
Abstract
Positional alkene isomerization is a powerful reaction for moving a C=C bond from one position to another. This transformation, as a high atom-economy and easy-to-handle process, has gained increasing prominence in both organic and material chemistry. Despite these advances, the stereospecific positional alkene isomerization to achieve bidirectional chirality transfer remains challenging. We report herein a bidirectional stereospecific positional alkene isomerization of chiral exocyclic alkene analogues by achiral Lewis base catalysis. By using this central-to-axial chirality transfer strategy, the axially chiral N-indolylquinolinones can be readily obtained from one configuration to two different configurations. Mechanistic studies indicated that the competitive alkene isomerization and Michael/retro-Michael addition would affect the conformation of exocyclic alkenes, thus achieving the bidirectional central-to-axial chirality transfer. In addition, combining the asymmetric allylic substitution-isomerization and photocatalytic Z/E isomerization, all eight stereoisomers of diaxially chiral quinolinones could be easily obtained in high enantioselectivities and diastereselectivities.
Similar content being viewed by others

Introduction
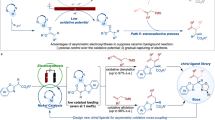
Alkenes are fundamental building blocks in organic synthesis due to their versatile reactivity and widespread applications in the production of polymers, pharmaceuticals, and fine chemicals1. Recent research in alkene chemistry has focused on developing selective and sustainable methods for their functionalization. Alkene isomerization represents one of the most important strategies widely used for the synthesis of different alkene isomers. Generally, alkene isomerization can be categorized into two types: geometric isomerization2,3,4,5,6,7,8 and positional isomerization9,10,11,12 (Fig. 1A). Both approaches enable atom-economical synthesis of alkenes, and significant efforts have been devoted to advancing this field13,14.

A Two types of alkene isomerization. B Achiral catalysts enable stereospecific positional alkene isomerization. C Our hypothesis of bidirectional central-to-axial chirality transfer. D Stereospecific positional alkene isomerization enables bidirectional central-to-axial chirality transfer.
Positional isomerization of alkenes is a powerful transformation for relocating a C=C bond from one position to another, thereby generating different alkene isomers with high inherent efficiency (Fig. 1A, bottom). Despite its utility, this transformation presents significant challenges in controlling the location and geometry of the resulting alkenes. Nevertheless, regioselective and stereoselective isomerizations have been successfully achieved through metal catalysis, organocatalysis, and photocatalysis15,16,17,18,19,20,21,22. Among these, enantioselective positional alkene isomerization has emerged as a powerful strategy for accessing chiral molecules23,24,25,26,27,28,29,30,31,32,33,34,35. In contrast, stereospecific positional alkene isomerization remains less explored, even though it offers an efficient route to synthesize challenging chiral molecules that are difficult to access via conventional methods (Fig. 1B)36. For example, Martín-Matute and others developed a series of achiral-base-catalyzed reactions focused on central-to-central chirality transfer in acyclic alkenes (Fig. 1B, top)37,38,39,40,41. On the other hand, our group recently reported the Asymmetric Allylic Substitution-Isomerization (AASI) strategy, which features a key step of enantiospecific positional alkene isomerization42,43,44,45,46. This approach enabled central-to-axial chirality transfer47,48,49,50 through transition-metal catalysis or achiral base catalysis, assisted by noncovalent interactions (Fig. 1B, bottom). Despite these advancements, the inherent suprafacial 1,3-hydrogen transfer mechanism of the isomerization restricts the process to yielding only one product configuration. The stereospecific positional alkene isomerization to achieve bidirectional chirality transfer remains an unresolved challenge that requires further exploration.
Indeed, during the central-to-axial chirality transfer process, the highly efficient stereospecific positional alkene isomerization relies on excellent conformational control of the substrate. Inspired by this characteristic, we envisioned that stereospecific bidirectional chirality transfer could be achieved using substrates with controllable conformations. As illustrated in Fig. 1C, exocyclic alkenes I, which possess specific configuration, can serve as suitable substrates. During the positional isomerization from exocyclic to endocyclic alkenes, different configurations of axially chiral products II can be obtained by controlling the reaction conditions, thereby enabling the synthesis of axially chiral molecules in totally different configurations51,52,53,54,55,56,57,58. However, the key challenge for this transformation lies in how to control the conformation of exocyclic alkenes I under similar reaction conditions.
Here, we report a stereospecific bidirectional chirality transfer for the synthesis of axially chiral N-indolylquinolinones. Starting from the same centrally chiral compound (R)-1, the base catalyst 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) yields 2 in the P configuration, whereas 1,4-diazabicyclo[2.2.2]octane (DABCO) produces axially chiral N-indolylquinolinones 2 in the M configuration. Mechanistic studies reveal the underlying mechanism of this stereodivergent transformation.
