
Abstract
The composition of muscle fibers, characterized by distinct contractile and metabolic properties, significantly influences meat quality and glucose homeostasis. However, the mechanisms by which three-dimensional (3D) genome topology integrates with epigenetic states to regulate muscle fiber specification and transformation remain poorly understood. Here, we present an integrative analysis of the transcriptome, epigenome, and 3D genome architecture in the slow-twitch glycolytic extensor digitorum longus (EDL) and fast-twitch oxidative soleus (SOL) muscles of the pig (Sus scrofa). Global remodeling of enhancer-promoter (E-P) interactions emerged as a central driver of transcriptional reprogramming associated with muscle contraction and glucose metabolism. We identified tissue-specific super-enhancers (SEs) that regulate muscle fiber-type specification through cooperation of chromatin looping and transcription factors such as KLF5. Notably, the SE-driven activation of STARD7 facilitated the transformation of glycolytic fibers into oxidative fibers by mitigating reactive oxygen species levels and suppressing ERK MAPK signaling. This study elucidates the principles of 3D genome organization in the epigenetic regulation of muscle fiber specification and transformation, providing a foundation for novel therapeutic strategies targeting metabolic disorders and enhancing meat quality.
Similar content being viewed by others

Introduction
Skeletal muscle comprises 40–50% of total body mass in humans and plays a pivotal role in insulin-stimulated glucose disposal, making it essential for maintaining systemic glucose homeostasis. Dysregulated skeletal muscle metabolism impairs insulin sensitivity and contributes to metabolic disorders such as obesity and Type 2 diabetes1,2. Muscle fibers exhibit metabolic diversity, with distinct properties shaped by their plasticity and fiber-type composition. Myofibers are classified based on the expression of myosin heavy chain (MyHC) isoforms: slow-twitch fibers predominantly express MYH7 (MyHC I), while fast-twitch fibers express MYH1 (MyHC IIa) and MYH4 (MyHC IIb)3. Slow fibers favor oxidative metabolism and endurance, whereas fast fibers rely on glycolysis and contract more rapidly4.
Although the total number of muscle fibers remains stable in adults2, fiber-type composition can adapt to external stimuli such as aging, exercise, or disease. In obesity and Type 2 diabetes, a shift from oxidative slow fibers to glycolytic fast fibers contributes to impaired insulin sensitivity, reduced metabolic flexibility, and insulin resistance5. In livestock production, the slow-fiber-rich soleus (SOL) muscle offers superior meat quality, characterized by higher redness, intramuscular fat, water-holding capacity, and lower tenderness, compared to the fast-fiber-rich extensor digitorum longus (EDL) muscle6,7. Understanding the regulatory mechanisms underlying muscle fiber specification and transformation could provide therapeutic insights for human metabolic diseases and strategies to enhance meat quality.
Advances in genome-wide 3D chromatin interaction mapping have revolutionized our understanding of spatial genome organization and its regulatory impact on gene expression. The mammalian genome is hierarchically structured into chromatin loops, topologically associating domains (TADs), and A/B compartments within the nucleus8. TAD boundaries, primarily defined by the CCCTC-binding factor (CTCF) and cohesin complexes9, are largely conserved across cell types and species10. However, chromatin loops and epigenetic landscapes within TADs exhibit cell-type specificity, driving the precise spatiotemporal expression of lineage-specific genes11. Proper 3D chromatin architecture is critical for transcriptional regulation during processes such as myogenesis12, prenatal muscle development13, and muscle stem cell aging14. These findings underscore the importance of 3D genome remodeling in shaping transcriptional programs during muscle fiber differentiation.
cis-regulatory elements (CREs), including enhancers and promoters, are defined by specific epigenetic features and regulate gene expression through cell-type-specific chromatin looping. Super-enhancers (SEs), large enhancer clusters, are key regulators of lineage-specific gene expression15. SEs coordinate with chromatin regulators to form extensive intra-TAD loops, enabling precise gene activation16. Previous studies revealed enhancer repertoires in distinct muscles, and highlighting the crucial role of transcription factors (TFs) in fiber-type specification17. Moreover, chromatin looping bound by SEs has been shown to activate fast-type myosin gene expression18. However, the genome-wide dynamics of enhancer-promoter (E-P) interactions between muscles, particularly their role in shaping muscle-specific phenotypes, remain unclear.
The pig (Sus scrofa) serves as a valuable biomedical model for studying energy metabolism and muscle diseases due to its physiological, anatomical, and metabolic similarities to humans19. In this study, we provide a comprehensive multi-omics dataset from pig EDL and SOL muscles to clarify the regulatory mechanisms underlying their developmental and morphological differences. We identified numerous active CREs and demonstrated how tissue-specific E-P interactions and TFs orchestrate transcriptional programs in distinct muscles. Furthermore, we identified key SEs that regulate slow- and fast-type myosin gene expression via chromatin interactions. Notably, we validated that SE-driven activation of STARD7 induces the transformation of glycolytic to oxidative fibers by reducing reactive oxygen species (ROS) and inhibiting ERK MAPK signaling. Together, our findings highlight the critical role of activated CREs and 3D genome organization in muscle fiber specification and transformation.
Results
Transcriptome dynamic and genome-wide analysis of CREs in SOL and EDL
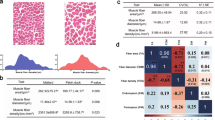
In this study, we collected SOL and EDL muscles from three 6-month-old Duroc pigs to compare their muscle characteristics (Fig. 1a). Slow-twitch fibers predominated in the SOL, while fast-twitch fibers were more abundant in the EDL, as evidenced by differential expression of myosin genes (Fig. 1b, c and Supplementary Fig. 1a–c). RNA-seq analysis of SOL and EDL muscles identified 788 differentially expressed genes (DEGs) including 366 SOL-induced DEGs and 421 SOL-induced DEGs (Supplementary Fig. 1d and Supplementary Data 1). Gene Ontology (GO) analysis and Gene Set Enrichment Analysis (GSEA) further revealed that glycolysis and muscle contraction pathways were more active in the EDL-induced DEGs, while oxidative phosphorylation and fatty acid β-oxidation pathways were more prominent in the SOL-induced DEGs (Supplementary Fig. 1e, f and Supplementary Data 1). To evaluate the rationality of using bulk tissues to present the status of myonuclei, we compared the epigenetics signal of bulk tissue and purified-myonuclei. We found that genome-wide H3K27ac signal showed a high similarity (R > 0.8) between bulk tissue and purified-myonuclei (Supplementary Fig. 2a, b). The majority of H3K27ac peaks (over 80%) identified in myonuclei are common in corresponding bulk tissue (Supplementary Fig. 2c). These findings confirmed that SOL and EDL can be classified as typical oxidative and glycolytic muscles, respectively. Notably, the epigenetic profiles of bulk tissues closely resembled those of purified myonuclei, suggesting that bulk tissues can effectively reflect the epigenetic state of myonuclei. Therefore, SOL and EDL tissue could serve as suitable models for studying the regulatory mechanisms of muscle fiber-type specification and transformation.

a Isolated EDL and SOL muscles. b Representative immunofluorescent staining of slow-twitch MyHC I (red) and fast-twitch MyHC IIb (green) in SOL and EDL sections. Magnification: ×20, Bar = 200 μm. c Heatmap of muscle fiber-type marker gene expression. d Classification and annotation of chromatin states, with each row representing a chromatin state and each column denoting histone marks, chromatin accessibility, genomic annotation, and gene expression levels. TssAFlank1 (n = 56, median = 1.076648): high H3K4me3 and ATAC-seq signal; TssAFlank2 (n = 214, median = 1.129079): high H3K4me3 but weak ATAC-seq signal. EnhA1 (n = 55, median = 1.476194): high H3K27ac and ATAC-seq signal; EnhA2 (n = 161, median = 1.460204): weak H3K27ac and ATAC-seq signal. RepPC (n = 65, median = 0.5147669): high H3K27me3 signal. Quiescent (n = 192, median = 0.6704646): lack epigenetics signal. e Tissue specificity of identified CREs. f Alluvial diagram depicting changes in promoter chromatin states of DEGs. g Expression changes of DEGs with switched chromatin states. Boxes represent the interquartile range with the median as a horizontal line; whiskers extend to the 5th and 95th percentiles. Significance was tested using a two-sided Wilcoxon test. h Functional enrichment analysis of tissue-specific enhancers using GREAT. Significance was tested using binomial test. i ATAC-seq tracks, chromatin states, and histone modifications at SLN and UQCR10 loci and their associated enhancers in SOL and EDL.
To investigate the relationship between chromatin epigenetic states and transcriptional regulation, we conducted ChIP-seq for H3K27me3, CUT&Tag for H3K27ac and H3K4me3, and Assay for Transposase-Accessible Chromatin (ATAC-seq) to assess chromatin accessibility in both SOL and EDL muscles. Principal component analysis (PCA) of each epigenetics mark datasets demonstrated high reproducibility between biological replicates (Supplementary Fig. 3a and Supplementary Data 2). Profiles of histone modifications and chromatin accessibility revealed that transcriptional activation was associated with increased chromatin accessibility and enrichment of active histone marks (H3K27ac and H3K4me3) (Supplementary Fig. 3b).
We applied ChromHMM to these epigenetics datasets, and categorized the six distinct chromatin states, each associated with specific gene expression patterns (Fig. 1d). The active state consisted of active transcription start site (TSS) proximal promoter states (TssFlank1 and TssFlank2), enhancer-related states (EnhA1 and EnhA2). The repressed polycomb (RepPC) state enriched with H3K27me3 are defined as repressive state. The remaining of genome was denoted as quiescent state with low epigenetics mark signals. Based on the genome state classification, we identified 18,610–19,961 candidate promoters, 21,081–24,128 candidate enhancers, and 16,473–18,564 candidate silencers in SOL and EDL muscles (Fig. 1e and Supplementary Data 3). The transition of promoter chromatin states from repressive or quiescent to active was accompanied with the activation of gene transcription, which was associated with the tissue-specific activation of glycolysis-related and contraction-related genes in SOL and EDL, such as BPGM and CACNA2D3 (Fig. 1f, g and Supplementary Fig. 3c).
