
Abstract
Disulfide bond shuffling (DBS) critically influences protein aggregation and stability, yet its spatial constraints and biological implications remain poorly understood. Here, we demonstrate that insulin undergoes DBS within an extended spatial range up to ~19 Å, generating heterogeneous crosslinked oligomers that alter aggregation pathway. While DBS products initially delay aggregation by inhibiting primary nucleation and elongation steps, they ultimately promote the formation of distinct fibrillar structures with enhanced β-sheet content. Native ion mobility-mass spectrometry reveals molecular crosstalk between DBS products and native insulin via both covalent and non-covalent interactions. Notably, DBS-modified insulin fibrils exhibit significantly increased neurotoxicity in neuronal and pancreatic cells through mitochondrial apoptosis activation as supported by proteomic and biophysical analysis. Our findings underscore the importance of controlling DBS in insulin for therapeutic applications and provide insights into the role of disulfide dynamics in protein aggregation and cytotoxicity, with implications for insulin and broader protein misfolding contexts.
Similar content being viewed by others


Introduction
Insulin, a protein characterized by its intricate disulfide bond network, exhibits a propensity to aggregate under certain conditions. This aggregation, often associated with loss of function and potential cytotoxicity, presents a significant challenge in both therapeutic applications and our understanding of protein misfolding diseases. Accumulation of insulin amyloid-like proteins is frequently observed at injection sites in type 2 diabetes patients, and in the brain has been linked to the promotion of Tau protein accumulation, contributing to neurodegenerative diseases1,2,3. Additionally, Parkinson’s patients show abnormally high levels of insulin oligomers in serum and present potential therapeutic target against insulin signaling4,5. Generally, the formation of amyloid-like protein fibrils involves several steps, including oligomers, protofibrils, and mature fibrils6. Each form of insulin amyloid-like aggregation exhibits distinct molecular conformations and varying degrees of toxicity to neuronal cells7,8. Understanding the intricate mechanisms governing insulin aggregation and its associated cytotoxicity, and developing strategies for disrupting its oligomerization is crucial for mitigating these above-mentioned challenges, including accelerating the onset of action of native insulin in body9.
Insulin contains three disulfide bonds that play a crucial role in maintaining its native structure and biological activity. Recent research has highlighted the importance of disulfide bonds in protein stability and aggregation propensity10,11. In vivo, disulfide bonds can reduce protein configurational entropy to stabilize proteins; conversely, the absence of ordered crosslinking can lead to significant diseases in organisms. In vitro, the proper formation of disulfide bonds plays a decisive role in the efficacy of monoclonal antibody drugs12. Changes in the physiological environment, such as temperature, pH, and cascading reactions involving relevant enzymes during disease progression, may disrupt disulfide bonds in insulin upon dissociation.
Disulfide bond shuffling (DBS), a dynamic process of disulfide interchange, has emerged as a significant factor in modulating protein assembly and function13,14,15. This phenomenon can lead to the formation of various oligomeric states, potentially altering the aggregation kinetics and the biological activity of proteins. Importantly, disordered crosslinking of protein disulfide bonds has been reported to promote the amyloid-like aggregation of native proteins, potentially accelerating or exacerbating the onset of diseases16. While numerous synthetic methods for disulfide bond formation have been extensively reported17,18,19,20, the manner of bicarbonate heating21 offers an alternative for simple, fast, and efficient formation of disulfide bonds, free of complicate chemistry and multistep redox reactions. Prior research demonstrated that bicarbonate buffer solutions facilitate crosslinking among protein subunits through the formation of disulfide bond in a spatial distance-constrained manner, where the attainable distance between two cystines is likely confined to be ~9 Å, comparable to that of classic amine-reactive crosslinkers21,22,23,24.
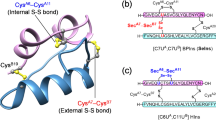
In this study, we explore the potential of distance-constrained DBS on regulating insulin aggregation and cytotoxicity. We employ a combination of advanced ion mobility-mass spectrometry (IM-MS) and other biophysical techniques, to elucidate the structural and kinetic aspects of insulin oligomerization and aggregation under the influence of disulfide bond dynamics. DBS was induced via gentle heating of freshly reduced insulin in the presence of certain contents of ammonium bicarbonate, featuring oxidative chemical-free creation of disulfides in a spatial distance-constrained manner (Fig. 1a, b). Direct evidence from native IM-MS have shown insulin DBS promotes the formation of a variety of disulfide-crosslinked oligomers, facilitating the noncovalent and covalent interactions between monomeric insulin and DBS products. Our findings reveal that DBS-provoked molecular crosstalk not only delays insulin aggregation via primarily targeting the primary nucleation and elongation microscopic steps, but also exacerbates the neurotoxicity of insulin amyloid aggregates in N2a, PC12 and PANC1 cells through selective upregulation and activation of the apoptotic pathway (Fig. 1c). Understanding these mechanisms is crucial for developing strategies to mitigate insulin aggregation, thereby improving the therapeutic management of diabetes. Furthermore, the implications of our findings extend beyond insulin, offering broader perspectives on the role of disulfide bonds in protein aggregation and cytotoxicity in various biological contexts.

a Bicarbonate heating as a spatial distance-constrained alternative to induce disulfide bond shuffling. b Selected 3D structure models for insulin DBS products predicted by AlphaFold, indicating the DBS working distance ranging from 2 Å to ~19 Å. c DBS-provoked molecular crosstalk between native insulin and DBS products delays insulin aggregation likely through a distinct pathway of heterogeneous secondary nucleation, leading to increased neurotoxicity via upregulation of the apoptotic pathway in cells.
Results
Spatial distance-confined Insulin DBS
We previously have demonstrated the distance confinement for bicarbonate heating-induced DBS products likely ranges from 2 Å (the length for disulfide bond) to over 9 Å21. For insulin, the crystal structure and primary structure with abundant disulfide sites clearly supports the feasibility of this type of disulfide bond shuffling25. Intriguingly, as shown by the selected 3D structure models of DBS products from AlphaFold (Figs. 1b, 2a, Supplementary figs. 1, 2), the attainable spatial distance for DBS products has been effectively extended from 9 Å to ~19 Å, a value significantly higher than that of previous report21. To experimentally test the DBS of insulin, we first employed reducing reagent TCEP to convert cysteine residues into free, highly reactive thiol states. Distinct from our previous disulfide crosslinking within nanoelectrospray microdroplets21, the insulin DBS in this study was carried out in bulk solution with elevated incubation temperature and extended reaction time to ensure the overall yield and to span the attainable distance for DBS crosslinking (Figs. 1b, 2b).
SDS-PAGE images revealed that the number of high molecular weight bands gradually increase along with prolonged heating of reduced insulin in bicarbonate buffer (Fig. 2c, Supplementary figs. 3, 4), indicative of the successful generation of a series of DBS products with distinct crosslinking levels. Conversely, when the cysteine thiol groups of reduced insulin chains were blocked with IAA, no bands for DBS products were observed even with four-hour incubation. Similarly, neither the reduced insulin without ammonium bicarbonate group nor the intact insulin group showed high molecular weight bands. These observations further demonstrate that ammonium bicarbonate solution thermodynamically promotes disordered covalent reshuffling among free thiols of insulin, leading to the reformation of disulfide bonds.