Results and discussion
Identification of a system for the preparation of enantioenriched exocyclic alkenes
Inspired by downstream cascade reactions based on asymmetric allylic substitution (AAS)59,60,61,62,63,64,65,66,67,68, we envisioned that this approach could be feasible for synthesizing enantioenriched exocyclic alkenes. To our delight, initial studies demonstrated that exocyclic alkene 1a could be efficiently generated via a one-pot synthesis involving DABCO-catalyzed allylic substitution followed by Fe/HOAc-promoted reductive amidation, without the hydrolysis of -CN group (Fig. 2A). Encouraged by this result, we first screened chiral Lewis base catalysts for the synthesis of compound Int-1a. As shown in Fig. 2B, the catalyst (DHQD)2PYR delivered the best performance, affording Int-1a in 96% enantiomeric excess (ee) and 92% yield after further optimization. Next, to investigate the effect of substituents at the C2 position of indoles 4, we employed various indoles as substrates in the reaction system (Fig. 2C). Electron-withdrawing groups, such as esters and sulfonyl groups, resulted in lower enantioselectivities. While high enantioselectivity was achieved using 1H-indole-2-carbaldehyde as the substrate, the allylic substitution product was incompatible with the Fe/HOAc reductive system, preventing the formation of the corresponding exocyclic quinolin-2(1H)-one (see Supplementary Information for details). Additionally, no satisfactory results were obtained when 1-(1H-indol-2-yl)ethan-1-one, 2-methylindole, or 2-phenylindole were used as substrates.

A One-pot synthesis of exocyclic alkene frameworks. B Screened chiral Lewis base catalysts for the synthesis of 1a. C The -CN group of 4a was changed by other groups in the presence of optimal catalyst at 0 °C. MBH, Morita–Baylis–Hillman.
Substrate scope of enantioenriched exocyclic alkenes
With suitable substrates and optimal reaction conditions in hand, we next explored the substrate scope for the synthesis of exocyclic quinolin-2(1H)-ones (Fig. 3). 2-Cyanoindoles bearing electron-neutral, electron-withdrawing, and electron-donating groups at the C3–C7 positions including halogen, alkyl, aryl, alkynyl or methoxy group all reacted smoothly, affording the corresponding exocyclic quinolin-2(1H)-ones in 93–98% ee and 51–91% yields (1a–1u). The absolute configuration of 1b was determined as R by single-crystal X-ray analysis of its dimeric form, and the configurations of other products were assigned by analogy. Notably, 5-methyl-1H-pyrrole-2-carbonitrile was also a suitable substrate, yielding 1v with 92% ee and 70% yield. In addition, ortho-NO2-substituted aryl MBH carbonates with various substituents at different positions of the phenyl ring were compatible with the reaction system, providing 1w–1e’ in 85–95% ee with moderate to excellent yields.

Reaction conditions: 3 (0.2 mmol), 4 (0.1 mmol), (DHQD)2PYR (20 mol%), DME (1.0 mL), -10 °C, 72 h. Then, iron powder (1.0 mmol), AcOH (1.0 mL), 30 °C for 8 h. Isolated yield, the ee values were determined by chiral high performance liquid chromatography (HPLC).
Optimized conditions and substrate scope for stereospecific positional alkene isomerization
With chiral exocyclic quinolin-2(1H)-ones 1 in hand, we next examined the positional isomerization of 1a (see Supplementary Information for detailed optimization studies). As shown in Table 1, organic bases such as DBU and DBN efficiently promoted the isomerization within minutes, affording (P)-2a in excellent yields and ee with 91% and 92% enantiospecificity (es), respectively. Solvent screening revealed that 1,4-dioxane was the optimal choice, yielding (P)-2a in 83% yield with 90% ee and 94% es (entries 3–7). Surprisingly, when the reaction was conducted in methanol (MeOH), the isomerization proceeded to generate 2a in the opposite configuration, albeit with moderate ee (entry 8). Encouraged by this result, we further optimized the reaction in MeOH using different bases (entries 9–11). In this context, DABCO demonstrated the best performance, yielding (M)-2a in 81% yield with 83% ee and 86% es (entry 9). When DABCO was employed as the catalyst, (M)-2a was also obtained in other solvents, such as acetonitrile (MeCN), 1,4-dioxane, or dimethoxyethane (DME). However, none of these solvents provided better results compared to MeOH (entries 12–14).
