
Abstract
CD4+ T cells are crucial for protective immunity to intracellular pathogens. In addition to secreting cytokines, CD4+ T cells promote control of Mycobacterium tuberculosis infection through cognate interactions with macrophages, but the mechanism has been unclear. Here, we show that SLAMF1/CD150 is highly and uniquely induced in macrophages by antigen-specific interactions with CD4+ T cells. In macrophages, SLAMF1 enhances the generation of reactive oxygen species and restricts Mtb replication. Mtb-infection of mice promotes SLAMF1 expression specifically on infected macrophages, not uninfected bystanders. SLAMF1 expression depends on adaptive immunity and also autophagy. Moreover, Slamf1−/− mice have higher Mtb burden and more rapid disease progression than wild type mice. Using Slamf1fl/fl conditional knock-out mice, we show that in vivo Slamf1 is specifically required in macrophages to restrict mycobacterial growth and limit IL-1β production. In macaques, macrophage SLAMFI expression also correlates with T cell responses and protection. Combined, these data demonstrate that SLAMF1 is a marker of macrophage-T cells interactions, and it promotes protection against Mtb.
Similar content being viewed by others

Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is the leading cause of death from an infectious disease worldwide1. Mtb is able to establish infection and persist by evading innate and adaptive immune defenses. Mtb first infects alveolar macrophages (AMs), which fail initially to restrict Mtb growth. Over time, Mtb infects additional myeloid cell populations, including monocytes, a variety of non-alveolar macrophages, dendritic cells, and polymorphonuclear neutrophils (PMNs)2,3,4. CD4+ T cells are crucial for protective immunity against TB5,6,7, although the mechanisms by which they enhance bacterial control by macrophages is not fully understood8. Their ability to secrete interferon (IFN)-γ has long been implicated as a key factor9, however, IFN-γ responses do not correlate with host protection. Tumor necrosis factor (TNF) is another important cytokine that mediates protection against Mtb, but CD4+ T cells that lack both IFN-γ and TNF still confer protection10,11. A member of the TNF superfamily, CD153 (CD30L), and its ligand, CD30, are expressed on CD4+ T cells and contribute to protection by regulating multiple cytokines and chemokines11,12. Interestingly, previous work using mixed bone marrow chimeras showed that infected macrophages must express major histocompatibility complex (MHC) class II antigen (MHCII) to optimally restrict intracellular Mtb growth, even when other macrophages in the same mouse express MHCII13. More recent work demonstrates that MHCII is required in recruited macrophages for T-cell mediated control of infection14. Thus, in addition to secreting diffusible cytokines, CD4+ T cells have to directly recognize Mtb-infected macrophages to promote Mtb control. How macrophages respond to cognate CD4+ T cell interactions and how that contributes to protection is not well understood.
CD4+ T cells are important to prevent disseminated TB, and at the same time, Mtb employs a variety of mechanisms to undermine effective macrophage-CD4+ T cell interactions8,15. In well-formed granulomas of non-human primates, CD4+ T cells are restricted to the lymphocytic cuff, away from the central core of infected macrophages16. In addition, granulomas generate an immunosuppressive environment17. Mtb also impairs the ability of infected macrophages to present antigen to T cells18,19,20,21,22,23,24. Thus, approaches to improve effective interactions between Mtb-infected macrophages and CD4+ T cells might offer therapeutic benefits and might be critical to vaccine-mediated protection. However, strategies to enhance macrophage-T cell interactions are hindered by the inability to assess which macrophages have presented antigen to T cells in vivo. Here, we sought to identify factors regulated by macrophage-T cell contact that contribute to protection against Mtb. Using transcriptional profiling, we found that antigen-specific CD4+ T cells markedly impact gene expression in both infected and bystander macrophages. Our study identified Signaling Lymphocyte Activating Molecule (SLAM) family member (SLAM/SLAMF1/CD150) as a key molecule of interest in the context of macrophage- T cell interactions. SLAMF1 was the most highly T-cell induced cell surface molecule. Furthermore, SLAMF1 was the only gene expressed at substantially higher levels in infected macrophages compared to bystander macrophages, specifically when T cells were present, suggesting that SLAMF1 plays a unique role in the macrophage-T cell interface during TB infection.
SLAMF1 is a member of a large family of transmembrane receptors from the immunoglobulin superfamily. In most cases, SLAM receptors are thought to signal between hematopoietic cells through homotypic SLAM-SLAM interactions. Some family members, including SLAMF1, also interact directly with microbes. SLAM receptors function as activating or inhibitory receptors based on their associations with the SLAM-associated protein family of adaptors and other effector molecules25,26,27,28,29. SLAMF1 was initially identified as a lymphocyte activation molecule due to its upregulation in activated T cells, and monoclonal antibodies directed against SLAMF1 promote CD4+ T cell proliferation and IFN-γ production30,31,32, including in response to Mtb antigens33,34. Thus, SLAMF1 was considered a coreceptor that potentiates Th1 responses, although more recent data demonstrate that SLAMF1 can negatively regulate T cell responses35. Additionally, in myeloid cells, SLAMF1 serves as a receptor for measles virus36, and it can modulate responses to LPS and outer membrane proteins from gram-negative bacteria, promoting phagosome maturation, bacterial killing, and production of reactive oxygen, nitric oxide, IL-12, and TNF25,35,37,38,39. Here, we show that macrophage-CD4+ T cell interactions promote SLAMF1 expression on macrophages in mice and non-human primates (NHP). Moreover, we show that Slamf1 is required in myeloid cells to promote T-cell driven stress on the bacilli and to control bacterial replication in mice. Thus, our findings suggest that direct contact between macrophages and CD4+ T cells mediates protection to Mtb through SLAMF1.
Results
Antigen-specific CD4+ T cells shape macrophage gene expression
To better understand how antigen-specific T cells enhance protection against Mtb, we used RNAseq to examine the impact of T cells on macrophage gene expression. First, we treated bone marrow-derived macrophages (BMDMs) with IFN-γ to induce expression of MHCII, enabling them to effectively present antigen. Thereafter, we infected the macrophages with mCherry-expressing Mtb (H37Rv strain) or left them uninfected and added Th1-polarized CD4+ T cells (Fig. 1A). The CD4+ T cells were from T cell receptor (TcR) transgenic (Tg) mice and recognize peptide-25 (P25), an epitope from Mtb Antigen 85B40,41. The next day, we sorted macrophages for analysis by bulk RNAseq. We defined three different macrophage types: uninfected macrophages were from an uninfected sample; bystander (mCherry-) and infected (mCherry + ) macrophages were sorted from an infected sample (Fig. 1A). Principal component analysis separated macrophages based upon whether they came from an infected or uninfected sample and the presence/absence of T cells, whereas infected and bystander macrophages were clustered together (Fig. 1B).

A Graphical representation of experimental design. B PCA plot of RNAseq expression based on 12000 genes with the greatest variance. C–F Volcano plots of Gene set 1 (C), Gene set 2 (D), Gene set 3 (E), and Gene set 4 (F). Data points correspond to all of the DE genes with fold-change > 2 and FDR ≤ 0.1. P values are adjusted for multiple testing following the Benjamini-Hochberg procedure. (C–E) A subset of the top fifteen most highly upregulated genes are shown in red. A subset of IFN-regulated genes is shown in purple. F Selected genes are shown in red. G SLAMF1 normalized read counts from infected, bystander, and uninfected macrophages co-cultured with or without T cells, as indicated. (UI) Uninfected; (Inf) Infected; (Bys) Bystander. Data are presented as mean values ± SD. Each data point is a biological replicate. P values from Brown Forsythe and Welch ANOVA test with Dunnett T3 multiple comparison test. H Heat map of 25 genes that were significantly DE (FDR < 0.1) in the generalized linear model with ‘T cell x infection status’ interaction term and also upregulated by Mtb infection and CD4+ T cells. log2FC for the respective comparisons (A-D) is indicated. Empty, white boxes indicate no significant difference. I, J Slamf1 expression values in BAL macrophages from scRNAseq scaled gene expression. Data is represented as heat maps, with each line representing expression from a single macrophage. BAL were obtained from NHPs that were unvaccinated (Unvac) or vaccinated with BCG by aerosol (AE), high dose intradermal (IDhigh), low dose intradermal (IDlow), or intravenous routes (IV) as described42. BALs were obtained 13 weeks (I) and 25 weeks post-vaccination (J) and either unstimulated or stimulated with PPD prior to scRNAseq, as indicated. FDR comparing the IV vaccinated group to all other groups combined is shown. ns- not significant. A Created in BioRender. Philips, J. (https://BioRender.com/x9c1725). C–F Elements created in BioRender. Philips, J. (https://BioRender.com/we24srw). Source data are provided as a Source Data file.
We performed differential expression (DE) analysis on the macrophage populations by normalizing gene expression across all experimental conditions and using a statistical model with an interaction term to isolate specific comparisons (with or without T cells, infected or bystander cells, infected or uninfected sample; (Supplementary Fig. 1A, and Supplementary Data 1). To determine how macrophages respond to antigen-stimulated CD4+ T cells, we compared the infected macrophages co-cultured with CD4+ cells to: (1) infected macrophages without CD4+ T cells, and (2) uninfected macrophages with CD4+ T cells. The first comparison identified 4627 DEGs (Supplementary Data 1; Gene set 1; Fig. 1C), while the second comparison identified 4274 DEGs (Supplementary Data 1; Gene set 2; Fig. 1D) (fold change ≥2, FDR < 0.1). There were 3487 DEGs in common, of which 3458 were differentially regulated in the same direction (Supplementary Fig. 1B), which defined a set of Regulated by T cells and Infection (RTI) genes (Supplementary Data 1; RTI Gene set). Amongst the most differentially expressed genes was Slamf1 as well as genes with established roles in protection from Mtb, including IL1a, IL1b, and Nos2 (Fig. 1C–D). GSEA of the RTI gene set revealed terms involving positive regulation of cytokine production, cell chemotaxis, and response to molecule of bacterial origin. Specific signaling pathways that were enriched included STAT cascade (Supplementary Fig. 1C). The data revealed a dramatic alteration in the macrophage cell surface, as the most enriched cellular components terms were extracellular matrix, external side of the plasma membrane, receptor complex, and membrane region (Supplementary Fig. 1C). The DEGs included many receptors implicated in innate and adaptive immunity including nine members of the TNF receptor superfamily (tnfrsf), seven c-type lectin receptors, co-stimulatory receptors for T cells (Cd86, Cd80, Cd200), three formyl peptide receptors, four SLAM family of receptors (Slamf1, Slamf6, Slamf7, and Slamf9), and Fc receptor family members. Notably, Slamf1 was one of the most highly induced genes overall, increasing more than 100-fold in Mtb-infected macrophages in the presence of CD4+ T cells (Fig. 1C, D), and it was the most highly induced cell surface molecule. Using qRT-PCR, we verified that Slamf1 expression was upregulated when Mtb-infected macrophages were co-cultured with CD4+ T cells (Supplementary Fig. 1D).
