
Abstract
Epi-immunotherapy appears promising for hepatocellular carcinoma (HCC) treatment, but immunosuppressive macrophages limit the capacity of epigenetic regulation to activate T cell-mediated tumoricidal immunity. Here we report an epi-immune nanosatellite (stEiNS) that co-delivers siRNA targeting the YTH N6-methyladenosine RNA binding protein 1 (YTHDF1) alongside the histone deacetylase IIa inhibitor TMP195, enabling epigenetic reprogramming of HCC tumor cells and M2 macrophages to enhance the immunotherapeutic response. stEiNS assembles size-mismatched nanoparticles via dynamic locks in a satellite-like structure, enabling deep tissue penetration. Knockdown of YTHDF1 by stEiNS in HCC cells, along with stEiNS-driven antitumor macrophage phenotype induction, intensifies macrophages-cytotoxic T lymphocytes interactions with tumor cells. stEiNS suppresses TNF/NF-κB signaling in tumor cells to inhibit CCL2-driven recruitment of myeloid-derived suppressor cells while activating the IFNγ/STAT1 pathway in M2-phenotype macrophages to promote their polarization toward an M1 phenotype. Collectively, these effects trigger robust tumoricidal immunity, leading to efficient tumor eradication, as validated in patient-derived tumor organoids, orthotopic HCC models, and recurrence models. In summary, we establish a dual-targeting stEiNS with promising epi-immunotherapeutic potential against advanced HCC and diverse malignancies.
Similar content being viewed by others
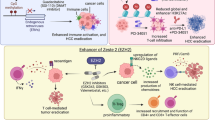


Introduction
Hepatocellular carcinoma (HCC), a highly lethal malignancy accounting for 85–90% of liver cancer cases, has a poor prognosis and high recurrence rate1,2. Clinically, combining vascular endothelial growth factor inhibitors with immune checkpoint inhibitors (ICIs) is pivotal for the treatment of advanced HCC3. However, very few patients benefit from these treatments. This limited efficacy of immunotherapy is largely attributed to epigenetic changes within the tumor immune microenvironment (TIME)4. In the TIME, tumor cells employ epigenetic mechanisms to diminish immune cell recognition and immunogenicity, thereby preventing immune-mediated cell death. Hence, reversible regulation by epigenetic therapy may be a potential strategy for clinical prevention and treatment of HCC.
The N6-methyladenosine (m6A) modification, the most prevalent mRNA modification, regulates mRNA splicing, transport, and translation, thereby influencing gene expression and key cellular processes. YTH m6A RNA-binding protein 1 (YTHDF1) preferentially binds methylated RNA to promote translation5,6, and overexpression of YTHDF1 can lead to the cancer immune desert phenotype and resistance to ICIs7. Furthermore, sustained neoantigen-specific immunity is also regulated by mRNA m6A methylation via YTHDF18. Hence, targeting YTHDF1 may offer strategies for overcoming immune evasion and enhancing immunotherapy efficacy in HCC.
However, immunosuppressive macrophages in HCC tumors hinder the effectiveness of m6A epigenetic regulation in T cell-mediated tumoricidal immunity, especially for immune-tolerant liver9,10,11,12. Macrophages (MΦs), essential components of innate immunity, promote tumor angiogenesis and mediate immunosuppression, closely correlated with a poor prognosis, especially when biased toward M2-type macrophages (M2Φs)13. Macrophage expression profiles are primarily regulated by epigenetic modifications, with rare somatic mutations14,15. Given the prevalence of these “renegade” MΦs, co-editing the epigenetic landscapes of both tumor cells and MΦs may be conducive to enhancing the immune efficacy of HCC treatments.
Here, we report an epi-immune nanosatellite (stEiNS) for effective delivery of siRNA targeting YTHDF1 and histone deacetylase (HDAC) IIa inhibitor TMP195 to epigenetically reprogram HCC tumor cells and M2Φs, which significantly increases the immunotherapeutic response against HCC. Specifically, we integrate two distinct nanoparticles with dynamic chemical bonds, locking smaller ones onto the surface of larger carriers to form a nanosatellite, termed stEiNS. These nanoparticles include M2Φ-targeting polypeptide (M2Φpep) and dopamine-modified TMP195 lipid nanoparticles (tDML), along with phenylboronic acid (PBA)-modified PAMAM dendrimers complexing siYTHDF1 (sPPN). Upon entering the tumor microenvironment (TME), smaller sPPN nanoparticles detach from stEiNS in response to hydrogen peroxide (H2O2), allowing deep tumor infiltration and then selectively binding to sialic acid (SA) on HCC cells via phenylboronic acid to enhance cellular uptake. Simultaneously, the M2Φpep on larger tDML nanoparticles exhibits high selectivity for M2Φs, effectively modulating histone acetylation to induce macrophage reprogramming. The locked stEiNS enables prolonged systemic circulation, while unlocking in the TME ensures dual-targeted drug delivery and facilitates effective therapeutic intervention. We demonstrate that stEiNS orchestrates the epigenetic modulation of the m6A reader YTHDF1, inducing HCC tumor cell apoptosis and driving macrophage phenotypic switching through histone acetylation (Fig. 1f). The cotargeting epi-immune strategy restores liver immune homeostasis, fostering a fully activated tumor immunity niche to effectively suppress HCC progression and recurrence.

a A boxplot of YTHDF1 expression in HCC patients and healthy people isolated from TCGA and Genotype-Tissue Expression determined by Gene Expression Profiling Interactive Analysis. The data were transformed as log2(TPM + 1). Normal: maxima 4.7099, minima 2.0076, center 3.4694; Tumor: maxima 5.6854, minima 2.3564, center 3.9079. Color key: red, HCC tissues; blue, normal liver tissues. YTHDF1 displays negative associations with (b) disease-free survival in HCC patients. c The prognostic significance of YTHDF1 in HCC patients. Number at risk refers to the number of patients in each experimental group who remain event-free at the specified time points and are still at risk of reaching the study endpoint. d The correlation between YTHDF1 and immune score. e YTHDF1 expression at the mRNA and protein levels in Huh7 and Hepa1-6 cells, with hepatocytes freshly isolated from C57BL/6 mice serving as the control (n = 3 independent experiments). f Schematic illustration of the role of the dynamic locking stEiNS in HCC tumor, which regulates the epigenetic modifications of both tumor cells and M2Φs to enhance the effectiveness of immunotherapy against HCC. Data are expressed as mean ± SD and were determined using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. ****P < 0.0001.
Results and discussion
YTHDF1 overexpression was associated with decreased immune infiltration and a poor prognosis in HCC
In the Cancer Genome Atlas (TCGA) dataset, YTHDF1 mRNA expression increased significantly in HCC tissues compared to normal tissues (Fig. 1a). This pattern was further confirmed in Huh7 and Hepa1-6 cell lines, where YTHDF1 expression at the mRNA and protein levels was significantly higher than that in the freshly isolated C57BL/6 hepatocytes (Fig. 1e). Furthermore, YTHDF1 expression negatively correlated with disease-free survival (Fig. 1b). Kaplan-Meier Plotter (KMP) analysis revealed a correlation wherein patients with HCC exhibiting heightened YTHDF1 expression demonstrated elevated recurrence rates (Fig. 1c), via GEPIA and Kaplan-Meier plotter (http://gepia.cancer-pku.cn, https://kmplot.com). An inverse relationship emerged between YTHDF1 expression and immune score, whereby increased expression of YTHDF1 aligned with low immune scores (Fig. 1d). This dichotomy was accentuated by the observation that the samples with low YTHDF1 expression showed substantial immune cell infiltration, which was indicative of an immune-inflammation phenotype. Conversely, the samples with high YTHDF1 expression exhibited lower levels of immune cell infiltration, suggesting an immune desert phenotype. Overall, elevated YTHDF1 expression in HCC was correlated with decreased immune infiltration and a poor prognosis.
Preparation and characterization of stEiNS
Clinically, elevated YTHDF1 expression in HCC, linked to poor prognosis and low immune score, highlights its potential in epi-immunotherapy, whereas immunosuppressive macrophages restrict m6A-targeted strategies. Meanwhile, small-molecule drugs often fail to achieve the desired spatiotemporal distribution in vivo, resulting in clinical outcomes that fall short of expectations16. Thus, a co-delivery system that epigenetically reprograms both tumor cells and M2Φs may be crucial for improving epi-immunotherapy efficacy in HCC. Cationic amphiphilic dendrimers, with strong affinity for siRNAs, protect RNA from degradation and extend their half-life, with PBA-modified G2 dendrimer nanoclusters and PEGylated G5-PBA dendrimers, stabilized by reversible boronate esters, demonstrating significant potential for optimized siRNA delivery17,18,19. Notably, we observed that PEG-modified G5 PAMAM dendrimers readily dispersed into uniform, small nanoparticles at pH 7.4 PBS. Leveraging this property, we selected small PBA-PEG-modified G5 dendrimers for siRNA delivery. Although reduced particle size enhances deep tumor penetration, it may limit efficient tumor accumulation, necessitating a balance between these factors.
Lipid nanoparticles (LNPs), a leading choice for systemic drug delivery, particularly for small molecules and genetic drugs, were selected for TMP195 delivery. Leveraging a transformable core-shell nanocarrier model20, we constructed a co-delivery system by integrating TMP195-loaded tDML and siYTHDF1-loaded sPPN into a nanosatellite (stEiNS) via an H2O2-controlled dynamic covalent bond. As depicted in Fig. 2a, unlike previous strategies, stEiNS dynamically locks two size-varied nanoparticles, offering great flexibility and precise control over drug ratios, representing an advancement over the previous approaches that modify one nanoparticle onto the structural components of another. Its nanosatellite structure ensures prolonged circulation and efficient tumor targeting, while rapid unlocking at the tumor site enhances deep penetration and robust epigenetic regulation, presenting a versatile platform for assembling two distinct nanoparticles in drug delivery. The nanosatellites co-loaded with TMP195 and siYTHDF1 are designated as stEiNS, whereas those loaded with siYTHDF1 alone are termed sEiNS, and those loaded with TMP195 alone are referred to as tEiNS.

a schematic illustration of the preparation of dynamic-locking stEiNS. b Transmission electron microscopy imaging of stEiNS, enlarged stEiNS, and H2O2-treated stEiNS (n = 3 independent experiments). c Particle size changes of tDML, sPPN, and stEiNS determined by dynamic light scattering (DLS) with the photographs of EiNS and stEiNS (n = 3 independent experiments). d Zeta potential alterations of tDML, sPPN, and stEiNS determined with DLS (n = 3 independent experiments). e Agarose gel electrophoresis of sPPN (n = 3 independent experiments). f UV absorption spectra of TMP195, DML, and tDML, sPPN, and stEiNS. g 1H NMR changes of PPN under an in vitro simulated tumor local high concentration H2O2 environment. h In vitro drug release profiles of free TMP195, tDML, and tEiNS (n = 3 independent experiments). (i) Storage stability of stEiNS in PBS at 4 °C. Data are expressed as mean ± SD.