With the optimal conditions established, we next explored the substrate scope. As shown in Fig. 4, substrates 1 bearing indolyl moieties with C3, C4, and C5 substituents—regardless of whether they were electron-withdrawing or electron-donating groups—all reacted smoothly, delivering the corresponding products in good yields and enantioselectivities. Reactions promoted by DBN generated (P)-(2b–2q) with 91–96% es and 82–91% yields, while reactions catalyzed by DABCO afforded (M)-(2b–2q) with 81–90% es and 71–81% yields. The absolute configuration of (P)-2b was confirmed as a dimer by single-crystal X-ray analysis, and the configurations of other products were assigned by analogy.

Reaction conditions: DBN (0.1 mmol, 0.1 mmol/mL) was rapidly added to 1a (0.1 mmol) in 1,4-dioxane (5 mL), or DABCO (0.025 mmol, 0.1 mmol/mL) was rapidly added to 1a (0.1 mmol) in MeOH (5 mL) at rt for 5–10 min. Isolated yield, the ee values were determined by chiral HPLC.
Moreover, (P)-(2r–2t) were obtained with relatively lower es (74–86%) when substrates 1 bearing C6-substituted indolyl moieties were used. In these cases, (M)-(2r–2t) were also obtained with excellent es (84–87%). However, only (P)-2u, derived from (R)-1u, was obtained, regardless of whether the reaction was promoted by DBN or DABCO. Notably, the bidirectional stereospecific isomerization of (R)-1v was also achieved under the optimal conditions, albeit with relatively lower es for the (M)-2v. Finally, under different reaction conditions, stereospecific isomerization of (R)-(1w–1e’) delivered (P)-(2w–2e’) with 80–95% es and 81–92% yields, as well as (M)-(2w–2e’) with 74–88% es and 73–82% yields. Notably, the model one-pot, three-step reaction of 3a and 4a afforded axially chiral N-indolylquinolinones (P)-1a (68% yield, 83% ee) and (M)-1a (63% yield, 80% ee) successfully, albeit with reduced enantioselectivities compared to the stepwise system. (See Supplementary Information for details).
Mechanistic studies
Next, we investigated the mechanism of bidirectional stereospecific positional alkene isomerization of 1. Given the highly reactive Michael acceptor nature of 1, we hypothesized that, during base-promoted positional alkene isomerization, competitive positional alkene isomerization and Michael/retro-Michael reactions may occur, potentially influencing the conformation of 1 (Fig. 5A). To explore this hypothesis, we performed density functional theory (DFT) calculations, using the isomerization of 1a catalyzed by DABCO in MeOH or DBN in dioxane as representative examples. A1 (or B1) was selected as the reference point for the Gibbs free energy profiles due to its relatively higher stability compared to its conformer A4 (or B5) (Fig. 5B, C).

A Proposed mechanism for the conformational control of 1a. B Free energy diagrams for the positional alkene isomerization enabled by DABCO in MeOH. Gibbs free energy obtained at the M06-2X/def2-TZVPP(SMD, MeOH)// M06-2X/def2-SVP(SMD, MeOH) level. C Free energy diagrams for the positional alkene isomerization enabled by DBN in 1,4-dioxane. Gibbs free energy obtained at the M06-2X/def2-TZVPP(SMD, 1,4-dioxane)// M06-2X/def2-SVP(SMD, 1,4-dioxane) level. Critical intermediate and transition state structures, after optimization, are illustrated in panels B and C.
For the DABCO-catalyzed isomerization of 1a in MeOH (Fig. 5B), our calculations indicated that the DABCO-participated aza-Michael/retro-Michael reaction proceeded readily, requiring an overall energy of only 15.6 kcal/mol, significantly lower than the energy required for deprotonation of the benzylic hydrogen by DABCO (24.8 kcal/mol). This result suggests that interconversion between conformers A1 and A4 via rotation about the C-N bond can readily occur. Furthermore, due to the lower energy barrier of TSA4 (23.7 kcal/mol) compared to TSA6 (24.8 kcal/mol), the reaction preferentially proceeds via the aza-Michael/retro-Michael pathway, followed by deprotonation of the benzylic hydrogen to form a persistent chiral ion pair A5 (green line). This process preserves the chiral information, enabling effective chirality transfer. Finally, rapid reprotonation of A5 via TSA5 generates 2a in the M configuration.
