
Abstract
Drug-resistance (DR) in bacteria often develops through the repetitive formation of drug-tolerant persisters, which survive antibiotics without genetic changes. It is unclear whether Mycobacterium tuberculosis (Mtb), the bacterium that causes tuberculosis (TB), undergoes a similar transitioning process. Recent studies highlight changes in trehalose metabolism as crucial for persister formation and drug resistance. Here, we observe that mutants lacking trehalose catalytic shift activity exhibited fewer DR mutants due to decreased persisters. This shift enhances Mtb survival during antibiotic treatment by increasing metabolic heterogeneity and drug tolerance, facilitating drug resistance. Rifampicin (RIF)-resistant bacilli display cross-resistance to other antibiotics linked to higher trehalose catalytic shift, explaining how multidrug resistance (MDR) can follow RIF-resistance. In particular, the HN878 W-Beijing strain exhibits higher trehalose catalytic shift, increasing MDR risk. Both genetic and pharmacological inactivation of this shift reduces persister formation and MDR development, suggesting trehalose catalytic shift as a potential therapeutic target to combat TB resistance.
Similar content being viewed by others

Introduction
The World Health Organization (WHO) estimated that between 2000 and 2020, over 200 million people contracted tuberculosis (TB), and more than 35 million died from the disease1. As of 2023, approximately one-quarter of the global population remained infected with Mycobacterium tuberculosis (Mtb), the bacterium that causes TB. Conventional TB treatment typically involves administering multiple antibiotics over an extended period, sometimes up to two years. This lengthy duration is largely due to the broad antibiotic tolerance exhibited by Mtb, which allows some bacteria to survive antibiotic exposure without acquiring genetic resistance2,3. This phenotypic heterogeneity primarily arises from the stochastic formation of a slowly replicating, metabolically distinct subpopulation known as persisters. These persisters are often triggered by antibiotic treatment4,5,6,7, and can survive lethal antibiotic doses, typically only being eliminated through extended, multi-drug regimens. The lengthy treatment duration significantly contributes to patient noncompliance, which in turn fosters the emergence of multidrug-resistant (MDR) TB. The unchecked spread of MDR-TB poses a major challenge to global efforts to control the disease. Therefore, it is urgent to understand the intrinsic factors within Mtb that promote MDR development and to discover new treatments aimed at preventing resistance from emerging during chemotherapy.
Persisters are phenotypic variants formed through metabolic remodeling rather than genetic changes8. Their formation and ability to withstand antibiotic effects are well-known intrinsic factors that Mtb employs to survive antibiotic pressure. Intriguingly, the level of antibiotic susceptibility remains unaltered when persisters regrow under antibiotic-free conditions. Evidence indicates that persisters surviving lethal doses of TB antibiotics are responsible for chronic and recurrent infections9,10,11. Furthermore, persisters are a source of genetic mutation-mediated MDR, as they often exhibit resilience to DNA damage caused by reactive oxygen species (ROS) generated within the bacilli during bactericidal antibiotic treatment8,12,13,14,15.
Extensive research has focused on elucidating the molecular mechanisms underlying persister biology, aiming to advance TB chemotherapeutics. Environmental cues such as nutrient starvation, redox stress, low pH, hypoxia, and uncured DNA damage have been identified as triggers for persister formation11,16,17,18. Bacterial intrinsic factors that help adapt to these stresses, including toxin-antitoxin (TA) modules in Escherichia coli19,20, also regulate persistence. Recently, metabolic remodeling has emerged as a pivotal strategy used by bacterial pathogens, including Mtb, to generate genetic mutation-free persisters and escalate antibiotic tolerance7,8,13,21,22,23,24. Metabolomics studies revealed that Mtb phenotypic heterogeneity is attributed to the formation of subpopulations with altered central carbon metabolism (CCM). This includes the capacity to co-catalyze multiple carbon sources, such as glycolytic and gluconeogenic substrates, to support full replication. It also involves the catabolic remodeling of cell wall glycolipids to enhance Mtb persister biology, bypassing the oxidative branch of the TCA cycle to downregulate NADH production. Additionally, Mtb can reroute carbon flux through the glyoxylate shunt or methylcitrate cycle, enhancing succinate biosynthesis and secretion to optimize membrane bioenergetics under diverse stresses8,13,22,25. Thus, metabolic remodeling is a crucial intrinsic factor that enables Mtb persisters to survive antibiotic effects. Mtb persisters rely on a distinct metabolic network that provides a phenotypic advantage, allowing them to withstand antibiotic stresses and associated DNA damage21,22,24,26,27,28,29,30,31.
Phenotypic heterogeneity has increasingly garnered attention as an additional intrinsic factor that promotes adaptive evolution in Mtb32. Expanding bacterial subpopulations with diverse metabolic activities can lead to the emergence of genotypes that differ from those of the original population. These heterogenous subpopulations serve as reservoirs for bacilli capable of withstanding bactericidal stresses. Recent mathematical simulations have demonstrated that even slight increases in mutation rates considerately accelerate the development of drug resistance, primarily driven by induced phenotypic heterogeneity33,34. A proof-of-concept study in yeast supported these findings, showing that initial drug-resistance mutations in a small fraction resulted in higher minimal inhibitory concentration (MIC) and faster progression of MDR. Moreover, elevated expression of efflux pumps, mediated through mechanisms involving mutS, further heightened mutation rates and phenotypic heterogeneity35,36. An example of how metabolic network remodeling fosters phenotypic heterogeneity is the random emergence of persisters, often described as a “gambler” subpopulation33. When exposed to antibiotics, only a subset of bacilli activate their DNA repair systems and stress responses37, leading to increased ROS levels. Since both the SOS response and general stress responses are essential for mutagenic DNA repair, persisters exhibit a significantly higher mutation rate. Furthermore, ample evidence indicates that adaptive metabolic remodeling enhances phenotypic heterogeneity, providing an evolutionary advantage through increased DNA mutagenesis.
We recently identified trehalose synthase (TreS) as a key mediator of metabolic remodeling and phenotypic heterogeneity that promote the formation of Mtb persisters13,22. Trehalose, a non-reducing glucose disaccharide abundant in Mtb, functions as both a carbohydrate store and a core component of cell wall glycolipids such as trehalose monomycolate (TMM) and trehalose dimycolate (TDM)38. Metabolomics analyses of Mtb bacilli collected from in vitro biofilm cultures revealed that a TreS-centered catalytic shift redirects free trehalose toward the biosynthesis of CCM intermediates. This provides an alternate source of energy and antioxidants, while diverting trehalose away from TMM and TDM production. As expected, a treS-deficient mutant (ΔtreS), lacking this catalytic shift, exhibited increased sensitivity to TB antibiotics, underscoring the importance of TreS-mediated metabolic remodeling in Mtb persister formation and antibiotic tolerance. We have also developed TreS-specific inhibitors and confirmed their potential as adjunctive therapeutic candidates39. Notably, the trehalose catalytic shift activity appears to be higher in DR-TB clinical isolates compared to drug-sensitive (DS)-TB clinical isolates, suggesting that this pathway may facilitate the emergence of drug resistance.
In this study, we demonstrate that inactivating the trehalose catalytic shift via CRISPRi-dCas9 technology not only limits persister formation and phenotypic heterogeneity but also reduces the rates of resistance development against key first-line TB antibiotics, rifampicin (RIF) and isoniazid (INH). We employed mathematical modeling to show that mycobacterial bacilli enhance their chances of developing resistance by increasing trehalose catalytic shift activity and phenotypic heterogeneity. The models further indicate that the transition rate from persisters to drug-resistant mutants is similar in both wildtype and ΔtreS, implying that the metabolic remodeling involved in persister formation plays a significant role in the emergence of drug resistance. Additionally, we identified a subpopulation of Mtb, termed pre-resistant bacilli, which appear prior to the development of drug resistance. Unlike persisters, these bacilli can grow even under antibiotic stress, and their formation is largely driven by enhanced trehalose catalytic shift activity. This phenotypic heterogeneity, stemming from persisters or pre-resistant bacilli, is a critical intrinsic factor in Mtb’s ability to acquire drug resistance. Hence, the trehalose catalytic shift represents a potential target for adjunctive therapy, not only to deepen our understanding of Mtb phenotypic heterogeneity but also to prevent the emergence of MDR-TB.
Results
Recent reports indicate that trehalose acts as a growth-permissive carbon source for DR-TB clinical isolates13. However, the trehalose-mediated growth can be reversed when co-treated with a TreS-specific inhibitor, validamycin A (ValA)13. Metabolomics profiling further supports the central role of the TreS-centered trehalose catalytic shift in the metabolic networks of DR-TB clinical isolates. The catalytic conversion of trehalose into intermediates of glycolysis and the pentose phosphate pathway (PPP) suggests that DR-TB clinical isolates preferentially utilize trehalose as a substrate for biosynthesis of CCM intermediates, rather than for producing cell wall glycolipids like TDM. These findings led us to hypothesize that the trehalose catalytic shift not only contributes to transient antibiotic tolerance but also plays a critical role in the emergence of multidrug-resistant mutants.
Trehalose metabolism differs between DR-TB and DS-TB clinical isolates
To investigate trehalose metabolism networks in DR-TB and DS-TB clinical isolates, we collected a total of 75 TB isolates from the TB clinical isolate library at the International Tuberculosis Research Center (ITRC). This collection included 15 DS-TB, 15 rifampicin single-resistant (RSR)-TB, 15 MDR-TB, 15 extensively drug-resistant (XDR)-TB, and 15 totally drug-resistant (TDR)-TB clinical isolates (Supplementary Data file 1). All strains were cultured in Middlebrook 7H9 liquid medium (m7H9) supplemented with sodium butyrate (SB), a known permissive carbon source for TB isolates13,23. The growth of both DS-TB and DR-TB clinical isolates was enhanced by the addition of 20 mM trehalose; however, co-treatment with ValA reduced growth in DR-TB, but not in DS-TB, clinical isolates (Fig. S1A). Although the heterogeneity in growth kinetics of DR-TB clinical isolates complicated precise statistical analysis, the observed impact of ValA indicated that DR-TB clinical isolates rely more heavily on TreS activity to utilize exogenous trehalose compared to DS-TB clinical isolates. To explore their metabolic networks, we extracted the total metabolome after culturing the isolates in m7H9 containing trehalose. We determined the trehalose-induced metabolic networks of TB clinical isolates by profiling ~200 TB metabolites in DR-TB and DS-TB clinical isolates. Bioinformatics analysis using MetaboAnalyst (v.6.0) identified metabolic networks uniquely altered in DR-TB clinical isolates, particularly those involved in trehalose consumption. Hierarchical clustering and heatmaps revealed distinct metabolomics patterns between DR-TB and DS-TB clinical isolates (Fig. S1B). Principal Component Analysis (PCA) further confirmed the divergence in metabolomics patterns (Fig. 1A, S1B). Pathway analysis highlighted significant alterations in trehalose metabolism, the D-alanine pathway, and the PPP. Targeted metabolomics indicated that trehalose abundance was significantly higher across all DR-TB clinical isolates (Fig. 1B, left). Furthermore, the biosynthesis of glycolytic and PPP intermediates, such as glucose 6-phosphate (Glc6P), pentose 5-phosphate (Pen5P), and sedoheptulose 7-phosphate (S7P) was comparable to or elevated in DR-TB clinical isolates relative to DS-TB clinical isolates (Fig. 1B, middle). This suggests that a substantial portion of exogenous trehalose in DR-TB clinical isolates is directed toward the biosynthesis of glycolytic and PPP intermediates13. Consistent with previous findings23, levels of phosphoenolpyruvate (PEP), the most downstream glycolytic intermediate, remained unchanged across clinical isolates (Fig. 1B, right). Conversely, TCA cycle metabolites were unaltered or downregulated in DR-TB clinical isolates (Fig. S1C). To validate these observations, we performed isotope tracing experiment using fully 13C labeled trehalose ([U-13C] trehalose). We randomly selected three DS-TB and six DR-TB clinical isolates, grew them to mid-log phase, then transferred cultures to m7H10 containing 20% [U-13C] trehalose mixed with 80% unlabeled trehalose. After 24 h, cultures were harvested, revealing significantly higher labeling in Glc6P and S7P, but not in malate (a TCA cycle intermediate). This indicates that exogenous trehalose is preferentially catabolized through glycolysis and PPP in DR-TB clinical isolates compared to DS-TB clinical isolates (Fig. S1D). The metabolic flux appears to have minimal impact on the biosynthesis of TCA cycle intermediates. Notably, treS mRNA expression remained unaltered across all DR-TB clinical isolates, with a slight induction in TDR-TB isolates, likely reflecting associated genetic mutations (Fig. 1C). Collectively, these findings demonstrate that the metabolic networks involved in trehalose consumption differ between DS-TB and DR-TB clinical isolates, with regulation occurring independently of transcriptional changes.

