is to count the number of tissues showing a bimodal distribution of expression levels for each gene. However, even if a gene genuinely exhibits bimodal expression across all tissues, our methodology may fail to recognize it as such if the mean expression levels (\({\bar{x}}_{g,i}\)) of the gene are low in some tissues. This is because our effect size threshold in the first round of dip tests penalizes gene-tissue pairs with low \({\bar{x}}_{g,i}\). Moreover, if gene expression follows a bimodal distribution across all tissues, then it does so likely due to a genetic polymorphism affecting expression. Thus, the expression of such genes would be highly correlated between tissues. Given this insight, discovering universally bimodal genes is more tractable using tissue-to-tissue co-expression of each gene.
For each gene \(g\), we construct the co-expression matrix \({{{\bf{R}}}}_{g}\), wherein each element is the Pearson's correlation coefficient of the expression levels of the gene between two tissues. To calculate the co-expression between a pair of tissues, we need to use the samples for which the TPM is measured for both tissues74. In general, even if the number of samples is large for both tissues, it does not imply that there are sufficiently many common samples. Therefore, using the sample information described in GTEx_Analysis_v8_Annotations_SampleAttributesDD.xlsx in the GTEx data portal, we counted the number of samples shared by each tissue pair and excluded the 41 tissue pairs that share fewer than 40 samples. We calculated the co-expression for each of the remaining 27 × 26/2 − 41 = 310 tissue pairs. We denote by \({S}_{i,j}\) the number of samples shared by the two tissues \(i\) and \(j\). Then, we calculated the Pearson correlation coefficient between \({\hat{\varepsilon }}_{g,i,s}\) and \({\hat{\varepsilon }}_{g,j,s}\) across the \({S}_{i,j}\) samples. Specifically, we calculate
where
and
We note that both \({m}_{g,i}\) and \({m}_{g,j}\) are close to 0 since they represent means of residuals from the generalized additive models. For each gene g, we define matrix \({{{\bf{R}}}}_{g}\) as a 27 × 27 tissue-to-tissue co-expression matrix with the \((i,j)\)-th element equal to \({r}_{g}\left(i,j\right)\) if \({S}_{i,j}\) ≥ 40; otherwise, that entry is recorded as missing (NA). We then vectorize the correlation matrix, \({{{\bf{R}}}}_{g}\), into the vector \({{{\bf{r}}}}_{g}\). Since the correlation matrix is symmetric, we only vectorize the strict upper-triangular part of \({{{\bf{R}}}}_{g}\). This region contains all unique off-diagonal correlations (351 in total). 41 tissue pairs have \({S}_{i,j}\) < 40, so their entries are discarded. Thus, \({{{\bf{r}}}}_{g}\) is a 310-dimensional vector. A gene, g, was removed from the downstream analysis if \({\hat{\varepsilon }}_{g,i,s}\) or \({\hat{\varepsilon }}_{g,j,s}\) was 0 across all \({S}_{i,j}\) samples for any of the 310 tissue pairs. In this process, one out of 473 switch-like genes was removed. We ran a PCA, using the prcomp() function in R, on vectors \({{{\bf{r}}}}_{g}\), for all genes for which we could calculate \({r}_{g}\left(i,j\right)\) for all 310 tissue pairs. In parallel, we ran PCA on only the set of vectors (genes) characterizing only the 472 ( = 473 − 1) switch-like genes.
In the space spanned by the first two principal components, we calculated the pairwise distance between genes using the dist() function in R with method = “euclidean”. We then performed hierarchical clustering using the hclust() function with method = “complete”. Finally, we used the cuttree() function with \(k\)=2 and \(k\)=3 to obtain two and three clusters, respectively.
Identifying the genetic basis of universal bimodality
To identify the genetic basis of bimodality for switch-like genes in cluster 2 A, we obtained the coordinates of the genes for both hg19 and hg38 using their Ensembl IDs as keys through Ensembl BioMart. We obtained coordinates of common structural variants using both the 1000 genomes project (hg19)75 and the HGSV2 dataset (hg38)76. We performed an overlap analysis using BedTools77 to identify polymorphic deletions or insertions into these genes. Additionally, we obtained the GTEx dataset for the expression quantitative trait loci (eQTL). We identified genes in cluster 2 A that had at least one eQTL, which was consistently associated with either increased or decreased expression of a given gene across all 27 tissues analyzed.