In contrast, for the DBN-catalyzed reaction in dioxane (Fig. 5C), the energy barrier for the aza-Michael reaction (17.0 kcal/mol) is higher than that for deprotonation of the benzylic hydrogen (14.5 kcal/mol). Although conformer interconversion between B1 and B5 could occur without DBN participation, the relatively higher energy barrier of TSB6 (compared to TSB5) prevents deprotonation from conformer B5. In this case, deprotonation proceeds directly from B1 to the chiral ion pair B2 via TSB1, followed by reprotonation to afford 2a in the P configuration. Combined with the results of the DABCO-catalyzed isomerization of 1a, we conclude that the stereospecific positional alkene isomerization proceeds through a competitive mechanism involving deprotonation and Michael/retro-Michael reactions. This mechanism herein facilitates a bidirectional central-to-axial chirality transfer, enabling precise control over the stereochemical outcome. In addition, the reason of relatively higher energy profiles of TSA4 (vs. TSB6) and TSA6 (vs. TSB1) may primarily arise from differences in the geometric configurations of their respective bases. Structural analysis shows that the ∠N1-C-N2 angles in A1/B1 and A4/B5 are 154.7° and 151.3°, respectively. Upon deprotonation, the transition states (TSA4, TSA6, TSB1, TSB6) exhibit ∠N1-C-N2 angles of 145.8°, 146.6°, 164.2°, and 162.9°, respectively. This suggests that the cage-like DABCO imposes greater steric hindrance during deprotonation, leading to significant structural distortion. In contrast, the DBN offers reduced spatial constraints, allowing for easier structural expansion.
Z/E geometrical and positional alkene isomerization to all eight stereoisomers
Considering the free amide group in axially chiral N-indolylquinolinones 2, we explored whether further functionalization could be employed to synthesize diaxially chiral compounds. Inspired by our previous report on the stereodivergent synthesis of axially chiral alkenes8, we anticipated that all eight stereoisomers could be generated, with both Z/E geometrical and positional alkene isomerization serving as key steps. Starting from (R)-1c, (P)-2c and (M)-2c were readily obtained through bidirectional stereospecific positional alkene isomerization. Subsequently, stepwise asymmetric allylic substitution-isomerization (AASI) of (M)-2c or (P)-2c provided access to four stereoisomers—(M1, P2, Z)-5, (M1, M2, Z)-5, (P1, P2, Z)-5 and (P1, M2, Z)-5 with high enantioselectivities and good Z/E ratios, respectively (Fig. 6). The remaining four stereoisomers were then obtained in the corresponding E-configuration through photocatalytic stereodivergent Z/E isomerization, without any loss of enantioselectivity.

A Reaction conditions for stepwise AASI, (P)-2c or (M)-2c (0.1 mmol), phenyl MBH carbonates (0.2 mmol), (DHQD)2PYR or (DHQ)2PYR (20 mol%), DME (1.0 mL), rt; then MeONa, toluene (1.0 mL), rt. B Reaction conditions for photocatalytic Z/E isomerization, (Z)-5 (0.1 mmol), Ir(ppy)3 (1 mol%), 420 nm LED, THF (1.0 mL), rt.
Synthetic transformations
Finally, we conducted synthetic transformations of axially chiral N-indolylquinolinones to explore their reactivity and functional group compatibility. Treatment of (P)-2a with various methylation reagents selectively afforded N-methylation and O-methylation products, both in high es (Fig. 7a, b). Meanwhile, the -CN group present in (P)-2i was readily converted to a -COOMe group under strong acidic conditions (Fig. 7c). Hydrolysis of (P)-2i yielded compound 9 in 81% yield with 90% ee (Fig. 7d). The -CN group was selectively reduced to a -CHO group using DIBAL-H as the reductant, whereas the Raney Ni/H₂ reduction system transformed the -CN group into a -CH₂NH₂ group (Fig. 7e, f). Reaction of (P)-2i with sodium azide produced tetrazole 12 in 85% yield and 91% ee (Fig. 7g). Additionally, condensation of (P)-2i with a chiral amino alcohol yielded compound 13 in 90% yield with 92% ee, which holds potential as a chiral ligand for asymmetric synthesis (Fig. 7h). Notably, in all transformations, the bromo group on the indole ring of (P)-2i remained intact, providing a handle for further functionalization.

All the reactions were performed at 0.1 mmol scale. a MeI (0.15 mmol), NaH (0.1 mmol), THF (1.0 mL), 0 °C to rt, 12 h. b Me3O+BF4- (0.15 mmol), DCM (1.0 mL), rt for 4 h. c H2SO4 aq. (6 N, 1.0 mL), MeOH (1.0 mL), 110 °C for 24 h. d NaOH aq. (4N, 1.0 mL), MeOH (1.0 mL), 40 °C for 6 h. e Diisobutylaluminium hydride (DIBAL-H, 1 M in THF, 0.2 mmol), THF, 0 °C for 18 h. f Raney Ni (30 mg), H2 ballon, MeOH (1.0 mL), 50 °C for 8 h. g NaN3 (0.4 mmol), NH4Cl (0.1 mmol), DMF (1.0 mL), 120 °C for 12 h. h (S)-tert-leucinol (0.15 mmol), ZnCl2 (0.1 mmol), chlorobenzene (1.0 mL), 140 °C for 18 h.
