
Abstract
Electrochemical CO2-to-ethanol conversion is challenged by sluggish C-C coupling kinetics and wide products distribution. Although Cu+ has been demonstrated to enhance multi-carbon (C2+) formation, the stabilization of Cu+ under reduction conditions is difficult. Here, we report a hydrogen-ethanol pretreatment strategy to obtain Cu nanoparticles covered by highly dispersed and disordered ZnO1-x clusters. Ethanol-induced ZnO1-x redispersion gives rise to abundant Cu+ on the subsurface. The optimal catalyst delivers a 73.0% ethanol Faradaic efficiency (FE) and 86.0% total C2+ FE at −0.9 V, with a 2.3 mmol cm−2 h−1 ethanol formation rate and single-pass ethanol yield of 18.0%. The catalyst also exhibits stability beyond 500 h, attributed to the stabilization of Cu+ by the ZnO1-x shield that requires a high energy barrier for lattice oxygen removal. In situ X-ray spectroscopy and calculations reveal a volcano relationship between Cu+ ratio in Cu species and ethanol FE. Optimal Cu+ density not only facilitates *OC-COH coupling but also optimizes the adsorption energy of *CH2CH2O on catalyst for ethanol electrosynthesis.
Similar content being viewed by others

Introduction
The electrochemical reduction of carbon dioxide (CO2RR) to ethanol powered by green electricity presents a promising strategy for carbon neutrality to mediate the greenhouse effect and store renewable energy into easily transportable liquid chemicals. However, the slow C-C coupling kinetics demanding 12-electron transfer and wide product distribution limit the synthesis of ethanol1,2,3,4.
Cu-based materials have been extensively studied and recognized as the most efficient catalysts for the C2+ production thus far albeit with low Faradaic efficiency (FE) obtained on pure copper surface5,6. A tandem reaction involving CO2 to *CO and *CO to C2+ has been generally demonstrated as *CO is a common intermediate probed in the operando Raman or infrared spectroscopy7,8,9. Meanwhile, the recent experimental evidences highlight two dominant pathways for C-C coupling: (1) the *CO-COH pathway, where a second proton-electron transfer to adsorbed *CO forms *COH10, and (2) the *CO-CHO pathway, where *CO undergoes hydrogenation to *CHO prior to coupling with another *CO11,12. The dominance of either pathway depends on the local microenvironment and catalyst topology. In particular, the critical *OC-COH intermediate formation during the synthesis of ethanol or ethylene implies that the C-C coupling requires two Cu sites with distinct properties13. Hence, many attempts regarding geometric and chemical tailoring of Cu sites have been performed to pursue the asymmetric reaction centers, such as physical mixing of different Cu domains14, surface doping1, interface engineering15, and alloying16, etc.
Among the aforementioned efforts, more and more works have gradually realized the role of Cu+ in enhancing FE for C2+ products (FEC2+) because the Cu+···Cu0 pairs17 reduce the energy barrier of C-C coupling compared to pure Cu. However, quantitative understanding of the correlation between Cu+ fraction in total Cu species and the selectivity of ethanol (versus ethylene) during the reaction remains ambiguous. Moreover, according to the Pourbaix diagram shown in Fig. 1a, Cu0 species should be the most stable phase under the typical working potentials and alkaline electrolyte conditions for CO2RR. The stabilization of Cu+ during the reaction is significantly challenging and deserves scrutiny. Therefore, some strategies, e.g., the confinement of organic framework18,19, pulsed electrolysis20, and single atom catalysis21,22 have been tried; however, either the ethanol FE still remains below 70%, or the stability of Cu-based catalyst can not be sustained beyond 200 h.
On the other side, Lian et al.23 carried out extensive molecular dynamics simulations using a neural network potential trained on first-principle data to investigate the lifetime of trapped oxygen in Cu oxide during CO2RR, revealing that the oxygen atoms tend to aggregate to form Cu2O instead of being distributed uniformly. It actually takes a considerable amount of time (~hours) to reduce oxidized copper in the electrolysis measurement. In this regard, the integration of a refractory metal oxide (i.e., more resistant to reduction than copper) with oxygen-derived Cu is expected to modulate the copper surface affinity and retard the reduction of Cu-O24,25,26,27. However, it is extremely difficult to precisely control the interface between such two metal domains to achieve a high ethanol FE with long-term stability.
Herein, we have proposed a stepwise synthesis approach (Fig. 1a) involving sequential hydrogen and ethanol atmosphere treatments of ZnO/CuO. Such treatment led to the formation of a controlled amount of Cu+ at the interface of obtained catalyst, significantly enhancing the selectivity of ethanol (73.0% at −0.9 V vs RHE) and demonstrating remarkable stability during long-term reactions (>500 h). The operando X-ray absorption and diffused reflection infrared spectroscopies demonstrated that the pretreatment with ethanol induced and promoted the redispersion of ZnO1-x and the concomitant formation of Cu+ beneath the ZnO1-x surface. Moreover, the highly dispersed ZnO1-x clusters served as a critical protective layer to stabilize Cu+ at an optimal percentage during the electrochemical reaction, which favored the C-C coupling and selective formation of ethanol.
Results and discussion
Synthesis and characterization of ZnO1-x-Cu2O/Cu
The passive ZnO/CuO precursor (ZnO: 14.1 wt%) was prepared using a ball milling method (Fig. S1). This material was then subjected to a pure hydrogen atmosphere at 300 °C for 1 h and followed by treatment with ethanol for another hour, as noted in Fig. 1a (i.e., H2-C2H5OH treatment). After the pretreatment, the sample was purged with helium at 300 °C for 2 h to remove any residual H2, ethanol or intermediates that evolved throughout the process.

a Schematic illustration of the synthetic route with the Pourbaix diagram (Orange represents Cu0, magenta represents Cu2+, pink represents the mixture of Cu+ and Cu0, and blue represents ZnO). b EDS-mapping of Zn and Cu elements for ZnO/CuO. c EDS-mapping of Zn and Cu elements for ZnO1-x/Cu. d EDS-mapping of Zn and Cu elements for ZnO1-x-Cu2O/Cu. e The Zn-O-Zn and Cu-Cu coordination numbers of the ZnO1-x-Cu2O/Cu, ZnO1-x/Cu, and ZnO/CuO characterized by extended X-ray adsorption fine structure analysis. f Cu LMM AES spectra of the ZnO1-x-Cu2O/Cu, ZnO1-x/Cu and ZnO/CuO. g Zn LMM AES spectra of the ZnO1-x-Cu2O/Cu, ZnO1-x/Cu and ZnO/CuO. Relevant source data are provided as a Source Data file.
X-ray diffraction (XRD) was employed to examine the crystal phase transition at three stages (Fig. S2). The diffraction patterns of the initial ZnO/CuO sample confirmed the coexistence of monoclinic copper oxide (PDF: #65-2309) and hexagonal zinc oxide (PDF: #65-3411). After H2 reduction, the peaks corresponding to ZnO persisted but were attenuated, while the prominent diffraction peaks of CuO disappeared and new peaks associated with the cubic Cu (PDF: #65-9026) emerged, confirming the reduction of copper oxide. The reduced sample was denoted as ZnO1-x/Cu. In sequence, upon the ethanol treatment, an additional peak at 29.6° corresponding to the cubic Cu2O (110) plane suggested the formation of Cu+ species; thus, the final sample was denoted as ZnO1-x-Cu2O/Cu.
High-resolution transmission electron microscopy (HRTEM) (Fig. S3) and energy-dispersive X-ray (EDX) spectroscopy (Fig. 1b–d) were employed to compare the sizes and morphologies of the aforementioned three samples. No aggregation or fragmentation occurred on Cu species, as similar diameters of 20–25 nm were observed. However, ZnO nanoparticles underwent significant changes during the treatment. In ZnO/CuO, ZnO existed as relatively large particles (10–20 nm). After H2 reduction, the ZnO1-x particles were dispersed on the Cu surface as smaller nanoparticles (~4–8 nm). Subsequent treatment in an ethanol atmosphere led to further dispersion of ZnO1-x clusters that were well-distributed and formed as the shell over Cu with a thickness of less than 2 nm. The trend in particle size changes was also proved by XRD results calculated by the Scherrer equation (Table S1). Moreover, it is anticipated that the core-shell structure of ZnO1-x-Cu2O/Cu would lead to the strong chemical interaction between ZnO1-x clusters and Cu nanoparticles28, resulting in the enhanced electron migration and the evolution of Cu+, which could be evidenced by a more careful characterization of ZnO1-x-Cu2O/Cu catalyst (Fig. S3). An additional clear interfacial plane with a lattice spacing of 0.301 nm assigned to Cu2O(110) was identified between Cu(111) and ZnO1-x(101) planes indicated by the specific lattice spacings of 0.208 nm and 0.248 nm, respectively29.
More specifically, the coordination numbers of Cu and Zn species in three samples were further investigated through extended X-ray absorption fine structure (EXAFS) curve fitting analysis (Fig. 1e and Table S2). The average coordination number (CN) of Cu–Cu exhibited a relatively minor change, increasing from 7.9 in ZnO/CuO to 8.7 in ZnO1-x-Cu2O/Cu. In comparison, the average CN of Zn-O-Zn significantly decreased from 10.1 in ZnO/CuO to 5.1 in ZnO1-x-Cu2O/Cu, indicating a clear redispersion of ZnO particles during the H2-C2H5OH treatment.
