
Abstract
Protein-ligand interactions play central roles in myriad biological processes and are of key importance in drug design. Deep learning approaches are becoming cost-effective alternatives to high-throughput experimental methods for ligand identification. Here, to predict the binding affinity between proteins and small molecules, we introduce Ligand-Transformer, a deep learning method based on the transformer architecture. Ligand-Transformer implements a sequence-based approach, where the inputs are the amino acid sequence of the target protein and the topology of the small molecule to enable the prediction of the conformational space explored by the complex between the two. We apply Ligand-Transformer to screen and validate experimentally inhibitors targeting the mutant EGFRLTC kinase, identifying compounds with low nanomolar potency. We then use this approach to predict the conformational population shifts induced by known ABL kinase inhibitors, showing that sequence-based predictions enable the characterisation of the population shift upon binding. Overall, our results illustrate the potential of Ligand-Transformer to accurately predict the interactions of small molecules with proteins, including the binding affinity and the changes in the free energy landscapes upon binding, thus uncovering molecular mechanisms and facilitating the initial steps in drug design.
Similar content being viewed by others


Introduction
Recent reports are revealing that deep learning approaches exhibit capabilities beyond their original function of predicting protein structures, including the possibility to infer protein dynamics and protein interactions1,2,3,4,5,6,7,8,9,10,11,12,13. These developments have opened the way to the use of deep learning in early-stage drug design14,15,16,17,18,19,20 to help address the problem of the progressive inefficacy of drug discovery pipelines21,22. One important aspect of this problem is the identification of ligands using high-throughput screening, which is resource-intensive, requiring substantial financial investment, extensive time, and specialized equipment. Additionally, the process can be inefficient due to the high rate of false positives and negatives, necessitating further validation steps that add to the overall cost and time burden. These limitations have driven the development of computational methods for drug design in the last several decades23,24,25,26. The advent of deep learning is now creating novel opportunities for more efficient, accurate, and cost-effective approaches to predict protein-ligand interactions by significantly reducing the experimental workload.
Here, we explore the idea of sequence-based drug design to use deep learning to predict how a protein target explores its conformational space when in complex with a candidate ligand. The goal with sequence-based drug design is to go beyond structure-based18,23,25,26 and ligand-based27,28 drug design methods, which largely rely on the conformations of the interaction partners in their free states. By taking as input the amino acid sequence of the protein target and the structure of the ligand, this approach offers as output a prediction of the conformational space of the complex and the corresponding binding affinity. We thus report Ligand-Transformer, a deep learning approach designed to model protein-ligand interactions.
The architecture of Ligand-Transformer is presented in Fig. 1. Briefly, Ligand-Transformer is based on the transformer framework of AlphaFold26 to generate protein representations from their sequences, and the Graph Multi-View Pre-training (GraphMVP) framework29 to generate ligand representations. Instead of using the final predicted protein structure, we leverage the intermediate outputs. For the ligands, during pre-training, GraphMVP injects the knowledge of 3D molecular geometry into a 2D molecular graph encoder, allowing downstream tasks to benefit from the implicit 3D geometric prior. By leveraging the prior knowledge encoded in these high-dimensional representations, we capture the structural features of protein-ligand interactions, resulting in an accurate modelling of the bound states. The representations are further processed by the main structure of Ligand-Transformer, which consists of three parts (Fig. 1). The first part is the feature encoders to re-process the representations of proteins and ligands. The second part is the cross-modal attention network to exchange the information between representations of the protein and ligand. The third part is composed of two downstream predictors, the first head for affinity predictions and the second head for distance predictions.

Ligand-Transformer represents a protein-ligand complex as a heterogeneous graph, incorporating residue and atom sets from the protein and ligand, respectively, along with pairwise features. The graph is formed from inputs generated by AlphaFold2 for proteins and GraphMVP for ligands, which are then re-encoded into an initial complete graph. This graph is subsequently refined through a 12-layer transformer-like network. This network updates both node and edge representations via self-attention with pair bias. The output is processed by the affinity head for binding affinity prediction and by the distance head for distance distribution prediction. Components within dotted-line boxes have fixed parameters, whereas those within solid lines are trainable.
Here, we describe Ligand-Transformer, a sequence-based deep learning framework that models protein-ligand interactions by predicting both the binding affinity and the conformational space of protein-ligand complexes. We apply it to identify inhibitors of the drug-resistant EGFRLTC kinase, achieving a hit rate of 58%, with two ligands exhibiting low-nanomolar affinity in validation binding experiments. We further show that Ligand-Transformer accurately predicts ligand-induced conformational population shifts of ABL kinase, consistent with experimentally determined conformational states. These results illustrate how Ligand-Transformer enables out-of-distribution predictions and captures the free energy landscape upon binding, offering a scalable and efficient alternative to high-throughput experimental assays in early-stage drug discovery.
Results
Performance comparison against state-of-the-art affinity prediction methods
We conducted a performance comparison of Ligand-Transformer against other affinity prediction methods30,31,32 utilizing the PDBbind2020 dataset (Table S1). Our results indicate that Ligand-Transformer achieves comparably better correlations with experimentally-measured values when compared to baseline methods.
To evaluate the predictive accuracy of Ligand-Transformer on both binding affinity and distance matrices, we used the PDBbind2020 dataset. We curated a subset of 13,420 complexes (Supplementary Data 1), ensuring manageable computational loads by limiting the maximum length of protein sequences to 384 residues, and the maximum number of atoms in each ligand to 128. Each complex in the dataset has an experimentally measured binding affinity (pKd), allowing us to compare predicted values directly against the measured data. We randomly split the dataset into training (10,375 complexes), validation (640), and test (936).