We also analyzed the impact of T cells on bystander cells and found that bystanders were impacted much like the infected cells. There were 4772 DEGs between bystanders co-cultured with CD4+ T cells compared to those without CD4+ T cells (Supplementary Data 1; Gene set 3; Fig. 1E). The overlap between the expression changes induced by T cells in infected and bystander macrophages was ~80%, with the direction and magnitude highly correlated (r = 0.97, p < 0.01) (Supplementary Fig. 1B). For example, although we pre-treated the macrophages with IFN-γ prior to infection, in both infected and bystander macrophages, the addition of T cells resulted in further induction of IFN-γ−regulated genes, such as Gbp4, Socs1, Irf1, and Ido1 (Fig. 1C–E). However, there was no significant difference in their induction between infected and bystander cells (Supplementary Data 1; Gene set 4), reflecting that infected and bystander macrophages both experience IFN-γ made by the T cells. Only 82 genes were differentially expressed between infected and bystander cells when T cells were present (Supplementary Data 1; Gene set 4; Fig. 1F), while there were 286 DEGs when T cells were absent (Supplementary Data 1; Gene set 5; Supplementary Fig. 1E). Slamf1 was significantly induced in infected macrophages compared to bystanders only when T cells were present (Fig. 1F–G). Additionally, when we used a generalized linear model with a ‘T cell x infection status’ interaction term, only 25 genes were induced by the combination of Mtb infection and CD4+ T cells (Fig. 1H). Of these, only 2 genes were more highly expressed in infected macrophages than bystanders specifically when T cells were present (Slamf1 and Rabgap1l) (Fig. 1H), with Slamf1 being much more markedly upregulated than Rabgap1l. To conclude, antigen-specific CD4+ T cells dramatically impact gene expression of both infected and bystander macrophages in a similar manner. Unexpectedly, SLAMF1 uniquely stood out in our analysis: it was not upregulated by infection alone; it was the most highly T-cell induced cell surface molecule; and it was the only gene expressed at substantially higher levels in infected macrophages compared to bystanders, specifically when T cells were present (Fig. 1F–H), suggesting that it plays a unique role in macrophage-T cell interactions.
Macrophage SLAMF1 expression is regulated by the combination of antigen and T cells in NHPs
To determine whether SLAMF1 expression was regulated in macrophages by T cell interactions in non-human primates (NHPs), we analyzed data from two published studies. The first compared macaques that had been vaccinated with BCG intradermally, intravenously (IV), or by aerosol42. The IV-BCG-vaccinated NHPs were markedly protected from Mtb challenge. In this study, thirteen- and 25-weeks post-vaccination, investigators obtained bronchoalveolar lavage (BAL) from NHPs. The BAL samples were either stimulated overnight with mycobacterial antigens (purified protein derivative; PPD) or left unstimulated and subsequently analyzed using single cell RNAseq (scRNAseq). We examined Slamf1 expression in macrophages in the scRNAseq data. We found that macrophages from animals in the IV vaccinated group had significantly higher Slamf1 expression (both in terms of average fold-change in expression and percentage of cells in which Slamf1 was detected) than macrophages from animals in the other arms of the study. Slamf1 expression was enhanced in the macrophages from IV vaccinated animals treated with PPD, but not the unstimulated samples (Fig. 1I–J). A major difference in the BALs from the IV vaccinated animals was significantly more CD4+ T cells compared to other arms of the study42. Thus, Slamf1 upregulation in BAL macrophages required antigen and correlated with the presence of abundant T cells, similar to the findings in our RNAseq analysis of murine macrophages. The second study we analyzed isolated granulomas from Mtb-infected NHPs. In that study, they compared granulomas that favored bacterial persistence from those that drove bacterial control using scRNAseq43. The protective granulomas were associated with numerous T cell populations. There were nine different macrophage subclusters identified (Mac1-9), and the Mac7 subcluster was associated with low-bacterial burden granulomas. We found that Mac7 was the only subcluster enriched for Slamf1 expression. To determine if our RTI gene signature was significantly enriched in particular Mac subclusters, we converted the RTI gene set to Macaca fascicularis orthologs and performed GSEA with affinity redundancy reduction44. The Mac7 gene signature was the most positively enriched for the RTI gene signature (Supplementary Fig. 1F–G), suggesting that the Mac7 macrophages may have had direct macrophage-T cell interactions. Thus, in two studies from NHPs, elevated macrophage Slamf1 expression was correlated with the presence of abundant T cells. In both cases, SLAMF1 expression was also associated with protective responses, suggesting that SLAMF1 might be involved in the mechanism by which T cells enhance macrophage control of infection.
SLAMF1 is induced in macrophages by CD4+ T cell contact
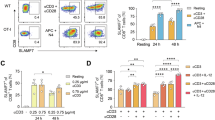
Because SLAMF1 is a cell surface receptor, we examined SLAMF1 surface expression on macrophages co-cultured with antigen specific CD4+ T cells by flow cytometry. We used F4/80 and CD4 to distinguish macrophages from CD4+ T cells (Supplementary Fig. 2A), and we examined the mean fluorescence intensity (MFI) of SLAMF1 on macrophages. We found no impact of IFN-γ treatment alone on SLAMF1 expression (Supplementary Fig. 2B). In the IFN-γ treated macrophages, SLAMF1 surface expression increased slightly with Mtb-infection or CD4+ T cell co-culture alone, and significantly more by the combination of both Mtb infection and CD4+ T cell co-culture (Fig. 2A). To determine whether infection was necessary or if antigen alone was sufficient to induce SLAMF1 expression, we treated the macrophages with the P25 peptide (Ag85b240-254), which contains the epitope recognized by the transgenic CD4+ T cells (Fig. 2B). We found that SLAMF1 surface expression increased significantly by the combination of P25 treatment and co-culture with T cells. SLAMF1 expression was also induced when we co-cultured macrophages with CD4+ T cells that recognize the Mtb protein, ESAT-6, or Ovalbumin, along with the cognate peptide (Fig. 2C, and Supplementary Fig. 2C). Thus, SLAMF1 is upregulated on the surface of macrophages when they present antigen to CD4+ T cells, and mycobacterial pathogen associated molecular patterns (PAMPs) are not essential for the upregulation. When we used macrophages that lacked MHCII, SLAMF1 was not induced (Fig. 2D).

A–I SLAMF1 expression was assessed by flow cytometry in BMDMs treated with IFN-γ, followed by infection with Mtb (A), treatment with P25 peptide (B, D, E, G, I), or ESAT-6 peptide (C), and subsequent co-culture for 24hrs with CD4+ T cells specific for P25 (A, B, D, E, G, I) or ESAT-6 (C). A, B Offset histograms show SLAMF1 fluorescence and corresponding SLAMF1 expression graphs. Data in the offset histograms are normalized to mode. A–G, I SLAMF1 MFI is expressed as the percent increase relative to IFN-γ-treated cells (without infection, peptide, or T cells). A n = 7 biological replicates; (B, C) n = 5 biological replicates from 3–5 independent experiments; A–C P values from matched one-way ANOVA with Holm-Sidak’s multiple comparison test. D SLAMF1 expression was compared in WT and MhcII−/− BMDMs. n = 7 biological replicates from 4 independent experiments. P value from Two-way ANOVA. E SLAMF1 expression in BMDMs after co-culture with WT or Ifng−/− P25 TcR Tg CD4+ T cells. n = 7 biological replicates from 4 independent experiments. P value from Two-way ANOVA. F BMDMs were grown in transwells in the indicated configuration, treated with P25 peptide, and co-cultured with CD4+ T cells and analyzed by flow cytometry. F, G Macrophages that contacted CD4+ T cells were compared to cells that did not make physical contact. n = 5 biological replicates from 5 independent experiments. P value from matched one-way ANOVA with Holm-Sidak’s multiple comparison test. H, I Macrophages that contacted CD4+ T cells were compared to macrophages grown in the same well without contact (conditioned). n = 5 biological replicates from 4 independent experiments, P value from matched one-way ANOVA with Holm-Sidak’s multiple comparison test. (A–E, G, I) Data are presented as mean values ± SEM. F, H Created in BioRender. Philips, J. (https://BioRender.com/w8kz9zz); Philips, J. (https://BioRender.com/negeejn). Source data are provided as a Source Data file.
In our experimental set-up, we pre-treated macrophages with IFN-γ because it upregulates MHCII expression. When we used BMDMs that lacked the IFN-γ receptor (Ifngr1−/−), they had both reduced MHCII expression and impaired SLAMF1 induction (Supplementary Fig. 2D–E). Although we pre-treated the macrophages with IFN-γ, it is possible that additional IFN-γ from the T cells contributes to SLAMF1 induction. To test the role T cell derived IFN-γ, we compared SLAMF1 expression on macrophages co-cultured with P25-specific, IFN-γ-deficient T cells to those that produce IFN-γ. We found no significant difference in macrophage SLAMF1 expression based upon the ability of the T cells to make IFN-γ (Fig. 2E). To conclude, IFN-γ alone is not sufficient for SLAMF1 induction; nor is it required from the T cells if the macrophages have already experienced IFN-γ, suggesting the involvement of an additional signal from the T cells. To specifically evaluate the impact of T cell contact on SLAMF1 expression, we turned to transwell assays using inserts with 1 μm pores through which cells physically interact and soluble factors are exchanged (Fig. 2F–I). In the contact setup, we cultured P25-treated macrophages on the underside of the insert, then inverted the insert, and added CD4+ T cells to the top, which allowed direct contact between cells (Fig. 2F). In the “no-contact” setup, macrophages were cultured on top of the insert, and CD4+ T cells were added to the bottom chamber (Fig. 2F). We found that SLAMF1 surface expression was significantly enhanced in macrophages that were in direct contact with the CD4+ T cells compared to the macrophages that lacked contact (Fig. 2G). Finally, in a third configuration, macrophages were cultured on top of the insert, and macrophages and CD4+ T cells were added to the bottom chamber where they could directly interact (Fig. 2H). In this set up, macrophages grown on the insert were exposed to cytokines secreted from macrophages and T cells below, but they did not have direct contact with CD4+ T cells and, therefore, mimicked bystander macrophages. We observed that the macrophages experiencing conditioned media did not induce SLAMF1 expression (Fig. 2I). Thus, SLAMF1 induction occurs when macrophages directly interact with antigen-specific CD4+ T cells. There could be soluble factors that contribute to SLAMF1 upregulation, but our data suggest that those factors are insufficient and direct cell-cell contact is required.
SLAMF1 is more highly expressed on infected macrophages than bystanders in vivo
To examine the expression pattern of SLAMF1 during Mtb infection in vivo, we infected WT mice with ~200 CFU of mCherry-expressing Mtb by aerosol. We analyzed lung immune cell populations at four weeks post-infection (wpi) using flow cytometry and published gating strategies (Supplementary Fig. 3A–B)45. We identified three macrophage subsets: AMs (Siglec-F+ CD11c+) and two different non-AM types (Siglec-F-) that differed in their expression of CD11c (CD64+ CD11b+ MHCIIvar CD11c- Ly6C- and CD64+ CD11b+ MHCIIhigh CD11c+). These non-AMs include tissue resident, interstitial macrophages and monocyte-derived macrophages, and we refer to them as CD11c- and CD11c+ non-AMs (or collectively as non-AMs). When we compared SLAMF1 expression in infected and uninfected bystander cells at 4 wpi, we found a significantly higher fraction of infected monocytes and non-AMs expressed SLAMF1 compared to uninfected bystanders from the same mice (Fig. 3A; and Supplementary Fig. 3C–D). In addition, for the monocytes and CD11c- non-AMs, which vary in their expression of MHCII, a greater percentage of MHCIIhigh cells expressed SLAMF1 compared to the MHCIIlow cells (Fig. 3B). As previously described2,45, at 4 wpi there were few infected AMs, making them difficult to assess. When we infected with a higher dose of Mtb ( ~ 1000 CFU), there were more infected AMs, and, as in the non-AMs, a higher fraction of infected AMs expressed SLAMF1 compared with uninfected AMs (Fig. 3C). The correlation between MHCII expression and SLAMF1 expression was also apparent at high dose in AMs, monocytes, and non-AMs (Fig. 3D). The MFI of SLAMF1+ cells was also significantly higher in infected AMs and non-AMs, compared to bystanders in the same mice (Fig. 3E). To conclude, SLAMF1 expression in vivo was higher in Mtb-infected monocytes and macrophages than uninfected bystander cells in the same mice, and its expression correlated with MHCII expression.