Furthermore, the chemical nature of copper and zinc species during the treatment was studied using in situ X-ray photoelectron spectroscopy (XPS). Figure S4 presents the Cu 2p edge spectra for the three catalysts. The ZnO/CuO catalyst exhibits two split peaks at binding energies of 933.1 eV (Cu 2p3/2) and 953.1 eV (Cu 2p1/2) with prominent satellite peaks, suggesting the feature of Cu2+. In comparison, the ZnO1-x/Cu and ZnO1-x-Cu2O/Cu catalysts display binding energies of 932.1 eV (Cu 2p3/2) and 951.9 eV (Cu 2p1/2) without satellite peaks, confirming the metallic state of Cu. The existence of Cu+ species was further confirmed using Cu LMM Auger spectroscopy (Figs. 1f and S5). The ZnO/CuO catalyst shows a single Cu2+ characteristic peak at 917.8 eV. The ZnO1-x/Cu catalyst shows a single peak at 918.9 eV, consistent with metallic Cu. While the ZnO1-x-Cu2O/Cu catalyst has two peaks at 916.8 eV and 918.8 eV, attributed to Cu+ and Cu0 species, respectively. This indicates that Cu2+ was fully reduced to the metallic form after H2 treatment, while some was re-oxidized to Cu+ during ethanol treatment30. On the other side, Zn 2p edge spectra (Fig. S6) display split peaks at ~1044.8 eV (Zn 2p1/2) and 1021.8 eV (Zn 2p3/2) for all three catalysts, indicating the absence of metallic Zn31. More specific results are obtained from the Zn LMM Auger spectra (Fig. 1g). All three spectra can be deconvoluted into two additional peaks with kinetic energies of ~988.6 eV and ~992 eV, corresponding to Zn2+ and Znδ+ (δ < 2), respectively. The existence of reduced Zn species suggests the existence of oxygen vacancies (Fig. S7). Notably, the proportion of Znδ+ increases from 25.8% in ZnO/CuO to 28.0% in ZnO1-x/Cu and further to 33.2% in ZnO1-x-Cu2O/Cu. This suggests the redispersion of ZnO particles during H2-C2H5OH treatment was accompanied by the formation of defects, consistent with the decrement in CN of Zn-O-Zn.
According to the above analysis, the H2 and C2H5OH treatment promoted the notable fragmentation of ZnO nanoparticles with partial reduction. In contrast, the Cu phase exhibits negligible change associated with particle size but mostly reduced. These phenomena can be attributed not only to different physicochemical properties of ZnO and CuO (e.g., higher oxygen affinity for ZnO32) but also to the selective adsorption and reaction of H2/C2H5OH with two metal oxides during the treatment. The underlying chemical transition will be investigated in the following section. In particular, it is also interesting to elucidate the formation mechanism of Cu+ during ethanol treatment and its relationship with the dispersion of the external ZnO1-x layer.
Formation mechanism of ZnO1-x-Cu2O/Cu upon H2-C2H5OH treatment
To gain a deep insight into the dynamic changes of the chemical states and coordination environment of the catalysts during the H2-C2H5OH treatments, ZnO/CuO, ZnO1-x/Cu, and ZnO1-x-Cu2O/Cu were characterized by the operando X-ray absorption spectroscopy (XAS) (Figs. 2 and S8). The Cu K-edge X-ray absorption near-edge structure (XANES) spectrum of ZnO/CuO exhibited a small shoulder at ~8984 eV, closely matching the CuO reference (Fig. 2a), indicating the Cu species existed primarily as Cu2+. During H2 reduction (ZnO/CuO@H2), the Cu K-edge XANES absorption edge shifted rapidly towards that of Cu foil, with the ZnO1-x/Cu sample displaying a peak at ~8981 eV, which is characteristic of Cu0, confirming the reduction of Cu2+ to its metallic state33. During C2H5OH treatment (ZnO1-x/Cu@C2H5OH), the absorption edge shifted back towards the Cu2O reference, indicating partial oxidation of Cu species. Ultimately, the absorption edge of the C2H5OH-treated ZnO1-x-Cu2O/Cu sample settled between those of Cu foil and Cu2O, suggesting a mixed oxidation state of Cu0 and Cu+, consistent with the Auger spectra, XRD, and TEM results.

a Operando normalized XANES spectra of ZnO1-x-Cu2O/Cu catalysts during H2-C2H5OH treatment and references of CuO, Cu2O and Cu foil at the Cu K edge, where ZnO/CuO@H2 represents the sample during H2 treatment, and ZnO1-x/Cu@C2H5OH represents the sample during C2H5OH treatment. b Edge-shift quantification of Cu oxidation states during H2-C2H5OH treatment. c Operando normalized XANES spectra of ZnO1-x-Cu2O/Cu catalysts during H2-C2H5OH treatment and references of ZnO and Zn foil at the Zn K-edge. d In situ DRIFTS Spectra of C2H5OH on ZnO1-x-Cu2O/Cu catalyst. e Bader charge calculation and charge density analysis of ZnO/Cu in H2 and C2H5OH atmosphere, respectively. f Energy changes associated with the ethoxy-induced the dispersion of ZnO1-x and the formation mechanism of Cu+. g Schematic diagram of the catalyst structural evolution during C2H5OH treatment. Relevant source data are provided as a Source Data file.
To quantify the Cu species with varying oxidation states at various stages, normalized difference edge analysis (Fig. 2b) and linear combination fitting33,34 (LCF, Fig. S9) were implemented. The proportion of Cu2+ species in ZnO/CuO decreased from ~100% to <5% after H2 reduction and disappeared completely after C2H5OH treatment in ZnO1-x-Cu2O/Cu. However, the proportion of Cu0 species increased rapidly to >95% after H2 reduction and then decreased to ~57% after C2H5OH treatment due to the emergence of Cu+ species during C2H5OH treatment (~26.4%), then increased and maintained at 43.4% in ZnO1-x-Cu2O/Cu.
Comparative k³-weighted Fourier transform EXAFS spectra of the above samples were fitted and analyzed with Cu foil, CuO, and Cu2O as references (Figs. S10, S11 and Table S2). The EXAFS spectrum of ZnO/CuO sample displayed one peak at ~1.56 Å, attributed to Cu–O coordination (CN = 3.8), and another peak at around 2.52 Å, attributed to Cu–Cu coordination (CN = 8). In ZnO1-x/Cu sample, the Cu–O scattering peak was absent, replaced by a Cu–Cu scattering peak at around 2.25 Å (CN = 11), indicating the reduction of Cu2+ into metallic form. In the ZnO1-x-Cu2O/Cu sample, a Cu–O scattering peak appeared but shifted to ~1.46 Å, matching with Cu2O reference, indicating the formation of Cu+ species. The wavelet transform results (Fig. S12) revealed a feature related to Cu–O pairs (~1.5 Å−1 versus 1.6 Å−1 for CuO) in k-space, supporting the generation of Cu+ species after C2H5OH treatment. Moreover, the average CN (Cu-O) = 1.6 and CN (Cu-Cu) = 8.7 demonstrated that the Cu+ percentage in total Cu species in ZnO1-x-Cu2O/Cu was ~40%, consistent with the quantitative analysis of XANES results (Fig. 2b).
The near-edge structures and coordination environments of Zn were probed for the aforementioned five samples during H2-C2H5OH treatments (Fig. 2c). The XANES spectra of all samples resembled that of the ZnO reference in shape and edge position rather than Zn foil, indicating Zn was in an oxided state. However, the sequential shifts from ~9661 eV (ZnO/CuO@H2) to 9660 eV (ZnO1-x-Cu2O/Cu) were observed for the edges of five samples with the exposure to H2 and subsequent C2H5OH, but still far away from 9657 eV for Zn foil, suggesting the partial reduction of Zn oxide. Similarly, the relative changes of the Zn–O–Zn multiple scattering intensity at ~9678 eV of these materials also suggested the fragmentation of large Zn oxide particles35, consistent with the XRD and HRTEM data. Moreover, the normalized difference edge analysis of Zn species in these samples (Fig. S13) revealed the proportion of Zn2+ species decreased to 64%, and 36% reduced Znδ+ existed in ZnO1-x-Cu2O/Cu.
Comparative k³-weighted Fourier transform EXAFS spectra of the above samples were fitted and analyzed with Zn foil and ZnO as references (Figs. S14, S15). All samples exhibited a first-shell scattering peak at ~1.49 Å corresponding to Zn–O coordination and a second-shell scattering peak at around 2.89 Å attributed to Zn–O–Zn coordination36. The fitting results in Table S3 showed the Zn–O–Zn coordination number in the ZnO1-x/Cu catalyst decreased from 12 (ZnO reference) to 7.6 ± 0.6, and further to 5.1 ± 0.5 in the C2H5OH-treated ZnO1-x-Cu2O/Cu, consistent with the wavelet transform results (Fig. S16). HRTEM analysis of ZnO1-x-Cu2O/Cu (Fig. S17) revealed a unique disordered structure for ZnO1-x, which has been reported to exhibit long-term stability due to high configurational entropy37.
Based on these quantitative analyses of Cu and Zn species for ZnO/CuO, ZnO1-x/Cu and ZnO1-x-Cu2O/Cu, it can be found that Zn2+ was continuously reduced by H2 and C2H5OH with the dispersion of Zn oxide, however, its thermodynamic stability suppressed such change (only ~36% Znδ+ at final stage). In contrast, Cu2+ was easily reduced to metallic Cu, but ~40% of them were re-oxidized to Cu+ in ethanol, which was rarely reported in previous literature, as ethanol was generally regarded as a reducing agent. In addition, the fractions of Znδ+ and Cu+ were very close, implying the chemical transitions related to these two phases are highly relevant.