To compare Ligand-Transformer to competing approaches, we also trained three other deep learning-based affinity prediction models30,31,32 on the same data partitions. Table S1 and Fig. S1 summarize these comparisons, showing that Ligand-Transformer achieves higher or on-par correlation with experimentally measured affinities relative to all three baseline methods. Furthermore, Ligand-Transformer effectively predicts protein-ligand distances. We found that approximately 95% of the residue-residue distance errors were below 0.5 Å, while ~95% of the residue-ligand atom distance errors were within 2 Å (Fig. S2). These results suggest that Ligand-Transformer is able to capture structural aspects of protein-ligand complexes that are useful for both binding affinity and distance matrix predictions. Additional details of these comparisons, as well as the ability of the model to estimate its own error and to generalize to unseen protein-ligand combinations (Figs. S3 and S4), are provided in the “Methods” and the Supplementary Methods.
Identification of EGFRLTC ligands
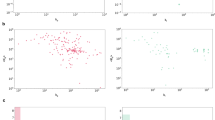
We illustrate the potential of Ligand-Transformer for the identification of initial hits in drug discovery pipelines by screening ligands targeting EGFRLTC, a mutant form of the EGFR kinase. EGFR is a key target in cancer therapy due to its role in cell growth33. As mutations in EGFR, such as L858R/T790M/C797S (LTC), can lead to resistance against all current EGFR inhibitors, there is a need for novel drugs to target this triple-mutant34. We first collected a dataset, called EGFRLTC-290 (Supplementary Data 2), consisting of 290 existing inhibitors with their measured half maximal inhibitory concentration (IC50) values, along with annotations indicating whether they are allosteric or orthosteric (see “Methods”). We found that Ligand-Transformer could predict the binding affinity with a Pearson’s correlation coefficient (R) value of 0.57 (Fig. 2a). To fine-tune the model on this specific dataset to achieve higher accuracy, we randomly split the EGFRLTC-290 dataset into ten parts, and conducted a tenfold cross validation to evaluate the performance and obtain an ensemble of fine-tuned models (Model FT1 to FT10, see “Methods”). The value of R of this test increased to 0.88 after fine tuning (Fig. 2b).

a, b Correlation between the experimental pIC50 values and the binding parameters (pKd and pIC50 values) using Ligand-Transformer for complexes in the EGFRLTC-290 dataset; all binding parameters are in molar units: a predicted pKd values without transfer learning (base model), and b predicted pIC50 values post tenfold validation with transfer learning. c Distribution of predicted distances between E762 and G857 of EGFRLTC in various ligand-binding states. y-axis: predicted distance between the E762 Cβ and G857 Cα atoms; x-axis: experimental pIC50 values of the ligands. Data points are colored based on their reported binding sites in the literature: orthosteric (blue), allosteric (green), and dual (orange). The kernel density estimations (KDE) plots for the distributions of predicted distances are indicated adjacent to y coordinates. d Comparison of the EGFR kinase domain X-ray structures in the active (PDB ID 2ITX) and inactive (PDB ID 2GS7) states. The αC-helix is shown as a ribbon in light blue and gray, representing the active and inactive states, respectively. The AMP-PNP ligand bound to PDB ID 2ITX is visualized as a stick-and-ball structure. Residues E762 and G857 of EGFR (corresponding to residues E738 and G833 in PDB ID 2GS7) are shown as sticks. Nitrogen, oxygen, phosphorus, and carbon atoms are colored dark blue, red, orange, and light blue, respectively. e Line graph of the predicted binding modes of an orthosteric inhibitor (8r54, colored in red) and an allosteric inhibitor (4i55, colored in blue) with EGFRLTC. The line graph illustrates the predicted minimal distances between each residue and the ligand. The error bars represent the predicted confidence scores (pMAE, see Algorithm 8 in Supplementary Information). The bottom part shows sequence annotation of EGFR based on UniProt P00533. ATP binding sites are shown in red dots, active site is shown in green dot, and the region of αC-helix is shown in blue box. f In vitro kinase inhibition assays for selected compounds, depicted as a percentage of activity relative to a DMSO control. The graph presents the mean of duplicate measurements for each ligand. The corresponding IC50 values are presented for each compound. Source data are provided as a Source Data file.
We identified notable differences in the binding modes of the predicted orthosteric and allosteric inhibitors. Statistical analysis using the t-test revealed no significant differences in activity distribution between the orthosteric and allosteric groups (p = 0.66) and the dual group (p = 0.39) (Fig. 2c). However, the predicted distance distributions exhibited significant variations (p < 10−21 for allosteric, p = 0.0025 for dual). The predicted distance between residues E762 and G857 was significantly greater when binding to allosteric inhibitors compared to orthosteric inhibitors. This distance can serve as an indicator of the αC-helix-in and αC-helix-out states (Fig. 2d), consistent with previous studies35,36. Specifically, the αC-helix-in state represents the active conformation, while allosteric inhibitors tend to bind to αC-helix-out state, which is the inactive conformation. Moreover, the residue-wise distance between the kinase and ligand indicates that the binding sites of allosteric inhibitors are situated in closer proximity to the αC-helix region rather than the ATP-binding sites and the active site (Fig. 2e).
We then used the predicted affinities and distances obtained from Ligand-Transformer to screen a subset of TargetMol containing 9090 compounds in stock (“Methods” and Supplementary Data 3). We selected candidates with the criteria that they are predicted to have high binding affinity by all of the 11 models (Table S3). Finally, we obtained 12 candidates (Table S4) with predicted IC50 between 1 and 100 nM). These candidates included a compound (brigatinib) previously identified as effective inhibitor targeting EGFRLTC with a reported IC50 of 1–38 nM37. To our knowledge, among the remaining 11 compounds, C1, C4, C5, C10, and C11 have not been reported to target EGFR, while C7 is known to target EGFR but not to EGFRLTC.
