
Abstract
There is a pressing need for soft robots capable of rapid movement at extreme environments, necessitating the development of ionic conductors with high ionic conductivity, low Young’s modulus, high elasticity, and high stability at extreme environments. However, existing ionic conductor materials cannot meet all of above needs. The preparation of water-free ionic conductive organogels is expected to solve the above problems. In this work, we design a synergy strategy to combine ion-dipole interactions with hydrogen bonds and successfully obtain ethylene glycol (EG) gels at a low polymer concentration of 5 mol%. Our EG gels possess high ionic conductivity (1.31 mS cm−1), low Young’s modulus (7.3 kPa), high elasticity (~100%), and good extreme environmental stability. The EG gels can work stably for long time at extreme environments (−27 to 123 °C and 10% to 70% relative humidity (RH)). Furthermore, we successfully achieved dielectric elastomer actuator devices stable operation at extreme environments and with 42 times longer lifetime than commonly used ionogels. We also achieved a soft robot that can work stably at extreme environments with a high speed of 3.6 BL s−1 (body lengths per second).
Similar content being viewed by others


Introduction
Soft robots based on dielectric elastomer actuators (DEAs) have shown great potential in microfactories1 and medical health2,3. DEAs with camouflage4, flight5, stealth6, amphibious7, and deep-sea exploration8, have been demonstrated. There is an urgent need for soft robots capable of stable operation and rapid movement in extreme environments, such as deserts and polar regions. These requirements impose stringent demands on the electrode materials, which must simultaneously exhibit high ionic conductivity, low Young’s modulus, high elasticity, and good stability at extreme environments9,10. Additionally, for applications such as stealth and camouflage, the electrode materials must have high transparency.
However, electrode materials with all of these properties don’t yet exist. Commonly used carbon grease suffers from issues such as easy shedding, opacity, and significant thermal effects, while inorganic nanomaterials have poor tensile properties and low transparency9. Suo et al.11 used a hydrogel ionic conductor as the electrode. Wang et al.12 brought ionogel ionic conductors into DEAs and then further introduced water stable ionic conductors into DEAs and realized a camouflaged soft robot7. However, hydrogels, due to the evaporation of water, exhibit very poor stability in extreme environments13. Moreover, ionic liquids are prone to leakage at high temperatures, which will cause the failure of DEAs7.
Ethylene glycol (EG) is widely used as a coolant in automotive applications14. In 2017, Liu et al.15 introduced EG into hydrogels to create anti-freezing conductive organohydrogels, which can operate from −55 to 44.6 °C. Since then, EG has been extensively used to address the issue of low-temperature stable operation16,17,18,19,20,21,22. However, hydrogels lose water and invalid above 60 °C16. Thus, the presence of water in hydrogels or organohydrogels precludes the long-term stability at extreme temperatures.
Water-free ionic conductive organogels are a promising alternative. EG is a good candidate. However, due to the weak solvating ability, the interactions between EG and polymers are weaker compared to hydrogels. This makes it a challenge to gelate EG. To date, only a few studies have successfully gelated EG at high molar fractions (>20 mol%)23,24,25. But they don’t have ionic conductivity and have high Young’s modulus. Yan et al.26 reported that PVA can gelate EG at a low concentration (13.3 mol%) in the presence of LiCl. However, due to its high Young’s modulus and poor elasticity, this deep eutectic gel is unsuitable for soft robot applications. There is an urgent need to develop a gel ionic conductor with low Young’s modulus, high elasticity, high ionic conductivity, and good extreme environmental stability to meet the requirements for stable operation of soft robots at extreme environments.
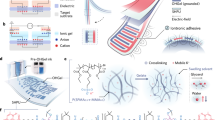
Herein, we designed a synergy strategy to enhance the interactions between polymers and solvents by introducing ion-dipole interactions and successfully gelated EG at a low molar fraction of 5 mol% (Fig. 1a). The gelation mechanism of EG gel and the source of extreme environmental stability were revealed by molecular dynamics (MD) and density functional theory (DFT) simulation. The obtained EG gels possessed high ionic conductivity (1.31 mS cm−1), low Young’s modulus (7.3 kPa), high elasticity (~100%) and good extreme environmental stability. These EG gels could work stably at extreme temperatures (−27 to 123 °C), extreme humidity (10% to 70% relative humidity (RH)) and high electric field (Fig. 1b). Even at −80 °C, the EG gel didn’t freeze. Utilized these extreme environmental stable EG gels, we successfully achieved extreme environmental stable DEAs with a high area strain of 54.9%. And the lifetime increased 42 times compared to commonly used ionogels (Table 1). We also achieved a soft robot that could work stably at extreme environments with a high average speed of 3.6 BL s−1 (body lengths per second). This study provides a general strategy for the preparation of extreme environmental stable gel ionic conductors for DEAs and soft robots.

a Diagram of intermolecular interactions in EG gels. The EG gel gelated at low concentration (5 mol%) by the ion-dipole interactions and H-bonds. b Schematic diagram of stable EG gel ionic conductors at extreme environments.
Results
Gelation mechanism of EG gels based on ion-dipole interactions and H-bonds synergy strategy
First, we selected EG as the gel electrolyte solvent (Supplementary Table 1). Then, we tried to use acrylamide (AAm), which could form multiple strong H-bonds with EG to gelate EG but failed (Fig. 2a). Conventional polymer monomers, including acrylic acid (AAc), N, N-dimethyl acrylamide (DMAAm), methyl acryloxyethyl trimethyl ammonium chloride (MATAC), etc., all failed to gelate EG (Supplementary Figs. 1,2). Another conventional polymer PVA cannot fully dissolve in EG due to the presence of numerous H-bonds, resulting in a brittle white opaque solid. Only when the LiCl concentration was increased to 2 M, the PVA could gelate EG but with a high Young’s modulus (Supplementary Fig. 3). These results showed that the previous hydrogen-bond-driven strategy used in hydrogels could not work in EG gels. Because of the significantly lower H-bond density in EG, the interactions between EG and polymers were significantly smaller than that of water (Supplementary Fig. 4).

a Polymerization process of AAm in EG and a schematic diagram of no gelation of AAm in EG. b Polymerization process of SBMA in EG and schematic diagram of EG gels. c The ESP of SBMA and schematic diagram of strong ion-dipole interactions and H-bonds between SBMA and EG. Unit: kcal mol-1. d EG coordination numbers of C12 and O15 of SBMA. e Comparison of van der Waals interactions, H-bonds and ion-dipole interactions between SBMA and EG and AAm and EG at 25 °C. f Comparison of van der Waals interactions, H-bonds, and ion-dipole interactions between SBMA and EG and AAm and EG at 100 °C. g Raman spectra of EG, SBMA and SBMA-EG-2 M gel. h 1H-NMR (600 MHz, DMSO-d6) of SBMA in SBMA-DMSO solution at 25, 50, 75 and 100 °C. i, j 1H-NMR (600 MHz, pure EG) of SBMA in SBMA-EG solution at 25, 50, 75 and 100 °C. Scale bars: 5 mm.
To enhance the solvent-polymer interactions, we introduced ion-dipole interactions into the polymer networks. For this, we chose a zwitterionic monomer, 2-(N-3-sulfopropyl-N, N-dimethylammonium) ethyl methacrylate (SBMA), which had both positive and negative charges in its side chain, as the gelation polymer. As shown in Fig. 2b, SBMA could gelate EG liquids quickly and get an EG gel with good mechanical properties because of the strong ion-dipole interactions and H-bonds with EG. We also calculated the electrostatic potential (ESP) of SBMA and AAm (Fig. 2c and Supplementary Fig. 5). The results showed that the cationic and anionic parts of SBMA had higher positive and lower negative potentials than that of AAm, respectively. This indicated that SBMA could form stronger ion-dipole interactions and H-bonds with EG.
To compare the strength of the solvent-polymer interactions, we conducted MD simulations (Fig. 2d, Supplementary Figs. 6–7 and Supplementary Table 2). These simulations revealed that SBMA had larger coordination numbers than AAm, indicating that SBMA formed stronger intermolecular interactions with EG (Supplementary Fig. 8). Additionally, we utilized the OPLS-AA force field to quantitatively assess the strength of ion-dipole interactions and H-bonds (Fig. 2e, f)27. The total interaction energy between SBMA and EG was approximately 2.403 kJ mol-1 at 25 °C, which was 2.5 times greater than AAm and EG. Importantly, ion-dipole interactions contributed 57.4% of the total interaction energy, indicating that strong ion-dipole interactions were the primary driving force for the gelation of EG. Additionally, at 100 °C, the H-bonds in AAm-EG-2 M are reduced to 64% of their 25 °C values, while the ion-dipole interactions remain largely unchanged, highlighting the stability of ion-dipole interactions at high temperatures (Supplementary Table 3).