Synthetic pathways for M2Φ pep-modified 1,2-distearoyl-sn-glycero-3-phosphate-ethanolamine (DSPE-M2), dopamine-modified DSPE (DSPE-DA), and PPN are shown in Supplementary Figs. S1, S2, along with their structural confirmation by 1H NMR and infrared spectroscopy (Supplementary Figs. S3–S8). The preparation methodology of stEiNS is shown in Fig. 2a. Lipid nanoparticles modified with DA and M2Φ peptide (tDML), composed of phospholipids, cholesterol, DSPE-DA, and DSPE-M2, were engineered for efficient TMP195 delivery. Meticulous optimization established a phospholipid concentration of 15 mg/mL, a phospholipid-to-cholesterol ratio of 8:1, and a drug-to-lipid mass ratio of 1:15 (Supplementary Fig. S9). Furthermore, the optimal mass ratio of PPN to siYTHDF1 was determined to be 25:1 to prepare sPPN, as corroborated by agarose gel electrophoresis (Fig. 2e).
Transmission electron microscopy revealed a characteristic round morphology of stEiNS (Fig. 2b). Its physical form (Fig. 2c) presented as a light blue opalescent liquid. tDML denoted M2Φ-targeted lipid nanoparticles loaded with TMP195, with a particle size of ~ 80 nm (Fig. 2c and Supplementary Fig. S10a). Conversely, sPPN comprised small nanoparticles loaded with siYTHDF1, featuring a particle size of ~ 20 nm (Fig. 2c and Supplementary Fig. S10b). Through the interaction of catechol and PBA, sPPN adhered to the surface of tDML upon coincubation, forming dynamic-locking stEiNS with a diameter of ~ 120 nm (Fig. 2c). The successful preparation of stEiNS was verified by changes in the zeta potential and UV absorption spectra (Fig. 2d, f). Furthermore, enlarged images of stEiNS revealed a surface densely decorated with numerous small nanoparticles, enabling the dynamic-locking switch (Fig. 2b), a feature absent in unmodified PAMAM combined with DML. stEiNS responded to 70 μM H₂O₂ which mimics the TME as confirmed by a hydrogen peroxide assay kit, and transformed into two distinct nanoparticle types (Fig. 2b). In the unique TME, the concentration of reactive oxygen species (ROS), particularly H2O2 is approximately 100 times higher than that in normal tissues. This elevated ROS level serves as an effective endogenous stimulus source, capable of freely diffusing across cellular membranes21. The disappearance of the characteristic benzene ring peak in the 1H NMR spectrum of PPN following 1 h of exposure to 70 µM H2O2 confirmed the cleavage of the amide bond and highlighted the rapid unlocking capability of stEiNS in the TME of HCC (Fig. 2g). Hence, we hypothesize that the extracellular TME harbors high levels of ROS, rendering the phenylboronic acid ester bond highly susceptible to cleavage. Consequently, stEiNS may circulate and accumulate at the tumor site, rapidly unlocking in the extracellular environment of the HCC tumor, which facilitates deep penetration into tumor tissue for dual-targeting of various cell populations.
As illustrated in Fig. 2h, free TMP195 served as a control to delineate the distinct release kinetics of the encapsulated versus unencapsulated drug, confirming the sustained release properties of the delivery system while excluding potential retention effects of the dialysis membrane. The drug exhibited an initial burst release, followed by a sustained release phase over an extended period. Free TMP195 was rapidly released, reaching near-complete release within 12 h. In contrast, tDML and tEiNS achieved approximately 70% TMP195 release over 48 h, significantly prolonging its release. Moreover, the drug release kinetics of H₂O₂-pretreated tEiNS and tDML showed no significant differences, indicating that PPN dissociation from tDML did not affect TMP195 release. Furthermore, the biocompatibility of stEiNS was evaluated using an MTT assay and a hemolysis test. The MTT assay confirmed that the blank formulation EiNS was highly safe, showing no significant impact on cell proliferation under the experimental concentrations (Supplementary Fig. S11a). In addition, less than 5% hemolysis was observed after incubation with various formulations at therapeutic concentrations in rat red blood cells, indicating the excellent biocompatibility of stEiNS (Supplementary Fig. S11b). Moreover, stEiNS maintained excellent stability in PBS, showing negligible differences in size and PDI for 7 days at 4 °C (Fig. 2i). Taken together, these results indicate that EiNS exhibits superior performance and has remarkable potential for the effective dual-targeting delivery of gene therapies and chemical drugs.
Improved dual-targeting of stEiNS
RNA drugs can theoretically manipulate virtually any gene of interest, whereas transmembrane crossing is limited by molecular weight, nuclease sensitivity and negative charge22. However, our investigations revealed robust Cy3 fluorescence in Huh7 and Hepa1-6 cells after administration of the sPPN and sEiNS formulations, with sEiNS subjected to preconditioning at 37 °C under H₂O₂ conditions. This highlights the remarkable capacity of both sPPN and sEiNS systems to facilitate rapid cytoplasmic internalization, with no discernible variance between the two nanocarriers (Fig. 3a–c). In Huh7 cells, all treatment groups achieved a Cy3-positive cell ratio of 99% (Fig. 3d). However, the mean fluorescence intensity (MFI) of sPPN and sEiNS were double those of the Lipofectamine 3000/s (Lipo/s) group (Fig. 3e). Notably, in Hepa1-6 cells, sPPN and sEiNS achieved a striking Cy3-positive cell ratio of 99%, in stark contrast to the modest ~ 61% observed with Lipo/s, accompanied by a corresponding 15-fold increase in MFI (Fig. 3d, e). To further assess the targeting efficiency of EiNS, we employed antibodies to block SA receptors23. Siglec-2/CD22 modulates immune responses by recognizing SA residues24, and can thus serve as a functional blockade for SA receptors to evaluate the specificity of SA-targeting agents. Blockade of Siglec-2/CD22 in Huh7 and Hepa1-6 cells markedly reduced s-Cy3EiNS uptake compared to untreated control (Supplementary Fig. S12a, b), indicating SA-dependent targeting. Our study underscores the superiority of sPPN and sEiNS over Lipofectamine 3000 in terms of transfection efficiency.

a, b Confocal images and (c) flow cytometric analysis illustrating the in vitro internalization of s-Cy3EiNS after 12 h of incubation with Huh7 and Hepa1-6 cells, with siYTHDF1 labeled with Cy3 (n = 3 independent experiments). Scale bar, 20 µm. d YTHDF1Cy3+ ratio in Huh7 and Hepa1-6 cells treated with various formulations (n = 3 independent experiments). e Mean fluorescence intensity (MFI) ratio of YTHDF1Cy3+ in Huh7 and Hepa1-6 cells across different treatment groups (n = 3 independent experiments). Confocal images showing subcellular localization in (f) Huh7 and (g) Hepa1-6 cells incubated with s-Cy3EiNS for 1, 3, and 6 h at 37 °C (n = 3 independent experiments). Nuclei were stained with DAPI (blue), endo/lysosomes with LysoTracker Green (green), and siYTHDF1 with Cy3 (red). h, i Quantitative co-localization analysis of S-Cy3 with endo/lysosomes labeled by LysoTracker Green (n = 3 independent experiments). j Schematic illustration of deep tumor penetration of s-Cy3EiNS in 3D tumorspheres. Created in BioRender. Y.Q. (2025) https://BioRender.com/r8jogdb. k Representative CLSM images showing enhanced penetration of sEiNS compared to Lipo/s in Huh7 and Hepa1-6 tumorspheres (n = 3 independent experiments). Scale bar, 100 µm. l The IVIS imaging and (m) fluorescence intensity quantification at various time points in mice with subcutaneous Hepa1-6 tumors (tumors marked with white circles, n = 3 mice per group). EiNS(-M2Φpep), EiNS without M2Φpep modification. Data are expressed as mean ± SD. Statistical significance was determined using one-way ANOVA followed by Tukey’s multiple comparisons test (d, e, h, i), or two-way ANOVA (m). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Huh7 and Hepa1-6 cells were treated with sEiNS, and the colocalization of the red fluorescence of Cy3 with the green fluorescence of LysoTracker Green gradually diminished over prolonged incubation periods, as evidenced by confocal fluorescence images (Fig. 3f, g). This decrease in colocalization indicated the effective escape of sEiNS from the endo/lysosomes. Quantitative evaluation of sEiNS colocalization with endo/lysosomes using Mander’s coefficients M1 and M2 further corroborated these observations (Fig. 3h, i). Taken together, these results underscore the potential biomedical applications of sEiNS in facilitating the intracellular delivery and lysosomal escape of siYTHDF1. PAMAM dendrimers escape into the cytoplasm via the proton sponge effect, with coarse-grained molecular dynamics simulations indicating that they promote endosomal rupture by increasing osmotic pressure and locally penetrating the membrane25.
The findings of the three-dimensional (3D) tumor sphere experiment demonstrated distinct fluorescence patterns at a depth of 120 μm. In particular, in the Lipo/s group, with an average particle size of ~ 60 nm, fluorescence was primarily clustered around the periphery of the tumor sphere. In contrast, the sEiNS group displayed a higher fluorescence intensity, characterized by a substantial distribution throughout the central region of the tumor sphere (Fig. 3j, k). These observations provide compelling evidence of the ability of sEiNS to penetrate deeply into the tumor tissue upon unlocking, effectively mitigating the diffusion constraints commonly encountered by nanomedicines within the intricate TME. Consistent with this, Chen et al.26 proposed that the 22-nm nanoparticles exhibit superior penetration capabilities compared to their 60-nm counterparts, as they can traverse both transcellular and paracellular pathways with remarkable efficacy.
The dual-targeting capability of EiNS was validated in vivo. As the liver is a major site for drug distribution and metabolism, Cy3 and Dir fluorescence signals were also detected in hepatic tissue, complicating accurate assessment of HCC-specific distribution (Fig. 3l). To evaluate the tumor-targeting capability of EiNS, a subcutaneous HCC model was established in C57BL/6 mice. Following intravenous administration, IVIS images indicated that both siYTHDF1-Cy3EiNS (s-Cy3EiNS) and DirEiNS exhibited significantly stronger fluorescence signals in the tumor region than siYTHDF1-Cy3 and DirEiNS(-M2Φpep), demonstrating improved in vivo targeted drug delivery capacity of EiNS (Fig. 3l, m). In addition, 24 h post-administration, mice were dissected to obtain tissue samples of the heart, liver, spleen, lung, kidney, and tumors. The fluorescence intensities of Cy3 and Dir in the tumor were significantly higher following EiNS delivery compared to siYTHDF1-Cy3 or DirEiNS(-M2Φpep) (Supplementary Fig. S13a).
