
Abstract
Glycosylation changes are closely related to various diseases, including cancer. The quantitative analysis of site-specific glycans at proteomics scale remains challenging due to low glycopeptide spectra interpretation. Here, we present GlyPep-Quant, a tool for sensitive quantification and identification of site-specific glycans. Using a well-trained machine learning model, GlyPep-Quant quantified 25.1%–178.9% more site-specific glycans without missing values than pGlycoQuant, MSFragger-Glyco, and Skyline. To utilize identified information from previous large-scale dataset, an MS1 feature library-based “virtual match-between-runs” quantification scheme was developed, enabling over eightfold more site-specific glycan identification/quantification than conventional MS2-based methods. Enhanced coverage prompted the development of a glycoproteomic biomarker discovery method, involving calculation of site-specific glycan abundances ratios at the same glycosylation site, minimizing individual expression and experimental condition variability. Two pairs of site-specific glycan ratios on sites P01011-N127 and P08185-N96, were selected as high-performance biomarkers to classify gastric cancer (GC) from healthy controls (AUC > 0.95). Moreover, the two ratios performed well in distinguishing GC using an independent cohort by the library-based quantification strategy with diagnostic accuracy up to 85%. GlyPep-Quant is poised for broader glycoproteomic applications.
Similar content being viewed by others

Introduction
Aberrant glycosylation is considered as a hallmark of cancer1,2,3. Sensitive quantitative and qualitative profiling of protein glycosylation is essential for identifying dysregulated glycosylation patterns, aiding the discovery of novel therapeutic targets and biomarkers4. However, the inherent macro- and microheterogeneity of protein glycosylation1, along with its low abundance, complicates the analysis of site-specific glycans and results in a low interpretation rate. Although some alterations in site-specific glycosylation associated with cancer have been identified, their further clinical translation is significantly hindered by challenges in reproducible quantitative analysis of site-specific glycan across large clinical cohorts, variability between sample batches, and limited identification and quantification capacity in single MS files.
Mass spectrometry (MS)-based analysis of intact glycopeptides provides both qualitative and quantitative information of site-specific glycans5. In glycopeptide analysis, the lower instrument and data analysis requirements keep data-dependent-acquisition (DDA) as a common MS acquisition method6,7,8. However, the low interpretation rate of glycopeptide spectra9 contributes to the missing value issues in glycopeptide quantification. To enhance glycopeptide spectrum identification, several advanced software, like pGlyco310, MSFragger-Glyco11, has been presented in the past years. In 2022, we devised the spectrum expansion strategy by leveraging peptide fragment patterns to assist data interpretation, and integrated it into Glyco-Decipher9 software, which largely improved glycopeptide identification performance. However, the shared identifications across different dataset files were relatively low, and the quantitation efficiency of individual files remained limited for large-scale glycoproteomic analysis.
In DDA quantitative analysis, label-free quantification (LFQ) is one of the most widely used strategies without the need of additional sample processing steps12. However, it suffers from missing values (MVs) problem13 because of the stochastic nature of DDA analysis14. Compared to various imputation methods15 to deal with the MVs problem, match-between-runs11,16,17,18 (MBR) quantification strategy mitigate the problem more directly. In MBR, by using features of identified spectra precursors, the most likelihood corresponding peaks of the same precursors are located in another run containing missing values16. Recently, MSFragger16 and pGlycoQuant18 applied MBR to the quantitative analysis of glycoproteomics data at intact glycopeptide level, and were able to acquire more quantitative results with high confidence compared to other widely-used methods18. However, due to the high expression heterogeneity of glycosylation among individuals, the identification level of a single glycoproteomic file is still relatively low, even MBR is adopted. Therefore, it is essential to generate enough amount of data files to achieve high quantification coverage of the whole dataset. And more importantly, the MS1 feature information of elution profiles from previously quantified dataset has not been fully utilized.
Here, we develop GlyPep-Quant, a software tool for large-scale glycoproteomic quantitative analysis. To enhance the performance of LFQ-MBR quantification, GlyPep-Quant utilizes a well-trained machine learning model to evaluate the contribution of different features and assess the confidence of quantitative results. Based on this, a two-step elution profile extraction method was developed to precisely and efficiently locate the matched profile. Compared to other state-of-the-art tools like Skyline19, MSFragger-Glyco11, and pGlycoQuant18, GlyPep-Quant quantified 25.1%–178.9% more glycopeptides without any MVs. Based on the excellent quantitative performance, a special strategy was developed in GlyPep-Quant to investigate the occupancy ratio changes of site-specific glycans for biomarker discovery. Traditionally, site-specific glycans with altered abundances (directly from the intensity of LFQ) are considered as biomarker candidates. However, peptide abundances can be affected by different sample preparation periods, different instrument conditions, and individual variances of total glycoproteomic expression level. Instead, we choose to investigate abundance ratios of site-specific glycans on the same site, and the relative changes between glycan abundances are elucidated, offering a perspective that is independent of the influences mentioned above and distinct from the abundance profiles of individual glycopeptides. By applying this strategy, two ratios were discovered as promising biomarkers through analysis of GC dataset7. To expand the quantification coverage for data acquired under similar conditions and fully utilize the information of previous quantifications, an MS1 feature library-based virtual MBR quantification algorithm was presented. It consists of a library of summarized MS1 features of different glycopeptide precursors and adopts a cluster matching strategy to perform a one-direction virtual MBR process to maximally obtain quantified results. Since it is performed from library to new runs, rather than “between runs” in LFQ-MBR, it is termed “virtual MBR”. The implementation of library-based quantification further enhanced quantitative coverage, achieving up to an eightfold increase compared to conventional quantification method. This improvement enables the accurate quantification of site-specific glycan abundance ratios across large sample cohorts, providing a valuable tool for early diagnostic applications.
Results
Development of GlyPep-Quant
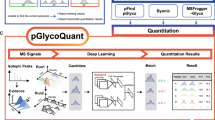
To enhance site-specific glycan quantification, which usually suffers from MV problem, we presented GlyPep-Quant, a comprehensive tool for large-scale glycoproteomic quantification analysis, and the analysis workflow consisted of four parts of novel methods and their applications (Fig. 1): (1) A new elution profile extraction strategy and a machine learning evaluation model for LFQ-MBR in GlyPep-Quant. (2) A biomarker discovery strategy, enabling the identification of abundance ratios of two glycans at the same glycosylation site as potential biomarkers. (3) An MS1 feature library-based quantification scheme in GlyPep-Quant to improve the identification and quantification of glycopeptides for new glycoproteome data acquired under similar conditions. (4) Direct disease diagnosis for individual samples based on the site-specific glycan ratios. GlyPep-Quant is integrated in our glycoproteomic data analysis software, Glyco-Decipher, and is also compatible with pGlyco310 identification.

The quantitative analysis in GlyPep-Quant includes two modules: label-free quantification with match-between-runs (LFQ-MBR) (left panel, red) and library-based identification and quantification (library-based virtual MBR, right panel, blue). In the LFQ-MBR process, MS files I and II in dataset A are utilized. Elution profiles are firstly extracted based on GPSM identification in MS file I, dataset A, and then the matched elution profiles are extracted from the matched data (MS file II, dataset A) using a two-step elution extraction method. The matched elution profiles include target (same precursor m/z value) and decoy elution profiles, and their confidence is evaluated by a well-trained machine learning model. For the generation of decoy elution profile, a random mass shift of 3–20 Th is applied to the original precursor ion m/z value, and the decoy elution profile is extracted within the same retention time range as the original. In library-based identification and quantification, an MS1 feature library is firstly constructed by summarizing MS1 features in a large dataset A acquired under the same conditions for a large cohort of samples, it is then applied to identify and quantify glycopeptides for a newly acquired MS file (or files) (from new dataset B) via a dedicated library-based virtual match-between-runs algorithm. LC-MS/MS runs in dataset B are newly acquired using the same sample types and experimental conditions as dataset A. Target and decoy elution profiles in the MS file(s) are extracted and matched based on the MS1 feature library. The decoy profile generation method is the same as MBR. The confidence of the matched result is evaluated by a separate machine learning model. Based on the quantitative results, a new biomarker discovery strategy is developed, which enables the screening of the site-specific glycan abundance ratios and allows diagnosis of individuals with newly acquired MS data.
The detailed workflow of GlyPep-Quant is elaborated in “Methods” section. Briefly, in LFQ-MBR, the extraction of glycopeptide elution profiles is parsed through a specially designed peak extraction method, which is inspired by the feature importance in the subsequently used machine learning model. Meanwhile, decoy profiles are extracted by adding 3–20 Th to glycopeptide precursor m/z in a ± 2 min retention time window in the data file to be matched. A pre-trained random forest machine learning model is employed for the confidence assessment of the extracted elution profiles. Results with a false quantification rate (FQR) of less than 1% are retained based on scores from the model. The FQR is calculated by the ratio of the number of decoy elution profiles to the total number of elution profiles in the result. To investigate the relationship between the stoichiometry of site-specific glycans and disease, and to fully utilize the deeper coverage of quantification results achieved by GlyPep-Quant, a novel disease biomarker discovery strategy is developed to screen the abundance ratios of different site-specific glycans on the same site as the biomarkers. This method provided an additional dimension to the expression level of glycopeptides and enabled the detection of stoichiometric alterations of site-specific glycans while excludes the influence of protein abundance changes. To better exploit the obtained quantified information, we introduced a library-based quantification method, library-based virtual MBR, to improve the quantification coverage of site-specific glycans in a new acquired single MS file. The methods of decoy elution profile extraction and FQR calculation is same with that of LFQ-MBR. Specific strategies, including dynamic programming to differentiate features with close RTs caused by glycopeptide isomers and to reduce unnecessary calculation of brute-force, were employed to summarize the MS1 feature of glycopeptide precursors and avoid duplicate matching of features with similar retention times, further enhanced the efficiency and accuracy of library-based quantification.
