
Abstract
Withanolides are steroidal lactones from nightshade (Solanaceae) plants with untapped drug potential due to limited availability of minor representatives caused by lack of biosynthetic pathway knowledge. Here, we combine phylogenomics with metabolic engineering to overcome this limitation. By sequencing the genome of the medicinal plant ashwagandha (Withania somnifera) and comparing it with nine Solanaceae species, we discover a conserved withanolide biosynthesis gene cluster, consisting of two sub gene clusters with differing expression patterns. We establish metabolic engineering platforms in yeast (Saccharomyces cerevisiae) and the model plant Nicotiana benthamiana to reconstitute the first five oxidations of withanolide biosynthesis, catalysed by the cytochrome P450 monooxygenases CYP87G1, CYP88C7, and CYP749B2 and a short-chain dehydrogenase/reductase, producing the aglycone of withanoside V. Enzyme functions are conserved within both sub gene clusters in W. somnifera and between W. somnifera and Physalis pruinosa. Our work sets the basis for biotechnological withanolide production to unlock their pharmaceutical potential.
Similar content being viewed by others


Introduction
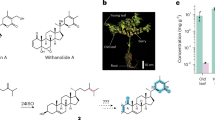
Plants are well-known for their extensive capabilities to produce structurally complex metabolites with potent biological activities. Even though many of these specialised metabolites have been studied extensively at a chemical level, the genetic basis for their biosynthesis is often still unknown, representing a major hurdle for biotechnological improvement of medicinal plants and development of microbial production systems alike. Such a lack of knowledge at the gene level also exists for withanolide biosynthesis. Withanolides are steroidal lactones occurring in several members of the nightshade family (Solanaceae). Named after the first discovery of these compounds in the medicinal plant Withania somnifera (ashwagandha), approx. 1200 withanolides are nowadays known from 22 genera of Solanaceae1,2. These metabolites have played a role in traditional medicine since ancient times3,4. For example, W. somnifera has been used in traditional Indian medicine, Ayurveda, as an anti-stress agent for millennia; these stress-relieving properties are also supported by modern placebo-controlled studies5,6,7. Detailed pharmacological studies underline the broad spectrum of biological activities of withanolides4,8,9,10,11,12 and withanolide-inspired drug candidates13. In stark contrast, only a single enzyme specific to withanolide biosynthesis has been identified and characterised so far14, hampering efforts to engineer withanolide metabolism in planta and produce withanolides biotechnologically15. This enzyme, sterol Δ24-isomerase (24ISO)14, catalyses the isomerisation of 24-methylenecholesterol (1), an intermediate of the general phytosterol pathway, to the withanolide-specific key intermediate 24-methyldesmosterol (2) (Fig. 1). As such, the 24ISO reaction represents the key committed step at which withanolide biosynthesis branches from phytosterol and brassinosteroid biosynthesis14. Several genes and enzymes in the general phytosterol pathway upstream of 24ISO were investigated in withanolide-producing plants16,17,18,19. In contrast, the biosynthetic pathway downstream of 24ISO, responsible for the conversion of 24-methyldesmosterol (2) to withanolides, is not yet known. Several putative withanolide biosynthesis gene candidates were tested by virus-induced gene silencing20,21, but no clear biochemical activity has been reported for them so far.

a 24-Methylenecholesterol (1) is converted by the only known withanolide biosynthetic enzyme sterol Δ24-isomerase (24ISO)14 into 24-methyldesmosterol (2). The enzymes for all subsequent biosynthetic steps are not known. b Representative genera and species of withanolide-producing plants and characteristic compounds from them. Photographs by Jakob Maximilian Horz, TU Braunschweig, Germany.
While the elucidation of plant biosynthetic pathways has been a slow and tedious process for a long time, advances in sequencing techniques over the past 20 years have dramatically accelerated the pace of gene function discovery in plants22. Most commonly, transcriptomic data is used to identify biosynthetic genes based on specific expression patterns in different growth stages, tissues or even cell types23,24,25,26. Genomic data was traditionally considered to be less important for pathway elucidation, because – in contrast to microorganisms27 – many biosynthetic genes in plants discovered early were not physically clustered28,29,30. However, this perspective is getting continuously challenged, as more plant genome sequences become available31. Indeed, many examples now show that biosynthetic gene clusters in plants are relatively common and enable efficient pathway elucidation30, as demonstrated recently for saponin, alkaloid, and terpenoid biosynthesis32,33,34,35.
During the discovery of the only withanolide-specific pathway gene, 24ISO, Knoch et al. reported possible clustering of this gene with other genes common for specialised metabolism14, but their analysis was still strongly limited by the lack of high-quality genome sequences of Solanaceae plants in 2018. A comparison of Capsicum annuum, Solanum melongena, and Petunia inflata indicated the co-occurrence of 24ISO with genes encoding cytochrome P450 monooxygenases and oxoglutarate-dependent dioxygenases14. However, none of the species of this comparison is known as a producer of canonical withanolides. In recent years, several high-quality genome sequences of withanolide-producing plants have been released. These include for example Physalis floridana (Physalis pubescens)36, Physalis grisea37, Physalis pruinosa37, Datura stramonium34,38, and Datura wrightii39.
In this work, we revisit the previous hypothesis of possible gene clustering in the context of withanolide biosynthesis. By sequencing the genome of the archetypical withanolide producer Withania somnifera and synteny analyses with other Solanaceae genome sequences, we reveal a conserved gene cluster in withanolide-producing plants that harbours the withanolide pathway gene 24ISO and multiple other genes typical for specialised metabolism. To overcome previous obstacles in functional validation of withanolide pathway genes, we employ metabolic engineering in the model organisms yeast (Saccharomyces cerevisiae) and Nicotiana benthamiana to successfully establish two independent platforms for withanolide pathway reconstitution. With these, we characterise three cytochrome P450 monooxygenases and a short-chain dehydrogenase/reductase that oxidise 24-methyldesmosterol (2) to construct the pivotal δ-lactone ring of withanolides. Our discovery of a conserved gene cluster for withanolide biosynthesis in Solanaceae plants and the development of synthetic biology systems will enable full elucidation and engineering of withanolide biosynthesis in the future, to further harness the drug potential of withanolides.
Results
Genome assembly of Withania somnifera
To explore the genomic context of withanolide biosynthesis, we generated a genome assembly of Withania somnifera, known as a prolific producer of withanolides. The genome size of W. somnifera was estimated to be 2.94 Gb. Building on Oxford Nanopore Technologies’ sequencing method, a total of 5.1 million reads (N50: 39.4 kb) corresponding to an estimated genome coverage of 34.6x were sequenced40. The de novo assembly comprised 93 contigs with an N50 length of 71 Mb and a total assembly size of 2.88 Gbp (Supplementary Table 1, Supplementary Table 2). The read coverage depth histogram shows a single peak around the estimated genome coverage suggesting a diploid genome with low heterozygosity (Supplementary Fig. 1). A total of 34,955 protein-encoding genes were predicted based on homology and transcriptome data, with an average gene length of 4978 bp and an average coding sequence (CDS) length of 1266 bp. BUSCO analysis41 using solanales_odb12 dataset revealed 96.2% complete homologues in the predicted proteins of W. somnifera. Additionally, we re-annotated the protein-coding genes of the chromosome-scale genome sequences of P. grisea and P. pruinosa, resulting in improved gene models and higher BUSCO completeness compared to the previous annotations37. The predicted genes of both genome sequences achieved 97.5% completeness in the BUSCO analysis.
The 12 pseudochromosomes from the chromosome-scale assembly of P. pruinosa37 were well represented by the top 36 largest contigs in our W. somnifera assembly (accounting for ~87% of the total assembly). A Circos plot revealed large overall synteny between both species (Fig. 2). Although the relative amount of repeats is comparable in P. pruinosa (77.59%) and W. somnifera (76.02%), W. somnifera has more terminal inverted repeats (TIR) (13.13% vs. 1.7%).

a Genomic landscape of the 36 W. somnifera pseudochromosomes (right) and 12 P. pruinosa pseudochromosomes (left). All density information is calculated in non-overlapping 1-Mbp windows. b Tandem repeats density. c Percentage of transposable elements (TEs), calculated in 10-Mbp non-overlapping windows. Total TEs in grey, long-terminal repeat (LTR) retrotransposons in red and terminal inverted repeats (TIR) in blue. d GC content. e Distribution of protein-coding genes. f Links between syntenic regions of both genomes.
Phylogenomics discovery of a putative withanolide biosynthetic gene cluster
With our high-quality assembly of the W. somnifera genome sequence in hands, we next set out to systematically explore the genomic context surrounding 24ISO, the only previously reported withanolide biosynthesis gene. To search for a possible conserved gene cluster, we analysed additional highly continuous genome sequences of the following withanolide-producing plants: Physalis floridana36, Physalis grisea37, Physalis pruinosa37, Datura stramonium34,38, and Datura wrightii39. For comparison, we included genome sequences of non-withanolide producing Solanaceae species, namely Solanum lycopersicum42 and Solanum tuberosum43 from the same subfamily Solanoideae as well as Nicotiana tabacum44. Using the experimentally characterised 24ISO genes14 as a bait, we identified their genomic positions and further orthologous genes as a starting point for synteny comparison. All genome sequences of withanolide-producing plants analysed here contained two copies of 24ISO in close proximity. Only in W. somnifera, a third 24ISO copy was additionally found on a different contig. No 24ISO orthologue was found in S. lycopersicum, S. tuberosum, and N. tabacum. Then, we compared the synteny of the genomic regions surrounding these 24ISO orthologues (Fig. 3, Supplementary Fig. 2, Supplementary Data 1). Genomes of withanolide producers contained a syntenic region that was absent in the non-producers S. lycopersicum and S. tuberosum. We then deduced the classes of encoded enzymes in this syntenic region by their Pfam domains. Strikingly, all genes in this syntenic region belong to gene families common in plant specialised metabolism, most importantly cytochrome P450 monooxygenases (CYPs), 2-oxoglutarate-dependent dioxygenases (ODDs), short-chain dehydrogenases/reductases (SDRs), and acyltransferases (AT). CYPs are particularly well-known for their central role in triterpenoid and steroid biosynthetic pathways26,45,46,47. The CYPs in the putative withanolide biosynthetic gene cluster fall into three different CYP families, namely CYP87, CYP88 (both part of the CYP85 clan) and CYP749 (part of the CYP72 clan). Both clans, particularly CYP85, are well-known as hotspots of CYPs involved in triterpenoid and steroid metabolism45. Less expected was the occurrence of sulfotransferase (ST) genes. While sulfotransferases are well-known in the biosynthesis of certain specialised metabolites such as glucosinolates48, a possible link to withanolide biosynthesis is not yet known. A few withanolides bearing 3-O-sulphate groups are known, however49,50, and their biosynthesis might involve a dedicated sulfotransferase. Phylogenetic analyses of the identified genes revealed that these genes from withanolide-producing species clustered together, forming a distinct clade; no orthologues are found in non-withanolide producing species like tomato, potato, and Nicotiana (Supplementary Figs. 3–8).

a Synteny plot. Gene lengths are unified for clarity. N. tabacum is included as an outgroup of the subfamily Solanoideae. W. somnifera 1 and W. somnifera 2 refer to the two gene clusters in W. somnifera on ctg090 and ctg003, respectively. The phylogenetic relationships shown at the left are adapted from ref. 131. b Heatmap summary of gene copy numbers of each gene family. Background colour is normalised to the highest number per column. Expression information about genes in the cluster are displayed in Supplementary Figs. 9–11.
Next, we analysed published RNA-seq datasets to verify that the genes in the gene cluster are not only physically clustered, but also co-expressed. Surprisingly, we observed two groups with distinct expression patterns, both in W. somnifera and in D. stramonium (Supplementary Figs. 9–11). This suggests that the withanolide gene cluster is separated into two sub gene clusters which are differentially regulated. Taken together, our phylogenomics analysis suggested that withanolide biosynthesis involves a conserved gene cluster.
Development of a yeast platform for production of withanolide pathway intermediates
Next, we wanted to support our discovery of a putative withanolide biosynthetic gene cluster by elucidating the biochemical functions of pathway enzymes. Reconstitution of withanolide biosynthesis in heterologous hosts has remained an unsolved problem up until now. The very low polarity and the limited accessibility of the last known intermediate 24-methyldesmosterol (2) prevents an efficient use as a substrate for enzyme assays in vitro. An alternative would be to produce 24-methyldesmosterol (2) in vivo, but no efficient metabolic engineering strategy has been reported so far. Previous studies showed that its precursor, 24-methylenecholesterol (1), can be produced in yeast (Saccharomyces cerevisiae) by deleting the ergosterol biosynthesis genes ERG4 and ERG5 and adding a gene encoding a Δ7 reductase from plants or animals, in order to hijack the sterol metabolism of yeast to produce plant-like sterols51,52,53. We decided to utilise and expand this strategy to set up a platform for functional evaluation of withanolide biosynthesis genes in yeast (Fig. 4a).

a Yeast engineering strategy to divert flux from ergosterol (5) biosynthesis to 24-methyldesmosterol (2). b GC-MS total ion current chromatograms showing production of 24-methyldesmosterol (2) and the shunt product ergosta-5,7,24-trien-3β-ol (6) in engineered yeast strain KMY23. c Comparison of electron impact mass spectra of ergosta-5,7,24-trien-3β-ol (6) and related compounds as TMS ethers supporting the proposed structure of 6. d Further yeast engineering to improve 24-methyldesmosterol (2) production. The bar plot shows means ± SD and data points of three independently grown yeast cultures. DCW: Dry cell weight. Budding_yeast icon by umasstr (https://github.com/umasstr) is licensed under CC0 (https://creativecommons.org/publicdomain/zero/1.0/) and was used without modifications. Source data are provided as a Source Data file.