a Primary structure of native human insulin. b Workflow for bicarbonate heating-driven insulin DBS. c Characterization of insulin DBS products with non-reducing, denaturing gel electrophoresis. Results in c were validated through three independent experimental replicates, establishing methodological reproducibility. d Representative IM-MS heatmap and isotopic distribution for insulin DBS products (50 mM bicarbonate, 50 °C, 4 h). Source data are provided as a Source Data file.
As a powerful tool for characterization of peptide and protein oligomers26, native IM-MS was employed and tuned with optimal conformation-resolving capability for capturing insulin DBS products. Representative 2D IM-MS heatmap for insulin DBS products under the intensive DBS condition (50 °C, 50 mM bicarbonate, 4 h), clearly supports the presence of various degrees of thiol-crosslinked insulin species (Fig. 2d). Accurate mass matching and charge state assignments based on isotopic distributions for each species allow one to reliably identify both the composition of subunits and the disulfide numbers connecting these chains (Fig. 2d, Supplementary fig. 5, Supplementary Table 1). Notably, ion mobility measurements further confirm the identification of disulfide formation as indicated by the shifts in their distinct drift time distributions for species with varied thiol crosslinking degrees (Supplementary fig. 6).
Collectively, above-mentioned evidence has experimentally demonstrated the effectiveness of spatial distance-confined DBS on the insulin system with bicarbonate heating. The insulin disulfide shuffling condition for subsequent exploration was also optimized (50 °C, 50 mM bicarbonate, 4 h) based on the level of crosslinking products.
DBS delays insulin aggregation
As reported by previous microscopic and macroscopic-level studies27,28,29,30,31,32,33,34,35,36,37, insulin aggregation generally proceeds through a series of microscopic steps: (1) destabilization of the native monomer, exposing hydrophobic patches; (2) rate-limiting formation of oligomeric nuclei via primary nucleation; (3) secondary nucleation, including surface-catalyzed nucleation and fragmentation, accelerating the aggregation; (4) elongation via monomer addition to fibril ends, forming β-sheets and laterally associating protofibrils; (5) formation of mature, twisted fibrils rich in cross-β structure. Notably, the aggregation process of native insulin is highly concentration-dependent and sensitive to environmental conditions such as pH, temperature, and ionic strength, influencing both the kinetics and morphology of the resulting fibrils, highlighting the complex nature of insulin aggregation.
We firstly used ThT profiles to track the macroscopic processes of insulin aggregation under agitation conditions. Data in Fig. 3a clearly indicate the decreasing in lag time occurs with the increasing of initial monomeric insulin concentrations. To further identify microscopic steps underlying protein aggregation32,33,34, we plotted the half time of aggregation (t1/2) against insulin concentrations and tentatively derived the linear dependence between them upon exponent transformation (Fig. 3d). Surprisingly, a negative curvature was observed from the half time-concentration dependence curves for 0% DBS group using scaling exponents, suggesting the presence of competition of multiple processes in parallel, including primary nucleation, elongation and fragmentation-involved secondary nucleation steps32,33,38. The presence of multiple microscopic steps during insulin aggregation has been widely acknowledged, featuring the primary nucleation and fragmentation competing with well-adopted secondary nucleation pathway especially under shaking conditions28,29,31,32,33.

a Aggregation of native insulin with varied initial concentrations (25 μM to 300 μM) monitored by ThT fluorescence. b Aggregation of insulin in the presence of 1% (weight percentage) DBS products. c Direct comparison of the aggregation behavior of insulin in the presence of varied levels (0%, 0.1%, 1% and 10%) of DBS products. Buffer conditions, 100 mM NaCl (HCl, pH 1.6); 50 °C, agitation at 200 rpm. d Log-log plots of the half time (t1/2) of aggregation in different concentrations of insulin with or without 1% and 10% DBS products. Data in (a–d) are presented as mean values +/- SD derived from three biological replicates. Raw ThT curves for 10% DBS group were shown in Supplementary fig. 7a. e CD and (f) ATR-FTIR measurements of insulin fibrils with varied extents of DBS products. g AFM images of insulin fibers at different time points during aggregation. Scale bar: 1 μm. h Distributions for height and length of insulin fibers. Significance level is marked with an asterisk (p value was estimated using the two-sided Wilcoxon test). Source data are provided as a Source Data file.
To elucidate the specific molecular steps influenced by DBS products during insulin aggregation, we performed a series of ThT fluorescence monitoring experiments on solutions containing varied levels of DBS products and 1 mg/mL native insulin (Fig. 3b–d). With 1% DBS products, it was observed with apparently prolonged lag phase while maintaining the half time-concentration dependence trends across the tested concentration ranges of monomeric insulin (Fig. 3b, Supplementary Table 2). Direct comparisons of ThT profiles for insulin aggregation between with 1% DBS and without DBS group throughout the concentration ranges (Supplementary fig. 7) suggest that, DBS primarily affects insulin primary nucleation, as the lag phase of the kinetic traces are prolonged by addition of the non-native DBS products while the slope for the ThT traces seems to be not significantly affected by 1% DBS products. Data with increasing DBS levels show that, the final ThT fluorescence increases as a function of the levels of insulin DBS products, featuring a fivefold elevation of overall fluorescence for the 10% DBS group (Supplementary fig. 8).
However, when normalizing ThT fluorescence (Fig. 3c), we can clearly observe the DBS-dependent prolonged lag phases and decreased growth rates for the elongation and secondary nucleation steps, as evidenced by the decreased ThT growth slopes and enlarged half time values (0% DBS: t1/2 = 12.42 ± 0.04 h, 0.1% DBS: t1/2 = 13.11 ± 0.05 h, 1% DBS: t1/2 = 15.34 ± 0.04 h, 10% DBS: t1/2 = 18.96 ± 0.06 h; insulin, 172 μM). This type of microscopic impact is likely linked to the inhibition of elongation steps34,38. Notably, even upon the addition of preformed insulin fibrils to the aggregation samples (Supplementary fig. 9), DBS continued to inhibit the aggregation rate of insulin, indicating alterations at the microscopic level of the aggregation mechanism. When fibrils were introduced into the insulin aggregation mixture (Supplementary fig. 9), the presence of DBS products prolonged the lag phase of aggregation to varying extents in a concentration-dependent manner. Moreover, the growth rate of the ThT fluorescence signal gradually decreased with increasing concentrations of DBS products (Supplementary fig. 9). These observations suggest that the influence of DBS on insulin aggregation extends beyond simple primary nucleation. The persistence of this effect in seeded experiments provides compelling evidence that elongation and/or secondary nucleation processes are also affected. Additionally, the observations of morphological changes in final aggregation nanostructures (Fig. 3g, h) further imply that DBS may exert an influence on the elongation and/or secondary nucleation process.
The variations in the scaling exponents in Fig. 3d are directly linked to the impact of DBS on the aggregation kinetics of insulin. As the scaling factor would be expected to be around 0.5 for purely secondary nucleation pathway32,33,38, data in Fig. 3d show significantly low values (~0.04) of scaling exponents at low insulin concentrations ( < 100 μM), we observe significantly low values (~0.04) for the scaling exponent. This near-zero scaling suggests a weak dependence on monomer concentration, which can arise from saturation within the composite steps of the aggregation process. As previously detailed39, aggregation processes such as elongation, secondary nucleation, and even primary nucleation can exhibit multi-step mechanisms, often involving an initial monomer-dependent step followed by a monomer-independent step. Saturation of these monomer-dependent steps leads to a reduced dependence of the overall aggregation rate on monomer concentration. Fragmentation, by its nature, is already monomer-independent. When multiple such saturated processes occur in parallel, scaling exponents approaching zero can be observed. Therefore, the observed scaling exponent of ~0.04 is consistent with a system where several aggregation steps are operating near saturation, minimizing the dependence on monomer concentration. The inhibition of insulin aggregation by DBS products can be further observed at elevated insulin concentrations (Supplementary fig. 10). Taken together, our findings demonstrate that the DBS product modulates several microscopic stages in the insulin aggregation pathway.