Gene-to-gene co-expression within tissues
We performed gene-to-gene co-expression analysis within the breast, colon, intestine, kidney and vagina tissues. In a given tissue \(i\), we denote the set of cluster-1 (tissue-specific switch-like) genes by \({C}_{i}\). Then, for \(i\) ∈ {breast, colon, intestine, kidney, vagina}, we calculated the Pearson correlation, across the \({S}_{i}\) samples, between \({\hat{\varepsilon }}_{g,i,s}\) and \({\hat{\varepsilon }}_{h,i,s}\) for every \(g\), \(h\) ∈ \({C}_{i}\) where \(g\) \(\ne\) \(h\).
Quantifying the effect of sex, age, and BMI on cluster-1 gene expression
For every gene-tissue pair (\(g,i\)), where \(g\) is a switch-like gene, and \(i\) is a tissue common to both sexes, we tested the hypothesis that the distribution of \({\hat{\varepsilon }}_{g,i,s}\) across male samples differed from that across female samples using the Wilcoxon rank-sum test. We applied the Benjamini-Hochberg procedure of multiple hypotheses correction with FDR = 5%. We quantified the effect size of the sex bias using Cohen’s d, using the threshold \({|d|}\) > 0.278. Additionally, for every gene-tissue pair (\(g,i\)), where \(g\) is a switch-like gene, and \(i\) can be any tissue, we tested the two hypotheses that \({\hat{\varepsilon }}_{g,i,s}\) is correlated with age and that \({\hat{\varepsilon }}_{g,i,s}\) is correlated with BMI. We applied the Benjamini-Hochberg procedure of multiple hypotheses correction with FDR = 5% to results from both analyses separately. We classified gene-tissue pairs as correlated with age or BMI using thresholds FDR < 5% and |\(r\)| > 0.3, where \(r\) is the Pearson correlation coefficient.
Analysis of methylation levels
Methylation data were annotated using the IlluminaHumanMethylationEPICanno.ilm10b4.hg19 library in R79. We retrieved CpG annotation information by invoking the getAnnotation() function, which provided genomic coordinates, probe IDs, and regulatory feature information for each CpG site for which we had methylation data available from GTEx. The extracted annotation included fields such as Name, chromosome (chr), genomic position (pos), rsID of SNPs at the CpG site (CpG_rs), UCSC_RefGene_Name, UCSC_RefGene_Group, and various enhancer and regulatory feature annotations.
We then performed correlation analyses between switch-like gene expression and methylation levels within a 50 kb window surrounding each gene, separately for cluster-1 and cluster-2 genes. Our analysis focused on six tissues (prostate, lung, muscle, ovary, colon, and breast) with at least 30 individuals having both gene expression and methylation data. For each analysis, we computed the Pearson correlation coefficient between the gene’s confounder-corrected expression values and the methylation levels at nearby CpG sites. We applied stringent thresholds (FDR < 5% and |\(r\)| > 0.5). For the analysis of cluster-2A genes in particular, we further required that a gene show a significant correlation with methylation in at least two tissues before concluding that methylation contributes to its multi-tissue bimodality.
Enrichment of switch-like genes among disease-linked genes
We performed enrichment analysis to test whether the top 40 genes differentially expressed in vaginal atrophy are overrepresented among cluster-1 vagina-specific switch-like genes. We denote the set of genes differentially expressed in vaginal atrophy as \(V\) and set of cluster-1 switch-like genes bimodally expressed in the vagina as \(C\)vagina. We calculated the fold enrichment of genes downregulated in vaginal atrophy among cluster-1 genes in the vagina by \(\frac{|{C}_{{{\rm{vagina}}}}\, \cap \, {V|}/|{C}_{{{\rm{vagina}}}}|}{{|G}\, \cap \,{V|}/{|G|}}\).