The dual-targeting drug delivery capacity of EiNS was further investigated for HCC tumor cells and M2Φs. M2-like macrophages were identified using a CD206 antibody and stratified into the low-expression area (tumor region) and high-expression area (M2-like macrophage region). Analysis of tumor sections revealed that, compared to free siYTHDF1-Cy3, intravenous administration of s-Cy3EiNS led to significantly higher Cy3 red fluorescence in tumor regions (Supplementary Fig. S13b). Notably, in the FITC-loaded EiNS (fEiNS) group, fluorescence signals were particularly enriched in M2-like macrophage-dense regions (Supplementary Fig. S13c). This suggested that EiNS disassembled into DML and PPN within the TME, promoting deeper penetration of PPN into tumor tissues and selectively targeting different cell types. EiNS can accumulate at the tumor site via both passive and active mechanisms, leveraging the enhanced permeability and retention effect, PBA-mediated recognition of SA-overexpressing HCC cells, and the high selectivity of M2Φpep for M2Φs23,27.
Therapeutic effect of sEiNS in vitro
The silencing efficacy of target genes is a critical parameter in evaluating gene delivery materials. YTHDF1 expression in Huh7 and Hepa1-6 cells was markedly suppressed at both mRNA and protein levels following transfection with Lipofectamine 3000 or treatment with engineered sEiNS (Fig. 4a, b). To assess the role of YTHDF1 in HCC, knockdown was induced using various formulations over 4 days. YTHDF1 silencing in Huh7 and Hepa1-6 cells potentiated the growth-inhibitory effects of sPPN and sEiNS, outperforming free siYTHDF1 or Lipo/s, and indicating superior delivery efficiency of engineered sEiNS (Fig. 4c, d). However, siYTHDF1 alone had a negligible effect on the proliferation of both cell lines. Furthermore, downregulation of YTHDF1 expression by sEiNS significantly suppressed colony formation, migration, and invasion of Huh7 and Hepa1-6 cells, and promoted cell apoptosis, thus inhibiting the progression of HCC tumors (Fig. 4e–h, Supplementary Figs. S14, and S15).

a YTHDF1 mRNA expression levels in Huh7 and Hepa1-6 cells following treatment with diverse nanomedicines (n = 3 independent experiments). b YTHDF1 protein expression levels in Huh7 and Hepa1-6 cells after exposure to various nanomedicines (n = 3 independent experiments). Cell viability of (c) Huh 7 and (d) Hepa1-6 cells assessed by MTT assay following treatment with various formulations (n = 3 independent experiments). e Representative images and (f) quantification of clonal assay in Huh7 and hepa1-6 cells with treatment of various formulations were presented (n = 3 independent experiments). g, h Apoptosis analysis and statistical evaluation of apoptosis rates in Huh7 and Hepa1-6 cells treated with sPPN, sEiNS, and Lipo/s for 24 h (n = 3 independent experiments). i Treatments of HCC patients-derived PDOs in different treatment groups (n = 3 independent experiments). Scale bar, 100 µm. Data are expressed as mean ± SD. Statistical significance was determined using one-way ANOVA followed by Tukey’s multiple comparisons test (a, f, g), or two-way ANOVA (c, d). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Building on the remarkable therapeutic efficacy of sEiNS, patients-drived organoids (PDOs) have been utilized to predict real-world drug responses in personalized treatment settings. Two PDOs with high YTHDF1 expression (Supplementary Fig. S16) were generated from individual primary liver cancer patients, and their morphologies after treatment with various formulations were observed using bright-field microscopy. Compared with other formulations, sPPN and sEiNS resulted in the smallest PDO volumes after treatment, demonstrating significant inhibition of HCC tumor progression (Fig. 4i).
Promoted MΦ phagocytosis of tumor cells by tEiNS
The immune-tolerant liver microenvironment, shaped by chronic antigen exposure and immunosuppressive non-parenchymal cells such as Kupffer cells, presents a major barrier to HCC immunotherapy. This tolerogenic landscape suppresses effector T cell responses and limits the efficacy of immune checkpoint blockade (ICB)9,10. Key immunosuppressive mechanisms include impaired immune synapse formation, T cell anergy, regulatory T cell induction, Fas–FasL-mediated apoptosis, and fibrinogen-like protein 1 (FGL1)-mediated immunosuppression11,12. The intricate crosstalk between tumor cells and the immune system profoundly influences disease progression and therapeutic outcomes28,29,30.
To assess macrophage-targeting efficiency, the fEiNS group exhibited markedly stronger fluorescence signals than both the DMSO control and the FITC lipid nanoparticles lacking M2Φpep modification (fDL) (Fig. 5a, b). Notably, FITC loaded-DML (fDML) and fEiNS achieved a ~99% FITC-positive cell ratio, in stark contrast to the ~85% and ~50% observed with fDL and fDMSO, respectively, accompanied by a corresponding 1.4-fold and 2.5-fold enhancement in MFI (Fig. 5c, d). In addition, blocking CD206 with mannose31 partially reduced fEiNS uptake compared to unblocked cells (Supplementary Fig. S12c, d), yet M2-like macrophages retained high uptake capacity despite inhibition. This is attributable to the M2Φpep ligand on EiNS, which binds M2Φs with 5.7-fold higher affinity than M1Φs, targeting multiple M2Φ surface markers and thereby internalization in these cells27,32.

a Confocal images (Scale bar, 15 µm) and (b) flow cytometry analysis depicting the uptake of FITC by RAW 264.7 cells using various delivery nanoparticles (n = 3 independent experiments). c FITC+ ratio in M2Φs treated with various formulations (n = 3 independent experiments). d Mean fluorescence intensity (MFI) ratio of FITC+ M2Φs across different treatment groups (n = 3 independent experiments). e Polarized RAW264.7 cells (M2-like phenotype) were treated with different formulations for 24 h, followed by flow cytometry analysis to determine the percentages of M1-like and M2-like macrophages (n = 3 independent experiments). f The ratio of M1-like/M2-like macrophages after treatment of varioius formulations (n = 3 independent experiments). g Phagocytic capacity of M2-polarized RAW264.7 cells pretreated with various formulations, assessed by flow cytometry and MTT assay in a coculture with GFP-labeled Hepa1-6 cells. Created in BioRender. Y.Q. (2025) https://BioRender.com/tait9j9. h Flow cytometry analysis of the phagocytic rate of M2-like RAW264.7 cells in a coculture system with GFP-labeled Hepa1-6 cells following incubation with different formulations (n = 3 independent experiments). i Differences in phagocytic rates of Hepa1-6 cells by RAW264.7 cells treated with different formulations (n = 3 independent experiments). j Hepa1-6 cell viability investigation using a coculture system of polarized M2-like RAW264.7 and GFP-labeled Hepa1-6 cells, incubated with different formulations (n = 6 independent experiments). k Co-culture of tEiNS-treated M2-like macrophages, Hepa1-6 cells, and isolated CD3+ T cells to assess CD8+ T cells activation. Created in BioRender. Y.Q. (2025) https://BioRender.com/tait9j9. l Representative scatter plots of flow cytometry data showing the percentage of GzmB+ and IFNγ+ cells within the CD8+ T cell population following co-culture with EiNS, tDL, tDML or tEiNS-treated M2-like macrophages, Hepa1-6 cells, and CD3+ T cells, as indicated (n = 3 independent experiments). m Quantitative results showing the numbers of GzmB+ and IFNγ+ cells as a percentage of the total CD8+ T cell population (n = 3 independent experiments). Data are expressed as mean ± SD and were determined using one-way ANOVA followed by Tukey’s multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001. ****P < 0.0001.
The epigenetic reprogramming of immune cells can mediate tumor immunity and influence cancer progression14. TMP195 induces the recruitment and differentiation of highly phagocytic and stimulatory MΦs within tumors by modifying epigenetic signatures and remodeling the TME33,34,35. MΦs enhance T cell-mediated antitumor responses by presenting tumor antigens to CD8+ T cells and secreting pro-inflammatory cytokines, thereby converting cold tumors into immunologically active hot ones. Polarization toward the M1 phenotype represents a promising immunotherapeutic strategy. To evaluate the potential of tEiNS to induce M2-to-M1 repolarization, we quantified CD86⁺ (M1-like) and CD206⁺ (M2-like) macrophages by flow cytometry, revealing that while EiNS had minimal impact, both tDML and tEiNS significantly increased the proportion of CD86⁺ macrophages compared to the control (Fig. 5e). This resulted in an M1-like/M2-like macrophage ratio 1.88-fold higher than that of the control group and 1.15-fold higher than that of the tDL group (Fig. 5f), highlighting the superior uptake capacity of engineered tEiNS over tDL for active targeting of M2Φs. Notably, there was no significant difference between the tDML and tEiNS treatment groups, indicating that the adsorption of PPN onto tDML to construct tEiNS did not impede the cellular uptake of tDML. These findings highlight the marked enhancement in the M1-like/M2-like macrophage ratio induced by tEiNS treatment. In addition, flow cytometry results revealed that treatment of M0 macrophages with tEiNS did not induce significant changes in the expression of the CD86, suggesting that TMP195 cannot directly promote the polarization of M0 macrophages toward the M1 phenotype (Supplementary Fig. S17). HDAC inhibition could induce phenotypic changes in both maturing macrophages and existing mature macrophages36. TMP195, as an HDAC inhibitor, can promote the polarization of M2 macrophages toward the M1 phenotype by altering histone acetylation, which could inhibit the expression of anti-inflammatory genes while enhancing the expression of pro-inflammatory genes. Polarization of M0 macrophages into M1 macrophages typically requires strong pro-inflammatory signals, such as interferon gamma (IFNγ) and Lipopolysaccharide (LPS), whereas differentiation into M2 macrophages is driven by IL-4 and IL-13.
MΦs regulate tumor progression through phagocytosis37. Compared to the EiNS group, tDML and tEiNS treatments enhanced macrophage phagocytic capacity by 1.92- and 1.36-fold, respectively (Fig. 5g–i), indicating a notable improvement in overall phagocytosis. Furthermore, coculture of Hepa1-6 cells with M2-like RAW264.7 macrophages pretreated with different formulations showed that tEiNS markedly reduced tumor cell viability (Fig. 5j). To investigate the effect of tEiNS-treated M2-like macrophages on CD8+ T cells cytotoxicity, we established a co-culture system comprising tEiNS-treated M2-like macrophages, Hepa1-6 cells, and isolated CD3+ T cells. Flow cytometry revealed a significant increase in granzyme B+ (GzmB+) and IFNγ+ CD8+ T cells in the tEiNS-treated co-culture group, with GzmB+ CD8+ T cells rising from 38.4% to 66.7% and IFNγ+ CD8+ T cells increasing from 29.9% to 52.9%, in contrast to the control. These findings highlight enhanced activation and functionality of cytotoxic T lymphocytes (CTLs) upon tEiNS treatment (Fig. 5k–m). Inhibiting Class IIa HDACs boosted the phagocytic and immunostimulatory functions of macrophages, steering them toward an antitumor phenotype and enhancing their capacity to activate CTL33. In summary, tEiNS reprograms M2-like macrophages toward an M1-like phenotype, enhancing phagocytic activity and CTL activation, underscoring its potential as a promising strategy for cancer immunotherapy.
stEiNS inhibited HCC progression and recurrence in vivo
As depicted in Fig. 6b, the diverse formulations relieved tumor burden to different degrees, and mice treated with stEiNS exhibited the lowest tumor burden at 20 days. H&E staining and ultrasound imaging further confirmed that the stEiNS treatment group had the smallest tumor area, indicating its superior efficacy in inhibiting HCC progression (Fig. 6c). Mice treated with stEiNS exhibited significantly prolonged survival, with a 60-day survival rate of 66.7% and continued survival in 50% of the mice beyond 80 days. By contrast, median survival in the EiNS and siYTHDF1 groups did not differ significantly from the control. Notably, the stEiNS group exhibited a median survival time 2.1-fold longer than the control, 1.89-fold longer than the TMP195 group, and 1.4–1.5-fold longer than the sPPN and tDML groups (Fig. 6d).