We considered the tissue specificity of candidate enhancers, and observed that over 26% enhancers show differential activity between SOL and EDL muscles (Fig. 1e). Comparative analyses across pig, human, and mouse genomes revealed significant differences in enhancer activity (Supplementary Fig. 3d). Genomic Regions Enrichment of Annotations Tool (GREAT) enrichment analysis indicated that tissue-specific enhancers were located near genes critical for muscle contraction and metabolic processes (Fig. 1h and Supplementary Fig. 3e). For instance, EDL-specific enhancers were found near the SLN, which regulates calcium transport essential for fast-twitch muscle contraction20. Similarly, SOL-specific enhancers were identified near the UQCR10, which is involved in the mitochondrial respiratory chain and oxidative phosphorylation (Fig. 1i)21. In summary, our results demonstrated that dynamic epigenetic states of promoters and enhancers may play pivotal roles in regulating transcriptomic changes associated with muscle fiber-type development.
Characterization of the 3D chromatin organization in SOL and EDL
The distinct epigenetic landscapes of SOL and EDL muscles highlight the importance of chromatin remodeling in defining oxidative and glycolytic muscle phenotypes. To investigate the multi-scale reorganization of chromatin structure and its effect on gene expression, we employed Bridge Linker-Hi-C (BL-Hi-C) to generate genome-wide chromatin interaction maps in SOL and EDL muscles. The Hi-C datasets of biological replicates with high correlation were merged, resulting in ~2.31 billion uniquely valid pairwise contacts (Supplementary Fig. 4a, b and Supplementary Data 2). These datasets achieved an ultra-deep resolution of 5 kilobase pairs (kb), enabling a detailed analysis of 3D genome organization (Supplementary Fig. 4c, d).
We analyzed genome compartmentalization into A and B compartments at a 50 kb resolution. Regions in the A compartment were characterized by higher GC content, greater gene density, elevated gene expression, and increased levels of active histone modifications and chromatin accessibility compared to regions in the B compartment (Supplementary Fig. 4e and Supplementary Data 4). Compartment segregation was largely conserved, with only ~4.67% of the genome undergoing compartment switching between SOL and EDL (Fig. 2a). These compartment changes were associated with gene expression shifts: regions transitioning from B to A compartments showed increased expression, while A to B transitions corresponded to decreased expression (Fig. 2b). Notably, the transcription factor SIM1, crucial for energy balance and obesity resistance22, showed increased expression in EDL in conjunction with a B to A compartment switch (Fig. 2c).

a Compartment switching between SOL and EDL. b The expression changes of genes located within regions that underwent the compartment switch. A-to-B (n = 576, median = 0.163); B-to-A (n = 536, median = −0.0529); stable (n = 11581, median = 0.0071). c SIM1 gene locus underwent a B-to-A switch from SOL to EDL, with higher expression in EDL than SOL. Tracks for RNA-seq and ATAC-seq signal, and histone modification are shown. d Overlap of TAD boundaries between SOL and EDL. e Pearson correlation coefficient was calculated between the insulation scores of TADs (n = 2981) in SOL and EDL. Statistical significance was assessed using a two-sided Pearson correlation test. f The distribution of DEGs and CREs in changed TADs and cTADs. g Gene expression levels in cTADs (n = 2376) with relatively low, medium, or high D-scores. h The enriched GO terms for genes within the cTADs with higher D-scores in EDL (EDL) and cTADs with higher D-scores in SOL (SOL). Significance was tested using binomial test. i Representative cTAD with different D-scores in two tissues. Top: Hi-C contact heatmaps of the genomic region containing MYH2 gene locus. D-scores of cTAD were marked. Middle: TAD boundaries and genome browser tracks of PC1 values. Bottom: tracks for RNA-seq and ATAC-seq signal, CTCF, and histone modification. For (b and g), boxes represent the interquartile range with the median as a horizontal line; whiskers extend to the 5th and 95th percentiles. Significance was tested using a two-sided Wilcoxon test (b) and two-sided Kruskal–Wallis test (g).
We identified 2999 TADs (3018 TAD boundaries) in SOL and 3041 TADs (3060 TAD boundaries) in EDL, with only 5.82–8.16% of TAD boundaries being tissue-specific (Fig. 2d, e; Supplementary Fig. 4f and Supplementary Data 5). The accuracy of TAD boundaries was validated using directionality index (DI) and insulation score (IS) analyses (Supplementary Fig. 4g). As expected, CTCF was enriched at TAD boundaries and contributed to TAD insulation strength (Supplementary Fig. 4h, i). Genome-wide comparison of insulation scores revealed high conservation of TAD structures between SOL and EDL, with a strong correlation (R = 0.98) and similar insulation profiles (Fig. 2e).
To assess intra-TAD interactions, we identified consensus TADs (cTADs) whose boundaries are stable in SOL and EDL and accounted for 73.50% of the genome. We found that DEGs and CREs with dynamic activity were primarily located within cTADs (Fig. 2f). To quantify cTAD’s tendency to self-interact, we calculated domain score (D-score) defined by the ratio of intra-TAD contacts to all cis contacts within a 2 Mb window (Supplementary Fig. 4j and Supplementary Data 5). D-scores positively correlated with the expression levels of genes within cTADs (Fig. 2g). GO enrichment analysis of genes within cTADs revealed that dynamic intra-TAD connectivity influenced essential genes for muscle contraction and metabolism (Fig. 2h). For instance, myosin light chain-2 (MYL2), a gene critical for myosin ATPase activity and predominantly expressed in slow-twitch fibers, exhibited both higher expression and D-scores in SOL compared to EDL (Fig. 2i).
In conclusion, while compartmentalization and TAD structures were largely conserved between SOL and EDL muscles, intra-TAD interactions showed significant changes and correlated with dynamic transcriptional activity.
Global remodeling of E-P interactions underpins functional divergence in SOL and EDL
To uncover the role of distal chromatin interactions among CREs in gene regulation, we conducted loop calling to identify chromatin loops in SOL and EDL muscles. This analysis revealed 16,086 chromatin loops in SOL and 12,923 in EDL (Fig. 3a and Supplementary Data 6), with most loops occurring within TAD boundaries (Fig. 3b). Chromatin loops were categorized based on CREs at their anchors, with E-P interactions constituting the largest proportion in both muscles (Fig. 3c). Aggregate peak analysis (APA) of different loop subtypes demonstrated that loop interaction strength varied by subtype and was positively correlated with CTCF enrichment, underscoring CTCF’s critical role in loop formation (Supplementary Fig. 5a–c). Motif analysis further identified significant enrichment of MEF2, KLF, and AP-1 TF families in different loop subtypes (Supplementary Fig. 5d, e).

a APA analysis of chromatin loops in SOL (n = 16,086) and EDL (n = 12,923). Bin size: 10 kb. b Proportion of inter-TAD and intra-TAD loops. c Distribution of loop interaction types: P-S (promoter-silencer), P-P (promoter-promoter), P-E (promoter-enhancer), and P-none (promoter with no other CREs). d Overlap of genes with distinct E-P interactions (top-left). Schematic representation of E-P rewiring for Groups I–III. e The proportion of DEGs in Groups I–III. f Transcriptional changes for genes in Groups I–III. Log2 (Foldchange) represents as the change of gene expression in SOL compared to EDL. ALL (n = 15125, median = 0.0093); I (n = 27, median = −1.0220); II (n = 36, median = −1.1904); III (n = 100, median = 1.2397). Boxes represent the interquartile range with the median as a horizontal line; whiskers extend to the 5th and 95th percentiles. Significance was tested using a two-sided Wilcoxon test. g The expression and binding enrichment of Top 10 TFs enriched in I–III loops. Color scale represent original values as indicated in the color bar. h Percentage of loops with TFBS for selected TFs. i The enriched GO terms for genes in Group I–III. II_activated: GO term that more activated in Group II; III_activated: GO term that more activated in Group III; Common: GO term common in three Groups. j Chromatin interactions around TNNI1 and TNNI2 locus. Top: Virtual 4 C profile of loop contact differences. Tracks for chromatin loops, RNA-seq, ATAC-seq, CTCF, histone modifications, and TFBS are shown below. Enhancers are marked in green.
To explore the functionality of loops, we compared the expression of genes with different loop subtypes, revealing that the genes with E-P interaction exhibited higher expression than those regulated by other CREs (Supplementary Fig. 5f). Approximately 86% of enhancers interacted with promoters other than the closest one (Supplementary Fig. 5g), and multiple enhancers exerted additive effects on gene transcription (Supplementary Fig. 5h). These findings highlight E-P interactions pivotal role in transcriptional regulation. Thus, we further classified enhancer target genes into three groups (I–III) based on distinct E-P rewiring patterns (Fig. 3d). Genes with SOL-specific interactions (Group III) were predominantly upregulated in SOL (69%, 69 of 100 DEGs), while those with EDL-specific interactions (Group II) were largely upregulated in EDL (66.7%, 24 of 36 DEGs) (Fig. 3e, f). These results suggested that the formation of E-P interactions was associated with the tissue-specific gene activation in SOL and EDL. Motif analysis of these looped enhancers revealed that AP-1 TFs (e.g., FOS, ATF3, FOSL2) were enriched in SOL-specific (Group III) loops, while KLF TFs (e.g., KLF3, KLF9, KLF5) were enriched in EDL-specific (Group II) loops (Fig. 3g). ATAC-seq footprint analysis supported these findings by identifying TFBS corresponding to these TFs (Fig. 3h). We also collected public TFs ChIP-seq datasets of C2C12 myotubes, and observed the enrichment of these TFs in the conserved region of enhancer (Supplementary Fig. 5i). These findings supported the binding of TFs in identified enhancers. To determine the functional processes of E-P interaction, we applied GO enrichment analysis to Group I–III genes, and found that Group I genes were involved in muscle structure development, while Group II and III genes were more enriched in muscle-contraction- and oxidative-metabolism-related processes, respectively (Fig. 3i). An example of tissue-specific E-P interactions is seen in the troponin complex, essential for the regulation of muscle contraction23. In this study, TNNI1 (troponin I1, slow-type) and TNNI2 (troponin I2, fast-type) were associated with SOL-specific and EDL-specific E-P interactions, respectively, aligning with their expression patterns (Fig. 3j).