A 2D (left panel) and 3D (right panel) principal component analysis (PCA) of metabolome profiles from DS- (red), RSR- (green), MDR- (blue), XDR- (magenta), and TDR-TB (light blue) clinical isolates cultured in m7H9 containing 20 mM trehalose. B Targeted metabolomics analysis focusing on intermediates in trehalose metabolism, glycolysis, and the pentose phosphate pathway. PEP, phosphoenolpyruvate. Data points represent mean values ± s.e.m. of 15 biological replicates and p-values were determined by one-way ANOVA with Bonferroni post-test correction (ns, not significant). IC ion counts. C qRT-PCR analysis of treS expression in TB clinical isolates. Data points represent mean values ± SD of 15 biological replicates and p-value was determined by Student’s unpaired t-test with Welch’s correction. Source data are provided as a Source Data File.
TreS-deficient M. smegmatis phenocopies Mtb mutants that lack trehalose catalytic shift
To study the role of the trehalose catalytic shift in the development of drug resistance in mycobacteria, we employed the CRISPRi-dCas9 technique to inducibly deplete treS gene expression in M. smegmatis (Fig. S2A)40,41. The CRISPRi treS mutant of M. smegmatis (termed ItreSSM) was cultured to mid-log phase, and treS knockdown was induced using varying concentrations of anhydrotetracycline (ATc). The efficacy of treS mRNA suppression was assessed by qRT-PCR, with 200 ng/mL ATc achieving ~90% knockdown (Fig. S2B left). We also generated IotsASM, targeting otsA, which encodes trehalose 6P synthase involved in Mtb trehalose metabolism but not directly implicated in the trehalose catalytic shift. Similar to treS-deficient Mtb (ΔtreS)13, ItreSSM produced persister-like bacilli within in vitro biofilm cultures (referred to as biofilm-persisters) at significantly lower levels than wildtype following ATc treatment. In contrast, both IotsASM and wildtype formed mature biofilm-persisters, without any apparent growth defects in Sauton media (Fig. S2C, D). ItreSSM without ATc served as a control and exhibited biofilm-persister formation comparable to wildtype (Fig. S2D). Targeted metabolomics revealed that the inability of ItreSSM to form intact biofilm-persisters was primarily due to impaired trehalose catalytic shift, which disrupted trehalose-mediated carbon flux through glycolysis and the PPP (e.g., Glc6P, glyceraldehyde 3P, and S7P) (Fig. S2E). Notably, depletion of PEP abundance and the PEP/pyruvate ratio has been identified as a metabolic strategy used by Mtb to induce persister formation, slow replication, and increase antibiotic tolerance23. Interestingly, ItreSSM accumulated PEP compared to wildtype (Fig. S2E). As a result, ItreSSM displayed increased susceptibility to antibiotics, such as RIF, INH, and BDQ relative to wildtype or IotsASM (Fig. S2F). These phenotypes closely mimic those observed in ΔtreS Mtb13. These findings collectively indicate that ItreSSM phenocopies ΔtreS Mtb.

A The rates at which the indicated mycobacterial strains acquired resistance to RIF (left panel) or INH (right panel) per generation were measured using the classical fluctuation assay. Values represent mean values ± s.e.m. of 25 biological replicates and p-values were determined by Student’s unpaired t-test with Welch’s correction. Data depict median (center bar), 25th and 75th percentile (lower and upper box bounds), and minimum and maximum values (lower and upper whiskers). B Colony formation assessed by spot assay. Ten colonies from RIF-resistant bacilli obtained in (A, left panel) were spotted onto m7H10 containing either no RIF or 25 µg/mL RIF. WT, naïve drug-sensitive M. smegmatis bacilli. C Co-culture of wildtype M. smegmatis expressing green fluorescence protein (GFP) and ItreSSM expressing red fluorescence protein (RFP) was subjected to intermittent exposure to RIF over 5 cycles, designated G0–G5 subcultures. The relative enrichment of wildtype and ItreSSM in each subculture was quantified by flow cytometry and expressed as percentage. Data points represent mean values ± s.e.m. of biological triplicates. Gray bars represent ItreSSM; black bars represent wildtype. Source data are provided as a Source Data File.
The trehalose catalytic shift is an adaptive strategy that promotes the emergence of drug-resistant mycobacterial mutants
If Mtb persisters survive antibiotic-induced oxidative stresses, such as ROS, which are a known DNA mutagen, their prolonged survival increases the likelihood of developing drug-resistant mutations. The metabolic strategies employed by Mtb persisters during this stage are directly or indirectly involved in the emergence of drug resistance42,43. To investigate whether the trehalose catalytic shift is functionally linked to the emergence of drug-resistant mutants, we performed a classical Luria-Delbrück fluctuation assay to measure the rates of spontaneous drug resistance in both wildtype and ItreSSM following ATc treatment44,45. We found that the mutation rate for RIF resistance in wildtype ranged from 5.1 × 10-7 to 1 × 10-6 mutations per generation (Fig. 2A, left). The RIF-resistance rates in ItreSSM without ATc were comparable to those of wildtype. RIF-resistant colonies were confirmed by spotting them on m7H10 containing high concentrations of RIF, up to 100 µg/mL (Fig. 2B and S3A). The fluctuation and spot assays indicated that the mean RIF-resistance rate in wildtype was ~6.6-fold higher than in ItreSSM. Similarly, we assessed INH-resistance, finding rates in wildtype ranging from 1.1 × 10-5 to 5.5 × 10-6 mutations per generation, whereas ItreSSM exhibited rates between 1.8 × 10-6 and 1.0 × 10-6 mutations per generation. The wildtype developed INH resistance at a rate ~5.4-fold higher than ItreSSM (Fig. 2A, right). These results suggest a functional link between the trehalose catalytic shift and the frequency of drug resistance development across first-line TB antibiotics, regardless of the modes-of-action.
To further explore this relationship, we performed a co-culture competition assay using wildtype expressing green fluorescent protein (GFP) and ItreSSM expressing red fluorescent protein (RFP) (Fig. S3B). In this setup, we measured relative viability following cyclic exposure to bactericidal concentrations of RIF or D-cycloserine (DCS), with intermittent washes in antibiotic-free PBS, and established subcultures labeled G1 through G5 (Fig. S3B). Flow cytometry tracked the relative abundance of GFP- and RFP-expressing bacilli within these subcultures (Fig. 2C). The iterative cycle of antibiotic treatment and regrowth in antibiotic-free m7H9 led to a gradual accumulation of wildtype bacilli within the populations. In G4 and G5 subcultures, GFP intensity saturated but did not reach 100%, suggesting the presence of drug-resistant bacilli from both strains (Fig. 2C). Spot assays confirmed that G3 was the earliest generation to exhibit a drug-resistant phenotype, and the lag phase period during regrowth in G4 and G5 was nearly identical to that of naïve bacilli (Fig. S3, D, E). These findings indicate that the trehalose catalytic shift represents an intrinsic strategy of Mtb, conferring a fitness advantage during intermittent antibiotic stress and driving natural selection. To support these findings, we conducted a fluctuation assay using M. smegmatis overexpressing treS (pTreS), which showed approximately a 2.0-fold increase in RIF resistance compared to wildtype (Fig. S3F).
DR mutants are metabolically heterogenous by forming bacilli exhibiting greater trehalose catalytic shift activity
Using the fluctuation assay and RIF spot assay, we isolated 10 RIF-resistant M. smegmatis colonies, designated as FluxRIF #1-#10 (Figs. 2A, B, and S3A). Consistent with previously reported DR-TB clinical isolates13, all FluxRIF and naïve bacilli displayed similar growth patterns in antibiotic-free m7H9 (Fig. S4A). However, while naïve bacilli failed to form colonies on m7H10 containing 25 µg/mL or higher RIF, all FluxRIF bacilli successfully grew on these plates (Figs. 2B and S3A). Whole genome sequencing (WGS) of FluxRIF #1-#10 was displayed in a genome-wide coverage and variant distribution with a Circos plot. ~2.18 Gbp of high-quality reads (phred score >2.0) was obtained per sample, with an average coverage of 99.97% of the genome at a minimum read depth of 10X (Fig. S3C, upper). Across all colonies, 125 variants including single nucleotide polymorphism, insertions, deletions, and complex mutations were identified relative to the M. smegmatis reference genome (NC_008596.1); 54.4% these were frameshift variants, and 36% were located in intergenic regions (Fig. S3C, lower). The consensus sequence at all examined positions matched the reference genome, indicating no significant mutations in coding regions, except for rpoB gene in FluxRIF #1 and #2. This suggests that all 10 FluxRIF bacilli belong to the same strain with no detectable mutations in known RIF resistance genes (Supplementary data file 2). Sequencing confirmed that FluxRIF #1 and #2 bacilli carried an L452P mutation within the RIF-resistance determining region (RRDR)46, a well-known RIF-resistant mutation in many DR-TB clinical isolates47,48. In contrast, FluxRIF #3–#10 may develop RIF resistance without the target-gene mutations. Thus, the presence of sequence variants in FluxRIF #3–#10 does not fully explain observed RIF resistance. To explore the role of the trehalose catalytic shift in these drug-resistant phenotypes, we monitored growth after supplementing cultures with 20 mM trehalose. The addition of trehalose enhanced growth rates in both groups. Since ValA has minimal impact on M. smegmatis TreS activity, we employed CRISPRi-dCas9 to knock down treS expression in FluxRIF bacilli. The suppression partially impeded trehalose-induced growth in FluxRIF bacilli but had little effect on naïve bacilli (Fig. S4A), suggesting that FluxRIF bacilli exhibit a more pronounced TreS-centered trehalose catalytic shift activity. Metabolomics profiling supported this, revealing that levels of Glc6P, fructose 1,6-bisphosphate (FBP), and S7P were significantly higher in FluxRIF bacilli than in naïve bacilli, despite similar trehalose levels (Fig. 3A). Conversely, TCA cycle intermediates showed no notable differences (Fig. S4B). These results indicate that FluxRIF bacilli have elevated catalytic activity for converting exogenous trehalose into glycolytic and PPP intermediates, aligning with observations in DR-TB clinical isolates (Fig. 1B)13. In addition, FluxRIF bacilli maintained high levels of PEP, likely reflecting continued replication even in the presence of RIF (Figs. 3A and S3D)23. Consistent with the metabolomics profile and drug-resistant phenotype, FluxRIF bacilli also exhibited higher treS mRNA expression compared to naïve bacilli, especially in RRDR mutation-free FluxRIF #3-#10 bacilli (Figs. 3B and S4C). Moreover, FluxRIF bacilli contained a larger subpopulation with lower membrane potential (ΔΨm), reduced ATP levels and an induced NADH/NAD+ ratio, resembling the bioenergetic state of Mtb persisters (Figs. 3C, D and S4D, E)13,21,23. As a result, RIF penetration into FluxRIF bacilli was significantly decreased compared to naïve bacilli, as supported by Ethidium bromide (EtBr) permeability assays (Fig. 3E). Taken together, these observations demonstrate that FluxRIF bacilli exhibit increased metabolic heterogeneity, characterized by expanded subpopulations with heightened trehalose catalytic shift and diminished bioenergetic function. This metabolic heterogeneity likely contributes to persister formation, antibiotic tolerance, and the development of drug resistance.