a Experimental scheme of the orthotopic Hepa1-6 model established in male C57BL/6 mice. Different formulations containing equivalent doses of siYTHDF1 (3 µg each) or/and TMP195 (10 mg/kg each) were intravenously (i.v.) administered via the tail vein. Created in BioRender. Y.Q. (2025) https://BioRender.com/9ze5vnk. b IVIS images of Luci+ Hepa1-6 tumor-bearing mice treated with diverse formulations. c B-ultrasound and H&E staining of mice livers in different groups (n = 3 independent experiments). d Survival analysis of mice in the indicated treatment groups (n = 6 mice per group). e IHC staining of YTHDF1 and MHC-I, and TUNEL staining in liver tumor sections from mice after indicated treatments, scale bar, 50 µm. f Flow cytometric analyses of the M1-like macrophages and M2-like macrophages after treatment with various formulations. Percentage of (g) M1-like phenotype (CD45+CD11b+F4/80+CD86+), (h) M2-like phenotype (CD45+CD11b+F4/80+CD206+), after treatment with different formulations (n = 5 independent experiments). i The ratio of M1-like/M2-like macrophages in vivo after treatment with different formulations (n = 5 independent experiments). j Percentage of CD8+T in CD3+T cells. Percentage of (k) GzmB+ T and (l) IFNγ+ in CD8+T cells (n = 5 independent experiments). m Heatmap of IL-6, TNF-α, and IFNγ expression profiles in HCC tumor tissues (n = 5 independent experiments). Data are expressed as mean ± SD and were determined using one-way ANOVA followed by Tukey’s multiple comparisons test. ns P ≥ 0.05, *P < 0.05, **P < 0.01, ***P < 0.001. ****P < 0.0001.
Downregulation of major histocompatibility complex class I (MHC-I) molecules, coupled with the immunosuppressive influence of tumor-associated MΦs, depleted systemic tumor-specific T cells, thus undermining the efficacy of immunotherapy38. Tumor cells evade immune surveillance by downregulating tumor-specific antigens and MHC-I, which are essential for T cell activation. Loss of MHC-I expression confers a selective advantage, enabling tumor cells to escape from cytotoxic T cell recognition and promoting the emergence of MHC-I–negative tumors39. stEiNS-mediated YTHDF1 knockdown significantly upregulated MHC-I expression compared to the control (Fig. 6e). As mentioned above, in vitro, sPPN and sEiNS demonstrated a comparable knockdown of YTHDF1 in Huh7 and Hepa1-6 cells, as well as similar inhibitory effects on the proliferation, migration, and invasion of HCC tumor cells (Fig. 4a–d and Supplementary Figs. S14, 15). However, in vivo, stEiNS, with its more optimal particle size compared to sPPN, showed a greater propensity for accumulation at the tumor site. Knockdown of YTHDF1 enhances IFNGR1 and IFNγ signaling in tumor cells, upregulating MHC-I expression and enabling direct antigen presentation, thereby activating CTLs40. In addition, YTHDF1 has been shown to promote the translation of lysosomal genes, its depletion impairs lysosomal proteolysis, stabilizes antigens, and further augments MHC-I expression—collectively restoring immune surveillance and eliciting a potent antitumor immune response7. In addition, down-regulation of YTHDF1 expression significantly induced tumor cell apoptosis and inhibited HCC progression (Fig. 6e).
Immunosuppressive MΦs in the HCC tumors hinder the effectiveness of epigenetic regulation in activating T cell-mediated antitumor responses. As pivotal orchestrators of immune suppression in the TME41, macrophages inhibit T cell function through multifaceted mechanisms, thereby driving tumor immune escape42. stEiNS markedly increased the proportion of M1-like macrophages (CD45⁺CD11b⁺F4/80⁺CD86⁺) in vivo from 37.4% to 75.8%, while concurrently reducing M2-like macrophages (CD45⁺CD11b⁺F4/80⁺CD206⁺) from 32.9% to 20.9% (Fig. 6f). Compared with the physiological saline group, both tDML and stEiNS treatments increased the infiltration of M1-like macrophages in tumors while decreasing M2-like macrophages, ultimately enhancing the M1-like/M2-like macrophage ratio of tumor-infiltrating MΦs by 3.07-fold (Fig. 6g–i). In addition, consistent with observations in other tumor models43,44, our results demonstrated that YTHDF1 knockdown via stEiNS decreased myeloid-derived suppressor cell (MDSC) infiltration (Supplementary Fig. S19). MDSCs are known to suppress antitumor immunity by inhibiting the functions of effector T cells and natural killer cells through multiple mechanisms, including intratumoral arginine depletion via Arginase-1, oxidative stress induction via iNOS, and TGF-β secretion45.
To verify whether elevated MHC-I expression, reduced MDSCs and repolarized M1-like macrophages can collectively activate the CTLs, we assessed the systemic T cell responses across treatment groups. Flow cytometry revealed minimal CD8⁺ T cell infiltration in the control, EiNS, and siYTHDF1 groups. In contrast, sPPN and stEiNS treatments markedly increased CD8⁺ T cells, with the stEiNS group exhibiting the highest frequencies of GzmB⁺ and IFNγ⁺ CTLs (Fig. 6j–l). Knockdown of YTHDF1 can enhance IFNγ activity in tumor cells, promote tumor cell antigen presentation, and regulate T cell receptor interactions and CTL activation7,40,46. In parallel, stEiNS reprogrammed macrophages toward an M1 phenotype, augmenting phagocytosis and contributing to TME remodeling. M1-like macrophages contribute to T cell mediated immunity by presenting tumor antigens via MHC-I to CD8⁺ T cells and secreting pro-inflammatory cytokines47. Consistently, IL-6, TNF-α, and IFNγ levels were significantly elevated in stEiNS-treated HCC tumors compared to other groups (Fig. 6m). Notably, TNF-α is the cytokine required for tumor toxicity mediated by effector T cells and macrophages48. In stEiNS-treated HCC, siYTHDF1 knockdown reduced TNF-α expression, whereas TMP195 significantly elevated it. Given that TNF-α is primarily secreted by activated macrophages, especially M1 macrophages, we speculate that the combined treatment enhances TNF-α secretion, thereby amplifying macrophage-driven reprogramming of the TME. These results confirm that stEiNS-mediated TME reprogramming markedly suppresses HCC progression and is critical for potentiating immunotherapeutic efficacy.
KMP data analysis revealed that HCC patients with elevated YTHDF1 expression experienced a higher recurrence rate (Fig. 1c). In our postoperative recurrence HCC model, stEiNS treatment achieved superior suppression of recurrence, as evidenced by the smallest tumor volume, lowest relative tumor weight, and the fewest Ki67-positive cells (Figs. 7b, c and f). Moreover, stEiNS increased the effector memory CD8+ T cell (TEM) population from 20% to 30%, thereby enhancing rapid recall responses and reinforcing durable antitumor immunity (Fig. 7d and Supplementary Fig. S22). Concurrently, stEiNS treatment significantly expanded the splenic CD8+ central memory T cell (TCM) population by 2.7-fold compared to the control (Fig. 7e and Supplementary Fig. S22), underscoring its potential to sustain long-term immunity and prevent relapse. As observed in acute infection, antigens drive naïve T cell differentiation into CTLs through epigenetic mechanisms, with a subset transitioning into memory T cells post-clearance to ensure durable immunity49. TEM cells, characterized by elevated migratory receptor expression, rapidly infiltrate inflamed tissues and exhibit potent effector functions, whereas TCM cells, despite requiring higher antigenic thresholds due to reduced surface T cell receptor (TCR) and elevated protein tyrosine phosphatase levels, persist long-term to maintain immune surveillance50. The concurrent expansion of CD8+ TCM and TEM highlights their crucial role in prolonging survival in tumor-bearing mice. We speculate that YTHDF1 knockdown in tumor cells upregulates antigen-presenting molecules (e.g., MHC-I), thereby enhancing tumor antigen recognition and T cell activation. Concurrently, TMP195 promotes macrophage polarization toward an M1-like phenotype, further enhancing antigen presentation and reinforcing T cell priming. Collectively, the tumor microenvironment shaped by YTHDF1 knockdown and M1-like macrophages may foster the differentiation and persistence of both TCM and TEM, thereby sustaining long-term antitumor immunity.

a Schematic representation of the Hepa1-6 postoperative recurrence model in male C57BL/6 mice. Ten days after subcutaneous tumor implantation, the tumor was surgically resected to establish the recurrence model. Different formulations containing equivalent doses of siYTHDF1 (3 µg each) or/and TMP195 (10 mg/kg each) were administered intravenously via the tail vein. Created in BioRender. Y.Q. (2025) https://BioRender.com/951b31b. b Tumor growth curves. c Relative tumor weight with photographs of the excised tumors (n = 5 independent experiments). The percentages of (d) CD8+ TEM (CD45+CD3+CD8+CD44+CD62L-) and (e) CD8+ TCM (CD45+CD3+CD8+CD44+CD62L+) gated from total CD8+ T cells in the spleen (n = 5 independent experiments). f IHC staining of Ki67 in HCC tumor tissues after various treatments. Scale bar, 50 µm. g Schema of experimental design of PD-1 plus stEiNS. A, the orthotopic Hepa1-6 mouse model. B, Hepa1-6 postoperative recurrence model. h IVIS images of Luci+ Hepa1-6 tumor-bearing mice treated with diverse formulations. i B-ultrasound of mice livers in different groups. Created in BioRender. Y.Q. (2025) https://BioRender.com/951b31b. j H&E staining of mice livers in different groups. Scale bar, 1 mm. k Tumor growth curves (n = 5 mice per group). l Relative tumor weight with photographs of the excised tumors (n = 5 mice per group). m IHC staining of Ki67 in HCC tumor tissues after PD-1 with or without stEiNS treatment. Scale bar, 50 µm. Data are expressed as mean ± SD. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test (c, d, e), two-way ANOVA (b, k), or two-tailed Student’s t test for pairwise comparisons (l). ns P ≥ 0.05, *P < 0.05, **P < 0.01, ****P < 0.0001.