In summary, our findings suggest that AP-1 and KLF families may contribute to the formation of SOL- and EDL-specific E-P interactions, respectively. This global remodeling of E-P interactions may drive tissue-specific transcriptional activation, shaping the distinct contractility of oxidative and glycolytic muscle types.
Super-enhancers control muscle fiber-type specification via proximity and chromatin looping
SEs are critical in driving lineage- and development-specific gene expression. To explore this, we utilized H3K27ac CUT&Tag datasets to identify SEs. A total of 554 and 592 SEs were identified in the SOL and EDL muscles, respectively (Fig. 4a and Supplementary Data 4). The majority of SEs were located in compartment A and within cTADs (Supplementary Fig. 6a). We compared genes with promoter overlapped with SEs (SE-proximal genes) in SOL and EDL muscles, and identified 177 EDL-specific SE-proximal genes and 140 SOL-specific SE-proximal genes. To elucidate the function of SEs, we categorized SE-proximal genes based on whether their promoters overlapped with SEs in SOL and EDL muscles, revealing that SEs in gene promoters might activate tissue-specific gene transcription (Supplementary Fig. 6b). GO analysis of SE-proximal genes demonstrated that SOL-specific and EDL-specific SE-proximal genes were respectively more activated in muscle contraction and oxidative metabolism (Fig. 4b), underscoring SEs’ roles in functional divergence between SOL and EDL muscles. Among these, a SOL-specific SE located in the MYH7 promoter (designated SE-MYH7) exhibited strong H3K27ac signal and chromatin accessibility, suggesting its potential role as a key regulatory element for MYH7 activation in slow-twitch SOL muscle (Fig. 4c). TF footprint analysis of differential ATAC peaks at SE-MYH7 revealed the potential binding of TFs, included MyoG24, Hoxa1325, and Vdr26 those have been reported to bind on the MYH7 promoter or activate MYH7 expression, as well as unreported TFs, including FOS (AP-1 family). FOS was highly expressed in SOL, and predicted to bound in most SOL-specific enhancers, suggesting its crucial role in activating SE-MYH7.

a Hockey stick plots based on input-normalized H3K27ac signals in SOL and EDL, highlighting SOL-specific genes (red) and EDL-specific genes (blue). b GO term enrichment for SE-proximal genes. c Tracks of RNA-seq, ATAC-seq, CTCF, histone modifications, and TFBS at the MYH7 locus. SE regions are marked in green. d Expression heatmap of SE-anchored genes. e Epigenetic states and chromatin interactions at MYH1 and MYH4 locus. Virtual 4 C plots show chromatin contact differences; tracks for RNA-seq, ATAC-seq, CTCF, and histone modifications are included. SE regions are marked in green. f 3D chromatin conformation models around the gene locus (bin size: 10 kb). g MYH7 and MYH4 expression changes after SE deletion in differentiated PSCs (n = 3). h Immunofluorescence staining of KLF5, Fast-type MyHC, and Slow-type MyhC in the EDL muscle. (Green: Slow-MyHC; Red: Fast-MyhC; Purple: KLF5; Blue: Nuclei) (n = 3). Fast-MyHC antibody detects total fast skeletal myosin heavy chain isoforms including MYH2, MYH4, and MYH8. The percentages indicate the proportion of KLF5-positive nuclei within slow-type or fast-type fibers. i Aggregate TF footprint plots for KLF5 in SOL and EDL muscles. j Track of KLF5 ChIP-seq in C2C12 myotubes differentiated for 0 day, 2 days, and 5 days. KLF5 peak was filled in green. k qPCR showing the expression change after KLF5 overexpression in differentiated PSCs (n = 3). l ChIP-qPCR showing the enrichment change of H3K27ac in SE-MYH1/4 after KLF5 overexpression in differentiated PSCs (n = 3). The enrichment level is shown as a percentage of input. IgG was used as a negative control. m Dual-reporter assay detecting the enhancer activity of SE-MYH1/4 after KLF5 overexpression in differentiated PSCs (n = 4). n 3C-qPCR showing the interaction frequencies of SE-MYH1/4 with gene promoters after KLF5 overexpression in differentiated PSCs (n = 3). For (g, k–n), significance was tested by a two-tailed unpaired student’s t-test. All values are presented as mean ± SEM.
To clarify the functionality of SE depending on distal looping, we annotated SEs to nearest loop anchors. In the SOL and EDL muscles, we identified 1352 and 817 SE-anchored loops, respectively, corresponding to 365 and 673 SE-promoter interactions (Supplementary Fig. 6c). Among 46 SE-anchored DEGs, the fast-type myosin MYH1 and MYH4 were found to interact with an EDL-specific SE (designated SE-MYH1/4) via chromatin looping (Fig. 4d). Virtual 4 C profiling and 3D chromatin modeling further supported these interactions (Fig. 4e, f).
To assess conservation across species, we analyzed H3K27ac ChIP-seq datasets from mouse EDL and SOL17. A conserved SE near the Myh3 gene (SE-fMyh) was identified, which interacts with and activates fast-type myosin genes, consistent with previous findings18. Approximately 50% of H3K27ac peaks within pig SE-MYH1/4 were sequence-conserved with SE-fMyh, with over 60% sequence identity (Supplementary Fig. 6d). However, the clustered enhancers in the myh7 promoter of mice were not classified as SEs by the ROSE algorithm27, despite exhibiting strong H3K27ac enrichment and chromatin accessibility (Supplementary Fig. 6d). To experimentally validate the functionality of theses SEs, we transfected the pGL3-enhancers vector into differentiated porcine satellite cells (PSCs), and confirmed their enhancer activity by performing Dual-luciferase reporter assays (Supplementary Fig. 6e). We also deleted conserved H3K27ac peaks within SE-MYH1/4 and SE-MYH7 in PSCs using CRISPR-Cas9, resulting in significant reductions in the expression of MYH1, MYH4, and MYH7, respectively (Fig. 4g and Supplementary Fig. 6f, g).
To explore the coordinated regulation of TFs with SE-MYH1/4, we ranked expressed TFs based on proportion of SEs with corresponding TFBS, revealing that KLF family as likely key regulators of SEs in EDL (Supplementary Fig. 6h). The gene expression of most KLFs was expectedly higher expressed in EDL than SOL (Supplementary Fig. 6i). Thus, we focused on KLF family binding at SE-MYH1/4, which is specifically activated in EDL. We validated that KLF5 is the most differentially expressed KLF transcription factors between SOL and EDL muscles, and higher expressed in fast-type fibers than in slow-type fibers (Fig. 4h). ATAC-seq footprint analysis indicated that KLF5 displayed distinct binding patterns, with higher binding scores in EDL compared to SOL (Fig. 4i; Supplementary Fig. 6j, k and Supplementary Data 7). Furthermore, we observed a significant enrichment of KLF5 in the SE-fMyh in C2C12 myotubes differentiated for 0 day, 2 days, and 5 days (Fig. 4j). These results suggested that KLF5 might be the key TFs to regulate the activity of SE-MYH1/4 in EDL. ChIP-qPCR results validated the binding of KLF5 and SE-MYH1/4 in EDL (Supplementary Fig. 6l). Additionally, we demonstrated that KLF5 induces the expression of MYH1 and MYH4 genes by increasing enrichment of H3K27ac and enhancer activity of SE-MYH1/4, and enhances the interaction frequency of SE-MYH1/4 with gene promoter (Fig. 4k–n). These results demonstrated that KLF5 can regulate the enhancer activity and interaction structure of SE-MYH1/4 to participate the formation of fast-type fiber.
The function of super-enhancers in muscle fiber-type transformation
The muscle fibers development includes two key processes: muscle fiber specification and transformation. GO analysis showed that SE-anchored genes are significantly enriched in the related process of oxidative phosphorylation and calcium ion transport, suggesting that SEs might also play crucial role in muscle fiber-type transformation (Supplementary Fig. 7a). The muscle-fiber transformation is accompanied with the expression change of MYH4 and MYH7 genes. To identify candidate SE-anchored genes essential for this process, we calculated the expression correlation of 46 SE-anchored DEGs with MYH4 and MYH7 across eight developmental stages by analyzing transcriptomic datasets of porcine skeletal muscle28. From this analysis, 23 SE-regulated DEGs were identified as candidate genes displaying opposite correlations in expression with MYH4 and MYH7 (|R| > 0.5) (Fig. 5a).

a Top: Pearson correlation coefficients between SE-regulated DEGs and MYH7 or MYH4. Bottom: Expression heatmap of SE-regulated DEGs in postnatal pig skeletal muscle at eight developmental stages. b Heatmap of 22 SE-regulated DEGs that conserved between pig and mouse SOL and EDL muscles. c Virtual 4 C plot of chromatin interactions around the STARD7 locus. Enhancers are marked in green. d 3D chromatin models of the STARD7 locus based on Hi-C data. e 3C-qPCR analysis of STARD7 promoter interactions in SOL and EDL. Interaction frequency is calculated relative to the R1 site. f Dual-luciferase reporter assay validating enhancer activity of TE-STARD7 and SE-STARD7 in PSCs (n = 3 per group). g, h Expression changes of STARD7 after TE-STARD7 (g) and SE-STARD7 (h) deletion in PSCs (n = 3 per group). For (e–h), significance was tested by a two-tailed unpaired student’s t-test. All values are presented as mean ± SEM.