A Targeted metabolomics profiles focusing on intermediates in trehalose metabolism, glycolysis, and the pentose phosphate pathway in naïve M. smegmatis bacilli and ten FluxRIF bacilli. Data points represent mean values ± s.e.m. of 10 biological replicates. B Fold change of treS mRNA expression in naïve M. smegmatis and FluxRIF bacilli under RIF treatment conditions relative to untreated conditions. Data points represent mean values ± s.e.m. of 10 biological replicates. C Membrane potential (ΔΨm) of naïve M. smegmatis and FluxRIF bacilli measured after treatment with 30 µg/mL RIF by flow cytometry. The percentage of bacilli exhibiting high and low ΔΨm for each strain is shown relative to the untreated condition. Data points represent mean values ± s.e.m. of biological triplicates of naïve bacilli and 10 biological replicates of FluxRIF bacilli. D Intrabacterial ATP concentrations in FluxRIF bacilli, expressed as a fold change relative to naïve bacilli. Data points represent mean values ± s.e.m. of biological triplicates of naïve bacilli and 10 biological replicates of FluxRIF bacilli. E Relative RIF permeability and EtBr permeability kinetics of naïve M. smegmatis or FluxRIF bacilli. Data points represent mean values ± s.e.m. of biological triplicates of naïve bacilli and 10 biological replicates of FluxRIF bacilli. p values were determined by Student’s unpaired t test with Welch’s correction, ns, not significant. Source data are provided as a Source Data file.
The trehalose catalytic shift confers mycobacterial cells with greater metabolic heterogeneity
Increasing metabolic heterogeneity within an isogenic population is a well-known strategy that promotes the emergence of persisters and drug-resistant mutants49,50,51. Recent studies have shown that DR-TB clinical isolates exhibit lower levels of TDM in their cell wall due to increased trehalose catalytic shift activity13,23,52. To explore the functional connection between the trehalose catalytic shift in FluxRIF bacilli and their ability to augment metabolic heterogeneity, we employed the fluorogenic dye Red Molecular Rotor-trehalose (RMR-tre). This dye specifically labels mycobacterial cell wall glycolipids containing trehalose, such as TDM53. We observed that RMR-tre labeling intensity in naïve bacilli during mid-log phase gradually decreased with increasing concentrations of free trehalose53, suggesting that RMR-tre acts as a substrate for Ag85, an enzyme involved in TDM biosynthesis, at levels comparable to free trehalose54,55,56. Using FACS, we quantified RMR-tre labeling before and after treatment with sublethal doses of RIF. In naïve bacilli, RMR-tre labeling was relatively homogenous prior to antibiotic treatment. However, after RIF treatment, a subfraction of RMR-trehigh bacilli emerged, indicating increased heterogeneity. This likely results from bacilli with induced trehalose catalytic shift activity preferentially consuming preexisting trehalose for generating CCM intermediates, leading to higher RMR-tre incorporation compared to endogenous trehalose. Notably, RMR-trehigh bacilli were absent in ΔtreS Mtb (Fig. S5A). To further investigate the role of the trehalose catalytic shift in forming the RMR-trehigh subfraction and associated metabolic heterogeneity, we repeated the assay using pTreSSM, M. smegmatis overexpressing treS, and ItreSSM. We found that the RMR-trehigh subfraction substantially overlapped with that of pTreSSM, whereas it was absent in ItreSSM, mirroring the phenotype in ΔtreS Mtb (Figs. 4A-C and S5A). This underscores the critical role of the trehalose catalytic shift in promoting metabolic heterogeneity in response to bactericidal antibiotics. Interestingly, the RMR-trehigh fraction was already significantly larger in FluxRIF bacilli compared to naïve DS-bacilli, even prior to antibiotic treatment, corroborating TLC-based quantification (Figs. S4F and S5B, C). To determine whether the RMR-trehigh subfraction in FluxRIF bacilli primarily consists of a viable population following treatment with bactericidal antibiotics, we tracked the dynamics of RMR-trehigh and RMR-trelow subfractions after exposure to bactericidal doses of RIF. We observed a profound decrease in the RMR-trelow subfraction, with the RMR-trehigh population becoming dominant (Fig. S5D). This suggests that the metabolic heterogeneity driven by expansion of the RMR-trehigh subfraction is largely due to enhanced trehalose catalytic shift activity, which correlates with antibiotic tolerance and the accumulation of drug-resistant mutations. Furthermore, this phenomenon was more pronounced in FluxRIF #3-#10 bacilli than in FluxRIF #1 and #2. The latter, which harbor the L452P mutation in the RRDR, maintained the RMR-trelow subfraction as a dominant population even after antibiotic treatment, with only a slight reduction (Fig. S5D). Whole-genome sequencing confirmed the absence of significant mutations in FluxRIF #3-#10 bacilli, indicating that their resistance is mainly driven by elevated trehalose catalytic shift activity rather than genetic mutations. In contrast, RIF resistance in FluxRIF #1 and #2 bacilli is likely mutation-mediated within the RIF target gene. RIF treatment rendered all FluxRIF #3-#10 bacilli more homogenous, either by inducing the trehalose catalytic shift in the RMR-trelow subfraction or by selectively killing less drug-tolerant RMR-trelow subfraction (Fig. S5D). This indicates that RMR-trehigh bacilli may represent a significant source of viable bacilli following treatment with bactericidal antibiotics. Overall, the trehalose catalytic shift is an intrinsic factor of Mtb that elevates metabolic heterogeneity by expanding the RMR-trehigh subfraction. This expansion facilitates persister formation and the development of pre-resistant bacilli – key factors in Mtb’s ability to survive prolonged antibiotic pressure.

RMR-tre fluorescence labeling patterns are shown for A wildtype M. smegmatis, B ItreSSM, and C pTreSSM (M. smegmatis overexpressing TreS) following treatment with a sublethal dose of RIF. The bacilli within the area labeled R1 are defined as the RMR-trehigh subfraction. Additionally, the RMR-tre fluorescence labeling patterns for D a representative FluxRIF bacillus, and E its CRISPRi treS knockdown strain after ATc treatment, are depicted.
The trehalose catalytic shift is necessary to increase drug-resistance frequency by expanding the persister subpopulation
Pathogenic bacteria can transiently acquire a drug-tolerant phenotype through a non-genetic mechanism by forming persisters. They can regrow once the effects diminish, creating a cycle that repeats until drug-resistant mutants emerge. The phenotypic reversibility between drug-sensitive bacilli and drug-tolerant persisters occurs when antibiotic priming is intermittent. However, continuous antibiotic pressure leads to the accumulation of genetic mutations that establish stable, resistant mutants (Fig. 5A)57,58.

A The schematic diagram illustrates a phenotypic transition model in which drug-sensitive population (gray) evolve into drug-resistant population (black) via the formation of drug-tolerant population (red). In this model, bacilli can reversibly switch between drug-sensitive and drug-tolerant states during the intermittent antibiotic treatment. Once a population becomes drug-tolerant, it remains in that state for multiple generations before reverting to a drug-sensitive state. After prolonged antibiotic exposure, each drug-tolerant bacillus irreversibly transitions to drug-resistant state and replicate. To assess the impact of the trehalose-catalytic shift on the frequency of each transition step, wildtype M. smegmatis and ItreSSM were utilized in a fluctuation assay as depicted in Fig. S6A. The reversibility and transition capacities were calculated as described in the Mathematical Modeling section in the “Methods”. Schematic diagram was created in BioRender. Lee, J. (2025) https://BioRender.com/otwhxsl. B The rates of formation of drug-resistant mutants in wildtype and ItreSSM against RIF were determined using the classical Luria-Delbrück fluctuation assay and the Lea-Coulson method (m/Nt, where m is the number of resistant colonies and Nt is the total input). Data points represent mean values ± s.e.m. of biological triplicates for the short-term exposure and 60 biological replicates for the long-term exposure. p-values were determined by Student’s unpaired t test with Welch’s correction. Source data are provided as a Source Data File.
To understand how the trehalose catalytic shift influences this process, we employed mathematical modeling and developed analytical formulas to predict its impact on the kinetics of phenotypic reversibility and clone-to-clone fluctuations within surviving populations. These populations serve as reservoirs for eventual drug-resistant bacilli (Fig. 5A)59,60. Our model captures the emergence of drug-tolerant persisters during population growth by allowing individual bacilli to reversibly switch between drug-sensitive and drug-tolerant states61. Once a bacillus becomes tolerant, it remains in that state for several generations before reverting to a drug-sensitive state (Figs. 5A and S6A, B). Using this framework, we previously modeled reversible switching in fluctuation assays, enabling analytical predictions of the statistical variation in tolerant bacilli across colonies derived from a single progenitor62. Analysis of the fluctuation assay data with this reversible switching model shows that ItreSSM generates roughly six-fold fewer drug-resistant colonies compared to wildtype (Fig. 5B right). Assuming equal mutation probabilities, this reduction in ItreSSM likely results from the production of less stable persisters that revert to a drug-sensitive state more rapidly. Furthermore, our findings indicate that the decreased number of resistant colonies in ItreSSM compared to wildtype is primarily due to a six-fold reduction in persister formation rate (Fig. 5B, left), as detailed in the Mathematical Modeling section of the Methods. Since the emergence of drug resistance is facilitated by a higher number of persisters8,50,63,64,65,66,67, we conclude that mycobacterial bacilli evolve into drug-resistant mutants through repeated cycles of persister and pre-resistant bacilli formation. The trehalose catalytic shift acts as a strategic mechanism to expand the subpopulation of persisters and pre-resistant bacilli, especially under conditions of high ROS-induced damage, thereby facilitating the emergence of drug-resistant mutants.