Advanced LFQ-MBR method in GlyPep-quant for glycopeptide quantification
To enable efficient LFQ-MBR computation, new methods are developed in GlyPep-Quant: A random forest machine learning model was trained to estimate the confidence of MBR match results. Ten features (Supplementary Table 1 and Supplementary Note 1) were selected and used in the machine learning model calculation, including but not limited to Pearson correlation coefficients for the total isotope distribution, monoisotopic peak, +1 and +2 peak distributions, and matrix of elution profiles for assessing the similarity of between initial elution profiles (extracted according to identification) and matched elution profiles (obtained from MBR).
One advantage of using machine learning model is that the contribution of different features can be obtained, thereby facilitates model understanding and identifying key variables. From the importance and target-decoy distributions of MS1 features illustrated in Supplementary Fig. 1, the similarity of isotopic distributions between initial and matched profiles has the most influence on the model’s predictions, with the most weight among all features, which means an extracted matched profile with a higher similarity to its corresponding initial profile tends to be correct and will be assigned with higher scores by the model. Therefore, this feature is primarily utilized in the profile extraction step to ensure the confidence of the acquired results before model prediction.
Next, we present a two-step elution profile extraction strategy to extract matched elution profiles with high confidence (Supplementary Figs. 2 and 3 and Supplementary Note 2). Within MBR process, GlyPep-Quant firstly extracts the elution profile that is closest to the apex of initial profile on retention time axis. Then, the Pearson correlation of isotopic intensity patterns between the initial and matched profile is calculated to assess the reliability. If the calculated similarity is lower than the threshold, each profile in the retention time window is matched to the initial profile to obtain the best matched one with highest similarity. Implementation of the quality control method in the initial profile extraction step is a key feature of our method to achieve reliable quantitative results.
Systematic evaluation of GlyPep-Quant quantification performance
The quantification performance of GlyPep-Quant was benchmarked using several previously published glycoproteome datasets from different aspects. To be noted, through glycan database-independent peptide matching algorithm in Glyco-Decipher9, two types of glycopeptides can be identified: (1) glycopeptides with glycans within the GlyTouCan database; (2) glycopeptides with modified glycans identified by the monosaccharide stepping method9. The modified glycans are denoted as glycan in the database and an additional moiety mass, and the structures are shown as “glycan + moiety mass”. Unless otherwise stated, the results from Glyco-Decipher consist of above two types of glycopeptides. The detailed glycan unit nomenclature20 of this manuscript is elaborated in Supplementary Note 3.
Firstly, we evaluated the quantitation confidence of GlyPep-Quant using different datasets with special treatments: a glycotransferase gene knockdown mouse dataset21 (Supplementary Figs. 4–6, Supplementary Table 2, and Supplementary Note 4), and a exoglycosidase treatment dataset22 (Supplementary Fig. 7, Supplementary Table 3, and Supplementary Note 5). In these datasets, specific types of glycopeptides (core-fucosed glycopeptides in the glycotransferase gene knockdown dataset, fucosylated or bisecting glycopeptides in the exoglycosidase treatment dataset) were removed in the experimental group while retained in the control group. We quantified all the glycopeptides identified in the experiment group by GlyPep-Quant, and then calculated the ratio of such glycopeptides that were quantitatively downregulated (<1 and <0.5) among all quantified glycopeptides in the experimental group and plotted the distribution of the quantitative ratios. It was found that the vast majority of experimental/control abundance ratios of such glycopeptides are closed to zero (Fig. 2a–c), which aligns well with the expectation. More detailed discussions are provided in Supplementary Notes 4 and 5. Above results indicate that the quantification achieved by are highly confident.

a Histogram of core-fucoslyated glycopeptides quantitative ratios between the experimental and control groups reported by GlyPep-Quant MBR for glycotransferase gene knockout sample dataset. b, c Histogram of fucosylated (b) and bisecting (c) glycopeptides quantitative ratios between the experimental and control groups reported by GlyPep-Quant based on Glyco-Decipher identification in the exoglycosidase-treatment sample dataset. d Heatmap of quantification values from the two-species proteome dataset. Quantitative values of glycopeptides for proteins that do not belong to the species are considered as entrapment values, which are shown in orange. True positive quantitative values are colored in blue. All quantification values are displayed in log2 scale. e Quantification results of different software tools on the two-species proteome sample. Above box plot: fold-change results of human serum and yeast glycopeptide. Boxes show the median values, which are also labeled near the boxes, and the interquartile ranges. The whiskers are 1.5-fold of interquartile and the outliers are hidden. Theoretical fold changes of organisms in different samples are marked in the figures. Below bar plot: Number of quantified glycopeptides without missing values from different tools. Source data are provided as a Source Data file. f Number comparison of quantification values and quantified glycopeptides between different tools in HeLa cell dataset. Standard deviations of different quantification results are listed in the right. “DB” means the glycopeptides with database glycans; “Modified” means glycopeptides with database glycans and with modified glycans identified by monosaccharide stepping.
It should be noted that in the experiment group, there are still a non-negligible number of specific types of glycopeptides could not be removed (Supplementary Notes 4 and 5) as the efficiency for the gene knockdown and enzymatic cleavage could not be 100%. It is not accurate to directly calculating the false discovery rate based on specific glycopeptide quantification values. Therefore, a two-species glycoproteome dataset18 was adopted to further assess the confidence of GlyPep-Quant MBR result because the glycoproteome in these species were different, and the presence of glycopeptides were not affected by the cleavage or knockout efficiency. Three liquid-chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) files from budding yeast and three from human serum were searched by Glyco-Decipher against corresponding protein databases, and the identification results were submitted to GlyPep-Quant quantification. Since these two samples have distinct proteomes, cross-species (human glycopeptide quantified in yeast samples, and vice versa) quantifications were regarded as false positive. The quantitation confidence can be approximately evaluated by the ratio of false positives in all quantification results (Fig. 2d and Supplementary Note 6). The high confidence of our glycopeptide quantification results was indicated by the calculated entrapment-based FQR of the GlyPep-Quant quantitative results, which was below 1% threshold (0.93%). GlyPep-Quant also has the ability to process quantification based on identification results from other software, like pGlyco3. The same evaluation workflow was performed for GlyPep-Quant quantification of the identification results yielded by pGlyco3, and the 0.81% entrapment-based FQR was obtained (Supplementary Fig. 8), which further indicates the high confidence of GlyPep-Quant analysis. We also evaluated the quantitation confidence of GlyPep-Quant using the PNGase F-treated deglycosylation dataset from pGlycoQuant18 study (Supplementary Figs. 9–19, Supplementary Tables 4–7, and Supplementary Note 7). The results demonstrated that GlyPep-Quant exhibited high confidence quantification performance comparable to that of pGlycoQuant.
Then, the accuracy of quantification results was evaluated by using a mixed-organism sample dataset23, containing glycopeptides from human serum and budding yeast with different quantities. For the data labeled as S10, S12, and S15, the abundances of human serum remained the same, while the relative abundances of yeast were 1, 1.2, and 1.5-fold. The precision of quantification was analyzed by comparison of mean quantities in different replicates of each sample to theoretical values. These mean values of all quantified glycopeptides, together with the number of quantified glycopeptides without MVs, are shown in Fig. 2e. It is found that compared to other quantitative analysis tools, GlyPep-Quant coupled to Glyco-Decipher identifications reported the most quantified glycopeptides, with 25.1%–178.9% more quantified glycopeptides without any missing value for human glycopeptides and yeast glycopeptides, respectively. The above results indicate the deep coverage performance of our software at both the identification and quantification stages. Meanwhile, GlyPep-Quant maintains a high level of quantification accuracy: the calculated quantitative fold-changes calculated by GlyPep-Quant is one of the closest to the theoretical values (Fig. 2e). Since the majority of these software tools can only identify glycopeptides with database glycans, we then compared the quantifications of this kind of glycopeptides in Glyco-Decipher with that of others (Supplementary Note 8), the results also indicate superior performance of our method. Moreover, based on the identification results of pGlyco3, GlyPep-Quant is able to quantify more glycopeptides (about 5.6% for human glycopeptides and 33.3% for yeast glycopeptides) without MVs than pGlycoQuant (Supplementary Fig. 20), which also demonstrates the strong performance of GlyPep-Quant when applied to identification results generated by other software.
Finally, a HeLa cell dataset18 was applied to evaluate the performance of GlyPep-Quant in MV elimination (Fig. 2f). The proportions of MV in line (PMVL) and in total (PMVT)18 were calculated and compared with those of other tools. The two values of GlyPep-Quant results are lower than Skyline and pGlycoQuant results (Fig. 2f) when all quantification were performed based on pGlyco3 identifications. Nonetheless, it should be noticed that the absolute number of quantified glycopeptides of GlyPep-Quant (based on Glyco-Decipher identifications) is significantly larger than pGlyco3-pGlycoQuant, which were adopted in their previous study, with 19.2% and 41.1% improvement in database and modified glycopeptides, respectively. The quantification precision can also be evaluated by the calculation of standard deviation17,18 after removing the MVs in quantification results. The standard deviations of GlyPep-Quant results are similar to those of other tools, indicating the high precision of our quantification results. The ability to precisely identify and quantify glycopeptides with modified glycans exhibit potential of GlyPep-Quant for more complicated glycoproteomic analysis. The above results indicate GlyPep-Quant is able to precisely quantify glycopeptides with high confidence and sensitivity.