With the help of established CRISPR/Cas techniques54, we inserted Physalis peruviana orthologues of the known plant genes sterol ∆7 reductase (7RED, also known as DWF5)55 and 24ISO14 into the genome of the prototrophic S. cerevisiae strain ST7574, which is derived from CEN.PK113-7D and contains a cas9 gene54. The resulting strain KMY14 only produced trace amounts of 24-methylenecholesterol (1) and 24-methyldesmosterol (2) (Fig. 4b). This result was in line with previous reports showing that under native conditions the ergosterol biosynthetic enzymes ERG4 and ERG5 efficiently consume the shared intermediate ergosta-5,7,24(28)-trien-3β-ol (3) via ergosta-5,7,22,24(28)-tetraen-3β-ol (4) to ergosterol (5)14,52,53. Upon deletion of ERG4 and ERG5 in the background strain ST7574 (strain KMY48), the required shared intermediate ergosta-5,7,24(28)-trien-3β-ol (3) was formed as the major sterol as reported in literature56,57. To redirect this intermediate to the desired product 24-methyldesmosterol (2), we deleted ERG4 and ERG5 in the 7RED/24ISO-expressing strain KMY14 to generate KMY23. Gratifyingly, KMY23 produced 24-methyldesmosterol (2) at a level of 7.6 mg/g dry cell weight (DCW) (Fig. 4b). Besides 24-methyldesmosterol (2), a major peak 6 at 14.5 min was observed in yeast, with a mass difference of −2 compared with 24-methyldesmosterol (2) (Fig. 4b). The mass spectrum of this compound 6 was almost identical to that of ergosterol (5) (Fig. 4c), but the difference in retention time indicated that 6 must be an isomer of ergosterol (5) (Fig. 4b). A striking difference in their mass spectra was the presence of a fragment at m/z 294 for 6 that was absent for ergosterol (5); in the mass spectrum of 24-methyldesmosterol (2), this fragment was shifted to m/z 296, matching the mass difference of the molecular ions. Previous studies reported that this fragment is indicative of sterols with a Δ24(28) or Δ24(25) double bond and is formed by a McLafferty rearrangement involving allylic cleavage of the C-22(23) bond57,58,59,60. The mass difference of −2 suggested that 6 must possess an additional double bond in the ABCD ring system. Ergosterol (5) and 6 both exhibit a major fragment at m/z 337 ([M − 131]+), which is characteristic of sterols with Δ5,7 dienes61,62; therefore, the additional double bond of 6 is most likely positioned at C-7,8. We therefore propose that compound 6 is ergosta-5,7,24-trien-3β-ol (Fig. 4c). This ∆7 analogue of 24-methyldesmosterol (2) would be formed if ergosta-5,7,24(28)-trien-3β-ol (3) is converted by 24ISO before reduction by 7RED can take place. This hypothesis was also supported by the fact that upon deletion of 24ISO the peak for ergosta-5,7,24-trien-3β-ol (6) completely disappeared (strain KMY77); instead, a mixture of the 7RED product 24-methylenecholesterol (1) and non-reduced ergosta-5,7,24(28)-trien-3β-ol (3) was observed (Fig. 4b). The occurrence of the non-reduced but isomerised product ergosta-5,7,24-trien-3β-ol (6) in KMY23 and non-reduced product ergosta-5,7,24(28)-trien-3β-ol (3) in KMY77 therefore indicates a misbalance between the very high activity of P. peruviana 24ISO and the limited activity of P. peruviana 7RED in our yeast system.
To facilitate subsequent gene discovery, we next wanted to improve the production of 24-methyldesmosterol (2) in yeast. In initial experiments, we observed that overexpression of mevalonate pathway genes only resulted in increased levels of squalene but not of downstream sterols. Therefore, to keep the metabolic burden on our strains as low as possible, no mevalonate pathway genes were overexpressed. Instead, we first added an additional copy of P. peruviana 7RED to improve the conversion of ergosta-5,7,24(28)-trien-3β-ol (3) to 24-methylenecholesterol (1); simultaneously, we overexpressed the upc2-1 allele, encoding transcription factor mutant UPC2G888D involved in the regulation of sterol metabolism and reported to boost the biosynthesis of sterols63,64. The resulting strain KMY50, however, did not show elevated levels of 24-methyldesmosterol (2). Previous studied showed that manipulation of sterol homeostasis, i.e., the balance of sterol acylation and sterol ester hydrolysis, can improve the production of sterols in yeast65. We therefore co-expressed the acyl-CoA:sterol acyltransferase gene ARE2 either with the sterol ester hydrolase gene YEH2 (KMY53) or with YEH1 (KMY54). In both cases, an improvement in 24-methyldesmosterol (2) levels was noted. KMY53 produced 9.3 mg/g DCW, whereas KMY54 reached 13.5 mg/g DCW (Fig. 4d). However, the final OD600 after cultivation for 96 h in shake flasks was severely reduced, particularly for strain KMY54. In summary, our data show that the key intermediate 24-methyldesmosterol (2) can be produced in engineered yeast, but further strain improvement is required to improve product levels, reduce the amounts of shunt product 6, and overcome growth defects.
Engineering of Nicotiana benthamiana for production of 24-methyldesmosterol
Due to these unresolved limitations of yeast as a platform for withanolide pathway reconstitution, we alternatively envisioned to generate a plant-based system using the popular model organism Nicotiana benthamiana66. In contrast to yeast, plants natively produce 24-methylenecholesterol (1) as a transient intermediate en route to phytosterols and brassinosteroids67. We therefore expected that usage of a plant host would enable us to circumvent the undesired yeast shunt product ergosta-5,7,24-trien-3β-ol (6) from the misbalance of 24ISO and 7RED activity. Furthermore, transient expression in N. benthamiana provides the added benefit of easy and flexible “mix-and-match” co-expression, which would drastically accelerate the pace of testing different gene combinations66. We first transiently expressed only 24ISO, the gene encoding sterol Δ24-isomerase14, in N. benthamiana; while this caused changes in the metabolic profile, no 24-methyldesmosterol (2) was detected. This suggested that insufficient amounts of 24-methylenecholesterol (1) accumulate under native conditions in N. benthamiana. We therefore sought to increase the formation of 24-methylenecholesterol (1) and hence 24-methyldesmosterol (2) by metabolic engineering. In principle, this could be achieved by two complementary strategies: Either by blocking the pathway downstream of 24-methylenecholesterol (1) controlled by the reductase DWF168 (strategy I), or overexpressing its upstream phytosterol pathway (strategy II) (Fig. 5a). We first attempted to silence DWF1 by virus-induced gene silencing69. In accordance with previous reports68, this caused a very strong dwarf phenotype and sick-looking plants (Supplementary Fig. 12a), probably due to the effects on brassinosteroid levels. After transient overexpression of 24ISO in DWF1-silenced N. benthamiana plants, small quantities of 24-methyldesmosterol (2) could be detected (Supplementary Fig. 12b). Nonetheless, the experimental challenges of coordinating the relatively slow systemic gene silencing with the relatively fast local transient overexpression and the severe phenotypic effects on the plants prompted us to test upstream pathway overexpression as an alternative strategy (Fig. 5a). First, we co-expressed 24ISO with a gene encoding truncated, feedback-insensitive hydroxymethylglutaryl coenzyme A reductase (tHMGR), which is known as a very efficient booster for the mevalonate pathway and triterpenoid production46. In these samples, we observed a very small peak below the limit of quantification corresponding to 24-methyldesmosterol (2) (Fig. 5b). We concluded that further reactions of the phytosterol pathway limit the 24-methyldesmosterol (2) yield.

a Overview of the two different engineering strategies that were attempted in this work. b Effect of phytosterol pathway gene overexpression (strategy II) on production of 24-methyldesmosterol (2). Total ion current (TIC) GC-MS chromatograms of representative samples from transient expression in N. benthamiana. The bottom bar plot indicates relative 24-methyldesmosterol (2) levels when a single gene of the 15-gene set from tHMGR to 24ISO was left out. Bar plots show means ± SEM and data points of three biological replicates. * P < 0.05, ** P < 0.01, NS not significant by Student’s two-tailed unpaired t test; exact P values are indicated. LOD: limit of detection, LOQ, limit of quantification. c GC-MS TIC chromatograms showing formation of two shunt products 7 and 8 dependent on 24ISO in N. benthamiana. d Proposed formation of shunt products 7 and 8 by background epoxidation of the Δ24 double bond in N. benthamiana. Nicotiana_benthamiana icon by Connor-Tansley (https://github.com/Ctansley) is licensed under CC-BY 4.0 Unported (https://creativecommons.org/licenses/by/4.0/) and was used without modifications. Source data are provided as a Source Data file.
As no comprehensive information is available which enzymes en route to 24-methylenecholesterol (1) are rate-limiting, we decided to transiently overexpress the full gene set for the phytosterol pathway reported from Arabidopsis thaliana (Supplementary Fig. 13) in addition to tHMGR and 24ISO in N. benthamiana. This comprised a total of 15 genes. Gratifyingly, this approach led to a drastic increase in 24-methyldesmosterol (2) formation to 0.9 mg/g dry weight (Fig. 5b). Notably, the byproduct ergosta-5,7,24-trien-3β-ol (6) was not observed, supporting our hypothesis that the 24ISO/7RED misbalance issue encountered in yeast can be circumvented by using a plant host. To understand which upstream pathway genes had the largest effect on 24-methyldesmosterol (2) titres, we performed a leave-one-out experiment (Fig. 5b). Statistically significant negative effects on 24-methyldesmosterol (2) levels were observed when tHMGR, SMO2-1 plus SMO2-2, DWARF5, or DWARF7 were not included, suggesting that these represent major bottlenecks towards 24-methylenecholesterol (1).
The chromatograms of N. benthamiana plants producing 24-methyldesmosterol (2) also contained two new product peaks dependent on the presence of 24ISO with a mass difference of +88 compared with 24-methyldesmosterol (2), which would fit to an extra trimethylsilyl (TMS)-bearing oxygen (Fig. 5c). Analysis of fragmentation patterns suggested that these compounds were oxidised in the side chain (Supplementary Fig. 14-16, Supplementary Data 2). The two compounds 7 and 8 were successfully isolated (isolated yield 0.03 and 0.03 mg/g dry weight, respectively) and fully characterised by NMR spectroscopy (Supplementary Fig. 17, Supplementary Data 3). Surprisingly, in comparison to 24-methyldesmosterol (2), the Δ24 double bond of 7 and 8 was shifted to Δ25 or Δ24(28), respectively. In agreement with the fragmentation analysis, NMR data showed that both products 7 and 8 contained an additional hydroxy group in allylic position of the double bond in the side chain. Hence, the systematic name of 7 is ergosta-5,25-diene-3β,24ξ-diol, while 8 is ergosta-5,24(28)-diene-3β,25-diol. Both compounds were isolated before from the withanolide-producing plant Physalis minima var. indica and named physalindicanol A (7) and B (8), respectively70. The occurrence of these regioisomeric allylic alcohols can be explained by background epoxidation of the Δ24 double bond of 24-methyldesmosterol (2)70 in N. benthamiana, generating hypothetical intermediate 9; either in vivo or during workup, this epoxide can then be opened to allylic alcohols 7 and 8 (Fig. 5d).
We concluded that, despite the undesirable background epoxidation, our metabolic engineering strategy in N. benthamiana reaching 0.9 mg/g dry weight of 24-methyldesmosterol (2) was suitable for further elucidation of withanolide biosynthesis.
Dihydroxylation of 24-methyldesmosterol in withanolide biosynthesis
After establishing heterologous platforms capable of producing the last known withanolide biosynthesis intermediate 24-methyldesmosterol (2), we turned our attention to screening genes from the gene cluster (Fig. 3) to identify the next steps in withanolide biosynthesis. Labelling studies demonstrated that withanolide biosynthesis proceeds by oxidative assembly of the side chain lactone71,72. Genes from the cytochrome P450 monooxygenase subfamilies CYP87G, CYP88C, and CYP749B were well conserved within all withanolide-producing species (Fig. 3, Supplementary Fig. 18) and therefore prioritised. To test if CYPs showed functional conservation between sub gene clusters and also between species, we selected CYP homologues not only from our main model system W. somnifera (Ws) but also from P. pruinosa (Pp) for a first rapid screening in our N. benthamiana platform producing 24-methyldesmosterol (2).
Of the tested CYP genes, only those from the CYP87G subfamily showed activity on 24-methyldesmosterol (2) detectable by GC-MS. The same activity was observed for the two W. somnifera homologues from the two sub gene clusters and for the tested homologue from P. pruinosa (Supplementary Fig. 19a). As all of these functionally equivalent CYPs belong to the same orthogroup, they were assigned the same systematic name CYP87G1 and were further distinguished by species codes (Ws or Pp) and internal names (see Supporting File). All subsequent experiments were carried out with the CYP87G1 homologue from P. pruinosa (PpCYP87G1). To test the function of PpCYP87G1 in yeast, the 24-methyldesmosterol (2) producing strain KMY23 in combination with a cytochrome P450 reductase gene from Arabidopsis thaliana was used (strain KMY55).