To further elucidate the evolutions of different components during this process, the interaction between ethanol molecules and the surface of the catalyst was probed by in situ Diffuse Reflectance Infrared Fourier Transform spectroscopy (DRIFTS) experiments under conditions identical to ethanol treatment at 300 oC (Fig. 2d). The spectra revealed a unidentate ν(CO) adsorption band at 1141 cm−1, along with asymmetric stretching vibrations of υas(CH3) at 2970 cm−1 and υas(CH) at 2915 cm−1, as well as bending vibrations of δas(CH3) and δs(CH3) at 1390 cm−1 and 1230 cm−1, respectively38,39. According to the literature40, these peaks suggested the cleavage of O-H in ethanol and primarily adsorbed as ethoxy species on the catalyst.
Bader charge calculation and charge density analysis (Fig. 2e) were further performed to investigate the charge distribution between ZnO and Cu under ethanol treatment. Based on the results of XRD and TEM, the model of ZnO/Cu was established and consisted of a pure Cu (111) surface with a 6 × 6 × 3 atomic layer and a ZnO cluster containing twenty ZnO molecular structures. As noted, the charges of Cu species in the initial state were close to Cu foil (the sample treated by H2, named as ZnO/Cu-H2). Subsequently, the ethanol adsorption was applied, which selectively interacts with ZnO (ZnO/Cu-C2H5OH). The charge transfer mainly occurs between ZnO and C2H5OH, however, it was intriguing to obtain Cu+ at the interface between ZnO and Cu phases, consistent with the experimental observation. To reveal the formation mechanism of Cu+ during ethanol treatment, the detailed process involved in the interaction between C2H5OH and ZnO/Cu was calculated and shown in Figs. 2f and S18. Compared to continuous copper surface, the favorable adsorption of C2H5OH happened on ZnO with the adsorption energy of −1.51 eV. Then a strong charger transfer between ZnO and C2H5OH was indicated by the stretching of O-H bond of C2H5OH from 0.998 Å to 1.002 Å, resulting in the breakage of O-H bond in ethanol with a small barrier of 0.27 eV, giving rise to the ethoxy group (*C2H5O) binding with Zn site, which was demonstrated in DRIFTS experiment. A similar phenomenon of ethanol dissociation on ZnO clusters has been previously reported in the reforming reaction using Cu/ZnO based catalysts38,41,42. Furthermore, the electron-donating effect of *C2H5O weakened the Zn-O bond in the cluster phase, resulting in a notably lower dissociation barrier of 0.59 eV in contrast to the Zn-O binding energy of 3.44–3.58 eV. The cleavage of Zn-O bonds facilitated the dispersion of ZnO clusters into smaller dimensions, forming the encapsulation of Cu nanoparticles by the ZnO1-x layer. Moreover, following the rupture of Zn-O bonds, the liberated oxygen atoms rearranged and spontaneously bonded with copper atoms, forming Cu⁺ ions at the interface between ZnO1-x and metallic Cu (Fig. 2g).
Electrochemical CO2RR to ethanol using ZnO1-x-Cu2O/Cu
These catalysts (ZnO/CuO, ZnO1-x/Cu, and ZnO1-x-Cu2O/Cu) were evaluated for the CO2RR using a gas diffusion electrode (GDE) in a flow cell with a 0.1 M KOH solution as the electrolyte. The GDE was pre-activated through linear sweep voltammetry (LSV), and Fig. S19 displays the LSV curves for the three catalysts in Ar and CO2 atmospheres, corrected for ohmic resistance (iR). The corresponding equivalent circuit and fitted electrochemical impedance spectroscopy (EIS) spectra were provided in Fig. S20. The current densities of all three catalysts were notably higher in the presence of CO2 compared to Ar, confirming the initiation of CO2 reduction reactions. Chronopotentiometric electrolysis was conducted over a potential range of −0.6 to −1.1 V vs RHE. The identified gas-phase products comprised CO, CH4, ethylene, and H2, while the liquid-phase products included formate, ethanol, acetate, and n-propanol.
Figure 3a presents the product current density and Faradaic efficiency (FE) over ZnO1-x-Cu2O/Cu at different potentials. Notably, the total FEC2+ reached 64% at −0.6 V, indicating significantly enhanced C-C coupling kinetics. As the potential became more negative, the FE and current density of total C2+ products further increased. It is evident that the total FEC2+ on ZnO1-x-Cu2O/Cu was clearly higher than that on two other catalysts (Figs. 3b and S21, S22). At −0.9 V, the FEC2+ reached a maximum of 86.1%, with a current density of ~226.0 mA cm−2, compared to 72.8% (92.0 mA cm−2) and 63.0% (59.0 mA cm−2) on ZnO1-x/Cu and ZnO/CuO. Among C2+ products, ZnO1-x-Cu2O/Cu catalyst also demonstrated a significant advantage for ethanol synthesis (Fig. 3c). At −0.9 V, the ethanol Faradaic efficiency (FEC2H5OH) on ZnO1-x-Cu2O/Cu reached 72.9%, compared to 41.6%, 52.2% on ZnO1-x/Cu and ZnO/CuO. Moreover, CO2 conversion was 24.7% by putting the carbon balance in consideration for ZnO1-x-Cu2O/Cu. Correspondingly, the formation rate and single-pass yield of ethanol were determined as 2.3 mmol cm−2 h−1 and 18%, respectively (Fig. S23), which were higher to the state-of-the-art results in the literature (0.6 ~ 1.1 mmol cm−2 h−1 and 16.5%)1,43.

a Faradaic efficiency (FE) values of all products and current density at various applied cathodic potentials for ZnO1-x-Cu2O/Cu. b FE values of C2+ products of different catalysts at different applied cathodic potentials. c FE values of C2H5OH of different catalysts at different applied cathodic potentials. d Long -term stability test of ZnO1-x-Cu2O/Cu at −0.9 V (versus RHE). e Comparison of the onset potential, FEC2+, FEC2H5OH, jC2H5OH, stability, and energy efficiency of the ZnO1-x-Cu2O/Cu catalyst with state-of-the-art results of related catalysts in literature. f FE values of C2+ products and C1 products of ZnO1-x-Cu2O/Cu at different applied cathodic potentials. g FE values of C2H5OH and C2H4 of ZnO1-x-Cu2O/Cu at different applied cathodic potentials. h FE ratio of C2+/C1 and C2H5OH/C2H4 of different catalysts at −0.9 V (versus RHE). Error bars in (a, b, c, f, g, h) represent standard deviations of three replicate measurements. Relevant source data are provided as a Source Data file.
The electrochemical active surface areas (ECSA) of the catalysts were measured using double-layer capacitance (Cdl) derived from cyclic voltammetry (CV) curves44 (Fig. S24). ZnO1-x-Cu2O/Cu exhibited a higher ECSA (Cdl = 2.02 mF cm−2) and faster interfacial electron transfer kinetics. Normalizing the partial current density for ethanol by ECSA (Fig. S25) clearly showed that ZnO1-x-Cu2O/Cu has a higher value than ZnO1-x/Cu and ZnO/CuO, indicating the enhancement in ethanol current density was primarily due to the generation of more active sites over ZnO1-x-Cu2O/Cu rather than its increased ECSA. Consistently, the double-layer capacitance (Fig. S26) determined by EIS fitting showed that the double-layer thickness on the ZnO1-x-Cu2O/Cu surface decreased by more than 50% compared to other two samples under reaction conditions45. These findings indicated that ZnO1-x-Cu2O/Cu effectively enhances interfacial electron transfer and electron-proton coupling kinetics, thereby strongly improving the catalyst activity.
The stability of ZnO1-x-Cu2O/Cu was evaluated through the potentiostatic test for over 500 h. The current density was maintained at ~240 mA cm−2, and the average FEC2H5OH remained around 72% (Fig. 3d). The maximum half-cell cathodic energy efficiency for ethanol at −1.0 V was calculated to be 42.4% for ZnO1-x-Cu2O/Cu, surpassing those of ZnO1-x/Cu (20.8%) and ZnO/CuO (10%). Key performance indicators over ZnO1-x-Cu2O/Cu, including onset potential, FEC2+, FEC2H5OH, jC2H5OH, stability, and energy efficiency, established a state-of-the-art performance for CO2RR to ethanol (Fig. 3e and Table S4).
The dependence of product distributions on applied potentials was shown in Fig. 3f, g. The FEC2+ exhibited a volcano-shaped curve between −0.6 and −1.1 V, while the FEC1 showed an inverse volcano trend, proving that the C-C coupling proceeds via C1 as intermediate, same as the tandem mechanism reported in literature6,46,47,48,49. Similar trends of FEC2+ and FEC1 over the potentials were also observed in the cases of ZnO1-x/Cu (Fig. S27) and ZnO/Cu (Fig. S28), suggesting that CO2RR on three catalysts proceeded similarly via C1 intermediate. FEC2H5OH also exhibited a volcano-shaped curve between −0.6 and −1.1 V, whereas FEC2H4 displayed an inverse trend (Fig. 3g). This suggested a competitive mechanism for the synthesis of ethanol versus ethylene, with the former as the dominant product, typically over 50% FE.
Figure 3h compares the FEC2+/FEC1 and FEC2H5OH/FEC2H4 ratios for three catalysts. At the optimal working potential (−0.9 V), ZnO1-x-Cu2O/Cu exhibited a FEC2+/FEC1 ratio of 10.2, which is 2.6 times and 3.2 times higher than ZnO1-x/Cu and ZnO/CuO, respectively. The FEC2H5OH/FEC2H4 ratio on ZnO1-x-Cu2O/Cu was also calculated to be 12.9, 2.7 times and 3.4 times higher than on ZnO1-x/Cu and ZnO/CuO, respectively. These results demonstrated that the presence of Cu+ in ZnO1-x-Cu2O/Cu, unlike the pure Cu metal in the other two catalysts, characterized by XAS for three spent samples (Figs. S29 and S30), not only promoted C-C coupling but also favored ethanol production over ethylene. More detailed mechanism study regarding the contribution of Cu+ during CO2RR will be discussed in the last section.