We then tested experimentally the inhibitory potency of these 11 candidates, and found six active compounds. Out of these six active compounds, three fell within the predicted IC50 range of 1 to 100 nM, with two of them, C1 and C10, exhibiting high potency, with IC50 values of 5.5 and 1.2 nM, respectively (Fig. 2f). C1 is a naphthyridine derivative, which is a non-traditional scaffold for EGFR inhibitors. Based on our predictions, the naphthyridine moiety may be capable of competing with the purine ring of ATP and forming hydrogen bonds with the backbone of M793, thereby stabilizing the ligand within EGFR pocket. C10 and C4 show Tanimoto similarities (based on Morgan fingerprints38, radius = 2, 2048 bits) of 0.77 and 0.35, respectively, to brigatinib. These compounds exhibit a high degree of pharmacophore overlap, particularly the aniline-pyrimidine scaffold. This structural motif may compete with the purine ring of ATP, occupying the EGFR pocket. Additionally, the pyrimidine moiety can form hydrogen bonds with backbones of M793 and P794, thereby enhancing its binding affinity. C5, C7, and C11 exhibit inhibitory activity against EGFRLTC in the range of 100 nM–10 μM, indicating relatively weak inhibitory potency. C11 is a pyridine compound, which consists of only two aromatic rings. From a structural perspective, it represents a novel EGFR inhibitor. C5 is a pyrimidine-2,4-dione derivative and features a quinoline ring, which is a classic structural motif commonly found in EGFR inhibitors. On the other hand, C7 has a complex structure and a relatively high molecular weight, which does not align with the characteristics of ATP-competitive inhibitors targeting EGFR.
To provide further insights into the binding modes of EGFRLTC inhibitors, we analyzed the predicted distances between residues E762 and G857 for the 11 newly identified candidate compounds (C1 to C11), as shown in Fig. S14a. Most positive inhibitors (C1, C4, C7, C10, and C11) exhibit distances below 5.3 Å, indicating that the αC-helix is in the “in” state. In contrast, C5 displays a slightly longer distance of 5.75 Å, suggesting a possible “out” or intermediate state of the αC-helix. This observation is further illustrated in the distance probability distributions (Fig. S14b), where two distinct peaks, corresponding to the “in” and “out” conformations of the αC-helix, are observed when the αC-helix binds to C5.
In addition to the E762–G857 distances, we analyzed the binding regions of these compounds to assess their interaction with the αC-helix region. As shown in Fig. S14c, all candidates are predicted to bind near the active site, consistent with our objective to identify inhibitors that directly target this region (“Methods”). Within this configuration, most positive inhibitors (C1, C4, C7, C10, and C11) are predicted to remain relatively distant from the αC-helix (minimum distance > 6 Å), aligning with the expected orthosteric binding mode. However, C5 is predicted to interact more closely with the αC-helix (minimum distance <5 Å), suggesting again that it may act as a dual inhibitor by interacting with both the active site and the αC-helix region (Fig. S14d). We modeled the complex structures using Protenix (v0.4.4)39 (Figs. S18–S23). The results confirm our prediction that C1, C4, C7, C10, and C11 predominantly occupy the orthosteric binding pocket, whereas C5 engages both the orthosteric and allosteric pockets. Additionally, when bound to C5, the αC-helix tends to adopt an “out” conformation (Fig. S24).
Conformational selectivity of ABL kinase inhibitors
Kinases are dynamic, interconverting between different conformational states40. Monitoring these transitions and characterizing the conformational states that a kinase populates has proven to be a significant challenge41. Kinases typically maintain highly conserved active states to ensure the accurate positioning of essential catalytic regions35. In contrast, their inactive states can be distinct among different kinases42. Examining how small molecules selectively bind individual kinases could be instrumental in developing inhibitors specifically targeting these unique inactive states42. Here, we investigated whether Ligand-Transformer can be used to predict the ligand binding-induced conformational population shift of the ABL kinase, which plays a pivotal role in several signalling pathways, governing crucial cellular processes such as growth, survival, invasion, adhesion, and migration43,44.
ABL has three major conformational states, one active state (A) and two inactive states (I1 and I2)41. To investigate the ability of Ligand-Transformer to capture the change of the conformation ensemble of ABL after binding, we collected 12 inhibitors of this kinase, each with a predominant state of ABL when bound, as determined by nuclear magnetic resonance (NMR) spectroscopy41. A total of 60 possible structures of ABL were obtained from the PDB IDs 6XR6, 6XR7, and 6XRG, with 20 structures corresponding to each of the three states A, I1 and I2 (Fig. 3a). We then used the predicted distances between the residues of ABL as constraints to reweight the conformational ensemble consisting of the 60 structures and calculate the population of each conformational state (see “Methods”). Upon binding, the predicted predominant states are in accordance with experimental measurements for 11 of 12 compounds (Fig. 3b). Furthermore, when grouping the inhibitors by their labelled predominant state, we found that each group had a significantly higher population within the corresponding state compared to the other two groups.

a Average distance maps and structural ensembles for ABL kinase states A (blue), I1 (orange), and I2 (green), corresponding to PDB IDs 6XR6, 6XR7, and 6XRG, respectively. b Conformational selectivity prediction of different inhibitors. Twelve molecules were divided into three groups based on their conformational selectivity as determined in the literature41: Group A (blue, n = 6), Group I1 (orange, n = 2), and Group I2 (green, n = 4). The grouped bar graph illustrates the differences in predicted population of binding state conformations of ABL when interacting with inhibitors from different groups. The red triangle represents the population of ABL conformations predicted by AlphaFold2 in the apo state. Population estimations are derived from predicted distance matrices (see “Methods” for details of calculations). The predicted populations of ABL state bound with each individual ligand are listed in Table S5. Source data are provided as a Source Data file.
We conducted a detailed exploration of the predicted distance distributions that illustrate the distinct conformation ensembles of ABL when bound with various inhibitors. Our analysis (Fig. 4) focuses on the spatial positioning of the αC-helix (residues 299–311), the phosphate-binding loop (P-loop, residues 267–275), and the activation loop (A-loop, residues 400–424) of ABL. These elements are pivotal in differentiating the three states of ABL41. We chose four representative residue pairs, selected for their high F-statistics indicating their effectiveness in distinguishing between the three states, which enabled us to investigate the distances between critical protein components.