In both N. benthamiana and yeast, a new product peak 10 with a mass shift of +88 was detected by GC-MS upon presence of PpCYP87G1 (Fig. 6b). Analysis of the mass spectrum from electron impact ionisation suggested that 10 might be a hydroxylation product of 24-methyldesmosterol (2) at C-22 (Supplementary Fig. 14-16, Supplementary Data 2). We isolated the shared product 10 from N. benthamiana (0.04 mg/g dry weight isolated yield) and elucidated its structure by NMR spectroscopy (Supplementary Fig. 17, Supplementary Data 3). In agreement with our GC-MS fragmentation analysis, 10 contained an additional hydroxy group at C-22. Due to the flexibility and free rotation of the side chain, the stereochemistry at C-22 could not be confidently deduced by nuclear Overhauser effect spectroscopy (NOESY). Instead, we compared the coupling pattern of H-22 of our isolated compound 10 with two pairs of structurally related C-22 epimers that were obtained by semi synthesis (Supplementary Fig. 20). For the two synthetic 22S epimers, doublets of doublets were observed at H-22, in contrast to apparent doublets of triplets for the two synthetic 22R epimers. As compound 10 also showed an apparent doublet of triplets for H-22, C-22 was assigned as 22R, which is also the configuration expected for natural withanolides. In conclusion, compound 10 was confirmed to be (22R)-ergosta-5,24-diene-3β,22-diol (22R-hydroxy-24-methyldesmosterol). Besides shared product 10, we also observed further compounds from PpCYP87G1 expression occurring exclusively either in yeast or in N. benthamiana. In yeast, a second compound 11 with a mass shift of −2 compared with 10 was produced, while in N. benthamiana further compounds 12 and 13 with mass differences of +88 and +16 compared with 10 were detected (Fig. 6b). As the exclusive occurrence in either yeast or N. benthamiana as well as the mass shifts were in excellent agreement with the occurrence of shunt products related to 24-methyldesmosterol (2) observed during our metabolic engineering efforts (Figs. 4b, 5c), we strongly suspected that these products reflected the same background reactions and not additional enzymatic activity of PpCYP87G1. To confirm this, we isolated the putative shunt products 12 and 13 from N. benthamiana (0.04 and 0.01 mg/g dry weight isolated yields). NMR spectroscopy indicated that both products contained a hydroxy group at C-22 (Supplementary Fig. 17, Supplementary Data 3). In addition, compound 12 possessed the same Δ24(28) double bond and C-25 hydroxy group as shunt product 8. Compound 13 did not contain any olefinic carbons in the side chain, but instead two carbons at 62.6 and 65.6 ppm, respectively, which imply that 13 contains an epoxy group. The isolation of epoxide 13 strongly supports our hypothesis that the rearranged allylic alcohols are derived from Δ24 epoxidation (Fig. 5d). Compound 12, named (22R)-ergosta-5,24(28)-diene-3β,22,25-triol was isolated before from Physalis minima and has been named phyministerol A73; our NMR data of 12 are in very good agreement with the published data (Supplementary Table 3). Compound 13 or (22R)-24,25-epoxyergost-5-ene-3β,22-diol has not been reported before, but steroids with matching side chains have been isolated from the nightshade plant Petunia hybrida (Supplementary Fig. 21)74.

a Proposed formation of intermediates and shunt products in N. benthamiana and S. cerevisiae during the dihydroxylation of 24-methyldesmosterol (2). Dashed arrows indicate alternative pathways to N. benthamiana shunt products. b Relative levels of identified compounds in GC-MS chromatograms of N. benthamiana (green) and S. cerevisiae (yellow) upon co-expression of genes from P. pruinosa (Pp). For each compound, peak areas were normalised to internal standard and sample dry weight (either N. benthamiana leaf dry weight or yeast dry cell weight) and converted to relative amounts by setting the highest mean value of each compound for each host organism to 100%. Bar plots show means ± SEM and data points of three biological replicates. W22H: withanolide biosynthesis 22-hydroxylase; W1H: withanolide biosynthesis 1-hydroxylase. Budding_yeast icon by umasstr (https://github.com/umasstr) is licensed under CC0 (https://creativecommons.org/publicdomain/zero/1.0/) and was used without modifications. Nicotiana_benthamiana icon by Connor-Tansley (https://github.com/Ctansley) is licensed under CC-BY 4.0 Unported (https://creativecommons.org/licenses/by/4.0/) and was used without modifications. Source data are provided as a Source Data file.
Next, we co-expressed PpCYP87G1 with the remaining CYP gene candidates in N. benthamiana to find the next step in the pathway. Only upon co-expression of CYPs of the orthogroup CYP88C7 we observed new product peaks by GC-MS (Supplementary Fig. 19b). Again, the same activity was observed for both CYP88C7 homologues from the W. somnifera sub gene clusters and for the tested homologue from P. pruinosa (Supplementary Fig. 19b). Notably, none of the other two tested CYP88C genes belonging to a different orthogroup showed any activity under these conditions (Supplementary Fig. 19b). We also introduced PpCYP88C7 into our yeast strain already harbouring PpCYP87G1 for further evaluation of the biochemical activity. One product 14 with a mass shift of +88 by GC-MS analysis compared with the PpCYP87G1 product 10 was found in both heterologous hosts. In N. benthamiana, extra peaks with an additional +88 mass shift were observed; no extra peak was detected in yeast. The mass spectra of all PpCYP88C7-dependent peaks showed a m/z 217 fragment, which has been reported as a diagnostic ion for 1,3-dihydroxylated cyclohexanes75 (Supplementary Figs. 14–16, Supplementary Data 2). The fragmentation pattern therefore suggested that CYP88C7 carries out hydroxylation of C-1. Although we could not purify sufficient amounts of 14 for NMR spectroscopy, we successfully obtained shunt product 15 from N. benthamiana (isolated yield 0.01 mg/g dry weight). NMR data indicated that the side chain of 15 was identical to shunt product (22R)-ergosta-5,24(28)-diene-3β,22,25-triol (phyministerol A) (12). However, one of the other carbons exhibited a drastic downfield shift to 73.1 ppm, indicating an additional hydroxy substituent. An HMBC correlation from methyl group H-19 and COSY correlations with H-2 unambiguously confirmed that the new hydroxy group was located at C-1 (Supplementary Fig. 17). A NOE correlation between methyl group H-19 and H-1 indicated an α configuration of C-1 (Supplementary Fig. 22). Previously isolated steroids from nightshade plants with a hydroxy group at C-1, such as withanosides, also possess a C-1 α configuration76.
Elucidation of oxidative lactone formation in withanolide biosynthesis
As the next step in the biosynthetic pathway, we speculated that the intermediate 14 would be oxidatively transformed to the corresponding lactone compound, which would correspond to the aglycone of the known withanolide glycoside withanoside V (16)76 (Fig. 7a). We therefore screened our remaining CYP candidate genes in combination with PpCYP87G1 and PpCYP88C7 in our N. benthamiana system. Only when PpCYP87G1, PpCYP88C7, and PpCYP749B2 were co-expressed, a new peak was observed by LC-MS that matched withanoside V aglycone (16) in terms of retention time, high-resolution mass and MS/MS fragmentation (Fig. 7b, Supplementary Fig. 23). This compound was not observed when samples were saponified before LC-MS analysis. The function of CYP749B2 from W. somnifera and P. pruinosa was conserved; surprisingly, though, only one of the two homologues from the W. somnifera gene cluster 1 showed activity (Supplementary Fig. 19c). Co-expression of PpCYP87G1 and PpCYP749B2 without PpCYP88C7 did not result in lactone formation, suggesting that lactone formation in the side chain cannot take place before C-1 hydroxylation.

a Proposed lactone formation from (22R)-ergosta-5,24-diene-1α,3β,22-triol (14) to withanoside V aglycone (16) by CYP749B2 (W26O) and SDR (W26DH) and semisynthetic conversion of 16 to (22R)-ergosta-5,24-diene-1α,3β,22,26-tetrol (17). b Co-expression of CYPs from P. pruinosa (Pp) in N. benthamiana leading to formation of withanoside V aglycone (16) in comparison to an authentic reference compound. Extracted ion LC-MS chromatograms of m/z 443 (low resolution) are shown with the respective ion levels. c Co-expression of CYPs from P. pruinosa (Pp) in yeast leading to formation of (22R)-ergosta-5,24-diene-1α,3β,22,26-tetrol (17). Extracted ion LC-MS chromatograms of m/z 469 are shown with the respective ion levels. Withanoside V aglycone (16) was not detected in these strains and was only produced when SDR was additionally included (Supplementary Fig. 24). d Increased formation of withanoside V aglycone (16) in N. benthamiana upon co-expression of SDR homologues with 24ISO and CYP87G1/CYP88C7/CYP749B2 genes. Comparable results were obtained when genes only from W. somnifera (left) or only from P. pruinosa (right) were used, with the exception of PpSDR-b. The bar plots show means ± SEM and data points of six biological replicates. e In vitro assays with PpCYP749B2 (obtained as yeast microsomes containing a cytochrome P450 reductase (CPR)) and PpSDR-a/b (purified soluble protein produced in E. coli). Semisynthetic intermediate 17 was used as a substrate. Extracted ion LC-MS chromatograms of m/z 443 to monitor formation of withanoside V aglycone (16) are shown with the respective ion levels. W26O: Withanolide biosynthesis 26-oxidase; W26DH: withanolide biosynthesis 26-dehydrogenase. Budding_yeast icon by umasstr (https://github.com/umasstr) is licensed under CC0 (https://creativecommons.org/publicdomain/zero/1.0/) and was used without modifications. Nicotiana_benthamiana icon by Connor-Tansley (https://github.com/Ctansley) is licensed under CC-BY 4.0 Unported (https://creativecommons.org/licenses/by/4.0/) and was used without modifications. Source data are provided as a Source Data file.
To further corroborate the role of CYP749B2 for lactone formation, we introduced PpCYP749B2 into our previous yeast strain already containing PpCYP87G1 and PpCYP88C7. Surprisingly, no withanoside V aglycone (16) was detectable in this system. We first suspected that weak CYP activity as often observed in yeast was the reason for this lack of activity. Strains were therefore cultured under modified conditions with larger culture vessels that offered better aeration. Nonetheless, we could still not detect the target lactone 16 in yeast. However, upon careful analysis of product profiles, we observed new peaks that were only present in yeast strains including all three CYP genes. One of them could be identified as (22R)-ergosta-5,24-diene-1α,3β,22,26-tetrol (17), the aglycone of cilistol v77, by comparison to an authentic standard obtained by chemical reduction of withanoside V aglycone (16) (Fig. 7a, c). These results suggest that PpCYP749B2 is indeed active in yeast and can oxidise C-26 of intermediate 14 but cannot complete the full oxidation to the lactone ring of 16 without support by other enzymes in yeast.
To find other enzymes that might complete the generation of the lactone moiety, we revisited the enzyme classes encoded by our withanolide gene clusters. As short-chain dehydrogenases/reductases are known to be capable of oxidising lactols to lactones, for example in the biosynthesis of nepetalactone78, their involvement seemed highly plausible. Gratifyingly, yeast strains that contained one of the SDR genes from W. somnifera or P. pruinosa in addition to the three CYP genes successfully produced withanoside V aglycone (16) (Supplementary Fig. 24). In N. benthamiana, co-expression of SDR together with the CYP genes led to a 3-7-fold increase of the peak area of withanoside V aglycone (16) (Fig. 7d).
To get further insights into the biochemical interplay of CYP749B2 and SDR, we performed in vitro assays with both enzymes; microsomes enriched with PpCYP749B2 were obtained from yeast, while two PpSDR homologues a/b containing His-tags were produced in E. coli and purified by affinity chromatography. In vitro assays were carried out with the tetrahydroxylated intermediate 17 obtained by semisynthesis as a substrate (Fig. 7e). Positive controls containing both PpCYP749B2 and PpSDR-a/b showed formation of withanoside V aglycone (16) as expected, confirming that all enzymes were active under assay conditions. Reactions only containing PpCYP749B2 did not result in formation of lactone 16, matching to our results for yeast strains lacking SDR. Surprisingly, assays containing only PpSDR-a without PpCYP749B2 reproducibly showed small amounts of the lactone product 16, but much less than when PpCYP749B2 was also included. This suggests that SDR can not only carry out the last oxidation from a putative aldehyde 18a or lactol 18b intermediate to lactone 16 but also – much less efficiently than CYP749B2 – the prior oxidation from alcohol 17 to aldehyde 18a.
Lastly, we performed virus-induced gene silencing in W. somnifera to support the role of CYP87G1, CYP88C7, CYP749B2, and SDR in planta. Fragments for silencing were designed to target all gene copies from the two sub gene clusters (Fig. 8a). Silencing constructs also contained a fragment for the phytoene desaturase (PDS) visual marker gene79 to serve as a guide for sample harvesting (Fig. 8a, b). Silencing of PDS alone caused a decrease of withaferin A levels compared to a nonspecific control targeting the gene of green fluorescent protein (which is absent in W. somnifera) (Supplementary Fig. 25); hence, all PDS co-silencing experiments were compared to a PDS-GFP co-silencing negative control. Silencing of 24ISO (together with PDS) was used as a positive control14. Successful silencing of target genes was also supported by qPCR (Fig. 8c). Silencing of all genes with the exception of SDR resulted in a drastic decrease of withaferin A levels to 4–22% mean values compared to the PDS-GFP silencing control; only for SDR no such decrease was observed (Fig. 8d). A similar metabolic effect was observed for two additional major compounds that were also classified as withanolides based on their high-resolution masses and retention times (Supplementary Fig. 26). This provided strong support that the withanolide gene cluster described here is indeed crucial for withanolide biosynthesis in planta.