Stabilization of Cu+ at appropriate fraction for ethanol electrosynthesis
The CO2RR performance over these catalysts (ZnO/CuO, ZnO1-x/Cu, and ZnO1-x-Cu2O/Cu) has recognized the critical importance of Cu+ on C-C coupling and selective synthesis of ethanol. However, as noted in Fig. 1a, the stabilization of Cu+ during the reaction is remarkably challenging under the working potentials of CO2RR, and the state of Cu species is highly dynamic in fact. In this regard, the actual fraction of Cu+ at the electrolysis conditions and the corresponding influence on CO2 conversion remain unknown. In particular, it is necessary to investigate the mechanism of Cu+ stability during CO2RR. Therefore, we conducted in-situ XAS measurements under conditions identical to CO2 electroreduction to track the changes in chemical states and local coordination environment of Cu and Zn in ZnO1-x-Cu2O/Cu during CO2RR.
As shown in Fig. 4a, the XANES spectra at the Cu K-edge of ZnO1-x-Cu2O/Cu at all reaction potentials displayed the low-energy peaks between Cu foil and Cu2O references, indicating an oxidation state between Cu0 and Cu+. Specifically, at open circuit potential, the absorption edge was closer to Cu2O reference. As the reaction potential increases, the absorption edge shifted slightly towards Cu foil, suggesting the gradual reduction of Cu+ during the reaction. However, no discernible shift was observed after −0.9 V vs RHE, but the spectra were still away from the spectrum of Cu foil, indicating no further reduction of Cu species happened with increasing reduction potential, and a certain fraction of Cu+ survived and resisted to reduction. LCF (Fig. S31) and normalized difference edge analysis (Fig. 4b) were then used to quantify the proportions of different Cu species during CO2RR. At open circuit potential, the proportion of Cu+ species in ZnO1-x-Cu2O/Cu was ~44.4%, consistent with the post-ethanol treatment results. With the potential applied, the proportion of Cu+ species decreased, dropping to ~33% at −0.7 V and continuously declining to a minimum value of ~28% at −0.9 V.

a Potential-depended Operando normalized XANES spectra of ZnO1-x-Cu2O/Cu catalysts during CO2RR and references of CuO, Cu2O, and Cu foil at the Cu K edge. b Average oxidation state of Cu in the ZnO1-x-Cu2O/Cu during CO2RR. c The corresponding k3-weighted FT-EXAFS spectra in R-space. d HRTEM image of ZnO1-x-Cu2O/Cu (the insets show the corresponding elemental mapping and high-resolution STEM-HAADF image). e EELS signals of Cu L at different tagged sites in (d). f The comparisons in term of the cross-sectional charge densities of ZnO1-x and Cu when applying charges on ZnO1-x-Cu2O/Cu. g Correlation of FEC2+ with the Cu+ fraction in total Cu species at different cathodic potentials. h Correlation of FEC2H5OH with the Cu+ fraction in total Cu species at different cathodic potentials. i Correlation of FEC2H4 with the Cu+ fraction in total Cu speices at different cathodic potentials. Relevant source data are provided as a Source Data file.
Subsequently, the k³-weighted Fourier transform EXAFS of ZnO1-x-Cu2O/Cu during the reaction was compared (Fig. 4c) and fitted without phase correction (Fig. S32). As the reaction potential increased, the scattering peak representing Cu–O coordination gradually got weakened, and stabilized after −0.9 V vs RHE, consistent with the information provided by XANES. In terms of the coordination environment (Table S5), the average CN of Cu-O bonds at open circuit potential was 1.6. The CN decreased to around 1.3 at −0.7 V vs RHE and slightly increased to about 1.2 at −0.9 V vs RHE, after which it remained stable. This suggested that the Cu structure reached a relatively steady state with the reaction proceeded.
Similar analysis was conducted for the transitions of the electronic configuration and coordination environment of Zn species during the electrochemical process. As shown in Fig. S33, the Zn K edge XANES spectra of ZnO1-x-Cu2O/Cu exhibit a shape and absorption edge position similar to that of ZnO reference, indicating its oxidation state. During CO2RR, the absorption edge of ZnO1-x-Cu2O/Cu shifted away from ZnO to Zn foil. This suggested the partial reduction of ZnO clusters distributed on the surface of Cu, which produced the local defects. However, the absorption edge at ~9666 eV and the pre-edge feature at ~9661 eV observed for ZnO1-x-Cu2O/Cu measured at −0.9-1.1 V proved that ZnO1-x still predominantly remained oxidized, as reported by the literature that these disordered ZnO1-x is very stable due to the high configurational entropic effect50,51,52. This unique structure of ZnO1-x may serve as the protective sheath for Cu⁺ species between Zn oxide and Cu metal phases, as ~28% Cu+ stayed even after the electroreduction treatment at −0.9 V. The k³-weighted Fourier transform EXAFS of ZnO1-x-Cu2O/Cu during CO2RR at the Zn K edge was shown in Fig. S33. The analysis of coordination numbers (Table S6) showed that the Zn-O-Zn coordination number in ZnO1-x-Cu2O/Cu was 5.1 ± 0.8 at open circuit potential, decreasing to 4.3 ± 0.5 after the reaction at −0.9 V, indicating a ~16% increment of reduced Zn oxide (i.e., ~43% ZnO1-x), which could promotes CO2 adsorption and activation. The reversed changes of Cu+ and ZnO1-x suggested that the redispersion of Zn oxide caused the direct exposure of some Cu+ to the reaction environment, leading to their reduction.
To further clarify the stabilization mechanism of Cu+ during the reaction, scanning transmission electron microscope (STEM) and corresponding electron energy loss spectroscopy (EELS) characterizations were performed on the ZnO1-x-Cu2O/Cu after CO2RR. As illustrated in the high-angle annular dark-field (HAADF) image (Fig. 4d) and the element mapping image shown as the inset, similar as the fresh sample, the post-reaction catalyst retained the well-dispersed structure of ZnO1-x surrounding Cu nanoparticles, and no separate phase was found, consistent with the XAS results. Furthermore, four positions (Fig. 4e) were selected for EELS elemental analysis to identify the oxidation states of copper. Site 1 (orange tagged) corresponded to Cu sites exposed to reaction environment without ZnO1-x cover, where the Cu L spectrum indicated a typical Cu0 species with the characteristic L3 and L2 peaks at 931, 956 eV53,54. Site 2 (magenta tagged) represented bulk Cu nanoparticles, also showing Cu0 species. In contrast, at the interface between Cu and ZnO1-x (Site 3, blue tagged), the Cu L spectrum revealed the presence of Cu⁺ species, as evidenced by a 0.9 eV shift of the L3 (at ~932 eV54) and L2 peaks towards higher energies (at ~958 eV53) and the absence of satellite peaks (the feature of Cu0)55,56. Similarly, Cu⁺ was also identified at another Cu and ZnO1-x interface (Site 4, purple-tagged). As noted, Cu+ species can only be maintained where it is covered by ZnO1-x shell or at the interface, attributing to the preservation of difficult-to-reduce ZnO1-x sites. The after-reaction XAS analysis (Fig. S34) also demonstrates the structural robustness of the ZnO1-x-Cu2O/Cu catalyst.
DFT calculations were then performed to unravel the underlying chemical transformations and electronic interactions between Cu+ and the adjacent ZnO1-x shell when the overpotential was applied. The optimized model of ZnO1-x cluster supported on Cu(111) surface after H2-ethanol treatment (ZnO1-x-Cu2O/Cu) was used, and we compared the cross-sectional charge density variations of ZnO1-x and Cu in Fig. 4f before and after being charged with additional electrons to simulate the electroreduction condition. The results revealed that these additional charges predominantly accumulated on the ZnO1-x clusters at the surface rather than at the subsurface Cu, indicated by the apparent increment of charge density observed at the Zn cations highlighted by the rectangle, compared to only a trace amount of Cu+ sites that were reduced. The protection effect of ZnO1-x actually could be ascribed to its greater accessibility to electrons at out layer and higher binding energy of Zn-O as 3.44–3.58 eV. This significantly decreases the kinetic reduction rate of Cu+ at the interface of Cu and ZnO1-x. In addition, Cu(111) surface-supported Cu2O model without ZnO1-x shell (named Cu2O/Cu) was also established in Fig. 4f, and the charge density of Cu2O cross section was compared with an equal amount of additional charges applied as ZnO1-x-Cu2O/Cu. It was noted that with the absence of ZnO1-x clusters, the extra charges are uniformly distributed at Cu2O, resulting in the conversion of Cu+ to Cu0, while bulk Cu remains in its metallic state. This further corroborated the protective role of ZnO1-x clusters in stabilizing Cu+ under reaction conditions.
As probed by in-situ XAS characterizations on ZnO1-x-Cu2O/Cu in Fig. 4a–c, the fraction of Cu+ was variable due to partial Cu+ species without the guard of Zn oxide. More importantly, the literature has rarely reported the correlations between Cu+ fraction in total copper species during CO2RR and the selective synthesis of ethanol versus ethylene. Therefore, the FEC2+ was first correlated with the fraction of Cu⁺ species and the applied reaction potential for ZnO1-x-Cu2O/Cu (Fig. 4g). It was observed that FEC2+ increased from 62.3% at −0.5 V vs RHE to a maximum of 86.1% at −0.9 V vs RHE. However, the fraction of Cu+ decreases from ~40% to ~28%. As the reaction potential was further increased to −1.1 V, the fraction of Cu+ remained stable, while the FEC2+ declined to 80.1%, caused by the competition of HER at high potential as the FE of H2 increased to 9.3%. The volcano-shaped dependence of FEC2+ on Cu+ fraction revealed that the enhanced C-C coupling on ZnO1-x-Cu2O/Cu was not solely related to the concentration of Cu+, but primarily ascribed to the synergy of -Zn-O-Cu+···Cu⁰ interface, which promoted the sequential process of CO2 → C1 → *CO-COH coupling, respectively57,58.