Immunotherapy enhances antitumor responses and has revolutionized cancer treatment by boosting immune surveillance to eradicate cancer. However, PD-1/PD-L1-based immunotherapy has shown limited success in many solid tumors51. Western blot analysis of membrane proteins from Huh7 and Hepa1-6 cells confirmed robust PD-L1 expression in tumor tissues (Supplementary Fig. S23a), further validated by immunohistochemical (IHC) of Hepa1-6 tumors (Supplementary Fig. S23b), consistent with prior reports52. Based on this, we evaluated the therapeutic potential of combining stEiNS with PD-1 monoclonal antibody (mAb) for HCC treatment. Specifically, Luc fluorescence changes, ultrasound imaging, and H&E staining confirmed that stEiNS significantly enhanced the antitumor immune efficacy of PD-1 in both orthotopic (Fig. 7g–j) and subcutaneous HCC models (Fig. 7g and 7k–m). As described above, stEiNS increased intratumoral M1-like macrophages and enhanced CD8⁺ T cell cytotoxicity, collectively activating antitumor immunity, suppressing HCC progression, and promoting tumor regression. IFNγ signaling is closely linked to antitumor immunity and responsiveness to ICB, whereas tumor MHC-I downregulation correlates with poor outcomes and resistance to ICIs7. As shown in Fig. 6e, m, stEiNS treatment markedly increased intratumoral IFNγ levels and upregulated MHC-I expression. Given the critical role of macrophages in PD-1/PD-L1 blockade efficacy, stEiNS promoted M1-like macrophage infiltration and activation, facilitating CTL-mediated tumor elimination via enhanced antigen presentation and pro-inflammatory cytokine release. By reprogramming the epigenetic landscape of both HCC cells and M2-like macrophages, stEiNS markedly potentiated PD-1 immunotherapy, amplifying both antitumor and anti-recurrence responses. Moreover, in vivo safety evaluations demonstrated no significant toxicity at administered doses, reinforcing the clinical potential of stEiNS (Supplementary Fig. S24).
Collective molecular mechanisms underlying stEiNS treatment
To elucidate the effect of stEiNS treatment on HCC microenvironment, we comprehensively investigated the mechanism underlying siYTHDF1 and TMP195 therapeutic strategies. Previous studies have confirmed the influence of siYTHDF1 on expression profiles in HCC tumor cells, identifying TNF/NF-κB signaling pathway as a critical downstream effector of YTHDF1 (Fig. 8a)53. In addition, the TCGA and GEO databases further demonstrated that high YTHDF1 expression in HCC was positively correlated with TNF/NF-κB signaling (Fig. 8b)54. As shown in Fig. 8c, EiNS treatment did not affect YTHDF1 expression, whereas sPPN and sEiNS treatment significantly knocked down YTHDF1, resulting in decreased p-p65 levels and increased caspase 3 levels in Huh7 and Hepa1-6 cells. The results of apoptosis flow cytometry and TUNEL staining also validated this conclusion (Figs. 4g and 6e). Moreover, confocal immunofluorescence confirmed the colocalization and positive correlation of YTHDF1 and p-p65 in PDOs (Fig. 8d, e). Overall, sEiNS-mediated YTHDF1 knockdown inhibited the activation of the TNF/NF-κB signaling pathway, thereby inducing tumor cell apoptosis.

a KEGG analysis of the differential expression genes under TYHDF1 deletion in HCC cells. b GSEA showing the association between YTHDF1 expression and NF-κB pathway activity in HCC. c Effects of YTHDF1 on the NF-κB/caspase 3 signaling pathway (n = 3 independent experiments). d, e Immunofluorescence showing YTHDF1 (pink) and p-p65 (green) localization in PDOs (n = 3 independent experiments). Nuclei were stained with DAPI (blue). Scale bar, 50 µm. f The sequence logo plot showed the sequence conservation of NF-κB, as well as predicted score and binding sequences of five chemokines with NF-κB. Quantitative analysis of the CCL2, CCL5, CCL20, CXCL2, and CXCL12 secreted by (g) Huh7 and (h) Hepa1-6 cells in the medium after treatment with different formulations for 24 h (n = 4 independent experiments). i CUT&RUN-qPCR analysis confirmed that NF-κB could bind to CCL2 promoter region in Huh7 and Hepa1-6 cells (n = 3 independent experiments). Data are expressed as mean ± SD and were determined using two-tailed Student’s t test. ns P ≥ 0.05, *P < 0.05, **P < 0.01.
Studies have demonstrated that activated NF-κB signaling stimulates tumor cells to secrete specific chemokines, thereby modulating the immune microenvironment43,44. Using the TCGA and the JASPAR databases (https://jaspar.elixir.no/), we analyzed chemokine expression and NF-κB binding affinity at their promotor regions, identifying CCL2, CCL5, CCL20, CXCL2, and CXCL12 as potential downstream targets of NF-κB (Fig. 8f and Supplementary Fig. S25). Subsequent ELISA confirmed that targeting YTHDF1 significantly reduced CCL2 expression, but not other chemokines, in HCC-conditioned medium (Fig. 8g, h). More importantly, CUT&RUN-qPCR analysis verified direct NF-κB binding to the CCL2 promoter (Fig. 8i). The CCL2-CCR2 axis influences the tumor microenvironment by recruiting CCR2+ immunosuppressive cells, including MDSCs55, consistent with our previous findings (Supplementary Fig. S19).
TMP195, a class IIa HDAC inhibitor, occupies the acetyllysine-binding site of class IIa HDACs, thereby inhibiting deacetylation33,34. To elucidate the mechanisms underlying the effects of tEiNS in RAW264.7 cells, we evaluated histone acetylation by measuring histone H3 and acetylated histone H3 levels via western blotting. tEiNS treatment significantly enhanced histone acetylation in M2-polarized RAW264.7 cells, indicating that, as an HDAC inhibitor, tEiNS promoted hyperacetylation and re-expression of epigenetically silenced genes (Fig. 9a). Subsequently, RNA sequencing identified 3921 differentially expressed genes (DEGs), 1946 upregulated and 1975 downregulated (fold change > 2 and P < 0.05), between cells with and without TMP195 treatment (Fig. 9b, c). KEGG analysis showed that downregulated DEGs were enriched in cytokine–cytokine receptor interaction, TNF signaling, and JAK/STAT1 pathways (Fig. 9d). Western blot and ELISA demonstrated that tEiNS increased phosphorylated STAT1 (p-STAT1) levels (Fig. 9e, f). GSEA revealed a negative correlation between YTHDF1 expression and IFNγ activity (Fig. 9g–i)56. As a key downstream effector of IFNγ, STAT1 drives antitumoral macrophage phenotype induction57. Notably, YTHDF1 knockdown elevated IFNγ levels, suggesting a collective effect with TMP195-induced STAT1 activation. Therefore, we propose that stEiNS-mediated YTHDF1 downregulation in HCC tumor cells modulates the TNF/NF-κB/caspase-3 axis to induce apoptosis and suppresses CCL2 secretion, thereby limiting MDSC infiltration and enhancing T cell recruitment and cytotoxicity. Concurrently, YTHDF1 knockdown promotes the repolarization of stEiNS-induced M2-like macrophages toward an M1 phenotype by increasing IFNγ release. Together, these effects activate the IFNγ/STAT1 pathway, promote pro-inflammatory cytokine secretion, and trigger tumoricidal immunity.

a M2-polarized RAW264.7 cells were treated with tEiNS for 24 h and histone H3 and acetyl-histone H3 levels were evaluated in the extracted proteins via western blotting with corresponding antibodies (n = 3 independent experiments). b Heatmap depicting the results of RNA-seq analysis in the control and TMP195-treatment groups. c Volcano plot displaying the distributions of upregulated and downregulated genes (≥ 2-fold difference, P value < 0.05) in the TMP195 and control groups. DEGs, differentially expressed genes; non-DEGs, non-differentially expressed genes. d KEGG analysis of biological process annotations of DEGs in TMP195-treated group, showing enrichment in immune-related pathways. e Effects of TMP195 on the IFNγ/STAT1 signaling pathway (n = 3 independent experiments). f TNF-α expression profiles in RAW264.7 cells after various treatment (n = 3 independent experiments). g GSEA showing pathways associated with YTHDF1 expression in HCC. h, i GSEA showing the association between YTHDF1 expression and IFNγ pathway in HCC. Data are expressed as mean ± SD and were determined using one-way ANOVA followed by Tukey’s multiple comparisons test. ns P ≥ 0.05, **P < 0.01, ****P < 0.0001.
In this study, we engineer a nanosatellite system, stEiNS, with selective tropism for tumor cells and macrophages upon dynamic unlocking at the tumor site, enabling effective epi-immunotherapy for HCC. The dual-targeting stEiNS achieves precise hierarchical targeted delivery of siYTHDF1 and TMP195, significantly inhibits proliferation of HCC organoids and tumors, and potentiates immunotherapy by activating of MΦs and T cells in an orthotopic HCC mouse model overexpressing YTHDF1. Mechanistically, we demonstrate that the stEiNS cascade inhibits HCC progression via three mechanisms: (I) YTHDF1 silencing mediates epigenetic regulation of HCC cells through the TNF/NF-κB/caspase-3 axis, enhancing antigen presentation and IFNγ expression while suppressing CCL2 secretion to restrict MDSC infiltration and increase CD8+ T cell proportion and cytotoxicity; (II) The repolarization of M2-like macrophages toward an M1-like phenotype induced by IFNγ in a stEiNS-dependent manner, through the IFNγ/STAT1 pathway, promotes increased CD8+ T cell cytotoxicity; and (III) amplifies PD-1 blockade efficacy. Our study presents an approach to augment antitumor immunity in patients with elevated YTHDF1 levels and could profoundly influence the clinical application of PD-1-based therapies for other cold tumors.
Methods
Ethics statement
This research complies with all relevant ethical regulations. All animal procedures were performed in accordance with the institutional guidelines of Scientific Research Ethics Committee of Qilu Hospital of Shandong University (ethical approval number: KYLL-2020(KS)-041) and Scientific Research Ethics Committee of the Second Hospital of Shandong University (ethical approval number: KYLL-2022-351). Human samples were obtained from all patients with written informed consent, and our study was approved by the Ethics Committee of Qilu Hospital, Shandong University, in accordance with the ethical guidelines of the Declaration of Helsinki. The study was approved by the Ethical Committee of Qilu Hospital of Shandong University (ethical approval number: KYLL-2020(KS)-041). The maximum tumor size in mice permitted by the Laboratory Animal Ethical and Welfare Committee of Shandong University Cheeloo College of Medicine was 2000 mm3, and the maximal tumor size/burden of mice was not exceeded.