We further examined whether these candidate DEGs with conserved expression patterns in pig and mouse EDL and SOL by accessing MuscleDB (Fig. 5b)29. Among of the conserved candidate genes, STARD7 is a lipid transport protein that delivers phosphatidylcholine to mitochondria, which has reported as a key gene for myoblast differentiation in mice and humans30,31. Consistent with its higher expression in SOL compared to EDL (Supplementary Fig. 7b), STARD7 expression positively correlated with MYH7 (R > 0.57) and negatively with MYH4 (R < −0.61) during muscle development, suggesting its critical role in muscle fiber-type transformation. Validation through virtual 4 C profiling, 3D chromatin modeling, and 3C-qPCR confirmed the interaction of a typical enhancer (TE-STARD7) and a super-enhancer (SE-STARD7) with the STARD7 promoter (Fig. 5c–e). Dual-luciferase reporter assays further validated the enhancer activity of both TE-STARD7 and SE-STARD7 in differentiated PSCs (Fig. 5f). CRISPR-Cas9 deletion experiments demonstrated reduced STARD7 expression upon enhancer deletion (Fig. 5g and Supplementary Fig. 7c). These findings suggest that SE could activate STARD7 transcription to drive muscle fiber-type transformation.
STARD7 induces the transformation of glycolytic fiber to oxidative fiber
To investigate the role of STARD7 in muscle fiber transformation, we conducted gain- and loss-of-function experiments both in vitro and in vivo. In PSCs, qPCR, cell immunofluorescence staining, and WB results confirmed that STARD7 promotes the transformation of glycolytic myotubes to oxidative myotubes in differentiated PSCs. (Fig. 6a–f). We measured extracellular acidification rate (ECAR) to detect glycolysis activity using Seahorse assays. Our results validated that ECAR decreases after STARD7-overexpression, while ECAR increased after STARD7-knockdown in living differentiated PSCs (Fig. 6g, h). Consistently, expression analysis revealed that STARD7 upregulates oxidative metabolism-related and slow-twitch troponin genes, while downregulating glycolysis-associated and fast-twitch troponin genes (Fig. 6i–j). These results demonstrated that STARD7 could induce the glycolytic to oxidative metabolism. For in vivo validation, LV3-shSTARD7 and LV3-shNC lentiviral particles were injected into the right and left gastrocnemius (GAS) muscles of mice, respectively, for two months (Fig. 6k). After 5 weeks of injection, Stard7-knockdown led to decreased MYH7 and increased MYH4 expression in mouse GAS muscles, with a concomitant metabolic shift toward glycolytic metabolism (Fig. 6l–n and Supplementary Fig. 8a). Immunofluorescence staining of GAS muscles further confirmed an increase in fast-type fibers and a decrease in slow-type fibers following Stard7-knockdown (Fig. 6o). Strad7 knockdown results in increased muscle weight and altered meat color characteristics, including higher L (lightness) and a (redness) values (Fig. 6p–r). Interestingly, overexpression of pig STARD7 gene in mouse also promoted a fiber-type switch from fast- to slow-type fibers and induced oxidative metabolic reprogramming (Supplementary Fig. 9a–f), suggesting the conserved role of STARD7 across species in regulating muscle fiber identity. Collectively, our findings demonstrate that STARD7 promotes glycolytic-to-oxidative muscle fiber transformation, reduces muscle weight, and improves muscle color by enhancing both redness and lightness.

a, b Immunofluorescence staining of fast-type MyHC and slow-type MyHC following STARD7 knockdown (a) and overexpression (b) in differentiated PSCs (n = 3). left panel: Representative images of immunofluorescence staining. Right panel: Statistical analysis of MYH7+ and MYH4+ fibers. Magnification, × 10, Bar = 200 µm. c, d qPCR and WB depicting the gene expression change between STARD7 knockdown and control group in differentiated PSCs (n = 3). e, f qPCR and WB showing the gene expression change between STARD7 overexpression and expression vector (EV) group in differentiated PSCs (n = 3). g, h The change of ECAR after STARD7-knockdown (g) and overexpression (h) in differentiated PSCs. Values represent the average of three independent experiments with four technical replicates for each assay. i, j The expression changes of maker genes (oxidative: COX5B, COX6B, CS, NDUFB4, PGC-1α, UQCR10; glycolytic: GPI, HK2, PKM, LDHA, LDHB; slow-type troponin: TNNC1, TNNI1, and TNNT1; fast-type troponin: TNNC2, TNNI2, and TNNT2) after STARD7 knockdown (i) and overexpression (j) in differentiated PSCs. k Representative images of GAS muscles from the left (shNC) or right (shSTARD7) hindlimbs. l, m qPCR and WB depicting of gene expression in GAS muscles from LV3-shSTARD7 and LV3-shNC groups (n = 3). n Expression changes of maker genes in GAS muscles from LV3-shSTARD7 and LV3-shNC group (n = 3 per group). o Immunofluorescence staining of fast-type myosin and slow-type myosin of GAS muscles from LV3-shSTARD7 and LV3-shNC group (n = 3 per group). Left: Representative images of immunofluorescence staining. Right: statistical analysis of the proportion changes of slow-type fibers (green) and fast-type fibers (red). Magnification, ×20, Bar = 50 µm. p–r The comparison of GAS muscle (n = 3 per group) in terms of weight, cross-sectional area, and meat color. L: lightness value; a*: redness value; b*: yellowness value. For (a–j), significance was tested by a two-tailed unpaired student’s t-test. two-sided unpaired t-test. For (l–r), significance was tested by a two-tailed paired student’s t-test. All values are presented as mean ± SEM.
STARD7 reduces intracellular ROS levels and ERK MAPK activity
To investigate the regulatory mechanism of STARD7 in muscle fiber-type transformation, we first examine its relationship with KLF5. qPCR analysis showed that overexpression of STARD7 in PSCs did not significantly alter the expression of most KLFs family members (Supplementary Fig. 10a), and conversely, overexpression of KLF5 has not effect on STARD7 expression in PSCs (Supplementary Fig. 10b). These results indicated that STARD7 is not involved in the KLFs-mediated regulatory networks.
We then performed RNA-seq analysis on STARD7-overexpressing (OV-STARD7) cells and control cells (Supplementary Data 8). The overexpression efficiency of STARD7 was validated, and its regulatory effects were confirmed by increased expression of MYH7 (a slow-twitch myosin isoform) and decreased expression of MYH4 (a fast-twitch myosin isoform) (Fig. 7a and Supplementary Fig. 10c). PCA analysis demonstrated high reproducibility across three biological replicates (Supplementary Fig. 10d). GO analysis of DEGs revealed significant enrichment of biological processes associated with muscle fiber-type transformation, such as muscle contraction (Fig. 7b and Supplementary Data 8). GSEA results revealed that pathways related to reactive oxygen species (ROS) metabolism and ERK-MAPK signaling were downregulated in OV-STARD7 cells (Fig. 7c). Consistently, RNA-seq analysis of pig STARD7-overexpressing GAS muscle also revealed a potential regulatory relationship between STARD7, ROS metabolism, and ERK-MAPK signaling (Supplementary Fig. 10e, f and Supplementary Data 8).

a RNA-seq tracks for STARD7, MYH4, and MYH7 loci in OV-STARD7 and empty vector (EV) PSC cells. b GO enrichment analysis of DEGs between OV-STARD7 and EV cells. c GSEA analysis of OV-STARD7 and EV cells. d Quantification of intracellular ROS levels in OV-STARD7 and EV cells (n = 3 per group). Excitation wavelength: 488 nm, emission wavelength: 525 nm. e Levels of phosphorylated ERK (p-ERK) and total ERK in OV-STARD7 and control cells (n = 3 per group). f, g qPCR and WB results showing the impact of exogenous H₂O₂ on STARD7-mediated muscle fiber-type transformation. h Schematic model of STARD7 regulation in muscle fiber-type transformation. For (d–g), significance was tested by a two-tailed unpaired student’s t-test. All values are presented as mean ± SEM.
Previous studies have shown that STARD7 reduces ROS levels in HepG2 cells32. Decreased ROS levels suppress MAPK signaling activity in various cell types33, and MAPK activation promotes slow-to-fast type fiber transformation in mouse C2C12 myoblasts34. In this study, we validated the preferential accumulation of p-ERK-MAPK in EDL compared to SOL (Supplementary Fig. 10g). We also evidenced that ROS level is higher in the EDL than SOL by using EMSA assay (Supplementary Fig. 10h). These results suggested that the reduction of ROS level and p-ERK activity might promote the formation of slow-fiber. To identify the association of ROS level and ERK-MAPK signaling with STARD7, we validated a significant reduction in intracellular ROS levels and ERK-MAPK activity in OV-STARD7 cells compared to controls (Fig. 7d, e). Furthermore, treatment with H₂O₂, which increased ROS levels, disrupted STARD7’s ability to mediate muscle fiber-type transformation, confirming the critical role of ROS in this process (Fig. 7f, g)35. Collectively, our study revealed that STARD7, activated by SE, could induce the transformation of glycolytic fibers into oxidative fibers by mitigating reactive oxygen species levels and suppressing ERK MAPK signaling (Fig. 7h).
Discussion
Although transcriptional mechanisms underlying muscle fiber-type specification and transformation have been extensively studied, the role of 3D genome architecture reorganization and chromatin state alterations in these processes remains insufficiently understood. In this study, we integrated multi-omics data to uncover how multiscale 3D genome architecture remodeling contributes to transcriptional reprogramming in distinct muscle types. On a large scale, we observed moderate compartment switches and subtle transcriptional changes. While most TAD boundaries remained stable, a subset exhibited dynamic interaction frequencies between muscle types, with reduced intra-TAD interactions correlating with transcriptional inactivation. This highlights the need for further exploration of chromatin loop rewiring within TADs.