a–c Difference distance matrices illustrating the structural variations between the three states of ABL: a Difference distance matrix of state I1 minus state A. b Difference distance matrix of state I2 minus state A. c Difference distance matrix of state I1 minus state I2. d F-statistic values derived from the one-way ANOVA to assess variations in distance distributions across each pair of residues among the three states (n = 20 per state). This test is two-sided. No adjustments were made for multiple comparisons. The color intensity correlates with the significance of the distance variation, with the accompanying scale indicating p-values. Source data are provided as a Source Data file.
The ability of Ligand-Transformer to characterise the conformational space of the protein target and its ligand in their complex state can be appreciated by analyzing the distance between V308 and F401, which represents the distance between the αC-helix and the DFG motif, comprising residues D400, F401, and G402, at the start of the activation loop (Fig. 5). The increased distance in states A, I1, and I2 is notable, reflecting the DFG-out conformation in states I1 and I2, and the DFG-in state in state A41. The distance between residues V275 and F401 represents the gap between the DFG motif and the P-loop (Fig. S15). This distance is significantly reduced in state I2, which adopts a P-loop stretched conformation, as opposed to the P-loop kinked conformation in states A and I141. Predictions from Ligand-Transformer corroborate this trend. The distance between residues H380 and G402 represents the span from the active site to the activation loop (Fig. S16). In state I2, which adopts an A-loop closed conformation41, this distance is larger than in the other states, and the measured structures show that the distance in state I1 is slightly shorter than in state A. In state I2, the A-loop-closed conformation results in a shorter distance between L403 and L406, indicating a more compact activation loop (Fig. S17).

a Structural overlay highlighting the distance between residues V308 and F401 in the conformations of states A (PDB ID 6XR6, blue), I1 (PDB ID 6XR7, orange), and I2 (PDB ID 6XRG, green) of ABL. Residues are represented by spheres at the Cβ atoms, and the proteins are depicted as ribbons. b Depiction of the spatial positioning of residues V308 and F401 within the ABL kinase state A (PDB ID 6XR6). The sequence annotation is based on UniProt entry P00519, with the ATP binding site, αC-helix, active site, and activation loop colored in teal, violet, red, and magenta, respectively. The protein backbone is rendered as a cartoon, the active site residues as sticks, and the Cβ atoms of residues V308 and F401 as spheres. c Distance distributions between residues V308 and F401 for ABL in complex with 12 different inhibitors. Kernel density estimations (KDE) plots for the distances from 20 measured structures are shown for state A (blue), state I1 (orange), and state I2 (green). Predicted distance probability distribution of the apo state, derived from AlphaFold2 (AF2), is depicted as grey bars with a bin width of 0.3 Å. The Ligand-Transformer predicted distance probabilities for the 12 inhibitors are displayed as colored bars with a bin width of 0.19 Å. The mean values of predicted distances are plotted as solid lines, with dashed lines representing the standard deviation. The symbol in the upper right corner denotes which conformational state of ABL the inhibitor selectively binds to as determined by NMR analysis41.
These conformational changes are all reflected in the distance predictions of Ligand-Transformer. Specifically, when ABL in the holo state is bound with an inhibitor selective to a specific state, it tends to favor a distance distribution pattern that aligns with the measured distribution of that particular state (Figs. 5 and S15–S17). Moreover, the predicted distance distributions from Ligand-Transformer exhibit a wide range with multiple peaks, differing from the apo state predictions by AlphaFold2. This suggests that Ligand-Transformer can provide information about the structural ensembles of a protein-ligand complex. These conformational changes upon binding are central to the biological regulation of kinase activity. For instance, the active conformation (state A) of ABL is associated with substrate phosphorylation and downstream signaling, while the inactive states (I1 and I2) serve as key regulatory checkpoints. Small molecules that preferentially stabilize specific conformations can either inhibit or activate the kinase, influencing cell fate decisions such as proliferation, apoptosis, and migration. Therefore, the ability to predict conformational population shifts has important implications for understanding drug mechanism of action and for the design of selective inhibitors that avoid off-target effects by exploiting unique features of inactive states. This capability is particularly relevant for kinases like ABL, where mutations can confer drug resistance by altering the accessible conformational landscape.
Discussion
We described a sequence-based virtual screening method of predicting the conformational space of a target protein and a ligand in their complex state, thus overcoming the limitations of relying on the structures of the binding partners in their free states. In this way, this approach provides the binding affinity and the corresponding binding mode, represented as distance matrices between the target protein and the ligand.
Through comparisons with baseline models30,31,32 and ablation experiments, we observed that Ligand-Transformer performs well in affinity predictions (Table S1, Supplementary Discussion). This result can be attributed to the protein and molecular representations provided by pre-trained AlphaFold26 and GraphMVP29, as well as the structural information learned from the distance matrices of protein-ligand complexes during the training.
We illustrated the use of Ligand-Transformer by predicting ligands for the mutant EGFRLTC kinase with a hit rate of over 50% in this particular case (7/12 positive compounds, Table S4), finding novel high-affinity ligands, showing that Ligand-Transformer enables out-of-distribution predictions for specific protein targets. Notably, Ligand-Transformer achieves this accuracy while being over two orders of magnitude faster than state-of-the-art co-folding (Boltz-1)45 and structure-based docking (Vina-GPU 2.1)46 methods (Fig. 6).

Estimated time to screen 10,000 molecules using Ligand-Transformer (a) and other popular docking methods (b). Observe that Ligand-Transformer is two orders of magnitude faster than Boltz-1.
Next, as the free energy landscape of a protein is crucial in determining its behavior47,48,49, we investigated the problem of predicting conformational ensembles, which remains an open problem in the development of next-generation structural prediction algorithms50. In this context, Ligand-Transformer offers an approach towards understanding the impact of ligand binding on conformational ensembles using predicted distance matrices. Specifically, the rational design of allosteric modulators of kinases, particularly type-4 inhibitors42 and activators51, often encounters challenges with the traditional lock-and-key paradigm. Our methodology proposes a potential solution, enabling the design of ligands that modulate kinase activity by influencing the population distribution across various conformational states.