a Construct design to co-silence all homologues of target genes and the visual marker gene phytoene desaturase (PDS). b Representative pictures of W. somnifera plants infiltrated with silencing constructs four weeks after infiltration. c Quantitative reverse transcription polymerase chain reaction analysis of target gene expression in silenced leaves. The bar plot shows mean ± SEM and data points of three biological replicates. d Relative quantification of withaferin A levels in silenced plants. The bar plot shows mean ± SEM and individual data points; the sample size of replicates showing successful silencing as judged by a photobleaching phenotype is shown above the bars. e, f Representative GC-MS chromatograms showing accumulation of intermediates in silenced plants (target gene plus PDS) in comparison to PDS-GFP-silenced plants. Chromatograms were normalised by leaf sample dry weight. Comparable data was obtained in two independent experiments. Compounds: 24-methylenecholesterol (1), 24-methyldesmosterol (2), ergosta-5,25-diene-3β,24ξ-diol (physalindicanol A) (7), ergosta-5,24(28)-diene-3β,25-diol (physalindicanol B) (8), (22R)-ergosta-5,24-diene-3β,22-diol (10), (22R)-ergosta-5,24(28)-diene-3β,22,25-triol (phyministerol A) (12), (22R)-ergosta-5,24-diene-1α,3β,22-triol (14), (22R)-ergosta-5,24(28)-diene-1α,3β,22,25-tetrol (15). Source data are provided as a Source Data file.
To further corroborate the biosynthetic roles of the pathway genes, we searched for accumulation of pathway intermediates in GC-MS samples from silenced plants. 24ISO silencing led to accumulation of 24-methylenecholesterol (1); CYP87G1-silenced plants accumulated 24-methyldesmosterol (2); plants with silenced CYP88C7 contained (22R)-ergosta-5,24-diene-3β,22-diol (10); and CYP749B2-silenced plants were enriched in intermediate 14 (Fig. 8e, f). Notably, the same shunt products that we observed in N. benthamiana also accumulated during gene silencing; shunt products 7 and 8 were major products when any of the CYP genes was silenced; shunt products 12 or 15, respectively, were detected when CYP88C7 or CYP749B2 were targeted. Accordingly, these metabolite profiles were in excellent agreement with our biosynthetic model derived from heterologous pathway reconstitution.
Taken together, our data provide support for a multistage oxidative assembly of the characteristic lactone ring of withanolides (Figs. 6a, 7a): First, C-22 is hydroxylated by CYP87G1; second, hydroxylation of C-1 is carried out by CYP88C7; lastly, the lactone ring is generated by step-wise oxidation of C-26 and lactonisation by CYP749B2 and SDR. CYP749B2 is crucial for the initial C-26 hydroxylation of intermediate 14 to intermediate 17, while SDR is solely responsible for the final conversion of aldehyde 18a or lactol 18b to lactone 16. In contrast, both CYP749B2 and SDR can carry out the oxidation of alcohol 17 to aldehyde 18a or lactol 18b. In N. benthamiana and W. somnifera, background enzymes can partially replace the dedicated withanolide gene cluster SDR, whereas in yeast the SDR is a crucial factor for successful lactone formation. In addition, shunt reactions by Δ24(25) epoxidation can occur in the heterologous host N. benthamiana and the native producer W. somnifera alike. Based on these results, we propose the trivial names “withanolide biosynthesis 22-hydroxylase” (W22H) for CYP87G1, “withanolide biosynthesis 1-hydroxylase” (W1H) for CYP88C7, “withanolide biosynthesis 26-oxidase” (W26O) for CYP749B2, and “withanolide biosynthesis 26-dehydrogenase” (W26DH) for SDR. Together, these three CYPs and the SDR are therefore responsible for the biosynthesis of the key δ-lactone ring and for preparing the enone formation in the A ring of withanolides.
Discussion
In this work, we identified a gene cluster for withanolide biosynthesis in Solanaceae plants. The gene cluster reported here adds to several previously reported cases of gene clustering in specialised metabolism in Solanaceae plants, for example for biosynthesis of glycoalkaloids, modified fatty acids, acyl sugars, or terpenoids23,47,80,81. As such, the Solanaceae family represents an increasingly intriguing model system to study gene clustering in plants. The function of the gene cluster described here for withanolide biosynthesis is supported by our phylogenomics analysis, the biochemical activity of CYP87G1, CYP88C7, CYP749B2 and SDR deduced by metabolic engineering in yeast and N. benthamiana, and virus-induced gene silencing in W. somnifera. Previously, several genes were already connected to withanolide biosynthesis. A CYP749 from W. somnifera identified by a transcriptome-based approach was associated with withanolide biosynthesis based on virus-induced gene silencing, which resulted in a strong change in the withanolide profile20. Employing a similar approach, a recent study in P. angulata implied a role of a CYP749 and a CYP88 in the biosynthesis of physalin-type withanolides82. The biochemical functions of these CYPs as well as the genomic locations of the underlying genes have been unknown so far. We located these previously suggested candidates in our assembly of W. somnifera. Indeed, the closest homologues of these CYPs are encoded in the gene clusters described here (Supplementary Table 4). However, other genes identified in previous studies to be involved in withanolide production in Withania somnifera like WsCYP93Id21, WsCYP71B1020, glycosyltransferases83 and others84 were not part of or located close to the gene cluster described in this study (Supplementary Table 5). Some of these genes were moderately co-expressed with 24ISO (Supplementary Table 6). Further work will be required to study the functional role of these genes in withanolide biosynthesis.
Similar to examples from other plants85, our synteny analyses highlight the plasticity of plant biosynthetic gene clusters. Even in closely related species of the same genus, changes in gene order and orientation as well as gene duplications are not unusual. How these differences are relevant for increasing the structural diversity of withanolides will need to be addressed in the future. A unique feature of the withanolide gene cluster is the occurrence of sub gene clusters, as supported by synteny analysis, gene expression analysis (Supplementary Figs. 9–11), and biochemical demonstration of functional conservation (Supplementary Fig. 19). Gene duplications followed by neofunctionalization are common drivers of evolution30. In the case of withanolide biosynthesis, it appears that such an evolutionary event has occurred not only on the single gene level, but at a whole gene cluster level. Future studies will need to carefully address functional differences between the sub gene clusters to gain further insights into the evolution and biological role of withanolides in nightshade plants.
Elucidating and engineering the biosynthetic pathways of specialised steroids from plants still represents a major challenge. While many other compound classes such as alkaloids or other terpenoids are nowadays readily accessible in yeast or N. benthamiana, progress for steroids has been lagging behind. Only recently, the first successful metabolic engineering examples of steroidal natural products derived from the more common precursor cholesterol were published86,87,88,89. These have been facilitated by the fact that cholesterol natively accumulates in N. benthamiana, albeit at low levels. In contrast, our data shows that the production of relevant levels of 24-methylenecholesterol (1) or 24-methyldesmosterol (2) in yeast or N. benthamiana requires substantial additional engineering of sterol metabolism. In yeast, lowering the formation of shunt products arising from the misbalance of 24ISO and 7RED activity will be critical. The production of withanolide pathway intermediates in N. benthamiana appeared more efficient, even though an extensive set of genes had to be co-expressed. For maximum production of 24-methyldesmosterol (2) in N. benthamiana, we had to transiently overexpress 15 genes at the same time. Maximum production of withanoside V aglycone (16) was achieved by co-expression of 19 genes. To our knowledge, this setup is amongst the most complex examples reported to date90,91,92. Once again, this highlights the tremendous power of efficient and facile co-expression in N. benthamiana for gene discovery and pathway engineering in plants66. Our metabolic engineering efforts now enable further elucidation of withanolide biosynthesis, in order to close the gap from traditional medicine to modern drug development and production.
Methods
Plant materials
W. somnifera (XX-0-STGAL—22/1985) plants were grown in the Botanical Garden of TU Braunschweig in a greenhouse with natural light at approximately 20 °C. Genes from P. pruinosa were obtained as synthetic genes from GeneWiz (Azenta Life Sciences) based on the published genome sequence37 and our re-annotation without handling of the plant.
Genome sequencing, assembly, and annotation
DNA extraction, quality control, short fragment depletion, and DNA repair for the Oxford Nanopore Technologies sequencing approach was performed with a CTAB-based protocol, Short Read Eliminator kit, and SQK-LSK10993. Sequencing was performed via MinION Mk1B on R9.4.1 flow cells. Basecalling was performed with Guppy v 6.4.6+ae70e8f in high accuracy mode. A genome sequence was constructed with NextDenovo2 v2.5.2 (read_cutoff = 1k, genome_size = 3 g, and seed depth = 30)94. RagTag v2.1.095 was used to reorder the contigs in the assembly based on homology with the close relative P. pruinosa genome assembly for direct comparison. Given the high N50 (71 Mb) of the assembly, nine contigs less than 100 kb in size were removed based on no BUSCO genes identified in those contigs. The reads were then mapped to the genome assembly using minimap296 to infer the ploidy and to estimate the genome size. Purge_haplotigs97 was used to generate a coverage histogram to assess the ploidy of the assembly. Mapping-based Genome Size Estimation (MGSE)98 was used to predict the genome size.
RNA was extracted and subjected to cDNA synthesis and sequencing (Supplementary Method 1). RNA-seq reads (Supplementary Table 7) were aligned with HISAT 2.2.199 using default parameters to generate hints for the gene prediction. A homology-based gene prediction of the W. somnifera genome was performed using GeMoMa v1.9100,101, based on annotations of six Solanoideae sub-family species (Physalis floridana36, Datura stramonium34, Iochroma cyaneum102, Atropa belladonna34, Lycium barbarum103, and Solanum lycopersicum42). For each of these six species, extracted coding sequences were aligned to the W. somnifera genome sequence with MMseqs2104 (version 15-6f452) using default parameters suggested by GeMoMa. For predicting gene models, GeMoMa utilised protein-coding gene alignments, intron position conservation, and RNA-seq data for refining intron boundaries. The resulting six gene annotation sets were filtered and merged using GeMoMa Annotation Filter (GAF) with parameters f = “start = =‘M’ and stop = =‘*’ and (isNaN(score) or score/aa > =3.50)” and atf = “tie = =1 or sumWeight > 1”. GeMoMa was also used to annotate the genome sequences of P. pruinosa and P. grisea using identical parameters, with a modified filtering step, f = “start = =‘M’ and stop = =‘*’ and (isNaN(score) or score/aa > =4.00)” and atf = “tie = =1 and sumWeight > 1” for both species. BUSCO v5.8.2105 with the solanales_odb12 dataset was used to assess the completeness of the genome assemblies and annotations. The sources of genomic data sets are listed in Supplementary Table 8.
Identification of transposable elements
For repeat sequence comparison, transposable elements (TEs) and tandem repeats were annotated in the genome sequences of Withania somnifera and Physalis pruinosa using Extensive de-novo TE Annotator106 (EDTA) v2.2.1 and Tandem Repeats Finder (TRF)107 v4.10.0, respectively, as described in Supplementary Method 2.
Synteny analysis
JCVI/MCscan108 v1.4.14 was employed to compare the local synteny among the genome sequences of known withanolide-producing species, P. floridana, P. pruinosa, P. grisea, W. somnifera, D. stramonium and D. wrightii. Non-withanolide producing species, S. lycopersicum, S. tuberosum and N. tabacum were also included. The sources of genomic data sets are listed in Supplementary Table 8. The region surrounding the P. pruinosa 24ISO gene was selected to identify microsynteny among these species. Connection of genes between species was validated and manually revised based on phylogenetic trees (Supplementary Fig. 3-8).
Phylogenetic analyses
Phylogenetic trees were constructed using a total of 32 Solanaceae species (Supplementary Table 8). For each gene family, Physalis pruinosa gene sequences from the biosynthetic gene cluster were used as queries in BLASTp109 searches against the polypeptide sequences of these 32 plant species, and the top BLAST hits were selected. This was performed using the Python script collect_best_blast_hits.py (https://github.com/bpucker/ApiaceaeFNS1)110. To differentiate between related gene families, additional outgroup sequences were included: SSR1 and SSR2 for 24ISO; flavonoid biosynthesis genes F3’H and F3’5’H for CYPs; DFR for SDRs; and FLS, F3H, and AOP for ODDs. Additional plant sequences for STs and ATs were also incorporated into their respective trees. Sequences were aligned using MAFFT111 v7.520 with default settings and the L-INS-i accuracy-oriented method. The amino acid alignments were translated back to codon alignments using pxaa2cdn from PHYX112. Alignments with occupancy below 10% were removed using pxclsq from PHYX112. The final phylogenetic trees were generated using IQ-TREE113 v2.3.6 with --alrt 1000 -B 1000 --seqtype CODON. The best-fit evolutionary models were inferred using ModelFinder114: MG + F3X4 + I + R5 for 24ISO, MG + F1X4 + R8 for CYPs, MGK + F3X4 + R7 for SDRs, MG + F3X4 + R7 for STs and ATs, and MG + F3X4 + R8 for ODDs. Phylogenetic trees were visualised and annotated using iTOL115 v6.8.
Expression analysis
To evaluate the expression of genes from identified gene clusters in withanolide-producing species analysed in this study, publicly available paired-end RNA-seq datasets were collected. However, sufficient datasets with diverse tissue samples were only available for D. stramonium and W. somnifera. The FASTQ files for both species were retrieved from the Sequence Read Archive (SRA) using prefetch and fasterq-dump from NCBI SRA toolkit v3.1.0. Transcript abundance was quantified using Kallisto v0.50.1116 based on the coding sequences. The individual count tables were then merged and filtered with customised Python scripts110,117. Expression heatmaps were generated in R to visualise the expression patterns of the identified gene clusters (https://github.com/NancyChoudhary28/Withanolide_biosynthesis/).