GlyPep-Quant allows the discovery of site-specific glycan ratios as the disease biomarkers
Since LFQ-MBR method in GlyPep-Quant enables deeper coverage of glycoproteomic quantification compared to other tools, it was then applied to quantify site-specific glycans for a large-scale gastric cancer (GC) human serum dataset, which was acquired previously7. The dataset consisted of a discovery cohort with 70 GC patients and 70 healthy controls (HCs) and a validation cohort with 30 GCs and 30 HCs. The detailed average sample conditions are described in Supplementary Table 8. LFQ-MBR was performed for the Glyco-Decipher identification result of the discovery cohort using GlyPep-Quant. As for the results of database glycans, 7867 site-specific glycans were quantified with 70% valid values in at least one group (Fig. 3a). It is worth mentioning that only 866 site-specific glycans remained after applying the same filter process to the quantification result without LFQ-MBR (Fig. 3a). That is an over eightfold improvement in quantification following MBR in GlyPep-Quant. As evidenced in Fig. 3b, serum carcinoembryonic antigen (CEA) levels, which is commonly used to monitor therapeutic response or tumor recurrence, and in some cases as an adjunct to radiological imaging or surgical biopsy for tumor diagnosis, demonstrated limited ability to independently classify GC and HC samples, emphasizing the need to discover novel biomarker candidates. It was reported24,25,26 that the relative abundance changes of different site-specific glycans on the same site are closely associated with disease-related biological processes, offering key insights into disease mechanisms. With the improved quantification coverage, here we developed an effective biomarker discovery method by calculating the ratios of site-specific glycan abundances on the same glycosylation site. This strategy not only minimizes the impact of total glycoprotein expression differences among individuals (individual variance), but also offers a more scalable approach compared to traditional single site-specific glycan abundance biomarker. Ratio biomarkers are particularly suited for application across different cohorts and large-scale experiments, with data collected at different times and under varying instrument conditions.

a Number of quantified site-specific glycans with different thresholds by GlyPep-Quant and by quantification only from MS2 identification. b AUC values and ROCs of serum CEA in discovery cohort (red) and validation cohort (blue). Source data are provided as a Source Data file. c Structures of glycans in two pairs of the screened site-specific glycan quantitative ratios with the top performance. d Representative MS2 spectra of glycopeptide (with sialyl-Lewis structure): “TLNQSSDELQLSMGNAMFVK linked with H7N6S4F2”, which corresponds to P01011-N127 H7N6S4F1; B/Y fragment ions (green/yellow) and b/y fragment ions (blue/red) are labeled in the spectrum. e The abundance distributions of these ratios in discovery and validation cohorts. Source data are provided as a Source Data file. f–i AUC values and ROCs of site-specific glycan ratios and their respective quantification values in discovery cohort and validation cohort. Source data are provided as a Source Data file.
The ratio calculation was first performed on the discovery cohort dataset. After data imputation (details in “Methods” section), for each site where at least two site-specific glycans were quantified (368 glycosites, 5575 site-specific glycans), ratios of quantification values of each two glycans were calculated. For example, if there were three site-specific glycans (A, B, C) quantified on site S, 3 kinds of ratios (Q(A)/Q(B), Q(A)/Q(C), Q(B)/Q(C)) will be computed. Receiver operating characteristic (ROC) curve analysis was conducted to assess the performance of binary classification (healthy vs. disease) using ratio biomarkers as features. The areas under the curves (AUC) for these ratios were calculated to quantify their effectiveness in detecting GC. Ratios with AUC values higher than 0.90 were considered as candidate biomarkers with good classification ability. Consequently, 278 such ratios were obtained, corresponding to 20 different glycosylation sites on 16 glycoproteins (Supplementary Fig. 21). As for the validation cohort with 30 GCs and 30 HCs, LFQ-MBR was also performed by GlyPep-Quant. And AUC values are calculated for the 278 sets of ratios mentioned above using quantification result in the validation cohort. Ratios with AUC values of at least 0.95 in the discovery cohort and 0.90 in the validation cohort were retained, resulting in 31 distinct ones, corresponding to two glycosylation sites on two different proteins (Supplementary Table 9). Next, for each glycosylation site, only the ratio with the highest AUC from both the discovery and validation cohorts was retained. This process resulted in two sets of ratios on two sites (P01011-N127 and P08185-N96), corresponding to four different site-specific glycans (Fig. 3c).
The two glycoproteins with dysregulated site-specific glycan occupancy ratios, are alpha-1-antichymotrypsin (AACT) (UniProt27 accession: P01011) and corticosteroid-binding globulin (CBG) (UniProt accession: P08185). AACT is a serine protease inhibitor involved in modulating inflammatory responses28. Studies have demonstrated that serum levels of AACT are significantly elevated in patients with GC, correlating with disease progression and histological grading29,30. Notably, studies have shown that AACT levels are elevated in advanced stages and grade III gastric cancers compared to earlier stages and grade II, suggesting its potential utility as a biomarker for GC diagnosis and staging7,30. CBG is the primary transport protein for glucocorticoids, binding approximately 80–90% of circulating cortisol with high affinity, thereby regulating its bioavailability to tissues31,32. Although specific studies on CBG in GC are limited, CBG has been shown to play a crucial role in the pathophysiology of various cancers, including ovarian cancer33 and lung cancer34, potentially through its influence on steroid hormone regulation and inflammatory processes. CBG influences various physiological processes, including immune response and inflammation. Alterations in CBG glycosylation patterns could potentially impact these processes, thereby affecting GC pathophysiology.
These selected site-specific glycan abundance ratios related to the two glycoproteins are then fully investigated. Interestingly, we found that among the four site-specific glycans, one was capped with sialyl-Lewis structures (P01011-N127-H7N6S2F2). Sialyl-Lewis structure is a glycosylation modification that is usually modified to the surface of protein or lipid molecules7. It plays an important role in the occurrence and development of many diseases, including cancer. Present on glycoproteins or glycolipids of tumor cells, it facilitates cell-cell interactions, adhesion, and signaling, contributing to cancer invasion and metastasis35. Therefore, studying the relationship between sialyl-Lewis structure and disease is helpful for advancing cancer diagnosis and treatment strategies. Here in this study, the presence of sialyl-Lewis structure on glycosite P01011-N127-H7N6S2F2 was confirmed by the detection of its diagnostic oxonium ions NHAF+ (m/z = 803.31) in the mass spectra (Fig. 3d). Additionally, its expression was upregulated in GC compared to HCs (Supplementary Figs. 22 and 23), aligning with the known biological relevance of the sialyl-Lewis structure in cancer progression.
We further plotted the ROC curves of the quantitative ratios, and those of the individual site-specific glycans, as shown in Fig. 3e–i. We found that across cohorts, the AUC of the ratio was significantly higher than the AUC of the corresponding single site-specific glycans. This suggests that the relative changes in site-specific glycan abundance performed better for GC detection than that of a single site-specific glycan abundance. We also illustrated the distribution of these ratios in each data of the discovery and validation groups (Fig. 3e), significant differences between HCs and GC samples were observed. All the p-values calculated for these ratios between the two groups were less than 0.001, indicating that these ratios effectively reflect the differences in site-specific glycan occupancy between healthy and disease samples, which are associated with physiological changes in organisms. In fact, these good diagnostic ratios for healthy and GC samples may not be obtained through the conventional screening process of disease biomarkers based on glycoproteomics. In conventional methods, for a single site-specific glycan, the abundance difference between disease and healthy samples is calculated, and the significance of the difference is usually evaluated using p-values obtained from t-tests. As shown in Supplementary Figs. 22 and 23, for certain site-specific glycan that makes up the ratios, P01011-N127-H6N5S3 in the validation cohort, the p-values are higher than 0.1. If the p-value threshold for significant differences is set to 0.1, this site-specific glycan will be filtered out. Compared to serum CEA levels, a widely adopted marker for monitoring therapeutic response or tumor recurrence and a supplementary tool to radiological imaging and surgical biopsy, the two selected site-specific glycan ratios exhibited markedly higher accuracy in GC diagnosis. The AUCs for these ratios were notably higher, exceeding 0.987 in the discovery cohort and 0.954 in the validation cohort (Fig. 3f–i), whereas the AUCs for CEA ranged from 0.509 to 0.542 (Fig. 3b). The two glycan quantification ratios have the potential as novel GC biomarkers to differentiate healthy and GC samples.
As a result, by re-analysis of the large-scale dataset, we found two site-specific glycan ratios which have excellent performance on distinguishing GC and healthy samples, and revealed the dynamic variation of glycosylation occupancy in GC disease process.
MS1 feature library-based virtual MBR quantification improves the identification and quantification of glycopeptides in newly acquired datasets
Based on the evaluation results from the previous section, advanced LFQ-MBR with GlyPep-Quant achieved an over eightfold improvement in quantification sensitivity compared to quantification based solely on identification. This enhancement arises from the annotation of numerous elution profiles without high quality MS2 scans by leveraging abundant profile features from the large-scale dataset. We expect that the elution profiles of the quantified glycopeptides from a large cohort of samples could be leveraged to substantially improve sensitivity in the identification and quantification of glycopeptides in newly acquired data under similar conditions.