The results that we presented indicate that Ligand-Transformer can accurately predict the interactions of small molecules with proteins, including binding affinities and population shifts, thus helping understand molecular mechanisms, and offering a tool to replace high-throughput experimental assays in the initial steps in drug design pipelines.
Methods
Protein representations generated by AlphaFold2
We utilized AlphaFold2, a pre-trained model to extract protein representations6. The protein representation consists of three key components. Firstly, we generated the single sequence representation \(\{{{{\bf{f}}}}_{i}^{{{\rm{msa}}}}\}\), where \({{{\bf{f}}}}_{i}^{{{\rm{msa}}}}\in {{\mathbb{R}}}^{{c}_{{{\rm{msa}}}}}\), \({c}_{{{\rm{msa}}}}=384\), and \(i\in \{1...{N}_{{{\rm{res}}}}\}\). This representation is derived by linearly projecting the first row of the multiple sequence alignment (MSA) representation. The MSA representation is the output of the final layer of the Evoformer and serves as the input to the structure module in AlphaFold26. Secondly, we established pair representations \(\{{{{\bf{f}}}}_{{ij}}^{{{\rm{pair}}}}\}\) for each pair of residues i and j, where \({{{\bf{f}}}}_{{ij}}^{{{\rm{pair}}}}\in {{\mathbb{R}}}^{{c}_{{{\rm{pair}}}}}\), \({c}_{{{\rm{pair}}}}=128\). The pair representations \(\{{{{\bf{f}}}}_{{ij}}^{{{\rm{pair}}}}\}\) are also outputs of the Evoformer and the input to the structure module in AlphaFold2 and are used in predicting inter-residue distances in AlphaFold2. Lastly, the structure representation \(\{{{{\bf{f}}}}_{i}^{{{\rm{struc}}}}\}\) was obtained from the final layer of the structure module in AlphaFold2, where \({{{\bf{f}}}}_{i}^{{{\rm{struc}}}}\in {{\mathbb{R}}}^{{c}_{{{\rm{struc}}}}}\), \({c}_{{{\rm{struc}}}}=384\), and \(i\in \{1...{N}_{{{\rm{res}}}}\}\). This structure representation is employed in AlphaFold2 for predicting side-chain dihedral angles and model confidence prediction. The MSA searching was conducted by MMseqs2 (default setting) on BFD/MGnify and Uniclust30 (2021_03). No structural template was fed during the prediction. Model 1.1.1 of AlphaFold2 was used for the inference, and no structural template was used.
Ligand representations generated by GraphMVP
For obtaining a stereospecific molecular geometry representation, we used GraphMVP, a pre-trained molecule encoder that transforms 2D ligands into graph representations29. GraphMVP is trained via self-supervised learning (SSL) using auxiliary tasks. Here, both the input atoms and chemical bonds underwent a one-hot encoding process before being input into GraphMVP. The outputs generated include the atom representation, denoted as \(\{{{{\bf{f}}}}_{k}^{{{\rm{atom}}}}\}\), where \({{{\bf{f}}}}_{k}^{{{\rm{atom}}}}\in {{\mathbb{R}}}^{{c}_{{{\rm{atom}}}}}\), \({c}_{{{\rm{atom}}}}=100\), and \(k\in \{1...{N}_{{{\rm{atom}}}}\}\), and the bond representation, represented as \(\left\{{{{\bf{f}}}}_{{kl}}^{{{\rm{bond}}}}\right\}\), where \({{{\bf{f}}}}_{{kl}}^{{{\rm{bond}}}}\in {{\mathbb{R}}}^{{c}_{{{\rm{bond}}}}}\), \({c}_{{{\rm{bond}}}}=100,\) for each chemical bond formed between atom k and atom l.
Datasets
PDBbind2020-subset
We utilized the publicly accessible PDBbind v2020 dataset37, which provides the structures of 19,443 protein-ligand complexes along with their experimentally measured binding parameters. We eliminated structures with more than one ligand and those whose ligand structures could not be read by RDKit (http://www.rdkit.org/). Additionally, to enhance the training efficiency, we initially limited the dataset to complexes where the ligand atom count was 128 or fewer, and the protein sequence length was restricted to 384 amino acids or fewer. For sequences exceeding 384 amino acids in length, we applied a truncation strategy (see Supplementary Methods and Discussion) by eliminating domains that were distant from the binding site. This resulted in a subset dataset (PDBbind2020-subset) consisting of 13,420 data points. We subsequently randomly divided the dataset into training, validation, and testing sets, containing 10,375, 640, and 936 data points, respectively (see Supplementary Data 1). During model training, we initially conducted warm-up training with 4480 complexes (PDBbind_4k) that only included single chains with lengths less than 384. Following this, we proceeded to train with the complete training set (PDBbind_10k) of 10,375 data points (see Supplementary Methods and Discussion for training details).
For activity data, the PDBbind dataset includes dissociation constant (Kd), half maximal inhibitory concentration (IC50), and inhibition constant (Ki) measurements. As prediction labels, we took the negative logarithm of these values (in molar units), resulting in the pKd, pIC50, and pKi values, respectively. We considered Ki and Kd measurements without distinction to train Ligand-Transformer to predict Kd values. The IC50 data are assay-specific and only comparable under certain conditions. Based on literature research52, augmenting mixed public IC50 data with public Ki data does not deteriorate the quality of the mixed IC50 data if the Ki is corrected by an offset. Therefore, we did not explicitly distinguish among the three types of activity data. Instead, we corrected all IC50 values by dividing them by a factor 2.3. That is, we added 0.35 log units to all pIC50 values to obtain their pKd equivalents for use as prediction labels. Ligand-Transformer can thus predict Kd, IC50, and Ki values.
For the distance data, the ground truth distances are computed from the PDB files using Biopython. As the distance prediction has a lower limit of 1 Å and an upper limit of 20 Å, all labels are truncated within this range when calculating the absolute error between the predicted distances and true distances.
