
Abstract
Cancer metastasis and recurrence remain the leading causes of cancer-related mortality, and lung is a major metastatic anatomical location. Chimeric antigen receptor macrophages (CAR-M) represent promising candidates for cancer therapy owing to their superior tumour-infiltrating and antigen-specific phagocytotic abilities, and to being professional antigen presenting cells. However, broader applications of CAR-Ms face challenges such as complex manufacturing processes and predominant accumulation in the liver following intravenous administration. Here we present an inhalable engineered small extracellular vesicle (sEV), which contains mesothelin-specific CAR messenger RNA (CARmRNA@aCD206 sEVs) for in situ generation of CAR-Ms. The sEVs are surface-integrated with anti-CD206 single-chain variable fragments (scFv) to target CD206-expressing, immunosuppressive (M2 phenotype) macrophages. The results in mouse models suggest that inhaled CARmRNA@aCD206 sEVs could accumulate in lung tissue and deliver CAR mRNA specifically to macrophages, facilitating in situ CAR-M production. In a lung metastasis model, inhaled CARmRNA@aCD206 sEVs effectively inhibit tumor growth and prime long-term memory immunity to prevent tumour recurrence. Collectively, our engineered sEV delivery platform demonstrates capability to selectively deliver CAR mRNA to macrophages in lung tissue, providing a promising immunotherapy strategy to effectively combat lung metastasis and recurrence via generation of CAR-Ms in situ.
Similar content being viewed by others


Introduction
Cancer is one of the leading causes of death worldwide, with nearly one in six deaths in 2020 alone (https://gco.iarc.fr/). Despite therapeutic advances, cancer metastasis and recurrence remain major obstacles to cancer treatment, accounting for over 90% of cancer-related deaths1,2,3,4. In particular, the lung is a frequent site of metastasis for prevalent solid tumors such as breast cancer, melanoma, and liver cancer3,5. Recently, chimeric antigen receptor (CAR)-T cell therapy has achieved great success in treating hematological malignancies, spurring ongoing trials in solid tumors6. However, the application of CAR-T therapy in solid tumors still faces challenges such as high cytotoxicity and insufficient tumor infiltration7. Therefore, novel therapeutic strategies are urgently needed to improve the outcomes of patients with lung metastatic cancer.
In contrast to T cells, macrophages exhibit strong phenotypic plasticity, superior tumor infiltrating abilities and represent the most abundant innate immune population within the tumor microenvironment8,9,10. Recent studies suggest that infusion of CAR macrophages (CAR-M) results in CAR-M-mediated cancer cell phagocytosis, specific tumor antigen presentation, pro-inflammatory cytokine secretion, and subsequent effective tumor growth inhibition11,12,13. This positions CAR-M therapy as the vanguard in solid tumor treatment. However, the CAR-M therapeutic frontier is still nascent and faces challenges. Due to the complex culture procedures and limited proliferative capacity of macrophages, large-scale CAR-M generation remains challenging14. Moreover, intravenously administered CAR-Ms are largely sequestered in the liver, limiting their therapeutic efficacy against extrahepatic tumors15,16. Consequently, utilizing the abundance of macrophages, in vivo reprogramming of macrophages into CAR-Ms to enhance and redirect their antigen-specific phagocytosis to exert their anti-tumor properties appears promising.
In vivo genetic engineering involves vector-mediated delivery of genes to target cells, enabling reprogramming without ex vivo handling17,18,19. Compared to conventional CAR therapies, in vivo genetically engineered CAR cell therapy transforms autologous cell-based drugs into universal, off-the-shelf drugs, simplifying preparation steps, avoiding isolation of cells to transform genes in the absence of primary environmental conditions, and reducing costs20,21,22. Importantly, these drugs for in vivo CAR cell therapy can be easily mass-produced, stored and transported in a stable form, and used directly when needed23,24,25. A notable advance in this field by Purik et al. involved the use of liposomes to introduce mRNA encoding CAR proteins into T cells, generating Fab CAR-T cells capable of specifically targeting fibroblasts26. Inspired by this, we hypothesized that vector delivery of CAR mRNA to macrophages in lung metastatic cancer could generate CAR-Ms in situ, addressing the challenges of laborious ex vivo preparations and hepatotropism of CAR-Ms.
Messenger RNA (mRNA), a critical molecule for the translation of genetic information into proteins, has garnered considerable attention for cancer immunotherapy applications due to its high translational efficiency and no risk of insertional mutagenesis27,28. However, its susceptibility to nuclease-mediated degradation in vivo necessitates efficient delivery systems29. While liposomes have been instrumental in this regard, their non-specific targeting and potential immunogenicity underscore the need for alternatives30. In contrast, biologically derived small extracellular vesicles (sEV) are considered promising drug delivery vehicles due to their favorable biocompatibility and modifiability31,32,33,34,35. It is worth noting that the role of sEVs as innate mRNA carriers was elucidated as early as 200736. To further improve the loading efficiency of macromolecular mRNA in sEVs, Kojima et al. developed a platform called EXOtic37. This system, exploiting the RNA-binding protein L7Ae peptide, facilitates the abundant packaging of C/D box-linked mRNA into budding sEVs, paving the way for mRNA delivery.