To calculate the \(p\)-values associated with these enrichments, we obtained 10,000 uniformly random samples (with replacement) of size |\(C\)vagina| from \(G\). The \(p\)-value for the enrichment of genes linked to vaginal atrophy among cluster-1 switch-like genes in the vagina is then given by the fraction of random samples among the 10,000 samples for which \(|{q}_{j}\cap {V|}\ge |{C}_{{\mathrm{vagina}}}\cap {V|}\). Here, \({q}_{j}\) is the set of genes in random sample \(j\) where \(j\) ∈ {1, …, 10000}.
Discretizing expression levels
We performed kernel density estimation using the density() function in R on the distributions of \(\log (1+{x}_{g,{\mathrm{vagina}},s})\) across the \({S}_{{\mathrm{vagina}}}\) samples for \(g\) ∈ \({C}_{{\mathrm{vagina}}}\) \(\cap\) \(V\).
We used the minimum of the estimated density as the switching threshold; if an individual had an expression level above the threshold in a given tissue, the gene was considered “on” in the individual in that tissue. The gene was considered “off” otherwise. We then calculate the concordance of expression among genes in the set \({C}_{{\mathrm{vagina}}}\) \(\cap\) \(V\) in the vagina as follows:
where 1(⋅) is the indicator function.
Immunohistochemistry
Vaginal biopsies, taken by use of punch biopsies from postmenopausal women, were fixed and stained by use of anti-ALOX12 antibodies70,80. This polyclonal rabbit anti-ALOX12 antibody (catalog no. HPA010691; Prestige Antibodies® powered by Atlas Antibodies, Sigma-Aldrich) was diluted 1:50 in 0.1% bovine serum albumin. Sections were incubated overnight at 4 °C. Negative controls, prepared by omitting the primary antibody, were included for all samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Bulk RNA-seq expression data (TPM, GTEx v8) is available from the GTEx open-access portal (https://gtexportal.org/home/downloads/adult-gtex/bulk_tissue_expression). Controlled-access GTEx data, including full metadata, can be requested through dbGaP accession phs000424.v10.p2 at [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000424.v10.p2]. DNA-methylation data were obtained from GTEx_v9_EPIC_data_noob_final_BMIQ_all_tissues_987.txt in the AnVIL workspace AnVIL_GTEx_V9_hg38 [https://anvil.terra.bio/#workspaces/anvil-datastorage/AnVIL_GTEx_V9_hg38]. All results generated for this study are provided as Supplementary Data 1–10 and archived on Figshare [https://doi.org/10.6084/m9.figshare.28848356], where they are freely available without restriction81. Source data are provided with this paper.
Code availability
The code used to generate the results in this study is publicly accessible on GitHub [https://github.com/AlberAqil/Switch_like_gene_expression_modulates_disease_risk_2025/]. A permanent version of all code is deposited on Zenodo [https://doi.org/10.5281/zenodo.15270596]82.
References
Jacob, F. & Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3, 318–356 (1961).
Banerji, J., Rusconi, S. & Schaffner, W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308 (1981).
Gillies, S. D., Morrison, S. L., Oi, V. T. & Tonegawa, S. A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 33, 717–728 (1983).
Serfling, E., Jasin, M. & Schaffner, W. Enhancers and eukaryotic gene transcription. Trends Genet 1, 224–230 (1985).
Allfrey, V. G., Faulkner, R. & Mirsky, A. E. Acetylation and methylation of histones and their possible role in the regulation of RNA Synthesis. Proc. Natl. Acad. Sci. USA 51, 786–794 (1964).
Riggs, A. D. & Jones, P. A. 5-methylcytosine, gene regulation, and cancer. Adv. Cancer Res. 40, 1–30 (1983).
Bestor, T., Laudano, A., Mattaliano, R. & Ingram, V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 203, 971–983 (1988).
Jenuwein, T. & Allis, C. D. Translating the histone code. Science 293, 1074–1080 (2001).
Levine, M. & Tjian, R. Transcription regulation and animal diversity. Nature 424, 147–151 (2003).
Muntoni, F., Torelli, S. & Ferlini, A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2, 731–740 (2003).