Materials
siYTHDF1 was obtained from GenePharma. TMP195 was sourced from Bidepharm. Lecithin and cholesterol were purchased from Aladdin and J&K, respectively. LysoTracker Green and DAPI were obtained from Meilunbio and Yuanye, respectively. DSPE, HOOC-PEG-NH2, maleimide-terminated 1,2-distearoyl-sn-glycero-3-phosphate-ethanolamine (DSPE-Mal), and the M2Φ pep (YEQDPWGVKWWY) were acquired from Chongqing Yusi Pharmaceutical Technology. 4-Carboxyphenylboronic acid, 4-(hydroxymethyl)phenylboron pinacol acid ester, and p-nitrophenyl chloroformate were obtained from Energy Chemical. polyamide-amine dendrimer (PAMAM) was sourced from Sigma. Antibodies used in this study are provided in Supplementary Table 2. ELISA kits for TNF-α, IFNγ, IL-6 were procured from Animal Union Biotechnology. Annexin V-FITC/PI apoptosis kit was purchased from Beyotime Biotechnology. Anti-mouse PD-1 antibody was purchased from BioXCell. MojoSortTM Mouse CD3 T Cell Isolation Kit and MojoSortTM Buffer were ordered from Biolegend. IL-4 and IL-13 cytokines were obtained from peprotech. Ultrapure water was used throughout the experiment. All other chemicals and solvents of analytical grade were obtained locally.
Cell culture and animals
DMEM medium and fetal bovine serum were procured from Gibco (Thermo Fisher Scientific). Hepa1-6 and Huh7 cell lines were obtained from ATCC. RAW264.7 cells were purchased from FENGHUISHENGWU. All cell lines used in this study were authenticated by short tandem repeat (STR) profiling. Cells were routinely tested for mycoplasma contamination using a PCR-based method and found to be negative. C57BL/6 mice (male, 5-weeks-old) were sourced from Beijing Vital River Laboratory Animal Technology. Mice were housed in a temperature-controlled environment (25 ± 2 °C) under a 12-hour light–dark cycle with 40–70% relative humidity, and given ad libitum access to a standard commercial diet and water. Tumor volumes were measured using a digital caliper by determining the length (L) and width (W) of subcutaneous tumors. The volume was calculated using the standard formula: volume = (L × W²)/2, where L represents the longest axis and W denotes the perpendicular shorter axis. Fresh human HCC and adjacent nontumour liver tissue samples were randomly collected from consecutive HCC patients undergoing curative resection at Qilu Hospital, Shandong University (Jinan, China). This study does not include analyses based on sex or gender, focusing primarily on a cohort of hepatocellular carcinoma patients with high YTHDF1 gene expression.
PDOs
Patient-derived tumor tissues were mechanically minced into small pieces, filtered through a 100 μm strainer, and centrifuged for 3 min at 300 × g. After discarding the supernatant, the organoids were mixed with Matrigel (Corning) and embedded in a low-adhesion 24-well plate. Once the Matrigel solidified, PDO culture medium (Biogenous, K2105-HCC) was added. Organoids were passaged using Organoid Dissociation Solution (Biogenous, E238001). For cryopreservation, the organoids were resuspended in Recovery Cell Culture Freezing Medium (Gibco) and frozen following standard protocols.
Bioinformatics analysis
The Cancer Genome Atlas (TCGA) datasets were downloaded from (http://gdac.broadinstitute.org/). The effect of YTHDF1 on prognosis was analyzed via GEPIA and Kaplan-Meier plotter (http://gepia.cancer-pku.cn, https://kmplot.com). Datasets of GSE were downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo)58,59,60. The gene expression data was analyzed for enrichment of biological themes using Gene Set Enrichment Analysis (GSEA) (http://software.broadinstitute.org/gsea/index.jsp). IFNγ induced genes signature was obtained from a protein-interaction network of interferon-stimulated genes, expanding the landscape of the innate immune system.
Synthesis of DSPE-M2, DSPE-DA and PPN
To synthesize DSPE-M2, 11.05 mg of DSPE-Mal and 17.6 mg of M2 macrophage targeting peptide (M2Φpep, YEQDPWGVKWWY) were dissolved in a mixture of methanol and DMSO, stirred overnight at room temperature under nitrogen protection. The resulting product was dialyzed using a 1000 Da MWCO dialysis membrane for 24 h to obtain DSPE-M2. For NBC synthesis, 0.5 g M1 was dissolved in 20 mL dichloromethane, followed by the addition of 0.6 mL triethylamine and 0.47 g M2. The mixture was stirred at room temperature for 1 h, and NBC was obtained by recrystallization. To prepare PNBC, HOOC-PEG-NH2 was dissolved in a sodium bicarbonate solution (reaction 1), while NBC was dissolved in DMSO (reaction 2). Reaction 2 was slowly added dropwise to reaction 1 under continuous stirring at room temperature. After overnight reaction, the mixture was dialyzed using a 3500 Da MWCO dialysis membrane for 24 h and lyophilized to obtain PNBC. Subsequently, 36 mg of PAMAM, 32.48 mg of PNBC, 5.21 mg of NHS, and 8.63 mg of EDC were dissolved in a 2:1 methanol:methylene chloride solution and stirred for 24 h. The mixture was then dialyzed using a 3500 Da MWCO membrane for 24 h to yield the final product, PPN.
Preparation of stEiNS
TMP195-loaded liposomes (tDML) were prepared by dissolving phospholipids, cholesterol, DSPE-DA, DSPE-M2, and TMP195 in ethanol, followed by the addition of ultrapure water and stirring at 30 °C for 4 h to evaporate ethanol. To generate small nanoparticles (sPPN), PPN was incubated with siYTHDF1 (the sequence of the human cell line siYTHDF1 is 5′-GGAACAACATCTATCAGCA-3′, the sequence of the mouse cell line siYTHDF1 is 5′-GCACACAACCTCTATCTTTA-3′, GenePharma) for 20 minutes and dispersed in PBS (pH 7.4). The final hybrid nanostructure (stEiNS) was formed by incubating 200 μL of tDML with sPPN at pH 7.4 for 2 h. The preparation scheme is illustrated in Fig. 2. In in vitro studies, stEiNS, tEiNS, or sEiNS were typically pretreated with H₂O₂ before experimentation. The synthesis of DSPE-M2, DSPE-DA, NBC, PNBC, PPN were confirmed by 1H NMR or infrared spectrum. The successful preparation of stEiNS was confirmed by transmission electron microscope (TEM, ZEISS).
In vitro drug release studies
In vitro release studies of TMP195 from TMP195 solution, tDML and tEiNS formulations were conducted using a dialysis membrane with a molecular weight cutoff of 3500 Da. Samples were enclosed in dialysis bags with both ends securely tied. These bags, containing either the TMP195-loaded tEiNS, tDML or solution, were then incubated in 40 mL of release medium (PBS, pH 7.4, with 5% Tween 80) within 50 mL centrifuge tubes. The tubes were placed in a thermostatic shaker and agitated continuously at 150 rpm at a constant temperature of 37 °C. At predetermined intervals, 1 mL of the release medium was withdrawn for analysis and replaced immediately with 1 mL of fresh release medium maintained at 37 °C. The concentration of TMP195 in the samples was quantified using high-performance liquid chromatography (HPLC).
TMP195 was quantitatively analyzed by using a 1260 Infinity II LC system (Agilent) with an ACE C18 column (100 × 4.6 mm). The mobile phase consisted of methanol/water (85/15, v/v) with a flow rate of 0.8 mL/min. The detection wavelength was set at 260 nm while the injection volume was 20 μL.
Gel retardation assay
A mixture of siYTHDF1 (200 ng) and PPN, at weight ratios ranging from 1:2 to 1:30, was prepared with a 6 × loading buffer. Electrophoresis was then performed in 1 × Tris-acetate EDTA (TAE) buffer at 60 V for 40 min. The gels were subsequently imaged with a UV illuminator (Tanon) to visualize the distribution of siYTHDF1.
The cellular uptake of EiNS to tumor cells or M2Φs in vitro
Cy3-labeled siYTHDF1 (s-Cy3) was used to prepare fluorescently labeled s-Cy3PPN, s-Cy3EiNS and Lipo/s-Cy3. Huh7 and Hepa1-6 cells were inoculated into a 24-well plate, followed by the addition of various formulations and co-culture for 12 h. Cellular uptake was observed under a Confocal Laser Scanning Microscope (CLSM, LSM780, ZEISS) and quantified by flow cytometry (CytoFLEX S, Beckman). In addition, RAW264.7 cells were seeded into confocal plates at a density of 5 × 105 cells per well and treated with 20 ng/mL IL-4 for 24 h to polarize macrophages to the M2Φs. Various formulations were then added and cultured for 4 h, and intracellular fluorescence was observed using CLMS and quantified by CytoFLEX S flow cytometry.
Endolysosomal escape
Huh7 and Hepa1-6 cells were treated with s-Cy3EiNS and stained with LysoTracker Green and DAPI at 1, 3, and 6 h. The cells were then fixed with 4% paraformaldehyde and imaged using CLSM. For quantitative analysis of the co-localization of s-Cy3 and LysoTracker Green within the cells, ImageJ was used to calculate the Manders co-localization coefficient.
Deep tumor penetration
The hanging drop method was used to construct 3D tumor spheroids of Huh7 and Hepa1-6 cells. s-Cy3EiNS and Lipo/s-Cy3 were added to the spheroids. The tumor spheroids were cultured in 24-well plates for 8 h, washed with PBS, and observed under CLSM.
In vitro cytotoxicity assay
siYTHDF1, sPPN, sEiNS, and Lipo/s were added to 96-well plates containing Huh7 and Hepa1-6 cells (with a siYTHDF1 concentration of 2 pmol). Subsequently, 20 μL of MTT solution (0.5 mg/mL in PBS) was added to each well, and the cells were incubated for an additional 4 h. Formazan formed from MTT was then extracted by adding 200 μL of DMSO and rocking for 15 min. Absorbance at 570 nm was measured daily using a microplate reader (Tecan) from day 0 to day 4.
Hemolysis test
Whole blood was collected from rabbit, and erythrocytes were isolated by centrifugation. The erythrocytes were washed twice with PBS and then co-cultured with tDML, sPPN, EiNS, stEiNS, normal saline, or H2O for 3 h. Subsequently, the supernatants were collected, and UV-vis absorbance at 570 nm was measured using a microplate reader to determine the hemolysis rate.
qRT-PCR analysis
Total RNA was extracted using TRIzol reagent (Invitrogen). Subsequently, 1 μg of total RNA served as the template for first-strand cDNA synthesis with the ReverTra Ace qPCR RT Kit (TOYOBO). All primer sequences were listed in Supplementary Table 1. qPCR reactions were performed using the FastStart Universal SYBR Green Master Mix (Roche). The expression levels were normalized to β-actin for each sample.
Western blot and immunohistochemistry (IHC)
Cells were collected, counted, and lysed in 2 × Laemmli buffer. Samples were heated to 95 °C for 7 min and passed through an insulin syringe. Immunoblotting was performed using the following antibodies: YTHDF1, p-p65, p-STAT1, caspase 3, PD-L1, GAPDH and NaK-ATPase. The details of the primary antibodies used in this study are provided in Supplementary Table 2. Detection was performed using HRP-conjugated anti-mouse IgG (Dako) and visualized with RapidStep™ ECL Reagent (Millipore). The IHC slides were blindly evaluated by two pathologists (F.S. and B.H.) to validate the expression of YTHDF1 and Ki67.