We further experimentally demonstrated the existence of ion-dipole interactions and H-bonds in SBMA-EG. We first used FTIR to prove the presence of H-bonds in SBMA-EG (Supplementary Fig. 9). Then, we conducted a Raman test. As shown in Fig. 2g, the O-S-O symmetric stretching peak shifted from 1035 cm-1 in SBMA to 1039 cm-1 in the SBMA-EG gel28. This blueshift was indicative of the formation of strong ion-dipole interactions and H-bonds between the O of SBMA and H of EG, leading to an increased electron cloud density around the O-S-O group. Additionally, the peak at 866 cm-1, corresponding to the C-C stretching vibration of EG, remained unchanged29,30. To further characterize the presence of ion-dipole interactions and H-bonds, we employed variable-temperature H-NMR. As shown in Fig. 2h and Supplementary Fig. 10, in the non-protonic solvent DMSO, as the temperature increases from 25 °C to 100 °C, the H-bonds are gradually disrupted. This leads to an increase in the electron cloud density around the H atoms. As a result, the shielding effect experienced by the H atoms increases. Consequently, the effective magnetic field strength felt by the H nuclei decreases, resulting in a decrease in the chemical shift. Therefore, the water peak shifts to a higher field31. In contrast, in the protic solvent EG, the hydroxyl hydrogen of EG significantly moves upfield with increasing temperature, indicating that the disruption of H-bonds causes the corresponding hydrogen to move upfield, while ion-dipole interactions cause a slight downfield shift in hydrogen. If ion-dipole interactions were absent, H5 to H9 would exhibit a larger upfield shift. But there are only a minor upfield shift in the chemical shifts of H5 to H9 in SBMA due to the combined effect of these two interactions (Fig. 2i, j and Supplementary Fig. 11). This indicates that there were ion-dipole interactions between SBMA and EG. Therefore, the ion-dipole interactions and H-bonds collectively play a crucial role in promoting the gelation of EG molecules.
Mechanical properties and extreme environmental stability of EG gels and microscopic interpretation of stability
Using this strategy, we successfully obtained EG gels with different concentrations, from 5 mol% (1 M) to 30 mol% (6 M). As shown in Supplementary Fig. 12, the fracture stress of the SBMA-EG gels increased from 0.02 MPa to 0.1 MPa, while the maximum tensile strain decreased from 1750% to 1000%. Cyclic tensile tests indicated that SBMA-EG-2 M maintained good elasticity, retaining over 90% of its initial elastic properties, and exhibited a relatively low hysteresis of 15.7%. The samples recovered within seconds after stretching to 900% (Supplementary Fig. 13).
We compared the SBMA-EG-2 M gel with a hydrogel gelated with 2 M AAm. Our EG gels showed significantly superior mechanical and weight stability at both low and high temperatures compared to the hydrogels (Supplementary Figs. 14–20 and Supplementary Movies 1,3,4). Experimental results revealed that the evaporation rate of hydrogels at room temperature was approximately 62 times higher than that of EG gels (Supplementary Fig. 16d,20c). As shown in Supplementary Note 1 and Supplementary Table 4, the evaporation process of solvents in hydrogels and EG gels has been investigated theoretically. Our findings revealed that the evaporation rate of the solvent is directly proportional to its saturated vapor pressure. This discovery not only elucidated the high stability of EG gels at extreme environments, but also provided a robust theoretical foundation for the development of more extreme environmental stable gel ionic conductors. The exceptional extreme environmental stability of EG gels was primarily attributed to the low volatility of EG (Supplementary Fig. 21,22). To further elucidate this, we conducted MD simulations using Gromacs32 and Multiwfn33,34 to compare the evaporation properties of EG and water. Our results indicated that the evaporation behaviors were strongly influenced by intermolecular H-bonds and Mean Square Displacement (MSD)35. Specifically, EG exhibited a larger average H-bonds number, higher H-bond lifetime, higher H-bond energy, and lower MSD, which collectively contributed to its longer lifetime and slower evaporation rate compared to water at high temperatures (Supplementary Figs. 23–25 and Supplementary Table 5). Furthermore, variable-temperature H-NMR experiments confirmed that the H-bonds in EG were more stable than those in water at high temperatures (Supplementary Fig. 26). Furthermore, SBMA-EG-2 M exhibited lower decrease in average H-bonds number and H-bond energy and longer H-bond lifetime compared to EG due to the strong and stable ion-dipole interactions and H-bonds between SBMA and EG (Supplementary Fig. 27). Therefore, the good extreme environment stability of the EG gel is not only due to the stable H-bonds in EG but also attributed to the presence of strong ion-dipole interactions and H-bonds between SBMA and EG.
Mechanical properties of EG gel ionic conductors and applicability of the gel mechanism
To prepare EG gel ionic conductors, we added different concentrations of LiCl into SBMA-EG system. We found that the LiCl didn’t affect the gelation process of SBMA in EG, increased the Young’s modulus of EG gels and introduced more ion-dipole interactions, further improved the extreme environmental stability of EG gels (Supplementary Figs. 28–31). The addition of LiCl disrupted the phase-separated structure in SBMA-EG system due to self-association, increasing the transparency of the EG gel from nearly 0% to 78%, which was further demonstrated by SEM (Supplementary Fig. 32)36. This significant enhancement in transparency is crucial for optical applications such as camouflage4 and stealth6. As the concentration of SBMA decreased, the Young’s modulus and stretchability both decreased and reached the minimum at 2 M (10 mol%) (Fig. 3a and Supplementary Fig. 33). When the concentration was reduced to 1 M (5 mol%), the stretchability increased to 1760% due to the disruption of aggregated structures. But too low a concentration can led to lack of self-supporting and difficulties in preparation. Therefore, we selected 2 M (10 mol%) for further demonstration.

a The tensile curves of SBMA-EG-LiCl gels with different concentrations of SBMA. b Cyclic tensile curve of SBMA-EG-2 M-LiCl for 1000 times at the loading rate of 400 mm min−1 with no relaxation time between two cycles under 300% strain. c Tensile curves of SBMA-EG-2 M-LiCl at different temperatures. d DSC curves of SBMA-EG-2 M-LiCl and AAm-H2O-2 M-LiCl from −80 to 25 °C. e DMA curves of SBMA-EG-2 M-LiCl from −80 to 20 °C. f The temperature-dependent sweep measurements of SBMA-EG-2 M-LiCl with the range from −20 to 100 °C. g Weight change of SBMA-EG-2 M-LiCl and AAm-H2O-2 M-LiCl after 1 day at −20 and 1 h at 100 °C. Data are presented as mean values +/− SD, n = 3. h Tensile curves of SBMA-EG-2 M-LiCl at 10% and 70% RH for 7 days and 40% RH for 4 months. i Weight change of SBMA-EG-2 M-LiCl and AAm-H2O-2 M-LiCl after 7 days at 10% and 40% RH at 25 °C. Data are presented as mean values +/− SD, n = 3. j Stretched display of AAm-H2O-2 M-LiCl at −20 °C after 24 h using camera. k Stretched display of SBMA-EG-2 M-LiCl at –20 °C after 24 h using a camera. l Stretch display of AAm-H2O-2 M-LiCl at 100 °C after 1 h using a camera. m Stretch display of SBMA-EG-2 M at 100 °C after 2 h using a camera. n Stretched display of SBMA-EG-2 M-LiCl in the refrigerator at −27 °C using infrared thermal imaging camera. o Stretch display of SBMA-EG-2 M-LiCl on a heating table at 123 °C using infrared thermal imaging camera. p Stretched and elasticity display of SBMA-EG-2 M-LiCl near a flame. Scale bars: 1 cm.