Sakai, T. et al. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 48, 880–888 (1991).
Cutting, G. R. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat. Rev. Genet. 16, 45–56 (2015).
Ertel, A. Bimodal gene expression and biomarker discovery. Cancer Inf. 9, 11–14 (2010).
Bessarabova, M. et al. Bimodal gene expression patterns in breast cancer. BMC Genomics 11, S8 (2010).
Justino, J. R. et al. An integrated approach to identify bimodal genes associated with prognosis in câncer. Genet. Mol. Biol. 44, e20210109 (2021).
Moody, L., Mantha, S., Chen, H. & Pan, Y.-X. Computational methods to identify bimodal gene expression and facilitate personalized treatment in cancer patients. J. Biomed. Inform. 100S, 100001 (2019).
Mason, C. C. et al. Bimodal distribution of RNA expression levels in human skeletal muscle tissue. BMC Genomics 12, 98 (2011).
Hartigan, J. A. & Hartigan, P. M. The dip test of unimodality. Ann. Stat. 13, 70–84 (1985).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Schriml, L. M. et al. Human Disease Ontology 2018 update: classification, content and workflow expansion. Nucleic Acids Res 47, D955–D962 (2019).
Jangravi, Z. et al. A fresh look at the male-specific region of the human Y chromosome. J. Proteome Res. 12, 6–22 (2013).
Cheung, S., Parrella, A., Rosenwaks, Z. & Palermo, G. D. Genetic and epigenetic profiling of the infertile male. PLoS One 14, e0214275 (2019).
Turjya, R. R., Khan, M. A.-A. -K. & Mir Md Khademul Islam, A. B. Perversely expressed long noncoding RNAs can alter host response and viral proliferation in SARS-CoV-2 infection. Future Virol. 15, 577–593 (2020).
Talotta, R., Bahrami, S. & Laska, M. J. Sequence complementarity between human noncoding RNAs and SARS-CoV-2 genes: What are the implications for human health?. Biochim. Biophys. Acta Mol. Basis Dis. 1868, 166291 (2022).
Arman, K., Dalloul, Z. & Bozgeyik, E. Emerging role of microRNAs and long non-coding RNAs in COVID-19 with implications to therapeutics. Gene 861, 147232 (2023).
Chia, D. J. Minireview: mechanisms of growth hormone-mediated gene regulation. Mol. Endocrinol. 28, 1012–1025 (2014).
Mayne, B. T. et al. Large scale gene expression meta-analysis reveals tissue-specific, sex-biased gene expression in humans. Front. Genet. 7, 183 (2016).
McEwen, B. S. & Milner, T. A. Understanding the broad influence of sex hormones and sex differences in the brain. J. Neurosci. Res. 95, 24–39 (2017).
Goel, N., Workman, J. L., Lee, T. T., Innala, L. & Viau, V. Sex differences in the HPA axis. Compr. Physiol. 4, 1121–1155 (2014).
Fink, A. L. & Klein, S. L. The evolution of greater humoral immunity in females than males: implications for vaccine efficacy. Curr. Opin. Physiol. 6, 16–20 (2018).
Laffont, S. & Guéry, J.-C. Deconstructing the sex bias in allergy and autoimmunity: from sex hormones and beyond. Adv. Immunol. 142, 35–64 (2019).
Carrel, L. & Willard, H. F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434, 400–404 (2005).
Tukiainen, T. et al. Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248 (2017).
Wu, Q. et al. Comprehensive analysis of the expression and prognostic value of SPINT1/2 in breast carcinoma. Front. Endocrinol. 12, 665666 (2021).
Takan, I., Karakülah, G., Louka, A. & Pavlopoulou, A. In the light of evolution:” Keratins as exceptional tumor biomarkers. PeerJ 11, e15099 (2023).
Behtaji, S. et al. Identification of oxytocin-related lncRNAs and assessment of their expression in breast cancer. Sci. Rep. 11, 6471 (2021).
Fiaz, T. et al. Peripheral mRNA expression and prognostic significance of emotional stress biomarkers in metastatic breast cancer patients. Int. J. Mol. Sci. 23, 14097 (2022).