Next, we designed a series of experiments to explore the structural impact of DBS products on insulin aggregation. The increase in β-sheet structure is corroborated by CD spectroscopy (Fig. 3e), like cryoEM structure evidence25, which confirms the presence of β-sheet conformation of native insulin filament, the content of which showing general dependency on the level of coexisting disulfide cross-linked oligomers as demonstrated by the increased level of negative peak at around 220 nm. ATR-FTIR data (Fig. 3f, Supplementary Table 3) further demonstrated the increase in β-sheet content upon addition of DBS products. Specifically, the β-sheet level rises from 19.16% to 29.76% with the addition of 10% DBS products. Considering the fact that DBS products alone do not form observable fibrils (Supplementary fig. 11), which was seemingly in line with previous observations on BSA and lysozyme16, we thus suspect that insulin DBS products directly involve in native insulin aggregation, presumably through unique molecular crosstalk between DBS products and native insulin. In short, insulin DBS products delay the microscopic steps of insulin aggregation presumably via targeting the elongation process while enhancing β-sheet folding in insulin structure (Fig. 3a–f), providing structural and kinetic basis for sequential altering thermodynamic morphologies of the ultimate aggregation nanostructures (Fig. 3g, h).
Given that we have identified the kinetically inhibitory role of DBS products in native insulin aggregation as shown above, we next sought to further investigate the potential impact of disulfide crosslinking on the morphology of native insulin assembly nanostructures. Representative AFM images in Fig. 3g show the morphological changes of insulin nanostructures as formed in the presence of various degrees of DBS products at distinct aggregation stages (7 h, 11 h, 14 h, 24 h). Firstly, all groups exhibited amyloid-like fibrillar morphology except the group of 10% DBS at 7 h, the structure of which we captured with evidence of amorphous aggregates. Compared to the 0% group, the 10% group gradually formed longer, thinner fiber bundles with prolonged aggregation time close to the final assembly stages.
Furthermore, at later assembly stages, varying degrees of disordered cross-links of disulfide bonds were observed to reduce the height of insulin fibers (Fig. 3h), significantly differentiating between the 10% and 0% groups (p < 0.0001). However, concerning fiber length, disordered cross-links of disulfide bonds facilitated the formation of longer, thinner fibers (Fig. 3h), notably more pronounced in the 10% group compared to the 0% group (14 h, p < 0.0001). The compositional heterogeneity of DBS products—featuring monomers, oligomers, and co-aggregation-prone species—could modulate insulin aggregation through multifaceted interactions with primary nucleation, elongation, and secondary pathways, thereby diversifying the resultant fibril polymorphs. These morphological changes in insulin fibers were also evident in TEM experiments (Supplementary fig. 12). These findings correlate well with the trends in the ThT-based aggregation curves (Fig. 3c, Supplementary figs. 8–10), where disordered disulfide crosslinks effectively slow down the aggregation process but enhance the final degree of insulin fibrillation.
Collective evidence from ThT, AFM, TEM, CD, and ATR-FTIR spectra have thus pointed out a unique fibrillation pathway for insulin under the influence of DBS products. Our data suggest that DBS products affect processes beyond primary nucleation, such as elongation and/or secondary nucleation. Notably, the presence of considerable degrees (e.g., 10%) of DBS products might serve as a supplemental seed and surface for heterogeneous primary and/or secondary nucleation and downstream elongation, catalyzing the generation of finally enriched cross-β fibrils. The current data resolution does not allow us to definitively distinguish between the effects on these individual processes.
DBS-insulin molecular crosstalk
The key to above-mentioned fibrillation pathway of heterogeneous secondary nucleation and aggregation, notably, is the noncovalent interactions and molecular crosstalk between DBS products and native insulin molecules. To verify this putative molecular mechanism, we conducted a series of native IM-MS experiments combined with tandem MS activation by using the incubation sample of native insulin and freshly reduced insulin. Representative MS spectra revealed significant interaction peaks between reducing A/B chain and native intact insulin for the incubation group compared to control groups of insulin alone and A/B chain group (Fig. 4a). Accurate mass matching and isotopic distribution examination confirm the presence of a series of DBS products and noncovalent interacting complexes. For example, peaks at m/z 1638.36 and m/z 1848.32 stand for noncovalent complexes between insulin and the A (AB-A5+) and B (AB-B5+) chains, respectively. Additionally, peaks at m/z 1143.58 (chain B with one disulfide bond), m/z 1190.50 (chain A with one disulfide bond), and m/z 1810.06 (insulin complexing with BB, AB-BB7+) were observed. Conversely, no such complex peaks were detected in the control groups (Fig. 4a).

a Representative native IM-MS spectrum showing the noncovalent interactions between monomeric insulin and DBS products. Insulin – native, untreated group as a control (0.5 mg/mL); A/B – insulin treated with TCEP (0.5 mg/mL); (Insulin + A/B) – the incubation group between native insulin and TCEP reduction products. b Representative CID spectrum for characteristic crosstalk products (AB-A/AB-B/AB-BB). c Native IM-MS heatmap showing the disruption of native insulin oligomerization with DBS products. Source data for (a, b) are provided as a Source Data file.
To further ascertain the noncovalent interactions and the presence of thiol crosslinking between chains, various tandem MS experiments were performed in addition to collisional cross-section (CCS) measurements for the differentiation of disulfide linkage isomer. Confirmation of noncovalent interactions within a multichain complex relies on the characteristic dissociation pattern of separating noncovalent subunits at a relative gentle condition prior to the deep fragmentation of protein backbones. For insulin-DBS complexes, as shown in Fig. 4b, isolation and activation of putative noncovalent complexes [AB-A]5+, [AB-B]5+ and [AB-BB]7+ produce expected dominant ions for [AB], [A], [B] and [BB] subunit chains at low collisional voltages ( < 30 V), supporting the preferential stripping of noncovalent interactions among these subunits. Quantification of dissociation products at various voltages from these noncovalent complexes (Supplementary fig. 13) further confirms the weak noncovalent interactions between intact insulin [AB] and DBS products ([A], [B] and [BB]).
Survival yield curves for noncovalent [AB-A]5+, [AB-B]5+ and [AB-BB]7+ precursor ions feature a much steeper jump (precursor ion intensity with changing ratio > 40%/10 V CID voltage) at certain collision voltages compared to the control of covalent ion pairs (Supplementary figs. 14, 15). Notably, the intensities of [AB] and [BB] are significantly higher than that of [A] and [B] species across all tested conditions (Fig. 4b, Supplementary figs. 13, 14), suggesting the presence of covalent disulfide crosslinking in [AB] and [BB] species that would require much higher energy ( > 50 V) to break the covalent bridges and separate individual [A] and [B] chains. Furthermore, we also performed a series of CCS measurements for these noncovalent complexes, all of which show 3.2% to 8.4% increments in measured CCS values in comparison with their covalent counterparts (Supplementary fig. 16). These observations not only validated the successful DBS reactions of freshly reduced insulin but also demonstrated the presence of complex interplay between DBS products with native insulin through noncovalent and covalent interactions.