Moreover, SBMA-EG-2 M-LiCl exhibited good elasticity and showed insensitivity to the loading rate (Supplementary Fig. 34). The tensile curve remained stable, with only a minor decrease in stress after 1000 cycles due to dissipation caused by the viscosity of EG, while maintaining nearly 100% elasticity and a low hysteresis of 10% (Fig. 3b and Supplementary Fig. 35)37. This was comparable to that of well-known highly resilient hydrogels (94-98%) (Supplementary Figs. 36, 37)13. The samples recovered within seconds after stretching to 800% (Supplementary Fig. 38 and Supplementary Movie 4). In addition, the EG gel exhibited good cyclic compression stability, with a relatively weak dependence on compression rate and good cyclic performance. During the first 100 cycles, the compression curve showed a little hysteresis due to dissipation caused by the viscosity of EG. However, after 100 cycles, the curve became stable with no relaxation time (Supplementary Fig. 39).
Although AAm can form EG gels at high concentrations (6 M, 30 mol%), these gels exhibit a high Young’s modulus (12.8 kPa) and poor elasticity (<80%) (Supplementary Figs. 40, 41). However, while deep eutectic gels can gelate EG, they exhibit a high Young’s modulus (63.6 kPa) and poor elasticity (<50%) (Supplementary Fig. 42)26. Therefore, AAm-EG-6 M-LiCl and deep eutectic gels both detrimental to achieving large area strains in DEAs and rapid movement in soft robots.
This method is also applicable to other polyzwitterions, such as carboxybetaine methacrylate (CBMA) and 2-methylacryloxyethyl phosphate choline (MAPC). As demonstrated in Supplementary Figs. 43, 44, both CBMA and MAPC can gelate EG at 5 mol% and can self-supporting, regardless of the presence of LiCl, due to the strong interactions between CBMA/MAPC and EG, which are confirmed by DFT calculations. The materials prepared at 10 mol% display good stretchability and elasticity. This method can be readily applied to large-area preparation (Supplementary Fig. 45).
Extreme environmental stability of EG gel ionic conductors
SBMA-EG-2 M-LiCl demonstrated high stability over a broad temperature range. Tensile tests conducted at various temperatures confirmed that the EG gel maintained good stretchability and stability across the entire temperature range (Fig. 3c and Supplementary Fig. 46). As shown in Fig. 3d, SBMA-EG-2 M-LiCl didn’t exhibit any noticeable crystallization peak in the DSC curve from −80 to 25 °C, whereas AAm-H2O-2 M-LiCl displayed a distinct crystallization peak at −13.4 °C. The storage modulus (G’) and loss modulus (G”) of SBMA-EG-2 M-LiCl didn’t increase significantly until the temperature approached −40 °C (Fig. 3e). Rheological tests further revealed that the storage modulus of the EG gel increased by 37% at −20 °C and decreased by 20% at 100 °C compared to room temperature, demonstrating good thermal stability. At room temperature, the loss tangent was 0.31, indicating superior elastic properties (Fig. 3f and Supplementary Fig. 47). Indeed, SBMA-EG-2 M-LiCl exhibited good weight stability across a wide temperature range. It retained 100% of its initial mass after 1 day at −20 °C, 98% after 7 days at 25 °C, and 80% after 1 h at 100 °C. In contrast, AAm-H2O-2 M-LiCl showed significant water loss even at room temperature (Fig. 3g).
Furthermore, our EG gel demonstrated remarkable stability across a broad humidity range from 10% to 70% RH. Tensile tests conducted at various humidity confirmed that the EG gel maintained good stability across this wide humidity range (Fig. 3h and Supplementary Fig. 48). Specifically, SBMA-EG-2 M-LiCl demonstrated good weight stability from 10% to 40% RH and were almost free of any leakage during use (Fig. 3i and Supplementary Fig. 49). Although it has components such as LiCl which was easy to absorb water, EG gel still maintained good stability at high humidity (70% RH), and the quality is only increased by 40%. Notably, after being heated at 60 °C for 1 h, the mass of the EG gel recovered to its initial value, and its mechanical properties were largely restored (Supplementary Figs. 50, 51).
We further demonstrated the extreme environmental stability of our EG gels compared to hydrogels. At −20 °C, AAm-H2O-2 M-LiCl froze after 1 day and broke under stretch, while SBMA-EG-2 M-LiCl still recovered within seconds after stretching to 1500% (Fig. 3j, k, and Supplementary Movie 2). At 100 °C, AAm-H2O-2 M-LiCl became dry and brittle after 1 h, breaking under 100% strain, whereas SBMA-EG-2 M-LiCl recovered within seconds after stretching to 800% (Fig. 3l, m and Supplementary Movie 2). Furthermore, we demonstrated the good stretchability and elasticity of SBMA-EG-2 M-LiCl at extreme environments using an infrared thermal imaging camera (Fig. 3n, o and Supplementary Movie 3). Additionally, SBMA-EG-2 M-LiCl retained its good stretchability and elasticity even when exposed to a flame (Fig. 3p and Supplementary Movie 4).
Electrical properties and extreme environmental stability of EG gel ionic conductors
Additionally, by reducing the polymer concentration and increasing the LiCl concentration, the ionic conductivity can be further improved to 2.55 mS cm-1 (Supplementary Fig. 52). However, too low a concentration of polymer can lead to issues such as lack of self-supporting and difficulties in preparation. Therefore, we selected 2 M (10 mol%) for further validation in DEA and soft robot applications. At the same time, the maximum solubility of LiCl in SBMA-EG-2 M is about 2.1 M, so we choose 2 M LiCl for subsequent application display. Our SBMA-EG-2 M-LiCl had a high ionic conductivity of 1.31 × 10-3 S cm-1 with a very low water content (1.5 wt%) at 25 °C (Fig. 4a, Supplementary Figs. 53, 54 and Supplementary Table 6). The EG gel maintains an ionic conductivity higher than 0.1 mS cm-1 over the entire temperature range from −20 °C to 150 °C, which is superior to most gel-based ionic conductors. This performance fully meets the conductivity requirements for electrode layers in DEAs and soft robotics. Achieving this combination of properties in a single material is highly challenging and has not been commonly reported (Fig. 4b and Supplementary Tables 8–11)38,39,40,41,42,43,44,45,46.

a Ionic conductivity of SBMA-EG-2 M-LiCl at different temperatures. Error bars show s.d., n = 3. b Comparison of performance of our EG gels and previously reported organohydrogels, organogels, deep eutectic gels, and ionogels. More information is seen in Supplementary Tables 8–11. c Ionic conductivity change of SBMA-EG-2 M-LiCl and AAm-H2O-2 M-LiCl after different times at 100 °C. Data are presented as mean values +/− SD, n = 3. d Ionic conductivity change of SBMA-EG-2 M-LiCl and AAm-H2O-2 M-LiCl after 1 day at −20 °C, 0 °C, 25 °C, 40 °C and 60 °C and after 3 h at 80 °C and 100 °C as a function of time. Data are presented as mean values +/− SD, n = 3. e Conductivity change of SBMA-EG-2 M-LiCl and AAm-H2O-2 M-LiCl after 1 day at 10% RH, 40% RH, and 70% RH and 25 °C as a function of time. Data are presented as mean values +/− SD, n = 3. f Display of SBMA-EG-2 M-LiCl lighting LED light at 100 °C. g Display of SBMA-EG-2 M-LiCl lighting LED light at −20 °C. Scale bars: 1 cm.
At 100 °C, SBMA-EG-2 M-LiCl maintained high ionic conductivity for 3 h, whereas AAm-H2O-2 M-LiCl rapidly decreased to nearly 0 after 1 h (Fig. 4c). Additionally, the electrical conductivity of SBMA-EG-2 M-LiCl could be restored within 1 s upon contact after damage (Supplementary Fig. 55).
We further evaluated the electrical stability of SBMA-EG-2 M-LiCl and AAm-H2O-2 M-LiCl at extreme environments. As shown in Fig. 4d, Supplementary Fig. 56 and Supplementary Table 6, AAm-H2O-2 M-LiCl exhibited high ionic conductivity only at moderate temperatures (0 to 40 °C) and high humidity (>40% RH), with a rapid decrease to near zero in other environments. In contrast, SBMA-EG-2 M-LiCl maintained high ionic conductivity (above 0.1 mS cm-1) and good electrical stability across a wide temperature range (−20 to 100 °C) and humidity range (10% to 70% RH) (Fig. 4d, e). After 7 days at 70% RH, the ionic conductivity of SBMA-EG-2 M-LiCl increased to 3.3 times its initial value. But it restored it to the original level after drying the sample at 60 °C for 1 h (Supplementary Fig. 57). This demonstrates the good electrical stability of the EG gel at high humidity. Additionally, SBMA-EG-2 M-LiCl successfully powered LED lights stably at −20 °C and 100 °C, demonstrating its potential for ionic conduction at extreme environments (Fig. 4f, g).