RIF-resistant mycobacterial cells are also resistant to INH and BDQ
Clinical data from Taiwan Medical Center indicate that 94.6% of RIF-resistant Mtb strains are also resistant to INH, while only 0.5% are mono-resistant to RIF68. Similar patterns have been observed in retrospective TB case studies conducted in New York City between 2010 and 202169. These findings suggest that RIF resistance can serve as a predictive biomarker for MDR-TB. Based on this, we hypothesize that RIF-resistant strains could possess a metabolic advantage that confers increased tolerance to second antibiotics like INH, even without prior exposure. RMR-tre labeling patterns indicate that FluxRIF bacilli contain a high abundance of RMR-trehigh subfraction (Figs. 4D and S5B, C). To test this hypothesis, we performed minimum inhibitory concentration (MIC) shift assays using selected FluxRIF bacilli and their CRISPRi treS mutant, referred to as ItreSFlux, comparing their antibiotic sensitivities to naïve bacilli. FluxRIF bacilli exhibited significantly higher tolerance to INH, with MIC values ~3.82 µg/mL, compared to around 1.84 µg/mL in naïve bacilli. Notably, this elevated INH tolerance was reduced in ItreSFlux after treatment with ATc, dropping to around 1.49 µg/mL (Fig. 6A, left). No such reduction was observed in ItreSFlux without ATc, indicating that the elevated tolerance to second antibiotics in FluxRIF bacilli depends on active trehalose catalytic shift (Fig. 4D and S5D). Spot assays on m7H10 containing bactericidal doses of INH corroborated the MIC shift assay results (Figs. 6B and S7A). Additionally, FluxRIF bacilli demonstrated increased tolerance to BDQ as well, highlighting the role of the trehalose catalytic shift in cross-resistance to multiple TB antibiotics (Fig. 6A, right). Interestingly, among 1500 ITRC TB clinical isolates, only 15 (<1%) were RSR-TB clinical isolates, and an inverse relationship, where INH resistance confers RIF cross-resistance, was not clearly observed. To further explore this, we examined INH-resistant bacilli (referred to as FluxINH) obtained from fluctuation assays (Fig. 2A, right). Testing two randomly selected FluxINH bacilli revealed that they were significantly more sensitive to RIF than naïve bacilli (Fig. 6C, D), likely due to increased RIF uptake (Fig. S7B). This altered permeability was tested by the EtBr permeability assay and the SDS sensitivity assay (Fig. S7C). Higher EtBr uptake in FluxINH bacilli, combined with similar SDS uptake compared to naïve bacilli, suggests that reduced efflux pump activity in FluxINH bacilli contributed to increased RIF uptake. INH requires metabolic activation through NAD+ adduct formation to exert its antimicrobial effect70. As shown in Fig. 3, FluxRIF bacilli demonstrated distinct metabolic networks, characterized by higher trehalose catalytic shift and reduced bioenergetic markers such as NAD+, ΔΨm, and ATP (Figs. 3C, D and S4D). This metabolic state likely influences the formation of INH-NAD adducts. The observed cross-resistance in FluxRIF bacilli was significantly reduced when treS was inhibited via CRISPRi-dCas9 (Fig. 6A), supporting the hypothesis that the trehalose catalytic shift contributes to the emergence of MDR-TB cases.

A IC50 values for INH (left panel) and BDQ (right panel) were measured in naïve M. smegmatis, FluxRIF, and ItreSFlux bacilli, both with and without ATc treatment. Data points represent mean values ± s.e.m. of biological triplicates. B Spot assay on m7H10 containing 10X MIC of INH (isoniazid) or BDQ (bedaquiline), using naïve M. smegmatis, FluxRIF, and ItreSFlux bacilli. C IC50 values for BDQ were determined for naïve M. smegmatis and two selected FluxINH bacilli. Data points represent mean values ± s.e.m. of biological triplicates. D Spot assay on m7H10 containing 10X MIC of BDQ (left panel) with naïve M. smegmatis and FluxINH bacilli. Data points represent mean values ± s.e.m. of 6 biological replicates of naïve bacilli and 10 biological replicates of FluxINH bacilli. The right panel displayed the average colony diameters and standard deviations of colonies grown on m7H10 containing 10X MIC of BDQ. Source data are provided as a Source Data File.
The trehalose catalytic shift enables the HN878 W-Beijing strain to acquire a high frequency of multidrug resistance
Clinical Mtb strains are classified into phylogeographic lineages 1 through 7, each exhibiting varying capacities for acquiring MDR mutations45,71. Notably, lineage 2 strains, including the HN878 W-Beijing strain (HN878), are associated with a heightened risk of MDR-TB emergence on a global scale. Our findings suggest that the trehalose catalytic shift in Mtb contributes to this increased MDR propensity by promoting persister formation and cross-resistance to multiple antibiotics (Figs. 4, 5, and 6). We hypothesize that elevated trehalose catalytic shift activity in HN878 plays a key role in its tendency to accumulate MDR mutations more frequently than other lineages. To investigate this, we examined treS expression in HN878 following exposure to sublethal doses of RIF. Results showed that treS mRNA was significantly upregulated—by ~7.3-fold—compared to untreated controls, a much higher induction than the 2 to 3-fold increase observed in lineage 4 strains such as Erdman and CDC1551 (Fig. 7A). Additionally, HN878 exhibited faster growth in m7H9 containing trehalose as the sole carbon source (Fig. 7B). When co-treated with ValA, trehalose-mediated growth was restored to levels comparable to lineage 4 strains, suggesting that trehalose may serve as a more favorable carbon source for HN878, likely due to its higher TreS activity (Fig. 7B). Metabolomics analysis revealed that the conversion of trehalose into glycolytic and PPP intermediates, such as Glc6P, Pen5P, and S7P, was significantly higher in HN878 than in lineage 4 strains (Fig. 7C). Collectively, these findings suggest that HN878 undergoes a more pronounced trehalose catalytic shift, which likely contributes to its higher frequency of MDR mutations.

A treS mRNA expression levels in HN878 and lineage 4 strains (e.g., H37Rv, Erdman, and CDC1551) were measured before and after treatment with RIF. The closed black circles indicate fold changes relative to untreated controls. Data points represent mean values ± s.e.m. of biological triplicates. B Growth kinetics of HN878 and lineage 4 strains on m7H9 with trehalose as the sole carbon source. The effect of ValA on HN878 growth is also illustrated. Values represent mean values ± s.e.m. of triplicates. C RIF treatment induced changes in the levels of trehalose, glucose 6P, and pentose 5P in HN878 and lineage 4 strains, relative to the untreated controls (No RIF). FC, fold change. Values are mean values ± s.e.m. of biological triplicates. D The effects of ValA (left panel) or CRISPRi-mediated treS inactivation (right panel) on the rates of RIF-resistance acquisition per generation in HN878 and lineage 4 clinical strains, measured via the classical fluctuation assay. Values are mean values ± s.e.m. of biological triplicates. p values were determined by Student’s unpaired t-test with Welch’s correction, ns, not significant. In a box plot (right panel), data depict median (center bar), 25th and 75th percentile (lower and upper box bounds), and minimum and maximum values (lower and upper whiskers). E Impact of ValA on the IC50 values of RIF against the indicated Mtb clinical strains: HN878 (~62 ng/mL), ERD (~31 ng/mL), CDC1551 (~22 ng/mL) and HN878 treated with ValA (~22 ng/mL). Source data are provided as a Source Data File.
To further validate the role of the trehalose catalytic shift in HN878 for the emergence of drug-resistant mutants, we conducted fluctuation assays with HN878 and lineage 4 strains, both with and without ValA, as well as CRISPRi treS mutants of HN878 (ItreSHN), CDC1551 (ItreSCDC), or Erdman (ItreSErd) (Figs. S2A and S8A). Consistent with previous reports45, HN878 displayed roughly a 5.0-fold higher frequency of developing RIF resistance compared to lineage 4 strains (Fig. 7D, left). Treatment with ValA significantly reduced this mutation rate to levels comparable to lineage 4 strains. Similarly, mutation frequencies of ItreSHN and ItreSCDC were comparable, confirming the involvement of trehalose catalytic shift (Fig. 7D, right). Furthermore, HN878 exhibited a higher MIC for RIF (~0.06 µg/mL) than lineage 4 strains (~0.03 µg/mL), due to its enhanced trehalose catalytic shift activity. When co-treated with ValA or in ItreSHN, the MIC value decreased to ~ 0.02 µg/mL (Figs. 7E and S8B, C). To link this metabolic activity to persister formation and drug tolerance, we utilized the most probable number (MPN) assay, which quantifies the frequency of total persisters including both traditional persisters and differentially detectable (DD) bacilli under RIF treatment and nutrient-starved conditions72,73. We found that the frequency of persister formation in HN878 was the highest among all clinical strains tested (Fig. S8D). The reduction in persister formation following co-treatment with ValA (Fig. S8D, left) or using ItreSHN (Fig. S8D, right) was most pronounced, indicating that the high frequency of MDR development in HN878 is largely attributed to its elevated trehalose catalytic shift activity and subsequent persister formation. Based on our mathematical modeling (Fig. 5), the frequent emergence of MDR-TB cases linked to infections with HN878 is primarily driven by increased trehalose catalytic shift activity and persister formation. Therefore, targeting this pathway offers a promising strategy for developing adjunctive therapies to prevent the emergence of MDR-TB.
Discussion
Persister formation is a widespread adaptive strategy among bacterial pathogens including Mtb, allowing survival under antibiotic effects for extended periods without developing genetic resistance8,74. The pathogenic cycle of TB includes a latent infection stage, during which Mtb bacilli often enter a persister state. In this state, they can opportunistically recur, increasing bacterial burden and serving as reservoirs for genetic mutations that lead to drug resistance33,75,76. Compared to heritable drug resistance, the biology of Mtb persisters remains in the early stages of investigation. Our study indicates that mycobacterial persisters are indeed an adaptive method that plays a crucial role in the pathogenic lifecycle of TB. This process is triggered by metabolic remodeling, such as trehalose catalytic shift, and is directly or indirectly linked to the evolutionary traits that promote the emergence of drug-resistant mutants8,11,77,78,79,80. Beyond enabling intermittent antibiotic tolerance and opportunistic relapse, our findings support the notion that Mtb persisters act as reservoirs for the development of multidrug resistance and contribute to the global spread of MDR-TB52,64,65,76,81,82,83. Notably, the accumulation of ROS resulting from antibiotic effects has been identified as a primary factor that kills invading bacilli. However, when pathogens survive this oxidative stress, ROS can also induce DNA mutagenesis30,84,85. Prolonged survival in the persister state, followed by regrowth through metabolic remodeling, is directly associated with the emergence of populations harboring genetic mutations conferring drug resistance12,86. These adaptive metabolic strategies underpin the accelerated development and dissemination of MDR-TB.
A handful of investigations have begun to elucidate the key metabolic remodeling strategies for persister formation. Our metabolomics studies, using Mtb persisters collected from in vitro biofilm cultures or under hypoxic stress, have validated the functional importance of preexisting Mtb cell wall glycolipids as alternative carbon sources13,22. Bioinformatic analysis of the metabolomics data revealed that trehalose metabolism is among the most significantly altered pathways compared to replicating Mtb. These findings suggest that Mtb persisters shift the catalytic direction of trehalose metabolism to biosynthesize intermediates in glycolysis and the PPP, a process we term the trehalose catalytic shift. Indeed, ΔtreS of Mtb, which lacks this catalytic shift activity, exhibited hypersensitivity to first-line TB antibiotics such as INH and RIF. Trehalose serves as a structural component of Mtb cell wall glycolipids, including TDM, which modulates host immune responses. Additionally, trehalose is a core carbohydrate in sulfolipid-1 (SL-1), a cell wall component recently reported to be linked to the opportunistic transmission of Mtb bacilli to new hosts87. Therefore, the trehalose catalytic shift plays multiple roles, including carbon storage, essential components for persister biology, antibiotic tolerance, immune evasion, and transmission38,88.