In conclusion, we have developed a stereospecific positional alkene isomerization enabled a bidirectional central-to-axial chirality transfer. From substrates (R)-1, the reaction delivered a series of axially chiral N-indolylquinolinones in both P and M configurations with good yields and enantiospecificities. DFT studies indicated that the DABCO leads to the Michael/retro-Michael reaction to change the conformation of (R)-1, followed by a stereospecific 1,3-proton transfer. In contrast, DBN-promoted stereospecific 1,3-proton transfer directly to achieve alkene isomerization, thus enabling the bidirectional stereospecific alkene isomerization. Combining the Z/E geometrical and positional alkene isomerization, all eight stereoisomers of diaxially chiral 1,4-disubstituted quinolinones could be easily obtained in high enantioselectivities and diastereoselectivities. It is anticipated that the bidirectional stereospecific positional alkene isomerization herein will facilitate the development of organic synthetic methodology especially for the asymmetric synthesis of axially chiral molecules.
Methods
General procedure for the synthesis of enantioenriched exocyclic alkenes 1
The reaction was conducted under air. A 2-dram scintillation vial equipped with a magnetic stir bar was charged with (DHQD)₂PYR (17.6 mg, 0.02 mmol, 0.2 equiv), 3 (0.2 mmol, 2.0 equiv) and 4 (0.1 mmol, 1.0 equiv). Dry DME (1.0 mL) was then added, and the mixture was stirred at −10 °C for 72 h. The reaction progress was monitored by thin layer chromatography (TLC). Upon completion, acetic acid (50 μL) was added to quench the reaction, and the solvent was removed in vacuo. To the residue, iron powder (55 mg, 10.0 equiv) and acetic acid (1 mL) were added in one pot, and the mixture was stirred at 30 °C for 8 h. After completion, the crude mixture was diluted with EtOAc, washed with water, and extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous Na₂SO₄, filtered, and concentrated. The residue was purified by column chromatography to afford compound 1.
General procedure for the synthesis of (P)-2
To a solution of 1 (0.1 mmol) in 1,4-dioxane (5 mL) was rapidly added a solution of DBN (0.1 mmol) in 1,4-dioxane (1 mL). The mixture was stirred at room temperature for 5–10 min (monitored by TLC). Upon completion, the reaction was quenched with 1 M HCl (50 μL), diluted with EtOAc, and washed with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with brine, dried over anhydrous Na₂SO₄, filtered, and concentrated in vacuo. The residue was purified by column chromatography to afford (P)-2.
General procedure for the synthesis of (M)-2
To a solution of 1 (0.1 mmol) in MeOH (5 mL) was rapidly added a solution of DABCO (0.025 mmol) in MeOH (0.25 mL). The mixture was stirred at room temperature for 5–10 min (monitored by TLC). Upon completion, the reaction was quenched with 1 M HCl (50 μL), diluted with EtOAc, and washed with water. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with brine, dried over anhydrous Na₂SO₄, filtered, and concentrated in vacuo. The residue was purified by column chromatography to afford (M)-2.
Data availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers CCDC 2392482 (1b-dimer), 2362721 [(P)-2b-dimer] and 2450980 (13). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Experimental procedures, characterization of new compounds, and all other data supporting the findings are available in the Supplementary Information. All data were available from the corresponding author upon request. Source data of DFT studies are provided with this paper. Source data are provided with this paper.
References
Davarnejad, R. (ed). Alkenes: Recent Advances, New Perspectives and Applications (IntechOpen, 2021).
Neveselý, T., Wienhold, M., Molloy, J. J. & Gilmour, R. Advances in the E → Z isomerization of alkenes using small molecule photocatalysts. Chem. Rev. 122, 2650–2694 (2022).
Corpas, J., Mauleón, P., Arrayás, R. G. & Carretero, J. C. E/Z photoisomerization of olefins as an emergent strategy for the control of stereodivergence in catalysis. Adv. Synth. Catal. 364, 1348–1370 (2022).
Hao, T. et al. Trace mild acid-catalysed Z → E isomerization of norbornene-fused stilbene derivatives: intelligent chiral molecular photoswitches with controllable self-recovery. Chem. Sci. 12, 2614–2622 (2021).
Molloy, J. J. et al. Boron-enabled geometric isomerization of alkenes via selective energy-transfer catalysis. Science 369, 302–306 (2020).
Wienhold, M. et al. Geometric isomerisation of bifunctional alkenyl fluoride linchpins: Stereodivergence in amide and polyene bioisostere synthesis. Angew. Chem. Int. Ed. 62, e202304150 (2023).