Additionally, the correlation between the FE of C2H5OH (Fig. 4h) or C2H4 (Fig. 4i) with the fraction of Cu+ species and the reaction potential was analyzed for ZnO1-x-Cu2O/Cu. With increasing reaction potential, the FEC2H5OH rised from 39.8% at −0.5 V vs RHE to 72.9% at −0.9 V, then decreased to 64.6% at −1.1 V, exhibiting a similar trend as FEC2+. In contrast, the FEC2H4 declined from 9.5% at −0.5 V to 5.6% at −0.9 V vs RHE, then slightly increased to 6.5% at −1.1 V, showing an opposite trend as FEC2H5OH. Given the variation in Cu+ concentration with potential, the distinct dependencies of FEs on reaction potentials for C2H5OH compared to C2H4 demonstrated that the optimal Cu+ concentration was essential to produce ethanol59,60.
Mechanism study of CO2RR to ethanol using ZnO1-x-Cu2O/Cu
The aforementioned structure-reactivity analysis preliminarily disclosed the CO2RR proceeded via CO2 → C1 → C2+ and emphasized the Cu+ sites in ZnO1-x-Cu2O/Cu apparently affected the selectivities of ethanol and ethylene. However, the related reaction mechanisms and intrinsic understandings remain unclear.
Hence, we employed in situ electrochemical attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) to identify potential intermediates in the CO2RR process catalyzed by ZnO1-x-Cu2O/Cu and ZnO1-x/Cu, with only the former having Cu+. Figure 5a displays the ATR-FTIR spectra for the ZnO1-x-Cu2O/Cu working electrode tested at different potentials. The peak at ~2064 cm−1 corresponds to the stretching vibrations of *CO adsorbed on the copper surface, providing evidence for CO2 reduction. The intensity of this peak increases with the potential initially and then decreases, verifying CO2RR proceeds via *CO as the intermediate (Fig. S35). The characteristic peaks at ~1627 cm−1 can be attributed to the feature of *OCCOH intermediates, confirming the asymmetrical C-C coupling. Moreover, the key intermediate *OC2H5 for ethanol was detected at around 1328 cm−1, validating the ethanol formation pathway and reaching its maximum peak intensity at the optimal working potential (−0.9 V).

a Potential-depended in situ ATR-FTIR spectra for ZnO1-x-Cu2O/Cu. b Potential-depended in situ ATR-FTIR spectra for ZnO1-x/Cu. c Free energy diagrams of the *CO intermediate reduction after CO2 activation to C2H5OH and C2H4. d The energy barrier comparisons of C-C coupling and the critical transition states involved the synthesis C2H5OH or C2H4 on ZnO1-x/Cu with different Cu+ fractions in total Cu species. e The binding analysis of *CH2CH2O with Zn site in ZnO1-x/Cu with 25% and 50% Cu+ fraction in total Cu species. Relevant source data are provided as a Source Data file.
To investigate the influence of Cu+ on the evolution of different intermediates, the ATR-FTIR spectra of CO2RR on ZnO1-x/Cu were shown in Fig. 5b as well. As depicted, ZnO1-x/Cu has similar intermediates, indicating same reaction pathway occurred as ZnO1-x-Cu2O/Cu. However, the intensity of *CO peak on ZnO1-x/Cu is significantly stronger than that on ZnO1-x-Cu2O/Cu, while the intensities of *OCCOH and *OC2H5 are quite weak throughout the working potential range (Figs. S36, S37). This comparison suggested that CO2 was effectively activated on the surface of ZnO1-x/Cu, but due to the lack of the synergy of Cu+···Cu0 pairs (Figs. S38–S40), the kinetic rate of C-C coupling became sluggish. This phenomenon was particularly apparent at −0.9 V, consistent with previous electrochemical performance results. These findings demonstrated that the Cu+ sites stabilized by Zn oxide promote the hydrogenation of *CO and the rapid C-C coupling, thereby accelerating ethanol formation as indicated by the enhanced adsorption of oxygen-containing intermediates (*OCCOH, *OC2H5). This observation aligned well with the in situ XAS results.
Subsequently, we simulated the reaction pathway by theoretical calculation based on the mechanism disclosed by in situ ATR-FTIR above to gain the quantitative insights into the enhancements of Cu+ on C-C coupling and selectivity of ethanol at the molecular level. According to the aforementioned structure analysis of ZnO1-x-Cu2O/Cu during the reaction, the model of ZnO1-x clusters supported on Cu(111) surface of 6 × 6 × 3 atomic layers was adopted here as previously. Meanwhile, 25% Cu+ was built at the interface between ZnO1-x and metallic Cu, which was similar to the experimental value of ~28% obtained by in situ XAS characterizations. As shown in Figs. S41 and 5c, the reaction pathway consisted of three stages, i.e., CO2 electroreduction into *CO, C-C coupling, and the following synthesis of ethanol or ethylene. Initially, CO2 molecule was preferentially adsorbed at ZnO1-x clusters as reported by our previous study28, then the *COOH was produced by proton-coupled electron transfer (PCET) with the energy barrier of 0.43 eV, which gives *CO via another PCET step and *CO was stabilized by Cu+ with a high adsorption energy of −0.73 eV, while the second *CO intermediate was derived by the same procedure and adsorbed on the adjacent Cu0 site with a lower adsorption energy of −0.27 eV. Such a different binding strength with *CO highlighted Cu+ favored the enrichment of *CO for the generation of multi-carbon products61, which could be ascribed to the low d band center and more abundant empty orbitals of Cu+ compared to Cu0 62,63. Besides, the low CN (8.7 ~ 9.1) of Cu as revealed by in situ XAS (Table S5) compared to pure Cu metal surface (12) has also been reported to be advantageous for *CO accumulation.
Prior to the C-C coupling, because of the strong Cu-C binding, *CO at Cu+ was hydrogenated to form *COH. As a consequence, the next step proceeded through *OC-*COH coupling (Fig. S42) at Cu0···Cu+ with a barrier of 0.21 eV. To have a better understanding of the dependence of FEC2+ on the fraction of Cu+, we adjusted the density of Cu+ at the interface of ZnO1-x/Cu model and computed the barriers of C-C coupling (ΔEC-C). Figure 5d exhibited an inverted volcano-type relationship between Cu+ concentration in total Cu species and the C-C coupling energy barrier. In the model of Cu metal, a high ΔEC-C as 0.69 eV was found due to the lack of stabilization effect for *CO intermediates. On the other side, when the concentration of Cu+ increased beyond 25%, the proton transfer on Cu surface was impeded by the spatial hindrance caused by high coverage of *CO, which reduced the PCET rate, leading to a high barrier of 0.72 eV at the case of 75% Cu+.
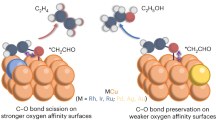
After the formation of *OC-COH, the CO2RR went through a succession of hydrogenation and deoxygenation steps. Herein, the potential determining step was probed as *CH2CHO + e− + H+ → *CH2CH2O with an energy change of 0.81 eV. The evolved *CH2CH2O intermediate was observed to connect with Zn site via Zn-O binding. Following this step, the reaction bifurcated into two pathways, i.e., the production of ethanol or ethylene, which was determined by the strength of Zn-O binding that can be modulated by the Cu+ density (Fig. 5d). As noted, Fig. 5d showed a reversed volcano correlation between the Cu+ ratio in total Cu species and the energy barriers to obtain transition states for ethanol (ΔECH3CH2O) or ethylene (ΔECH2CH2OH) synthesis, consistent with the current density trends of ethanol and ethylene (i.e., the reactivity) demonstrated in Fig. S43. Meanwhile, the difference between ΔECH3CH2O and ΔECH2CH2OH transition states was also plotted in Fig. S44, which represented the competition of ethanol and ethylene pathways, i.e., the selectivity. For instance, when this value was inclined to zero, the superiority of ethanol production would gradually disappear. As shown in Fig. 5d, all ΔECH3CH2O were lower than ΔECH2CH2OH at the cases of different Cu+ fractions, aligning with the experimental finding that the CO2RR to ethanol was dominated in this work. Moreover, the difference of these two values became significant only at optimal Cu+ fraction of ~25% (Figs. S45, S46), as demonstrated by the experimental results (~28%) in Figs. 4h, i. Compared to pure Cu metal surface, the addition of an appropriate amount of Cu+, with its more empty orbits, was beneficial to withdrawal electrons from ZnO1-x. This strengthened the binding energy of *CH2CH2O with Zn site via Zn-O bond, leading to the hydrogenation occurred at the *C end to produce *CH3CH2O with a lower barrier of 0.08 eV compared to 0.21 eV on metallic Cu, which produced ethanol ultimately. However, when more Cu+ formed, the improved chemical interactions between ZnO1-x and Cu substrate induced the partial reduction of Zn oxide as indicated by the in situ XAS results (Table S6), which increased the electron density of Zn site (Fig. S42) and reduced the binding energy of *CH2CH2O with Zn site, as evidenced by the higher ICOHP value increases from −1.431 (25% Cu+) to −1.400 (50% Cu+, Fig. 5e). The weak binding of *CH2CH2O with Zn sites promoted the hydrogenation occurred at the *O end to produce *CH2CH2OH, giving rise to the production of ethylene via dehydration, evidenced by the gradually decreased gap between ΔECH3CH2O and ΔECH2CH2OH. As a consequence, the selectivity of C2H4 would be progressively enhanced, matching with the dependence of FEC2H4 on potentials (Fig. 4i).