Zhang, H., Zhang, C. & Zhang, S. Single-cell RNA transcriptome of the human endometrium reveals epithelial characterizations associated with recurrent implantation failure. Adv. Biol. (Weinh.) 8, e2300110 (2024).
Pathare, A. D. S. et al. Endometrial receptivity in women of advanced age: an underrated factor in infertility. Hum. Reprod. Update 29, 773–793 (2023).
Yeivin, A. & Razin, A. Gene methylation patterns and expression. EXS 64, 523–568 (1993).
Naveh-Many, T. & Cedar, H. Active gene sequences are undermethylated. Proc. Natl. Acad. Sci. USA 78, 4246–4250 (1981).
Bird, A. P. CpG-rich islands and the function of DNA methylation. Nature 321, 209–213 (1986).
Oliva, M. et al. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat. Genet. 55, 112–122 (2023).
Zhou, W., Triche, T., Laird, P. & Shen, H. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res 46, e123 (2018).
Hawe, J. S. et al. Genetic variation influencing DNA methylation provides insights into molecular mechanisms regulating genomic function. Nat. Genet. 54, 18–29 (2022).
Chundru, V. K. et al. Rare genetic variants underlie outlying levels of DNA methylation and gene-expression. Hum. Mol. Genet. 32, 1912–1921 (2023).
Goldstein, I., Dicks, B., Kim, N. N. & Hartzell, R. Multidisciplinary overview of vaginal atrophy and associated genitourinary symptoms in postmenopausal women. Sex. Med. Today 1, 44–53 (2013).
Szymański, J. K., Słabuszewska-Jóźwiak, A. & Jakiel, G. Vaginal aging—what we know and what we do not know. Int. J. Environ. Res. Public Health 18, 4935 (2021).
Kim, H.-K., Kang, S.-Y., Chung, Y.-J., Kim, J.-H. & Kim, M.-R. The recent review of the genitourinary syndrome of menopause. J. Menopausa Med 21, 65–71 (2015).
Hummelen, R. et al. Vaginal microbiome and epithelial gene array in post-menopausal women with moderate to severe dryness. PLoS One 6, e26602 (2011).
Faubion, S. S., Sood, R. & Kapoor, E. Genitourinary syndrome of menopause: management strategies for the clinician. Mayo Clin. Proc. 92, 1842–1849 (2017).
Buchanan, D. L. et al. Role of stromal and epithelial estrogen receptors in vaginal epithelial proliferation, stratification, and cornification. Endocrinology 139, 4345–4352 (1998).
Anderson, D. J., Marathe, J. & Pudney, J. The structure of the human vaginal stratum corneum and its role in immune defense. Am. J. Reprod. Immunol. 71, 618–623 (2014).
Greenman, C. et al. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 (2007).
Ho, M. et al. Update of the keratin gene family: evolution, tissue-specific expression patterns, and relevance to clinical disorders. Hum. Genomics 16, 1 (2022).
Szegedi, A. et al. Protein kinase C isoenzymes differentially regulate the differentiation-dependent expression of adhesion molecules in human epidermal keratinocytes. Exp. Dermatol. 18, 122–129 (2009).
Pontén, F., Jirström, K. & Uhlen, M. The human protein Atlas-a tool for pathology. J. Pathol. 216, 387–393 (2008).
Cotreau, M. M. et al. A study of 17β-estradiol-regulated genes in the vagina of postmenopausal women with vaginal atrophy. Maturitas 58, 366–376 (2007).
Miyagawa, S. & Iguchi, T. Epithelial estrogen receptor 1 intrinsically mediates squamous differentiation in the mouse vagina. Proc. Natl. Acad. Sci. USA. 112, 12986–12991 (2015).
Zhang, X. et al. Identification of key pseudogenes in nasopharyngeal carcinoma based on RNA-Seq analysis. BMC Cancer 21, 483 (2021).
Gao, L. et al. Gene expression profile of THZ1-treated nasopharyngeal carcinoma cell lines indicates its involvement in the inhibition of the cell cycle. Transl. Cancer Res. 10, 445–460 (2021).