By mapping chromatin loops, we precisely linked enhancers to their target genes. We found that Enhancer-promoter (E-P) interactions play a pivotal role in activating genes associated with muscle contraction and oxidative metabolism. For instance, the troponin (Tn) complex, comprising TNNI1, TNNC1, TNNI2, and TNNT1, controls Ca²⁺-dependent muscle contraction and adapts to exercise or disuse36. We identified spatiotemporal remodeling of E-P interactions governing Tn complex transcription, underscoring the role of enhancers and chromatin looping in establishing distinct contractile properties across muscles. While TF cooperativity at enhancers is recognized as critical for developmental gene activation, its role in muscle-specific transcription remains poorly understood. Previous studies revealed that AP-1 TFs partner with PGC-1α to regulate hypoxic gene programs in muscle cells37. PGC-1α is a key regulator of metabolism, and can increase oxidative capacity and trigger a shift of fast-type to slow-type fibers38. KLF15 cooperates with transcriptionally factor MRTFB to promote contractile gene expression in vascular smooth muscle cells39. Our findings indicate that the AP-1 and KLF families may shape SOL- and EDL-specific E-P interactions, respectively. Collectively, our study provides a valuable resource for elucidating the relationship between transcriptional reprogramming and 3D genome remodeling, particularly the role of TFs and E-P interactions in tissue-specific gene activation essential for muscle contraction and glucose metabolism.
In this study, we demonstrated for the first time that STARD7 can drive the transformation of fast-type fibers to slow-type fibers both in vivo and in vitro. Using CRISPR/Cas9 genome editing, we showed that in situ deletion of SE-STARD7 effectively decreases the expression of the STARD7 gene. Previous studies have established individual relationships between STARD7 and intracellular ROS levels32, ROS level and MAPK signaling33, and MAPK signaling and muscle fiber-type transformation35. However, the coordinated regulation of these factors in muscle development had not been reported. Our findings validate that STARD7 promotes fast-to-slow fiber transformation by reducing ROS levels and inhibiting ERK MAPK signaling activity. In summary, we identified and characterized a previously unrecognized SE that activates STARD7 expression, elucidating its function and regulatory mechanism in muscle fiber-type transformation.
A key finding of this study is the elucidation of regulatory mechanisms specifying muscle fiber types at the 3D genome level. In adult muscles, the contraction and metabolic properties of specialized myofibers are governed by the expression of slow-type myosin genes (MYH7) and fast-type myosin genes (MYH1, MYH2, MYH4)40. In mice, fast-type Myh genes (fMyh) are regulated by SE-fMyh through competitive chromatin interactions18,24. We firstly identified SE-MYH1/4 as an EDL-specific SE with stronger H3K27ac enrichment and chromatin accessibility in EDL than in SOL. While SE-fMyh functions in both mouse SOL and EDL, SE-MYH1/4 shows EDL-specific activity and drives fast-type fiber specification through chromatin interactions. This highlights the species-specific epigenetic states of SEs controlling fast-type myosin expression.
SE activity depends on TF cooperation and changes in TF enrichment41. Prior studies demonstrated that Myog and Myod bind to the promoter of Myh7, enhancing its transcriptional activity in mice24. Consistent with this, our result revealed that the TF binding sites for Myog and Myod are detected in SE-MYH7 in pig SOL. Notably, we validated that KLF5 can bind at SE-MYH1/4 to promote its enhancers activity and frequency of interaction with the fast-type Myh genes. Additionally, not common with the other KLFs, our results suggested that KLF2 expressed higher in SOL than EDL, and might be activated by a SOL-specific SE. This suggested that KLF2 might be involved in the formation of slow-type fibers. Collectively, these findings establish SE-mediated chromatin interactions and coordinated TF regulation as key determinants of muscle fiber phenotype.
Adult muscle fibers, despite their specified types, can undergo transformation in response to functional demands. These transformations are accompanied by various physiological and biochemical adaptations in skeletal muscle, including mitochondrial biogenesis, angiogenesis, ATP metabolism, and muscle contraction42. Such adaptations are tightly regulated by key genes and gene families associated with muscle development, particularly the well-known MRFs family, Pax family, and the MSTN gene. Additionally, signaling pathways such as the FoxO signaling pathway, PGC-1α pathway, Wingless/Integrated (Wnt), and Sonic Hedgehog (Shh) signaling pathways play critical roles in facilitating muscle fiber-type transformations43.
Muscle is a heterogeneous tissue, with over 68% myonuclei originating from myofibers44. The epigenetics status of CREs and 3D chromatin structure in purified-myonuclei is crucial for investigate muscle fiber-type development, but remains poorly understand due to small sample sizes and complex isolation procedures. Previous studies reported that the trend of chromatin accessibility differences of enhancers between purified myonuclei of slow-type and fast-type muscles is coordinated with the H3K27ac signal between bulk tissues18. In this study, we also revealed a high similarity of H3K27ac signal of myonuclei and bulk tissue of SOL and EDL muscles. These results suggest that it would be feasible to use bulk tissue to reflect the information of epigenetics and 3D chromatin organization of purified-myonuclei. Thus, our multi-omics datasets can provide valuable resources to study the coordination regulation of CREs and distal chromatin looping to shape distinct muscle fibers.
While our mechanistic validation was primarily conducted in porcine-derived cell models, in vivo confirmation of the STARD7–ROS-MARK regulatory axis remains to be established. Nevertheless, our findings from both pig and mouse models support the conserved role of STARD7 in muscle fiber-type transformation. Future studies will include in vivo experiments in mice to further validate the regulatory mechanism of STARD7 on oxidative metabolism reprogramming and muscle development.
In conclusion, our study provides critical insights into the principles of 3D genome organization as they relate to epigenetic regulation of gene expression in distinct muscle types. We demonstrated that chromatin rewiring at multiple genomic scales and the epigenetic states of CREs underlie transcriptome reprogramming, ultimately dictating the contractile and metabolic properties of specialized muscles. Furthermore, we uncovered the pivotal role of SEs in muscle fiber-type specification and validated KLF5 as a key transcription factor driving the specification of myotubes into fast-type fibers. Lastly, we established that STARD7 is activated by a distal SE, enabling its role in inducing the transformation of fast-type fibers into slow-type fibers through ROS reduction and ERK MAPK signaling inhibition.
Methods
Ethics statement
Animal maintenance and experimental procedures were conducted according to Regulations for the Administration of Affairs Concerning Experimental Animals (Ministry of Science and Technology, China, revised in March 2017) and approved by the animal ethical and welfare committee of South China Agricultural University (Permit Number 2021F036, Permit Date 2 March 2021).
Animals and sample collection
Three 6-month-old female Duroc pigs were obtained from the breeding pig farm of Guangdong Wen’s Foodstuffs Group Co., Ltd. (Yunfu, China). All mouse experiments were performed using male C57BL/6J mice (6 weeks old), purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Mice used in the lentivirus infection were maintained in SPF animal room, with five mice per cage, on a 12/12-h light-dark cycle condition at a controlled ambient temperature of 22 ± 2 °C and relative humidity of 55 ± 10%. Food and water were provided ad libitum. Muscle samples from different anatomical sites were rapidly manually separated from each carcass, immediately flash-frozen in liquid nitrogen, and stored at −80 °C or fixed in 4% paraformaldehyde for histology staining.
RNA-seq library construction and sequencing
The total RNA of tissues or cells was extracted using TRIzol reagent (Invitrogen, 15596026). RNA integrity was qualified by Bioanalyzer 2100 (Agilent, USA) and confirmed by agarose gel electrophoresis. Extracted RNA was used to construct strand-specific libraries with NEBNext® Ultra™ Directional RNA Library Prep Kit (NEB, USA). The libraries were sequenced on the Illumina Novaseq™ 6000 (PE150) platform.
ChIP-seq library construction and sequencing
Approximately 25 mg of muscle tissues were minced with a scalpel, washed in cold PBS buffer, cross-linked by formaldehyde, and quenched with Glycine Solution. Afterward, the samples were lysed and sonicated to obtain soluble sheared chromatin. Half of the soluble chromatin was collected as input control at –20 °C for DNA sequencing, and the remaining was used for immunoprecipitation reacting with anti-H3K4me3 (Millipore, 07-473) and anti-H3K27ac (Millipore, 07-360). Chromatin was de-cross-linked, purified, and used to generate libraries. For both input DNA and immunoprecipitated DNA, each library was sequenced on the Illumina HiSeq X Ten (PE150) platform by Newblue Technology Co., Ltd., Guangzhou, China.
CUT&Tag library construction and sequencing
The muscle tissue was placed into a pre-chilled Dounce homogenizer containing cold lysis buffer. The tissue was homogenized using the A pestle until no resistance was felt, indicating thorough disruption of the tissue. The homogenate was then pre-cleared by filtration through a 100 μm nylon mesh filter to remove debris. Subsequently, the filtered homogenate was further homogenized using the B pestle for 20 strokes to ensure complete lysis. The supernatant was carefully transferred to a low-binding tube. Nuclei were pelleted by density gradient centrifugation at 3000 × g for 30 min in a solution composed of 60% Percoll and 2.5 M sucrose. The collected nuclei were incubated with concanavalin A-coated magnetic beads. Cell-bound beads were resuspended and then immunoprecipitated with anti-H3K27me3 (Millipore, 07-449) and anti-CTCF (Millipore, 07-729) with rotation at 4 °C overnight. The enzyme pA-Tn5 Transposase precisely binds the DNA sequence near the target protein under the antibody guidance. The DNA sequence is tagmented, with adapters added at the same time at both ends, which can be enriched by PCR to form the sequencing-ready libraries. After the PCR reaction, libraries were purified with the AMPure beads (Beckman, A63881), and library quality was assessed on the Agilent Bioanalyzer 2100 system. The constructed library was sequenced on the Illumina Novaseq™ 6000 (PE150) platform by Seqhealthy Technology Co., Ltd., Wuhan, China.