Clone assays
Cells are seeded at low density and treated with various formulations, followed by incubation until colonies emerge. After 10 days, colonies were fixed and stained with crystal violet. The number of colonies were then quantified to evaluate cell proliferation and clonogenic potential.
The wound healing analysis
Cells were cultured to near confluence, and a straight scratch is made using a sterile pipette tip. After washing with PBS, fresh medium with or without various formulatons was added. Cell migration into the scratch area was monitored at 0 h and 24 h, and wound closure was analyzed to assess migratory capacity.
In vitro invasion assays
Transwell assays for invasion were conducted using chambers with Matrigel coating. Huh7 and Hepa1-6 cells were seeded in the upper chambers in DMEM, while the lower chambers contained DMEM supplemented with 10% FBS as a chemoattractant. After 24 h, invaded cells were fixed with 4% paraformaldehyde and stained with crystal violet. Transmembrane cells were counted in three randomly selected fields under a light microscope.
Apoptosis
An Annexin V-FITC/PI apoptosis kit was used to assess the ability of each formulation to induce tumor cell apoptosis. Huh7 and Hepa1-6 cells were seeded in 6-well plates and incubated in a CO2 incubator for 24 h. The medium was then replaced with fresh medium containing sPPN, sEiNS, or Lipo/s with a blank control. Subsequently, the cells were collected, transferred to flow cytometry tubes, and stained according to the kit instructions. Apoptosis was quantified by flow cytometry.
PDOs study
To evaluate the cell killing effect of stEiNS, the PDOs were seeded in 96-well plates and incubated for further 48 h. Afterwards, the PDOs were cultured with fresh medium containing various formulations and coincubated for another 7 days. Afterward, the morphology of PDOs was then imaged.
CUT&RUN
The Hyperactive pG-MNase CUT&RUN Assay Kit for PCR/qPCR (Vazyme, HD101-01) was used to conduct the CUT&RUN assay. Briefly, 1 × 10⁵ HCC cells were collected and bound to ConA beads. The cells were then incubated with 2 µL of NFκB1 antibody at 4 °C overnight. On the following day, after washing the cells twice with DIG washing buffer, they were incubated with pG-MNase enzyme for 1 h at 4 °C. After two rounds of DIG washing, the cells were treated with CaCl₂ for 1 h at 4 °C. The reaction was stopped by adding 100 mL of stop buffer, and the cells were resuspended and incubated for 30 min at 37 °C. After incubation, the cells were placed on a magnet, and the unattached liquid was removed. DNA was then eluted from the beads by gently washing them twice with 80% ethanol and resuspending them in double-distilled water. Finally, qRT-PCR was performed according to the manufacturer’s protocol.
Phenotypic switching of MΦ
RAW264.7 cells were seeded into a 24-well plate at a cell density of 1 × 105 per well, and treated with 20 ng/mL IL-4 for 24 h to polarize the cells to M2 phenotype. After PBS washing, the cells were incubated with EiNS, tDL, tDML, or tEiNS (100 nM TMP195) for 24 h, followed by antibody staining and flow cytometry analysis.
MΦ Phagocytosis
RAW264.7 cells, induced by IL-4, were treated with EiNS, tDL, tDML, and tEiNS (100 nM TMP195) for 24 h. GFP-labeled Hepa1-6 cells were seeded in 6-well plates at a density of 5 × 104 cells per well and then mixed with RAW264.7 cells at a ratio of 2:1. After co-culturing in fresh medium for 4 h, the wells were washed with PBS to collect RAW264.7 cells, which were then stained using immunofluorescence. Flow cytometry was subsequently employed to detect the number of double-positive cells and calculate the phagocytosis rate.
Detection of MΦ-regulated T cell cytotoxicity in vitro
Murine spleens were carefully removed from C57BL/6 mice and washed three times with precooled sterile PBS to isolate primary CD3+ T cells. The spleens were gently fragmented on a steel wire mesh in precooled sterile PBS, and the splenocyte suspension was filtered through 70 μm nylon mesh. CD3+ T cells were isolated using the MojoSortTM Mouse CD3 T Cell Isolation Kit (BioLegend, USA). For T cell activation, isolated CD3+ T cells were incubated with plate-bound anti-CD3 antibodies (2 μg/mL, BioLegend) and soluble anti-CD28 antibodies (5 μg/mL, BioLegend) for 24 h. RAW264.7 cells, polarized by IL-4 and IL-13, were treated with EiNS, tDL, tDML, and tEiNS (100 nM TMP195) for 24 h. Subsequently, the treated macrophages, Hepa1-6 cells, and isolated CD3+ T cells were co-cultured for 24 h. The cells were then labeled with antibodies, including anti-CD45-BV510, anti-CD3-FITC, anti-CD8-PerCP-Cy5.5, anti-GzmB-Alexa Fluor 647, and anti-IFNγ-Brilliant Violet 650TM, and analyzed by flow cytometry.
RNA sequencing (RNA-seq) and analysis of RNA-seq data
RAW264.7 cells, induced by IL-4 and IL-13, were treated with or without TMP195 for 24 h. Total mRNA was extracted from macrophage cells using a spin column kit (Fastagen Biotech) and evaluated using an RNA 6000 kit (Agilent Technologies). Then, RNA libraries were established with a TruSeq RNA library preparation kit (Illumina), quantified via quantitative real-time PCR, normalized, pooled, and sequenced using an Illumina HiSeq 4000 instrument. Statistical analysis and ggplot2 (v3.3.2) were performed using the R program v4.0.3, and a P < 0.05 was considered statistically significant.
Distribution of EiNS in vivo
The HCC subcutaneous tumor model was established using Luciferase-labeled Hepa1-6 (Luc Hepa1-6) cells in C57BL/6 mice. Dir and Cy3 were used to label DML and PPN, respectively. siYTHDF1-Cy3, s-Cy3EiNS, DirEiNS(-M2Φpep), and DirEiNS were administered via tail vein injection (n = 3 mice per group). An in vivo imaging system (IVIS, PerkinElmer) was employed to capture Dir and Cy3 signals in the mice at various time points. Major tissues and tumors were dissected for ex vivo fluorescence imaging. Tumor sections were prepared, and cell nuclei were stained with DAPI and mouse anti-CD206 (M2-like macrophages) to evaluate the dual-targeted drug delivery capability of EiNS to HCC tumor cells and M2-like macrophages in vivo.
Treatment protocols in orthotopic HCC models
We established an orthotopic HCC model using Luc Hepa1-6 cells in C57BL/6 mice. Various formulations including normal saline (Control), EiNS, siYTHDF1, TMP195, sPPN, tDML, and stEiNS were administered every two days through the tail vein (siYTHDF1 at 3 μg, TMP195 at a dose of 10 mg/kg, n = 11 mice). IVIS imaging was used to monitor tumor progression. The antitumor effects of the various formulations were evaluated using an ultrahigh-resolution photoacoustic imaging system (VisualSonics). The mice were euthanized after 20 days (n = 5 mice). Tumor tissues were collected for IHC or immunofluorescence staining to evaluate YTHDF1 and MHC-I expression, while TUNEL staining assessed antitumor effects. Mouse tumors were filtered through a copper mesh, and lymphocyte types were labeled with antibodies for flow cytometry analysis of immune cell content. In addition, ELISA kits were used to analyze cytokine concentrations. Survival curves of the mice in each group were recorded for 80 days (n = 6 mice).
Suppression of HCC recurrence
We established a subcutaneous tumor model by implanting Luc Hepa1-6 cells in C57BL/6 mice, in the right armpit (n = 5 mice per group). Upon reaching a volume of 100 mm³, the tumors were surgically excised, and various formulations were administered every two days for five cycles over 15 days. Throughout the study period, we monitored the mouse body weight and tumor volume every three days. Tumor tissues were subjected to a comprehensive analysis that included H&E staining for morphology, and Ki67 immunohistochemistry for proliferation evaluation. In addition, immune cells were isolated, probed with antibodies, including anti-CD45-BV510, anti-CD3-FITC, anti-CD8-PerCP-Cy5.5, anti-CD44-PE-Cy7, and anti-CD62L-APC antibodies, and analyzed via CytoFLEX S flow cytometry.
PD-1 plus stEiNS administration
PD-1 monoclonal antibodies (PD-1 mAb, BioXCell), and stEiNS were administered once every three days to two distinct tumor models. For the combination treatment group, mice received stEiNS treatment (siYTHDF1 3 μg and TMP195 10 mg/kg) via tail vein injection on day 1, followed by PD-1 mAb (200 μg)61 via tail vein injection on the subsequent day, repeated for five cycles. IVIS imaging was used to monitor tumor progression in the orthotopic HCC model, whereas mouse body weight and tumor volume were measured every three days in the recurrence models. The mice were euthanized after 20 days in the orthotopic HCC model and after 15 days in the recurrence model, respectively. Histological examination included H&E staining for morphological analysis, and Ki67 immunohistochemistry for proliferation evaluation.
Statistics and reproducibility
No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publication62,63. The researchers were blinded to the experimental groups during data collection when possible. All mice were randomly assigned to the experimental groups. All experiments have been reproduced at least three times, and all attempts at replication were successful with self-consistent results. Data are expressed as the mean ± standard deviation (SD). Statistical significance was determined using the Student’s t test for comparisons between two groups, or one-way ANOVA or two-way ANOVA followed by Tukey’s multiple comparison test for comparisons among multiple groups, using Prism version 9.5.1 (GraphPad Software, Inc.). To assess survival differences between groups, Kaplan–Meier survival curves were compared using the log-rank test. Significance levels were defined as n.s. (not significant, P > 0.05), *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated from this study are available within the article, supplementary information, or Source Data file. The RNA-seq data have been deposited in the Genome Sequence Archive (GSA) under accession number PRJNA1263123, and are publicly available at: https://www.ncbi.nlm.nih.gov/sra/PRJNA1263123. Source data are provided in this paper.
References
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 7, 6 (2021).
Li, Z. et al. Neoadjuvant tislelizumab plus stereotactic body radiotherapy and adjuvant tislelizumab in early-stage resectable hepatocellular carcinoma: the Notable-HCC phase 1b trial. Nat. Commun. 15, 3260 (2024).
Ducreux, M. et al. The management of hepatocellular carcinoma. Current expert opinion and recommendations derived from the 24th ESMO/World Congress on Gastrointestinal Cancer, Barcelona, 2022. ESMO Open 8, 101567 (2023).
Xu, Y. et al. Epi-immunotherapy for cancers: rationales of epi-drugs in combination with immunotherapy and advances in clinical trials. Cancer Commun. 42, 493–516 (2022).
Liu, X. et al. YTHDF1 facilitates the progression of hepatocellular carcinoma by promoting FZD5 mRNA translation in an m6A-dependent manner. Mol. Ther. Nucleic Acids 22, 750–765 (2020).
Zhu, Y. et al. The potential role of m6A reader YTHDF1 as diagnostic biomarker and the signaling pathways in tumorigenesis and metastasis in pan-cancer. Cell Death Discov. 9, 34 (2023).