Inspired by the “spectrum library” used in DIA data analysis, here we devised an MS1 feature library-based virtual MBR quantification strategy (hereinafter abbreviated as “library-based quantification” or “library-based MBR”), to make full use of quantified precursor features from the large-scale datasets acquired from a large cohort of samples under the same experimental procedures. The quantification library is composed of different glycopeptide precursor MS1 features from result of previous datasets. Through a virtual one-direction MBR process from the library to the newly acquired LC-MS/MS run(s) for a different sample under the same experimental condition, glycopeptides in the new sample would be identified and quantified even without MS2 interpretation, potentially enhancing the analysis sensitivity.
Library construction and virtual MBR with a newly acquired LC-MS/MS file are two key steps in this library-based quantification method. When building the quantification library derived from a dataset, the MS1 features of glycopeptide precursors from identifications, including elution profiles, retention time of apexes, intensity distributions of isotopic peaks in the elution profiles, are extracted and recorded in the library. Different elution profiles with the same precursor are firstly separated into several clusters based on the retention time of peak apexes, using the Density-Based Spatial Clustering of Applications with Noise (DBSCAN)36,37 unsupervised clustering method. Then for each elution profile in one cluster, the intensity distribution of isotopic peaks is normalized and summed up to form a “combined” MS1 feature. The retention time of this feature is the time point obtained from clustering. Finally, these “combined” MS1 features are recorded in the quantification library.
Different from the traditional LFQ-MBR between MS files, a one-direction virtual MBR process was then performed only from the constructed quantification library to the newly acquired MS file (details in “Method” section). There are occasions where the same precursor has more than one elution profiles with different retention times present in both the library and the newly acquired data. These elution profiles are formed by glycopeptide and their isomers, as they share the same precursor m/z but exhibit different retention behaviors towards reversed-phase separation. Although part of the glycosylation structure can be analyzed through MS2 spectra identification9,22, it is impossible to distinguish all these isomers by MS1 features. Therefore, GlyPep-Quant performs glycoproteomic quantification at the glycopeptide level (peptide and glycan composition). Another issue is that the MS1 features in the library formed by the isomers may have closely spaced but different retention times, resulting in the overlap of retention time windows used for matching. Therefore, MS1 features which belong to glycopeptide isomers can locate in an overlapped time window, leading to interference in the library-based quantification process (Supplementary Note 9). To address this problem, we proposed an “MS1 feature cluster matching” method (simplified as FC method). For those MS1 features whose corresponding time windows overlap, they are treated as a “MS1 feature cluster”. Together with the merged time window, this cluster, rather than individual features, is used for matching with the profiles. The detailed method is shown in Fig. 4, Supplementary Tables 10 and 11 and explained in Supplementary Note 9. To avoid unnecessary calculations in a brute-force method, where all possible MS1 feature-profile match combinations should be considered, FC algorithm applies a special dynamic programming (DP) method. This method identifies the best-scored path across the match table from the bottom right to the top left, based on the predicted scores of feature-to-profile matches. It can reduce the time complexity from O (n × mn) to O (n × m2), where m and n represent the number of potential elution profiles in the retention time window and the number of MS1 features in the specific cluster, respectively.

a Construction workflow of library used in library-base identification and quantification by GlyPep-Quant. b Isotopic distributions and glycopeptide precursor information used in this example. Source data are provided as a Source Data file. c Extracted elution profiles in the whole retention time window of the precursor in (b). Source data are provided as a Source Data file. d Scores, DP and match indexes matrices used in the MS1 feature cluster matching algorithm, and the best matching path selected by the matching algorithm. The cluster contains MS1 features I, II, and III, their corresponding matched profiles are profile 3, 5, and 6. Since feature II-profile 5 and III-profile 5 have the highest score among feature II and III matches, if FC algorithm is not utilized, then feature III-profile 6 cannot be obtained for its lower score. However, the score of feature III-profile 6 match (0.896) is also high, and more importantly, profile 6 is the one which can be extracted based on GPSM identification, meaning it be correct profile. These results show that FC algorithm can acquire more accurate and comprehensive quantification, minimizing quantitative loss caused by precursors of the same m/z eluting in different time regions.
After establishing the library-based quantification method, we assessed its performance from different aspects. The library built from the discovery dataset (termed “discovery library”) recorded approximately 40,000 MS1 features at different charge states, corresponding to about 22,000 site-specific glycans (Fig. 5a). Then 20 new human serum glycoproteomic MS files including 10 healthy control and 10 GC samples (termed “verification dataset”) were acquired. The information of this new dataset is elaborated in “Methods” section, Supplementary Table 8 and Supplementary Note 10–all the experiment procedures, instrument settings are remained the same as those of the discovery cohort dataset7.

a Workflow of the evaluation process. An MS1 feature library is built using the features from the discovery dataset identifications, and applied to library-based quantification method on the verification dataset. b Bar plot of library-based quantification sensitivity evaluation result. Upper: number of quantified site-specific glycans (SSG) from library-based quantification. Bottom: number of quantified site-specific glycans from conventional identified-MS2-based quantification method. c Bar plot of library-based quantification confidence evaluation result. Blue: the number of “theoretically matchable profiles” (profiles that can be matched in both library-based quantification and MS2 identification). Orange: the number of un-matched theoretically matchable profiles (profiles whose features are preserved in the library and could also be directly identified by MS2 spectra, but not matched from library-based quantification).
Firstly, the identification sensitivity of the library-based virtual MBR strategy was assessed. This new method enabled the quantification of 14091 to 16758 site-specific glycans across the 20 samples (Fig. 5b). In contrast, only 1331 to 1770 site-specific glycans were identified and quantified by the conventional identified-MS2-based method (elution profiles were extracted only based on identified MS2 spectra) using Glyco-Decipher (Fig. 5b). The identification and quantification coverage were over 8 times by using this new method than the conventional identified-MS2-based method, which significantly enhanced the depth of qualitative and quantitative analysis of site-specific glycans in individual samples. On the other hand, more than 73% of the site-specific glycans in the library (containing 22743 site-specific glycans) were identified and quantified in the MS1 spectra of the new data (verification dataset), which indicates the great potential of the quantitative libraries in the qualitative and quantitative analysis of MS1 spectra. By summarizing and applying the site-specific glycans MS1 feature information from existing results, the qualitative and quantitative analysis of a single new run can be elevated to a level previously attainable only through match-between-run operations on large-scale sample datasets.
We then assessed the confidence of the library-based quantification results. After performing the library-based quantification, some matched profiles are associated with high quality MS2 spectra, which can be identified directly by Glyco-Decipher. We denoted glycopeptide elution profiles which correspond to identified MS2 spectra and whose features were also recorded in the quantification library as “theoretically matchable profiles”. “Recorded” indicates that the retention time ranges of the library feature (cluster) and the elution profile should be overlapped, with the precursor information remaining the same (Supplementary Figs. 25 and 26). These profiles can be matched through library-based quantification theoretically, and how many of them are actually matched can indicate the confidence of results, or say, the ability of library-based quantification method to retrieve true positive results in new LC-MS/MS run. We then calculated the ratio of “actually matched profiles by library-based quantification” to the “theoretically matchable profiles”.
The number of “theoretically matchable profiles” varied from 1418 to 1812 across the 20 samples. Among these elution profiles, about 95.7% “theoretically matchable profiles” was actually matched, which means a vast majority of these profiles could be matched through library-based quantification method (Fig. 5c), indicating high confidence of the method. It should be noted that the remaining 4.3% of matches are not all incorrect; rather, they may be attributed to special circumstances, such as: (1) For some glycopeptide precursors, the number of identification profiles in the LC-MS/MS run exceeds the number of MS1 features in the library; (2) Some MS1 features are matched to a profile different from the identification profile, but the matched profile is derived from an isomer of the same precursor as the identification profile. More detailed discussion is presented in Supplementary Figs. 27–29 and Supplementary Note 10, suggesting that the actual false positive rate is lower than 4.3% (Fig. 5c), and the library-based quantification method can obtain reliable results.
Site-specific glycan ratio biomarkers allow the diagnosis of disease for individual samples
Two site-specific glycan abundance ratios (corresponding to four site-specific glycans) are determined as the high-performance biomarkers to classify the GC and HC based on the discovery and validation dataset. The 20 individuals (10 HCs and 10 GC patients) for the acquisition of the verification dataset are different with the 200 individuals in the discovery and validation cohorts. It’s of interest to investigate whether these two ratio biomarkers could correctly diagnose the GC from the 20 individuals. For this purpose, the four site-specific glycans should be quantified from the MS data of these samples. We first investigated if these 4 glycans could be identified and quantified from the 20 samples by using conventional identified-MS2-based methods. As shown in Fig. 6a, two site-specific glycans (P01011-N127-H7N6S2F2, P08185-N96-H7N6S3) were challenging to quantify, with only 1 and 7 quantifiable values, respectively, across the 20 samples. Thus, the ratio calculation relies on interpolated values, which may deviate significantly from the actual values and lead to inaccurate diagnosis. We then examined whether these 4 glycans could be quantified by the new method across 20 samples. The application of library-based quantification led to a significant improvement in quantitative performance, effectively filling most of the MVs present in the original quantifications. The aforementioned two glycans were quantified in 16 data samples, and the other two glycans in the ratios were quantified in all data samples, making the calculation of abundance ratios more accurate and reducing the impact of MVs.