EGFRLTC-290
We collected 400 inhibitors targeting the L858R/T790M/C797S triple-mutant EGFR from ChEMBL53 and SciFinder (as of November 2022). These inhibitors were obtained from about 90 literature sources, and all data were double-checked with the original publications to ensure some important information, e.g., tested with human protein, and L858R/T790M/C797S triple-mutant EGFR form. For duplicates, we retained the one with higher bioactivity, resulting in a final set of 290 inhibitors with IC50 values ranging from 0.08 nM to 150 μM. We classified the inhibitors into orthosteric inhibitors, allosteric inhibitors, or those that bind to both sites, based on information from their originally published literature.
TargetMol library for screening EGFRLTC inhibitors
The library used for virtual screening for EGFRLTC inhibitors in this study contained a total of 9090 compounds, all sourced from TargetMol. This collection encompassed 2040 approved drugs, 5370 bioactive compounds, and 1680 natural compounds. Following the deduplication process, the final count of unique compounds in the library was reduced to 5600, as detailed in Supplementary Data 3.
Screening strategy of EGFRLTC inhibitors
When selecting candidate ligands from the TargetMol library for binding to EGFRLTC, we first considered the predicted binding activity. We normalized the predicted affinities from 11 models, including the base model (training with hyperparameters described in Table S3) and fine-tuned models (Model FT1 to FT10), and defined an overall affinity score as the minimal predicted affinity (normalized) among the 11 models (see Supplementary Methods and Discussion). In screening the candidate ligands, we also took into account the binding location of the ligands. We calculated the predicted distances of the ligand to residues K745, E762, D855, the three functional residues of EGFR, and normalized these distances to obtain a distance score. We screened molecules based on the following criteria: overall affinity score > 0.3 (i.e., top 50 results) or affinity score of any model rank <10; and all distance scores <0.5, to ensure that the binding location of the small molecule is not too far from the target region. Following these criteria, we obtained 12 candidates (Table S4).
Reweighting state populations of the ABL kinase
ABL exists in three different states, with state A corresponding to PDB ID 6XR6, state I1 corresponding to PDB ID 6XR7, and state I2 corresponding to PDB ID 6XRG. Each PDB file contains a set of \({N}_{{{\rm{conf}}}}=20\) conformations. We use the average residue distance among the 20 conformations in each state as the distance matrix {\({d}_{{ij}}^{s}\)}. Specifically, \({d}_{{ij}}^{s}=\frac{1}{{N}_{{{\rm{conf}}}}}{\sum }_{n=1}^{{N}_{{{\rm{conf}}}}}{d}_{{ij}}^{s(n)}\), where state \(s\in \left\{{{\rm{A}}},{{{\rm{I}}}}_{1},{{{\rm{I}}}}_{2}\right\}\), and \(i,j\in \{1,\ldots,{N}_{{{\rm{res}}}}=287\}\). The conformational ensemble of the ABL kinase can be simplified and represented by the weights \({{\bf{w}}}=[{w}^{{{\rm{A}}}},{w}^{{{{\rm{I}}}}_{1}},{w}^{{{{\rm{I}}}}_{2}}]\) of the three states. Consequently, the distance matrix \(\left\{{\bar{d}}_{{ij}}({{\bf{w}}})\right\}\) of the conformational ensemble can be expressed as a weighted average: \({\bar{d}}_{{ij}}\left({{\bf{w}}}\right)={\sum }_{s}{d}_{{ij}}^{s}{w}^{{{\rm{s}}}}\). During the reweighting process, we optimize the weights w to minimize the mean squared error (MSE) between the protein distance matrix \(\{{\hat{d}}_{{ij}}\}\) predicted by Ligand-Transformer and the distance matrix \(\{{\bar{d}}_{{ij}}\left({{\bf{w}}}\right)\}\) of the conformational ensemble. The optimized weights w* are considered to represent the conformational population corresponding to the Ligand-Transformer predicted distance matrix. Specifically, we use Sequential Least Squares Programming (SLSQP) from SciPy to solve the following optimization problem with constraints and bounds:
Baselines
HAC-Net
We obtained the code for HAC-Net from the official repository at https://github.com/gregory-kyro/HAC-Net, and used their pre-processed input HDF files for the PDBbind2020 dataset. We adjusted the dataset split to align with the division we utilized for training and testing. The training process and hyperparameters adhered to their default settings.
TankBind
We sourced the code for TankBind from https://github.com/luwei0917/TankBind. Using their dataset construction script, we constructed a dataset split for PDBbind2020 consistent with what we used in our study. We adhered to their default training settings to retrain their model on our version of the dataset.
MONN
We obtained the code from https://github.com/lishuya17/MONN. Their dataset pre-processing discards complexes with molecules like nucleic acids and polypeptides. Therefore, we made certain modifications to their data handling code to accommodate all the data we required. We rebuilt the dataset to align with the split we used in our paper and retrained their model using their default parameters.
In vitro EGFRLTC inhibition assays
The HTRF KINASE-TK assay kit (Cat#62TK0PEJ) was purchased from PerkinElmer for evaluating compound inhibition against EGFR L858R/T790M/C797S kinase. Compounds were serially diluted in DMSO to achieve a final assay concentration 200-fold lower than the detection concentration, maintaining a consistent final DMSO content of 0.5% in the assay system. A total of 25 nL of each compound was transferred to a 384-well reaction plate (Greiner, Cat#784075) using an Echo655 acoustic dispenser. The EGFR L858R/T790M/C797S kinase was used at a working concentration of 0.7 nM, chosen within the linear range where the reaction rate remains constant to ensure accurate activity measurements. The kinase solution was prepared in 1× kinase reaction buffer (comprising 5 mM MgCl₂, 1 mM DTT, and 1 mM MnCl₂), and 2.5 μL of this solution was added to each well. The plate was centrifuged at 140 g for 1 min and incubated at 25 °C for 10 min. To initiate the reaction, a substrate-ATP mixture was prepared in the same kinase reaction buffer, containing TK substrate at a saturating working concentration of 1 μM and ATP at 0.2 μM, a concentration approximately equal to the Km value. This ATP level was selected in accordance with the Cheng-Prusoff equation (where ATP = Km, IC50 = 2Ki) to accurately reflect the inhibitory potency of test compounds. Then, 2.5 μL of the substrate-ATP mixture was added to each well, followed by centrifugation at 140 g for 1 min. The plate was sealed and incubated at 25 °C for 50 min. Subsequently, a 2× detection reagent mixture containing XL665 and anti-phospho-tyrosine antibody in detection buffer was prepared, and 5 μL of this solution was added to each well. The plate was centrifuged again (140 g, 1 min) and incubated at 25 °C for 60 min.