This study has reported an approach to synthesize ZnO1-x-Cu2O/Cu catalysts via a H2-C2H5OH successive pretreatment. A unique geometry of Cu nanoparticles covered by highly dispersed and disordered ZnO1-x shell was obtained, and an ethoxy-induced mechanism was demonstrated to be responsible for the formation of abundant Cu+ at the interface of ZnO1-x and Cu. The obtained ZnO1-x-Cu2O/Cu achieved FEC2+ over 86% with current density of 226 mA cm−2 and FEC2H5OH reaching 73% at −0.9 V vs RHE. Moreover, this catalyst exhibited an outstanding stability over 500 h of continuous operation, which was attributed to the stabilization of Cu+ under electroreduction conditions by the adjacent out-layer Zn oxides that were more resistant to reduction due to the high binding energy of Zn-O. Ultimately, a quantitative analysis regarding the correlation between Cu+ fraction in total Cu species and the synthesis of ethanol revealed that the presence of appropriate amount of Cu+ could strengthen the binding energy of *CH2CH2O with Zn and promote the ethanol formation. This work provided a strategy to design an efficient and stable Cu-based catalyst for CO2 electroreduction to ethanol.
Methods
Catalysts reparation
Materials
All chemicals were analytical reagents and were used without further purification. Anhydrous copper acetate was purchased from Shanghai Maclean Biochemical Technology Co., Ltd, and PVP was purchased from Aladdin. All other reagents were purchased from Sinopharm Chemical Reagent Co. Ultra-pure water was used throughout the experiments.
Synthesis of Cu NPs
Anhydrous copper acetate was added in 50 mL ethanol containing 2 g PVP to form a 40 mmol L−1 copper acetate solution. Under the condition of vigorous stirring, 1600 mg ascorbic acid aqueous solution was gradually added dropwise to the copper acetate solution, and kept at a constant temperature of 60 °C in a water bath, then stirred for 1 h. The obtained orange-red solution was centrifuged to obtain Cu particles, which were alternately washed three times with deionized water and ethanol.
Synthesis of ZnC12N2H8O4
The synthesis of ZnC12N2H8O4 involved the preparation of three solutions. Solution A: 1.6 mmol of Zn(NO3)2·6H2O was ultrasonically dispersed in 20 mL of deionized water. Solution B: 1.6 mmol of 2,2′-bipyridine was ultrasonically dispersed in 10 mL of methanol. Solution C: 0.8 mmol of oxalic acid was ultrasonically dispersed in 10 mL of deionized water. Solutions B and C were added to Solution A, and the mixture was stirred at 25 °C for 1 h. The resulting layered organozinc complexes were obtained by centrifugation, followed by washing three times with water and methanol, and drying under vacuum.
Synthesis of ZnO/CuO catalyst
The ZnO/CuO catalyst was synthesized using a mechanochemical process. The high-energy planetary ball milling was performed using an XQM series (zirconia milling jar, 80 mL capacity; zirconia milling balls, 2.5 mm diameter) from Changsha Tianchuang Powder Co., Ltd Typically, 200 mg of ZnC12N2H8O4 powder and 200 mg of Cu NPs powder, along with various numbers of milling balls, were milled at 870 rpm for 1 h to obtain the ZnO/CuO catalyst.
Synthesis of ZnO1-x/Cu catalyst
The obtained ZnO/CuO catalyst was further treated in a fixed-bed reactor. Specifically, the catalyst with a particle size of 125–250 µm was loaded into a quartz tube and subjected to in-situ treatment under 1 bar and 300 °C with a H2 flow rate of 100 mL min−1 for 1 h to obtain the ZnO1-x/Cu catalyst.
Synthesis of ZnO1-x-Cu2O/Cu catalyst
The obtained ZnO1-x/Cu catalyst was further treated in a fixed-bed reactor under atmospheric pressure. Specifically, the catalyst was loaded into a quartz tube and subjected to in situ treatment with an ethanol vapor for 1 h. Liquid ethanol (99.8% purity) was evaporated into a continuous vapor stream using a temperature-controlled evaporation system. The evaporation chamber was maintained at 150 °C (well above the boiling point of ethanol: 78.4 °C) to ensure complete vaporization and avoid condensation. The vapor flow rate was precisely regulated to 30 mL min−1 using a calibrated mass flow controller (MFC) designed for organic vapors. The MFC was calibrated against ethanol vapor density at 150 °C, ensuring accurate volumetric flow control. The reactor and gas lines were uniformly heated to 300 °C during pretreatment, ensuring ethanol remained in the gaseous state (critical temperature: 243 °C). Subsequently, the system was purged with He gas to remove all residual ethanol and other intermediates, resulting in the ZnO1-x-Cu2O/Cu catalyst.
Electrochemical experiments
All electrocatalytic experiments were conducted using an electrochemical workstation (CH Instruments 660, Inc., USA). The flow cell configuration included a gas diffusion electrode (GDE, 1 × 3 cm) loaded with the catalyst as the working electrode, an ion exchange membrane, and an iridium-coated titanium plate (1 × 3 cm) as the anode. A Hg/HgO electrode served as the reference electrode. The catalyst ink was formulated by dispersing 10 mg of ZnO1-x-Cu2O/Cu powder in a mixture of 300 μL isopropanol, 660 μL deionized water, and 40 μL of 5 wt% Nafion® D521 dispersion, followed by 30-minute sonication in an ice bath to ensure homogeneity. The ink was uniformly drop-coated onto a hydrophobic carbon paper substrate (Sigracet 28BC, 20 wt% PTFE) to achieve a catalyst loading of 1.0 mg cm−2. The cathodic compartment was continuously supplied with CO2 gas (99.999% purity) at a flow rate of 15 sccm, regulated by a calibrated mass flow controller (Sevenstar Electronics). The 0.1 M KOH electrolyte was circulated through the cathodic chamber at 10 mL min−1 via a peristaltic pump (Ditron Technology), with a water-jacketed reservoir maintaining the electrolyte temperature at 25 ± 2 °C. A Sustainion® anion-exchange membrane (Dioxide Materials) separated the cathodic and anodic compartments to prevent product oxidation, and the electrolyte pH was monitored.
Before the electrocatalytic experiments, 20 cyclic voltammetry (CV) scans were performed at a scan rate of 50 mV s−1 between 0 V and −2.0 V (vs. Hg/HgO). Linear sweep voltammetry (LSV) curves were recorded at a scan rate of 50 mV s−1. The ECSA was estimated by measuring the capacitive current associated with double-layer charging from the scan-rate dependence of CVs. The potential window of CVs was 0.091 V ~ −0.09 V vs. Hg/HgO, and the scan rates were 20, 40, 60, 80, 100, and 120 mV/s. The linear slope can be used to represent the electrochemically active surface area.
All potentials were converted to the reversible hydrogen electrode (RHE) using the following formula:
all the potentials were not corrected by iR compensation. Gas-phase and liquid-phase samples were collected after stabilizing each potential for 15 min. Gas-phase products were analyzed using a gas chromatograph (GC; Superlab Smart GC) equipped with a packed column (Porapak N 80/100 mesh), a thermal conductivity detector (TCD; for H2), and a flame ionization detector (FID; for CO, CH4, and C2H4). Liquid-phase products were analyzed by proton nuclear magnetic resonance spectroscopy (1H NMR, Bruker 600 MHz). To achieve high-rate CO2 reduction in a flow cell reactor, it is essential to perform a comprehensive carbon balance analysis. The total carbon input, supplied as CO2 from the inlet of the reactor, must be meticulously accounted for across all possible pathways. This includes carbon converted into CO2 reduction products, carbon sequestered in the form of carbonates via reactions with the electrolyte, and carbon associated with any unreacted CO2 that exits the reactor as residual gas.
Faradaic efficiency (FE) of the products was calculated using the following formula:
where Z is the number of electrons required to form 1 mol of product, F is the Faraday constant (96,485 C mol−1), n is the total amount of product for each sampling time (in mol), I is the current, and t is the running time for reaction.
The formation rate (R) of each product was calculated using the following formula:
where S was the geometric area of the electrode (cm2).
The CO2 conversion rate was determined using the following formula:
where fCO2 was the CO2 flow rate (mol s−1).
The energy efficiency (EE) was calculated on the basis of the cathodic CO2RR coupled with anodic water oxidation reaction (\({O}_{2}+4{H}^{+}+4{e}^{-}\leftrightarrow 2{H}_{2}O\)) by following literature with the equation:
where \({E}_{{{ox}}^{0}}\) and \({E}_{{{red}}^{0}}\) are the thermodynamic potentials for H2O oxidation (1.23 V versus RHE) and CO2RR to ethanol (0.09 V versus RHE), respectively. Eox and Ered are the applied potentials at anode and cathode.
Catalysts characterization
The crystal structure of the as-prepared catalysts was determined with a Bruker (APEXII) diffractometer using Cu Kα radiation with a wavelength of 1.5406 Å. The X-ray diffraction (XRD) patterns were recorded between 20 and 80° with a step size of 0.02° and a scan rate of 0.5 s/step.
The morphology of the as-prepared catalysts was studied by transmission electron microscopy (TEM) using a JEOL JEM-2100F instrument operating at an acceleration voltage of 200 kV. The elemental distribution for reduced catalysts was determined by scanning TEM combined with energy dispersive X-ray analysis (STEM-EDX) using a FEI cubed Cs-corrected Titan instrument operating at an acceleration voltage of 300 kV.