Metabolic engineering of S. cerevisiae
For metabolic engineering of yeast (S. cerevisiae), the EasyClone-MarkerFree system was used54. All yeast strains generated in this study were derived from S. cerevisiae ST7574 (CEN.PK background) expressing cas9 available on Euroscarf. Biobricks were assembled via USER cloning118. Amplification of all genes and promoters was performed using Phusion U polymerase (Thermo Fisher Scientific) with primers containing uracil overhangs. Resulting biobricks were assembled into AsiSI/Nb.BsmI (NEB) digested integrative vectors by USER cloning. Assembled synthetic constructs were transformed into NEB 5-alpha competent E. coli cells (NEB). Positive clones as judged by colony PCR were verified through Sanger sequencing (Microsynth Seqlab). Linear integration fragments containing the target genes flanked by homology arms for genomic integration were generated by NotI-HF (NEB) digestion. Linearised synthetic constructs were introduced into S. cerevisiae using the standard lithium acetate method119. Site-specific integrations were confirmed using standardised primers included in the EasyClone-MarkerFree Vector Set. Yeast strains generated in this study are listed in Supplementary Table 9, primers in Supplementary Data 4, plasmids in Supplementary Table 10, and coding sequences of inserted genes in Supplementary Data 5.
For gene deletions, homology-directed repair templates were prepared with High-Fidelity Q5 polymerase (NEB) using a PCR with 10 cycles and primers containing overlapping sequences. Guide RNAs (gRNAs) for deleting ERG4, ERG5 and 24ISO were designed using CHOPCHOP120. The required 20 nt gRNA sequences (Supplementary Table 11) were introduced into pCfB8792 (Addgene Plasmid # 126910) by inverse PCR using primers containing gRNA sequences as overhang. The resulting amplicons containing modified gRNA sequences were gel purified (Macherey-Nagel) and treated with DpnI (NEB) for 30 min at 37 °C. DpnI-digested product was ligated using T4 DNA ligase (NEB) for 2 h at room temperature and transformed into NEB 5-alpha competent E. coli cells. Sequencing was done to confirm the gRNA sequence from isolated plasmids.
Cultures for yeast strains were set in triplicates for extraction of metabolites. Primary cultures were cultivated at 30 °C under continuous shaking at 210 rpm in YPD broth (Carl Roth) containing peptone (20 g/L), yeast extract (10 g/L) and glucose (20 g/L) supplemented with 200 mg/L G418. Overnight primary cultures were used to set secondary cultures with a starting OD600 ~ 0.2 in YPD medium without antibiotics. Secondary cultures of synthetic constructs harbouring galactose-inducible promoters were induced with YP medium containing peptone (20 g/L), yeast extract (10 g/L) supplemented with filter-sterilised galactose (20 g/L). Secondary cultures were grown for 4 days and OD600 was measured just before harvesting; then, metabolite profiles were analysed by GC-MS and LC-MS as described below.
Transient expression in Nicotiana benthamiana
Transient expression in Nicotiana benthamiana was performed with N. benthamiana LAB strain121, which was grown from seeds either in a greenhouse with 11 to 16 h of illumination per day at 21–23 °C or in a phytochamber with 16.5/7.5 h photoperiod at 100 μmol m−2 s−1 light intensity and a temperature of 22 °C during the day and 20 °C during the night. When possible, genes were amplified from cDNA (W. somnifera and A. thaliana) or alternatively obtained as synthetic genes (W. somnifera and P. pruinosa) and cloned into the plasmid pHREAC122 by Golden Gate cloning or In-Fusion cloning as reported121. If necessary, Golden Mutagenesis123 was used to remove undesired BsaI sites with silent mutations. All primers for metabolic engineering and transient expression in N. benthamiana are listed in Supplementary Data 4. The resulting plasmids were transformed into Agrobacterium tumefaciens GV3101 by electroporation. The A. tumefaciens strains were cultured in LB medium with antibiotics (25 μg/mL gentamicin, 50 μg/mL rifampicin, 50 μg/mL kanamycin) for 2 days at 28 °C with shaking. Cells were harvested, resuspended in MMA infiltration buffer (10 mM MgCl2, 10 mM MES, 100 μM acetosyringone), and adjusted to OD600 0.1. For co-expression of multiple genes, the corresponding strains were mixed so that each strain had a final OD600 0.1. The mixtures were syringe-infiltrated into the abaxial side of 4-week-old N. benthamiana leaves. For compound purification, vacuum infiltration was used instead as described below. After infiltration, plants were maintained in a greenhouse or phytochamber until further analysis. Leaf samples were harvested 7 days post-infiltration for GC-MS and LC-MS analysis as described below.
GC-MS sample preparation
GC-MS analysis of yeast strains was performed with cell pellets. Cells were harvested at 5000 × g for 5 min and washed twice with sterile water. Harvested cells (1.5 mL) were saponified at 90 °C for 10 min with 500 µL of 20% aqueous KOH in 50% ethanol containing 5 µg/mL of 5α-cholestane as internal standard. To the saponified extracts, 0.5 mm glass beads (Carl Roth, Germany) and either 500 µL hexane or 500 µL ethyl acetate were added depending on the expected polarity window of metabolites. Samples were then lysed in a FastPrep-24TM 5 G homogeniser (MP Biomedicals, CA, USA) with predefined settings for S. cerevisiae. The organic layer was transferred to a glass vial, and the extraction process repeated a second time. Both organic layers were pooled in a glass vial and concentrated in vacuo using an RVC 2-25 CDplus (Christ, Osterode am Harz, Germany) rotary vacuum concentrator with a cooling trap. Dried extracts were derivatised using 1:1 pyridine: (BSTFA + 1% TMCS) at 70 °C for 1 h. GC-MS analysis was carried out as described below.
For GC-MS analysis of N. benthamiana leaf samples after transient expression, 10 leaf disks were excised using a cork borer no. 5 (ø = 10 mm) and lyophilised until their weight remained constant ( ≈ 20 mg). Then, a 5 mm steel bead was added, and the disks were ground in a MM 400 ball mill (Retsch, Haan, Germany) at 30 Hz for 20 s. Ground leaves were saponified at 70 °C for 1 h with 500 µL of 10% aqueous KOH in 90% ethanol, containing 10 µg/mL of 5α-cholestane as internal standard. Afterwards, 300 µL deionised water was added. The saponified mixture was extracted twice with either 500 µL hexane or 500 µL ethyl acetate depending on the expected polarity window of metabolites. Before extraction with ethyl acetate, ethanol from the saponification solution was evaporated in a rotary vacuum concentrator with a cooling trap. After addition of solvent, samples were vortexed well and centrifuged for 1 min at 10,000 × g. Both organic layers were pooled in a glass vial and concentrated in vacuo as for yeast samples. Derivatisation was performed for 1 h at 70 °C using 75 µL of 1:1 pyridine: (BSTFA + 1% TMCS). Afterwards, the samples were diluted with 100 µL ethyl acetate and centrifuged for 10 min at 10,000 × g. The supernatant was subjected to GC-MS analysis as described below.
Semi-quantification of compounds from GC-MS data (shown in Fig. 6b) was performed by integration of peak areas from compound-specific extracted ion chromatograms (EIC). For each compound, the EIC of the most abundant ion was chosen. Compounds 2, 6, 7, 8, 10, and 11 were extracted more efficiently with hexane and therefore quantified from hexane extracts. Ethyl acetate extracts were used for integration of compounds 12, 14, and 15. The following EICs were chosen for integration of each compound: 5α-cholestane (internal standard), m/z 217; 2, m/z 129; 6, m/z 363; 7, m/z 157; 8, m/z 157; 10, m/z 295; 11, m/z 293; 12, m/z 183; 14, m/z 383; 15, m/z 183. Peak areas were normalised to internal standard and sample dry weight and converted into relative amounts with the highest mean value set to 100%.
LC-MS sample preparation
LC-MS was used for the detection of oxidised withanolide pathway intermediates. N. benthamiana leaf samples and yeast cells were harvested in the same way as described for GC-MS analysis. 600 µL of ethyl acetate were added to the ground leaf powder or yeast cell pellets and vortexed vigorously. For quantification of withanoside V aglycone (16), 0.1 mg/mL emodin was added to ethyl acetate as an internal standard. The mixture was subsequently centrifuged at 14,000 × g for 15 min. The supernatant was transferred to a glass vial and concentrated in vacuo. Afterwards, the dried extracts of N. benthamiana leaf samples were redissolved in 600 µL ethanol, the dried extracts of yeast cell pellets were redissolved in 200 µL acetonitrile and analysed by LC-MS as described below.
General chemical methods and chemicals
The controls and numbers of biological replicates used for GC-MS and LC-MS analyses are described in the figures and figure legends; no technical replicates were analysed. GC-MS analyses were carried out on a Hewlett Packard HP6890N GC system connected to a mass selective detector 5973 N with an OPTIMA 5 MS column (30 m × 0.25 mm i.d., 0.25 µm film, Macherey-Nagel, Düren, Germany). Helium was used as carrier gas at constant flow rate of 1.5 mL/min. Injection volume was 1 µL with a split of 1:5. For the quantification of 24-methyldesmosterol (2), the initial oven temperature was set to 100 °C followed by a ramp of 30 °C/min until 275 °C. Thereafter, the temperature was further raised to 290 °C with a ramp of 3 °C/min and held for 4 min, then raised to 300 °C with a ramp of 3 °C/min and held for 3.83 min. The total run time was 22 min. For all other measurements, the initial oven temperature was set to 100 °C followed by a ramp of 30 °C/min until 275 °C. The temperature was then further raised to 300 °C with a ramp of 3 °C/min and held for 15.83 min. The total run time was 30 min. Mass spectra were obtained with a scan range of m/z 43 to 800, with a solvent delay of 8 min after injection. GC-MS data was analysed using Agilent MSD ChemStation F.01.03.2357.
Analytical and semipreparative LC-MS measurements were performed on an Agilent Infinity II 1260 system consisting of a G7167A autosampler, G7116A column thermostat, G7111B quaternary pump, G7110B make-up pump, G7115A diode array detector, G1364F fraction collector, and G6125B single quadrupole mass spectrometer equipped with an ESI source (positive mode, 4000 V, 12 L min−1 drying gas, 350 °C gas temperature). Analytical LC-MS measurements were carried out either using a C8 (data shown in Fig. 7b) or a C18 column (all other LC-MS measurements). C8 measurements were carried out using a Phenomenex Kinetex 2.6 µm C8 100 Å 4.6 × 150 mm column at 20 °C with 5 mM ammonium acetate in water (mobile phase A) and 5 mM ammonium acetate in MeOH (mobile phase B) and the following gradient: 0–5 min, 10-60% B; 5–12 min, 60–75% B; 12–15 min, 75–100% B; 15–21 min, 100% B; 21–21.1 min, 100-10% B; 21.10–24 min, 10% B. The flow rate was 0.8 mL/min. The injection volume was 5 µL and the total run was 24 min. C18 measurements were carried out using a Poroshell 120 EC-C18, 100 × 4.6 mm, 2.7 μm column at 40 °C with 0.1% (v/v) formic acid in water (mobile phase A) and acetonitrile (mobile phase B) and the following gradient: 0–8 min, 20–60% B; 8–10 min, 60–95% B; 10–12 min, 95% B; 12–12.10 min, 95–20% B; 12.10–14 min, 20% B. The flow rate was 1 mL/min. The total run was 14 min. LC-MS data was analysed using Agilent OpenLab CDS ChemStation C.01.10. For the quantification of withanoside V aglycone (16), peak areas were integrated using the following EICs: 16, EIC 443; emodin (internal standard), EIC 271.
High resolution MS/MS measurements for identification of withanoside V aglycone (16) were carried out on a Vanquish LC (Thermo Fisher) using a Phenomenex Kinetex 2.6 µm C8 100 Å 4.6 × 150 mm column at 30 °C with a flow rate of 0.8 mL/min and 5 mM ammonium acetate in water (mobile phase A) and 5 mM ammonium acetate in MeOH (mobile phase B) as well as the following gradient: 0-5 min, 70- 100% B; 5–5.1 min, 100–70% B; 5.1–7, 70% B. Mass spectra were obtained on an Orbitrap Q Exactive Plus mass spectrometer (Thermo Fisher) operated in PRM mode isolating the precursor with a m/z of 443 with an isolation window of 1 m/z followed by fragmentation at 10, 35 and 60 eV and detection of product ions at a resolution of 17,500. The AGC (automatic gain control) target and maximum injection time were set to 2e5 and 100 ms, respectively. The heated ESI (electrospray-ionisation) source was operated at 0 eV CID (collision-induced dissociation), sheath gas flow 60, auxiliary gas flow 17, sweep gas flow 4, spray voltage 3.5 kV, capillary temperature 288 °C, S-lens RF level 50.0 and aux gas heater 475 °C. High-resolution MS measurements for identification of withanolides in VIGS samples were performed using a Waters QTof Premier mass spectrometer connected to a Waters Acquity UPLC system.
Flash chromatography was performed on an Biotage Isolera One with columns and solvents as described in the Supplementary Information. All methods and gradients for compound purification are described in detail in Supplementary Table 12-17.
NMR spectra of isolated compounds were recorded using Bruker spectrometers operating at 400, 500, and 600 MHz for 1H NMR, and at 100, 126, and 151 MHz for 13C NMR. Experiments were conducted at 298 K with the specified deuterated solvents. Chemical shifts (δ) were referenced relative to the residual solvent signal (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm; C5D5N: δH = 8.74, 7.58, and 7.22 ppm, δC = 150.35, 135.91, and 123.87 ppm) and expressed in ppm, with coupling constants reported in Hz. Analysis was conducted using TopSpin (version 4.1.3) or MestReNova (version 14.3.1).
An authentic sample of 24-methylenecholesterol (1) was provided as a kind gift by Prof. Dr. Hans-Joachim Knölker (TU Dresden, Germany). 24-Methyldesmosterol (2) was synthesised according to a procedure by Edwards et al.124. Withanoside V aglycone (16) was prepared by enzymatic hydrolysis of withanoside V (PhytoLab) as described below.