Besides, native IM-MS was used to capture the self-assembly oligomers of insulin and compare the impact of DBS products on insulin oligomerization behavior. Representative heatmap (Fig. 4c) for native insulin oligomerization present clear signal for dimer, tetramer and hexamers, which are indispensable intermediates for insulin fibrillation9,25,40,41. Interestingly, larger oligomers like tetramer and hexamer significantly diminished by the incubation with both the freshly reduced insulin (A/B) and with mature DBS products, albeit with slightly higher levels of small oligomers of insulin observed upon mature DBS product treatment compared to freshly reduced insulin chains (Fig. 4c).
Taken together, native IM-MS measurements and CID tandem experiments have successfully enabled the validation of noncovalent and covalent interactions between native intact insulin and DBS products, which is vital for the elucidation of DBS-doped insulin aggregation pathway. We thus suspect that, it is the molecular crosstalk induced via noncovalent and covalent interactions between DBS and monomeric and oligomeric insulin species during the primary nucleation step that competing with the formation of native insulin hexamer for on-pathway oligomerization, which potentially contributes to the delayed aggregation process.
DBS enhances insulin neurotoxicity
Finally, we investigated the neurotoxicity induced by insulin fibers with varying concentrations of disordered disulfide-linked aggregates. Our findings demonstrated a concentration-dependent increase but varied levels in neurotoxicity in mouse neuroblastoma (N2a) cells, rat pheochromocytoma cells (PC12), and human pancreatic cancer cells (PANC1) (Fig. 5a–c, Supplementary Table 4). Specifically, insulin with increased DBS (10% DBS) showed significantly higher neurotoxicity compared to control insulin (0% DBS) at various concentrations, which was most pronounced at 50 μM in N2a cells and at 0.01 μM and 0.05 μM in PC12 cells (p < 0.001).

a–c MTT assay for the cellular viability of PC12, PANC1 and N2a cell lines treated with insulin fibers. Significance level is marked with an asterisk (p-values estimated using the Two-way ANOVA with multiple comparisons test, *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001, ****p-value < 0.0001, n = 5 biological replicates. Error bars indicate SDs). d, e Western blotting images and quantification of Bax/Bcl-2 associated with apoptotic N2a cells (insulin, 50 μM). f, g Flow cytometric analysis and quantification of N2a apoptosis (insulin, 50 μM). Significance level for (e, g) is marked with an asterisk (p values estimated using two-sided unpaired t-test), *p-value < 0.05, **p-value < 0.01. Data are presented as mean values +/- SD derived from three biological replicates.). h Hierarchical clustering analysis of 887 quantified proteins from label-free bottom-up proteomics experiments. i Chord plot analysis of GO terms for up-regulated proteins in the 10% DBS products group compared to control group (no DBS treatment). Fisher’s exact test (one-tailed) with Benjamini-Hochberg multiple testing correction was applied for GO term enrichment analysis. Source data are provided as a Source Data file.
We further examined whether insulin fiber-induced neurotoxicity involved the apoptosis pathway. Western blot analysis (Fig. 5d, e, Supplementary fig. 17c, d) revealed a significant increase in the Bax/Bcl-2 ratio in cells treated with 10% DBS insulin compared to the control group (p < 0.01). This finding was further supported by flow cytometry (Fig. 5f, g, Supplementary figs. 17a, b, 18), which demonstrated a significantly higher apoptotic rate in the 10% DBS group (p < 0.05).
Proteomic experiments with bottom-up LC-MS/MS setups identified 2,876 proteins, with 887 quantified (Fig. 5h, Supplementary fig. 19a–d). Hierarchical clustering and Gene Set Variation Analysis (GSVA) revealed functional alterations in protein expression levels between groups. Pairwise comparisons identified differentially expressed proteins, with 34 up-regulated and 68 down-regulated proteins in the 10% vs Control group. GO and KEGG pathway analyses indicated that up-regulated proteins were involved in energy metabolism, mitochondrial membrane permeability alteration, apoptotic pathways, and neurodegenerative diseases (Fig. 5i, Supplementary fig. 19e–g, Supplementary fig. 20). Down-regulated proteins were associated with protein-containing complex assembly regulation, cell meiotic division cycle, protein catabolic process regulation, and DNA geometric and conformational changes (Supplementary fig. 21). In the 10% vs 0% and 0% vs Control groups, GO and KEGG enrichment analyses revealed unique and shared pathways related to organelle functions, protein metabolism, cholesterol synthesis and metabolism, oxidation-reduction processes, and neuronal cell vesicle transport (Supplementary figs. 22–26).
In summary, our study provides compelling evidence that DBS in insulin enhances its aggregation propensity, leading to increased neurotoxicity via the induction of mitochondrial apoptosis. These findings highlight the importance of controlling DBS during insulin production and storage to minimize potential risks associated with neurotoxicity.
Discussion
This study provides insights into the impact of DBS on insulin aggregation and its associated neurotoxicity. Our findings demonstrate that DBS products significantly alter the aggregation pathway of insulin, leading to the formation of morphologically distinct fibrils with enhanced neurotoxic properties. We demonstrate that DBS in insulin occurs within a spatially confined regime, likely facilitated by the close proximity of cysteine residues within the insulin structure. This process generates a heterogeneous mixture of disulfide-crosslinked insulin oligomers, which exhibit a unique molecular crosstalk with native insulin molecules. This crosstalk, evidenced by native IM-MS and tandem MS analysis, disrupts the formation of native insulin hexamers and delays the overall aggregation kinetics. Despite the delayed kinetics, DBS ultimately enhances insulin aggregation, leading to the formation of longer, thinner fibrils enriched with β-sheet content. These observations demonstrate the primary inhibition of nucleation and secondary inhibition of elongation steps during insulin aggregation microscopic steps induced by the addition of DBS products.
Importantly, our study highlights the significant biological implications of DBS in insulin. We demonstrate that insulin with increased DBS exhibits significantly enhanced neurotoxicity in various cell lines, including neuronal and pancreatic cells. This enhanced toxicity is attributed to the induction of mitochondrial apoptosis, evidenced by increased Bax/Bcl-2 ratios and apoptotic rates in cells treated with DBS-rich insulin. Proteomic analysis further supports the apoptotic mechanism, revealing the upregulation of proteins associated with energy metabolism, mitochondrial membrane permeability, and apoptotic pathways in cells exposed to DBS-rich insulin. Additionally, we observed the downregulation of proteins involved in cell cycle regulation, protein metabolism, and neuronal vesicle transport, indicating a global cellular response to DBS-induced insulin aggregation.
Many prior reports have explored the general principles of protein aggregation and the influence of molecular chaperones and non-native protein structures on amyloid formation30,33,34,42,43,44. However, this study advances beyond these general principles by directly examining the implications of DBS in insulin for human health. Our findings might bear significant implications for insulin production, storage, and therapeutic applications, as similar DBS products can also be observed in more clinically relevant buffer conditions (Supplementary fig. 27). However, it is important to emphasize that a reducing environment is a prerequisite for the formation of these species, as demonstrated by the critical role of DTT in Supplementary fig. 27. While reducing conditions are not typically maintained during standard insulin storage or therapeutic use, they can arise in specific contexts, such as during certain manufacturing processes, stability testing, or in localized physiological environments. Additionally, our data show that increased temperature accelerates DBS formation, but these reactions can also occur at lower temperatures, including those close to room temperature. These observations highlight the potential relevance of our findings to real-world scenarios, while also underscoring the importance of considering the specific environmental conditions under which DBS formation may occur.