This study uncovers an additional role of the trehalose catalytic shift in accelerating the development of permanent MDR in Mtb. The bacilli maintain viability by forming persisters through this shift activity, even under bactericidal levels of oxidative stress. This process induces DNA mutagenesis via activation of the trehalose catalytic shift. Notably, RIF-resistant bacilli, harboring high levels of trehalose catalytic shift, tend to exhibit increased antibiotic tolerance to a second antibiotic, even without prior exposure, highlighting how this adaptive strategy can facilitate the progression to MDR-TB. Furthermore, our findings demonstrate that the trehalose catalytic shift enhances phenotypic heterogeneity within the population. By inhibiting treS expression through CRISPRi or chemically deactivating TreS with ValA, a key enzyme in this pathway, we observed a significant reduction in the emergence of DR mycobacterial mutants against clinically relevant TB antibiotics. Fascinatingly, our mathematical modeling clarifies that the trehalose catalytic shift uniquely facilitates persister formation and confers phenotypic stability, preventing persisters from reverting to the DS-state. This suggests that the frequency of DR mutant emergence is predominantly influenced by the extent of persister formation, phenotypic heterogeneity and stability. Our findings also show that the transition rates from persisters to permanent DR mutants are nearly identical between wildtype and ΔtreS. Therefore, the metabolic remodeling strategies that promote Mtb persister formation represent promising targets for therapeutic intervention. Targeting these pathways could not only eradicate Mtb persisters but also prevent the onset of MDR-TB.
We have developed a technique to monitor mycobacterial phenotypic heterogeneity resulting from active trehalose catalytic shift by labeling with an RMR-tre fluorogenic dye combined with FACS analysis. In our recent report53, we demonstrated that RMR-tre serves as a substrate of Ag85, an enzyme involved in the biosynthesis of TDM, at levels comparable to free trehalose. In this work, we found that RMR-tre labeling intensity correlates with TreS activity (Fig. A-C). This is likely because mycobacterial bacilli with higher TreS channel more trehalose toward the biosynthesis of CCM intermediates, limiting internal trehalose availability for TDM biosynthesis. Consequently, when exogenous RMR-tre is supplied, bacilli with greater TreS utilize more RMR-tre as a substrate for TDM production, whereas TreS-deficient bacilli rely more on endogenous trehalose. In response to antibiotic treatment, Mtb exhibited an accumulation of RMR-trehigh bacilli, a subfraction displaying labeling patterns similar to those of pTreS, but absent in ItreSSM or ΔtreS Mtb. This suggests that activation of the trehalose catalytic shift increases metabolic and phenotypic heterogeneity, resulting in the formation of a population that exhibits tolerance to antibiotics. Notably, FluxRIF bacilli showed a higher proportion of RMR-trehigh subfraction compared to naïve DS-bacilli, underscoring the essential role of the trehalose catalytic shift in the metabolic networks of DR-bacilli and thus providing insight into mechanisms that contribute to their cross-resistance to other antibiotics. Treatment with bactericidal concentrations of antibiotics caused a notable increase in the proportion of bacilli with high TreS activity. This effect may stem from the greater antibiotic susceptibility of the subfraction with low TreS activity or from antibiotic-induced alterations in metabolic heterogeneity, which promotes the induction of TreS activity.
The exploration of metabolic strategies beyond the trehalose catalytic shift is warranted, as a substantial subset of ItreSSM or ΔtreS Mtb strains continue to develop drug-resistant mutations, albeit at a significantly reduced rate. Consistent with data obtained from FluxRIF bacilli, metabolomics analyses of DR-TB clinical isolates revealed distinct metabolic activities involved in trehalose catalysis compared to DS-TB and some RSR-TB clinical isolates. These DR-TB clinical isolates exhibited biochemical features similar to Mtb persisters, characterized by dysregulated membrane bioenergetics and active glycolysis and PPP, which serve as alternate sources of energy and antioxidants. Additionally, they showed a reduced abundance of cell wall TDM, a proinflammatory ligand of Mtb, as part of an immune evasion strategy13,22,89. The catabolic remodeling of TDM provides infected Mtb bacilli with a spatiotemporal advantage, allowing them to maintain their latent state without relying on host nutrients or provoking excessive immune responses. TB clinical isolates often utilize host fatty acids or cholesterol as primary carbon sources, which require endergonic pathways such as the TCA cycle, glyoxylate shunt, methylmalonyl CoA pathway, and methylcitrate cycle, followed by gluconeogenic reactions. Gluconeogenesis involves primarily energy-consuming reactions that biosynthesize carbohydrate intermediates. Therefore, the trehalose catalytic shift offers a catalytic advantage by enabling the exploitation of largely exergonic metabolic networks to support the energy demands and antioxidant needs critical for Mtb persister biology, antibiotic tolerance, and the eventual emergence of MDR mutants.
Labeling FluxRIF bacilli with RMR-tre dye revealed that those carrying RRDR mutations exhibited significantly fewer RMR-trehigh bacilli compared to the RRDR mutation-free FluxRIF bacilli (Fig. S5B, C). Additionally, treatment with bactericidal antibiotics selectively eliminated the RMR-trelow subfraction, a phenomenon more pronounced in RRDR mutation-free FluxRIF bacilli (Fig. S5D). This suggests that RRDR mutation-free FluxRIF bacilli may serve as a primary reservoir for the future development of DR mutations and the emergence of permanent MDR mutants. Consequently, these pre-resistant subpopulations likely require higher levels of trehalose catalytic shift to sustain their drug-resistant phenotype. Collectively, Mtb can attain a permanent DR phenotype through the formation of Mtb persisters or pre-resistant bacilli by inducing the trehalose catalytic shift activity. Similar to carbapenem-resistant Enterobacteriaceae (CRE) clinical isolates90, Mtb persisters may revert to a DS-state once antibiotic effects diminish (Fig. 5A). In contrast, pre-resistant bacilli were found to be phenotypically stable, consistently managing their metabolic networks with high levels of trehalose catalytic shift. The functional relevance of the trehalose catalytic shift in the formation of Mtb persisters and/or pre-resistant bacilli was confirmed by genetic inactivation of treS in FluxRIF bacilli (Fig. 4D, E).
Consistent with previous findings, we observed that the higher frequency of MDR mutations in lineage 2 clinical strains such as HN878 is largely attributed to their increased trehalose catalytic shift activity. This elevated activity is linked to the induced formation of persisters and pre-resistant bacilli. Our mathematical modeling suggests that the greater propensity of persister formation correlates with an increased likelihood of phenotypic heterogeneity and the emergence of MDR-TB. Additionally, our cross-resistance studies revealed that Mtb bacilli with elevated trehalose catalytic shift activity are more prone to develop MDR-TB mutations. This research illuminates the metabolic basis underlying the higher incidence of MDR-TB cases in infections with HN878. Targeting the trehalose catalytic shift in HN878 presents a therapeutic strategy to improve the efficacy of existing TB antibiotics by preventing both the formation of persisters and the emergence of MDR-TB cases. Recently, we demonstrated that certain trehalose structural analogues can disrupt Mtb persister formation and antibiotic tolerance by inhibiting TreS-centered trehalose catalytic shift activity, thereby enhancing the antimicrobial effects of INH or RIF39. The potential for these compounds to synergize with clinically relevant TB antibiotics against HN878 infection warrants further investigation.
Methods
Bacterial strains, culture conditions, and chemicals
Mycobacterium smegmatis, including drug-sensitive and drug-resistant strains, along with their CRISPRi strains including ItreSSM and IotsASM were cultured at 37 °C in Middlebrook 7H9 broth (m7H9) (Difco) or on Middlebrook 7H10 agar (m7H10) (Difco). The media were supplemented with 0.04% Tyloxapol (for planktonic growth in m7H9 only), 0.5 g L−1 BSA (Fraction V), 0.2% glycerol, 0.2% dextrose, and 0.085% NaCl. Kanamycin (50 µg/mL) was used to select CRISPRi mutants. Mycobacterium tuberculosis strains including HN878, Erdman, H37Rv, and CDC1551, along their corresponding CRISPRi strains (ItreSHN, ItreSErd, ItreSCDC, and ItreSRv) were cultured in a biosafety level 3 (BSL-3) facility. The TB clinical isolates, including drug-sensitive (DS), rifampicin single-resistant (RSR), MDR, extensively drug-resistant (XDR), and totally drug-resistant (TDR) TB clinical isolates, were isolated from sputum samples of patients with active pulmonary TB at the National Masan Hospital (NMH), South Korea. All procedures adhered to relevant ethical regulations, and the isolates were collected as part of a prospective observational cohort study (ClinicalTrials.gov NCT00341601) conducted between 2005 and 2008. The study was approved by the ethics review boards of both NMH and the National Institute of Allergy and Infectious Diseases (NIAID), with all participants providing written informed consent.
When appropriate, cultures were supplemented with 10 mM sodium butyrate, 20 mM trehalose, 200 µM Validamycin A (ValA), 200 ng/mL anhydrotetracycline (ATc), or varying MIC of rifampicin (RIF), isoniazid (INH), bedaquiline (BDQ), or d-cycloserine (DCS). These compounds were purchased from Sigma and Advanced ChemBlocks Inc. Experiments involving CRISPRi mutants were conducted with or without ATc; untreated cultures served as controls.
Metabolite extraction and LC-MS analysis
M. smegmatis- or Mtb-laden filters were generated and incubated at 37 °C for 5 days to reach mid-log phase of growth21. To prepare for filter culture-based metabolomics, cultures on agar-supported filters were treated with trehalose and/or ValA. M. smegmatis or Mtb-laden filters were metabolically quenched by immersion in a precooled mixture of acetonitrile:methanol:H2O (40:40:20, v:v:v) at −40 °C. Metabolites were extracted via mechanical lysis using 0.1-mm zirconia beads in a Precellys tissue homogenizer for 4 min at 6000 rpm, repeated twice under continuous cooling at or below 2 °C. The lysates were clarified by centrifugation and filtered through a 0.22-µm Spin-X column. The residual protein content was measured with a BCA protein assay kit (Thermo Scientific) to normalize metabolite levels to cell biomass.
Extracted metabolites were separated using a Cogent Diamond Hydride type C column (gradient 3) with mobile phase comprising solvent A (ddH2O with 0.2% formic acid) and solvent B (acetonitrile with 0.2% formic acid). An Agilent 6230 TOF mass spectrometer (MS) was coupled to an Agilent 1290 Liquid Chromatography (LC) system. Dynamic mass axis calibration was maintained via continuous infusion of a reference mass solution through an isocratic pump with a 100:1 splitter. This setup achieved mass errors of ~5 ppm and a mass resolution between 10,000 to 25,000 over the m/z range of 62–966, with a dynamic range of 5 log10. Ions were identified based on unique accurate mass and retention time identifiers corresponding to expected isotopomer distributions. Data processing was conducted using Agilent Qualitative Analysis B.07.00 and Profinder B.07.00 software (Agilent Technologies), with a mass tolerance of <0.005 Da. Clustered heatmaps, hierarchical clustering, principal component analysis (PCA), and pathway enrichment analysis were performed using MetaboAnalyst (ver. 6.0). All metabolomics data represent the average of at least two independent triplicates.