a Heatmap of quantification values of site-specific glycan abundance ratios from library-based quantification and identified-MS2-based quantification. b Scatter plot of CEA concentrations and site-specific glycan ratios quantification values in different samples. CEA concentrations are acquired from sample information, and site-specific glycan ratio quantification values are obtained from library-based quantification. Source data are provided as a Source Data file. c Sensitivity, specificity and diagnostic accuracy of CEA and the two site-specific glycan ratios on these samples.
Then, the diagnosis performance of the selected ratios was evaluated using the new data from the verification cohort, using the best diagnostic threshold (corresponding to the point in the upper left corner of the blue ROC curve in Fig. 3f and h) obtained from the discovery group results. The result indicated that the two abundance ratios demonstrated excellent performance in detecting GC when used individually—with 100% sensitivities—indicating that these two ratios can make a completely correct diagnosis of disease samples (Fig. 6b, c). On the other hand, the specificity and diagnostic accuracy of ratio 2 (70% and 85%) is higher than those of ratio 1 (50% and 75%), showing a better performance on diagnosis as GC biomarker. Oppositely, serving as a surveillance and diagnostic aid in GC, CEA is ineffective in accurately diagnosing the health status of the samples (Fig. 6 and Supplementary Table 12). Specifically, 19 out of 20 samples had CEA concentration values below the diagnostic threshold, leading to their classification as healthy samples. However, 9 of them are actually cancer sample. If the diagnostic results are based solely on CEA concentration standards, almost all disease samples would be incorrectly classified as negative, hindering their timely treatment.
It is worth noting that this new dataset was acquired nearly a year after the discovery and validation datasets, with different instrument states, utilizing the same type but different enrichment columns and analytical columns, which indicated that some of the elution behavior of glycopeptide precursors may differ significantly. However, the library-based quantification still obtained deep coverage and confident results, and the selected site-specific glycan abundance ratios also demonstrated strong diagnostic abilities.
Several studies38,39 also conducted glycoproteomic analysis to discover biomarker candidates for the diagnosis of cancer, and the diagnostic efficacy of the marker candidates were evaluated by calculating the AUC values in a dataset-dependent way using the discovery and validation cohorts. That is, the optimal diagnostic results of marker candidates were achieved by using distinct diagnostic thresholds in different datasets. On the contrary, here in this study, the diagnostic thresholds of marker candidates were derived from a large-scale dataset and applied directly in verification samples. This strategy offers greater practical value for marker’s clinical application. Also, the high diagnostic accuracy of the discovered ratio biomarkers in detecting GC using a fixed diagnostic threshold underscores the robustness of the library-based virtual MBR quantification method, and provides another aspect in investigating the protein glycosylation alterations associated with disease.
Discussion
Due to the low interpretation rate of glycopeptide spectra and the stochastic nature of DDA, the sensitivity for the qualitative and quantitative analysis of site-specific glycans at proteomics scale remains poor. To address this problem, we developed GlyPep-Quant, a software using a match-between-run (MBR) method to eliminate the MVs between runs and a library-based virtual MBR strategy to enhance the identification of glycopeptides without MS2 spectra for the newly acquired dataset. We demonstrated that an over eightfold increase in site-specific glycan identifications were achieved by using above methods, comparing to conventional identified-MS2-based method.
For MBR, we employed a well-trained random forest model to assess importances among individual features across different elution profiles, assigning scores for evaluating result confidence. Being different from existing LFQ methods that indiscriminately extract all peaks within a time window, GlyPep-Quant adopts the interpretability of the random forest model over deep learning models, enabling us to clearly discern the contributions of each selected feature. Based on the model feature importance, we devised a two-step peak extraction strategy that effectively implements an initial quality control step during the peak extraction process, enhancing sensitivity and precision in quantification. The performance of GlyPep-Quant LFQ method was benchmarked using several publicly available datasets and compared to other state-of-the-art quantitative software tools, including MSFragger-Glyco, Skyline and pGlycoQuant. GlyPep-Quant yielded a higher number of quantified glycopeptides without any MVs.
For the library-based virtual MBR strategy, MS1 precursor features are summarized according to their m/z values and retention times, and recorded in a library. Apex retention time of elution profiles from the same precursors across different dataset were classified and clustered by an unsupervised machine learning method, along with their isotopic distributions. Then, an MS1 feature library-based virtual match-between-run process was conducted from the library to the newly acquired single-file data. Since the features in the library were extracted from already identified glycopeptides from existing MS2 spectra through database search, the search space of library-based MBR was not as large as some other MS1-based identification methods40,41. Moreover, to address the challenge of close yet discrete elution profiles generated by glycopeptide precursor isomers within a single retention time window, we developed a dedicated MS1 feature cluster matching algorithm using dynamic programming. This algorithm enables precise and accurate matching of elution profiles from the library. Following evaluation, the library-based quantification achieves high confidence result and greatly increases the quantification coverage for single-file analysis.
Library construction from MS2 identification and virtual MBR with a newly acquired LC-MS/MS file are two key steps in this library-based quantification method. To allow effective and accurate matching, all LC-MS/MS runs for library construction and for the following identification through library-based virtual MBR should be acquired under identical conditions, and the sample type (e.g., serum, cell lysate, tissue lysate…) should be the same. To increase the library coverage, LC-MS/MS runs for the same type of samples but with different physiological or pathological conditions (e.g., healthy and disease) could be aggregated to increase the types of glycopeptide identifications. It should be stressed that currently GlyPep-Quant does not predict elution profiles or support library from predicted elution profiles, and the developed library-based MBR scheme only works for experimentally built library. And the dataset used for library construction should be representative, and the size of datasets should be controlled. A more detailed discussion is provided in the Supplementary Note 11.
For isomers of glycopeptides, especially when the peptides are the same but the structures of N-glycans are different, their masses are basically the same and they may elute at similar retention times; it is difficult to distinguish such glycopeptides using only MS1 information, so we quantify glycopeptides at the level of “glycan composition + peptide”, and the DP algorithm is also proposed to solve the problem that information of such glycopeptides may be missed or repeated during quantification. However, it cannot be denied that the structural analysis of glycosylation is a research direction of concern in glycoproteomics22,42,43, and we will continue to work on the quantification of glycopeptides and the analysis of glycan structures in glycopeptides, including the incorporation of retention time prediction models44,45. If it is necessary to confirm and analyze the structure of a glycopeptide in the quantitative results, we currently recommend that researchers use targeted mass spectrometry scanning technology, such as selected reaction monitoring46 or parallel reaction monitoring47 to fragment specific precursor ions and then use MS2 spectra for in-depth analysis.
Based on the accurate and sensitive quantifications, we reanalyzed datasets from previous large-scale glycoproteomic studies. Through calculating site-specific glycan abundance ratios, we found certain ratios with potential as disease biomarkers for diagnostic purposes. The two selected glycan abundance ratios demonstrated much better performance than the widely adopted marker for cancer surveillance, CEA, on the diagnosis of GC across two different datasets. Additionally, by computing relative differences between site-specific glycans, this approach has the potential to mitigate biases introduced by variations in experimental/instrumental conditions and protein abundance levels. Next, the diagnostic performances of the selected site-specific glycan abundance ratios were validated on a newly generated dataset, where missing value issues were addressed using the library-based virtual MBR strategy. Consequently, the two ratio biomarker candidates also exhibited good performance in detecting disease in the new sample cohort, with 100% sensitivity and up to 85% diagnosis accuracy. These results emphasized the robustness of the library-based virtual MBR quantification method, as well as the efficacy of the discovered ratio biomarker candidates.
Currently, GlyPep-Quant is equipped with both LFQ-MBR and library-based virtual MBR quantification strategies, and has been integrated into our flagship software, Glyco-Decipher. We expect GlyPep-Quant to have broad applications in large-scale glycoproteomic studies.
Methods
This clinical sample collection project was approved by the Ethic Committee of the First Affiliated Hospital of the Fourth Military Medical University, Xi’an, China (Approved NO. of ethic committee: KY20192088-F-1). All participants received the written informed consent. The research design and implementation were completely adhered to the principles outlined in the Declaration of Helsinki.
Glycopeptide quantification by label-free quantification with match-between-runs using GlyPep-Quant
Extraction of elution profiles. Firstly, elution profiles of identification results (denote as “initial profiles”) are directly extracted using the same method described in our previous article9. The elution profiles are extracted after tracing the monoisotopic peak and up to +6 isotopic peaks.
Then, GlyPep-Quant navigates through the results, focusing on the elution profile matches between those files. GlyPep-Quant initially identifies and selects results uniquely obtained in file A, considering glycan, peptide, glycosylated site, retention time, and charge for quantification. Subsequently, a two-step method (see “extraction method” below) is used to trace peaks with identical m/z values in File B (as target elution profiles). Decoy profiles are generated by adding 3–20 Th to MS1 peaks in file B in a ± 2 min retention time window. The tolerance in extraction is ±10 ppm in this work except stated.