Specifically, the bicarbonate buffer and temperature ramping settings, although distinct from typical insulin formulations, offer a controlled environment relevant to challenging industrial conditions (e.g., fluctuating temperatures in hot climates or emergency settings) that might promote the formation of DBS products33,45,46. The current experimental design simply accelerates the reactions, enabling observation and characterization of the DBS process and its effects within a feasible experimental timeframe. Consequently, our study suggests that controlling DBS during these processes is crucial to minimize the formation of neurotoxic aggregates. Further research is warranted to develop strategies to inhibit DBS in insulin and mitigate its potential risks. This study provides a framework for understanding the complex interplay between protein misfolding, aggregation, and neurotoxicity, paving the way for the development of safer and more effective protein therapeutic biosimilars more than the native insulin system.
Methods
Chemicals
Human insulin standard was purchased from TargetMol Co., Ltd. (Shanghai, China). Analytical grade ammonia bicarbonate (NH4HCO3) was obtained commercially from RHΛWN reagent (Shanghai, China). Thioflavin T (ThT), Tris (2-carboxyethyl) phosphine (TCEP), and iodoacetamide (IAA) were purchased from Sigma-Aldrich (St. Louis, MO). Dithiothreitol (DTT) and BCA assay kit were provided commercially from Beyotine Biotech Co., Ltd. (Shanghai, China). Purified water (resistivity of 18.2 MΩ.cm) was obtained from a Milli-Q® Reference System (Millipore Corp., Bedford, MA, USA). The peptide quantitation analysis assay kit was commercially achieved from ThermoFisher (Pittsburgh, PA, USA). Trypsin and LysC were purchased from Beijing Shengxia Proteins Scientific Ltd. (Beijing, China). Annexin V-FITC/PI apoptosis detection kit was purchased from Dalian Meilun Biotechnology Co., Ltd. (Dalian, China). Anti-Bcl-2 antibody and anti-Bax antibody were obtained from Abcam Trading Co., Ltd (Shanghai, China). Mouse-β-actin antibody was provided commercially by Beyotine Biotech Co., Ltd. (Shanghai, China). Goat anti-rabbit IgG, goat anti-mouse IgG and horseradish enzyme tags were purchased from Beijing Zhongshan Jinqiao Co., Ltd. (Beijing, China). Fetal bovine serum (FBS) and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Gibco (Carlsbad, CA). The mouse neuroblastoma N2a cell line (N2a), Rat adrenal medullary pheochromocytoma cell line (PC12) and human pancreatic cancer cell PANC1 were obtained from the American Type Culture Collection (Manassas, VA).
Insulin disulfide bond shuffling
Firstly, native insulin was reduced through adding 20 mM TCEP to the stock solution, where the final reaction concentration of insulin is 0.5 mg/mL. After 1 h incubation under room temperature, the disulfide bonds of insulin were completely reduced. Subsequently, Sep-Pak C18 (Waters) was utilized to eliminate TCEP. Vacuum concentration dryer condensed into A/B chain powder. The powder of mixed A/B chain was redissolved in 0, 5, 10 and 50 mM ammonium bicarbonate buffer, respectively. The total concentration of A/B chain was 1 mg/mL. The solution was then heated at 50 °C for 0, 0.25, 0.5, 1, 2 and 4 h to facilitate disordered cross-linking of the disulfide bonds between A and B chains. Meanwhile, parallel preparation of 5 mM IAA blocked A/B chain (1 mg/ml) sulfhydryl group and the intact insulin group. The mixed of A/B chain with IAA was incubated at room temperature and dark for 2 h.
Predicting DBS products with AlphaFold3
To predict potential disulfide bond formation between segments of insulin peptides, we employed the AlphaFold3 structure prediction tool47. The crystal structure of human insulin was obtained from the Protein Data Bank (PDB ID: 1A7F), corresponding to UniProt entry P01308. The amino acid sequences of the A and B chains were extracted as follows: A chain, GIVEQCCTSICSLYQLENYCN; B chain, FVNQHLCGSHLVEALYLVCGERGFFYTPK. All cysteine residues in both chains were maintained in their reduced state, with free thiol groups. Under the “Protein” input type option, the sequences of the A and B chains were entered in various combinations into the input fields, and separate predictions were generated. The resulting structural models were downloaded in.cif format for further analysis. The model was obtained by AlphaFold3 on July 18, 2024. We assessed the confidence of the predicted structures using the predicted Local Distance Difference Test (pLDDT) scores provided by AlphaFold. Our analysis of cysteine proximity primarily focused on residues within regions pLDDT scores > 70. To evaluate the potential for disulfide bond formation, we used PyMOL to measure inter-cysteine distances within the predicted structures. Thiol-thiol distances were analyzed to identify spatial arrangements conducive to DBS formation, providing insights into the structural feasibility of such interactions. It is important to note that this in silico analysis provides a preliminary assessment of potential disulfide bond formation and does not definitively confirm the presence of such bonds. Further experimental validation, like native IM-MS measurements shown in this study, is required to confirm these predictions.
Non-reducing denaturing gel electrophoresis
Take 10 μL of samples incubated at each time point in the above groups and boil 5 min at 2 × 10 μL loading buffer, 95 °C for electrophoretic analysis. Loading buffer (2×) contains 100 mM Tris, 30% glycerol, 0.1% bromophenol blue, SDS 4%, and lacks disulfide reducing agents such as DTT. The anode Buffer contains 200 mM Tris, pH 9.0, the cathode buffer is 100 mM Tris, 52.7 mM Tricine and 3.47 mM SDS, pH 8.25. The boiled samples of each group with 10 microliters were added to the concentrated glue channel and samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). All electrophoresis experiments were conducted at 100 V for 20 min through the stacking gel and at 150 V for 60 min through the separating gel. The gels were stained with Coomassie blue stain, and the image was acquired using a scanner.
ThT fluorescence assays
Monomeric insulin (1 mg/mL) and cross-linking (0.1%, 1%, 10%) were incubated alone or together in 100 mM NaCl buffer (HCl, pH 1.6) to study the impact of disulfide cross-linking disorder on insulin aggregation. In addition, to investigate the nucleation mechanism of disordered disulfide cross-linked aggregates on insulin aggregation, insulin at different concentrations was co-incubated with disordered disulfide cross-linked aggregates at varied levels in 100 mM NaCl buffer (HCl, pH 1.6). All samples of each group were incubated in the metal bath at 50 °C for 200 rpm for a certain time. Each group of samples was taken 3 µL at intervals of 1 h and put into a black 96 well plate, and then 200 μL of ThT (10 μM) solution was added to incubate for 10 min. ThT fluorescence measurements were carried out on a multifunctional microplate reader (Molecular Devices Co., Ltd.) at the excitation/emission wavelength of 430/490 nm. The signals from the sample wells were corrected by subtracting the signal from the blanked wells containing the buffer and ThT but no sample. All reactions were performed with three replicates. All data were plotted using GraphPad Prism 6.01, with the mean and SD (SD, n = 3) of the mean displayed as points and connecting lines with error bars. The aggregation curves were fitted by Boltzmann Sigmoidal model.
Circular Dichroism Spectroscopy (CD)
The CD spectra were recorded in the region from 190 to 260 nm on a Biologic MOS-500 spectrometer. All spectra were recorded with a wavelength interval of 1.0 nm and an acquisition time of 1 s. CD samples with a concentration of 0.1 mg/mL were prepared by diluting from the stock solution of the insulin fiber.