DEAs that work stably at extreme environments based on EG gel ionic conductors
Previous studies have demonstrated that an ionic conductivity greater than 0.01 mS cm-1 is sufficient to drive DEA devices and soft robots47. Our EG gel exhibits an ionic conductivity of up to 1.31 mS cm-1, which fully meets the required criteria. We further utilized our EG gel ionic conductors to construct a transparent DEA capable of operating at extreme environments48. The DEA’s working principle is based on the Maxwell stress generated by the electric field applied to the electrode layers (Fig. 5a)9. Through derivation, we found that the area strain of DEA is related to the Young’s modulus of the electrode layer (Supplementary Note 2). As shown in Supplementary Fig. 58, with the increase in the thickness of the EG gel, both the area strain and frequency response characteristics of the DEA device decreased. However, a thickness that is too low would bring problems of self-supporting and processing, so we chose an EG gel with a thickness of 0.8 mm as the electrode layer. As shown in Fig. 5b, Supplementary Fig. 59, Supplementary Table 7, and Supplementary Movie 5, SBMA-EG-2 M-LiCl achieved an actuation area strain of up to 54.9% at 38 MV m-1. This is higher than the 52.5% for carbon grease electrodes, 3 times that of hydrogel electrodes, and 2.4 times that of ionogel electrodes. The large strain can be attributed to the high stretchability (>700%), high ionic conductivity (1.31 mS cm-1), and low Young’s modulus (7.3 kPa) of SBMA-EG-2 M-LiCl (Supplementary Fig. 60). Due to the good elasticity of SBMA-EG-2 M-LiCl and Ecoflex 00-30 (Supplementary Fig. 61), the actuator exhibited good frequency response characteristics. At a high frequency of 100 Hz, the actuation area strain maintained 67.9% of its initial value, similar to the performance of carbon grease (Supplementary Fig. 62). Hydrogels completely dried out after 20 min at 100 °C and were unable to actuate at −20 °C (Fig. 5d and Supplementary Fig. 63). In contrast, DEAs based on SBMA-EG-2 M-LiCl could function in both high and low temperature environments. At 100 °C, the SBMA-EG-2 M-LiCl-based DEAs achieved 92.7% of initial area strain after 20 min (Fig. 5e and Supplementary Movie 6). After 3 h at 100 °C, they still maintained 40.8% of initial area strain, resulting in a 9-time longer lifetime compared to hydrogel-based DEAs (Supplementary Fig. 64a and Supplementary Movie 6). At −20 °C, the DEA device with SBMA-EG-2 M-LiCl gels achieved an area strain of 31.4% at 36 MV m-1 (Fig. 5f, Supplementary Fig. 64b, and Supplementary Movie 5).

a The working principles of DEAs which can work at extreme environments. b The actuation area strain of DEAs using SBMA-EG-2 M-LiCl, carbon grease, AAm-H2O-2 M-LiCl and PVDF-50 as electrode layer, f = 200 mHz. c The actuation area strain of DEAs at 100 °C before and after 20 min using AAm-H2O-2 M-LiCl as electrode layer. d The actuation area strain of DEAs at −20 °C before and after 1 day using AAm-H2O-2 M-LiCl as electrode layer. e The actuation area strain of DEAs at 100 °C before and after 20 min using SBMA-EG-2 M-LiCl as electrode layer. f The actuation area strain of DEAs at −20 °C before and after 1 day using SBMA-EG-2 M-LiCl as electrode layer. g Lifetime and lifetime ratio of DEAs with SBMA-EG-2 M-LiCl and PVDF-50 electrode layer at 25, 60 and 100 °C. h Area strain and lifetime of DEAs with SBMA-EG-2 M-LiCl and PVDF-50 electrode layer at 60 °C. i Comparison of performance of our DEAs based on EG gels and previously reported DEAs.
To evaluate the stability of DEA devices under long-term operation and to demonstrate the effect of ion-dipole interactions on device lifetime, we tested the lifetime of DEAs based on EG gel and ionogel electrodes at different temperatures. As shown in Fig. 5g, EG gels exhibited a lifetime of up to 300 min at 25 °C and 10% RH, which is 42 times longer than that of ionogels, due to the strong LiCl-polymer interactions and LiCl-EG interactions (Supplementary Fig. 29). At 60 °C, the lifetime of the DEA device is slightly reduced to 258 min and the area strain remains 86% after working at 60 °C for 4 h due to the weakening of these interactions and a small amount of volatilization of EG (Fig. 5h). At 100 °C, the LiCl-polymer interactions and LiCl-EG interactions were severely disrupted, making it easier for LiCl to leakage from electrodes to dielectric layer, leading to DEA failure7. Combined with the increased likelihood of thermal failure in the dielectric layer, the lifetime of DEAs was significantly reduced to 15.1 min, but still much higher than the 0.4 min for ionogels. Across all tested temperatures, the lifetime of the EG gel was over 40 times longer than that of the ionogel, primarily due to the greater propensity of the ionogel to undergo ionic leakage compared to the EG gel. The amperometric I-t tests revealed that the ionogel exhibits significant ionic leakage even at room temperature, which becomes more pronounced and rapid at 80 °C. In contrast, the EG gel showed no ionic leakage under either room temperature or 80 °C (Supplementary Fig. 65)49. Additionally, we measured the effect of humidity on the lifetime of EG gels. The lifetime of EG gels at 40% humidity was notably lower than at 10% humidity due to the absorption of water from the air, but it was still 25 times longer than that of ionogels (Supplementary Fig. 66)50,51. DEA devices can also work stably in humidity environments up to 70% RH and maintain almost 100% area strain of initial after 14 days at 70% RH (Supplementary Fig. 67).
To further investigate the lifetime of our DEA devices under more extreme environmental conditions, we tested them at 85 °C and 60% RH. As shown in Supplementary Fig. 68, our EG gels could still function normally after being placed under the stringent conditions of 85 °C and 60% RH for 5 h, achieving a relatively large strain of 9.6%, retaining 46.8% of the initial strain, and showing no obvious morphological changes. In contrast, under the same conditions, the ionogels only achieved a small strain of 3.5%, retained only 41.2% of the initial strain, and the electrode layer visibly detached, shrank towards the middle. This demonstrates that our EG gels possess more good stability under extreme environments than ionogels and can even operate stably after being stored for 5 h at 85 °C and 60% RH.
The area strain and operating temperature range of our DEA devices exceed most of the previous work (Fig. 5i and Supplementary Table 12)7,12,39. Furthermore, SBMA-EG-2 M-LiCl exhibited minimal heat generation during operation, whereas carbon grease electrodes showed rapid temperature increases (Supplementary Fig. 69 and Supplementary Movie 7). Our EG gels can operate stably over the long term on other dielectric layers, such as VHB and PDMS, and it also functions normally in a double-layer DEA devices, demonstrating the broad applicability of our EG gels (Supplementary Figs. 70 and 71).
Soft Robots that work stably at extreme environments based on EG gel ionic conductors
To verify the feasibility of our materials in soft robots, we fabricated soft robots based on DEAs without additional encapsulation7 (Fig. 6a and Supplementary Fig. 72) and demonstrated their fast crawling performance at extreme environments (Fig. 6b). To simplify, the locomotion speed (V) of soft robots is related to the frequency (f) and span (δ, the distance advanced by the foot in one cycle) by the equation: \(V=f\times \delta \)47. Our soft robots based on SBMA-EG-LiCl achieve a large span at 30 Hz, due to the large gtgarea strain and great high-frequency response characteristics (Fig. 6c). As shown in Fig. 6d, the crawling speed of the soft robot increased with frequency, reaching a maximum speed of 3.5 BL s-1, or approximately 15.4 cm s-1, at 30 Hz and 28 MV m-1. The soft robot also maintained high speeds at −20 °C and 100 °C (Supplementary Fig. 73 and Supplementary Movie 8). This is the first demonstration of a soft robot that can stably operate from −20 to 100 °C with an average speed of 3.6 BL s-1. AAm-EG-LiCl gels and deep eutectic gels cannot drive soft robots effectively due to their low area strains and poor high-frequency response characteristics, resulting in low span at 30 Hz (Supplementary Fig. 74). The operating temperature range (−20 to 100 °C) and speed (3.5 to 3.7 BL s-1) of our soft robots are significantly wider and higher, respectively, than most reported soft robots. Additionally, the required electric field intensity (28 to 32 MV m-1) is much lower than that of most reported soft robots (Fig. 6e, f)7,8,52,53,54,55,56,57,58. Figure 6g, h showed the fast crawling of soft robots at extreme temperatures, indicating that our soft robots could work stably at extreme environments.