Confidence assessment of extracted profiles. Different features (Supplementary Note 1) are computed for the target and decoy elution profiles after the extraction process. A random forest machine learning model that has been trained beforehand applies these features to scoring. For each target and decoy elution profiles, a score between 0 and 1 is calculated to estimate the confidence. All profiles are sorted in the descending order of these scores, and the division of the number of decoy elution profiles by the total number of elution profiles yields the false quantitative rate (FQR). The FQR is calculated by:
Target elution profiles in quantification results with FQR within 1% are retained for further analysis.
Similarly, for the library-based MBR, the FQR is calculated by:
Summary of quantification results. For the same data file, final glycopeptide quantification results, which are written in the output file “glycopeptide.csv”, are calculated by summing up the areas of initial profiles from step (1) and target profiles obtained from step (2) for glycopeptides with same sequences, modifications, glycans. The results in “site.csv” are the quantification results for glycopeptides with same glycans and glycosites.
A random forest machine learning model for confidence evaluation
Glyco-Decipher identification results from four MS data files of human serum were used for the establishment and training of machine learning model. If one glycopeptide was identified in two data files (A and B) at the same charge state, with retention time difference less than 2 min, the elution profiles of this glycopeptide were extracted in these two files. Then the decoy profiles were obtained by the extraction of decoy peaks, which are generated by mass shifts (+3–20 Th) of the isotopic peaks of glycopeptide precursor within a ± 2 min retention time window. Similar decoy generation methods have been adopted in other researches16,18.
Ten features, including different types of similarities (Supplementary Note 1), from these target-target and target-decoy elution profile pairs were calculated to assess the similarities of extracted elution profiles to initial profiles and theoretical isotopic distributions.
For the calculation of similarities, “stats.pearsonr” function in SciPy48 Python package is used to obtain the Pearson Correlation Coefficient. For two one-dimensional arrays of equal length, such as isotopic distributions in a pair of elution profiles, where the similarity between mono-peak and +1 or +2 peak distributions within an individual profile is to be assessed, Pearson correlation coefficient can directly be computed using this method. However, for two one-dimensional arrays of different lengths, e.g., total intensity distributions along time axes for two profiles, prior to computing the Pearson correlation coefficient, the shorter array is first resized to match the length of the longer array using the “ndimage.zoom” function from the SciPy package, which is primarily designed for image resizing and scaling operations.
In the case of two two-dimensional arrays with matching lengths along one dimension but differing lengths along the other (such as similarity assessment between elution profile matrices), the same procedure applies. First, the shorter array is interpolated to match the length of the longer one by employing the “ndimage.zoom” method. Subsequently, both arrays are flattened into one-dimensional arrays before calculating their Pearson correlation coefficient.
In our analysis, we compute a multitude of similarity measures between various isotopic peak distributions as features, primarily focusing on the similarity in intensity distribution along the retention time axis for different isotopic peaks. For instance, “iso_single_similar0” assesses the similarity in intensity profiles of mono-peaks across the initial elution profile and its matched counterpart. However, when the matched elution profile is derived from decoy peaks (simulation of random matches), it often contains numerous zero values, which can lead to one array being entirely composed of zeros. This scenario poses a problem when computing Pearson correlation coefficients because, during the calculation, the denominator involves the standard deviation, which becomes zero when all array values are zero, resulting in an undefined correlation coefficient (due to division by zero), typically represented as Nan values. To circumvent this issue and prevent it from affecting subsequent calculations, we assign a value of −1 to these cases, signifying very low similarity within the context of Pearson correlation coefficients.
Furthermore, to mitigate the influence of substantial zero values on the accurate assessment of similarity, when calculating features such as “iso_single_similar0”, “iso_single_similar1”, “iso_single_similar2”, “mono&+1_similar”, “mono&+2_similar”, and “evidence_similar”, we apply penalties to the computed Pearson correlation coefficient between two arrays, arr1 and arr2, using the following formula:
Where
Specially, for the model feature “matrix_similar”, the calculation process is:
For each glycopeptide precursor, we extract the intensity values of its monoisotopic peak along with up to six additional isotopic peaks (+1 to +6) from all MS1 spectra within the retention time window of the GPSM identification. If signals are successfully extracted across m MS1 spectra, the result is a two-dimensional matrix of shape (m × 7).
Since the number of MS1 scans (i.e., retention time points) may differ between the two elution profiles–one derived from the GPSM identification and the other from the MBR-matched result–the matrices typically differ in their first dimension. However, if the two profiles correspond to the same glycopeptide precursor, the underlying signal patterns should be highly similar. To align them, we apply the “ndimage.zoom” function from the SciPy48 library, which was originally developed for image resizing and interpolation.
This function is used to rescale the matrix with fewer time points so that its first dimension matches that of the longer matrix, while preserving the overall signal pattern. After rescaling, both matrices are flattened along the column (i.e., isotopic peak) dimension to produce two one-dimensional vectors.
For example, consider two elution profile matrices with shapes (9 × 7) and (8 × 7), representing 9 and 8 time points and 7 isotopic peaks, respectively. We first rescale the (8 × 7) matrix to (9 × 7) using ndimage.zoom function, and then flatten both matrices column-wise to obtain two (1 × 63) vectors. We then compute the Pearson correlation coefficient between these vectors to quantify their similarity.
This operation is analogous to the flattening step in convolutional neural networks, where 2D feature maps are passed into fully connected layers, or a pooling layer is converted to a fully connected layer49. By applying this process, we can approximate the similarity between two elution profiles. Importantly, this correlation is only one of the ten features we use to evaluate the similarity between matched elution profiles. Our model integrates these diverse features to capture similarity from multiple perspectives, and our results demonstrate the robustness and reliability of this approach.
After the calculation of these features, a random forest regressor model in Scikit-Learn50 was trained to predict the confidence with a vector in 1 × 10 size as input and a score in range (0,1) was given for each profile. All target and decoy profiles were scored and sorted in descending order. FQR was calculated as the ratio of decoy profiles in total result under specific threshold. The final result was kept with calculated FQR less than 1%.
For library-based MBR, the attributes: “time_diff”, “theo_similar”, “iso_similar”, “iso_single_similar0”, “iso_single_similar1”, were used trained another random forest machine learning model to evaluate the confidence of matched profiles.
A two-step elution profile selection method based on feature importance in machine learning model
The elution profile extraction and selection method in Glyco-Decipher is as follows, and Supplementary Note 2:
-
(1)
GlyPep-Quant finds the MS1 spectrum where the maximum summed intensities of extracted precursor isotopic peaks in the initial profile, and obtain the retention time t1 of this MS1 spectrum. Then, for the MS1 spectra in a ± 2 min retention time window in the matched MS data, our software finds the one whose retention time closest to t1, and where summed intensities of isotopic peaks higher than 0. Suppose the retention time of this MS1 spectrum be t2.
-
(2)
From two directions (before or after t2), precursor isotopic peaks in MS1 spectra in a ± 2 min retention time window are matched and selected, and the summed intensity of these peaks for each MS1 spectrum is calculated and recorded. This selection process ends when the summed intensity drops below than 15% of the maximum of these intensities, or there are over 5 continuous MS1 spectra with precursor peaks are missing.
-
(3)
The isotopic similarity between the initial profile and extracted profile from step (2) is calculated. If this similarity is higher than 0.15 (average of decoy profiles in the training set of random forest model), this profile is applied to subsequent model prediction, otherwise, this profile is discarded and new profile will be selected from step (4).
-
(4)
In the retention time window, the method in step (2) is used to generate all possible extracted profiles from MS1 spectrum with start retention time to the end. Then the similarities between initial profile and these extracted profiles are calculated. The extracted profile with highest similarity to the initial one is retained.
Benefit from the two-step elution profile extraction method and well-trained machine learning model, confident quantitative results are obtained, and the estimated FQR (0.93%) is lower than 1% threshold and similar to that of pGlycoQuant (0.88%) result using the same dataset and method.
Imputation method used in quantification results of large-scale data
Similar to previous researches17,51, values for the imputation of MVs are obtained from a different Gaussian distribution compared to the Gaussian distribution of all log2 valid quantification values. The Gaussian function is generated using “stats” function in Scipy Python package, and the all the quantification values in the same file are fitted into a Gaussian distribution using “optimize” function. After fitting, the standard deviation and the mean value of the Gaussian function can be obtained, and a new distribution is generated with 0.5 in standard deviation and a down-shift of 3.6 in mean compared to the previous distribution. For all the MVs in the same file, values picked randomly from the new Gaussian distribution are imputed. This imputation method is applied to each file in this work.
Indicators used to measure the proportion of missing values: PMVL and PMVT
PMVL (%) is defined as the percentage of quantified glycopeptides that contain more than one missing value out of the total number of quantified glycopeptides. PMVT (%) indicates the percentage of MVs relative to the entirety of quantitation values. These indicators were firstly proposed in the pGlycoQuant18 article.
Library construction in library-based quantification
The quantification library used in library-based quantification is composed of different glycopeptide precursor MS1 features from identification and quantification of another before similar datasets.
For a previously acquired dataset, all elution profiles of glycopeptide precursors are extracted based on Glyco-Decipher identifications. The retention time of elution apex, together with the m/z values and intensity distributions of isotopic peaks in the elution profiles, are recorded in the library. The elution profiles with the same glycopeptide and charge state are grouped together. Since precursors with same m/z and charge values can generate elution profiles in different time area (discussed in Supplementary Note 9), to restore the original features to the greatest extent, elution profile clustering algorithm was developed: different elution profiles with same precursor are firstly separated into several clusters based on the retention time apex, using the DBSCAN unsupervised clustering method. Then for each elution profile in one category formed by clustering, the intensity distribution of isotopic peaks is normalized to 0–1 scale, and normalized again after merging them together. The retention time of the “combined” library elution profile is the time point obtained from clustering, and the m/z values and glycopeptide information remained same with before. Finally, these “combined” library elution profiles are recorded in the quantification library for further analysis.