Fluorescence emissions at 620 nm (Cryptate) and 665 nm (XL665) were measured using a microplate reader. Each assay condition was tested in duplicate. The percentage of kinase inhibition induced by the compounds was quantified using the equation:
To evaluate the potency of the inhibitors, IC50 values and dose-response curves were generated using GraphPad Prism 7.0 software. This was achieved by fitting the calculated percentage inhibition and the logarithm of the compound concentrations to a variable slope (four-parameter) nonlinear regression model. The model is expressed by the equation:
where X denotes the log of the inhibitor concentration and Y represents the percentage inhibition. Furthermore, the percentage of enzymatic activity in the presence of inhibitors, expressed as % Control, is derived by subtracting the % Inhibition from 100%.
Screening efficiency
To evaluate the computational efficiency of Ligand-Transformer compared to other state-of-the-art docking methods, we estimated the runtime required for a typical virtual screening task of 10,000 molecules against protein targets of varying lengths. For Ligand-Transformer, we screened a subset of FDA-approved drugs (n = 2500) against all proteins in the human proteome with fewer than 565 residues, while for Boltz-1 we evaluated a reduced set of 11 proteins ranging from 50 to 550 amino acids in length (sampled every 50 amino acids) with 100 molecules due to its substantial computational cost; in both cases, we extrapolated these results to estimate the time required for screening 10,000 molecules. For traditional docking methods such as Vina, runtime depends primarily on docking box size rather than protein length. Therefore, we used the average runtime across proteins in the AutoDock-GPU 140 benchmark46 set as an estimate of typical performance, treating this as a constant independent of protein length.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All structural models predicted by Protenix (v0.4.4, https://protenix-server.com) are available at the ModelArchive Database (https://modelarchive.org) with the identifiers ma-2zc19, ma-e6x11, ma-sjtz3, ma-7o3rc, ma-jzm4l, and ma-3jjn5. Additional data generated in this study are provided in the Supplementary Information. Source data are provided with this paper.
Code availability
The code used to develop the model, perform the analyses, and generate the results in this study is Ligand-Transformer (v0.1.0, https://github.com/zshengyu14/LigandTransformer), publicly available and free for academic use under the MIT License (snapshot archived at Zenodo, https://doi.org/10.5281/zenodo.15467622). Third-party software used includes ColabFold (v1.3.0, https://github.com/sokrypton/ColabFold)—MIT License; GraphMVP (https://github.com/chao1224/GraphMVP)—MIT License; HAC-Net (v1.4.2, https://github.com/gregory-kyro/HAC-Net)—MIT License; TankBind (v0.5.0, https://github.com/luwei0917/TankBind)—MIT License; and MONN—available for non-commercial research use only. Original license headers and copyright statements have been retained in all source files.
References
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with alphafold3. Nature 630, 493–500 (2024).
Del Alamo, D., Sala, D., Mchaourab, H. S. & Meiler, J. Sampling alternative conformational states of transporters and receptors with AlphaFold2. eLife 11, e75751 (2022).
Evans, R. et al. Protein complex prediction with AlphaFold-multimer. Preprint at https://www.biorxiv.org/content/10.1101/2021.10.04.463034v1 (2021).
Guo, H.-B. et al. AlphaFold2 models indicate that protein sequence determines both structure and dynamics. Sci. Rep. 12, 10696 (2022).
Heo, L. & Feig, M. Multi‐state modeling of G‐protein coupled receptors at experimental accuracy. Proteins 90, 1873–1885 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Krishna, R. et al. Generalized biomolecular modeling and design with RoseTTAFold all-atom. Science 384, eadl2528 (2024).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Robertson, A. J., Courtney, J. M., Shen, Y., Ying, J. & Bax, A. Concordance of X-ray and AlphaFold2 models of SARS-CoV-2 main protease with residual dipolar couplings measured in solution. J. Am. Chem. Soc. 143, 19306–19310 (2021).
Stein, R. A. & Mchaourab, H. S. SPEACH_AF: sampling protein ensembles and conformational heterogeneity with alphafold2. PLoS Comp. Biol. 18, e1010483 (2022).
Van Der Maaten, L. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 15, 3221–3245 (2014).
Wayment-Steele, H. K. et al. Predicting multiple conformations via sequence clustering and AlphaFold2. Nature 625, 832–839 (2024).
Brotzakis, Z. F., Zhang, S., Murtada, M. H. & Vendruscolo, M. AlphaFold prediction of structural ensembles of disordered proteins. Nat. Comm. 16, 1632 (2025).
Du, Y. et al. Machine learning-aided generative molecular design. Nat. Mach. Intell. 6, 589–604 (2024).
Sadybekov, A. V. & Katritch, V. Computational approaches streamlining drug discovery. Nature 616, 673–685 (2023).
Schneider, P. et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 19, 353–364 (2020).
Thomas, M., Bender, A. & de Graaf, C. Integrating structure-based approaches in generative molecular design. Curr. Op. Struct. Biol. 79, 102559 (2023).
Isert, C., Atz, K. & Schneider, G. Structure-based drug design with geometric deep learning. Curr. Op. Struct. Biol. 79, 102548 (2023).
Ren, F. et al. A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models. Nat. Biotech. 43, 63–75 (2025).
Ren, F. et al. AlphaFold accelerates artificial intelligence powered drug discovery: efficient discovery of a novel CDK20 small molecule inhibitor. Chem. Sci. 14, 1443–1452 (2023).