Preparation of withanoside V aglycone (16)
Withanoside V aglycone (16) was prepared by enzymatic hydrolysis of withanoside V (PhytoLab) following a modified protocol by Matsuda et al.76. A solution of withanoside V (1.2 mg, 1.56 µmol) in sodium acetate buffer (0.4 mL, 0.2 M, pH 5) was treated with cellulase (from T. reesei, 4 µL, ≥ 700 U/g, ρ 1.1-1.3 g/mL). The mixture was stirred gently at 50 °C for 24 h and then subjected to an SPE column (Oasis PRiME HLB 1cc (30 mg)). After loading and washing with 5% MeOH (H2O:MeOH 95:5), steroids were eluted with 4 mL of 75% acetonitrile (H2O:acetonitrile 25:75). The eluate was concentrated under reduced pressure to yield a mixture of the desired aglycone of withanoside V (16) with remaining or partially deglycosylated starting material (1.0 mg). This mixture was then purified by semipreparative HPLC to yield 0.4 mg (0.90 µmol, 58%) of withanoside V aglycone (16). The identity of the isolated compound was confirmed by comparison of 1H NMR spectral data with literature125. Semipreparative purification was achieved with a Phenomenex Luna 5 µm C8 100 Å 250 × 100 mm column and a gradient of 5 mM ammonium acetate in water (A) and methanol (B) at a flow rate of 4 mL/min: 0–4 min, 80–100% B; 4–8 min, 100% B; 8–8.1 min, 100–80% B, 8.1–10 min, 80% B.
Purification of pathway intermediates heterologously produced in N. benthamiana
For compound purification, N. benthamiana plants were vacuum-infiltrated126 in a 9.2 L ROTILABO desiccator (Carl Roth, Karlsruhe, Germany) connected to a MZ 2 NT membrane pump (Vacuubrand, Wertheim, Germany) at 30 mbar for 1 min. 120 and 90 plants were used for the isolation of PpCYP87G1 and PpCYP88C7 (shunt) products, respectively. Leaves were harvested 7 days post infiltration and lyophilised for 3 days until the dry weight remained constant. The dried leaves were ground at room temperature into powder with a blender and extracted with hexane for 24ISO and PpCYP87G1 products and with ethyl acetate for PpCYP87G1 + PpCYP88C7 products. The crude extracts were concentrated in vacuo and purified by consecutive rounds of chromatography as described in Supplementary Tables 12–17. NMR spectra of isolated compounds are shown in Supplementary Figs. 27–66.
Chemical synthesis
Synthetic procedures used in the context of determining the C-22 stereochemistry of (22R)-ergosta-5,24-diene-3β,22-diol (10) and for preparing (22R)-ergosta-5,24-diene-1α,3β,22,26-tetrol (17) are described in Supplementary Method 3. The full synthetic routes are shown in Supplementary Fig. 67. NMR spectra of synthetic compounds are shown in Supplementary Figs. 68–99. X-ray crystallographic data from this work is shown in Supplementary Table 18.
Heterologous protein production
For in vitro assays, codon-optimised PpCYP749B2 was produced in yeast (strain KMY95) together with a cytochrome P450 reductase (CPR) and obtained as a microsomal fraction. Microsomes were isolated from yeast strains BSY1127 as a negative control and KMY95 (this work). Yeast strains were grown in YPD medium at 30 °C for 48 h; then, cells were harvested by centrifugation at 5000 × g at RT for 5 min, washed with 10 mL TEK buffer (50 mM Tris, pH 7.4, 1 mM EDTA, 0.1 M KCl), and centrifuged again with the same conditions. The supernatant was discarded and washed cells were then lysed with glass beads (⌀ = 0.5 mm, 1/3 volume of lysate) with a bead homogeniser (FastPrep-24Tm 5 G) shaking at 6.0 m/s for 40 s with two cycles in 7 mL TEB buffer (50 mM Tris, pH 7.4, 1 mM EDTA, 0.6 M sorbitol). The volume was adjusted to 20 mL with TEB buffer and the cell debris was removed by centrifugation at 10,000 × g at 4 °C for 10 min. Microsomes were isolated from the supernatant by ultracentrifugation at 100,000 × g at 4 °C for 1 h. Microsomes were finally resuspended in 800 μL of TEG buffer (50 mM Tris, pH 7.4, 1 mM EDTA, 20% glycerol), aliquoted to 200 μL per tube and stored at −80 °C until further use.
SDR proteins were produced in E. coli and purified by affinity chromatography based on a His tag. PpSDR-a and PpSDR-b sequences were cloned into the protein overexpression vector pET-28a(+) containing a C-terminal 6xHis tag (Supplementary Data 4). The vectors were transformed into E. coli BL21 (DE3) for heterologous expression. A single colony for each strain was picked from LB agar plate with 50 μg/mL kanamycin and cultured overnight at 37 °C with shaking at 250 rpm. Starter cultures were diluted 100-fold into 1 L LB cultures with 50 μg/mL kanamycin and cultured at 37 °C, with shaking at 250 rpm until OD600 reached 0.6-0.8. Cultures were then induced with 0.1 mM IPTG and cultured at 18 °C with shaking at 250 rpm for 16 h. Cell pellets were harvested by centrifugation at 3200 × g for 10 min and then resuspended in 10 mL ice-cold loading buffer [50 mM Tris-HCl, 500 mM NaCl, 5% glycerol, 50 mM glycine, 20 mM imidazole, cOmplete EDTA-free protease inhibitors (Roche), pH 8]. After sonication in an ice bath for 8 min (5 s on, 10 s off), cell debris was removed by centrifugation at 25,000 × g for 20 min at 4 °C. The supernatant was transferred to a 15 mL centrifuge tube; then, 800 μL Ni-NTA agarose (Cube Biotech) were added and gently mixed on a rocking platform for 1 h at 4 °C. The mixture was centrifuged at 3200 × g for 1 min at 4 °C, the supernatant was removed, and the Ni-NTA agarose was washed twice with 10 mL ice-cold loading buffer, then transferred to a 2 mL tube, washed three times with 800 μL elution buffer [50 mM Tris-HCl, 500 mM NaCl, 5% glycerol, 50 mM glycine, 500 mM imidazole, pH 8], and the supernatant was collected after centrifugation at 2400 × g for 1 min at 4 °C. The eluates were combined and concentrated using an Amicon® Ultra centrifugal filter, 10 kDa MWCO. The final protein concentration was measured using NanoDrop.
In vitro assays with PpCYP749B2 and PpSDR
Assays with yeast microsomes alone were performed in 250 μL total volume containing 25 μL yeast microsomes, 22 μM of the substrate (22R)-ergosta-5,24-diene-1α,3β,22,26-tetrol (17), and 2 mM NADPH in 50 mM Tris-HCl buffer, pH 7.5. In vitro assays with PpSDR alone were performed in 250 μL total volume containing 76 μg/mL enzyme, 22 μM of 17, and 2 mM NADP+ in 50 mM Tris-HCl buffer, pH 7.5. In vitro assays with yeast microsomes and PpSDR together were performed in 250 μL total volume containing 25 μL yeast microsomes, 76 μg/mL PpSDR-a or -b, 22 μM of the substrate 17, 1 mM NADP+, and 2 mM NADPH in 50 mM Tris-HCl buffer, pH 7.5. All reactions were incubated at 30 °C with shaking at 400 rpm for 16 h.
All reactions were stopped by addition of 250 µL ethyl acetate. After vortexing and centrifugation, 200 μL of the ethyl acetate layer were collected. The extraction was repeated twice. Ethyl acetate layers were combined and dried in vacuo. The dried extracts were then redissolved in 200 µL of acetonitrile and analyzed by LC-MS on a C18 column as described above.
Virus-induced gene silencing
Virus-induced gene silencing in N. benthamiana is described in Supplementary Method 4. To evoke virus-induced gene silencing in W. somnifera, a short gene fragment of 145-400 bp was selected for each target gene, based on the leaf-expressed homologues from W. somnifera gene cluster 1. To target all homologues of target genes in the two gene clusters, VIGS fragments were chosen from conserved regions (Fig. 8a). Possible off-targets outside the two gene clusters and VIGS fragment efficiency were predicted using si-Fi128. Fragments were designed in this way for CYP87G1, CYP88C7, CYP749B2 and SDR. A second fragment targeting phytoene desaturase (PDS) was chosen as a visual marker (Fig. 8a)79. As a positive control, the 24ISO construct from Knoch et al.14. was adjusted to our W. somnifera 24ISO sequence. As a negative control, a GFP fragment was chosen with no potential targets in W. somnifera. All fragments were obtained as synthetic gene fragments including the required overhangs for cloning (Azenta Life Sciences or Twist Biosciences). VIGS fragments are listed in Supplementary Table 19.
Vectors used for VIGS were pTRV1 and pTRV2-MCS (pYL156129). Using In-Fusion cloning (Takara Biosciences), the PDS fragment was cloned into pTRV2-MCS linearised with EcoRI-HF and BamHI-HF to give pTRV2-PDS. A pTRV2-GFP vector was prepared in the same way as an extra negative control. The vector pTRV2-PDS was further linearised with KpnI-HF and XhoI and either the GFP or a target gene fragment inserted by In-Fusion cloning. After confirmation by Sanger sequencing, all resulting plasmids were transformed into Agrobacterium tumefaciens GV3101 by electroporation.
W. somnifera plants were grown in a phytochamber with 16.5/7.5 h photoperiod at 100 μmol m−2 s−1 light intensity and a temperature of 22 °C during the day and 20 °C during the night. Agrobacterium strains containing pTRV1 or a pTRV2-derivative were grown and harvested as described in section “Transient expression in Nicotiana benthamiana”. Cells were resuspended in infiltration buffer and diluted to an OD600 of 1.0. Cell suspensions containing pTRV1 and a pTRV2-derivative were mixed in a 1:1 ratio and syringe-infiltrated into the leaves of 3–4-week-old W. somnifera seedlings. For each vector combination, six replicates were infiltrated. First signs of photobleaching on PDS-silenced plants appeared after two to three weeks post infiltrations.
Leaf samples were harvested after 4-5 weeks post infiltration for expression and metabolite analysis. For plants infiltrated with a pTRV2-PDS derivative, white (photobleached) leaf areas were chosen if available. The number of biological replicates showing photobleaching as an indicator of successful silencing is provided in the respective figures. Six to ten leaf disks were excised with a cork borer no. 4 (ø = 8 mm), transferred to a 2 mL tube and frozen in liquid nitrogen. For metabolite analysis, samples from all six biological replicates were collected and lyophilised. For analysis of withaferin A levels, the samples were ground and extracted as described in section “LC-MS sample preparations”. Ethyl acetate used during LC-MS sample preparation contained 0.1 mg/mL emodin as internal standard. LC-MS samples were analysed on a C18 column as described above. Withaferin A was identified based on an authentic standard. Peak integration was done using the following EICs: withaferin A (A), EIC 471; unidentified withanolide at 7.6 min (B), EIC 493; unidentified withanolide at 8.2 min (C), EIC 495; emodin (internal standard), EIC 271. Peak areas were normalised by internal standard and dry weight. To check for accumulation of biosynthetic intermediates, samples were processed as described in section “GC-MS sample preparations”.
For expression analysis, samples from white leaves of three replicate plants were collected as described above for metabolite samples. Leaves were ground in a pre-cooled ball mill and RNA extracted using the GeneJET Plant RNA purification kit (Thermo Fisher Scientific). After gDNA digestion, 100 ng of RNA were used for cDNA synthesis with the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). qPCR was performed using the 2× qPCRBIO SyGreen Mix Lo-ROX (Nippon Genetics) with a QuantStudio3 qPCR cycler (Thermo Fisher Scientific), according to the manufacturer’s instructions. 1 µL of 1:10 diluted cDNA was used as template. Primers used during qPCR are listed in Supplementary Data 4. Data was analysed using the 2(−∆∆Ct) method130 and expression levels calculated in relation to EF-1a and PDS-GFP control plants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequences of genes from the withanolide gene clusters shown here are provided as a Supplementary Data 1. Raw sequencing reads generated in this study have been deposited in the European Nucleotide Archive (ENA) with accession ERP150021 under BioProject PRJEB64854. RNA-seq data has been deposited in the ENA with accession ERR13615536. The assembled genome sequence and the structural annotation of W. somnifera have been deposited in GenBank with the assembly accession code GCA_965601375 [http://www.ebi.ac.uk/ena/browser/view/GCA_965601375] and are also provided via LeoPARD [https://doi.org/10.24355/dbbs.084-202503200640-0]. Re-annotations of the Physalis genome sequences are provided via LeoPARD [https://doi.org/10.24355/dbbs.084-202409130931-0] and also via GitHub [https://github.com/NancyChoudhary28/Withanolide_biosynthesis/tree/main/data]. Source data are provided with this paper.
Code availability
Scripts and commands of the Circos plot generation, heatmap construction, and input sequences used to construct phylogenetic trees are described and uploaded in Zenodo [https://doi.org/10.5281/zenodo.15632852] and GitHub [https://github.com/NancyChoudhary28/Withanolide_biosynthesis].
References
Xia, G., Cao, S., Chen, L. & Qiu, F. Natural withanolides, an update. Nat. Prod. Rep. 39, 784–813 (2022).
Schirmer Pigatto, A. G., Mentz, L. A. & Gonçalves Soares, G. L. Chemotaxonomic characterization and chemical similarity of tribes/genera of the Solanoideae subfamily (Solanaceae) based on occurrence of withanolides. Biochem. Syst. Ecol. 54, 40–47 (2014).
Huang, M. et al. Withanolides from the genus Physalis: a review on their phytochemical and pharmacological aspects. J. Pharm. Pharmacol. 72, 649–669 (2020).