A–E C57Bl/6 mice were infected with ~200 CFU (A, B) or ~ 1000 CFU (C–E) of mCherry Mtb for 4 weeks. A, C SLAMF1 positivity on infected (mCherrypos) and uninfected (mCherryneg) macrophages and monocytes was analyzed by flow cytometry. B, D SLAMF1 positivity of macrophages and monocytes from infected mice separated by MHCII status. A, B n = 19 mice from 6 independent experiments. A **** p = 4 × 10−15, 1.4 × 10−11, and 3.5 × 10−7 for pairwise comparisons for monocytes, CD11c- non-AMs and CD11c+ non-AMs, respectively. B ****p = 9 × 10−10 and 7 × 10−9 for pairwise comparison for monocytes and CD11c- non-AMs, respectively. A, B P value from matched Two-way ANOVA with Sidak’s multiple comparisons test. C, D n = 17 mice from 4 independent experiments. C **** p = 3 ×10−6; 1.2 ×10−5, and 2 ×10−6 for pairwise comparisons for monocytes, CD11c+ non-AMs, and AMs, respectively. C, D P value from matched Two-way ANOVA. E MFI of SLAMF1 in infected macrophages and monocytes compared to uninfected cells from the same mice. n = 22 mice from 5 independent experiments. P value from non-parametric multiple Wilcoxon t tests (F, G) C57Bl/6 mice were infected with ~150 CFU Mtb for 2, 4, and 8 weeks. F Representative lung SLAMF1 immunohistochemistry with higher magnification inset at 8 weeks post-infection. Quantitative image analysis showing weak (yellow), moderate (orange), and strong (red) SLAMF1 intensity. Hematoxylin staining is in blue. The arrow indicates high expression in macrophages adjacent to lymphocyte aggregate. Scale bar = 500 μm, inset enlarged scale bar = 100 μm (G) Total number of SLAMF1+ lung cells quantified by digital image analysis. n = 5 mice from one experiment, P value from Brown-Forsythe and Welch One-way ANOVA with Dunnett’s multiple comparison test. Data presented as mean values ± SEM. A–E Each data point represents one mouse; lines connect data from the same mouse. Source data are provided as a Source Data file.
SLAMF1 expression on infected macrophages depends upon T cells and autophagy
To further examine SLAMF1 expression in vivo, we stained lungs for SLAMF1 at 2, 4, and 8 weeks post-infection (wpi) by immunohistochemistry and performed quantitative image analysis. Infected lungs from Slamf1−/− mice were negative controls. SLAMF1 staining increased over time (Fig. 3F–G; and Supplementary Fig. 3E), and regions of interstitial expansion by macrophages and alveolitis had the highest SLAMF1 expression (Fig. 3F). Foci of high expression within macrophages were observed near lymphocyte clusters in well-defined regions of granulomatous inflammation (Fig. 3F). To evaluate whether SLAMF1 induction in macrophages depends upon T cells, we infected Rag1−/− mice, which lack B cells and T cells, and Tcra−/− mice, which lack CD4+ and CD8+ T cells. SLAMF1 induction in Mtb-infected monocytes and non-AMs was significantly reduced in Rag1−/− and Tcra−/− mice compared to WT mice (Fig. 4A–C). Importantly, despite the high level of Mtb infection in these mice (Supplementary Fig. 4A), SLAMF1 expression remained low. In addition, in Rag1−/− mice SLAMF1 expression was significantly lower in MHCIIhigh monocytes and CD11c- non-AMs compared to WT mice (Fig. 4D–E). Tcra−/− mice also had significantly lower SLAMF1 positivity in MHCIIhigh CD11c- non-AMs compared to WT mice (Fig. 4E). Next, we adoptively transferred naïve T cells from WT mice into Tcra−/− mice 6 days post-infection (dpi) and evaluated SLAMF1 expression 4 weeks post-T cell transfer. T cell transfer increased SLAMF1 expression on Mtb-infected monocytes and non-AMs (Fig. 4F–H; and Supplementary Fig. 4B). T cell transfer also increased the proportion of SLAMF1+ cells in MHCIIhigh cells relative to their MHCIIlow counterparts (Fig. 4I–J). Thus, cell surface expression of SLAMF1 in monocytes and macrophages depends not only upon infection but also on adaptive immunity and αβ T cells. Combined with our ex vivo studies, we conclude that macrophage SLAMF1 expression is driven by direct T cell interactions.

A–E WT, Rag1−/−, and Tcra−/− mice were infected with ~100 CFU mCherry Mtb for 4 weeks. A–C SLAMF1 positivity on infected and uninfected monocytes and non-AMs is shown. P value from two-way ANOVA Mixed-effects model with Tukey’s multiple comparisons test. D, E SLAMF1 positivity of infected and uninfected monocytes and CD11c- non-AMs separated by MHCII status. P value from two-way ANOVA with Dunnett test. (A-E) n = 9 mice in each group. Data from 3 independent experiments. F–H Tcra−/− mice were infected with ~100 CFU mCherry Mtb, and naïve T cells from WT mice were transferred 6 dpi. Lungs were harvested 5 wpi; F–H SLAMF1 positivity in Mtb-infected and uninfected monocytes and non-AMs. (I, J) SLAMF1 positivity of infected and uninfected monocytes and CD11c- non-AMs separated by MHCII status. F–J Tcra−/− n = 9 mice; Tcra−/− + WT/T cells n = 10 mice. Data is from 2 independent experiments. P values from two-way ANOVA. K, L Atg5fl/fl, Atg7fl/fl, Atg14fl/fl Cre + , and Cre- littermate controls infected with ~1000 CFU GFP-Mtb for 17 days. K SLAMF1 positivity in Mtb-infected (GFPpos) and uninfected (GFPneg) monocytes and macrophages from Atg Cre- mice. n = 15 mice from 4 independent experiments. P value from two-way ANOVA with Sidak multiple comparisons test. Each data point represents one mouse; lines connect data from the same mouse. L SLAMF1 positivity in Mtb-infected and uninfected monocytes and macrophages from Cre- and Cre+ mice, as indicated. Cre- mice (Atg5fl/fl, Atg7fl/fl, Atg14fl/fl) were combined for the analysis. Cre- n = 15 mice; ATG5 cKO n = 10 mice; ATG7 cKO n = 9 mice; ATG14 cKO n = 8 mice from 2–3 independent experiments. P value from two-way ANOVA with Dunnett multiple comparisons test. (A–J, L) Each data point represents one mouse. A–J, L Data presented as mean values ± SEM. Source data are provided as a Source Data file.
These findings suggested that SLAMF1 might be useful to assess macrophage- T cell interactions in vivo. Since autophagy is required for optimal MHCII antigen presentation46, including in the context of mycobacterial infection22,47, we examined mice that are defective in autophagy in myeloid cells (Atg7fl/fl LysM-Cre + , Atg5fl/fl LysM-Cre + , and Atg14fl/fl LysM-Cre+) and their Cre-littermate controls. We infected mice with GFP-expressing Mtb and examined lung cells at 17 dpi, shortly after antigen specific T cells arrived in the lung. In the Cre- animals, SLAMF1 expression was induced on infected monocytes and macrophages relative to bystanders as expected (Fig. 4K). In the Atg5, Atg7, and Atg14 Cre+ mice, there were significantly fewer SLAMF1+ CD11c+ non-AMs compared to controls (Fig. 4L). There was no significant impact of autophagy on SLAMF1 expression in AMs, monocytes, and CD11c− non-AMs (Supplementary Fig. 4C–E). This suggests that Atg-dependent processes are particularly important for antigen presentation by CD11c+ non-AMs during Mtb infection. The non-AM populations are a heterogenous population of cells that differ in lysosomal biogenesis, proteolytic activity, and MHCII expression4,48,49, all of which could lead to differences in antigen presentation and SLAMF1 expression. The role of autophagy in CD11c+ non-AMs fits our recent findings that autophagy-related processes appear to play less of a role in AMs than CD11c+ non-AMs early during infection45,50. Overall, we conclude that SLAMF1 expression in lung macrophages in vivo is driven by macrophage-T cell interactions and, therefore, its expression might be a useful in vivo indicator to assess such interactions, potentially reflecting differences in antigen presentation by cell type, host genetics and immune status, and bacterial strain.
SLAMF1 controls Mtb in the lungs
CD4+ T cells enhance the ability of macrophages to control Mtb. To establish whether SLAMF1 is important in protection against Mtb, we infected WT and Slamf1−/− mice with low dose ( ~ 100–300 CFU) and high dose ( ~ 1000 CFU) H37Rv Mtb and evaluated bacterial burden and inflammatory cell infiltrate. At the lower dose, we assessed the lungs at 2, 4, and 8 wpi by H&E. The lungs of WT and Slamf1−/− mice looked similar at 2 wpi, but by 4 and 8 weeks, there was notably more inflammatory infiltrate (Fig. 5A–B). At the lower dose, the bacterial burden was slightly higher in Slamf1−/− mice at 4 wpi, but the difference did not reach statistical significance over repeated experiments (Supplementary Fig. 5A). This is not unexpected because even CD4+ T cell depletion does not significantly impair host control at this early time point in the low dose model of infection with H37Rv51. High dose infection has been shown to impose different requirements for immune control in the mouse model52,53, and recent studies demonstrate that autophagy is required specifically during high dose infection for protection by promoting T cell responses54. Therefore, next we assessed whether the requirement for SLAMF1 was more pronounced during high dose infection. When we infected mice with ~1000 CFU of H37Rv Mtb, Slamf1−/− mice had significantly more Mtb in the lungs compared to WT mice 4 wpi (Fig. 5C). We had a similar finding when we infected mice with the Erdman Mtb strain (Supplementary Fig. 5B). Slamf1−/− mice also succumbed earlier than WT mice to infection with H37Rv and HN878 Mtb (Fig. 5D; and Supplementary Fig 5C), demonstrating that SLAMF1 confers protection against various Mtb strains. To further assess how SLAMF1 modulates immune responses, we analyzed lung cells by flow cytometry 4 wpi. We observed a significant difference between the overall myeloid cell distribution in the lungs of Slamf1−/− mice compared to WT mice (Fig. 5E; p < 0.0001, Kruskal-Wallis), with more monocytes, non-AMs and PMNs, although the pairwise comparisons did not all reach statistical significance (and we saw no difference in AMs and DCs; Fig. 5E; and Supplementary Fig. 5D). There were also overall significantly more infected monocytes, non-AMs and PMNs (p < 0.0001, Kruskal-Wallis), with significantly more infected neutrophils (Fig. 5F; and Supplementary Fig. 5E). The Slamf1−/− mice also had more lung CD4, CD8, γ/δ T cells and NK cells, with no difference in the number of B cells (Fig. 5G; and Supplementary Fig. 5F). We found no difference in the frequency of ESAT6-specific CD4+ T cells or TB10.4-specific CD8+ cells based on tetramer staining (Fig. 5H). We measured cytokine levels in lung homogenates and found that Slamf1−/− mice had significantly more IL-1β, a trend towards less TNF, and unchanged IFN-γ and IL-6 compared to WT mice (Fig. 5I). To conclude, Slamf1−/− mice have higher mycobacterial burden in the lungs despite the presence of more macrophages, more CD4+ and CD8+ T cells, and more IL-1β, consistent with reduced antimicrobial effector function and an ineffective inflammatory response.