a Structure diagram of the soft robot. b Schematic illustration of soft robot crawling at extreme environments. c Demonstration of the drive of soft robots in one cycle. d The speed of the soft robot varied with the frequencies and reached a maximum speed of 3.5 BL s−1, about 15.4 cm s−1 at a frequency of 30 Hz. Data are presented as mean values +/− SD, n = 3. e, f Comparison of our soft robot: e Crawl speed versus temperature range. f Crawl speed versus electric field. The data and references involved in the charts are presented in Supplementary Table 13. g The soft robot locomotion at a low temperature environment of −20 °C and reached a maximum speed of 3.7 BL s−1, about 16.3 cm s−1 at 32 MV m-1 and 30 Hz. h The soft robot locomotion at a high temperature environment of 100 °C and reached a maximum speed of 3.6 BL s−1, about 15.8 cm s−1 at 28 MV m-1 and 15 Hz. Scale bars: 1 cm.
Discussion
In this study, we successfully combined ion-dipole interactions with H-bonds to develop EG gels with a low polymer concentration of 5 mol%. The addition of LiCl will not affect the gelation process of EG. Our EG gel exhibits high ionic conductivity (1.31 mS cm-1), low Young’s modulus (7.3 kPa), high elasticity (~ 100%), and good stability at extreme environments. This strategy is versatile and can be easily extended to other zwitterionic monomers. We then fabricated DEAs that demonstrated large area strain and good extreme environmental stability. The lifetime of DEAs with EG gel electrodes was significantly longer than those with hydrogel and ionogel electrodes. Furthermore, we developed a soft robot that can work stably and fast crawling at extreme environments. This study offers an effective approach to address the challenge of gelation at low concentrations in organic solvents. This study also provides a reference for the development of next-generation soft robots that can operate stably and rapidly at extreme environments (Supplementary Note 3).
Methods
Materials
Acrylamide (AAm, 99%), 2-(N-3-sulfopropyl-N, N-dimethyl ammonium) ethyl methacrylate (SBMA, 98%), 3-sulfopropyl acrylate potassium salt (98%), methyl acryloxyethyl trimethyl ammonium chloride (MATAC, 50 wt% in water), N, N-dimethyl acrylamide (DMAAm, 98%) and lithium chloride (LiCl, 99%) were purchased from Meryer. Acrylic acid (AAc, 99%) and poly (vinyl alcohol) (PVA, 12.0-16.0 mPa‧s) was purchased from Aladdin. Ethylene glycol (EG, 99.5%) was purchased from Alfa Aesar. Carboxybetaine methacrylate (CBMA, 98%), 2-methylacryloxyethyl phosphate choline (MAPC, 98%), and 1-hydroxycyclohexyl phenyl ketone (HCPK, Irgacure 184, 99%) was purchased from Bide Pharmatech. Poly (ethylene glycol) diacrylate (PEGDA, Mw = 400, 98%) and 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)mide (EMITFSI, 98%) were purchased from Energy Chemical. Ecoflex 00-30 and Sil-Poxy™ were purchased from Smooth-on. Carbon grease (NyoGel 756 G) was purchased from Nye Lubricants. Poly (vinylidene fluoride-co-hexafluropropylene) (PVDF-HFP-4455) and VHB 4910 (0.5 mm) was purchased from 3 M company. PDMS was purchased from Dow Corning. All of the above materials were used as received.
Preparation of normal hydrogels and conductive hydrogels
First, 0.2824 g (4 mmol) of AAm and 0.1697 g (4 mmol) of LiCl were dissolved in 2 mL of water by stirring overnight. Then, 0.1 mol% PEGDA (1.4 μL) and 0.25 mol% HCPK (2.1 mg) relative to AAm were added to the solution and stirred until homogeneous. The mixture was poured into a dumbbell-shaped Teflon mold and exposed to UV light (320-400 nm, 36 W, 6 cm distance) for 1 h. The resulting hydrogel was then soaked in 2 M LiCl solution for 24 h to reach swelling equilibrium. These conductive hydrogels were denoted as AAm-H2O-2 M-LiCl. For the preparation of normal hydrogels, the same procedure was followed, except that LiCl was not added, and the hydrogels were soaked in deionized water for 24 h to reach swelling equilibrium. These normal hydrogels were denoted as AAm-H2O-2 M.
Preparation of normal EG gels and conductive EG gels
First, 0.1697 g (4 mmol) of LiCl was dissolved in 2 mL of EG by stirring overnight. Then, 1.117 g (4 mmol) of SBMA, 0.1 mol% PEGDA (1.4 μL), and 0.25 mol% HCPK (2.1 mg) relative to SBMA were added to the solution and stirred until homogeneous. The mixture was poured into a dumbbell-shaped Teflon mold and exposed to UV light (320-400 nm, 36 W, 6 cm distance) for 1 h. The resulting conductive EG gel samples were denoted as SBMA-EG-2 M-LiCl. Other concentrations of SBMA-EG-x M-LiCl were prepared using the same method by changing only the concentration of SBMA (x = 1, 2, 3, 4, 6). For the preparation of normal EG gels, the same procedure was followed, but without adding LiCl. These normal EG gels were denoted as SBMA-EG-x M, x = 1, 2, 4, 6. AAm-EG-6 M-LiCl, CBMA-EG-2 M, CBMA-EG-2 M-LiCl, MAPC-EG-2 M, and MAPC-EG-2 M-LiCl were prepared using the same methods. AAm, AAc, DMAAm, 3-sulfopropyl acrylate potassium salt, and MATAC were used to prepare EG gels using the same methods but all failed.
Preparation of PVA-EG-LiCl
The preparation way followed a previously reported method26. Firstly, 0.1/0.5/2/3.6 M LiCl was dissolved in EG at 100 °C to obtain a transparent clarified solution. Then added 10 wt% PVA, heated and stirred at 110 °C until uniform. Then cooled at room temperature, and frozen at −20 °C overnight, then thawed at room temperature for 3 h, and finally baked at 60 °C for 3 h. Finally, we only obtained PVA-EG-2 M LiCl and PVA-EG-3.6 M LiCl.
Preparation of dielectric layer of DEAs
The dielectric layer was prepared as follows: Parts A and B of Ecoflex 00-30 (1.4 g each) were thoroughly mixed in a 6 cm diameter plastic box. The mixture was degassed in a vacuum for 15 min to remove air bubbles, then cured at 25 °C for 2 days. The resulting dielectric layer had a diameter of 5.5 cm and a thickness of 1 mm.
Preparation of electrodes of DEAs
The preparation of hydrogel and EG gel electrodes for DEAs followed a similar procedure to that of conductive hydrogels and EG gels, but using a round Teflon mold. The resulting electrode layers had a diameter of 15 mm and a thickness of 0.8, 1.0, and 1.2 mm, respectively. A 0.8 mm electrode was selected for subsequent testing. For ionogel electrodes, 1 g of [EMI][TFSI] and 1 g of PVDF-HFP were dissolved in 3 mL of acetone and stirred for 24 h at 25 °C to achieve a well-dispersed solution. The solution was then slowly poured into a glass petri dish to allow the solvent to evaporate. The mixture was dried in a vacuum oven at 60 °C for 24 h. Finally, the ionogel was pressed at 60 °C for 15 min using a specific mold to obtain a round ionogel electrode, denoted as PVDF-50.
Preparation of PDMS
To obtain PDMS films with low Young’s modulus, we selected a polymer monomer to crosslinker ratio of 1:40. The mixture was thoroughly stirred in a circular plastic container with a diameter of 5.5 cm and then degassed under vacuum for 10 min to completely remove any bubbles. Subsequently, the mixture was cured overnight at 80 °C to ensure full crosslinking, resulting in a PDMS film with a final thickness of approximately 1 mm.
Preparation of DEAs
The Ecoflex 00-30 film was pre-stretched by 200% × 200% and mounted onto a rigid acrylic ring (D = 11 cm) using clips. Hydrogel and EG gel electrodes were placed at the center of each side of the pre-stretched film. Copper tapes were used to provide electrical connections to the high-voltage power supply (Model 615-10, TREK). PDMS and VHB 4910 were each pre-stretched to 150% × 150% and 280% × 280% times their original dimensions, respectively. Subsequently, DEA devices were assembled using the same method described above.