Attenuated Total Reflection Fourier-Transform Infrared Spectrophotometer (ATR-FTIR)
The secondary structure of fibrils formed after insulin aggregation in different groups was analyzed using ATR-FTIR (ThermoFisher Scientific Nicolet iS50). Following established protocols, all ATR-FTIR samples were centrifuged at 14,000 x g for 15 min at 4 °C after aggregation, and the supernatant was discarded. The resulting pellets were resuspended in D2O, thoroughly mixed, and washed three times to eliminate residual salts or aggregation buffer. Peak deconvolution and curve fitting in the 1600–1700 cm−1 range were performed using OMNIC software (version 8.3). The deconvoluted peaks were further processed via second derivative analysis and numerical fitting using Peakfit software (version 4.12), with the iteration threshold set to 0.07 and an R² value greater than 0.99. The peak areas corresponding to specific secondary structure components were then extracted for statistical analysis.
Atomic Force Microscopy (AFM)
AFM images were taken by the BenYuan instrument under the tapping mode. Dilute all sample (Insulin fiber) solutions to 0.1 mg/mL with deionized water. 10 μL of the diluted sample on a clean mica slide. Allow it to stand at room temperature for 20 min, then the retained liquid on the mica surface was removed by filter paper. Conduct experiments after the mica slide has dried at room temperature.
Transmission Electron Microscope (TEM)
TEM images were taken by a Tecnai G2 F20 microscope with an accelerating voltage of 200 kV. TEM samples of all the insulin fiber were prepared by diluting the stock solution to a concentration of 0.1 mg/mL. A volume of 10 µL of the diluted solution was pipetted onto the surface of a carbon-coated copper grid for 10 min and removed by filter paper. After that, the droplet was removed with filter paper. The grid was dried in the fume cupboard before measurement.
Native IM-MS
The disulfide bond cross-linked samples of each group for 4 h were diluted to an appropriate concentration with 10 mM ammonium acetate buffer, and the dilution multiple was consistent among the samples of each group. Approximately 8 μL sample was loaded into a homemade nanospray source and MS instrument was run in positive ion mode to represent the reformation of inter-chain disulfide bonds between the A and B insulin chains. To investigate the interaction between insulin and disordered disulfide bond crosslinked or free A/B chains during aggregation, solutions of 1 mg/mL insulin and 1 mg/ml A/B chains or DBS products were separately prepared. In the experimental group, equal volumes (25 μL) of these solutions were mixed, and control groups received 25 μL of deionized water each. The samples were subjected to 2 min of sonication at room temperature (300 W), following a 1-hour equilibration period at room temperature before analysis. All reactions were performed with three replicates. Nanospray voltages range between 1.0 ~ 1.8 kV and the sampling cone was used at 50 V. All IM-MS data were collected using Waters Synapt XS instruments. The MS cone temperature was 75 °C. The traveling-wave ion mobility separator was operated at a pressure of ∼2.85 mbar, and DC voltage waves (40 V wave height, traveling velocity of 700 m/s) to generate ion mobility separation. The CCS calibration was carried out with Insulin, Melittin, Bradykinin and Bovine Serum Albumin with mass ranges of m/z 100 ~ 5000. Each sample was prepared three replicates and obtained stable and reproducible signal. CCS calibration curves were generated using a previously described protocol48, and using literature CCS values with nitrogen (He) derived for use with the Synapt instrument platform. To improve the accuracy, in our study, we have employed an updated calibration method48 for CCS measurements. Similar to previous studies49,50,51, triplicate measurements were taken to obtain uncertainty values using the following equation:
where σ is the relative standard deviation of triplicate measurements (was 0.58%), cal_error is the uncertainty range for CCS calibration (was 0.27%), and database_error is the uncertainty in database values (set as 0.3%). The total uncertainty for TWIMS CCS of insulin is thus estimated to be ~0.71%. All IM-MS data were processed using the MassLynx v4.2 and Driftscope software affiliated with the instrument.
Cell culture and cytotoxicity assay
N2a (mouse neuroblastoma) and PC12 (rat pheochromocytoma) cells serve as established neuronal and neuroendocrine models, respectively, with well-characterized responses. These cell lines were used to assess neurotoxicity via MTT assays and cellular responses to protein aggregates using western immunoblotting for apoptotic markers. In addition, PANC1 cells (human pancreatic cancer) were selected to investigate potential pathological implications of insulin aggregation in its native environment. Given that insulin transitions from a hexameric storage form to monomers upon secretion, we hypothesized that this physiological transition could, under pathological conditions, increase the risk of amyloid aggregation and subsequent cytotoxicity within pancreatic tissue. PANC1 cells allowed us to explore this hypothesis using cell viability assays.
MTT assays were performed on PC12, N2a and PANC1 cells and was utilized to test the cytotoxicity induced by insulin aggregates. In the presence of 10% FBS and 1% penicillin/streptomycin, PC12, N2a and PANC1 cells were cultured in a sterile DMEM medium at 37 °C in a humid atmosphere with 5% CO2. The three kinds of cultured cells were inoculated in 96 well plates with a density of 5 × 103 cells/well and incubated for 24 h (n = 5). Insulin fibers of each group in the plateau stage were dialyzed with sterilized PBS solution for 12 h. After that, it was concentrated to an appropriate volume and diluted to the required concentration with PBS (cell culture grade). The initial monomeric insulin concentrations (10, 25, and 50 µM for N2a; 0.005, 0.01, and 0.05 µM for PC12; and 0.5, 1.0, and 5 µM for PANC1) were determined using a NanoDrop spectrophotometer (Nano-300, Hangzhou Allsheng Instruments Co., Ltd), measuring absorbance at 280 nm. These concentrations were chosen based on preliminary cell viability assays to ensure that the monomeric insulin itself did not induce significant cytotoxicity before fibril formation. All insulin fibers were incubated with cell lines for 24 h. Subsequently, MTT solution (10 μL, 5 mg/mL) was added to each well and incubated for 4 h. To dissolve the formazan crystals, DMSO (200 μL) was added to each well after removing the medium. The absorbance at 570 nm and 690 nm were measured using a microplate reader (Molecular Devices Co., Ltd.), and cell viability was calculated accordingly. Measure the relative viability (RV) by following formula:
Western blot analysis
Protein lysates were prepared from N2a and PC12 cells treated with insulin fiber. Protein concentrations were measured using a BCA kit, and the absorbance was measured at 562 nm using a miniature flat-panel reader. Equal amounts of protein samples were separated by electrophoresis and transferred to polyvinylidene difluoride (PVDF) membranes. Subsequently, the PVDF membrane was blocked with 5% skimmed milk powder (1 h, room temperature), followed by overnight incubation at 4 °C with primary antibodies anti-Bax (1: 2000), anti-Bcl-2 (1: 2000), and β-actin (1: 3000). The membrane was washed with TBST and then incubated with secondary antibodies at room temperature for 1.5 h. Finally, protein bands were detected using Azure C600 (Azure Biosystems, USA) instrument, and the density of each band was analyzed using ImageJ (version 1.54) software.