A, B WT and Slamf1−/− mice were infected with ~150 CFU H37Rv Mtb. Representative lung histopathology at 2, 4, and 8 weeks post-infection (wpi). Slamf1−/− mice had prominent neutrophilic inflammation at 4 and 8 wpi. Scale bars = 200 μm. n = 5 mice per group. Data from one experiment. B Representative H&E and acid-fast staining at 4 and 8 wpi shows increased inflammation in Slamf1−/− as compared to WT mice. Scale bars = 20 μm (H&E); 10 μm (AFB). C–H WT and Slamf1−/− mice were infected with ~1000 CFU H37Rv Mtb. C Lung Mtb burden was assessed 4 wpi. n = 22 mice. P value from two-tailed nonparametric Mann-Whitney t test. Data presented as mean values ± SEM. Different colors indicate independent experiments. D Survival was monitored (n = 15 mice per group). P value from Kaplan-Meier simple survival analysis. E–G Lungs were harvested and analyzed by flow cytometry. E Total number of different myeloid cells in the lung. F Number of Mtb-infected myeloid subsets. G Number of lymphoid cells. H Total number and percentages of antigen-specific CD4+ and CD8+ T cells. I Cytokine levels in the lung homogenates. E–G, I Each data point represents one mouse. Data consists of 12 mice from 3 independent experiments. E **** p = 2 × 10−15, F **** p = 1.3 × 10−7, and G **** p = 7.5 × 10−13 comparing WT to Slamf1−/− using Kruskal-Wallis. E–G, I Pairwise comparisons used the multiple Mann-Whitney tests. H n = 10–18 mice. P value from Mann-Whitney t test. E–I Data presented as mean values ± SEM. Source data are provided as a Source Data file.
SLAMF1 is required in myeloid cells to promote Mtb control
SLAMF1 is expressed in a variety of hematopoietic cells; it was originally identified because it is induced on activated T cells30. Consistent with this, we found that there was a higher percentage of SLAMF1+ CD4+ and CD8+ T cells in the lungs of Mtb-infected compared to uninfected mice and constitutive expression in B cells (Supplementary Fig. 6A–B). Also in the NHP vaccine study by Darrah et al.42, the IV vaccinated animals had significantly higher expression of SLAMF1 in T cells compared to other groups, consistent with the strong Th1 response in these animals (Supplementary Fig. 6C). Thus, to distinguish whether SLAMF1 is required specifically in myeloid cells to enhance Mtb control, we generated Slamf1fl/fl mice by inserting loxP sites within intron 1 and intron 6 of the Slamf1 locus by CRISPR. We backcrossed them to C57Bl/6 LysM-Cre+ mice, which express the Cre recombinase in monocytes, macrophages, some dendritic cells, and neutrophils55, and infected with mCherry-expressing Mtb. We examined SLAMF1 surface expression by flow cytometry to assess loss of SLAMF1 in Mtb-infected myeloid cells. SLAMF1 expression was significantly reduced in infected non-AMs and PMNs in the Cre+ mice compared to the Cre- littermate controls. SLAMF1 was also reduced in monocytes and AMs in the Cre+ mice, but the differences were not statistically significant, perhaps reflecting inefficient excision of Slamf1 in those cells (Fig. 6A). As expected for a LysM-Cre strain, there was no impact of Cre status on Slamf1 expression in lymphoid cells (Supplementary Fig. 6D). Unlike the mice with germline deletion of Slamf1, which had more CD4, CD8, γ/δ T cells and NK cells in the lung, there were no significant differences in lymphocyte numbers in the cKO mice compared to their littermate controls, suggesting that those differences are due to a lymphocyte intrinsic role of SLAMF1 (Supplementary Fig. 6E). The Slamf1fl/fl LysM-Cre+ mice had no significant differences in the number of myeloid cells in the lung compared to Cre- controls (Supplementary Fig. 6F). However, the absence of Slamf1 in myeloid cells lead to increased bacterial burden in the lungs and significantly more infected monocytes, CD11c+ non-AMs, PMNs and a trend toward more highly infected CD11c- non AMs (Fig. 6B-C). Similar to the germline mutants, the cKO mice also had more IL-1β, less TNF, and no difference in IFN-γ and IL-6, demonstrating that the cytokine differences can be explained by a role of SLAMF1 in myeloid cells (Fig. 6D). We conclude that SLAMF1 is required in myeloid cells for optimal control of Mtb infection in myeloid cells.

A–D Slamf1fl/fl LysM-Cre+ and Slamf1fl/fl Cre- mice were infected with ~1000 CFU Mtb (mCherry-H37Rv), and lungs were harvested 4 wpi for flow cytometry, cytokine analysis, and CFU. (A) SLAMF1 positivity on infected myeloid cells. B Lung Mtb burden. C Number of Mtb-infected myeloid subsets. D Cytokine levels in the lung homogenates. A–D n = 17 mice in each group. Each data point represents one mouse. Data from 3 independent experiments. A **** p = 7.7 × 10−6 for pairwise comparison of CD11c+ non-AMs. C **** p = 1 × 10−15 comparing Cre- to Cre+ mice using Kruskal-Wallis. A–D P values from multiple Mann-Whitney t tests. E Fluorescence microscopy images showing bacteria (smyc’::mCherry-red), reporter (hspX::GFP-green), nuclei (DAPI-blue) and heatmap images of GFP intensity. BMDMs infected with reporter Mtb and co-cultured with or without T cells. Scale bars = 10 μm. F MFI of hspX::GFP signal for each bacterium measured from 10–15 images in each group from 2 independent experiments. Each data point is from one bacteria or a tightly clustered group of bacteria. **** p = 5 × 10−9. P values are from one-way ANOVA with Tukey multiple comparisons test. G Intracellular ROS production was compared between WT and Slamf1−/− BMDMs. Macrophages were co-cultured with or without T cells upon P25 treatment. Each data point represents one biological replicate. n = 5-6 biological replicates from 3 independent experiments. P values from two-way ANOVA. H, I WT or Slamf1−/− BMDMs were treated with IFN-γ, infected with H37Rv Mtb, and co-cultured with P25 TcR Tg T cells. CFU were calculated 4 hpi (day 0) and 3 dpi. I Samples were treated with or without NAC as indicated from five (H) or two (I) independent experiments, each with 6 replicates. P values from paired Student’s t test (H), One-way ANOVA (I). A–D, F–I, Data presented as mean values ± SEM. Source data are provided as a Source Data file.
Macrophage SLAMF1 imposes T-cell induced stress on the bacilli
Within macrophages, Mtb encounters various stresses including redox stress, nitric oxide (NO), and pH variations depending on the subcellular compartments in which Mtb localizes. Reporter Mtb strains serve as valuable tools for studying the dynamic changes that occur within the intracellular environment of macrophages, allowing for analysis of Mtb’s response to the host environment at the level of an individual bacterium. Previous studies with gram negative bacilli have demonstrated that upon recognizing outer membrane proteins, SLAMF1 is internalized and positively regulates NADPH oxidase activity, leading to ROS generation, phagosome maturation, and enhanced bacterial killing37,38. SLAMF1 has also been shown to regulate nitric oxide and TNF production35,56,57. To determine the role of SLAMF1 in modulating the intracellular environment experienced by the bacilli, we infected BMDMs from WT and Slamf1−/− mice with a reporter Mtb strain (hspX’::GFP, smyc’::mCherry Mtb). This reporter strain expresses GFP (controlled by hspX promoter) and mCherry (constitutively expressed by the smyc promoter)58. hspX is a component of the DosR regulon, an Mtb signaling pathway that responds to environmental stresses, including hypoxia, nitric oxide (NO), and carbon monoxide. Upon infection, we examined GFP fluorescence in the macrophages co-cultured with or without T cells. We found a significant increase in GFP expression in infected macrophages co-cultured with T cells compared to macrophages without T cells (Fig. 6E–F). This indicated that macrophages generate an intracellular environment that is unfavorable for Mtb upon interaction with T cells. Importantly, Mtb residing in Slamf1−/− macrophages exhibited significantly less GFP expression than Mtb within WT macrophages, particularly in the presence of T cells (Fig. 6E–F). Next, we examined whether SLAMF1 promoted NO or ROS production by macrophages. Using the Greiss reagent to measure nitrite, we found that NO production increased when macrophages were co-cultured with antigen specific T cells upon cognate peptide treatment or Mtb infection. However, there was no difference between WT and Slamf1−/− macrophages (Supplementary Fig. 6G–H). We examined ROS production in macrophages using the fluorescent indicator, CellRox, and flow cytometry. We found that Slamf1−/- macrophages elicited less ROS in response to peptide and T cells than the WT controls (Fig. 6G). The ability of macrophages to control Mtb replication was also lower in Slamf1−/− macrophages compared to controls (Fig. 6H). However, upon the addition of N-acetylcysteine (NAC), a ROS scavenger, bacterial growth was higher in wild-type macrophages, while no difference was observed in Slamf1−/− macrophages (Fig. 6I). Thus, SLAMF1 contributes to generating a stressful intracellular environment, elevated ROS, and enhanced microbial control by macrophages.
Discussion
A direct cognate (pMHCII-TCR) interaction between CD4+ T cells and infected macrophages appears to be important for the control of intracellular Mtb13,14, but the molecular details of how cognate interactions modulate macrophage functions have not been well described. We found that CD4+ T cells cause dramatic changes in the expression of macrophage cell surface proteins, including numerous receptors involved in innate and adaptive immunity, an effect that is seen in both infected and bystander macrophages. In our transcriptomic analysis, SLAMF1 stood out as a uniquely and significantly induced cell surface molecule. Using ex vivo and in vivo studies, we showed that SLAMF1 expression in macrophages is driven by the combination of infection (or antigen) and the presence of T cells, in both NHPs and mice. The notion that SLAMF1 is regulated by effective macrophage-T cells interactions is consistent with an observation previously made in people with leprosy: SLAMF1 expression is high in tuberculoid lesions, which have an effective cell mediated immune response, and low in lepromatous leprosy, characterized by ineffective cell mediated immunity59. Others have shown that SLAMF1 is induced by gram-negative bacilli, their outer membrane proteins, and/or LPS37,57,60. Reports also describe SLAMF1 induction in macrophages in response to Mtb lysate and BCG56,61. We found, using live Mtb, that SLAMF1 surface expression was markedly higher with the addition of antigen specific T cells. Importantly, in vivo Mtb infection alone did not significantly induce SLAMF1 expression on monocytes and macrophages in the absence of adaptive immunity. Altogether, our data strongly support the idea that during Mtb infection, SLAMF1 expression in macrophages is primarily upregulated by their interaction with T cells and, as such, may provide a useful marker for assessing effective macrophage- T cell interactions. Correspondingly, the failure of macrophages to induce SLAMF1 points to a defect in macrophage-T cell interactions, as we found in autophagy-deficient mice. The failure of robust T cell immunity in lepromatous leprosy may also explain the reduced SLAMF1 expression seen there. Importantly, a fundamental aspect of how Mtb subverts host immunity is by impairing macrophage- T cell interactions18,19,20,21,22,23,24. Thus, host directed therapies and vaccines that restore the functionality of the interaction are needed. However, there have not been tools to assess this in vivo. We suggest that SLAMF1 may serve as a guidepost in pre-clinical animal models where tissue macrophages can be readily accessed. In vaccinated or TB infected patients, its expression on BAL macrophages may be evidence of cognate T cell interactions.