Preparation of soft robots
The preparation method of soft robots is as follows: 1) The Ecoflex 00-30 film was pre-stretched by 200% × 200% and mounted onto a rigid acrylic ring using clips. 2) PET frames with thicknesses of 0.2 mm and 0.6 mm were cut into specific shapes using a laser cutting system (Epilog Legend 36 EXT, Chinese Laser Systems) and glued to both sides of the Ecoflex 00-30 film with a specialized silicone binder (Sil-Poxy™, Smooth-on). The assembly was left to solidify in air for 1 day. 3) EG gel electrodes, prepared using a custom Teflon mold, were attached to the PET frames on both sides of the Ecoflex 00-30 film. 4) Excess Ecoflex 00-30 membrane was removed with a scalpel, and the pre-stretched film was allowed to bend into a specific curvature while restrained by a PET plate. 5) Customized feet were fitted at the four vertices of the soft robot. 6) The EG gel electrodes were connected to the high voltage power supply using 0.1 mm diameter copper wire.
Tensile mechanical test
Mechanical tests were performed using Sunstest UTM2502. Unless otherwise noted, all tensile tests were performed at room temperature (24 ± 1 °C) and ambient humidity (30 ± 10%) at the loading rate of 40 mm min−1, the size of all the specimens for tensile tests was 6 mm × 1.9 mm ± 0.1 mm. The resilience tests and cyclic compression performance were performed for 1000 times at the loading rate of 400 mm min−1 with no relaxation time between two cycles. Young’s modulus was calculated from the initial slope of the stress-strain curves. Each mechanical test was repeated with at least three individual samples.
Characterization
Transmission data was obtained using HITACHI UH4150 spectrophotometer. Differential scanning calorimetry (DSC) measurements were recorded on a TA Instruments DSC 250 with a heating speed of 5 °C min−1 from −80 to 25 °C. Dynamic thermomechanical analyzer (DMA) tests were carried on with TA Instruments DMA 850 with a heating speed of 3 °C min−1 from −80 to 20 °C with a strain of 20 μm and frequency of 1 Hz. Fourier transform infrared spectra (FTIR) was recorded in the wavenumber range from 400 to 4000 cm−1 measured using a Fourier transform infrared spectrometer (Bruker, Horiba, Germany). Raman spectroscopy was performed utilizing a Raman imaging microscope (LabRAM HR Evolution, HORIBA Jobin Yvon, Japan). The wavelength of the excitation laser was 532 nm. Raman mappings (scan range 22 μm × 22 μm; depth of scanning, 22 μm) were collected using the condition that the laser power was 8.0 mW, the exposure time was 0.1 s (10 Hz), and the scan time was 200, and the image pixel size was 1.0 μm. Nuclear magnetic resonance (NMR) spectra was acquired on a JEOL JNM-ECA600 spectrometer (600 MHz). To study the H-bond in water and EG, we performed NMR measurements using water and EG without deuteration. Rheology measurements were conducted using an Aaton Paar Physica MCR302 with a parallel plate of 8 mm diameter. The thickness of the samples was fixed to 1.0 mm. The temperature-dependent sweep measurements were performed at 1.0% strain and frequency of 6.28 rad s-1 from room temperature to 100 °C with a heating rate of 4.5 °C min-1 and continued measuring for 500 s after reaching 100 °C.
SEM imaging
The morphologies of all the gel samples were characterized by cold field emission scanning electron microscope (CFESEM, Hitachi SU8600) at an operating voltage of 3 kV. EG gels were frozen in liquid nitrogen and sliced to observe the cross-section while retaining the solvent.
Electrochemical properties
The electrochemical properties of the hydrogels and EG gels were measured using an electrochemical workstation (CHI 660E, Chenhua). The impedance data of hydrogels and EG gels were obtained using the electrochemical AC impedance spectroscopy (EIS) method with a frequency range from 1 to 106 Hz and 0.1 V voltage, under a variety of temperatures of −20, 0, 25, 50, 75, 100, 125, and 150 °C. Hydrogels and EG gels were sandwiched between two thin stainless-steel sheets for measurement. The ionic conductivity of hydrogels and EG gels was calculated according to the following equation:
where L is the distance between the two probes, R is the electrical resistance of the hydrogels and EG gels, and A is the cross-sectional area of the hydrogels and EG gels.
Ionic leak test
The degree of ion leakage can be characterized by the amount of ions entering the dielectric layer, i.e., the magnitude of the leakage current49. To characterize the degree of ionic leakage in EG gels and ionogels, we sandwiched VHB between two layers of PVDF-50 ionogel and EG gel, respectively. We then tested their amperometric I-t curves at room temperature and 80 °C for 1 h with an applied voltage of 5 V.
Stability test
To assess the weight stability under different environments, AAm-H2O-2 M, AAm-H2O-2 M-LiCl, SBMA-EG-2 M, and SBMA-EG-2 M-LiCl samples were subjected to the following environments: 1) Stored in a refrigerator at −20 °C and 40% RH for 7 days. 2) Exposed to air at 25 °C with 10% and 40% RH for 7 days. 3) Placed in an oven at 100 °C and 16% RH for 1 h. The weight changes of the samples were measured as a function of time. The weight retention was calculated according to the following equation:
where ω is the weight retention, m0 is the initial weight and m is the weight at different time.
To measure the stability of mechanical properties at different temperatures, SBMA-EG-2 M-LiCl was placed at different temperatures (−20, 60, 80, 100, 120 °C) and then quickly removed for tensile test. Consider the temperature error during loading, the actual temperature is approximately −10, 40, 60, 80, 100 °C, respectively. To evaluate the stability of mechanical properties at different humidity, SBMA-EG-2 M-LiCl was exposed to 10%, 40%, and 70% RH for varying durations, followed by tensile testing. To characterize the stability of electrical properties, AAm-H2O-2 M-LiCl and SBMA-EG-2 M-LiCl were subjected to different environmental environments: 1) Stored in a refrigerator at −20 °C and 0 °C for 1 day. 2) Exposed to air at 25 °C with 10% and 40% RH. 3) Placed in an oven at 40 °C and 60 °C for 1 day, and at 80 °C and 100 °C for 3 h. EIS was then used to measure the ionic conductivity of the gels under these environments, and the ionic conductivity changes were calculated as a function of time. We characterized the stability of the EG gel at 70% RH after different time, including weight stability, mechanical property stability, and electrical property stability. Additionally, we measured the changes in weight, mechanical properties, and electrical properties after exposure to 60 °C for 1 h.
Infrared thermal imaging measurements
An infrared thermal imaging camera (Fluke Ti450) was used to demonstrate the stretchability and rapid elastic recovery of SBMA-EG-2 M and SBMA-EG-2 M-LiCl at both high and low temperatures. The low-temperature environment was achieved by freezing the samples in a refrigerator at −20 °C for 15 min, while the high-temperature environment was achieved by heating the samples on a hot plate at 120 °C.
Performance characterization of DEAs
The lateral expansion of the DEAs was recorded using a video camera (Canon M50) at drive voltages ranging from 3 to 10 kV in 0.5 kV increments. The electric field intensity (E) was calculated using the formula:
where V is the drive voltage and D is the thickness of the dielectric layer after pre-stretching. Each voltage was held for 10 s. The frequency response of the DEAs was also captured by the same video camera at frequencies ranging from 0.1 to 100 Hz with a drive voltage of 8 kV. Image analysis software (ImageJ, NIH) was used to analyze the video images and calculate the change in the overlapping area of the top and bottom electrodes. The actuation area strain was determined with the following relation:
where α is the actuation area strain, A1 is the area in the actuated state and A0 is the initial area. High-temperature thermal stability was evaluated by placing the DEA devices in an oven at 100 °C. The devices were removed and tested immediately after different time intervals. Low-temperature drive performance was tested by placing the DEA devices in a refrigerator at −20 °C for 1 day, followed by immediate testing upon removal. For the lifetime test, DEA devices operated continuously at different temperatures (25, 60 and 100 °C) and humidity (10% and 40% RH) until failure with a voltage of 7 kV and a frequency of 200 mHz. To test the lifetime of DEAs assembled with our EG gel at more extreme environments, we assembled DEAs using both our EG gels and ionogels. These devices were then subjected to different durations of exposure at 85 °C and 60% RH and then test their area strains.