Metternich, J. B. & Gilmour, R. A bio-inspired, catalytic E → Z isomerization of activated olefins. J. Am. Chem. Soc. 137, 11254–11257 (2015).
Wang, J. et al. Photocatalytic Z/E isomerization unlocking the stereodivergent construction of axially chiral alkene frameworks. Nat. Commun. 15, 3254 (2024).
Fiorito, D., Scaringi, S. & Mazet, C. Transition metal-catalyzed alkene isomerization as an enabling technology in tandem, sequential and domino processes. Chem. Soc. Rev. 50, 1391–1406 (2021).
Massad, I. & Marek, I. Alkene isomerization through allylmetals as a strategic tool in stereoselective synthesis. ACS Catal. 10, 5793–5804 (2020).
Hassam, M., Taher, A., Arnott, G. E., Green, I. R. & van Otterlo, W. A. L. Isomerization of allylbenzenes. Chem. Rev. 115, 5462–5569 (2015).
Liu, X., Li, B. & Liu, Q. Base-metal-catalyzed olefin isomerization reactions. Synthesis 51, 1293–1310 (2019).
Molloy, J. J., Morack, T. & Gilmour, R. Positional and geometrical isomerisation of alkenes: the pinnacle of atom economy. Angew. Chem. Int. Ed. 58, 13654–13664 (2019).
Obeid, A.-H. & Hannedouche, J. Iron-catalyzed positional and geometrical isomerization of alkenes. Adv. Synth. Catal. 365, 1100–1111 (2023).
Larsen, C. R. & Grotjahn, D. B. Stereoselective alkene isomerization over one position. J. Am. Chem. Soc. 134, 10357–10360 (2012).
Meng, Q.-Y., Schirmer, T. E., Katou, K. & König, B. Controllable isomerization of alkenes by dual visible-light-cobalt catalysis. Angew. Chem. Int. Ed. 58, 5723–5728 (2019).
Kapat, A., Sperger, T., Guven, S. & Schoenebeck, F. E-Olefins through intramolecular radical relocation. Science 363, 391–396 (2019).
Occhialini, G., Palani, V. & Wendlandt, A. E. Catalytic, contra-thermodynamic positional alkene isomerization. J. Am. Chem. Soc. 144, 145–152 (2021).
Crossley, S. W. M., Barabé, F. & Shenvi, R. A. Simple, chemoselective, catalytic olefin isomerization. J. Am. Chem. Soc. 136, 16788–16791 (2014).
Rubel, C. Z. et al. Stereodivergent, kinetically controlled isomerization of terminal alkenes via nickel catalysis. Angew. Chem. Int. Ed. 63, e202320081 (2024).
Tortajada, A. et al. Alkene isomerisation catalysed by a superbasic sodium amide. Angew. Chem. Int. Ed. 63, e202407262 (2024).
Blank, L. et al. Deconjugative photoisomerization of cyclic enones. J. Am. Chem. Soc. 147, 10023–10030 (2025).
Morack, T., Onneken, C., Nakakohara, H., Mück-Lichtenfeld, C. & Gilmour, R. Enantiodivergent prenylation via deconjugative isomerization. ACS Catal. 11, 11929–11937 (2021).
Liu, X. & Liu, Q. Catalytic asymmetric olefin isomerization: Facile access to chiral carbon-stereogenic olefinic compounds. Chem Catal. 2, 2852–2864 (2022).
Zhang, X.-X. et al. Catalytic asymmetric isomerization of (homo)allylic alcohols: recent advances and challenges. ChemCatChem 14, e202200126 (2022).
Liu, C., Yuan, J., Zhang, Z., Gridnev, I. D. & Zhang, W. Asymmetric hydroacylation involving alkene isomerization for the construction of C3-chirogenic center. Angew. Chem. Int. Ed. 60, 8997–9002 (2021).
Vasseur, A., Bruffaerts, J. & Marek, I. Remote functionalization through alkene isomerization. Nat. Chem. 8, 209–219 (2016).
Romano, C. & Mazet, C. Multicatalytic stereoselective synthesis of highly substituted alkenes by sequential isomerization/cross-coupling reactions. J. Am. Chem. Soc. 140, 4743–4750 (2018).
Liu, X., Rong, X., Liu, S., Lan, Y. & Liu, Q. Cobalt-catalyzed desymmetric isomerization of exocyclic olefins. J. Am. Chem. Soc. 143, 20633–20639 (2021).
Huang, R.-Z., Lau, K. K., Li, Z., Liu, T.-L. & Zhao, Y. Rhodium-catalyzed enantioconvergent isomerization of homoallylic and bishomoallylic secondary alcohols. J. Am. Chem. Soc. 140, 14647–14654 (2018).