Isotope tracing analysis using 13C12 trehalose
To investigate trehalose metabolism in DS- and DR- TB clinical isolates, we performed isotope tracing experiments using fully 13C labeled trehalose (13C12 trehalose), purchased from Cambridge Isotope Laboratory (CIL). Nine TB clinical isolates were randomly selected, three DS- and six DR- strains, and cultured in m7H9 until mid-log phase. The cultures were then transferred to fresh m7H9 containing 20 mM trehalose, composed of 20% 13C12 trehalose and 80% 12C unlabeled trehalose. After 1 day, the cultures were harvested, washed with PBS, and rapidly quenched by immersion into a precooled mixture of acetonitrile/methanol/H2O (40:40:20) at −40 °C. The extent of isotopic labeling in key metabolites such as glucose 6-phosphate (glycolysis), sedoheptulose 7 phosphate (PPP), and malate (TCA cycle) was quantified by dividing the summed peak height of all labeled isotopologue species by the total peak heights of both labeled and unlabeled isotopologues, expressed as a percentage. To correct for naturally occurring 13C isotopologues (i.e., [M + 1] and [M + 2]), label-specific ion counts were adjusted accordingly.
qRT-PCR analysis
M. smegmatis or Mtb strains were cultured in m7H9 until reaching mid-log phase. Bacilli were harvested by adding an equal volume of guanidine thiocyanate buffer. Total RNA was extracted using TRIzol reagent and the PureLink RNA Mini Kit (Invitrogen), following the manufacturer’s instructions. Genomic DNA contamination was eliminated using the Turbo DNA-free kit (Invitrogen). cDNA was synthesized from 500 ng of RNA using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR was performed on a C1000 Thermal Cycler (Bio-Rad). Primers and probes were designed using the PrimerQuestTM Tool (Integrated DNA Technologies), with sequences provided in Supplementary data file 2. Gene expression levels were quantified by calculating the ΔΔCt values, normalized to the housekeeping gene sigA. Fold changes were expressed as log2 values relative to control samples.
Lipid extraction and TLC analysis
TDM and TMM from mycobacterial bacilli were prepared as previously described13. Bacilli were transferred to a 15 mL amber glass bottle and incubated overnight with 3 mL of chloroform: methanol (2:1, v:v) to sterilize the bacteria and extract total lipids. Subsequently, 10 mL acetone was added, and the mixture was incubated for 24 h at − 80 °C. After centrifugation, the lipid-containing supernatant was decanted. The lipids were then resuspended in 1 mL of chloroform: methanol (2:1, v:v), and equal amounts from each condition were loaded onto thin-layer chromatography (TLC) plates. TMM and TDM were resolved using TLC in a solvent system of chloroform: methanol: H2O (90:10:1, v:v:v). Lipids were visualized by spraying the plates with 1% molybdophosphoric acid in ethanol, followed by charring.
CRISPRi knockdown generation
Single guide RNA (sgRNA) sequences were designed to target the 3’-end of the non-template strand within the open reading frame of the target genes. Each sgRNA consists of ~20 nucleotides located upstream of an effective protospacer adjacent motif (PAM) sequence. The PLJR962 M. smegmatis CRISPRi backbone plasmid was amplified in E. coli, selected with kanamycin (50 µg/mL), and subsequently digested with BsmBI restriction enzymes (NEB). The digested plasmid was then purified. Designed oligonucleotide primers (see Supplementary Table 1) were annealed and ligated into the BsmBI-digested plasmid. Competent M. smegmatis cells were prepared by washing mid-log phase cultures multiple times with ice-cold 15% glycerol. The recombinant plasmid was introduced into these competent cells via electroporation using a Pulse Controller II and Gene Pulser II (BioRad). Transformed cultures were grown to mid-log phase and plated on m7H10 containing 50 µg/mL kanamycin to select for successful mutants. Selected colonies were regrown in m7H9 with kanamycin and incubated with ATc (200 ng/mL) for at least 1 day to induce target gene repression. Knockdown of the target genes was confirmed by qRT-PCR.
Luria-Delbrück fluctuation assay and analysis
The classical fluctuation assay was modified for this study45. To generate single-cell suspensions of M. smegmatis wildtype and ItreSSM, cultures were diluted to an OD595 of 0.00005 in 200 µL within 96-well plates. The cultures were then incubated and allowed to grow until reaching an OD595 of 0.7–1.0. From each of 60 randomly selected cultures, 100 µL was plated on m7H10 containing either 100 µg/mL RIF or 200 µg/mL INH. Spontaneous resistant colonies were counted after incubation for up to 10 days. To determine the total viable cell input, the remaining 100 µL of each culture was serially diluted and plated on antibiotic-free m7H10. Mutation rates were calculated using the Lea-Coulson method: m/Nt, where m is number of resistant colonies, and Nt is the total number of input cells91.
Co-culture competition assay
The plasmids pTE-OX-GCT5 and pGMEH-p38-mRFP (Addgene) were transformed into wildtype M. smegmatis and ItreSSM, respectively, to generate wildtype::GFP and ItreSSM::RFP strains. Using these strains, we evaluated their relative viability following cyclic exposure to bactericidal concentrations of RIF or D-cycloserine (DCS). Briefly, equal volumes of mid-log phase cultures of wildtype::GFP and ItreSSM::RFP, each at an OD595 of ~0.7, were mixed to create the initial untreated culture (G0). This mixture was then treated with 100 µg/mL RIF or DCS for 1 day, followed by washing with PBS and resuspension in antibiotic-free m7H9. The resulting culture, adjusted to an OD595 of 0.05, was allowed to grow until reaching an OD595 of ~0.7, referred to as G1 subculture. This cycle of treatment, washing, and regrowth was repeated sequentially until G5. Flow cytometry was employed to determine the relative abundance of wildtype::GFP and ItreSSM::RFP during each generation (G0 to G5), allowing to track changes in strain viability over successive cycles of antibiotic exposure.
Spot assay
Cultures of FluxRIF and FluxINH at mid-log phase were diluted to an OD595 of 0.1. Five 10-fold serial dilutions were then prepared from each culture, and 2 µL of each dilution were spotted onto m7H10 containing various concentrations of antibiotics: 8–100 µg/mL RIF, 4–32 µg/mL INH, or 0.0075–0.03 µg/mL BDQ. The plates were incubated at 37 °C until colonies became visible.
Whole genome sequencing of RIF resistant M. smegmatis
Genomic DNA was extracted from FluxRIF #1-#10 bacilli and sequenced using the Illumina HiSeq platform (Novogen Corp). To ensure high-quality reads, raw sequences were trimmed with Trimmomatic (v0.39.2)92 using the parameters “SLIDINGWINDOW:4:20 MINLEN:50.” Variant analysis across the ten samples was performed using the Snippy pipeline (v4.6.0, default options; https://github.com/tseemann/snippy). The M. smegmatis reference genome (accession: NC_008596.1) was downloaded in GenBank format from NCBI and used for alignment. The pipeline involved: I. Mapping high-quality reads to the reference genome with BWA-MEM (v0.7.17-r1188), II. Calling variants with FreeBayes (v1.3.6)93, III. Filtering variants with BCFtools (v1.18)94, and IV. Annotating variant effects with SnpEff (v5.0e)95. Given the stringent criteria of the Snippy pipeline in variant calling, we manually inspected the mapped bases at each position using SAMtools (v1.18) mpileup on the BAM files generated by BWA-MEM. The SnpEff results from all samples were organized by variant position and effect, then visualized using the ComplexUpset package (v1.3.3) in R. The reference bases and variant alleles were plotted with ggplot2 (v3.5.1), based on sequencing depth.
RRDR sequencing of lab-made RIF resistant M. smegmatis
The rpoB gene sequences of the FluxRIF mutants were compared to the reference rpoB gene sequences for wildtype M. smegmatis96. ApE plasmid-editing software was utilized to identify mutations. FluxRIF strains were streaked onto antibiotic-free LB agar plates and incubated overnight at 37 °C to isolate single colonies for sequencing. The RRDR (RIF Resistance Determining Region) of the rpoB gene was amplified and sequenced using Sanger Sequencing (Quintara Biosciences) using the following primers: Msmeg-rpoB-fwd (5’-gctgatccagaaccagatcc-3’) and Msmeg-rpoB-rev (5’-gatgacaccggtcttgtcg-3’).
Membrane bioenergetics—membrane potential, NAD/NADH, and energy charge
For membrane potential (ΔΨm) measurement, cultures were grown in m7H9 to mid-log phase and concentrated to an OD595 of ~1.0 in fresh m7H9. Cultures were stained with 15 µM DiOC2 and incubated at 37 °C for 40 min. Following incubation, cultures were washed with PBS to remove excess dye. As a positive control for membrane depolarization, one culture was treated with 5 µM of the carbonyl-cyanide 3-chlorophenylhydrazone (CCCP; Invitrogen), while PBS served as the vehicle control. The assay was performed in black, clear-bottom 96-well plates (Costar). Fluorescence was measured using a SpectraMax M4 spectrofluorimeter (Molecular Devices), recording green fluorescence (excitation 488 nm, emission 530 nm) and shifts to red fluorescence (excitation 488 nm, emission 610 nm). ΔΨm was calculated as the ratio of red to green fluorescence, with each condition measured in triplicate.
M. smegmatis-laden filters generated for the metabolomics profiling were used to measure intrabacterial ATP and NADH/NAD levels. ATP concentrations were determined using the BacTiter Glo Microbial Cell Viability Assay kit (Promega), following the manufacturer’s instructions. NAD and NADH levels were measured with the FluroNAD/NADH detection kit (Cell Technology), also according to the provided instructions. Bacterial metabolism was rapidly quenched by immersing the filters into the respective assay buffer or reagent, ensuring immediate stabilization of metabolic states. ADP and AMP levels were quantified using LC-MS metabolomics, with calibration curves generated from chemical standards spiked into homologous mycobacterial extracts to correct for matrix-associated ion suppression effects.
RMR-tre labeling and flow cytometry
RMR-tre was synthesized as previously described and characterized by nuclear magnetic resonance (NMR) spectroscopy53. Wildtype M. smegmatis, ItreSSM, pTreSSM, FluxRIF, or ItreSFlux cultures in mid-log phase were treated with 1X MIC of RIF for 1 day. Following treatment, cultures were stained with the fluorogenic dye RMR-tre at a final concentration of 10 µM and incubated at 37 °C for 1 h. After incubation, the cultures were analyzed using a flow cytometer (Attune NxT, Thermo Fisher Scientific). The reported values represent the gated cell fractions. Data were exported from the flow cytometer and analyzed using FlowJo software (BD Biosciences). Error bars indicate the standard deviation from biological replicates.
Mathematical modeling
The classical Luria-Delbrück fluctuation assay was adapted for this study (Fig. S6A). To generate single-lineage bacilli of M. smegmatis wildtype and ItreSSM, cultures were diluted to an OD595 of 0.00005 in 200 µL and dispensed into 96-well plates. Cultures were incubated until reaching an OD595 of 0.7–1.0. From 60 randomly selected wells, 100 µL was plated on m7H10 containing 100 µg/mL RIF, and the number of spontaneous RIF-resistant colonies was counted after 10 days. To estimate the total viable input cell number, the remaining 100 µL of each culture was serially diluted and plated on antibiotic-free m7H10. To infer the reversible switching rates between drug-sensitive and drug-tolerant states (Fig. 5A), we utilized recent mathematical models that relate fluctuation assay data to these rates60. The calculation is based on the fluctuation assay results, where single-cell clones are subjected to lethal stress, and the clone-to-clone variation in surviving bacilli is used to estimate switching rates. This method has successfully been applied to decipher reversible phenotypic transitions between drug-sensitive and drug-tolerant states in both bacterial pathogens and cancer cells61,97,98. The basic mathematical model encompasses the following ingredients:
-
Single cells exist in two phenotypic states: drug-sensitive and drug-tolerant, and reversibly switch between them with certain kinetic rates (Fig. 5A).
-
These transitions occur spontaneously prior to drug exposure.
-
Cells proliferate at a given rate, assumed equal in both states.
-
During division, both daughter cells inherit the mother’s cell state just before division.