CD4+ T cells exert diverse effects to enhance control of Mtb, including secreting cytokines, promoting myeloid cell recruitment, and enhancing CD8+ T cell functionality51,62,63,64. CD4+ T cell-derived IFN-γ is important for recruiting monocyte-derived macrophages to the site of infection, promoting their extravasation from the vasculature, and enhancing MHCII expression14,64,65,66. Thus, IFN-γ is important in establishing conditions in the lung to promote cognate interactions between macrophages and T cells. The direct contact between CD4+ T cells and macrophages is also essential for optimal control13,14. Our data suggest that macrophage SLAMF1 plays a role in mediating the response to direct control. We show that SLAMF1 is specifically required in myeloid cells for their control of Mtb, demonstrating that it plays a cell intrinsic role in mice. Ex vivo experiments in macrophages demonstrated that SLAMF1 generates a stressful intracellular environment for Mtb and contributes to ROS generation consistent with its known role in promoting NADPH oxidase activity and phagosome maturation37,38. Our findings are supported by the observations that SLAMF1 is associated intracellularly with mycobacterial antigens and promotes control of the vaccine strain, BCG56,61. Thus, we propose that macrophage- T cell interactions enhance Mtb control by driving SLAMF1 expression on macrophages and thereby enhancing their effector functions and antimycobacterial capacity. At the same time, Mtb has virulence factors that undermine macrophage ROS production and phagosome maturation67 potentially blunting SLAMF1 efficacy. In people living with HIV or those treated with cancer chemotherapy who have impaired CD4+ T cells, reduced SLAMF1 expression on infected macrophages might contribute to poor antimycobacterial control. Even in WT mice with normal T cell responses, we detect SLAMF1 surface expression in only ~50–80% of infected monocytes and macrophages. This suggests that SLAMF1-mediated immune control is not fully deployed during Mtb infection, which may be explained by the ability of Mtb to interfere with antigen presentation8,15. Since SLAMF1 is a homotypic cell surface receptor, it is amenable to activation by monoclonal antibodies and soluble SLAMF1. It will be important to determine whether targeting SLAMF1 has therapeutic benefit.
There are limitations to our study. We used Tcra−/− mice to show that T cells are required for SLAMF1 expression in vivo. Since Tcra−/− KO mice lack both CD4+ T cells and CD8+ T cells, both cell types may be able to drive SLAMF1 expression on macrophages in vivo. In addition, the stronger phenotype of Rag1−/− compared to Tcra−/−mice on SLAMF1 expression suggests that additional lymphocytes can also contribute to macrophage SLAMF1 induction in vivo. Although SLAMF1 is induced in AMs, we did not find evidence that it plays a role in mycobacterial control in AMs. AMs are particularly permissive to Mtb infection during the first two weeks of infection and become restrictive after the onset of adaptive immunity48. One possibility is that SLAMF1 is not required in AMs because cytokine responses are sufficient to drive AM control51. We also did not assess the impact of SLAMF1 in DCs, as they constituted a minor infected population in our study. Also, Slamf1 has been shown to promote autophagy in PMNs39, but we did not establish whether the elevated bacterial burden in Slamf1-deficient PMNs is due to a cell intrinsic requirement. Lastly, we observed reduced TNF and elevated Il-1β production in Mtb-infected Slamf1−/− and Slamf1 cKO mice. However, we did not investigate the impact of these cytokine differences on disease outcome. TNF is vital for granuloma formation, macrophage activation and polarization, cell death regulation, and cellular metabolic shifts68. Il-1β can promote Mtb killing but also exacerbate disease by increasing inflammation and tissue damage69. More recently, cognate interaction between macrophages and T cells was proposed to upregulate glycolytic metabolism14. Therefore, further investigation is necessary to establish how SLAMF1’s ability to regulate cytokine production, macrophage polarization, and cellular metabolism affects bacterial control.
SLAMF1 is required to control other Th1-inducing intracellular pathogens, including Leishmania major and Blastomyces dermatitidis35,70. In those studies, Slamf1−/− mice had a higher pathogen burden than WT animals. However, since conditional KO mice were not previously available, it was difficult to parse the myeloid from lymphocyte requirements of SLAMF1. By generating Slamf1fl/fl LysM-Cre+ mice, we were able to establish that myeloid expression of SLAMF1 is specifically required to control Mtb in myeloid cells. Germline absence of Slamf1 leads to additional defects including increased lymphocyte and myeloid cell recruitment. Collectively, we propose that SLAMF1 plays an important role linking innate and adaptive responses; the dynamic interplay between macrophages and T cells enhances SLAMF1 expression to augment the antimycobacterial ability of infected macrophages. This immune mechanism of protection against Mtb likely extends to a spectrum of diverse intracellular pathogens.
Methods
Ethics consideration
NYU School of Medicine and Washington University School of Medicine Institutional Animal Care and Use Committees approved all mouse work (Protocol# 21-0245 and 24-0201). Euthanasia was performed in accordance with the 2013 AVMA Guidelines for the Euthanasia of Animals. Mouse were euthanized by CO2 asphyxiation.
Bacterial strains and plasmids
All Mtb strains were cultured in Middlebrook 7H9 (BD Biosciences) with BBL OADC enrichment (BD Biosciences), 0.05% Tween 80 (Fisher) or tyloxapol (Millipore Sigma), and 0.2% glycerol (Sigma). M. tuberculosis (Mtb) H37Rv strain was from William R Jacobs Jr (AECOM). Mtb-mCherry was cultured with 50 μg/ml hygromycin (Gold Biotechnology); Erdman-GFP and H37Rv-GFP were grown with 25 μg/ml Kanamycin. Reporter strain (hspX’::GFP smyc’::mCherry) was cultured with both kanamycin and hygromycin. Strains (Erdman-GFP and HN878) and plasmids (mCherry and GFP) were provided by Christina Stallings (WUSM). The hspX’::GFP, smyc’::mCherry plasmid was provided by Shumin Tan (Tufts University School of Medicine)58.
Mice
All mice were in the C57BL/6 J background. WT (000664), Rag1−/− (002216), Tcra−/−(002116), MHCII−/− (H2dlAb1-Ea; 003584), and Ifng−/− (002287)71 mice were from The Jackson Laboratory and bred in house. Slamf1−/− mice35 were provided by Idit Shachar (Weizmann Institute) and re-derived. Nur77-GFP P25 TCR Tg mice were provided by Joel Ernst (UCSF)40,72. To obtain P25-specific, IFN-γ-deficient T cells, the P25 TCR Tg mice were crossed to Ifng−/− mice. Genotyping by Transnetyx confirmed the homozygous Ifng gene knockout and P25 transgenes. For the initial RNAseq data, P25 TCR Tg T cells were derived from mice that contained a Nur77-GFP transgene72. In subsequent experiments, T cells without the transgene were used. Atg5flox/flox-LysM-Cre, Atg7flox/flox-LysM-Cre and Atg14flox/flox-LysM-Cre were previously described50. ESAT-6 Tcr transgenic (C7PLTg/THY1.1) mice were provided by Christina Stallings (WUSM) and originally from Michael Glickman (MSKCC). OT-II mice (B6.Cg-Tg(TcraTcrb)425Cbn/j bred to B6.SJL mice and maintained as OT-II CD45.1+ were provided by Kenneth Murphy (WUSM). Ifngr1−/− mice (B6.129S7-Ifngr1tm1Agt/J) were provided by David Sibley (WUSM). Slamf1fl/fl LysM-Cre+ mice were generated in this study and described below. Both male and female mice between the ages 8-14 weeks old were used in the experiments.
Macrophage differentiation and culture
Bone marrow cells from tibia and femurs of 8-12 weeks old mice were differentiated for 7 days in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS), 20% L929 conditioned media, and 1% penicillin-streptomycin. Cells were harvested in phosphate-buffered saline (PBS) with 5 mM EDTA. The cells were washed once and resuspended in macrophage culture medium (DMEM, 10% FBS, 10% L929).
RNAseq screen
BMDMs cultured for 24 h with 100 U/ml of IFN−γ were uninfected or infected with a single cell suspension generated by slow speed spin of mCherry-expressing Mtb at an MOI 1. Three hpi, Nur77-GFP P25TCR-Tg effector T cells were added at a 1:1 ratio for 24 h. Cells were harvested, stained with antibodies, and sorted using a Sony Synergy cell sorter. BMDMs were identified by CD11b-APC (clone M1/70; BioLegend) and sorted into infected and bystander cells based upon mCherry fluorescence. P25TCR-Tg T cells were identified by CD4-APC/Cy7 (L3T4; BioLegend). RNA was extracted with miRNAeasy Micro Kit (Qiagen, Valencia, CA).
PolyA enriched RNA libraries were sequenced on an Illumina HiSeq 4000 at the NYU Langone’s Genome Technology Center (RRID: SCR_017929). Raw reads were preprocessed with Trimmomatic (v0.36) to trim trailing/leading (+/− 20) bases with a quality score (phred+33) < 20, remove Illumina specific sequences (i.e. sequencing adapters and barcodes), and drop fragments with length < 30. Remaining reads were aligned to the Ensembl mouse reference genome (GRCm38.97) using HiSat2 (v2.1.0). Read counts for all protein coding genomic features were summarized with featureCounts (v1.5.1), incorporating all sequence fragments with a mapping quality score > 10. Based upon principal component analysis and hierarchical clustering, two sample outliers were identified and removed from all subsequent analyses; both samples were bystander BMDMs. Read count normalization, gene outlier removal, and differential gene expression analyses were carried using the DESeq2 R package (Bioconductor.org). In brief, DESeq2 performs variance stabilizing transformation on raw read counts, employs an empirical Bayes procedure to estimate dispersion and fold-change estimates that are used to fit a generalized linear model with a negative binomial distribution for each gene. A Wald test is used to compare contrasts of model coefficients, and P values are adjusted for multiple testing following the Benjamini-Hochberg procedure. Subsequently, over-representation and gene-set enrichment analyses of Gene Ontology terms were performed using the WebgestaltR package (FDR < 0.05). Data has been deposited in the NCBI Gene Expression Omnibus (GSE224054).
Macrophage infection and peptide treatment
Prior to bacterial infection or antigen peptide stimulation, macrophages were cultured for 24 h with 100 U/ml of IFN−γ. A mid-log phase culture of mCherry H37Rv was resuspended in macrophage culture medium. The bacterial suspension was sonicated for 2 cycles of 5–10 s pulse with 5 s interval at 75% power using a water bath sonicator. Single cell suspension was made by repeatedly centrifuging the sonicated bacterial suspension at 132 × g for 5 min until the OD600 measured ≤0.05. Bacterial concentration was estimated by considering 1 OD600 = 3 × 108 bacteria/ml. Macrophages were washed once with culture medium and bacterial suspension was added to the macrophages to achieve an MOI of 10. For antigen peptide stimulation, BMDMs were treated with 0.5 μM peptides (Ag85240-254, ESAT61-20, or Ova323-339). Following bacterial infection or peptide stimulation, the macrophages were washed twice with culture medium and co-cultured with antigen specific CD4+ T cells at a ratio of 1:5 (macrophage: T cells) for 24 h.