Wu, Y., Singh, R. P. & Deng, L. Asymmetric olefin isomerization of butenolides via proton transfer catalysis by an organic molecule. J. Am. Chem. Soc. 133, 12458–12461 (2011).
Golec, J. C. et al. BIMP-catalyzed 1,3-prototropic shift for the highly enantioselective synthesis of conjugated cyclohexenones. Angew. Chem. Int. Ed. 59, 17417–17422 (2020).
Bizet, V., Pannecoucke, X., Renaud, J.-L. & Cahard, D. Ruthenium-catalyzed redox isomerization of trifluoromethylated allylic alcohols: mechanistic evidence for an enantiospecific pathway. Angew. Chem. Int. Ed. 51, 6467–6470 (2012).
Piva, O., Mortezaei, R., Henin, F., Muzart, J. & Pete, J. P. Highly enantioselective photodeconjugation of.alpha.,.beta.-unsaturated esters. Origin of the chiral discrimination. J. Am. Chem. Soc. 112, 9263–9272 (1990).
Piva, O. & Pete, J.-P. Highly enantioselective protonation of photodienols an unusual substituent effect on the induced chirality. Tetrahedron Lett. 31, 5157–5160 (1990).
Liu, Q., Xu, W.-Y., You, C.-C., Zhou, R.-G. & He, Y. Enantiospecific 1,3-hydrogen transfer of alkenes and alkynes. Trends Chem. 6, 627–640 (2024).
Martinez-Erro, S. et al. Base-catalyzed stereospecific isomerization of electron-deficient allylic alcohols and ethers through ion-pairing. J. Am. Chem. Soc. 138, 13408–13414 (2016).
Martinez-Erro, S., García-Vázquez, V., Sanz-Marco, A. & Martín-Matute, B. Stereospecific isomerization of allylic halides via ion pairs with induced noncovalent chirality. Org. Lett. 22, 4123–4128 (2020).
García-Vázquez, V., Martínez-Pardo, P., Postole, A., Inge, A. K. & Martín-Matute, B. Synthesis of α,γ-chiral trifluoromethylated amines through the stereospecific isomerization of α-chiral allylic amines. Org. Lett. 24, 3867–3871 (2022).
Postole, A., Martínez-Pardo, P., García-Vázquez, V. & Martín-Matute, B. Chiral trifluoromethylated enamides: synthesis and applications. Chem Catal. 3, 100813 (2023).
Han, Z. et al. Catalytic asymmetric allylic substitution/isomerization with central chirality transposition. Org. Lett. 24, 4246–4251 (2022).
Wang, J. et al. Tandem iridium catalysis as a general strategy for atroposelective construction of axially chiral styrenes. J. Am. Chem. Soc. 143, 10686–10694 (2021).
Sun, C. et al. Asymmetric allylic substitution-isomerization to axially chiral enamides via hydrogen-bonding assisted central-to-axial chirality transfer. Chem. Sci. 11, 10119–10126 (2020).
Wu, Y.-X., Liu, Q., Zhang, Q., Ye, Z. & He, Y. Asymmetric allylic substitution-isomerization for accessing axially chiral vinylindoles by intramolecular π…π stacking interactions. Cell Rep. Phys. Sci. 3, 101005 (2022).
Zhang, X.-L. et al. Stepwise asymmetric allylic substitution-isomerization enabled mimetic synthesis of axially chiral B,N-heterocycles. Angew. Chem. Int. Ed. 61, e202210456 (2022).
Zou, J.-Y. et al. Asymmetric allylic substitution-isomerization for the modular synthesis of axially chiral N-vinylquinazolinones. Angew. Chem. Int. Ed. 62, e202310320 (2023).
Min, X.-L., Zhang, X.-L., Shen, R., Zhang, Q. & He, Y. Recent advances in the catalytic asymmetric construction of atropisomers by central-to-axial chirality transfer. Org. Chem. Front. 9, 2280–2292 (2022).
Yang, H., Chen, J. & Zhou, L. Construction of axially chiral compounds via central-to-axial chirality conversion. Chem. Asian J. 15, 2939–2951 (2020).
Campolo, D., Gastaldi, S., Roussel, C., Bertrand, M. P. & Nechab, M. Axial-to-central chirality transfer in cyclization processes. Chem. Soc. Rev. 42, 8434–8466 (2013).
Liu, S., Pang, Y. L., Xu, H. & Zheng, C. Chirality transfer strategy in asymmetric total syntheses. Trends Chem. 4, 969–972 (2022).