The X-ray photoelectron spectroscopy (XPS) tests were performed on a Thermo Scientific ESCALAB Xi+ spectrometer equipped with a high-pressure reactor using Al Kα radiation (1486.6 eV). The carbon peak at 284.8 eV was used as a reference.
The in-situ attenuated total reflection-surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) experiments were conducted in a modified electrochemical cell integrated into a Nicolet 6700 Fourier transform infrared spectrometer equipped with a liquid nitrogen-cooled MCT detector. Catalyst ink was drop-cast onto a Ge ATR crystal coated with an Au film. A Pt foil served as the counter electrode, and a Hg/HgO electrode as the reference electrode. Prior to measurements, the detector was cooled with liquid nitrogen for at least 30 min to ensure signal stability. The electrolyte used was 0.1 M KOH. Before testing, CO2 gas was bubbled through the electrolyte and continuously during measurements. Spectra were recorded while stepwise varying the potential from OCP to −1.8 V vs. RHE, using the spectrum collected at open circuit potential as the background.
The operando XAS spectra were measured in transmission mode at 20 kV and 40 mA using the RapidXAFS 2 M system (Anhui Absorption Spectroscopy Analysis Instrument Co., Ltd.). A Si (533) spherically bent crystal analyzer with a 500 mm curvature radius was employed for Cu and Zn analysis. The operando XAS experiments were conducted using a custom-designed electrochemical flow cell fabricated from PEEK (polyether ether ketone). The X-ray window comprised a 14 mm-diameter Kapton film (100 μm thickness) mounted on a threaded flange, enabling precise control over the liquid film thickness between the working electrode and the window. The working electrode consisted of a 1.5 × 3 cm2 carbon paper substrate. A Hg/HgO reference electrode (1 M KOH) reference electrode was employed. During operando XAS collection, the CO2 electroreduction reaction was conducted using an electrochemical workstation (CHI 760E). Potentiostatic electrolysis was performed in the range from open circuit potential to −1.2 V vs. RHE. The acquired XAS data were processed in Athena (version 0.9.26) for background, pre-edge, and post-edge calibration, followed by Fourier transform fitting in Artemis (version 0.9.26). For wavelet transform analysis, χ(k) data exported from Athena were imported into the Hama Fortran code with the following parameters: sample R range of 1–4 Å, k range of 0–15 Å−1, k-weighting of 2, and the Morlet function (κ = 10, σ = 1) as the mother wavelet to provide the overall distribution64.
Density functional theory calculations
All the density functional theory calculations were completed using the Vienna ab initio simulation package (VASP)65,66. The projector augmented wave (PAW) method was adopted to describe the ion-electron interactions67. The generalized gradient approximation in the Perdew-Burke-Ernzerhof (PBE) function was used, and the cut-off energy of 500 eV was set for the plane waves basis68,69. The thickness of the vacuum space was set to 20 Å to prevent interactions between the different atomic layers, and the Brillouin zone was sampled using Monkhorst-Pack 3 × 3 × 1 k-points for the structure calculations. Grimme’s DFT-D3 scheme of dispersion correction was adopted to describe van der Waals (vdW) interactions in the systems70. The energy and force convergence thresholds for the iteration in self-consistent field (SCF) were set to 10−5 eV and 0.02 eV·Å−1, respectively. The model of ZnO1-x/Cu was established according to the results of XRD and TEM electron microscopy. The model consists of Cu and ZnO1-x two sections. The Cu section was the pure Cu (111) surface containing a 6 × 6 × 3 atomic layer. The ZnO1-x section was a cluster containing ten ZnO molecule structures. Then the ZnO1-x/Cu model was constructed by the Cu surface as the support of ZnO1-x cluster. In all models, cyan represents Cu0, blue represents Cu+, pink represents Zn, red represents O, brown represents C, and white represents H, respectively. The atomic coordination of model is provided as a Supplementary Data 1 file.
The adsorption energies (\({E}_{{\mbox{ad}}}\)) were obtained by the following equation:
In the above equation, the \({E}_{{\mbox{total}}}\), \({E}_{{\mbox{clean catalysts}}}\), and \({E}_{{\mbox{adsorbent}}}\) represent the total energies of the adsorbent-adsorbed catalysts system, clean catalysts, and the adsorbent in vacuum, respectively.
The Gibbs free energy change (\(\Delta G\)) for each reaction step was calculated by the computational hydrogen electrode (CHE) model proposed by Nørskov and co-workers, which uses one-half of the chemical potential of hydrogen as the chemical potential of the proton-electron pairs71. The \(\Delta G\) was obtained by the following equation:
In the above equation, the \(\Delta E\) is reaction energy obtained directly from the DFT calculation. The \(\Delta {E}_{{\mbox{ZPE}}}\) and \(T\Delta S\) are the contributions of the zero-point energy and entropy for the \(\Delta G\). \(T\) represents the temperature, which was set as 298.15 K. The parameters \(e\) and \(U\) represent the number of electrons and the applied electrode potential, respectively. \({\Delta G}_{{\mbox{pH}}}\) is the free energy correction of pH, which could be calculated by \({\Delta G}_{{\mbox{pH}}}={k}_{{\mbox{B}}}T\times {\mbox{pH}}\times {{\mathrm{ln}}}\, 10\). Notably, the pH value was set to be zero in this work.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information file. The data generated in this study for main manuscript are provided in the Source Data file. Other raw data of the presented figures and tables are available from the corresponding authors upon request. Source data are provided with this paper.
References
Ma, W. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
An, B. et al. Cooperative copper centres in a metal–organic framework for selective conversion of CO2 to ethanol. Nat. Catal. 2, 709–717 (2019).
Wang, P. et al. Boosting electrocatalytic CO2–to–ethanol production via asymmetric C–C coupling. Nat. Commun. 13, 3754 (2022).
Li, Y. C. et al. Binding site diversity promotes CO2 electroreduction to ethanol. J. Am. Chem. Soc. 141, 8584–8591 (2019).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Luc, W. et al. Two-dimensional copper nanosheets for electrochemical reduction of carbon monoxide to acetate. Nat. Catal. 2, 423–430 (2019).
Zhan, C. et al. Revealing the CO coverage-driven C–C coupling mechanism for electrochemical CO2 reduction on Cu2O nanocubes via Operando Raman spectroscopy. ACS Catal. 11, 7694–7701 (2021).
Gao, J. et al. Selective C–C coupling in carbon dioxide electroreduction via efficient spillover of intermediates as supported by Operando Raman spectroscopy. J. Am. Chem. Soc. 141, 18704–18714 (2019).
Kim, Y. et al. Time-resolved observation of C–C coupling intermediates on Cu electrodes for selective electrochemical CO2 reduction. Energy Environ. Sci. 13, 4301–4311 (2020).
Gao, W., Xu, Y., Fu, L., Chang, X. & Xu, B. Experimental evidence of distinct sites for CO2-to-CO and CO conversion on Cu in the electrochemical CO2 reduction reaction. Nat. Catal. 6, 885–894 (2023).
Liu, K. et al. Transient pulsed discharge preparation of graphene aerogel supports asymmetric Cu cluster catalysts promote CO2 electroreduction. Nat. Commun. 16, 1203 (2025).
Du, C. et al. Cascade electrocatalysis via AgCu single-atom alloy and Ag nanoparticles in CO2 electroreduction toward multicarbon products. Nat. Commun. 14, 6142 (2023).
Xia, C. et al. Highly selective electrocatalytic CO2 conversion to tailored products through precise regulation of hydrogenation and C–C coupling. J. Am. Chem. Soc. 146, 20530–20538 (2024).
Wang, Y. et al. The role of phase mixing degree in promoting C−C coupling in electrochemical CO2 reduction reaction on Cu-based catalysts. Angew. Chem. Int Ed. 63, e202400952 (2024).
Zhang, Z. et al. Two ships in a bottle” design for Zn–Ag–O catalyst enabling selective and long-lasting CO2 electroreduction. J. Am. Chem. Soc. 143, 6855–6864 (2021).
Shen, H. et al. Asymmetrical C–C coupling for electroreduction of CO on bimetallic Cu–Pd catalysts. ACS Catal. 12, 5275–5283 (2022).
Chu, Y. et al. Dynamic (sub)surface-oxygen enables highly efficient carbonyl-coupling for electrochemical carbon dioxide reduction. Adv. Mater. 36, 2400640 (2024).
Yue, K. et al. Stabilized Cu0-Cu1+ dual sites in a cyanamide framework for selective CO2 electroreduction to ethylene. Nat. Commun. 15, 7820 (2024).
Chen, X. et al. Multivalent Cu sites synergistically adjust carbonaceous intermediates adsorption for electrocatalytic ethanol production. Nat. Commun. 15, 7691 (2024).
Herzog, A. et al. Operando Raman spectroscopy uncovers hydroxide and CO species enhance ethanol selectivity during pulsed CO2 electroreduction. Nat. Commun. 15, 3986 (2024).
Xu, H. et al. Highly selective electrocatalytic CO2 reduction to ethanol by metallic clusters dynamically formed from atomically dispersed copper. Nat. Energy 5, 623–632 (2020).
Ding, J. et al. A tin-based tandem electrocatalyst for CO2 reduction to ethanol with 80% selectivity. Nat. Energy 8, 1386–1394 (2023).
Lian, Z., Dattila, F. & López, N. Stability and lifetime of diffusion-trapped oxygen in oxide-derived copper CO2 reduction electrocatalysts. Nat. Catal. 7, 401–411 (2024).