Performance characterization of soft robots
A video camera (Canon M50) was used to record the soft robot crawling on a customized substrate at frequencies of 2, 5, 10, 20, 30, and 40 Hz at room temperature. The crawling performance of the soft robot was also recorded at −20 °C in a refrigerator and at 110 °C on a hot plate, using the optimal frequency. The crawling speed was calculated using the following formula:
where V is the crawling speed, t is the crawl time and ∆x is the displacement of the soft robot’s back foot from its initial position.
Computational methods
All the DFT calculations were performed with the Gaussian 16 package on the chemistry department server at Tsinghua University. The B3LYP hybrid functional used a 6-311 + G** level of basis set for anioniccite and 6-311 G** level of basis set for other parts. And including the atom-pairwise dispersion (DFT-D3) correction with Becke-Johnson (BJ) damping to optimize geometry. The optimized geometry, frequency calculation and electrostatic potentials (ESPs) of every structure were carried out considering the implicit solvent model.
MD simulations were carried out using the Gromacs 2020.6 program32. For systems containing zwitterionic molecules with positive and negative charges, the Cl&P force field59 was selected to simulate the SBMA-EG system. For non-charged systems (H2O, EG, AAm-EG), the Optimized Potentials for Liquid Simulations All Atom27 (OPLS-AA) force field was employed. The parameters for SBMA were modeled using the Cl&P force field, and the topol and gro files for SBMA were generated using fftool60. The parameters for EG were taken from the OPLS-AA force field in fftool. The topol and gro files of AAm and EG were generated using the LigParGen server27,61,62,63. The Restrained ElectroStatic Potential (RESP)64 of AAm was calculated using the Multiwfn33 as the atomic partial charges. The water model was the SPC/E model65. The nonbonded potential energy was calculated by a Lennard-Jones (L-J) 12 − 6 potential and a Coulomb potential.
Where rij is the distance between atoms i and j, qi is the partial charge assigned to atom i, and εij and σij are energy and size parameters obtained by Jorgensen combining rules.
The H2O model contained 560 H2O molecules, the EG model contained 360 EG molecules, the SBMA-EG model contained 20 SBMA and 180 EG molecules, and the AAm-EG model contained 20 AAm and 180 EG molecules. The 50000-step steepest descent method was used to avoid unreasonable contact with the system. NPT ensemble was used to pre-equilibrate the system, and Berendsen temperature coupling and Berendsen pressure coupling were used to control the temperature to 298 K and 398 K, the pressure was maintained at 1 atm, the non-bonding cutoff radius was 1.4 nm, and the integration step was 2 fs. The NPT simulation used 10 ns for the EG model and 1 ns for the H2O model. Finally, a 10 ns simulation was performed by the GROMACS 2020.6 program. The bond length and angle were constrained by the LINCS algorithm. The Nose-Hoover temperature coupling and Parrinello-Rahman pressure coupling were used to control the temperature to 298 K and 398 K and the pressure was maintained at 1 atm. The two-way intercept was set to 1.2 nm, van der Waals interaction and the long-distance electrostatic interaction were set by the particle-mesh Ewald method. The trajectory file during the simulation was saved every 1.0 ps. Periodic boundary environments were applied in all directions. H-bonds were constrained. The last 5 ns were used for analysis. RDFs, intermolecular nonbonding interactions, and H-bonds were generated using Gromacs, visualizations were generated with VMD66.
The intermolecular nonbonding interactions between SBMA and EG included van der Waals interactions, ion-dipole interactions and H-bonds67. Since the interactions between all solute molecules and all EG molecules in the simulated system were calculated by gromacs, the average van der Waals interactions were calculated using the following formula:
Where Totalvan der Waals is the van der Waals interactions between all solute molecules and all EG molecules in the simulated system, V is the volume of the simulated system, ns is the number of the solute molecules, nEG is the number of EG molecules.
We used an order of gmx hbond for hydrogen bonds (H-bonds) analysis. The H-bond program used geometric criteria to determine the H-bonds, which was considered to be a H-bond when the H-bond donor-acceptor distance was less than 3.5 Å and the H-bond donor-acceptor angle was less than 30 degrees. We used the H-bond program to calculate the average existence time of H-bonds to obtain the autocorrelation function of H-bonds, and further describe the average lifetime and bonding energy of H-bonds. The average H-bond strength was calculated using the following formula:
where nH-bonds is the number of H-bonds between all solute molecules and all EG molecules in the simulated system, ns is the number of solute molecules, nEG is the number of EG molecules, EH-bonds is the H-bond energy, which is about 20 kJ mol-1, V is the volume of the simulated system. The average ion-dipole interactions were calculated using the following formula:
We calculate the volume H-bond density (VHB) by dividing the total number of H-bonds (nHB) by the total volume of the simulated system (V).
Data availability
The data that support the findings of this study are available within this article and its Supplementary Information. Molecular structure coordinates are provided with this article. All data are available from the corresponding author upon request.
References
Chung, S. E., Dong, X. & Sitti, M. Three-dimensional heterogeneous assembly of coded microgels using an untethered mobile microgripper. Lab Chip 15, 1667–1676 (2015).
Sitti, M. Miniature devices: voyage of the microrobots. Nature 458, 1121–1122 (2009).
Sitti, M. et al. Biomedical applications of untethered mobile milli/microrobots. P IEEE 103, 205–224 (2015).
Li, P. et al. Transparent soft robots for effective camouflage. Adv. Funct. Mater. 29, 1901908 (2019).
Ren, Z. et al. A high-lift micro-aerial-robot powered by low-voltage and long-endurance dielectric elastomer actuators. Adv. Mater. 34, e2106757 (2022).
Christianson, C., Goldberg, N. N., Deheyn, D. D., Cai, S. & Tolley, M. T. Translucent soft robots driven by frameless fluid electrode dielectric elastomer actuators. Sci. Robot. 3, eaat1893 (2018).
Cheng, Z. et al. A highly robust amphibious soft robot with imperceptibility based on a water-stable and self-healing ionic conductor. Adv. Mater. 35, 2301005 (2023).
Li, G. et al. Self-powered soft robot in the mariana trench. Nature 591, 66–71 (2021).
Qiu, Y., Zhang, E., Plamthottam, R. & Pei, Q. Dielectric elastomer artificial muscle: materials innovations and device explorations. Acc. Chem. Res. 52, 316–325 (2019).
Chen, B. et al. Highly stretchable and transparent ionogels as nonvolatile conductors for dielectric elastomer transducers. ACS Appl. Mater. Interfaces 6, 7840–7845 (2014).
Keplinger, C. et al. Stretchable, transparent, ionic conductors. Science 341, 984 (2013).
Cao, Y. et al. A Transparent, self-healing, highly stretchable ionic conductor. Adv. Mater. 29, 1605099 (2016).
Yang, C. & Suo, Z. Hydrogel ionotronics. Nat. Rev. Mater. 3, 125–142 (2018).
Yue, H., Zhao, Y., Ma, X. & Gong, J. Ethylene glycol: properties, synthesis, and applications. Chem. Soc. Rev. 41, 4218–4244 (2012).
Rong, Q. et al. Anti-freezing, conductive self-healing organohydrogels with stable strain-sensitivity at subzero temperatures. Angew. Chem. Int. Ed. 56, 14159–14163 (2017).
Han, L. et al. Mussel-inspired adhesive and conductive hydrogel with long-lasting moisture and extreme temperature tolerance. Adv. Funct. Mater. 28, 1704195 (2018).
Rong, Q., Lei, W., Huang, J. & Liu, M. Low Temperature tolerant organohydrogel electrolytes for flexible solid-state supercapacitors. Adv. Energy Mater. 8, 1801967 (2018).
Chen, F. et al. Rational fabrication of anti-freezing, non-drying tough organohydrogels by one-pot solvent displacement. Angew. Chem. Int. Ed. 57, 6568–6571 (2018).
Sui, X. et al. Zwitterionic osmolyte-based hydrogels with antifreezing property, high conductivity, and stable flexibility at subzero temperature. Adv. Funct. Mater. 30, 1907986 (2019).
Zhang, X. F. et al. Inorganic salts induce thermally reversible and anti-freezing cellulose hydrogels. Angew. Chem. Int. Ed. 58, 7366–7370 (2019).