ATAC library construction and sequencing
Approximately 5 mg of tissue was cut into small pieces and crushed into a fine powder in liquid nitrogen. The pulverized tissues were resuspended in lysis buffer (50 mM HEPES with pH 7.5 (Life Technologies, 15630-080), 140 mM NaCl (Ambion, AM9760G), 1 mM EDTA with pH 8.0 (Ambion, AM9260G), 10% glycerol (Sigma, G7757), 0.5% NP-40 (Roche, 11754599001), and 0.25% Triton X-100 (Amresco, 0694), followed by isolation of 50,000 nuclei using a previously published protocol with minor modifications45. Then, the isolated nuclei were incubated in the transposition reaction mix (12.5 μL TD buffer, 10 μL ddH2O, and 2.5 μL TDE (Illumina, FC-121-1030) at 37 °C for 1 h, and then purified with a QIAGEN MinElute PCR Purification Kit (QIAGEN, 28006). Next, an average of 11 cycles of PCR was performed with transposed DNA and amplified libraries were run on an Agilent TapeStation 2200 (Agilent Technologies, USA) using a D5000 DNA ScreenTape to assess their quality via visualizing nucleosomal laddering. The final libraries were quantified and sequenced on the Illumina HiSeq X Ten (PE150) platform.
BL-Hi-C library construction and sequencing
Approximately 5 × 106 isolated cells were resuspended in 1 ml of 1% SDS lysis buffer (50 mM HEPES-KOH, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% SDS) at room temperature for 15 min and centrifuged at 500 × g for 2 min. The pellet was washed with 1 ml 0.1% SDS lysis buffer and centrifuged at 500 × g for 2 min. The supernatant was discarded, and the cells were incubated with 50 µl of 0.5% SDS for 30 min at 37 °C. The SDS was quenched by the addition of 145 µl H2O and 12.5 µl 20% Triton X-100, and the genome was digested overnight at 37 °C with 100U AluI (NEB, R0137V) into blunt-end fragments. Cleaved chromatin was A-tailed by adding 10 mM dATP solution (NEB, N0440S) and Klenow Fragment (3′ → 5′ exo-) (NEB, M0212L) with rotation at 37 °C for 40 min. Then, chromatin was treated with adenine and ligated with a biotinylated bridge linker (F: CGCGATATC/bio-T/TATCTGACT; R: GTCAGATAAGATATCGCGT) at 16 °C for 4 h. After ligation, the nuclei with in situ proximity ligation were treated with l-exonuclease (NEB, M0262S) and exonuclease I (NEB, M0293V) for 1 h at 37 °C to remove noise DNA contain no ligation DNA or bridge linker. The DNA was obtained by proteinase K digestion at 55 °C overnight and purified by phenol-chloroform extraction with ethanol precipitation. Then, the DNA was fragmented into 300 bp on average by sonication, and the biotin-labeled DNA fragments were pulled down with streptavidin-coated M280 beads. The DNA library was sequenced using the BGISEQ DNBSEQ-T7 (PE150) platform by Yingzi Technology Co., Ltd., Wuhan, China.
RNA-seq data analysis
Clean reads were aligned to the pig reference genome with chain-specific parameters: “rna-strandness RF” by Hisat246. The gene counts were calculated by featurecounts47 and then normalized to transcripts per kilobase million (TPM). Only genes with TPM ≥ 0.5 in at least one sample were considered as expressed genes and used for further analysis. Differentially expressed genes were identified with FDR < 0.05 and fold change > 1 using DESeq248. GO analysis was assessed using PANTHER, and GSEA was implemented by the R package Clusterprofiler49,50. KEGG pathway analyses were carried out in KOBAS-I51.
ChIP-seq and CUT&Tag data analysis
Clean reads were mapped to the pig reference genome (susScr11) or mouse reference genome (mm39) using BWA52. Picard was used to obtain unique mapped reads without PCR duplicates (http://broadinstitute.github.io/picard/). For the H3K27me3 dataset, peaks were called using MACS253 with parameters (-g 2.8e + 9 -q 0.05 –keep-dup all –nomodel –broad). For H3K4me3, H3K27ac, and CTCF datasets, peaks were called using MACS2 with parameters (-g 2.8e9 -p 0.00001 –shift 0 –extsize 150 –nomodel -B –SPMR –keep-dup all –call-summits). Peaks were further processed by following four steps as previous study54: (i) narrow peaks with P < 10–5 were remained; (ii) read coverage of 2 kb region centered at the midpoint of peaks was calculated by Deeptools55 and normalized with read depth (IPRPM = IP peak region total reads∕IP total mapped reads per million; INPUTRPM = INPUT peak region total reads∕INPUT total mapped reads per million); (iii) the enriched regions for H3K4me3 and H3K27ac were defined with more than a two-fold change of normalized read coverage (IPRPM > 2 × INPUTRPM) and with normalized read coverage change (IPRPM-INPUTRPM) > 1; (iv) the overlap of enriched regions were merged, and then the 2 kb regions centered at the midpoint of merged regions were acquired to identify enhancers and promoters. Motif analysis was performed using HOMER’s findMotifsGenomDe.pl program with parameters (-len 8,10,12 -gc)56.
ATAC-seq data analysis
ATAC-seq datasets were processed with reference to the ENCODE ATAC-seq pipeline (https://github.com/kundajelab/atac_dnase_pipelines). After checking and trimming the adapter with cutadapt57, the clean reads were mapped to the pig genome (susScr11) using Bowtie2 with parameters (-very-sensitive -X 2000)58. The reads with low MAPQ reads ( <25), reads mapped to mitochondrial chromosome, and PCR duplicated reads were removed using samtools59 and Picard. The peak for each sample was called individually with MACS2 (-g 2.8e + 9, -p 0.01, –nomodel, –shift -75, –extsize 150, -B, –SPMR, –keep-dup all, –call-summits). The significant peaks (P < 10−5) of each sample were used for further analysis. Then, the filtered peaks of replicates were merged with BEDtools (at least 50% overlap between two replicates).
Chromatin state analysis and characterization of CREs
ChromHMM (version 1.22) was used to learn and characterize chromatin states27. Candidate enhancers were defined as H3K27ac peaks not overlapping with H3K4me3 peaks or TSS regions (3.0 kb upstream to 1.5 kb downstream of TSS) overlapped with S3 or S4 regions. Candidate promoters were defined as H3K4me3 peaks overlapped with S5 or S6 regions. Candidate silencers were defined as H3K27me3 peaks overlapped with S1 regions. SEs were identified using the ROSE algorithm with default parameters, and all enhancers were used as input constituent enhancers. The CREs with no overlap in other tissues were defined as tissue-specific CREs. Candidate CREs (promoters and enhancers) were converted from pig genomic (susScr11) to human/mouse genomic locations (hg19/mm9) by using the LiftOver tool with the parameter “minMatch = 0.5”. The CRE that could be converted to human/mouse genomic locations was considered as sequence-conserved CREs. Furthermore, the CREs were considered usage-conserved if the corresponding homologous sequence was annotated as human/mouse CREs in the FANTOM5 database60.
Footprint analysis
The vertebrates TF motifs were downloaded from JASPAR (2024)61. The binding score of each TFs and differential TF binding analysis were performed by TOBIAS with default parameters62. Only TFs with TPM ≥ 0.5 in at least one sample were considered as expressed TFs and used for further analysis.
BL-Hi-C data analysis
The adapter and linker sequence of raw Hi-C reads was trimmed using cutadapt57. These processed clean data were used to generate contact matrices using the HiC-Pro pipeline63. Chromatin A/B Compartment was identified using cooltools at 50 kb resolution64. TAD and TAD boundaries were identified using HICExplorer at 50 kb resolution65. The definition of cTADs and their intra-TAD interaction strength (D-score) was calculated as reported in the previous study66. Loop calling was performed by HICCUPS in 5, 10, and 25 kb resolution67. The aggregate enrichment of TADs and loops was generated by coolpup68. Genome-wide interaction and heatmap were visualized using the WashU EpiGenome browser69. Genomeflow is set 3DMax model to construct 3D chromatin conformation model with sparse contact matrix as input at 10 kb resolution70. The contact frequency of loops was calculated by hicPlotViewpoint function in HICExplorer. The interactions were then normalized by the number of interactions in the whole chromosome, and used to generate virtual 4 C profiles.
Isolation of PSCs
For PSCs isolation, hindlimb muscles of newborn piglets within one week were cut into pieces and digested with 2 mg/ml type II collagenase (Sigma, 9001-12-1) at 37 °C for 2.5 h. The tissue suspension was then filtered through 100-, 200-, and 400-mesh sieves and washed in DPBS (Gibco, 14190144) with 1% penicillin–streptomycin (Gibco, 15140122). Single cells were collected by centrifugation at 1800 × g. Subsequently, cells were differentially adhered for 2 times to obtain purified PSCs.
Cell culture
For proliferation, PSCs were cultured in RPMI-1640 medium (Gibco, 11875119) with 20% FBS (Gibco, 10099141), 1% non-essential amino acids (Gibco, 11140050), 0.5% chicken embryo extract (GEMINI, 0928501), 1% GlutaMax (Gibco, 35050061), 1% antibiotic–antimycotic (Gibco, 15140122), and 2.5 ng/ml bFGF (Gibco, 13256029) under moist air with 5% CO2 at 37 °C. For differentiation, the cells that reached 80–90% confluence were switched to differentiation in RPMI 1640 medium containing 2% horse serum (HyClone, SH30074.03).
Cell transfection
All small interfering RNA (siRNA) sequences used in this study are presented in Supplementary Data 9. Lipofectamine 3000 (Invitrogen, L3000150) and jetPRIME reagent (Polyplus transfection, 114-15) were respectively used for the transfection of plasmids and siRNA according to the instructions.