Singh, A., Raza, A., Amin, S., Damodaran, C. & Sharma, A. K. Recent Advances in the Chemistry and Therapeutic Evaluation of Naturally Occurring and Synthetic Withanolides. Molecules 27, 886 (2022).
Mukherjee, P. K. et al. Withania somnifera (L.) Dunal - Modern perspectives of an ancient Rasayana from Ayurveda. J. Ethnopharmacol. 264, 113157 (2021).
Lopresti, A. L., Smith, S. J., Malvi, H. & Kodgule, R. An investigation into the stress-relieving and pharmacological actions of an ashwagandha (Withania somnifera) extract: A randomized, double-blind, placebo-controlled study. Med. (Baltim) 98, e17186 (2019).
Chandrasekhar, K., Kapoor, J. & Anishetty, S. A Prospective, Randomized Double-Blind, Placebo-Controlled Study of Safety and Efficacy of a High-Concentration Full-Spectrum Extract of Ashwagandha Root in Reducing Stress and Anxiety in Adults. Indian J. Psychol. Med. 34, 255–262 (2012).
Santagata, S. et al. Using the Heat-Shock Response To Discover Anticancer Compounds that Target Protein Homeostasis. ACS Chem. Biol. 7, 340–349 (2012).
Zhang, H. et al. Cytotoxic Withanolide Constituents of Physalis longifolia. J. Nat. Prod. 74, 2532–2544 (2011).
Crane, E. A. et al. Profiling withanolide A for therapeutic targets in neurodegenerative diseases. Bioorg. Med. Chem. 27, 2508–2520 (2019).
Jana, C. K. et al. Synthesis of Withanolide A, Biological Evaluation of Its Neuritogenic Properties, and Studies on Secretase Inhibition. Angew. Chem. Int. Ed. 50, 8407–8411 (2011).
Vanden Berghe, W., Sabbe, L., Kaileh, M., Haegeman, G. & Heyninck, K. Molecular insight in the multifunctional activities of Withaferin A. Biochem. Pharmacol. 84, 1282–1291 (2012).
Cigler, M. et al. Orpinolide disrupts a leukemic dependency on cholesterol transport by inhibiting OSBP. Nat. Chem. Biol. https://doi.org/10.1038/s41589-024-01614-4 (2024).
Knoch, E. et al. Third DWF1 paralog in Solanaceae, sterol Δ24-isomerase, branches withanolide biosynthesis from the general phytosterol pathway. Proc. Natl Acad. Sci. USA. 115, E8096–E8103 (2018).
Dhar, N. et al. A Decade of Molecular Understanding of Withanolide Biosynthesis and In vitro Studies in Withania somnifera (L.) Dunal: Prospects and Perspectives for Pathway Engineering. Front. Plant Sci. 6, 1031 (2015).
Jadaun, J. S. et al. Over-expression of DXS gene enhances terpenoidal secondary metabolite accumulation in rose-scented geranium and Withania somnifera: active involvement of plastid isoprenogenic pathway in their biosynthesis. Physiol. Plant. 159, 381–400 (2017).
Singh, S. et al. Sterol partitioning by HMGR and DXR for routing intermediates toward withanolide biosynthesis. Physiol. Plant. 152, 617–633 (2014).
Agarwal, A. V. et al. Virus-Induced Silencing of Key Genes Leads to Differential Impact on Withanolide Biosynthesis in the Medicinal Plant, Withania somnifera. Plant Cell Physiol. 59, 262–274 (2018).
Singh, A. K. et al. Virus-induced gene silencing of Withania somnifera squalene synthase negatively regulates sterol and defence-related genes resulting in reduced withanolides and biotic stress tolerance. Plant Biotechnol. J. 13, 1287–1299 (2015).
Shilpashree, H. B., Sudharshan, S. J., Shasany, A. K. & Nagegowda, D. A. Molecular characterization of three CYP450 genes reveals their role in withanolides formation and defense in Withania somnifera, the Indian Ginseng. Sci. Rep. 12, 1602 (2022).
Srivastava, S. et al. Light and auxin responsive cytochrome P450s from Withania somnifera Dunal: cloning, expression and molecular modelling of two pairs of homologue genes with differential regulation. Protoplasma 252, 1421–1437 (2015).
Jacobowitz, J. R. & Weng, J.-K. Exploring Uncharted Territories of Plant Specialized Metabolism in the Postgenomic Era. Annu. Rev. Plant Biol. 71, 631–658 (2020).
Jeon, J. E. et al. A Pathogen-Responsive Gene Cluster for Highly Modified Fatty Acids in Tomato. Cell 180, 176–187 (2020).
Nett, R. S., Dho, Y., Low, Y.-Y. & Sattely, E. S. A metabolic regulon reveals early and late acting enzymes in neuroactive Lycopodium alkaloid biosynthesis. Proc. Natl Acad. Sci. Usa. 118, e2102949118 (2021).
Li, C. et al. Single-cell multi-omics in the medicinal plant Catharanthus roseus. Nat. Chem. Biol. 19, 1031–1041 (2023).
Chuang, L., Liu, S. & Franke, J. Post-Cyclization Skeletal Rearrangements in Plant Triterpenoid Biosynthesis by a Pair of Branchpoint Isomerases. J. Am. Chem. Soc. 145, 5083–5091 (2023).
Scherlach, K. & Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 12, 3864 (2021).
Nützmann, H.-W., Huang, A. & Osbourn, A. Plant metabolic clusters – from genetics to genomics. N. Phytol. 211, 771–789 (2016).
Nützmann, H.-W. & Osbourn, A. Gene clustering in plant specialized metabolism. Curr. Opin. Biotechnol. 26, 91–99 (2014).
Smit, S. J. & Lichman, B. R. Plant biosynthetic gene clusters in the context of metabolic evolution. Nat. Prod. Rep. 39, 1465–1482 (2022).
Marks, R. A., Hotaling, S., Frandsen, P. B. & VanBuren, R. Representation and participation across 20 years of plant genome sequencing. Nat. Plants 7, 1571–1578 (2021).
Reed, J. et al. Elucidation of the pathway for biosynthesis of saponin adjuvants from the soapbark tree. Science 379, 1252–1264 (2023).
Wu, S. et al. Convergent gene clusters underpin hyperforin biosynthesis in St John’s wort. N. Phytol. 235, 646–661 (2022).
Zhang, F. et al. Revealing evolution of tropane alkaloid biosynthesis by analyzing two genomes in the Solanaceae family. Nat. Commun. 14, 1446 (2023).
Liu, C. et al. A chromosome-level genome assembly reveals that a bipartite gene cluster formed via an inverted duplication controls monoterpenoid biosynthesis in Schizonepeta tenuifolia. Mol. Plant 16, 533–548 (2023).
Lu, J. et al. The Physalis floridana genome provides insights into the biochemical and morphological evolution of Physalis fruits. Hortic. Res. 8, 244 (2021).
He, J. et al. Establishing Physalis as a Solanaceae model system enables genetic reevaluation of the inflated calyx syndrome. Plant Cell 35, 351–368 (2023).
Rajewski, A., Carter-House, D., Stajich, J. & Litt, A. Datura genome reveals duplications of psychoactive alkaloid biosynthetic genes and high mutation rate following tissue culture. BMC Genom. 22, 201 (2021).
Goldberg, J. K. et al. A de novo long-read genome assembly of the sacred datura plant (Datura wrightii) reveals a role of tandem gene duplications in the evolution of herbivore-defense response. BMC Genomics 25, 15 (2024).
Pucker, B., Irisarri, I., Vries, J. D. E. & Xu, B. Plant genome sequence assembly in the era of long reads: Progress, challenges and future directions. Quant. Plant Biol. 3, e5 (2022).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Hosmani, P. S. et al. An improved de novo assembly and annotation of the tomato reference genome using single-molecule sequencing, Hi-C proximity ligation and optical maps. 767764 Preprint at https://doi.org/10.1101/767764 (2019).
Pham, G. M. et al. Construction of a chromosome-scale long-read reference genome assembly for potato. GigaScience 9, giaa100 (2020).
Sierro, N. et al. The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 5, 3833 (2014).
Malhotra, K. & Franke, J. Cytochrome P450 monooxygenase-mediated tailoring of triterpenoids and steroids in plants. Beilstein J. Org. Chem. 18, 1289–1310 (2022).
Chuang, L., Liu, S., Biedermann, D. & Franke, J. Identification of early quassinoid biosynthesis in the invasive tree of heaven (Ailanthus altissima) confirms evolutionary origin from protolimonoids. Front. Plant Sci. 13, 958138 (2022).
Itkin, M. et al. Biosynthesis of Antinutritional Alkaloids in Solanaceous Crops Is Mediated by Clustered Genes. Science 341, 175–179 (2013).
Hirschmann, F., Krause, F. & Papenbrock, J. The multi-protein family of sulfotransferases in plants: composition, occurrence, substrate specificity, and functions. Front. Plant Sci. 5, 556 (2014).
Shingu, K., Furusawa, Y. & Nohara, T. New Withanolides, Daturametelins C, D, E, F and G-Ac from Datura metel L. (Solanaceous Studies. XIV). Chem. Pharm. Bull. 37, 2132–2135 (1989).
Xu, Y. et al. 2,3-Dihydrowithaferin A-3β-O-sulfate, a new potential prodrug of withaferin A from aeroponically grown Withania somnifera. Bioorg. Med. Chem. 17, 2210–2214 (2009).
Yang, J., Li, C. & Zhang, Y. Engineering of Saccharomyces cerevisiae for 24-Methylene-Cholesterol Production. Biomolecules 11, 1710 (2021).
Sawai, S. et al. Sterol Side Chain Reductase 2 Is a Key Enzyme in the Biosynthesis of Cholesterol, the Common Precursor of Toxic Steroidal Glycoalkaloids in Potato. Plant Cell 26, 3763–3774 (2014).
Xu, S., Chen, C. & Li, Y. Engineering of Phytosterol-Producing Yeast Platforms for Functional Reconstitution of Downstream Biosynthetic Pathways. ACS Synth. Biol. 9, 3157–3170 (2020).
Jessop-Fabre, M. M. et al. EasyClone-MarkerFree: A vector toolkit for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnol. J. 11, 1110–1117 (2016).
Choe, S. et al. Lesions in the sterol Δ7 reductase gene of Arabidopsis cause dwarfism due to a block in brassinosteroid biosynthesis. Plant J. 21, 431–443 (2000).
Barrero, A. F., Oltra, J. E., Poyatos, J. A., Jiménez, D. & Oliver, E. Phycomysterols and Other Sterols from the Fungus Phycomyces blakesleeanus. J. Nat. Prod. 61, 1491–1496 (1998).
Liu, G., Chen, Y., Færgeman, N. J. & Nielsen, J. Elimination of the last reactions in ergosterol biosynthesis alters the resistance of Saccharomyces cerevisiae to multiple stresses. FEMS Yeast Res. 17, fox063 (2017).
Nes, W. R., Joseph, J. M., Landrey, J. R., Behzadan, S. & Conner, R. L. Steric effects at C-20 and C-24 on the metabolism of sterols by Tetrahymena pyriformis. J. Lipid Res. 22, 770–777 (1981).
Wretensjö, I. & Karlberg, B. Characterization of sterols in refined borage oil by GC-MS. J. Am. Oil Chem. Soc. 79, 1069–1074 (2002).
Massey, I. J. & Djerassi, C. Mass spectrometry in structural and stereochemical problems. 252. Structural and stereochemical applications of mass spectrometry in the marine sterol field. Synthesis and electron impact induced mass spectral fragmentation of.DELTA.24- and.DELTA.24(28)-3.beta.-hydroxy-.DELTA.5-sterols. J. Org. Chem. 44, 2448–2456 (1979).
Kenny, P. T. M. & Wetzel, J. M. Letter: Fragmentation Studies of Ergosterol. The Formation of the Fragment Ion at m/z 337. Eur. Mass Spectrom. 1, 411–413 (1995).
Brooks, C. J. W., Horning, E. C. & Young, J. S. Characterization of sterols by gas chromatography-mass spectrometry of the trimethylsilyl ethers. Lipids 3, 391–402 (1968).
Lewis, T. L., Keesler, G. A., Fenner, G. P. & Parks, L. W. Pleiotropic mutations in Saccharomyces cerevisiae affecting sterol uptake and metabolism. Yeast 4, 93–106 (1988).
Xiu, X. et al. Modular remodeling of sterol metabolism for overproduction of 7-dehydrocholesterol in engineered yeast. Bioresour. Technol. 360, 127572 (2022).
Jiang, Y. et al. Manipulation of sterol homeostasis for the production of 24-epi-ergosterol in industrial yeast. Nat. Commun. 14, 437 (2023).
Kwan, B. D., Seligmann, B., Nguyen, T.-D., Franke, J. & Dang, T.-T. T. Leveraging synthetic biology and metabolic engineering to overcome obstacles in plant pathway elucidation. Curr. Opin. Plant Biol. 71, 102330 (2023).
Sonawane, P. D. et al. Plant cholesterol biosynthetic pathway overlaps with phytosterol metabolism. Nat. Plants 3, 16205 (2017).
Choe, S. et al. The Arabidopsis dwarf1 Mutant Is Defective in the Conversion of 24-Methylenecholesterol to Campesterol in Brassinosteroid Biosynthesis. Plant Physiol. 119, 897–908 (1999).
Velásquez, A. C., Chakravarthy, S. & Martin, G. B. Virus-induced Gene Silencing (VIGS) in Nicotiana benthamiana and Tomato. J. Vis. Exp. JoVE e1292 https://doi.org/10.3791/1292 (2009).
Sinha, S. C., Ali, A., Bagchi, A., Sahai, M. & Ray, A. B. Physalindicanols, New Biogenetic Precursors of C28-Steroidal Lactones from Physalis minima var. indica. Planta Med. 53, 55–57 (1987).