Lin, W. et al. Tumor-intrinsic YTHDF1 drives immune evasion and resistance to immune checkpoint inhibitors via promoting MHC-I degradation. Nat. Commun. 14, 265 (2023).
Han, D. et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274 (2019).
Ronca, V. et al. The liver as a central “hub” of the immune system: pathophysiological implications. Physiol. Rev. 105, 493–539 (2025).
Varanasi, S. K. et al. Bile acid synthesis impedes tumor-specific T cell responses during liver cancer. Science 387, 192–201 (2025).
Yu, J. et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 27, 152–164 (2021).
Li, J. J. et al. The liver microenvironment orchestrates FGL1-mediated immune escape and progression of metastatic colorectal cancer. Nat. Commun. 14, 6690 (2023).
Sangro, B., Sarobe, P., Hervás-Stubbs, S. & Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 18, 525–543 (2021).
Zhang, J., Huang, L., Ge, G. & Hu, K. Emerging epigenetic-based nanotechnology for cancer therapy: Modulating the tumor microenvironment. Adv. Sci. 10, e2206169 (2023).
Wang, T., Zhang, J., Hou, T., Yin, X. & Zhang, N. Selective targeting of tumor cells and tumor associated macrophages separately by twin-like core–shell nanoparticles for enhanced tumor-localized chemoimmunotherapy. Nanoscale 11, 13934–13946 (2019).
Li, Z. et al. A tumor microenvironments-adapted polypeptide hydrogel/nanogel composite boosts antitumor molecularly targeted inhibition and immunoactivation. Adv. Mater. 34, 2200449 (2022).
Chen, J., Zhu, D., Liu, X. & Peng, L. Amphiphilic dendrimer vectors for RNA delivery: State-of-the-art and future perspective. Acc. Mater. Res. 3, 484–497 (2022).
Liu, C., Shao, N., Wang, Y. & Cheng, Y. Clustering small dendrimers into nanoaggregates for efficient DNA and siRNA delivery with minimal toxicity. Adv. Healthc. Mater. 5, 584–592 (2016).
Liu, H. et al. Nanoassemblies with effective serum tolerance capability achieving robust gene silencing efficacy for breast cancer gene therapy. Adv. Mater.33, e2003523 (2021).
Hu, Q. et al. Tumor microenvironment-mediated construction and deconstruction of extracellular drug-delivery depots. Nano Lett. 16, 1118–1126 (2016).
Zhou, Z. et al. Tumor specific in situ synthesis of therapeutic agent for precision cancer therapy. J. Nanobiotechnology 22, 612 (2024).
Han, X. et al. An ionizable lipid toolbox for RNA delivery. Nat. Commun. 12, 7233 (2021).
Zhang, X. et al. NEU4 inhibits motility of HCC cells by cleaving sialic acids on CD44. Oncogene 40, 5427–5440 (2021).
Islam, M. et al. Suppressing immune responses using siglec ligand-decorated anti-receptor antibodies. J. Am. Chem. Soc. 144, 9302–9311 (2022).
Xu, E., Saltzman, W. M. & Piotrowski-Daspit, A. S. Escaping the endosome: assessing cellular trafficking mechanisms of non-viral vehicles. J. Control. Release 335, 465–480 (2021).
Chen, W. et al. Size-dependent penetration of nanoparticles in tumor spheroids: A multidimensional and quantitative study of transcellular and paracellular pathways. Small 20, 2304693 (2024).
Xiao, H. et al. M2-like tumor-associated macrophage-targeted codelivery of STAT6 inhibitor and IKKβ siRNA induces M2-to-M1 repolarization for cancer immunotherapy with low immune side effects. ACS Cent. Sci. 6, 1208–1222 (2020).
Ramirez, C. F. A. et al. Cancer cell genetics shaping of the tumor microenvironment reveals myeloid cell-centric exploitable vulnerabilities in hepatocellular carcinoma. Nat. Commun. 15, 2581 (2024).
O’Leary, K. Liver metastases cultivate an immune desert. Nat. Rev. Cancer 21, 143–143 (2021).
Gougelet, A. Epigenetic modulation of immunity: towards new therapeutic avenues in hepatocellular carcinoma?. Gut 68, 1727–1728 (2019).
Zhuang, J. et al. Redox-responsive AIEgen diselenide-covalent organic framework composites targeting hepatic macrophages for treatment of drug-induced liver injury. Small 20, e2402656 (2024).
Cieslewicz, M. et al. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl. Acad. Sci. USA 110, 15919–15924 (2013).
Guerriero, J. L. et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543, 428–432 (2017).
Lobera, M. et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 9, 319–325 (2013).
Yue, Y. et al. Biomimetic nanoparticles carrying a repolarization agent of tumor-associated macrophages for remodeling of the inflammatory microenvironment following photothermal therapy. ACS Nano 15, 15166–15179 (2021).
Li, X. et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene 40, 1836–1850 (2021).
Gholamin, S. et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 9, eaaf2968 (2017).
Nguyen, D.-H. et al. Reprogramming the tumor immune microenvironment using engineered dual-drug loaded Salmonella. Nat. Commun. 15, 6680 (2024).
Jin, Y. et al. IL-21 reinvigorates exhausted natural killer cells in patients with HBV-associated hepatocellular carcinoma in STAT1-depedent pathway. Int. Immunopharmacology 70, 1–8 (2019).
You, Q. et al. m6A reader YTHDF1-targeting engineered small extracellular vesicles for gastric cancer therapy via epigenetic and immune regulation. Adv. Mater. 35, 2204910 (2023).
Schupp, J. et al. Targeting myeloid cells in the tumor sustaining microenvironment. Cell. Immunol. 343, 103713 (2019).
DeNardo, D. G. & Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 19, 369–382 (2019).
Wang, L. et al. Targeting N6-methyladenosine reader YTHDF1 with siRNA boosts antitumor immunity in NASH-HCC by inhibiting EZH2-IL-6 axis. J. Hepatol. 79, 1185–1200 (2023).
Bao, Y. et al. Targeting m6A reader YTHDF1 augments antitumour immunity and boosts anti-PD-1 efficacy in colorectal cancer. Gut 72, 1497–1509 (2023).
Gabrilovich, D. I. & Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174 (2009).
Clement, M. et al. CD8 coreceptor-mediated focusing can reorder the agonist hierarchy of peptide ligands recognized via the T cell receptor. Proc. Natl. Acad. Sci. USA 118, e2019639118 (2021).
Muntjewerff, E. M., Meesters, L. D. & van den Bogaart, G. Antigen cross-presentation by macrophages. Front. Immunol. 11, 1276 (2020).
Huang, A. et al. Engineered apoptosis-bioinspired nanoparticles initiate immune cascade for cancer immunotherapy of malignant ascites. ACS Appl. Mater. Interfaces 15, 10371–10382 (2023).
Pan, X. & Zheng, L. Epigenetics in modulating immune functions of stromal and immune cells in the tumor microenvironment. Cell. Mol. Immunol. 17, 940–953 (2020).
Liu, Q., Sun, Z. & Chen, L. Memory T cells: strategies for optimizing tumor immunotherapy. Protein Cell 11, 549–564 (2020).
Zhang, P. et al. A PD-L1 Antibody-conjugated PAMAM dendrimer nanosystem for simultaneously inhibiting glycolysis and promoting immune response in fighting breast cancer. Adv. Mater. 35, 2305215 (2023).
Zhu, Y. et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 68, 1653–1666 (2019).
Zhang, X. et al. N6-Methyladenosine reader YTHDF1 promotes stemness and therapeutic resistance in hepatocellular carcinoma by enhancing NOTCH1 expression. Cancer Res. 84, 827–840 (2024).
Deng, Y. B. et al. Identification of genes preferentially methylated in hepatitis C virus-related hepatocellular carcinoma. Cancer Sci. 101, 1501–1510 (2010).
Liang, T. et al. Cancer-associated fibroblast-induced remodeling of tumor microenvironment in recurrent bladder cancer. Adv. Sci. 10, 2303230 (2023).
Woo, H. G. et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res. 70, 3034–3041 (2010).
Lawrence, T. & Natoli, G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761 (2011).
Désert, R. et al. Human hepatocellular carcinomas with a periportal phenotype have the lowest potential for early recurrence after curative resection. Hepatology 66, 1502–1518 (2017).
Park, Y. Y. et al. Yes-associated protein 1 and transcriptional coactivator with PDZ-binding motif activate the mammalian target of rapamycin complex 1 pathway by regulating amino acid transporters in hepatocellular carcinoma. Hepatology 63, 159–172 (2016).
Roessler, S. et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 70, 10202–10212 (2010).
Han, Y. et al. TMP195 exerts antitumor effects on colorectal cancer by promoting M1 macrophages polarization. Int. J. Biol. Sci. 18, 5653–5666 (2022).
Jing, W. et al. An in situ engineered chimeric IL-2 receptor potentiates the tumoricidal activity of proinflammatory CAR macrophages in renal cell carcinoma. Nat. Cancer 6, 838–853 (2025).
Liu, J. et al. Glypican-3-targeted macrophages delivering drug-loaded exosomes offer efficient cytotherapy in mouse models of solid tumours. Nat. Commun. 15, 8203 (2024).
Acknowledgements
This work was supported by the grants from the Taishan Scholars Program of Shandong Province (Grant No. tstp20221158, T.L.), National Natural Science Foundation of China (Grant No. 82073200 & 81874178, T.L.), Major basic research of Shandong Provincial Natural Science Foundation (Grant No. ZR2021ZD26, T.L.), the Natural Science Foundation of Shandong Province (ZR2023QH362, Y.Q. and ZR2023QH191, C.C.), the Fundamental Research Funds of Shandong Province (ZR2022ZD18 and ZR2022ZD17, X.J.), the China National Postdoctoral Program for Innovative Talents (BX20230204, C.C.), and the China Postdoctoral Science Foundation (2023M732091, C.C.). Thanks for the technical support from Y.Y., X.Y., J.Z., M.W., and L.W. in Advanced Medical Research Institute/Translational Medicine Core Facility of Advanced Medical Research Institute, Shandong University. We thank BioRender for support with schematics and visual presentations of the work.
Author information
Authors and Affiliations
Contributions
T.L., X.J., Y.Q., and C.C. conceptualized the study and designed the experiment. Y.Q. Synthesized and characterized the structure of engineered nanosatellites. Y.Q., F.S., S.L., G.Z., and C.L. performed the cell and animal experiments. Y.Q., C.C., F.S., G.Z., S.L. and Z.Z. Contributed to data analysis. Y.Q. crafted all the figures and wrote the manuscript. T.L., X.J., C.C., and F.S. edited and revised the manuscript and supervised the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wei Tao, Xin-Yuan Guan, Zhaohui Wang, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qu, Y., Chen, C., Sun, F. et al. Epigenetic modulation with nanosatellite triggers tumoricidal immunity for hepatocellular carcinoma treatment. Nat Commun 16, 7340 (2025). https://doi.org/10.1038/s41467-025-61974-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61974-w