Macrophage and T-cell co-culture in transwell inserts
Using 12-well transwell inserts (1 μm pore; Corning, USA), 2.5 × 105 macrophages were seeded either on the top or the underside of the inserts and incubated overnight at 37˚C in 5% CO2 in the presence of IFN-γ (100 U/ml). After overnight incubation, 500 μl of the culture media was added to both the transwell and the bottom well. Macrophages were treated with P25 peptide antigen for 4 h followed by co-culture with CD4+ T-cells at a ratio of 1:5 (macrophage: T cells). In the “no-contact” configuration, macrophages were seeded onto the top side of the membrane of the transwell inserts, and CD4+ T cells were added to the bottom well. In the “contact” configuration, macrophages were grown on the underside of the transwell membrane. Following overnight incubation, the inserts were inverted into the cell culture plate containing culture medium. After macrophage peptide stimulation, antigen-specific CD4+ T-cells were added in the transwell insert to allow interaction between the macrophages and T-cells.
Macrophage SLAMF1 analysis by flow cytometry
Mtb-infected or antigen-treated macrophages co-cultured with CD4+ T cells for 24 h were washed twice with PBS and harvested using 5 mM EDTA. Cells were surface stained with a combination of antibodies to stain for macrophages, T cells and SLAMF1 and either fixed with 4% PFA for 30 minutes or not. Antibodies used were A488 anti-CD150 (TC15-12F12.2, Biolegend, Stemcell technologies), anti-CD150 (TC15-12F12.2, Stemcell technologies), Rat IgG2a anti-CD150 (TC15-12F12.2, Stemcell technologies), PerCP/ PE-Cy5/ PE-Cy7 anti-F4/80 (BM8, Biolegend), A488 Donkey anti-Rat secondary antibody (Thermofisher Scientific), BV421/APC anti-CD4 (GK1.5, Biolegend). Cells were acquired on FACSCalibur (488 nm, 635 nm lasers, BD Biosciences) or LSR Fortessa X-20 flow cytometer (355 nm, 405 nm, 488 nm, 561 nm, and 633 nm lasers, BD Biosciences) or LSR Fortessa (405 nm, 488 nm, 561 nm, and 633 nm lasers, BD Biosciences) or Sony SH800 (405 nm, 488 nm, 561 nm, 638 nm laser, Sony). The data was analyzed using FlowJo software (BD Biosciences). The gating strategy is shown in Supplementary Fig. 2A.
T cell isolation and expansion
To generate antigen-specific T cells, lymph nodes (inguinal, brachial, axillary, mesenteric and superficial cervical) were isolated from transgenic mice euthanized with CO2 asphyxiation. Lymph nodes were transferred to a 70 μm cell strainer previously primed with RPMI-10 media (RPMI-1640 supplemented with 10% FCS, 2 mM L-glutamine, 10 mM HEPES, 100 μM non-essential amino acids (NEAA), 1 mM sodium pyruvate, 1 x β-mercaptoethanol). The lymph nodes were crushed using a sterile 3 cc syringe plunger, allowing the cells to pass through the strainer into complete RPMI media. The strainer was washed once with RPMI-10 media. Cells were centrifuged at 300 × g for 10 min at 4 °C; RBC were lysed with ACK lysis buffer followed by washing with RPMI-10 and centrifuging at 300 × g for 5 min at 4 °C. The pellet was resuspended in 300 μl MACS buffer (PBS + 2% FBS + 2 mM EDTA), and naïve CD4+ T cells were isolated using mouse CD4+ T cell isolation kit (Miltenyi Biotech #130-104-454) as per manufacturer’s protocol. Naïve CD4+ T cells isolated from magnetic sorting were expanded by co-culturing with irradiated splenocytes (30 gray) from C57Bl/6 mice at a ratio of 1:10. The splenocyte and T cell mixture was supplemented with IL-2 (5 ng/ml), IL-12p70 (10 ng/ml), anti-IL-4 antibody (50 ng/ml), and peptide antigen (0.5 μM). The cells were expanded for 9 days. Fresh RPMI-10 media supplemented with IL-2 (5 ng/ml) was added every 2–3 days, and cells were transferred from 6 well plates to 150 mm petri dishes (Genclone #32-106) and maintained at a cell density of 1 – 2 × 106 cells/ml.
qRT-PCR
Total RNA was extracted from the Mtb-infected or antigen peptide stimulated macrophages co-cultured with T cells using TRIzol reagent (Thermo Fisher Scientific, #15596026) and Direct-zol RNA Miniprep plus Kit (Zymo Research, #R202)) per manufacturers’ protocols. cDNA was synthesized using 100-500 ng of total RNA with High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, #4368813). Semi-quantitative real-time PCR (semi qRT-PCR) was performed on BioRad thermal cycler (CFX Connect Real-time PCR Detection system) using iTaqTM Universal SYBR Green supermix (BioRad, #1725122). The primers used were synthesized by Integrated DNA technologies. Primer sequences of the target genes Gapdh (Forward: 5’ AGGTCGGTGTGAACGGATTTG 3’, Reverse: 5’ TGTAGACCATGTAGTTGAGGTCA 3’) and Slamf1 (Forward: 5’ CAGAAATCAGGGCCTCAAGAG 3’, Reverse: 5’ CACTGGCATAAACTGTGGTGG 3’) were obtained from Harvard primer bank. The data was analyzed by ∆∆ threshold cycle method73. Relative changes in the gene expression levels were quantified by normalization with Gapdh.
scRNA Seq Analysis of IV BCG study
Single-cell RNAseq data from GEO-NCBI (GSE139598) was analyzed with the SEURAT software package (https://doi.org/10.1016/j.cell.2021.04.048) in R v4.2.1 (https://www.R-project.org/), using gene and cell-type annotations provided by the authors.
Mouse infections
Mtb was cultured to mid-log phase (OD600 0.6–0.8), and a single-cell suspension was generated through sequential low-speed centrifugation at 132 × g until stabilization of OD600 of the supernatant. Mice were infected by aerosol using a Glas-Col inhalation exposure system. Harvested tissues were homogenized using Bio-Gen PRO200 tissue homogenizer (PRO Scientific) and plated on Middlebrook 7H11 supplemented with BBL OADC enrichment (BD Biosciences) for CFU enumeration. In some cases, homogenized samples were passed through a 70 µm cell strainer (BD Biosciences) before plating. CFU were enumerated after 2–3 weeks of incubation at 37 °C.
Flow cytometry
Mouse lungs were perfused with sterile PBS (Thermo Fisher) before harvesting. Lungs were digested at 37 °C with 75 U/mL DNase I (Millipore Sigma) and 0.63 mg/mL collagenase D (Millipore Sigma). The cell suspension was washed with PBS containing 2% heat-inactivated FBS (Thermo Fisher) and 2 mM EDTA (Corning). Staining for surface markers was done at 4 °C for 20 min. The following reagents were used for staining: anti-CD16/32 Fc Block (93, Biolegend), LIVE/DEAD Fixable Blue Dead Cell Stain (Thermo Fisher), BD Horizon Fixable Viability Stain 780 (BD Biosciences), BUV395 anti-CD80 (16-10A1, BD Biosciences), BV421 anti-I-A/I-E (M5/114.15.2, Biolegend), BV510 anti-CD11c (N418, Biolegend), BV650 anti-CD150 (SLAM) (TC15-12F12.2, Biolegend), BV711 anti-CD11b (M1/70, Biolegend), BV786 anti-CD64 (X54-5/7.1, BD Biosciences), PE anti-Siglec-F (E50-2440, BD Biosciences), PE-Cy7 anti-Ly6G (1A8, Biolegend), Alexa700 anti-CD45 (30-F11, Biolegend), APC-Cy7 anti-Ly6C (HK1.4, Biolegend), BUV395 anti-NK1.1 (PK136, BD Biosciences), BV421 anti-TCR γδ (GL3, Biolegend), BV711 anti-CD19 (6D5, Biolegend), PE/FITC anti-CD44 (IM7, Biolegend), PE-Cy7 anti-CD8α (53-6.7, Biolegend), APC anti-CD4 (GK1.5, Biolegend), APC-Cy7 anti-TCR β (H57-597, Biolegend), BV605 anti-Ly6C (HK1.4, Biolegend), and 1:200 BV605 anti-TCR β (H57-597, BD Biosciences). After staining, cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 30 min at RT and acquired on a LSR Fortessa X-20 flow cytometer (BD Biosciences) with 355 nm, 405 nm, 488 nm, 561 nm, and 640 nm lasers running BD FACSDiva software. Flow cytometry data was compensated manually post-acquisition and analyzed using FlowJo software (BD Biosciences). Gating strategies are depicted in Supplementary Fig. 3A–D. Channel voltages were set using single-stained cell controls. Gates for mCherry were set using cells from uninfected mice, and gates for CD80, CD44, CD11c, NK and SLAMF1 were set using fluorescence minus one (FMO) controls. Absolute cell counts were calculated using Trucount tubes (BD Biosciences) or Precision count beads (Biolegend).
T cell adoptive transfer
T cells were isolated from C57BL/6 spleens. Harvested spleens were placed in a 70 μm cell strainer previously primed with complete RPMI media (RPMI-1640 supplemented with 10% FBS). Spleens were gently crushed using the flat end of a sterile 3 cc syringe plunger, allowing the cells to pass through the strainer. Collected cells were centrifuged at 300 x g for 10 min and resuspended in 5 ml of ACK lysis buffer, allowing RBCs to lyse, followed by washing twice with complete RPMI media. The pellet was suspended in 500 μl sterile MACS buffer and used for magnetic separation of naïve T cells using mouse Pan T cell isolation Kit II (Miltenyi Biotech #130-095-130) as per the manufacturer’s protocol. Enriched naïve T cells were washed and resuspended in complete RPMI media to a density of 1 × 108/ml. T cells were injected into Mtb-infected Tcra−/− mice via retro-orbital route. Each mouse received 2 × 107 cells.
Tetramer staining
MHC class II tetramer ESAT64-17 (I-A(b) QQWNFAGIEAAASA) conjugated to PE and MHC class I tetramer TB10.44-11 (H-2K(b) IMYNYPAM) conjugated to BV421 (obtained from NIH Tetramer Core Facility) were used to detect Mtb-specific CD4+ and CD8+ T cells, respectively. Tetramer staining was done prior to surface staining with antibodies. Lung single cell suspensions were incubated with 100 nM of freshly prepared Dasatinib (Sigma-Aldrich) for 30 min at 37 °C in a CO2 incubator. Thereafter, the cells were washed with FACS buffer and incubated with saturating concentrations of MHC tetramers. For MHCII tetramer staining, the cells were incubated with ESAT64-17 tetramer at 37 °C for 30 minutes in a CO2 incubator. For MHCI tetramer staining, the cells were incubated with TB10.44-11 tetramer for 30 minutes at 4 °C. Cells were washed once with FACS buffer and stained with surface antibodies.