Quantitative real-time PCR (qPCR)
Total RNA was reverse-transcribed to create cDNA by Evo M-MLV RT Kit (Accurate Biotechnology, AG11711) following the manufacturer’s manual. The qPCR reaction was performed on Quant Studio 7 Flex Real-Time PCR System (Thermo Fisher Scientific, USA). The relative expression level of one gene was determined by the 2–ΔΔCt method. All primers used in this study were presented in Supplementary Data 9.
Chromatin Immunoprecipitation qPCR (ChIP-qPCR)
Chromatin immunoprecipitation experiment was carried out by using ChIP kit (Beyotime, P2078) according to the recommended protocol. Briefly, 4 × 106 PSCs were cultured to 90% confluence and fixed for 10 min at room template with 1% formaldehyde. The crosslinking was stopped by adding 125 mM glycine. Cell lysates were sheared by sonication in 1% SDS lysis buffer to generate chromatin fragments, followed in sequence with immunoprecipitation, reversal of cross-links, and DNA purification. Two micrograms of anti-KLF5 (Active Motif, 61099) and H3K27ac anti-H3K27ac (Millipore, 07-360) or negative control IgG was used to each immunoprecipitated reaction. Fold enrichment was quantified using RT-qPCR. All primers used in this study were presented in Supplementary Data 9.
Western blotting (WB)
Tissue or cells were lysed in RIPA buffer (Beyotime Biotechnology, P0013C) with 1% (v/v) PMSF (Beyotime Biotechnology, ST505). Denatured protein samples were separated by SDS-PAGE and transferred to the 0.45 μm PVDF membranes (Millipore, IPVH00010). After blocking with 5% non-fat milk, the proteins in membranes were subjected to immunoblotting analysis with primary and secondary antibodies. The membranes were developed with an ECL Kit (Millipore, WBULS0500) for visualization. The antibodies used in this study included anti-MyhC IIb (ProteinTech, 20140-1-AP), anti-MyhC I (ProteinTech, 22280-1-AP), anti-STARD7 (ProteinTech, 15689-1-AP), anti-ERK1/2 MAPK (ProteinTech, 11257-1-AP), anit-phosphop-ERK1/2 MAPK (Proteintech, 28733-1-AP), anti-β-tubulin (Servicebio, GB11017), anti-GAPDH (Absin, abs132004), and secondary antibodies goat anti-rabbit IgG (Beyotime, A0208). All protein levels were normalized to corresponding β-tubulin or GAPDH, and densitometric quantification of the bands was performed using ImageJ software.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde and permeabilized using 0.5% Triton X-100 for 30 min. After three washes, they were blocked in QuickBlock™ Blocking Buffer (Beyotime, P0220) for 2 h, followed by incubation with the primary antibody overnight and the secondary antibody for 1 h. Nuclei were counterstained with DAPI. Multiplex immunofluorescence staining based on tyramide signal amplification was performed by using four-color multiple fluorescent immunohistochemical staining kit (Absin, abs50012) according to the manufacturer’s protocols. Immunofluorescence staining antibodies included anti-Fast Myosin Skeletal Heavy chain (Serviebio, GB112130-100), anti-Slow Skeletal Myosin Heavy chain (Serviebio, GB112131-100), anti-KLF5 (Active motif, 61099), and secondary antibodies biotinylated goat anti-mouse IgG (BIOSS, bs-0296G-Bio), and Alexa Fluor 488-labeled goat anti-rabbit IgG (H + L) (Beyotime, A0423). Images of at least three random fields of view were captured using a Nikon ECLIPSE Ti microscope (Nikon, Tokyo, Japan) and quantified using ImageJ. Succinate dehydrogenase (SDH) histochemistry was applied to assess mitochondrial oxidative enzyme activity in muscle fibers. Briefly, cryosections were air-dried at room temperature for 30 min, followed by incubation in a reaction buffer containing 1.5 mM nitroblue tetrazolium, 5 mM EDTA, 48 mM succinic acid, 0.75 mM sodium azide, and 30 mM methylphenylmethyl sulfate in phosphate buffer (pH 7.6) at 37 °C for 45 min. After incubation, sections were rinsed in deionized water for 5 min, briefly dehydrated in 50% ethanol for 2 min, and mounted using Eukitt for microscopic analysis.
CRISPR-cas9 experiment
The CRISPR/Cas9 plasmid pX330-U6-Chimeric_BB-CBh-hSpCas9 (pX330) was obtained from Addgene (Plasmid #42230). The sgRNAs against the selected enhancers were listed in Supplementary Data 9. The sgRNAs were designed, synthesized, annealed, and cloned into px330 plasmid to form the recombinant plasmids. After the confluence of PSCs reached 80–90%, we transfected the recombinant pX330-sgRNA plasmids, together with pcDNA3.1(+) plasmids (containing neomycin resistance) to cells by lipofectamine 3000 (Invitrogen, L3000150). After 36 h, G418 (100 μg/mL; Sigma, G8168) selection of the cells was performed for approximately 2 weeks. Then, the mixed population of cells was collected to extract RNA and DNA. The genomic DNA of collected cells was isolated according to the protocol of the MicroElute Genomic DNA Kit (OMEGA, D3096) for PCR. PCR was performed using the specific primers (Supplementary Data 9). The deletion efficiency was analyzed by 1% agarose gel electrophoresis and sanger sequencing of PCR product.
Validation of loops by 3C-qPCR
The isolated muscle cells from muscles tissues or PSCs were cross-linked in 1% formaldehyde for 10 min at room temperature and then quenched by adding glycine (final to 0.125 M). The samples were recovered by centrifugation and washed with 1 mL DPBS (Gibco, 14190144). Each library was cleaved with 0.2% NP40 (Sigma, 74385), transferred to the enzyme digestion reaction system, and incubated with 100U Mbol restriction enzyme (NEB, R0147S) at 37 °C for 2 h. Centrifuge at 3000 × g at 4 °C for 10 min and discard the supernatant. For ligation, T4 DNA ligase (NEB, M0202M) was treated for 5 h at 16 °C and for 2 h at room temperature. Ligated DNA was purified by phenol/chloroform/isoamyl-alcohol (25:24:1) extraction and ethanol precipitation. qPCR was performed to calculate the relative interaction frequencies. The 3C-qPCR primers were designed using unidirectional strategy (Supplementary Data 9).
Detection of intracellular ROS level
The ROS levels after overexpression of STARD7 in PSCs were detected using ROS assay kits (Beyotime, S0033M). Briefly, the PSCs were harvested and added to DCFH-DA solution diluted with serum-free medium. The cell suspension was incubated with the DCFH-DA for 30 min with shaking every 5 min in the dark. At the end of the incubation period, the fluorescence intensity was detected by using BioTek’s Synergy™ HT Reader.
Cell metabolism assays
Seanorse XF Glycolytic Rate Assay Kit (Seahorse, 103344-100) was used to detect the extracellular acidification rate (ECAR) reflected the glycolysis capacity of PSCs. Briefly, cells (4 × 104 per well) were seeded and cultured in Seahorse XFe24 cell plate (Seahorse, 102340-100) before conducting the assay. After transfection, the media was changed to Seahorse XF DMEM Medium (pH 7.4, Seahorse, 103575-100) supplemented with 10 mM glucose (Seahorse, 103577-100), 1 mM pyruvate (Seahorse, 103578-100) and 2 mM glutamine (Seahorse, 103579-100). After cultured at 37 °C in a CO2-free incubator for 60 min, the real-time ECAR of cells was first recorded under basal condition as data on basic glycolysis. Through the sequential addition of 0.5 μM rotenone/antimycin A (Rot/AA) followed by 50 mM of 2-depxy-d-glucose (2-DG), the ECAR was recorded and compensatory glycolysis were calculated by using a Seahorse XFe24 Extracellular Analyzer according to the manufacturer’s protocols.
Dual-Luciferase reporter assay
The pig genomic DNA was used as a template to amplify enhancers. The amplified products were separated on a 1.5% agarose gel, purified, and then cloned into the modified pGL3-Promoter plasmids. Constructed plasmids were transfected to PSCs using Lipofectamine 3000, and incubated for 24 h with three technical replicates for each construct. Luciferase activity was measured by Dual-Luciferase Assay (YEASEN, 11402ES60) according to the manufacturer’s manual, and Renilla luciferase activity was normalized against Firefly luciferase activity.
Knockdown of STARD7 in vivo by lentivirus infection
Six-week C57 male mice were injected with 100 μL final volumes of lentivirus containing small interfere STARD7 (LV3-shSTARD7) or control (LV3-shNC) at 2 × 107 TU/ml into the right and left GAS muscles of the hind legs, respectively. LV3-shSTARD7 and LV3-shNC were synthesized by GenePharma (China, Shanghai). The mice were injected weekly and killed after 8-weeks injection. GAS muscles were collected once a week. The meat color, including L*(lightness), a*(redness), and b* (yellowness), was measured by CR-400 Chroma meter (Konica Minolta Sensing Ins., Japan) at 45 min postmortem as a mean of three random readings calibrated against a standard white plate (8-mm diameter aperture, d/0 illuminant).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw ChIP-seq, ATAC-seq, and CUT&Tag data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under the accession number PRJNA1036842. The processed Hi-C data are available in Zenodo (accession: 10.5281/zenodo.15381487). The RNA-seq data from STARD7 overexpression experiment are deposited in the SRA under accession number PRJNA1112571. Publicly available datasets used in this study include: Mouse SOL and EDL muscle ChIP-seq and ATAC data (GSE123879), H3K27ac ChIP-seq datasets of purified-myonuclei and bulk tissues (GSE123879 and GSE182667), C2C12 myotubes-related ChIP-seq and ATAC-seq datasets (PRJNA305993, PRJNA844370, PRJNA320053, PRJNA260504, PRJNA185319, PRJNA63475, PRJNA268849, PRJNA734569, and PRJNA1081841). The data supporting the findings of this study are available from the corresponding authors upon request. Source data for the figures and Supplementary Figs. are provided as a Source Data file. Source data are provided with this paper.