A detailed formulation for this stochastic population dynamics model, which describes clonal expansion with state switching, has been recently reported60. Assuming colonies expand over \(T\) generations, the variability in the number of tolerant bacilli, measured by the squared coefficient of variation (standard deviation divided by the mean), \(C{V}_{{N}_{T}}^{2}\), is given by:
where \(T\) is the normalized expansion time (in bacterial doubling times), \({T}_{{on}}\) is the average duration (also normalized to bacterial doubling times) that a single bacillus spends in the drug-tolerant state, and \(f\) is the fraction of persisters59,60. Here, \(C{V}_{{N}_{T}}^{2}\) is the model-predicted squared coefficient of variation of the drug-tolerant population across clones. For a fixed \(f\), smaller \({T}_{{on}}\) (faster switching between cell states) yields reduced inter-clonal fluctuations, while larger \({T}_{{on}}\) enhances these fluctuations59,60.
During extended antibiotic exposure, each drug-tolerant bacillus irreversibly transitions to a drug-resistant state with a small probability \(p\ll 1\). Given the number of drug-tolerant bacilli \({N}_{T}\), the number of drug-resistant colonies \({N}_{R}\) follows a binomial distribution. The clone-to-clone variation in \({N}_{R}\) can be approximated as:
where \({\bar{N}}_{R}\) is the average number of drug-resistant colonies and \(C{V}_{{N}_{R}}\) denotes the coefficient of variation of \({N}_{R}\). Using equation (2), the coefficient of variation of \({N}_{R}\) allows estimation of \({{CV}}_{{N}_{T}}^{2}\):
Combining this with equation (1), we can estimate \({T}_{{on}}\). Applying this analysis to the fluctuation assay data for \({N}_{R}\) across 60 single-cell lineages, we obtained:
for the wildtype, where the \(\pm\) denoted the 95% confidence interval estimated by bootstrapping. Using this value in (1), with \(T=30\) (representing 30 bacterial divisions before plating on RIF-containing plates) and an average frequency of drug-tolerant persisters of \(f={10}^{-3}\), we estimate the transient heritability of the wildtype persister state as:
For ItreSSM, the number of colonies surviving antibiotic treatment was roughly six-fold lower than in wildtype (Figs. 2A, 5B right). Assuming the same mutation probability \(p\) for both genotypes, this reduction likely results from around six-fold decrease in persister-formation rate in ItreSSM following short-term treatment (Fig. 5B left), while \({T}_{{on}}\) remains unchanged. The fluctuation assay data for ItreSSM yields:
and, with \(f=\frac{{10}^{-3}}{6}\), the estimated \({T}_{{on}}\) from (1) is:
This represents approximately a 20% decrease compared to wildtype, suggesting that ItreSSM persisters are less stable and revert to a drug-sensitive state more rapidly. Overall, the primary reason for the six-fold reduction in drug-resistant colonies appears to be a corresponding decrease in persister formation rates in ItreSSM relative to wildtype.
Antibiotic permeability (RIF uptake)
M. smegmatis wildtype, FluxRIF, or FluxINH in mid-log phase were incubated with 1X MIC of RIF at 37 °C. Bacteria were harvested at 0, 2, 4, 24, and 48 h and CFUs were determined by serial dilution plating on m7H10. The cell-free supernatant was collected by filtering through a 0.22 µm filter. RIF was extracted by adding a precooled solution of LC-MS grade acetonitrile: methanol: H2O (40:40:20) −40 °C. RIF detection and quantification were performed by LC-MS, as previously described21,23,99. The intrabacterial RIF concentration was calculated as [RIF]drug only – [RIF]filtrate, with three biological replicates per group.
EtBr permeability assay
M. smegmatis wildtype and drug-resistant strains in mid-log phase were cultured in m7H9 until reaching an OD595 of 0.7. Cultures were centrifuged at 13,000 rpm for 3 min, the supernatant discarded, and the pellets washed with PBS. The OD595 was adjusted to 0.4, and glucose was added to a final concentration of 0.4%. Ethidium bromide (EtBr) was added at 8 mg/mL, and 100 µL aliquots were transferred into each well of black, clear-bottom 96-well plates (Costar). Fluorescence was measured every 60 s for 60 min using a SpectraMax M5 spectrofluorometer (excitation 530, emission 595 nm). This assay assesses EtBr uptake as an indicator of cell membrane permeability.
SDS susceptibility test
The susceptibilities of wildtype and FluxINH were evaluated following a method adapted from Banaei et al.100. Briefly, cultures in mid-log phase were diluted with growth medium to an O.D595 of 0.01 and incubated with 0.01% SDS in triplicate. At 0 and 8 h of incubation, CFUs were monitored by plating on m7H10 to assess viability.
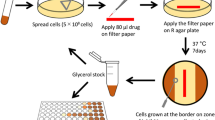
MPN (most probable number) assay to detect total persister bacilli
The MPN assay was performed as previously reported73. Briefly, HN878, H37Rv, Erdman, and CDC1551 strains in mid-log phase were grown in m7H9, harvested by centrifugation at 5000 rpm for 8 min, and washed twice with PBS. The cultures were then centrifuged at 800 rpm for 8 min to generate a single-cell suspension. The supernatant was transferred to a separate tube and diluted with PBS to achieve an OD595 of 0.1. CFU assay was used for the inoculum quantification. A total of 20 mL of the single-cell suspension was prepared for each condition, transferred to a vented flask, and incubated at 37 °C for 2 weeks. Starved cultures were split into two 8 mL cultures in vented flasks and treated with either 100 µg/mL RIF with or without 200 µM Validamycin A (ValA). These cultures were incubated at 37 °C for 5 days. After incubation, cultures were harvested by centrifugation at 5000 rpm for 8 min, washed with PBS, and resuspended in 1 mL PBS. For the MPN assay, 15 µL of this suspension was diluted into 135 µL of m7H9 in 96-well plates. Ten-fold serial dilutions were carried out across the wells, which were then incubated at 37 °C. OD595 was measured after 3 and 5 weeks to determine bacterial growth. The remaining culture from the assay was also used for CFU enumeration, with dilutions ranging from 10-5 and 10-1. ItreSHN and ItreSCDC, treated with or without ATc, were included in the assay.
Statistical analysis
All data were analyzed using Prism (v10.0; GraphPad Software). Statistical significance was determined using Student’s unpaired t-test with Welch’s correction or one-way ANOVA with Bonferroni post-test correction. P values less than 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this published article or its supplementary files, and available from the corresponding author upon request. The raw metabolomics datasets generated in this study have been deposited in the NIH metabolomics workbench repository under accession code ST003973. The whole genome sequencing datasets of FluxRIF #1-#10 have been deposited in the Genebank, NCBI repository under accession code PRJNA1273936. The Source data are provided with this paper.
References
WHO. Global Tuberculosis Report (WHO, 2024).
Nathan, C. Taming tuberculosis: a challenge for science and society. Cell Host Microbe 5, 220–224 (2009).
Nathan, C. Drug-resistant tuberculosis: a new shot on goal. Nat. Med. 20, 121–123 (2014).
Gold, B. & Nathan, C. Targeting phenotypically tolerant Mycobacterium tuberculosis. Microbiol. Spectr. 5, 10–1128 (2017).
Gold, B., Warrier, T. & Nathan, C. A multi-stress model for high-throughput screening against non-replicating Mycobacterium tuberculosis. Methods Mol. Biol. 1285, 293–315 (2015).
Rhee, K. Y. et al. Central carbon metabolism in Mycobacterium tuberculosis: an unexpected frontier. Trends Microbiol. 19, 307–314 (2011).
Schrader, S. M. et al. Multiform antimicrobial resistance from a metabolic mutation. Sci. Adv. 7, eabh2037 (2021).
Eoh, H., Liu, R., Lim, J., Lee, J. J. & Sell, P. Central carbon metabolism remodeling as a mechanism to develop drug tolerance and drug resistance in Mycobacterium tuberculosis. Front. Cell. Infect. Microbiol. 12, 958240 (2022).
Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 322, 107–131 (2008).
McDermott, W. Microbial persistence. Yale J. Biol. Med. 30, 257–291 (1958).
Zhang, Y., Yew, W. W. & Barer, M. R. Targeting persisters for tuberculosis control. Antimicrob. Agents Chemother. 56, 2223–2230 (2012).
Huemer, M., Mairpady Shambat, S., Brugger, S. D. & Zinkernagel, A. S. Antibiotic resistance and persistence-implications for human health and treatment perspectives. EMBO Rep. 21, e51034 (2020).
Lee, J. J. et al. Transient drug-tolerance and permanent drug-resistance rely on the trehalose-catalytic shift in Mycobacterium tuberculosis. Nat. Commun. 10, 2928 (2019).
Van den Bergh, B., Fauvart, M. & Michiels, J. Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol. Rev. 41, 219–251 (2017).
Vilcheze, C. et al. Enhanced respiration prevents drug tolerance and drug resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 114, 4495–4500 (2017).
Podlesek, Z. & Zgur Bertok, D. The DNA damage inducible SOS response is a key player in the generation of bacterial persister cells and population-wide tolerance. Front. Microbiol. 11, 1785 (2020).
Molina-Quiroz, R. C. et al. Cyclic AMP regulates bacterial persistence through repression of the oxidative stress response and SOS-dependent DNA repair in uropathogenic Escherichia coli. MBio 9, e02144-17 (2018).
Parbhoo, T., Mouton, J. M. & Sampson, S. L. Phenotypic adaptation of Mycobacterium tuberculosis to host-associated stressors that induce persister formation. Front. Cell Infect. Microbiol. 12, 956607 (2022).
Wang, X. & Wood, T. K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 77, 5577–5583 (2011).
Gerdes, K. & Maisonneuve, E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 66, 103–123 (2012).
Eoh, H. & Rhee, K. Y. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 110, 6554–6559 (2013).
Eoh, H. et al. Metabolic anticipation in Mycobacterium tuberculosis. Nat. Microbiol. 2, 17084 (2017).
Lim, J. et al. Phosphoenolpyruvate depletion mediates both growth arrest and drug tolerance of Mycobacterium tuberculosis in hypoxia. Proc. Natl. Acad. Sci. USA 118, e2105800118 (2021).
Quinonez, C. G. et al. The role of fatty acid metabolism in drug tolerance of Mycobacterium tuberculosis. MBio 13, e0355921 (2022).
de Carvalho, L. P. et al. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 17, 1122–1131 (2010).
Quinonez, C. G. et al. Fatty acid metabolism of Mycobacterium tuberculosis: a double-edged sword. Micro Cell 9, 123–125 (2022).
Borah, K. et al. Metabolic fluxes for nutritional flexibility of Mycobacterium tuberculosis. Mol. Syst. Biol. 17, e10280 (2021).
Mackenzie, J. S. et al. Bedaquiline reprograms central metabolism to reveal glycolytic vulnerability in Mycobacterium tuberculosis. Nat. Commun. 11, 6092 (2020).
Chakraborty, S. et al. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91 (2013).
Nandakumar, M., Nathan, C. & Rhee, K. Y. Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat. Commun. 5, 4306 (2014).
Wang, Z. et al. Mode-of-action profiling reveals glutamine synthetase as a collateral metabolic vulnerability of M. tuberculosis to bedaquiline. Proc. Natl. Acad. Sci. USA 116, 19646–19651 (2019).
Vega, N. M. & Gore, J. Collective antibiotic resistance: mechanisms and implications. Curr. Opin. Microbiol 21, 28–34 (2014).
Dewachter, L., Fauvart, M. & Michiels, J. Bacterial heterogeneity and antibiotic survival: understanding and combatting persistence and heteroresistance. Mol. Cell 76, 255–267 (2019).