Wang, H., Zhang, Q. & Zi, W. Synergistic catalysis involving palladium for stereodivergent Csp3–Csp3 coupling reactions. Acc. Chem. Res. 57, 468–488 (2024).
Wei, L., Fu, C., Wang, Z.-F., Tao, H.-Y. & Wang, C.-J. Synergistic dual catalysis in stereodivergent synthesis. ACS Catal. 14, 3812–3844 (2024).
Sun, H., Ma, Y., Xiao, G. & Kong, D. Stereodivergent dual catalysis in organic synthesis. Trends Chem. 6, 684–701 (2024).
Krautwald, S. & Carreira, E. M. Stereodivergence in asymmetric catalysis. J. Am. Chem. Soc. 139, 5627–5639 (2017).
Krautwald, S., Sarlah, D., Schafroth, M. A. & Carreira, E. M. Enantio- and diastereodivergent dual catalysis: α-Allylation of branched aldehydes. Science 340, 1065–1068 (2013).
Li, P. et al. Stereodivergent access to non-natural α-amino acids via enantio- and Z/E-selective catalysis. Science 385, 972–979 (2024).
Mi, R. et al. Rhodium-catalyzed atropodivergent hydroamination of alkynes by leveraging two potential enantiodetermining steps. Sci. Adv. 10, eadr4435 (2024).
Wang, P. et al. Sigma-bond metathesis as an unusual asymmetric induction step in Rhodium-catalyzed enantiodivergent synthesis of C–N axially chiral biaryls. J. Am. Chem. Soc. 145, 8417–8429 (2024).
Yang, P., Wang, R.-X., Huang, X.-L., Cheng, Y.-Z. & You, S.-L. Enantioselective synthesis of cyclobutane derivatives via cascade asymmetric allylic etherification/[2 + 2] photocycloaddition. J. Am. Chem. Soc. 145, 21752–21759 (2024).
Fu, C. et al. Modular access to chiral bridged piperidine-γ-butyrolactones via catalytic asymmetric allylation/aza-Prins cyclization/lactonization sequences. Nat. Commun. 15, 127 (2024).
Liu, J., Cao, C.-G., Sun, H.-B., Zhang, X. & Niu, D. Catalytic asymmetric umpolung allylation of imines. J. Am. Chem. Soc. 138, 13103–13106 (2016).
Moon, P. J., Wei, Z. & Lundgren, R. J. Direct catalytic enantioselective benzylation from aryl acetic acids. J. Am. Chem. Soc. 140, 17418–17422 (2018).
Chen, T. et al. Rational design and stereodivergent construction of enantioenriched tetrahydro-β-carbolines containing multistereogenic centers. J. Am. Chem. Soc. 146, 29928–29942 (2024).
Zhuang, H.-F., Gu, J., Ye, Z. & He, Y. Stereospecific 3-aza-Cope rearrangement interrupted asymmetric allylic substitution-isomerization. Angew. Chem. Int. Ed. 64, e202418951 (2025).
Liu, W.-B. et al. Enantioselective γ-alkylation of α,β-unsaturated malonates and ketoesters by a sequential Ir-catalyzed asymmetric allylic alkylation/Cope rearrangement. J. Am. Chem. Soc. 138, 5234–5237 (2016).
Rieckhoff, S., Meisner, J., Kästner, J., Frey, W. & Peters, R. Double regioselective asymmetric C-allylation of isoxazolinones: Iridium-catalyzed N-allylation followed by an aza-Cope rearrangement. Angew. Chem. Int. Ed. 57, 1404–1408 (2018).
Wei, L., Zhu, Q., Xiao, L., Tao, H.-Y. & Wang, C.-J. Synergistic catalysis for cascade allylation and 2-aza-Cope rearrangement of azomethine ylides. Nat. Commun. 10, 1594 (2019).
Yi, Z.-Y., Xiao, L., Chang, X., Dong, X.-Q. & Wang, C.-J. Iridium-catalyzed asymmetric cascade allylation/retro-Claisen reaction. J. Am. Chem. Soc. 144, 20025–20034 (2022).
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (22201131 for Y.H.), the Natural Science Foundation of Jiangsu Province (BK20220137 for Y.H.) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX25_0768 for H.-F.Z.).
Author information
Authors and Affiliations
Contributions
Y.H. designed and directed the project. Q.L. and H.-F.Z. performed the experiments. J.G. performed the DFT calculations. Y.H. wrote the manuscript with the revisions of all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Q., Gu, J., Zhuang, HF. et al. Stereospecific positional alkene isomerization enables bidirectional central-to-axial chirality transfer. Nat Commun 16, 6782 (2025). https://doi.org/10.1038/s41467-025-61990-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61990-w