Flow cytometric analysis
PC12 and N2a cells were seeded in the 6-well plates with a density of 8 × 105 cells/well. After adherent growth, 0.005 and 50 μM insulin fibers were added to the cell culture plate and incubated for 24 h. All experiments were performed with three replicates. The above cells were washed with PBS three times and digested with trypsin without EDTA. Staining the cells with Annexin V-FITC and PI for 10 min in the dark led to the samples for cellular apoptotic studies estimated by a flow cytometer (BD LSR Fortessa, USA) and analyzed by FlowJo software (version 10.10)
Sample preparation for proteomics
N2a cells were lysed in lysis buffer containing 8 M urea, 50 mM Tris-HCl, 5 mM CaCl2, 30 mM NaCl and protease inhibitor (v/v, 1:100), and add appropriate amount of HCl to adjust pH 8.0. Each sample was prepared in triplicate. Then use probe sonicator to extract proteins from cells, and further centrifuge at 14,000 x g for 15 min. Perform protein assay according to the instructions for BCA Reagent Kit manufacturer’s protocols. According to the measured protein concentration, take a certain volume of supernatant containing 100 μg of protein for subsequent experiments. The protein extract was reduced with 10 mM DTT at 37 °C for 30 min and then alkylated with 50 mM IAA for another 30 min in the dark. Add DTT again to a final concentration of 10 mM to react with excess IAA and incubate at room temperature for 5 min. All samples were subjected to digestion with LysC and trypsin (w/w, 1:100) and incubated at 37 °C for 4 h. Add tris buffer to dilute urea to <1 M, and incubate at 37 °C overnight. The samples were quenched with 10% TFA to a final concentration of 0.25% and centrifuged at 14,000 x g for 15 min. Desalting the supernatant using Sep-Pak C18 (Waters) according to the manufacturer’s protocols and dry down the sample. Resuspend samples with an appropriate volume of 0.1% formic acid (FA)-H2O, centrifuge at 14,000 x g for 10 min. Determine the peptide concentration of the supernatant using an assay kit and aliquot an appropriate number of samples for LC-MS/MS analysis.
LC-MS/MS analysis for proteomics
Samples were analyzed on an Orbitrap Eclipse Mass Spectrometer (ThermoFisher Scientific, Waltham, MA) coupled to a Dionex UltiMated 3000 UPLC system. Each sample was dissolved in 0.1% FA-H2O before loading onto a 75 µm inner diameter NanoViper microcapillary column that is packed with 25 cm of bridge Ethylene Hybrid C18 particles (2 µm, 100 Å, ThermoFisher Scientific). Mobile phase composed of 0.1% aqueous formic acid (A) and ACN containing 0.1% formic acid (B) was pumped into homemade microcapillary column with a gradient program as below: 0−20 min, 3%−15% B; 20−90 min, 15%−30% B; 90−105 min, 30%−45% B, 105−110 min, 45%−95% B, 110−120 min, 95% B; and flow rate, 300 nL/min. The injection volume was fixed at 3.0 μL. The instrument was run in data-dependent acquisition (DDA) mode, capturing survey scans of peptide precursors in the m/z range of 300 to 2000 using the orbitrap (OT) with a resolution of 120,000. Subsequently, MS/MS acquisition was performed in the orbitrap (OT). The automatic gain control (AGC) target for both MS1 and MS2 was set to Standards. Precursors underwent fragmentation continuously for 3 s, with stepped normalized collision energies of 25, 30, and 35. Maximum injection times were defined as 50 ms for MS1 scans and Dynamic for MS2 scans. A dynamic exclusion period of 45 s, with a tolerance of 10 ppm, was applied to the precursors. All acquisitions were conducted in positive polarity mode, and a critical step was the prior equilibration of each sample before injection.
Data analysis
Protein identification and quantification were conducted through MaxQuant (version 2.5.2.0)50 with Uniport Mus Musculus reviewed database (May 19, 2024) with trypsin/P as selected enzymes. Notably, MS1 scans utilized a precursor ion mass tolerance of 10 ppm and allowed for two missed cleavages. A false discovery rate (FDR) of 1% was applied for both protein and peptide identification via the target decoy strategy. Specific modifications, including carbamidomethyl of cysteine residues, oxidation of methionine residues, and acetylation at protein N-termini, were considered during analysis. Statistical analysis involved filtering for reverse proteins, site-specific proteins, and potential contaminants using Perseus (version 1.6.7.0). To address individual expression variations, proteins detected <2 times in one group were filtered post-grouping. Missing data points were imputed using the normal distribution method with specified parameters. Subsequent statistical analyses, including two-sample Student’s t-test and one-way ANOVA, were performed, with p-values adjusted using the Benjamini-Hochberg method. Visualization techniques such as hierarchical clustering and volcano plots were generated using R packages and Hiplot Pro online software. For protein intensity profiling, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were conducted through R packages. PPI analysis was accomplished on the STRING online website, and Cytoscape 3.9.1 software was used to analyze PPI network nodes.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The proteomics data in this study have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD053868. Unless otherwise stated, all data supporting the results of this study can be found in the article, supplementary, and source data files. Source data are provided in this paper. Source data are provided with this paper.
References
Wesseling, H. et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 183, 1699–1713.e1613 (2020).
Despres, C. et al. Identification of the Tau phosphorylation pattern that drives its aggregation. Proc. Natl. Acad. Sci. USA. 114, 9080–9085 (2017).
Vadukul, D. M. et al. α-Synuclein Aggregation Is Triggered by Oligomeric Amyloid-β 42 via Heterogeneous Primary Nucleation. J. Am. Chem. Soc. 145, 18276–18285 (2023).
Rodriguez-Rodriguez, P. et al. Tau hyperphosphorylation induces oligomeric insulin accumulation and insulin resistance in neurons. Brain 140, 3269–3285 (2017).
Nowell, J., Blunt, E. & Edison, P. Incretin and insulin signaling as novel therapeutic targets for Alzheimer’s and Parkinson’s disease. Mol. Psychiatry 28, 217–229 (2023).
Das, A., Shah, M. & Saraogi, I. Molecular Aspects of Insulin Aggregation and Various Therapeutic Interventions. ACS Bio. & Med. Chem. Au. 2, 205–221 (2022).
Kachooei, E. et al. Oligomeric Forms of Insulin Amyloid Aggregation Disrupt Outgrowth and Complexity of Neuron-Like PC12 Cells. PLoS One 7, e41344 (2012).
Akbarian, M. et al. Mechanisms behind the Fibrillation and Toxicity of Insulin Fibrils on Neuron Cells by Engineered Curcumin Analogs. ACS Chem. Neurosci. 13, 2613–2631 (2022).
Lieblich, S. A. et al. 4S-Hydroxylation of Insulin at ProB28 Accelerates Hexamer Dissociation and Delays Fibrillation. J. Am. Chem. Soc. 139, 8384–8387 (2017).
Fass, D. & Thorpe, C. Chemistry and Enzymology of Disulfide Cross-Linking in Proteins. Chem. Rev. 118, 1169–1198 (2018).
Fass, D. Disulfide Bonding in Protein Biophysics. Annu. Rev. Biophys. 41, 63–79 (2012).
Wang, Y., Lu, Q., Wu, S.-L., Karger, B. L. & Hancock, W. S. Characterization and Comparison of Disulfide Linkages and Scrambling Patterns in Therapeutic Monoclonal Antibodies: Using LC-MS with Electron Transfer Dissociation. Anal. Chem. 83, 3133–3140 (2011).
Iadanza, M. G. et al. The structure of a β2-microglobulin fibril suggests a molecular basis for its amyloid polymorphism. Nat. Commun. 9, 4517 (2018).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Radamaker, L. et al. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 10, 1103 (2019).
Yang, M., Dutta, C. & Tiwari, A. Disulfide-Bond Scrambling Promotes Amorphous Aggregates in Lysozyme and Bovine Serum Albumin. J. Phys. Chem. B. 119, 3969–3981 (2015).