MS1 feature cluster matching algorithm used in the library-based quantification
To precisely obtain matched profiles based on library features, in this library-based quantification method: suppose one library elution profile has a retention time T and m/z values [mz0, mz1, … mz6], GlyPep-Quant will extract all the elution profiles whose isotopic peak m/z values equals to those of library profile, in a specific time window T ± △t, and △t can be set by users. The one with the highest score predicted by the newly trained machine learning model will be retained and used for elution area calculation.
There are occasions where more than one elution profiles with same precursor and different retention time both in the library and in the actual data. These elution profiles may be formed by glycan and glycopeptide isomers, for their precursors have same m/z values and different retention behaviors. To deal with the problem that some right results may be omitted in library-based quantification, we proposed an “MS1 feature cluster matching” algorithm (simplified as FC algorithm). For those MS1 features whose corresponding time windows overlap, they are treated as a “MS1 feature cluster”. Together with the merged time window, this cluster, rather than individual features, is used in matching with the profiles. Before explain the method, it is necessary to claim some facts: though the retention time will shift in different data files, the elution order of specific glycopeptides is fixed; identification and quantification of glycoproteomic results differentiate in DDA data for acquisition characteristics and the low efficiency of glycoproteomic identification, causing some MS1 features cannot match with any profile, or some profiles might not be recorded in the library. Hence, we develop this strategy based on two rules: (1) The elution order of matched profiles must be consistent in MS1. (2) The profile corresponding to specific MS1 feature may not appear in the matched data, and there may be some profiles whose feature the library does not record.
The detailed algorithm is shown in Fig. 4 and explained in Supplementary Note 9. To avoid unnecessary calculation in brute-force method where all possible MS1 feature-profile match combinations should be considered, FC algorithm applied a special dynamic programming method. Based on the predicted scores of feature-profile matches, the best-scored path across the scoring table from bottom right to top left. As shown in Fig. 4, the cluster contains MS1 feature I, II, and III, their corresponding matched profiles are profiles 3, 5, and 6. In traditional process, since feature II-profile 5 and III-profile 5 have the highest score among feature II and III matches, if FC algorithm is not utilized, then feature III-profile 6 cannot be obtained for its lower score. However, the score of feature III-profile 6 match (0.896) is also high, and more importantly, profile 6 is one which can be extracted based on GPSM identification, meaning it be correct profile. These results show that FC algorithm can acquire more accurate and comprehensive quantification, minimizing quantitative loss caused by precursors of the same m/z eluting in different regions. The detailed computation process and pseudo code are provided in Supplementary Tables 10 and 11, and this method can reduce the time complexity from O(n × mn) to O(n × m2), where m and n are the number of potential elution profiles in the retention time window, and the number of MS1 features in the specific cluster, respectively.
Experimental procedure and sample information
Chemicals and reagents
Trifluoroacetic acid (TFA), formic acid (FA), Urea, ammonium bicarbonate (NH4HCO3), dithiothreitol (DTT), iodoacetamide (IAA), and trypsin (bovine, TPCK-treated) were purchased from Sigma (St. Louis, MO, USA). Acetonitrile (ACN, HPLC grade) was purchased from Merk (Darmstadt, Germany). Deionized water was purified by a Milli-Q system (Millipore, Milford, MA).
Clinical sample collection
This project was approved by the Ethic Committee of the First Affiliated Hospital of the Fourth Military Medical University, Xi’an, China (Approved NO. of ethic committee: KY20192088-F-1). All participants received the written informed consent. The research design and implementation were completely adhered to the principles outlined in the Declaration of Helsinki. All healthy individuals showed no signs of gastrointestinal tract cancer, confirmed by serum biomarker screening and CT scans. Clinical serum samples were collected from the biobank of Xijing Hospital. The GC patients and HCs in the three cohorts (discovery, validation, and verification) were carefully matched by age and gender. The clinical characteristics are outlined in Supplementary Table 8.
Intact N-Glycopeptide enrichment in the HRN platform
Five microliters of individual human serum from GC patients, HCs were diluted tenfold with 8 M urea/0.1 M NH4HCO3, followed by denaturation via reduction (20 mM DTT at 37 °C for 2 h) and alkylation (40 mM IAA at 25 °C for 40 min). The urea concentration in the mixture was reduced to below 2 M with 0.1 M NH4HCO3, and proteins were digested with trypsin at an enzyme-to-protein ratio of 1:50 (w/w) at 37 °C for 16 h. The resulting tryptic peptides were desalted using Oasis HLB C18 cartridges (Waters) and then lyophilized. Glycopeptide enrichment and MS analysis were performed on intra-batch samples in random order to minimize bias. Intact N-glycopeptides were enriched using an automated method6: Briefly, the lyophilized tryptic peptides from 5 μL serum were dissolved in 0.1% TFA/80% ACN and automatically injected onto a HILIC column at a flow rate of 0.2 mL/min. After washing away the non-glycopeptide fraction, the glycopeptide fraction was collected. System washing and re-equilibration were performed before each new sample injection, with the total enrichment cycle taking approximately 20 min per sample.
Microflow LC-MS/MS analysis in the HRN platform
The enriched glycopeptides obtained from 5 μL of serum were resuspended in 0.1% FA buffer and analyzed using microflow LC-MS/MS. This was conducted on an Ultimate 3000 LC system, coupled online to an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific, USA). Glycopeptide separation was performed on a commercial Acclaim PepMap 100 C18 column (2 μm particle size, 1 mm inner diameter, 150 mm, Thermo Fisher Scientific) with a flow rate of 50 μL/min. The column was kept at a temperature of 45 °C. The mobile phase consisted of solution A (0.1% FA) and solution B (0.1% FA/80% acetonitrile). The total 60 min LC gradient was described as follows: held at 4% B for 0.5 min, from 4% to 9% B for 0.5 min, from 9% to 45% B for 51 min, from 45% to 95% B for 2 min, held on 95% B for 4 min to clean the system, and finally return to 4% B for 2 min to re-equilibration. The mass spectrometer was operated in DDA mode. Full scan MS spectra were acquired over a mass range of 350 to 1800 m/z at a resolution of 60,000, with a normalized AGC target of 300 and a maximum injection time (IT) of 25 ms. MS/MS scans were carried out at a resolution of 30,000, using a 2 m/z isolation window, with a normalized AGC target of 200 and a maximum IT of 100 ms. Glycopeptides were fragmented using stepped higher-energy collisional dissociation with normalized collision energies of 20%, 30%, and 40%.
Global parameters of Glyco-Decipher analysis
Unless otherwise stated, the parameters of Glyco-Decipher used in this article are: precursor mass tolerance: 10 ppm; fragment mass tolerance: 20 ppm; enzyme: full trypsin digestion with three maximum missed cleavages; carbamidomethylation (+57.022 Da) at C was set as fixed modification; and oxidation (+15.995 Da) at M was set as variable modification. False discovery rate (FDR) of glycopeptide spectrum match (GPSM) was controlled to less than 1%.
The use of protein databases in various datasets: UniProt S.pombe protein database (5149 entries) and Homo sapiens protein database (20350 entries) are used for the identification search of yeast and human serum data in the two-glycoproteome dataset. The protein database consisting of proteins from Homo sapiens protein database (20350 entries) and UniProt S. cerevisiae protein database (6721 entries) is used for the analysis of the mixed-organism dataset. Homo sapiens protein database is used for the analysis of HeLa cell dataset.
The sources of data and software tools are listed in Supplementary Tables 13 and 14.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mixed-organism data from Yang et al. 23 were downloaded from iProX52 partner repository with the dataset identifier PXD023980. Raw data from Kong et al. 18 were downloaded from the MassIVE53 repository with accession code MSV000089484 [https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=1587813df5934fbc8c1d1a6fd8479caf] (the two-glycoproteome dataset, HeLa cell dataset, deglycan dataset). The glycotransferase gene knockout sample dataset from Shen et al. 22 was downloaded from PRIDE54 repository with identifier PXD025859. The exoglycosidase-treatment sample dataset from Chen et al. 21 was downloaded from PRIDE54 repository with identifier PXD035158. The gastric cancer dataset (discovery and validation dataset) from Liu et al. 7 were downloaded from JPOST55 repository with accession number PXD036733. The results of other software except Glyco-Decipher were also acquired in this repository. The information of other datasets, whose results were shown in Supplementary Information, were described in Supplementary Table 13. The gastric cancer verification dataset, all Glyco-Decipher and GlyPep-Quant results are available in PRIDE with accession number PXD057799. Source data are provided with this paper.
Code availability
GlyPep-Quant was developed in the Java and Python language, and has been integrated into Glyco-Decipher. The standalone software Glyco-Decipher package56 can be downloaded from: https://github.com/DICP-1809/Glyco-Decipher.
References
Mereiter, S., Balmaña, M., Campos, D., Gomes, J. & Reis, C. A. Glycosylation in the era of cancer-targeted therapy: where are we heading? Cancer Cell 36, 6–16 (2019).
Hart, G. W. & Copeland, R. J. Glycomics hits the big time. Cell 143, 672–676 (2010).