Histopathology and immunohistochemistry
Tissues were fixed in 10% neutral buffered formalin for 48 h, transferred to 70% EtOH, and underwent routine paraffin-embedding. Paraffin blocks were sectioned at 5 µm thickness. Slides were baked for 30–60 min at 65 °F and deparaffinized in xylene, rehydrated through a series of graded ethanol to distilled water, and stained with hematoxylin and eosin or Niehl-Neelsen acid-fast staining. For immunohistochemistry, heat-induced epitope retrieval (HIER) was performed using a pressure cooker on steam setting for 25 minutes in Tris-EDTA pH 9.0 (EMS; 64142-10) followed by 3% hydrogen peroxide. Slides were rinsed in distilled water and blocked (BioCare, BE965H) for 15 min, followed by rinses in 1× PBS, and incubation with rabbit monoclonal anti-SLAMF antibody (Abcam, Ab228978) diluted 1:100 in DaVinci Green Diluent (BioCare, PD900M) for 60 min at RT, followed by rabbit Mach-2 HRP-Polymer (BioCare; RHRP520L) for 30 min, and then counterstained with hematoxylin, followed by bluing using 0.25% ammonia water. Labeling was performed on a Biocare IntelliPATH autostainer. Slides were scanned using a Midi II Panoramic scanner (Epredia). Quantitative image analysis was performed using HALO software (v3.0.311.405; Indica Labs) on at least one lung lobe cross-section from each animal. For SLAMF quantification, the Area Quantification module (v2.1.11) was used. Lungs were annotated to exclude airways and large vessels. In all instances, manual inspection was performed on each sample to ensure that the annotations were accurate, and tissue artifacts were excluded.
Slamf1 conditional knock-out mice
Slamf1fl/fl mice were generated using reagents designed for CRISPR/Cas9 gene-editing by the Genome Engineering & Stem Cell Center (GESC@MGI) Center at WUSM as described previously74. Guide RNAs (gRNAs) were designed in intron 1: 5’- TTCCAAGTGCCCATCTATGTNGG and intron 6: 5’- GGAAAGTGGGCTAGATCACANGG. In silico analysis was performed to select guides with the fewest predicted off-target sites. A single-stranded oligo-deoxynucleotide (ssODN) was designed to pair with each gRNAs. Both gRNAs and ssODN were obtained from IDT. The gRNAs and ssODN were validated in mouse Nura2a cells for efficient loxP insertion before mouse sessions. Founders were genotyped by NGS at each insertion site as described74. Germline-targeted mice were generated by the Pathology Transgenic Core at WUSM in the C57Bl/6 background using standard protocols. Offspring were screened by PCR amplification and Sanger sequencing to identify edited founders. For two founder lines, loxP sites were confirmed to be in cis by backcrossing with C57Bl/6 LysM-Cre+ mice.
Enzyme linked-immunosorbent assay
Lung homogenates were centrifuged twice at 10,000 × g at 4 °C for 10 minutes. The supernatant was filtered using a 0.22 μm multiscreen-filter plate by centrifuging at 250 × g at 4 °C for 10 minutes. The flow-through obtained upon filtration was used to measure cytokine levels with sandwich ELISA kits as per manufacturer’s protocol (BD Biosciences, BD OptEIA murine IL-6 (#555240), TNF (#555268), IFNγ (#555138); R&D systems, mouse IL-1β/IL-1F2 DuoSet ELISA(#DY401), TNF (#DY410-05)).
NO measurements
The amount of nitrite, a stable end product of nitric oxide, present in the culture supernatant was determined using Griess reagent (Thermo fisher Scientific, #G7921) as per the manufacturer’s protocol. Briefly, cell culture supernatant was collected from macrophages either infected with H37Rv or P25 treated and co-cultured with or without T cells for 24 hrs. Equal volumes of culture supernatant and 1:1 mixture of N-(1-naphthyl)ethylenediamine and sulfanilic acid diluted in deionized water was added to a 96-well plate and incubated for 30 minutes. Using a microplate reader, absorbance at 548 nm was measured, and nitrite levels in the supernatant were estimated using a standard curve plotted with known concentrations of sodium nitrite solution.
ROS Assessment
Intracellular ROS production in macrophages upon antigen treatment was analyzed using ROS reactive dye Cellrox Red. After 7 days of differentiation, WT and Slamf1−/− BMDMs were rested in L929 free media for 1 day, followed by IFN-γ, P25 peptide treatment and co-culture with T cells (macrophage:T cells ratio= 1:3) as described above. Subsequently, cells were stained with Cellrox Red (5 μM) (Thermofisher Scientific, #C10422) in fresh L929 free media for 30 mins, harvested, surface stained with BV421 anti-CD4 (GK1.5, Biolegend), PE/Cy7 anti-F4/80 (BM8, Biolegend) and acquired on Sony Sorter SH800.
Macrophage infection with reporter Mtb strain and fluorescence microscopy
Single cell suspension of hspX’:: GFP reporter Mtb strain, grown in the presence of hygromycin and kanamycin, was made through sequential low-speed centrifugation at 132 × g until the OD600 measured ≤0.05. BMDMs cultured for 24 hrs with 100 U/ml IFNγ in a clear bottom 96 well plate were infected with reporter Mtb at an MOI of 10. After 4 hrs, the macrophages were washed with culture medium and co-cultured with antigen-specific CD4+ T cells at a ratio of 1:5 (macrophage: T cells) for 24 h. Subsequently, the cells were fixed with 4% PFA for 30 mins, stained with DAPI (300 nM), and mounted with Prolong Antifade Mountant. Images were captured using a Nikon Eclipse Ti fluorescence microscope equipped with 60X apochromat oil-objective lens and analyzed using NIS-Elements version 4.40. A region of interest (ROI) was selected around each mCherry-expressing bacterium and the corresponding GFP signal for that bacteria was measured.
Macrophage intracellular bacterial growth
Mtb (H37Rv) growth in WT (C57BL/6 J) and Slamf1−/− BMDMs was assessed using a single cell suspension generated by low speed centrifugation75. BMDMs were cultured for 24 h with 100 U/ml of IFN-γ and then infected at an MOI 5 with a single cell suspension of Mtb that was obtained through sequential low-speed centrifugation at 132 × g with the supernatant collected following each spin until the OD600 is stabilized. At 4 hpi, BMDMs were washed to remove extracellular bacteria. At 4 hpi (Day 0) and 3 dpi, BMDMs were lysed with 0.06% SDS in water, and lysates were plated on 7H11 agar. Bacterial CFUs were counted 15–20 days later. After 4 h of infection, BMDMs were co-cultured with P25 TcR Tg T cells at a 1:3 ratio, and some samples were incubated with 2 mM NAC.
Statistics
In vivo data is from mice randomly sorted into each experimental group. No blinding was performed during animal experiments. Statistical differences were calculated using Prism (GraphPad Software). For two groups with normal distributions, unpaired Student’s t test was used. For two groups with non-normal distributions, Mann-Whitney test was used. For more than two groups with normal distributions, one-way ANOVA was used to compare one variable, and two-way ANOVA. For more than two groups with non-normal distributions, Kruskal-Wallis test was used. Center values and error bars represent mean ± SEM or mean ± SD as detailed in figure legends. Some figures were created using BioRender.
Biological Materials
All unique material are available upon request to JAP. BSL3 strains are available to qualified investigators with appropriate containment facilities.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNAseq datasets generated during this study have been deposited in GEO under accession code GSE224054; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE224054. Processed data generated in this study are provided in the Source data files. All other data are available in the article and its Supplementary files or from the corresponding author upon request. Source data are provided with this paper.
References
WHO. Global tuberculosis report 2021. (World Health Organization, 2021).
Cohen, S. B. et al. Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell host microbe 24, 439–446 (2018).
Wolf, A. J. et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 179, 2509–2519 (2007).
Lee, J. et al. CD11cHi monocyte-derived macrophages are a major cellular compartment infected by Mycobacterium tuberculosis. PLoS Pathog. 16, e1008621 (2020).
Caruso, A. M. et al. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J. Immunol. 162, 5407–5416 (1999).
Flynn, J. L. et al. Tumor necrosis factor-α is required in the protective immune response against mycobacterium tuberculosis in mice. Immunity 2, 561–572 (1995).
Kwan, C. K. & Ernst, J. D. HIV and tuberculosis: a deadly human syndemic. Clin. Microbiol Rev. 24, 351–376 (2011).
Ernst, J. D. Mechanisms of M. tuberculosis immune evasion as challenges to TB vaccine design. Cell Host Microbe 24, 34–42 (2018).
Flynn, J. L. et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178, 2249–2254 (1993).
Sakai, S. et al. CD4 T cell-derived IFN-γ plays a minimal role in control of pulmonary mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog. 12, e1005667 (2016).
Sallin, M. A. et al. Host resistance to pulmonary Mycobacterium tuberculosis infection requires CD153 expression. Nat. Microbiol 3, 1198–1205 (2018).
Foreman, T. W. et al. CD30 co-stimulation drives differentiation of protective T cells during Mycobacterium tuberculosis infection. J. Exp. Med. https://doi.org/10.1084/jem.20222090 (2023).
Srivastava, S. & Ernst, J. D. Cutting edge: Direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J. Immunol. 191, 1016–1020 (2013).
Becker, S. H., Ronayne, C. E., Bold, T. D. & Jenkins, M. K. Antigen-specific CD4+ T cells promote monocyte recruitment and differentiation into glycolytic lung macrophages to control Mycobacterium tuberculosis. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1013208 (2025).
Ankley, L., Thomas, S. & Olive, A. J. Fighting persistence: how chronic infections with mycobacterium tuberculosis evade T cell-mediated clearance and new strategies to defeat them. Infect. Immun. 88, e00916–e00919 (2020).
Kauffman, K. D. et al. Defective positioning in granulomas but not lung-homing limits CD4 T-cell interactions with Mycobacterium tuberculosis-infected macrophages in rhesus macaques. Mucosal Immunol. 11, 462–473 (2018).
Gern, B. H. et al. TGFβ restricts expansion, survival, and function of T cells within the tuberculous granuloma. Cell Host Microbe 29, 594–606.e596 (2021).
Portal-Celhay, C. et al. Mycobacterium tuberculosis EsxH inhibits ESCRT-dependent CD4(+) T-cell activation. Nat. Microbiol 2, 16232 (2016).
Grace, P. S. & Ernst, J. D. Suboptimal antigen presentation contributes to virulence of mycobacterium tuberculosis in vivo. J. Immunol. 196, 357–364 (2016).
Srivastava, S. & Ernst, J. D. Cell-to-Cell Transfer of M. tuberculosis Antigens Optimizes CD4 T Cell Priming. Cell Host Microbe 15, 741–752 (2014).
Patankar, Y. R. et al. Limited recognition of Mycobacterium tuberculosis-infected macrophages by polyclonal CD4 and CD8 T cells from the lungs of infected mice. Mucosal Immunol. 13, 140–148 (2020).
Saini, N. K. et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol 1, 16133 (2016).
Srivastava, S., Grace, P. S. & Ernst, J. D. Antigen export reduces antigen presentation and limits T cell control of M. tuberculosis. Cell Host Microbe 19, 44–54 (2016).
Georgieva, M., Sia, J. K., Bizzell, E., Madan-Lala, R. & Rengarajan, J. Mycobacterium tuberculosis GroEL2 modulates dendritic cell responses. Infect. Immun. 86, e00387–17 (2018).
van Driel, B. J., Liao, G., Engel, P. & Terhorst, C. Responses to microbial challenges by SLAMF receptors. Front Immunol. 7, 4 (2016).
Wu, N. & Veillette, A. SLAM family receptors in normal immunity and immune pathologies. Curr. Opin. Immunol. 38, 45–51 (2016).