Laps, S., Atamleh, F., Kamnesky, G., Sun, H. & Brik, A. General synthetic strategy for regioselective ultrafast formation of disulfide bonds in peptides and proteins. Nat. Commun. 12, 870 (2021).
Jin, A. H. et al. Conotoxins: Chemistry and Biology. Chem. Rev. 119, 11510–11549 (2019).
Jiang, S., Hägglund, P., Carroll, L., Rasmussen, L. M. & Davies, M. J. Crosslinking of human plasma C-reactive protein to human serum albumin via disulfide bond oxidation. Redox. Biol. 41, 101925 (2021).
Baumann, A. L. et al. Chemically Induced Vinylphosphonothiolate Electrophiles for Thiol–Thiol Bioconjugations. J. Am. Chem. Soc. 142, 9544–9552 (2020).
Li, G. et al. Bicarbonate buffers can promote crosslinking and alternative gas-phase dissociation pathways for multiprotein complexes. Int. J. Mass Spectrom. 469, 116687 (2021).
Sinz, A. Cross-Linking/Mass Spectrometry for Studying Protein Structures and Protein–Protein Interactions: Where Are We Now and Where Should We Go from Here? Angew. Chem. Int. Ed. 57, 6390–6396 (2018).
Hanozin, E. et al. Covalent Cross-Linking as an Enabler for Structural Mass Spectrometry. Anal. Chem. 91, 12808–12818 (2019).
Iacobucci, C. et al. First Community-Wide, Comparative Cross-Linking Mass Spectrometry Study. Anal. Chem. 91, 6953–6961 (2019).
Wang, L. et al. Structural basis of insulin fibrillation. Sci. Adv. 9, eadi1057 (2023).
Guo, R.-C. et al. Artificial Peptide-Protein Necrosomes Promote Cell Death. Angew. Chem. Int. Ed. 62, e202314578 (2023).
Vestergaard, B. et al. A helical structural nucleus is the primary elongating unit of insulin amyloid fibrils. PLoS Biol. 5, e134 (2007).
Librizzi, F. & Rischel, C. The kinetic behavior of insulin fibrillation is determined by heterogeneous nucleation pathways. Protein Sci. 14, 3129–3134 (2005).
Eden, K., Morris, R., Gillam, J., MacPhee, C. E. & Allen, R. J. Competition between primary nucleation and autocatalysis in amyloid fibril self-assembly. Biophys. J. 108, 632–643 (2015).
Luo, J., Warmlander, S. K., Graslund, A. & Abrahams, J. P. Non-chaperone proteins can inhibit aggregation and cytotoxicity of Alzheimer amyloid beta peptide. J. Biol. Chem. 289, 27766–27775 (2014).
Selivanova, O. M. et al. How to determine the size of folding nuclei of protofibrils from the concentration dependence of the rate and lag-time of aggregation. II. Experimental application for insulin and LysPro insulin: aggregation morphology, kinetics, and sizes of nuclei. J. Phys. Chem. B. 118, 1198–1206 (2014).
Ziaunys, M., Sneideris, T. & Smirnovas, V. Self-inhibition of insulin amyloid-like aggregation. Phys. Chem. Chem. Phys. 20, 27638–27645 (2018).
Saha, S. & Deep, S. Glycerol inhibits the primary pathways and transforms the secondary pathway of insulin aggregation. Phys. Chem. Chem. Phys. 18, 18934–18948 (2016).
Arosio, P. et al. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 7, 10948 (2016).
Foderà, V., Librizzi, F., Groenning, M., van de Weert, M. & Leone, M. Secondary Nucleation and Accessible Surface in Insulin Amyloid Fibril Formation. J. Phys. Chem. B. 112, 3853–3858 (2008).
Akbarian, M., Yousefi, R., Farjadian, F. & Uversky, V. N. Insulin fibrillation: toward strategies for attenuating the process. Chem. Commun. 56, 11354–11373 (2020).
Chatani, E. et al. Early aggregation preceding the nucleation of insulin amyloid fibrils as monitored by small angle X-ray scattering. Sci. Rep. 5, 15485 (2015).
Meisl, G. et al. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 11, 252–272 (2016).
Meisl, G. et al. Scaling behaviour and rate-determining steps in filamentous self-assembly. Chem. Sci. 8, 7087–7097 (2017).
Ivanova, M. I., Sievers, S. A., Sawaya, M. R., Wall, J. S. & Eisenberg, D. Molecular basis for insulin fibril assembly. Proc. Natl. Acad. Sci. USA. 106, 18990–18995 (2009).
Carpenter, M. C. & Wilcox, D. E. Thermodynamics of Formation of the Insulin Hexamer: Metal-Stabilized Proton-Coupled Assembly of Quaternary Structure. Biochemistry 53, 1296–1301 (2014).
Wu, J. et al. Identifying the role of co-aggregation of Alzheimer’s amyloid-β with amorphous protein aggregates of non-amyloid proteins. Cell Rep. Phys. Sci. 3, 101028 (2022).
Bahmani, M., Akbarian, M., Tayebi, L. & Farjadian, F. The inhibitory effect of curcumin loaded poly (vinyl caprolactam) nanohydrogel on insulin fibrillation. Process Biochem. 117, 209–218 (2022).
Akbarian, M., Tayebi, L., Mohammadi-Samani, S. & Farjadian, F. Mechanistic Assessment of Functionalized Mesoporous Silica-Mediated Insulin Fibrillation. J. Phys. Chem. B. 124, 1637–1652 (2020).
Richter, B., Bongaerts, B. & Metzendorf, M. I. Thermal stability and storage of human insulin. Cochr. Database Syst. Rev. 11, CD015385 (2023).
Maikawa, C. L., d’Aquino, A. I., Lal, R. A., Buckingham, B. A. & Appel, E. A. Engineering biopharmaceutical formulations to improve diabetes management. Sci. Transl. Med. 13, eabd6726 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Richardson, K., Langridge, D., Dixit, S. M. & Ruotolo, B. T. An Improved Calibration Approach for Traveling Wave Ion Mobility Spectrometry: Robust, High-Precision Collision Cross Sections. Anal. Chem. 93, 3542–3550 (2021).
Ruotolo, B. T., Benesch, J. L. P., Sandercock, A. M., Hyung, S.-J. & Robinson, C. V. Ion mobility–mass spectrometry analysis of large protein complexes. Nat. Protoc. 3, 1139–1152 (2008).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Li, G. et al. Site-specific chirality-conferred structural compaction differentially mediates the cytotoxicity of Aβ42. Chem. Sci. 14, 5936–5944 (2023).
Acknowledgements
The project was financially supported by the National Key R&D Program of China (2022YFA1305200 to G.L.), the National Natural Science Foundation of China (22474062, 22293030, 22293032 to G.L.), joint funding support from the Fundamental Research Funds for the Central Universities (Nankai University, 63241610 to G.L.).
Author information
Authors and Affiliations
Contributions
G.L. conceived, designed, and supervised the project. W.Q. and Juan L. performed the MS and other experiment, collected and processed the data. R.L. completed proteomics experiment. G.L. and W.Q. wrote the manuscript. Jun L., X.W., B.H., Z.Z., and Z.Y. were involved in the data analysis, project discussion and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Georg Meisl and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qin, W., Li, R., Liu, J. et al. Spatially constrained disulfide bond shuffling delays insulin aggregation and enhances neurotoxicity. Nat Commun 16, 6841 (2025). https://doi.org/10.1038/s41467-025-62257-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62257-0