Pinho, S. S. & Reis, C. A. Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15, 540–555 (2015).
Cao, L. et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 184, 5031–5052.e26 (2021).
Bagdonaite, I. et al. Glycoproteomics. Nat. Rev. Methods Prim. 2, 1–29 (2022).
Liu, L. et al. Automated intact glycopeptide enrichment method facilitating highly reproducible analysis of serum site-specific N-glycoproteome. Anal. Chem. 93, 7473–7480 (2021).
Liu, L. et al. Robust glycoproteomics platform reveals a tetra-antennary site-specific glycan capping with Sialyl-Lewis antigen for early detection of gastric cancer. Adv. Sci. 11, 2306955 (2024).
Kawahara, R. et al. Glycoproteome remodeling and organelle-specific N-glycosylation accompany neutrophil granulopoiesis. Proc. Natl. Acad. Sci. USA 120, e2303867120 (2023).
Fang, Z. et al. Glyco-Decipher enables glycan database-independent peptide matching and in-depth characterization of site-specific N-glycosylation. Nat. Commun. 13, 1900 (2022).
Zeng, W.-F., Cao, W.-Q., Liu, M.-Q., He, S.-M. & Yang, P.-Y. Precise, fast and comprehensive analysis of intact glycopeptides and modified glycans with pGlyco3. Nat. Methods 18, 1515–1523 (2021).
Polasky, D. A., Yu, F., Teo, G. C. & Nesvizhskii, A. I. Fast and comprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco. Nat. Methods 17, 1125–1132 (2020).
Zhang, Y., Fonslow, B. R., Shan, B., Baek, M.-C. & Yates, J. R. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 113, 2343–2394 (2013).
Karpievitch, Y. V., Dabney, A. R. & Smith, R. D. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinforma. 13, S5 (2012).
Aebersold, R. & Mann, M. Mass spectrometry-based proteomics. Nature 422, 198–207 (2003).
Jin, L. et al. A comparative study of evaluating missing value imputation methods in label-free proteomics. Sci. Rep. 11, 1760 (2021).
Yu, F., Haynes, S. E. & Nesvizhskii, A. I. IonQuant enables accurate and sensitive label-free quantification with FDR-controlled match-between-runs. Mol. Cell. Proteom. 20, 100077 (2021).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Kong, S. et al. pGlycoQuant with a deep residual network for quantitative glycoproteomics at intact glycopeptide level. Nat. Commun. 13, 7539 (2022).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Varki, A. et al. Essentials of Glycobiology (Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2015).
Chen, Z. et al. Recognition of core-fucosylated glycopeptides based on the Y1+Fuc/Y1 ratio in low-energy HCD spectra. Anal. Chem. 94, 17349–17353 (2022).
Shen, J. et al. StrucGP: de novo structural sequencing of site-specific N-glycan on glycoproteins using a modularization strategy. Nat. Methods 18, 921–929 (2021).
Yang, Y. et al. GproDIA enables data-independent acquisition glycoproteomics with comprehensive statistical control. Nat. Commun. 12, 6073 (2021).
Watanabe, Y., Allen, J. D., Wrapp, D., McLellan, J. S. & Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 369, 330–333 (2020).
Zhang, Y. et al. Site-specific N-glycosylation characterization of recombinant SARS-CoV-2 spike proteins. Mol. Cell. Proteom. 20, 100058 (2021).
Pont, L. et al. Site-specific N-linked glycosylation analysis of human carcinoembryonic antigen by sheathless capillary electrophoresis–tandem mass spectrometry. J. Proteome Res. 20, 1666–1675 (2021).
The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212 (2015).
Bernacka, K., Kuryliszyn-Moskal, A. & Sierakowski, S. The levels of alpha 1-antitrypsin and alpha 1-antichymotrypsin in the sera of patients with gastrointestinal cancers during diagnosis. Cancer 62, 1188–1193 (1988).
Baker, C., Belbin, O., Kalsheker, N. & Morgan, K. SERPINA3 (aka alpha-1-antichymotrypsin). Front. Biosci. 12, 2821–2835 (2007).
Jin, Y. et al. Alpha-1-antichymotrypsin as a novel biomarker for diagnosis, prognosis, and therapy prediction in human diseases. Cancer Cell Int. 22, 156 (2022).
Bae, Y. J. & Kratzsch, J. Corticosteroid-binding globulin: modulating mechanisms of bioavailability of cortisol and its clinical implications. Best. Pr. Res. Clin. Endocrinol. Metab. 29, 761–772 (2015).
Henley, D. E. & Lightman, S. L. New insights into corticosteroid-binding globulin and glucocorticoid delivery. Neuroscience 180, 1–8 (2011).
Misao, R., Iwagaki, S., Sun, W. S., Fujimoto, J. & Tamaya, T. Levels of corticosteroid-binding globulin mRNA in human ovarian cancers. Tumour Biol. 20, 263–269 (1999).
Gulfo, J. et al. New roles for corticosteroid binding globulin and opposite expression profiles in lung and liver. PLoS ONE 11, e0146497 (2016).
Ugorski, M. & Laskowska, A. Sialyl Lewis(a): a tumor-associated carbohydrate antigen involved in adhesion and metastatic potential of cancer cells. Acta Biochim. Polonica 49, 303–311 (2002).
Ester, M., Kriegel, H.-P., Sander, J. & Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining 226–231 (AAAI Press, Portland, Oregon, 1996) https://dl.acm.org/doi/10.5555/3001460.3001507.
Schubert, E., Sander, J., Ester, M., Kriegel, H. P. & Xu, X. DBSCAN revisited, revisited: why and how you should (Still) use DBSCAN. ACM Trans. Database Syst. 42, 19:1–19:21 (2017).
Keser, T. et al. High-throughput and site-specific N-glycosylation analysis of human alpha-1-acid glycoprotein offers a great potential for new biomarker discovery. Mol. Cell Proteom. 20, 100044 (2021).
Nie, S. et al. Glycoprotein biomarker panel for pancreatic cancer discovered by quantitative proteomics analysis. J. Proteome Res. 13, 1873–1884 (2014).
Ivanov, M. V. et al. DirectMS1: MS/MS-free identification of 1000 proteins of cellular proteomes in 5 minutes. Anal. Chem. 92, 4326–4333 (2020).
Dai, Y., Yang, Y., Wu, E., Shen, C. & Qiao, L. Deep learning powers protein identification from precursor MS information. J. Proteome Res. 23, 3837–3846 (2024).
Zeng, W.-F., Yan, G., Zhao, H., Liu, C. & Cao, W. Uncovering missing glycans and unexpected fragments with pGlycoNovo for site-specific glycosylation analysis across species. Nat. Commun. 15, 8055 (2024).
Sun, W. et al. Glycopeptide database search and de novo sequencing with PEAKS GlycanFinder enable highly sensitive glycoproteomics. Nat. Commun. 14, 4046 (2023).
Bouwmeester, R., Gabriels, R., Hulstaert, N., Martens, L. & Degroeve, S. DeepLC can predict retention times for peptides that carry as-yet unseen modifications. Nat. Methods 18, 1363–1369 (2021).
Zeng, W.-F. et al. AlphaPeptDeep: a modular deep learning framework to predict peptide properties for proteomics. Nat. Commun. 13, 7238 (2022).
Shi, T. et al. Advances in targeted proteomics and applications to biomedical research. Proteomics 16, 2160–2182 (2016).
Bourmaud, A., Gallien, S. & Domon, B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: principle and applications. Proteomics 16, 2146–2159 (2016).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Lecun, Y., Bottou, L., Bengio, Y. & Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 86, 2278–2324 (1998).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Doerr, A. Proteomics data reuse with MassIVE-KB. Nat. Methods 16, 26–26 (2019).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Okuda, S. et al. jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res. 45, D1107–D1111 (2017).
Zhu, H., Fang, Z. & Ye, M. MS1 feature library-based virtual match-between-runs quantification improves site-specific glycan identification and occupancy ratio analysis. Zenodo https://doi.org/10.5281/zenodo.15675491 (2025).
Acknowledgements
This work was supported, in part, by funds from the National Key Research and Development Program of China (Grant Nos. 2022YFC3400800 [H.Q.], 2024YFA1306300 [Y.W.] and 2021YFA1302600 [M.Y.]), the National Natural Science Foundation of China (Grant Nos. 22034007 [M.Y.], 22274014 [M.D.] and 22304174 [Z.F.]) and China Postdoctoral Science Foundation (Grant No. 2023M743424 [Z.F.]).
Author information
Authors and Affiliations
Contributions
These authors contributed equally: He Zhu, Zheng Fang. M.D. and M.Y. conceived the project. H.Z. developed the algorithm, wrote the software, and analyzed the results. Z.F. assisted in the interpretation of the results and software design. Y.N. collected the sample of human serum. Under the supervision of M.D., L.L. and Y.W. prepared the sample and performed mass spectrometry analyses with MS parameter optimizations. M.D. and M.Y. supervised the project. H.Z., Z.F., H.Q., M.D. and M.Y. discussed the project and the manuscript. H.Z., Z.F., M.D. and M.Y. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Henrique Duarte, Yi Yang and Wen-Feng Zeng for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, H., Fang, Z., Liu, L. et al. Library-based virtual match-between-runs quantification in GlyPep-Quant improves site-specific glycan identification. Nat Commun 16, 6483 (2025). https://doi.org/10.1038/s41467-025-61673-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61673-6