Dragovich, M. A. & Mor, A. The SLAM family receptors: potential therapeutic targets for inflammatory and autoimmune diseases. Autoimmun. Rev. 17, 674–682 (2018).
Guo, H., Cruz-Munoz, M. E., Wu, N., Robbins, M. & Veillette, A. Immune cell inhibition by SLAMF7 is mediated by a mechanism requiring src kinases, CD45, and SHIP-1 that is defective in multiple myeloma cells. Mol. Cell Biol. 35, 41–51 (2015).
Wu, Y. et al. SLAMF7 regulates the inflammatory response in macrophages during polymicrobial sepsis. J. Clin. Invest 133, e150224 (2023).
Aversa, G., Chang, C. C., Carballido, J. M., Cocks, B. G. & de Vries, J. E. Engagement of the signaling lymphocytic activation molecule (SLAM) on activated T cells results in IL-2-independent, cyclosporin A-sensitive T cell proliferation and IFN-gamma production. J. Immunol. 158, 4036–4044 (1997).
Cocks, B. G. et al. A novel receptor involved in T-cell activation. Nature 376, 260–263 (1995).
Castro, A. G. et al. Molecular and functional characterization of mouse signaling lymphocytic activation molecule (SLAM): differential expression and responsiveness in Th1 and Th2 cells. J. Immunol. 163, 5860–5870 (1999).
Pasquinelli, V. et al. Expression of signaling lymphocytic activation molecule-associated protein interrupts IFN-gamma production in human tuberculosis. J. Immunol. 172, 1177–1185 (2004).
Pasquinelli, V. et al. Phosphorylation of mitogen-activated protein kinases contributes to interferon γ production in response to Mycobacterium tuberculosis. J. Infect. Dis. 207, 340–350 (2013).
Wang, N. et al. The cell surface receptor SLAM controls T cell and macrophage functions. J. Exp. Med 199, 1255–1264 (2004).
Tatsuo, H., Ono, N. & Yanagi, Y. Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J. Virol. 75, 5842–5850 (2001).
Berger, S. B. et al. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nat. Immunol. 11, 920–927 (2010).
Ma, C. et al. Receptor signaling lymphocyte-activation molecule family 1 (Slamf1) regulates membrane fusion and NADPH oxidase 2 (NOX2) activity by recruiting a Beclin-1/Vps34/ultraviolet radiation resistance-associated gene (UVRAG) complex. J. Biol. Chem. 287, 18359–18365 (2012).
Pellegrini, J. M. et al. Neutrophil autophagy during human active tuberculosis is modulated by SLAMF1. Autophagy, 17, 1–10 (2020).
Tamura, T. et al. The role of antigenic peptide in CD4+ T helper phenotype development in a T cell receptor transgenic model. Int Immunol. 16, 1691–1699 (2004).
Bold, T. D., Banaei, N., Wolf, A. J. & Ernst, J. D. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS Pathog. 7, e1002063 (2011).
Darrah, P. A. et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 577, 95–102 (2020).
Gideon, H. P. et al. Multimodal profiling of lung granulomas in macaques reveals cellular correlates of tuberculosis control. Immunity 55, 827–846.e810 (2022).
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z. & Zhang, B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47, W199–W205 (2019).
Grigsby, S. J. et al. CpsA mediates infection of recruited lung myeloid cells by Mycobacterium tuberculosis. Cell Rep. 43, 113607 (2024).
Münz, C. Canonical and non-canonical functions of the autophagy machinery in mhc restricted antigen presentation. Front Immunol. 13, 868888 (2022).
Jagannath, C. et al. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat. Med 15, 267–276 (2009).
Zheng, W. et al. Mycobacterium tuberculosis resides in lysosome-poor monocyte-derived lung cells during chronic infection. PLoS Pathog. 20, e1012205 (2024).
Pisu, D. et al. Single cell analysis of M. tuberculosis phenotype and macrophage lineages in the infected lung. J. Exp. Med 218, e20210615 (2021).
Mittal, E. et al. Mycobacterium tuberculosis virulence lipid PDIM inhibits autophagy in mice. Nat. Microbiol 9, 2970–2984 (2024).
Lai, R., Williams, T., Rakib, T., Lee, J. & Behar, S. M. Heterogeneity in lung macrophage control of Mycobacterium tuberculosis is modulated by T cells. Nat. Commun. 15, 5710 (2024).
Scott, H. M. & Flynn, J. L. Mycobacterium tuberculosis in chemokine receptor 2-deficient mice: influence of dose on disease progression. Infect. Immun. 70, 5946–5954 (2002).
Moreira-Teixeira, L. et al. Mouse transcriptome reveals potential signatures of protection and pathogenesis in human tuberculosis. Nat. Immunol. 21, 464–476 (2020).
Feng, S. et al. Autophagy promotes efficient T cell responses to restrict high-dose Mycobacterium tuberculosis infection in mice. Nat. Microbiol 9, 684–697 (2024).
McCubbrey, A. L., Allison, K. C., Lee-Sherick, A. B., Jakubzick, C. V. & Janssen, W. J. Promoter specificity and efficacy in conditional and inducible transgenic targeting of lung macrophages. Front Immunol. 8, 1618 (2017).
Song, T., Dong, C. & Xiong, S. Signaling lymphocyte-activation molecule SLAMF1 augments mycobacteria BCG-induced inflammatory response and facilitates bacterial clearance. Int J. Med Microbiol 305, 572–580 (2015).
Yurchenko, M. et al. SLAMF1 is required for TLR4-mediated TRAM-TRIF-dependent signaling in human macrophages. J. Cell Biol. 217, 1411–1429 (2018).
Sukumar, N., Tan, S., Aldridge, B. B. & Russell, D. G. Exploitation of Mycobacterium tuberculosis reporter strains to probe the impact of vaccination at sites of infection. PLoS Pathog. 10, e1004394 (2014).
García, V. E. et al. Signaling lymphocytic activation molecule expression and regulation in human intracellular infection correlate with Th1 cytokine patterns. J. Immunol. 167, 5719–5724 (2001).
Degos, C. et al. Omp25-dependent engagement of SLAMF1 by Brucella abortus in dendritic cells limits acute inflammation and favours bacterial persistence in vivo. Cell Microbiol 22, e13164 (2020).
Barbero, A. M. et al. SLAMF1 signaling induces Mycobacterium tuberculosis uptake leading to endolysosomal maturation in human macrophages. J. Leukoc. Biol. 109, 257–273 (2021).
Lu, Y. J. et al. CD4 T cell help prevents CD8 T cell exhaustion and promotes control of Mycobacterium tuberculosis infection. Cell Rep. 36, 109696 (2021).
Mayer-Barber, K. D. & Barber, D. L. Innate and adaptive cellular immune responses to mycobacterium tuberculosis infection. Cold Spring Harb. Perspect. Med 5, a018424 (2015).
Maciag, K., Plumlee, C. R., Cohen, S. B., Gern, B. H. & Urdahl, K. B. Reappraising the role of T cell-derived IFN-γ in Restriction of Mycobacterium tuberculosis in the Murine Lung. J. Immunol. 213, 339–346 (2024).
Green, A. M., Difazio, R. & Flynn, J. L. IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J. Immunol. 190, 270–277 (2013).
Sakai, S., Mayer-Barber, K. D. & Barber, D. L. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr. Opin. Immunol. 29, 137–142 (2014).
Chandra, P., Grigsby, S. J. & Philips, J. A. Immune evasion and provocation by Mycobacterium tuberculosis. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-022-00763-4 (2022).
Yuk, J. M., Kim, J. K., Kim, I. S. & Jo, E. K. TNF in human tuberculosis: a double-edged sword. Immune Netw. 24, e4 (2024).
Silvério, D., Gonçalves, R., Appelberg, R. & Saraiva, M. Advances on the role and applications of interleukin-1 in tuberculosis. mBio 12, e0313421 (2021).
Kohn, E. M. et al. SLAMF1 is dispensable for vaccine-induced T cell development but required for resistance to fungal infection. J. Immunol. 208, 1417–1423 (2022).
Dalton, D. K. et al. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science 259, 1739–1742 (1993).
Moran, A. E. et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med 208, 1279–1289 (2011).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta delta C(T)) method. Methods 25, 402–408 (2001).
Sentmanat, M. F., White, J. M., Kouranova, E. & Cui, X. Highly reliable creation of floxed alleles by electroporating single-cell embryos. BMC Biol. 20, 31 (2022).
Mittal, E. et al. Single cell preparations of Mycobacterium tuberculosis damage the mycobacterial envelope and disrupt macrophage interactions. Elife 12, e85416 (2023).
Acknowledgements
Dedicated to the memory of our dear colleague, Guozhe Yang. We thank the Genome Engineering and iPSC Center (GEiC) and the Pathology Transgenic Core at the Washington University in St. Louis for generating the Slamf1fl/fl mice. We thank Cox Terhorst (BIDMC/HMS), Idit Shachar (Weizmann), Christina Stallings (WUSM), Michael Glickman (MSKCC), Ray Ohara (WUSM), Kenneth Murphy (WUSM), David Sibley (WUSM), Joel Ernst (UCSF) and Shumin Tan (Tufts) for providing reagents. We thank members of the Pulmonary Morphology Core (WUSM), Bursky Center for Human Immunology and Immunotherapy Programs (CHiiPs; WUSM), Immunology & Pathology Flow Cytometry & Fluorescence Activated Cell Sorting Core Facility (WUSM), Division of Comparative Medicine (WUSM), and NYU Langone’s Genome Technology Center for assistance. Cox Terhorst, Patricia Darrah (NIH), Kevin Urdahl (University of Washington), and members of the Philips laboratory contributed to helpful discussions. The following sources provided funding and reagents: NIH R21 AI155380 (JAP), R01 AI184568 (JAP), R01 AI130454 (JAP), F31 AI152321 (SJG), K08 AI135098 (AJM), K08 AI119150 (CPC), P30 CA016087, R01 AI051242 (JDE); NIH Tetramer Core Facility; and Washington University School of Medicine Bursky Center for Human Immunology and Immunotherapy Programs at Washington University Immunomonitoring Laboratory.
Author information
Authors and Affiliations
Contributions
C.P.C., J.D.E., and J.A.P. conceived the study. G.V.R.K.P. and J.A.P. guided the overall direction. G.V.R.K.P., S.J.G., C.P.C., and J.A.P. designed experiments. G.V.R.K.P., S.J.G., C.P.C., E.M., G.Y., Y.L., S.F., F.C., T.K., and N.J. conducted experiments. G.V.R.K.P., S.J.G., G.A.E., C.P.C., E.M., S.F., T.K., N.J., M.M., A.J.M., J.D.E., J.A.P. analyzed data. G.A.E. performed bioinformatic analysis. The manuscript was written by G.V.R.K.P., S.J.G., and J.A.P., with reviewing and editing provided by G.V.R.K.P., S.J.G., C.P.C., T.K., S.F., E.M., N.J., G.A.E., G.Y., M.M., J.D.E., A.J.M., and J.A.P. C.P.C., S.J.G., J.D.E., A.J.M., and J.A.P. acquired funding. C.P.C., M.M., A.J.M., J.D.E., and J.A.P. provided supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Janice Endsley, Margarida Saraiva and Cristina Ulivieri for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Krishna Prasad, G.V.R., Grigsby, S.J., Erkenswick, G.A. et al. Macrophage-T cell interactions promote SLAMF1 expression for enhanced TB defense. Nat Commun 16, 6794 (2025). https://doi.org/10.1038/s41467-025-61826-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61826-7