Gill, H. K., Smith, R. W. & Whiting, D. A. Biosynthesis of the nicandrenoids: stages in the oxidative elaboration of the side chain and the fate of the diastereotopic 25-methyl groups of 24-methylenecholesterol. J. Chem. Soc. Chem. Commun. 1459 https://doi.org/10.1039/c39860001459 (1986).
Andrews-Smith, W., Gill, H. K., Smith, R. W. & Whiting, D. A. Stages in the biosynthesis of the epoxy lactol side chain of Nic-1, insect antifeedant steroid of Nicandra physaloides. J. Chem. Soc. Perkin 1, 291–296 (1991).
Wu, J. et al. New steroids with anti-inflammatory activity from the whole plants of Physalis minima. Nat. Prod. Res. https://doi.org/10.1080/14786419.2024.2340048 (2024).
Elliger, C. A. et al. Petuniasterones, novel ergostane-type steroids of Petunia hybridia Vilm. (Solanaceae) having insect-inhibitory activity. X-Ray molecular structure of the 22,24,25-[(methoxycarbonyl)orthoacetate] of 7α,22,24,25-tetrahydroxy ergosta-1,4-dien-3-one and of 1α-acetoxy-24,25-epoxy-7α-hydroxy-22-(methylthiocarbonyl)acetoxyergost-4-en-3-one. J. Chem. Soc. Perkin 1 711–717 https://doi.org/10.1039/P19880000711 (1988).
Harvey, D. J. & Vouros, P. Mass Spectrometric Fragmentation of Trimethylsilyl and Related Alkylsilyl Derivatives. Mass Spectrom. Rev. 39, 105–211 (2020).
Matsuda, H., Murakami, T., Kishi, A. & Yoshikawa, M. Structures of withanosides I, II, III, IV, V, VI, and VII, new withanolide glycosides, from the roots of Indian Withania somnifera Dunal. and inhibitory activity for tachyphylaxis to clonidine in isolated guinea-pig ileum. Bioorg. Med. Chem. 9, 1499–1507 (2001).
Zhu, X.-H., Ando, J., Takagi, M., Ikeda, T. & Nohara, T. Six New Withanolide-Type Steroids from the Leaves of Solanum cilistum. Chem. Pharm. Bull. 49, 161–164 (2001).
Lichman, B. R. et al. The evolutionary origins of the cat attractant nepetalactone in catnip. Sci. Adv. 6, eaba0721 (2020).
Yamamoto, K. et al. Improved virus-induced gene silencing allows discovery of a serpentine synthase gene in Catharanthus roseus. Plant Physiol. 187, 846–857 (2021).
Fan, P. et al. Evolution of a plant gene cluster in Solanaceae and emergence of metabolic diversity. eLife 9, e56717 (2020).
Matsuba, Y. et al. Evolution of a complex locus for terpene biosynthesis in solanum. Plant Cell 25, 2022–2036 (2013).
Hua, C. et al. Identification of P450 Candidates Associated with the Biosynthesis of Physalin-Class Compounds in Physalis angulata. Int. J. Mol. Sci. 24, 14077 (2023).
Anjali, P., Narayanan, A. K., Parihar, D., Patil, A. & Nagegowda, D. A. Characterization of two glycosyltransferases that modulate withanolide biosynthesis and defense in Withania somnifera. Mol. Biol. Rep. 52, 675 (2025).
Shilpashree, H. B., Narayanan, A. K., Kumar, S. R., Barvkar, V. & Nagegowda, D. A. The cytochrome P450 enzyme WsCYP71B35 from Withania somnifera has a role in withanolides biosynthesis and defence against bacteria. Physiol. Plant. 176, e14180 (2024).
Franke, J. et al. Gene Discovery in Gelsemium Highlights Conserved Gene Clusters in Monoterpene Indole Alkaloid Biosynthesis. ChemBioChem 20, 83–87 (2019).
Christ, B. et al. Repeated evolution of cytochrome P450-mediated spiroketal steroid biosynthesis in plants. Nat. Commun. 10, 3206 (2019).
Yin, X. et al. Deciphering the network of cholesterol biosynthesis in Paris polyphylla laid a base for efficient diosgenin production in plant chassis. Metab. Eng. 76, 232–246 (2023).
Winegar, P. H. et al. Verazine biosynthesis from simple sugars in engineered Saccharomyces cerevisiae. Metab. Eng. 85, 145–158 (2024).
Lucier, R. et al. Steroidal scaffold decorations in Solanum alkaloid biosynthesis. Mol. Plant 17, 1236–1254 (2024).
Schultz, B. J., Kim, S. Y., Lau, W. & Sattely, E. S. Total Biosynthesis for Milligram-Scale Production of Etoposide Intermediates in a Plant Chassis. J. Am. Chem. Soc. 141, 19231–19235 (2019).
Zhang, Y. et al. Synthetic biology identifies the minimal gene set required for paclitaxel biosynthesis in a plant chassis. Mol. Plant 16, 1951–1961 (2023).
Dudley, Q. M. et al. Reconstitution of monoterpene indole alkaloid biosynthesis in genome engineered Nicotiana benthamiana. Commun. Biol. 5, 949 (2022).
Horz, J. M., Wolff, K., Friedhoff, R. & Pucker, B. Genome sequence of the ornamental plant Digitalis purpurea reveals the molecular basis of flower color and morphology variation. 2024.02.14.580303 Preprint at https://doi.org/10.1101/2024.02.14.580303 (2024).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 107 (2024).
Alonge, M. et al. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 23, 258 (2022).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinf. 19, 460 (2018).
Natarajan, S., Gehrke, J. & Pucker, B. Mapping-based genome size estimation. BMC Genomics 26, 482 (2025).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinf. 19, 189 (2018).
Powell, A. F. et al. Genome sequence for the blue-flowered Andean shrub Iochroma cyaneum reveals extensive discordance across the berry clade of Solanaceae. Plant Genome 15, e20223 (2022).
Cao, Y.-L. et al. Wolfberry genomes and the evolution of Lycium (Solanaceae). Commun. Biol. 4, 671 (2021).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Tang, H., Krishnakumar, V. & Li, J. JCVI: JCVI utility libraries. Zenodo https://doi.org/10.5281/zenodo.31631 (2015).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinf. 10, 421 (2009).
Pucker, B. & Iorizzo, M. Apiaceae FNS I originated from F3H through tandem gene duplication. PLOS ONE 18, e0280155 (2023).
Katoh, K. & Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780 (2013).
Brown, J. W., Walker, J. F. & Smith, S. A. Phyx: phylogenetic tools for unix. Bioinformatics 33, 1886–1888 (2017).
Minh, B. Q. et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res 52, W78–W82 (2024).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Choudhary, N. & Pucker, B. Conserved amino acid residues and gene expression patterns associated with the substrate preferences of the competing enzymes FLS and DFR. PLOS ONE 19, e0305837 (2024).
Bitinaite, J. et al. USERTM friendly DNA engineering and cloning method by uracil excision. Nucleic Acids Res. 35, 1992–2002 (2007).
Gietz, R. D. & Schiestl, R. H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 (2007).
Labun, K. et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 (2019).
Chuang, L. & Franke, J. Rapid Combinatorial Coexpression of Biosynthetic Genes by Transient Expression in the Plant Host Nicotiana benthamiana. In Engineering Natural Product Biosynthesis: Methods and Protocols (ed. Skellam, E.) 395–420 (Springer US, New York, NY, 2022). https://doi.org/10.1007/978-1-0716-2273-5_20.
Peyret, H., Brown, J. K. M. & Lomonossoff, G. P. Improving plant transient expression through the rational design of synthetic 5′ and 3′ untranslated regions. Plant Methods 15, 108 (2019).
Püllmann, P. et al. Golden Mutagenesis: An efficient multi-site-saturation mutagenesis approach by Golden Gate cloning with automated primer design. Sci. Rep. 9, 10932 (2019).
Edwards, J. T. et al. Decarboxylative alkenylation. Nature 545, 213–218 (2017).
Lischewski, M. et al. Withanolides from Dunalia australis. Phytochemistry 30, 4184–4186 (1991).
Chuang, L., Enders, A., Offermann, S., Bahnemann, J. & Franke, J. 3D-printed autoclavable plant holders to facilitate large-scale protein production in plants. Eng. Life Sci. 22, 803–810 (2022).
Dias, S. L. et al. Biosynthesis of the allelopathic alkaloid gramine in barley by a cryptic oxidative rearrangement. Science 383, 1448–1454 (2024).
Lück, S. et al. siRNA-Finder (si-Fi) Software for RNAi-Target Design and Off-Target Prediction. Front. Plant Sci. 10, 1023 (2019).
Liu, Y., Schiff, M., Marathe, R. & Dinesh-Kumar, S. P. Tobacco Rar1, EDS1 and NPR1/NIM1 like genes are required for N-mediated resistance to tobacco mosaic virus. Plant J. 30, 415–429 (2002).
Livak, K. J. & Schmittgen, T. D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408 (2001).
Huang, J. et al. Nuclear phylogeny and insights into whole-genome duplications and reproductive development of Solanaceae plants. Plant. Commun. 4, 100595 (2023).
Acknowledgements
We are thankful to Dr. Christiane Wittmann (Botanical Garden Duisburg-Essen) for providing information about XX-0-STGAL-22/1985. We gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft (FR 3720/7-1 to J.F.; PU 718/2-1 to B.P.; INST 187/741-1 FUGG to C.-P.W.). In addition, work in the group of J.F. was supported by the SMART BIOTECS alliance between the Technische Universität Braunschweig and the Leibniz Universität Hannover, supported by the Ministry of Science and Culture (MWK) of Lower Saxony. J.P is funded by China Scholarship Council (202208320109). A.A. is funded by a scholarship from the Egyptian Ministry of Higher Education (call 2019/2020). P.H. received financial support provided by the European Research Council (ERC Consolidator Grant “RadCrossSyn”), Deutsche Forschungsgemeinschaft (Heisenberg-Program HE7133/8-1), and Boehringer Ingelheim Stiftung (Plus 3 Perspectives Programme). This work was supported by the BMBF-funded de.NBI Cloud within the German Network for Bioinformatics Infrastructure (de.NBI) (031A532B, 031A533A, 031A533B, 031A534A, 031A535A, 031A537A, 031A537B, 031A537C, 031A537D, 031A538A). We thank Dr. Annika Stein and Stephan Rohrbach (Leibniz University Hannover, Germany) for preliminary work on this project and Dave Biedermann (Leibniz University Hannover, Germany) for synthesis of 24-methyldesmosterol. Prof. Dr. Hans-Joachim Knölker (TU Dresden, Germany) is gratefully acknowledged for providing authentic 24-methylenecholesterol as a kind gift. Umicore is acknowledged for a generous gift of metathesis catalysts. We thank Dr. David Nelson (University of Tennessee, USA) and the P450 nomenclature committee for naming of CYPs. We thank Thorsten Marschall (Botanical Garden of TU Braunschweig, Germany) and Annette Kaiser for excellent technical support in handling Withania somnifera propagation. We thank Dr. Gerald Dräger (Leibniz University Hannover, Germany) for excellent X-ray crystallography support. We thank Dr. Lorenzo Caputi and Dr. Klaus Gase (Max Planck Institute for Chemical Ecology, Jena, Germany) for helpful advice regarding silencing construct design. We also thank all members of the research group Plant Biotechnology and Bioinformatics (TU Braunschweig, Germany) for discussion and support.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
J.F. and B.P. conceived and supervised the project. S.E.H., N.C., K.M., J.P., A.B., A.A., B.P. and J.F. designed the experiments. N.C. and B.P. performed the assembly and annotation of the W. somnifera genome sequence. N.C. performed re-annotations of Physalis genome sequences, synteny analyses, phylogenetic analyses, and gene expression analyses. K.M. designed and conducted yeast metabolic engineering experiments. J.P. designed and conducted metabolic engineering in N. benthamiana. S.E.H, J.P., A.B. and J.E. performed transient expression in N. benthamiana. S.E.H. and J.P. purified compounds. A.A. performed NMR analysis. S.E.H., K.M., J.P. and A.B. generated and interpreted chromatographic and mass spectrometric data. S.E.H. optimised and performed deglycosylation of withanoside V. J.P. performed heterologous protein production and in vitro assays. A.B. performed gene silencing in N. benthamiana and W. somnifera. R.F. performed high molecular weight DNA extraction and nanopore sequencing. C.-P.W. and M.H. designed and performed MS/MS and high-resolution MS measurements. M.B. and P.H. designed synthetic routes. M.B. performed synthesis. S.E.H., N.C., K.M., J.P., A.B., A.A., B.P. and J.F. wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xingtan Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hakim, S.E., Choudhary, N., Malhotra, K. et al. Phylogenomics and metabolic engineering reveal a conserved gene cluster in Solanaceae plants for withanolide biosynthesis. Nat Commun 16, 6367 (2025). https://doi.org/10.1038/s41467-025-61686-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61686-1
This article is cited by
-
Cookbook for plant genome sequences
BMC Genomics (2026)
-
Elucidation of gene clusters underlying withanolide biosynthesis in ashwagandha through yeast metabolic engineering
Nature Plants (2026)
-
Cyanobacterial community upregulate growth and stress response genes to boost withanolide biosynthesis in the roots of Withania somnifera (L.) Dunal
Journal of Plant Biochemistry and Biotechnology (2026)
-
High Salinity Induces Changes in the Leaf Transcriptome of Withania somnifera
Plant Molecular Biology Reporter (2026)
-
CoExpPhylo – a novel pipeline for biosynthesis gene discovery
BMC Genomics (2025)