Ringel, M. S., Scannell, J. W., Baedeker, M. & Schulze, U. Breaking Eroom’s law. Nat. Rev. Drug Discov. 19, 833–834 (2020).
Waring, M. J. et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 14, 475–486 (2015).
Kuntz, I. D. Structure-based strategies for drug design and discovery. Science 257, 1078–1082 (1992).
Schneider, G. Virtual screening: an endless staircase? Nat. Rev. Drug Discov. 9, 273–276 (2010).
Lyu, J. et al. Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229 (2019).
Sadybekov, A. A. et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601, 452–459 (2022).
Aparoy, P., Kumar Reddy, K. & Reddanna, P. Structure and ligand based drug design strategies in the development of novel 5-lox inhibitors. Curr. Med. Chem. 19, 3763–3778 (2012).
Giordano, D., Biancaniello, C., Argenio, M. A. & Facchiano, A. Drug design by pharmacophore and virtual screening approach. Pharmaceuticals 15, 646 (2022).
Liu, S. et al. Pre-training molecular graph representation with 3D geometry. Int. Conf. Learn. Represent. (2022).
Lu, W. et al. Tankbind: trigonometry-aware neural networks for drug-protein binding structure prediction. Adv. Neural Inf. Process. Syst. 35, 7236–7249 (2022).
Li, S. et al. Monn: a multi-objective neural network for predicting compound-protein interactions and affinities. Cell Syst. 10, 308–322. e311 (2020).
Kyro, G. W., Brent, R. I. & Batista, V. S. Hac-Net: a hybrid attention-based convolutional neural network for highly accurate protein–ligand binding affinity prediction. J. Chem. Inf. Model. 63, 1947–1960 (2023).
Hynes, N. E. & Lane, H. A. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 5, 341–354 (2005).
Lu, X. et al. Targeting EGFRL858R/T790M and EGFRL858R/T790M/c797s resistance mutations in NSCLC: current developments in medicinal chemistry. Med. Res. Rev. 38, 1550–1581 (2018).
Jura, N. et al. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 42, 9–22 (2011).
Zhang, X., Gureasko, J., Shen, K., Cole, P. A. & Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 (2006).
Liu, Z. et al. PDB-wide collection of binding data: current status of the PDBbind database. Bioinformatics 31, 405–412 (2015).
Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010).
ByteDance AML AI4Science Team et al. Protenix-advancing structure prediction through a comprehensive AlphaFold3 reproduction. Preprint at https://www.biorxiv.org/content/10.1101/2025.01.08.631967v1 (2025).
Taylor, S. S. & Kornev, A. P. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77 (2011).
Xie, T., Saleh, T., Rossi, P. & Kalodimos, C. G. Conformational states dynamically populated by a kinase determine its function. Science 370, eabc2754 (2020).
Attwood, M. M., Fabbro, D., Sokolov, A. V., Knapp, S. & Schiöth, H. B. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat. Rev. Drug Discov. 20, 839–861 (2021).
Witte, O. N., Dasgupta, A. & Baltimore, D. Abelson murine leukaemia virus protein is phosphorylated in vitro to form phosphotyrosine. Nature 283, 826–831 (1980).
Greuber, E. K., Smith-Pearson, P., Wang, J. & Pendergast, A. M. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat. Rev. Cancer 13, 559–571 (2013).
Wohlwend, J. et al. Boltz-1: democratizing biomolecular interaction modeling. Preprint at https://www.biorxiv.org/content/10.1101/2024.11.19.624167v2 (2024).
Tang, S. et al. Vina-gpu 2.1: towards further optimizing docking speed and precision of Autodock Vina and its derivatives. IEEE/ACM Trans. Comput. Biol. Bioinform. 21, 2382–2393 (2024).
Lindorff-Larsen, K., Best, R. B., DePristo, M. A., Dobson, C. M. & Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 433, 128–132 (2005).
Boehr, D. D., Nussinov, R. & Wright, P. E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 (2009).
Mittermaier, A. & Kay, L. E. New tools provide new insights in NMR studies of protein dynamics. Science 312, 224–228 (2006).
Lane, T. J. Protein structure prediction has reached the single-structure frontier. Nat. Methods 20, 170–173 (2023).
Gong, G. Q. et al. A small-molecule pi3kα activator for cardioprotection and neuroregeneration. Nature 618, 159–168 (2023).
Kalliokoski, T., Kramer, C., Vulpetti, A. & Gedeck, P. Comparability of mixed IC50 data - a statistical analysis. PLoS ONE 8, e61007 (2013).
Mendez, D. et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 47, D930–D940 (2019).
Guo, Y., Gao, B., Gao, P., Fang, L. & Gou, S. Novel anilinopyrimidine derivatives as potential EGFRT790M/C797S inhibitors: design, synthesis, biological activity study. Bioorg. Med. Chem. 70, 116907 (2022).
Karnik, K. S., Sarkate, A. P., Tiwari, S. V., Azad, R. & Wakte, P. S. Design, synthesis, biological evaluation and in silico studies of EGFR inhibitors based on 4-oxo-chromane scaffold targeting resistance in non-small cell lung cancer (NSCLC). Med. Chem. Res. 31, 1500–1516 (2022).
Acknowledgements
We would like to acknowledge funding from UKRI (10059436 and 10061100, M.V.).
Author information
Authors and Affiliations
Contributions
S.Z., S.P.O., and M.V. conceived the project. S.Z., D.H., R.I.H., and S.P.O., performed the computational work. D.H., R.I.H., and Y.Q. performed the experimental work. M.V. and A.Y. funded the project. All authors analyzed data and wrote the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, S., Huo, D., Horne, R.I. et al. Sequence-based virtual screening using transformers. Nat Commun 16, 6925 (2025). https://doi.org/10.1038/s41467-025-61833-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61833-8
This article is cited by
-
Navigating the labyrinth of drugging the disordered
Biophysical Reviews (2025)