Arepyeva, M. A. et al. A mathematical model for predicting the development of bacterial resistance based on the relationship between the level of antimicrobial resistance and the volume of antibiotic consumption. J. Glob. Antimicrob. Resist. 8, 148–156 (2017).
El Meouche, I. & Dunlop, M. J. Heterogeneity in efflux pump expression predisposes antibiotic-resistant cells to mutation. Science 362, 686–690 (2018).
Olivares Pacheco, J., Alvarez-Ortega, C., Alcalde Rico, M. & Martinez, J. L.Metabolic compensation of fitness costs is a general outcome for antibiotic-resistant pseudomonas aeruginosa mutants overexpressing efflux pumps. MBio 8, e00500–e00517 (2017).
Revitt-Mills, S. A. & Robinson, A. Antibiotic-induced mutagenesis: Under the Microscope. Front. Microbiol. 11, 585175 (2020).
Kalscheuer, R. & Koliwer-Brandl, H. Genetics of mycobacterial trehalose metabolism. Microbiol. Spectr. 2 (2014).
Kalera, K. et al. Targeting Mycobacterium tuberculosis Persistence through inhibition of the trehalose catalytic shift. ACS Infect. Dis. 10, 1391–1404 (2024).
Li, S. et al. CRISPRi chemical genetics and comparative genomics identify genes mediating drug potency in Mycobacterium tuberculosis. Nat. Microbiol 7, 766–779 (2022).
Rock, J. Tuberculosis drug discovery in the CRISPR era. PLOS Pathog. 15, e1007975 (2019).
Dwyer, D. J., Kohanski, M. A. & Collins, J. J. Role of reactive oxygen species in antibiotic action and resistance. Curr. Opin. Microbiol. 12, 482–489 (2009).
Kaufmann, B. B. & Hung, D. T. The fast track to multidrug resistance. Mol. Cell 37, 297–298 (2010).
Zheng, Q. A new practical guide to the Luria-Delbrück protocol. Mutat. Res. 781, 7–13 (2015).
Ford, C. B. et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat. Genet. 45, 784–790 (2013).
Goldstein, B. P. Resistance to rifampicin: a review. J. Antibiot.67, 625–630 (2014).
Li, M. C. et al. rpoB mutations and effects on rifampin resistance in mycobacterium tuberculosis. Infect. Drug Resist. 14, 4119–4128 (2021).
Miotto, P., Cabibbe, A. M., Borroni, E., Degano, M. & Cirillo, D. M. Role of disputed mutations in the rpob gene in interpretation of automated liquid mgit culture results for rifampin susceptibility testing of mycobacterium tuberculosis. J. Clin. Microbiol. 56, e01599–17 (2018).
Dhar, N. & McKinney, J. D. Microbial phenotypic heterogeneity and antibiotic tolerance. Curr. Opin. Microbiol. 10, 30–38 (2007).
Sanchez-Romero, M. A. & Casadesus, J. Contribution of phenotypic heterogeneity to adaptive antibiotic resistance. Proc. Natl. Acad. Sci. USA 111, 355–360 (2014).
Avery, S. V. Microbial cell individuality and the underlying sources of heterogeneity. Nat. Rev. Microbiol. 4, 577–587 (2006).
Safi, H. et al. Phase variation in Mycobacterium tuberculosis glpK produces transiently heritable drug tolerance. Proc. Natl. Acad. Sci. USA 116, 19665–19674 (2019).
Banahene, N. et al. A far-red molecular rotor fluorogenic trehalose probe for live mycobacteria detection and drug-susceptibility testing. Angew. Chem. Int. Ed. Engl. 62, e202213563 (2023).
Backus, K. M. et al. The three Mycobacterium tuberculosis antigen 85 isoforms have unique substrates and activities determined by non-active site regions. J. Biol. Chem. 289, 25041–25053 (2014).
Takayama, K., Wang, C. & Besra, G. S. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 18, 81–101 (2005).
Tahlan, K. et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 1797–1809 (2012).
Shaffer, S. M. et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017).
Chang, C. A. et al. Ontogeny and vulnerabilities of drug-tolerant persisters in HER2+ breast cancer. Cancer Discov. 12, 1022–1045 (2022).
Shaffer, S. M. et al. Memory sequencing reveals heritable single-cell gene expression programs associated with distinct cellular behaviors. Cell 182, 947–959.e917 (2020).
Singh, A. & Saint-Antoine, M. Probing transient memory of cellular states using single-cell lineages. Front. Microbiol. 13, 1050516 (2022).
Hossain, T., Singh, A. & Butzin, N. C. Escherichia coli cells are primed for survival before lethal antibiotic stress. Microbiol. Spectr. 11, e0121923 (2023).
Saint-Antoine, M. G. R. S. A. A fluctuation-based approach to infer kinetics and topology of cell-state switching. bioRxiv https://doi.org/10.1101/2022.03.30.486492 (2022).
Davies, J. & Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74, 417–433 (2010).
Levin-Reisman, I. et al. Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830 (2017).
Lewis, K. & Shan, Y. Why tolerance invites resistance. Science 355, 796 (2017).
Modi, S. R., Lee, H. H., Spina, C. S. & Collins, J. J. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 499, 219–222 (2013).
Munita, J. M. & Arias, C. A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 4:10.1128 (2016).
Su, W. J., Feng, J. Y., Huang, C. C. & Perng, R. P. Increasing drug resistance of Mycobacterium tuberculosis isolates in a medical center in northern Taiwan. J. Formos. Med. Assoc. 107, 259–264 (2008).
Lindsey, J. A. et al. Rifampin mono-resistant tuberculosis in New York City, 2010-2021: a retrospective case series. Open Forum Infect. Dis. 10, ofad534 (2023).
Rawat, R., Whitty, A. & Tonge, P. J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 100, 13881–13886 (2003).
Merker, M. et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet. 47, 242–249 (2015).
Saito, K. et al. Oxidative damage and delayed replication allow viable Mycobacterium tuberculosis to go undetected. Sci. Transl. Med. 13, eabg2612 (2021).
Saito, K. et al. Rifamycin action on RNA polymerase in antibiotic-tolerant Mycobacterium tuberculosis results in differentially detectable populations. Proc. Natl. Acad. Sci. USA 114, E4832–E4840 (2017).
Cohen, N. R., Lobritz, M. A. & Collins, J. J. Microbial persistence and the road to drug resistance. Cell Host Microbe 13, 632–642 (2013).
Schrader, S. M., Vaubourgeix, J. & Nathan, C. Biology of antimicrobial resistance and approaches to combat it. Sci. Transl. Med.12, eaaz6992 (2020).
Gomez, J. E. & McKinney, J. D. M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis 84, 29–44 (2004).
Allison, K. R., Brynildsen, M. P. & Collins, J. J. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473, 216–220 (2011).
Amato, S. M., Orman, M. A. & Brynildsen, M. P. Metabolic control of persister formation in Escherichia coli. Mol. Cell 50, 475–487 (2013).
Lewis, K. Persister cells. Annu. Rev. Microbiol. 64, 357–372 (2010).
Singh, R., Ray, P., Das, A. & Sharma, M. Role of persisters and small-colony variants in antibiotic resistance of planktonic and biofilm-associated Staphylococcus aureus: an in vitro study. J. Med. Microbiol. 58, 1067–1073 (2009).
Boldrin, F., Provvedi, R., Cioetto Mazzabo, L., Segafreddo, G. & Manganelli, R. Tolerance and Persistence to Drugs: A Main Challenge in the Fight Against Mycobacterium tuberculosis. Front. Microbiol. 11, 1924 (2020).
Brauner, A., Fridman, O., Gefen, O. & Balaban, N. Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320–330 (2016).
Windels, E. M., Michiels, J. E., Van den Bergh, B., Fauvart, M. & Michiels, J. Antibiotics: Combatting Tolerance To Stop Resistance. MBio 10, e02095–19 (2019).
Kohanski, M. A., Dwyer, D. J., Hayete, B., Lawrence, C. A. & Collins, J. J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810 (2007).
Yano, T. et al. Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: a pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 286, 10276–10287 (2011).
Windels, E. M. et al. Bacterial persistence promotes the evolution of antibiotic resistance by increasing survival and mutation rates. ISME J. 13, 1239–1251 (2019).
Ruhl, C. R. et al. Mycobacterium tuberculosis sulfolipid-1 activates nociceptive neurons and induces cough. Cell 181, 293–305.e211 (2020).
Vanaporn, M. & Titball, R. W. Trehalose and bacterial virulence. Virulence 11, 1192–1202 (2020).
Galagan, J. E. et al. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499, 178–183 (2013).
Band, V. I. et al. Antibiotic combinations that exploit heteroresistance to multiple drugs effectively control infection. Nat. Microbiol. 4, 1627–1635 (2019).
Foster, P. L. Methods for determining spontaneous mutation rates. Methods Enzymol. 409, 195–213 (2006).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Viehweger, A. et al. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 29, 1545–1554 (2019).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Ullah, I. et al. Rifampicin resistance mutations in the 81 bp RRDR of rpoB gene in Mycobacterium tuberculosis clinical isolates using Xpert MTB/RIF in Khyber Pakhtunkhwa, Pakistan: a retrospective study. BMC Infect. Dis. 16, 413 (2016).
Guha, M., Singh, A. & Butzin, N. C. Priestia megaterium cells are primed for surviving lethal doses of antibiotics and chemical stress. Commun. Biol. 8, 206 (2025).
Cross, T. et al. Prevalence and mechanisms of high-level carbapenem antibiotic tolerance in clinical isolates of Klebsiella pneumonia. Preprint at bioRxiv https://doi.org/10.1101/2025.02.19.639047 (2025).
Luna, B. et al. A nutrient-limited screen unmasks rifabutin hyperactivity for extensively drug-resistant Acinetobacter baumannii. Nat. Microbiol. 5, 1134–1143 (2020).
Banaei, N. et al. Lipoprotein processing is essential for resistance of Mycobacterium tuberculosis to malachite green. Antimicrob. Agents Chemother. 53, 3799–3802 (2009).
Acknowledgements
We thank Jeremy Rock for providing materials and for his valuable consultation on CRISPRi-dCas9 mutagenesis. We also extend our gratitude to all members in the Hyungjin Eoh lab for their assistance and valuable discussion throughout the work. In particular, we thank Brandon Do and Rhoeun Lee for their help with preliminary experiments. This research was supported by the Donald E. & Delia B. Baxter Foundation, NIH R21AI139386, R01AI168088 (H.E.), NSF grant 2117338 (B.M.S.), and NIH R35GM148351 (A.S.). Fig 5A, and Supplementary Fig. 2A, 3B, and 6A were made using BioRender and are used in this manuscript on a CC-BY license.
Author information
Authors and Affiliations
Contributions
J.J.L., A.S., S. E., S. C., B.M.S., and H.E. designed research studies; J.J.L., D.H.S., S.K.L., S. D., G.Y.L., S.G., H.J.S., E.R.M.B., Z.E.T., A.S., and J.S.L. conducted experiments; J.J.L., G.Y.L., S.G., A.S., M.S.V.N, B.M.S., and H.E. analyzed data; J.S.L. provided materials; L.J.J., A.S., B.M.S., and H.E. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, J.J., Swanson, D.H., Lee, SK. et al. Trehalose catalytic shift inherently enhances phenotypic heterogeneity and multidrug resistance in Mycobacterium tuberculosis. Nat Commun 16, 6442 (2025). https://doi.org/10.1038/s41467-025-61703-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61703-3