Long, Y. et al. Super-stretchable, anti-freezing, anti-drying organogel ionic conductor for multi-mode flexible electronics. Adv. Funct. Mater. 33, 2304625 (2023).
Zhao, Y. et al. Wide-humidity range applicable, anti-freezing, and healable zwitterionic hydrogels for ion-leakage-free iontronic sensors. Adv. Mater. 35, 2211617 (2023).
Lee, Y.-Y. et al. A strain-insensitive stretchable electronic conductor: Pedot:pss/acrylamide organogels. Adv. Mater. 28, 1636–1643 (2016).
Wang, Z. et al. TouGh, transparent, 3D-printable, and self-healing poly(ethylene glycol)-gel (PEGgel). Adv. Mater. 34, 2107791 (2022).
Zhao, Y. et al. A self-healing electrically conductive organogel composite. Nat. Electron. 6, 206–215 (2023).
Wang, J., Zhan, B., Zhang, S., Wang, Y. & Yan, L. Freeze-resistant, conductive, and robust eutectogels of metal salt-based deep eutectic solvents with poly(vinyl alcohol). ACS Appl. Polym. Mater. 4, 2057–2064 (2022).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Xu, S. et al. Force-induced ion generation in zwitterionic hydrogels for a sensitive silent-speech sensor. Nat. Commun. 14, 219 (2023).
Jindal, A. & Vasudevan, S. Ethylene glycol dihedral angle dynamics: relating molecular conformation to the raman spectrum of the liquid. J. Phys. Chem. B 125, 1888–1895 (2021).
Kuzmin, V. V. et al. Correlations among the Raman spectra and the conformational compositions of ethylene glycol, 1,2- and 1,3-propylene glycols. J. Mol. Struct. 1243, 130847 (2021).
Xu, M. et al. Bicontinuous-phase electrolyte for a highly reversible Zn metal anode working at ultralow temperature. Energ. Environ. Sci. 17, 8966–8977 (2024).
Páll, S. et al. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 153, 134110 (2020).
Lu, T. & Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2011).
Zhang, J. & Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 23, 20323–20328 (2021).
Rana, B., Fairhurst, D. J. & Jena, K. C. Investigation of water evaporation process at air/water interface using hofmeister ions. J. Am. Chem. Soc. 144, 17832–17840 (2022).
Peng, B. et al. Phase-Separated polyzwitterionic hydrogels with tunable sponge-like structures for stable solar steam generation. Adv. Funct. Mater. 33, 2214045 (2023).
Sun, B., Liu, K., Wu, B., Sun, S. & Wu, P. Low-hysteresis and tough ionogels via low-energy-dissipating cross-linking. Adv. Mater. 36, e2408826 (2024).
Liao, H., Guo, X., Wan, P. & Yu, G. Conductive MXene nanocomposite organohydrogel for flexible, healable, low-temperature tolerant strain sensors. Adv. Funct. Mater. 29, 1904507 (2019).
Lee, J. et al. Water-processable, stretchable, self-healable, thermally stable, and transparent ionic conductors for actuators and sensors. Adv. Mater. 32, 1906679 (2020).
Gao, Y. et al. Highly stretchable organogel ionic conductors with extreme-temperature tolerance. Chem. Mater. 31, 3257–3264 (2019).
Yao, P. et al. Environmentally stable, robust, adhesive, and conductive supramolecular deep eutectic gels as ultrasensitive flexible temperature sensor. Adv. Mater. 35, 2300114 (2023).
Lu, C. et al. Liquid-free, anti-freezing, solvent-resistant, cellulose-derived ionic conductive elastomer for stretchable wearable electronics and triboelectric nanogenerators. Adv. Funct. Mater. 32, 2207714 (2022).
Li, T., Wang, Y., Li, S., Liu, X. & Sun, J. Mechanically robust, elastic, and healable ionogels for highly sensitive ultra-durable ionic skins. Adv. Mater. 32, 2002706 (2020).
Yu, Z. & Wu, P. Underwater communication and optical camouflage ionogels. Adv. Mater. 33, 2008479 (2021).
Lei, Z. & Wu, P. A highly transparent and ultra-stretchable conductor with stable conductivity during large deformation. Nat. Commun. 10, 3429 (2019).
Lyu, X. et al. Multi-modal sensing ionogels with tunable mechanical properties and environmental stability for aquatic and atmospheric environments. Adv. Mater. 36, 2410572 (2024).
Feng, W. et al. A large-strain and ultrahigh energy density dielectric elastomer for fast moving soft robot. Nat. Commun. 15, 4222 (2024).
Zhang, Y. et al. Highly transparent, underwater self-healing, and ionic conductive elastomer based on multivalent ion-dipole interactions. Chem. Mater. 32, 6310–6317 (2020).
Jin, Z. et al. Multivalent design of low-entropy-penalty ion-dipole interactions for dynamic yet thermostable supramolecular networks. J. Am. Chem. Soc. 145, 3526–3534 (2023).
Albuquerque, F. B. & Shea, H. Influence of humidity, temperature and prestretch on the dielectric breakdown strength of silicone elastomer membranes for DEAs. Smart Mater. Struct. 29, 105024 (2020).
Albuquerque, F. B. & Shea, H. Influence of electric field, temperature, humidity, elastomer material, and encapsulation on the lifetime of dielectric elastomer actuators (DEAs) under DC actuation. Smart Mater. Struct. 30, 125022 (2021).
Li, T. et al. Fast-moving soft electronic fish. Sci. Adv. 3, e1602045 (2017).
Pei, Q., Rosenthal, M., Stanford, S., Prahlad, H. & Pelrine, R. Multiple-degrees-of-freedom electroelastomer roll actuators. Smart Mater. Struct. 13, N86–N92 (2004).
Jing, Z., Li, Q., Su, W. & Chen, Y. Dielectric Elastomer-Driven Bionic Inchworm Soft Robot Realizes Forward and Backward Movement and Jump. Actuators 11, 227 (2022).
Gu, G., Zou, J., Zhao, R., Zhao, X. & Zhu, X. Soft wall-climbing robots. Sci. Robot. 3, eaat2874 (2018).
Wang, D. et al. Dexterous electrical-driven soft robots with reconfigurable chiral-lattice foot design. Nat. Commun. 14, 5067 (2023).
Ji, X. et al. An autonomous untethered fast soft robotic insect driven by low-voltage dielectric elastomer actuators. Sci. Robot. 4, eaaz6451 (2019).
Duduta, M., Clarke, D. R. & Wood, R. J. A High speed soft robot based on dielectric elastomer actuators. In 2017 IEEE International Conference on Robotics and Automation (ICRA) 4346-4351 (IEEE, 2017).
Canongia Lopes, J. N. & Pádua, A. A. H. CL&P: A generic and systematic force field for ionic liquids modeling. Theor. Chem. Acc. 131, 1129 (2012).
Padua, A., Goloviznina, K. & GONG, Z. agiliopadua/fftool: XML force field files (v1.2.1). Zenodo https://doi.org/10.5281/zenodo.4701065 (2021).
Jorgensen, W. L. & Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. PNAS 102, 6665–6670 (2004).
Dodda, L. S., Vilseck, J. Z., Tirado-Rives, J. & Jorgensen, W. L. 1.14*CM1A-LBCC: Localized bond-charge corrected CM1A charges for condensed-phase simulations. J. Phys. Chem. B 121, 3864–3870 (2017).
Dodda, L. S., Cabeza de Vaca, I., Tirado-Rives, J. & Jorgensen, W. L. LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 45, W331–W336 (2017).
Bayly, C. I., Cieplak, P., Wendy D, C. & Kollman, P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 97, 10269–10280 (1993).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Kumar, R., Schmidt, J. R. & Skinner, J. L. Hydrogen bonding definitions and dynamics in liquid water. J. Chem. Phys. 126, 204107 (2007).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22425504 (C.W.)).
Author information
Authors and Affiliations
Contributions
C.W. and L.S. conceived the concept and designed the experiments. L.S. carried out experiments, collected the data, and wrote the paper. W.W.F, C.Y.L., L.L.C., T.C. and K.Z.J. provided advice to the manuscript. All authors reviewed and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yi Cao who co-reviewed with Bin Xue and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, L., Feng, W., Liu, Y. et al. Elastic and ultra stable ionic conductors for long-life-time soft robots working at extreme environments. Nat Commun 16, 6004 (2025). https://doi.org/10.1038/s41467-025-61217-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61217-y