Code availability
All custom code used in this study is publicly available at GitHub: https://github.com/tan5251/Super-ehancer-and-muscle-fiber-developmnet and has been archived in Zenodo with the https://doi.org/10.5281/zenodo.15504624. The code is released under the MIT license.
References
Wu, H. & Ballantyne, C. M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Invest. 127, 43–54 (2017).
Picard, B., Lefaucheur, L., Berri, C. & Duclos, M. J. Muscle fibre ontogenesis in farm animal species. Reprod. Nutr. Dev. 42, 415–431 (2002).
Tajsharghi, H. & Oldfors, A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 125, 3–18 (2013).
Schiaffino, S. & Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 91, 1447–1531 (2011).
Oberbach, A. et al. Altered fiber distribution and fiber-specific glycolytic and oxidative enzyme activity in skeletal muscle of patients with type 2 diabetes. Diabetes Care 29, 895–900 (2006).
Kim, G. D. et al. The influence of fiber size distribution of type IIB on carcass traits and meat quality in pigs. Meat Sci. 94, 267–273 (2013).
Lu, X. et al. The relationship between myofiber characteristics and meat quality of Chinese Qinchuan and Luxi cattle. Anim. Biosci. 34, 743–750 (2021).
Fujita, Y., Pather, S. R., Ming, G. L. & Song, H. 3D spatial genome organization in the nervous system: from development and plasticity to disease. Neuron 110, 2902–2915 (2022).
Lupianez, D. G. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025 (2015).
Zheng, H. & Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 20, 535–550 (2019).
Liu, Y. et al. CTCF coordinates cell fate specification via orchestrating regulatory hubs with pioneer transcription factors. Cell Rep. 42, 113259 (2023).
He, M. et al. Genome-wide chromatin structure changes during adipogenesis and myogenesis. Int. J. Biol. Sci. 14, 1571–1585 (2018).
Marti-Marimon, M. et al. Major reorganization of chromosome conformation during muscle development in pig. Front. Genet. 12, 748239 (2021).
Zhao, Y. et al. Multiscale 3D genome reorganization during skeletal muscle stem cell lineage progression and aging. Sci. Adv. 9, eabo1360 (2023).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Song, M. et al. Cell-type-specific 3D epigenomes in the developing human cortex. Nature 587, 644–649 (2020).
Ramachandran, K. et al. Dynamic enhancers control skeletal muscle identity and reprogramming. PLoS Biol. 17, e3000467 (2019).
Dos Santos, M. et al. A fast Myosin super enhancer dictates muscle fiber phenotype through competitive interactions with Myosin genes. Nat. Commun. 13, 1039 (2022).
Bassols, A. et al. The pig as an animal model for human pathologies: a proteomics perspective. Proteom. Clin. Appl 8, 715–731 (2014).
Valentim, M. A., Brahmbhatt, A. N. & Tupling, A. R. Skeletal and cardiac muscle calcium transport regulation in health and disease. Biosci. Rep. 42, BSR20211997 (2022).
Akarsu, S. et al. Mitochondrial complex I and III mRNA levels in bipolar disorder. J. Affect Disord. 184, 160–163 (2015).
Bonnefond, A. et al. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi-like features. J. Clin. Invest. 123, 3037–3041 (2013).
Cao, T., Thongam, U. & Jin, J. P. Invertebrate troponin: insights into the evolution and regulation of striated muscle contraction. Arch. Biochem. Biophys. 666, 40–45 (2019).
Long, K. et al. Identification of enhancers responsible for the coordinated expression of myosin heavy chain isoforms in skeletal muscle. BMC Genomics 23, 519 (2022).
Lin, X. et al. Hoxa11 and Hoxa13 facilitate slow-twitch muscle formation in C2C12 cells and indirectly affect the lipid deposition of 3T3-L1 cells. Anim. Sci. J. 92, e13544 (2021).
Gogulothu, R. et al. Disrupted expression of genes essential for skeletal muscle fibre integrity and energy metabolism in Vitamin D deficient rats. J. Steroid Biochem. Mol. Biol. 197, 105525 (2020).
Loven, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Yang, Y. et al. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 49, 1313–1329 (2021).
Terry, E. E. et al. Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues. Elife 7, e34613 (2018).
Horibata, Y. et al. The phosphatidylcholine transfer protein StarD7 is important for myogenic differentiation in mouse myoblast C2C12 cells and human primary skeletal myoblasts. Sci. Rep. 10, 2845 (2020).
Rojas, M. L. et al. Role of the lipid transport protein StarD7 in mitochondrial dynamics. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1866, 159029 (2021).
Flores-Martin, J., Reyna, L., Ridano, M. E., Panzetta-Dutari, G. M. & Genti-Raimondi, S. Suppression of StarD7 promotes endoplasmic reticulum stress and induces ROS production. Free Radic. Biol. Med. 99, 286–295 (2016).
Kulisz, A., Chen, N., Chandel, N. S., Shao, Z. & Schumacker, P. T. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am. J. Physiol. Lung Cell Mol. Physiol. 282, L1324–L1329 (2002).
Shi, H. et al. Modulation of skeletal muscle fiber type by mitogen-activated protein kinase signaling. FASEB J. 22, 2990–3000 (2008).
Li, J., Zhang, Z., Bo, H. & Zhang, Y. Exercise couples mitochondrial function with skeletal muscle fiber type via ROS-mediated epigenetic modification. Free Radic. Biol. Med. 213, 409–425 (2024).
Gomes, A. V., Potter, J. D. & Szczesna-Cordary, D. The role of troponins in muscle contraction. IUBMB Life 54, 323–333 (2002).
Baresic, M., Salatino, S., Kupr, B., van Nimwegen, E. & Handschin, C. Transcriptional network analysis in muscle reveals AP-1 as a partner of PGC-1alpha in the regulation of the hypoxic gene program. Mol. Cell Biol. 34, 2996–3012 (2014).
Zhang, L. et al. Skeletal muscle-specific overexpression of PGC-1alpha induces fiber-type conversion through enhanced mitochondrial respiration and fatty acid oxidation in mice and pigs. Int. J. Biol. Sci. 13, 1152–1162 (2017).
Fang, G. et al. KLF15 maintains contractile phenotype of vascular smooth muscle cells and prevents thoracic aortic dissection by interacting with MRTFB. J. Biol. Chem. 300, 107260 (2024).
Pette, D. & Staron, R. S. Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 50, 500–509 (2000).
Jiang, Y., Jiang, Y. Y. & Lin, D. C. Super-enhancer-mediated core regulatory circuitry in human cancer. Comput. Struct. Biotechnol. J. 19, 2790–2795 (2021).
Yan, Z., Okutsu, M., Akhtar, Y. N. & Lira, V. A. Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J. Appl. Physiol. 110, 264–274 (2011).
Mo, M. et al. Molecular mechanisms underlying the impact of muscle fiber types on meat quality in livestock and poultry. Front. Vet. Sci. 10, 1284551 (2023).
Dos Santos, M. et al. Single-nucleus RNA-seq and FISH identify coordinated transcriptional activity in mammalian myofibers. Nat. Commun. 11, 5102 (2020).
Buenrostro, J. D., Wu, B., Chang, H. Y. & Greenleaf, W. J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21 29 21–21 29 29 (2015).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Mi, H. et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45, D183–D189 (2017).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Bu, D. et al. KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 49, W317–W325 (2021).
H. Li, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Zhao, Y. et al. A compendium and comparative epigenomics analysis of cis-regulatory elements in the pig genome. Nat. Commun. 12, 2217 (2021).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 17, 10–12 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Lizio, M. et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 16, 22 (2015).
Rauluseviciute, I. et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 52, D174–D182 (2024).
Bentsen, M. et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 11, 4267 (2020).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Open2C, Abdennur, N. et al. Cooltools: enabling high-resolution Hi-C analysis in Python. PLoS Comput. Biol. 20, e1012067 (2024).
Wolff, J. et al. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 48, W177–W184 (2020).
Chen, L. et al. Dynamic 3D genome reorganization during development and metabolic stress of the porcine liver. Cell Discov. 8, 56 (2022).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Flyamer, I. M., Illingworth, R. S. & Bickmore, W. A. Coolpup.py: versatile pile-up analysis of Hi-C data. Bioinformatics 36, 2980–2985 (2020).
Zhou, X. et al. The human epigenome browser at Washington University. Nat. Methods 8, 989–990 (2011).
Trieu, T., Oluwadare, O., Wopata, J. & Cheng, J. GenomeFlow: a comprehensive graphical tool for modeling and analyzing 3D genome structure. Bioinformatics 35, 1416–1418 (2019).
Acknowledgements
This research was funded by the grant(2023ZD04047) from the Biological Breeding-National Science and Technology Major Project (STI2030-Major Project) and three grants from the Guangdong Province: the Science and Technology Program (2023B1212060057), the Key Area Research and Development Program (2022B0202090002) and the Natural Science foundation projects (2025A1515012313).
Author information
Authors and Affiliations
Contributions
B.T. and L.X. conducted the experiments. B.T. designed the analytical strategy and performed the bioinformatics analysis. L.H., Z.L., G.C., S.W., L.X., L.L., J.W., E.Z., and G.L. contributed to the experimental design and data interpretation. B.T. drafted the original manuscript. B.T., T.G., and Z.W. conceived the study and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Huating Wang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tan, B., Hong, L., Xiao, L. et al. Rewiring of 3D chromatin topology orchestrates transcriptional reprogramming in muscle fiber-type specification and transformation. Nat Commun 16, 5833 (2025). https://doi.org/10.1038/s41467-025-60866-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60866-3