Dudbridge, F. & Fletcher, O. Gene-environment dependence creates spurious gene-environment interaction. Am. J. Hum. Genet. 95, 301–307 (2014).
Vetr, N. G., Gay, N. R., MoTrPAC Study Group & Montgomery, S. B. The impact of exercise on gene regulation in association with complex trait genetics. Nat. Commun. 15, 3346 (2024).
Thomas, D. Gene–Environment-wide association studies: emerging approaches. Nat. Rev. Genet. 11, 259–272 (2010).
Gilliland, F. D. et al. Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. Am. J. Respir. Crit. Care Med. 166, 457–463 (2002).
Li, Q. et al. Genome-wide search for exonic variants affecting translational efficiency. Nat. Commun. 4, 2260 (2013).
Liu, L. et al. Mutation of the CDKN2A 5’ UTR creates an aberrant initiation codon and predisposes to melanoma. Nat. Genet. 21, 128–132 (1999).
Khan, Z. et al. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science 342, 1100–1104 (2013).
Wang, S. H., Hsiao, C. J., Khan, Z. & Pritchard, J. K. Post-translational buffering leads to convergent protein expression levels between primates. Genome Biol. 19, 83 (2018).
Kallak, T. K. et al. Vaginal gene expression during treatment with aromatase inhibitors. Clin. Breast Cancer 15, 527–535.e2 (2015).
Shalek, A. K. et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240 (2013).
Perez, L. V. et al. Principal Component Analysis To Address Multicollinearity. Whitman College: Walla Walla, WA, USA (2017).
Hellwig, B. et al. Comparison of scores for bimodality of gene expression distributions and genome-wide evaluation of the prognostic relevance of high-scoring genes. BMC Bioinforma. 11, 276 (2010).
Dobrin, R. et al. Multi-tissue coexpression networks reveal unexpected subnetworks associated with disease. Genome Biol. 10, R55 (2009).
1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, eabf7117 (2021).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Cohen, J. et al. Statistical Power Analysis for the Behavioral Sciences. (Routledge, London, England, 1988).
Hansen, K. D. IlluminaHumanMethylationEPICanno.ilm10b4.hg19: annotation for Illumina’s EPIC methylation arrays. R package version 0.6.0, https://bitbucket.com/kasperdanielhansen/Illumina_EPIC (2017).
Kallak, T. K. et al. Aromatase inhibitors affect vaginal proliferation and steroid hormone receptors. Menopause 21, 383–390 (2014).
Aqil, A. Switch-like genes modulate disease risk (supplementary data), FigShare. https://doi.org/10.6084/m9.figshare.28848356 (2025).
Aqil, A. Switch-like gene expression modulates disease risk (code), Zenodo. https://doi.org/10.5281/zenodo.15270596 (2025).
Acknowledgements
We thank Dr Vincent Lynch, Dr Eric Strobel, and Dr Charikleia Karageorgiou for carefully reading this manuscript. We also thank Hiba Hassan and Fatima Azeem for their help in creating schematics and selecting reader-friendly color schemes. O.G. and N.M. acknowledge support from the National Institute of General Medical Sciences (under grant no. 1R01GM148973-01). O.G. also acknowledges support from the National Science Foundation (under grant nos. 2049947 and 2123284) and National Institute of General Medical Sciences (R35-GM156519). N.M. also acknowledges support from the Japan Science and Technology Agency (JST) Moonshot R&D (under grant no JPMJMS2021), the National Science Foundation (under grant no. 2052720), and JSPS KAKENHI (under grant no. 24K14840). The funders had no role in study design, data collection and analysis, publication decision, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
O.G. and N.M. co-supervised the project. M.S., O.G., and N.M. conceived the project. Y.L. performed the initial computations with help from Z.W., S.I., M.R., and N.M. A.A. created the figures and performed the final computational analysis. T.K.K. performed the protein staining experiments on vaginal tissues. AA, OG, and NM wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Natalie Telis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Aqil, A., Li, Y., Wang, Z. et al. Switch-like gene expression modulates disease risk. Nat Commun 16, 5323 (2025). https://doi.org/10.1038/s41467-025-60513-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60513-x