Li, D., Li, X. & Gong, J. Catalytic reforming of oxygenates: State of the art and future prospects. Chem. Rev. 116, 11529–11653 (2016).
Peng, C. et al. 111) Facet-oriented Cu2Mg intermetallic compound with Cu3-Mg sites for CO2 electroreduction to ethanol with industrial current density. Angew. Chem. Int Ed. 63, e202316907 (2024).
Okatenko, V. et al. Alloying as a Strategy to boost the stability of copper nanocatalysts during the electrochemical CO2 reduction reaction. J. Am. Chem. Soc. 145, 5370–5383 (2023).
Tian, G. et al. Understanding the potential screening effect through the discretely structured ZnO nanorods piezo array. Nano Lett. 20, 4270–4277 (2020).
Zhang, F. et al. Revealing the dynamics of oxygen vacancy in ZnO1–x/Cu during robust methanol synthesis from CO2. ACS Catal. 14, 7136–7148 (2024).
Álvarez Galván, C. et al. Reverse water-gas shift reaction at the Cu/ZnO interface: influence of the Cu/Zn ratio on structure-activity correlations. Appl. Catal. B Environ. 195, 104–111 (2016).
Cai, Z. et al. Hierarchical Ag-Cu interfaces promote C-C coupling in tandem CO2 electroreduction. Appl. Catal. B: Environ. 325, 122310 (2023).
Morales, C., del, Campo, A., Méndez, J., Prieto, P. & Soriano, L. Re-oxidation of ZnO clusters grown on HOPG. Coatings 10, 401 (2020).
Yang, J., Zhang, M., Chen, M., Zhou, Y. & Zhu, M. Oxygen vacancies in piezoelectric ZnO twin-mesocrystal to improve peroxymonosulfate utilization efficiency via piezo-activation for antibiotic ornidazole removal. Adv. Mater. 35, 2209885 (2023).
Gaur, A., Shrivastava, B. D. & Joshi, S. K. Copper K-edge XANES of Cu(I) and Cu(II) oxide mixtures. J. Phys. Conf. Ser. 190, 012084 (2009).
DuBois, J. L. et al. A systematic K-edge X-ray absorption spectroscopic study of Cu(III) sites. J. Am. Chem. Soc. 122, 5775–5787 (2000).
Hsu, H. S. et al. Evidence of oxygen vacancy enhanced room-temperature ferromagnetism in Co-doped ZnO. Appl. Phys. Lett. 88, 242507 (2006).
Liu, X. et al. In situ spectroscopic characterization and theoretical calculations identify partially reduced ZnO1−x/Cu interfaces for methanol synthesis from CO2. Angew. Chem. Int Ed. 61, e202202330 (2022).
Mora-Fonz, D. et al. Why are polar surfaces of ZnO stable?. Chem. Mater. 29, 5306–5320 (2017).
Jin, S. et al. Alcohol-induced strong metal-support interactions in a supported Copper/ZnO catalyst. Angew. Chem. Int. Ed. 62, e202301563 (2023).
Rodríguez, C., Moreno, S. & Molina, R. Operando DRIFT-MS for studying the oxidative steam reforming of ethanol (OSRE) reaction. MethodsX 10, 102169 (2023).
Rodríguez, C., Moreno, S. & Molina, R. Operando DRIFT-MS study of oxidative steam reforming of ethanol (OSRE) on Ni-Co mixed oxides under non-equilibrium conditions. Chem. Eng. J. 480, 148243 (2024).
Bai, H. et al. The active site of syngas conversion into ethanol over Cu/ZnO/Al2O3 ternary catalysts in slurry bed. J. Catal. 380, 68–82 (2019).
Lorenzut, B. et al. Hydrogen production through alcohol steam reforming on Cu/ZnO-based catalysts. Appl. Catal. B Environ. 101, 397–408 (2011).
Zhang, L. et al. Oxophilicity-controlled CO2 electroreduction to C2+ alcohols over Lewis acid metal-doped Cuδ+ catalysts. J. Am. Chem. Soc. 145, 21945–21954 (2023).
Niu, Z.-Z., Chi, L.-P., Liu, R., Chen, Z. & Gao, M.-R. Rigorous assessment of CO2 electroreduction products in a flow cell. Energy Environ. Sci. 14, 4169–4176 (2021).
Rabiee, H. et al. Rational designing microenvironment of gas-diffusion electrodes via microgel-augmented CO2 availability for high-rate and selective CO2 electroreduction to ethylene. Adv. Sci. 11, 2402964 (2024).
Jouny, M., Hutchings, G. S. & Jiao, F. Carbon monoxide electroreduction as an emerging platform for carbon utilization. Nat. Catal. 2, 1062–1070 (2019).
Jouny, M. et al. Formation of carbon–nitrogen bonds in carbon monoxide electrolysis. Nat. Chem. 11, 846–851 (2019).
Montoya, J. H., Shi, C., Chan, K. & Nørskov, J. K. Theoretical insights into a CO dimerization mechanism in CO2 electroreduction. J. Phys. Chem. Lett. 6, 2032–2037 (2015).
Chen, C. et al. Exploration of the bio-analogous asymmetric C–C coupling mechanism in tandem CO2 electroreduction. Nat. Catal. 5, 878–887 (2022).
Paez-Ornelas, J. I. et al. Understanding the role of oxygen vacancies in the stability of ZnO(0001)-(1 × 3) surface reconstructions. J. Phys. Chem. C. 125, 7980–7989 (2021).
Guo, S.-T. et al. Stabilizing undercoordinated Zn active sites through confinement in CeO2 nanotubes for efficient electrochemical CO2 reduction. Angew. Chem. Int. Ed. 63, e202314099 (2024).
Wang, J. et al. Experimental and DFT study of electrochemical promotion of Cu/ZnO catalysts for the reverse water gas shift reaction. ACS Sustain. Chem. Eng. 12, 11044–11055 (2024).
Meng, H. et al. Designing Cu0−Cu+ dual sites for improved C−H bond fracture towards methanol steam reforming. Nat. Commun. 14, 7980 (2023).
Chung, K. et al. Non-oxidized bare copper nanoparticles with surface excess electrons in air. Nat. Nanotechnol. 17, 285–291 (2022).
Morales, M. R. et al. In-depth structural and analytical study of the washcoating layer of a Mn-Cu monolithic catalyst using STEM-FIB, EDX and EELS. Insights into stability under working conditions. Appl. Surf. Sci. 563, 150318 (2021).
Bogatyrenko, S. & Kryshtal, A. Thermal expansion coefficients of Ag, Cu and diamond nanoparticles: in situ TEM diffraction and EELS measurements. Mater. Charact. 178, 111296 (2021).
Xiao, H., Cheng, T., Goddard, W. A. & Sundararaman, R. Mechanistic explanation of the pH dependence and onset potentials for hydrocarbon products from electrochemical reduction of CO on Cu(111). J. Am. Chem. Soc. 138, 483–486 (2016).
Zhang, X. Y. et al. Direct OC-CHO coupling towards highly C2+ products selective electroreduction over stable Cu0/Cu2+ interface. Nat. Commun. 14, 7681 (2023).
Gu, Z. et al. Efficient electrocatalytic CO2 reduction to C2+ alcohols at defect-site-rich Cu surface. Joule 5, 429–440 (2021).
Rahaman, M., Dutta, A., Zanetti, A. & Broekmann, P. Electrochemical reduction of CO2 into multicarbon alcohols on activated Cu mesh catalysts: an identical location (IL) study. ACS Catal. 7, 7946–7956 (2017).
Garza, A. J., Bell, A. T. & Head-Gordon, M. Mechanism of CO2 reduction at copper surfaces: pathways to C2 products. ACS Catal. 8, 1490–1499 (2018).
Li, C.-G. et al. Insights into the structures and electronic properties of Cun+1μ and CunSμ (n = 1–12; μ = 0, ±1) clusters. Sci. Rep. 7, 1345 (2017).
Geoghegan, B. L. et al. Combining valence-to-core X-ray emission and Cu K-edge X-ray absorption spectroscopies to experimentally assess oxidation state in organometallic Cu(I)/(II)/(III) complexes. J. Am. Chem. Soc. 144, 2520–2534 (2022).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12, 537–541 (2005).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P. et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 46, 6671–6687 (1992).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244–13249 (1992).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput Chem. 27, 1787–1799 (2006).
Nørskov, J. K. et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 152, J23 (2005).
Acknowledgements
This work is supported by Zhejiang Provincial Key R&D Program (2024C03109), National Key Research and Development Program of China (2022YFE0128600), National Natural Science Foundation of China (22278365, 22178299), Natural Science Foundation of Zhejiang Province (LR22B060002), Research Funds of Institute of Zhejiang University-Quzhou (IZQ2023KJ4004), China Postdoctoral Science Foundation (2024M752806). The authors would also like to thank the shiyanjia lab (www.shiyanjia.com) for the support of XPS test.
Author information
Authors and Affiliations
Contributions
F.Z. and P.X. conceptualized and designed the experiments. F.Z. and C.W. conducted the experimental work, N.C. established the theoretical framework, designed, and performed the simulations. S.W. assisted with XAS characterizations. Y.H. and Y.S. essential material resources. M.Y. provided essential characterization resources. Y.B. contributed to the interpretation of the data. Z.L. contributed to the experimental designing and data analysis. P.X. supervised the project and took primary responsibility for drafting the manuscript, with support from F.Z. All authors offered valuable feedback, contributing to the research direction, analysis, and final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, F., Cao, N., Wang, C. et al. In situ stabilization of Cu+ for CO2 Electroreduction via Environmental-molecules-induced ZnO1-x shield. Nat Commun 16, 6082 (2025). https://doi.org/10.1038/s41467-025-61189-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61189-z