To improve the targeting of sEVs to specific cells, researchers typically use genetic engineering or chemical modification methods to modify sEVs38,39,40. Our previous research has revealed that sEVs, when modified to express single chain variable fragments (scFv), exhibit precise targeting to cells expressing congruent antigens41,42,43,44. Considering the mannose receptor CD206 as a marker for immunosuppressive macrophages, it could serve as an ideal cellular target for engineering sEVs to deliver their mRNA cargo specifically to macrophages within the tumor microenvironment45. However, traditional intravenous administration tends to limit the ability of sEVs as they primarily accumulate in the liver rather than in extrahepatic tissues such as the lung46. Interestingly, previous studies indicate that pulmonary administration via inhalation enables sEVs to concentrate in lung tissue and improves sEV bioavailability in the lung47,48,49,50. Taken together, we hypothesized that inhalation of CAR mRNA-loaded sEVs with surface expression of anti-CD206 scFv (CARmRNA@aCD206 sEV) could achieve targeted reprogramming of immunosuppressive macrophages to CAR-Ms in lung metastases.
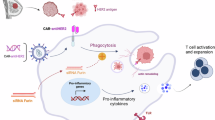
To validate this concept, we construct inhalable engineered sEVs loaded with CAR mRNA (CARmRNA@aCD206 sEVs) to generate mesothelin-specific CAR-Ms in situ, thereby simplifying CAR-M preparation (Fig. 1a). As anticipated, inhaled CARmRNA@aCD206 sEVs rapidly accumulate in the lung and delivered their CAR mRNA cargo specifically into macrophages via CD206 recognition, dramatically improving the efficiency of in situ CAR-M generation (Fig. 1b). The in situ generated CAR-Ms, polarize to an immunostimulatory phenotype with enhanced phagocytosis, increased pro-inflammatory cytokine secretion, and cytolytic capacity against tumors, potently reduce tumor burden, induce long-term immune memory to prevent recurrence, and prolong overall survival in a murine lung metastatic model. In conclusion, this in situ CAR-M generation platform provides a safe and effective immunotherapy strategy for treating lung metastatic cancer and preventing tumor recurrence, which may advance the widespread application of CAR technology for clinical cancer therapy.

a Schematic illustration of the preparation progress of CARmRNA@aCD206 sEVs. b Targeted delivery and anti-tumor mechanisms of CARmRNA@aCD206 sEVs in lung metastatic cancer. Inhaled CARmRNA@aCD206 sEV could accumulate in lung tissue and deliver CAR mRNA to macrophages for in situ CAR-M generation. Subsequently, the CAR-Ms generated by CARmRNA@aCD206 sEVs could trigger potent anti-tumor effects by enhanced antigen-specific tumor phagocytosis, specific tumor presentation and reprogramming of the tumor microenvironment. c Representative TEM image (inset) and size distribution of CARmRNA@aCD206 sEVs (n = 3 independent experiments). Scale bar: 500 nm. Expression of d sEV markers (Calnexin, HSP70, CD63, TSG101 and CD9) and e target proteins (anti-CD206 scFv and L7Ae) in the genetically engineered cells and sEVs. Experiment was repeated twice independently with similar results. f Flow cytometric analysis showing the expression of anti-CD206 scFv on the surface of CARmRNA@aCD206 sEVs (n = 5 independent experiments). g RT-qPCR analysis of CAR mRNA contained in the sEVs (n = 5 independent experiments). Not determined (ND) represents a Ct value greater than 37 or not detected. h RT-qPCR analysis of CAR mRNA contained in sEVs to evaluate the critical role of L7Ae-C/D box interaction in mRNA loading (n = 5 independent experiments). i RT-qPCR analysis of CAR mRNA contained in the sEVs in the RNA degradation assay (n = 5 independent experiments). Data are presented as mean ± SD. Data were analyzed using one-way ANOVA with Tukey’s test in (g). **** p ≤ 0.0001. Source data are provided as a Source Data file.
Results
Preparation and characterization of CARmRNA@aCD206 sEVs
To enable in situ generation of CAR-Ms within lung metastatic cancer, we first developed sEVs capable of delivering CAR mRNA cargo specifically to macrophages. The preparation of CARmRNA@aCD206 sEVs was adapted and refined from the EXOtic system37 (Supplementary Fig. 1). Specifically, to promote the packaging of C/D box-modified CAR mRNA into sEVs, we prepared a CD63-L7Ae fusion construct by genetically fusing the RNA-binding protein L7Ae to the protein CD63. To further improve the efficiency of mRNA release into the cytoplasm of target cells, we linked Cx43 S368A to the CD63-L7Ae fusion construct via a P2A peptide, thereby generating the CD63-L7Ae-P2A-Cx43 S368A construct. In parallel, we incorporated the C/D box sequence into the 3’-UTR of an anti-mesothelin CAR construct (anti-MSLN CAR-C/D box plasmid), permitting its abundant packaging into sEVs through L7Ae-C/D box interaction. To confer macrophage-selective targeting, we genetically fused anti-CD206 scFv to the transmembrane protein LAMP2, generating an anti-CD206-LAMP2 construct. Subsequently, the anti-CD206-LAMP2, CD63-L7Ae-P2A-Cx43 S368A, and anti-MSLN CAR-C/D box mRNA lentiviral plasmids were co-transfected with helper plasmids into HEK 293T cells to produce lentiviruses encoding the respective genes. These lentiviruses were then sequentially transduced into 293T cells and subjected to antibiotic selection to obtain monoclonal cell lines. Importantly, each round of lentiviral transduction and antibiotic selection was completed before proceeding to the next. Similarly, 293T cells producing sEVs without aCD206 expression (CARmRNA@sEV) or targeting HA (CARmRNA@aHA sEV) were prepared using similar methods.
To ensure consistent loading of mRNA into sEVs across different groups, we quantified the integration of the target gene in the established monoclonal cell lines using quantitative polymerase chain reaction (qPCR) and DNA fluorescence in situ hybridization (FISH). Together, the qPCR and DNA FISH results showed that all three of our selected monoclonal 293T cells that produced EVs containing CAR mRNA had a vector copy number (VCN) of four for the CAR mRNA expression cassette (Supplementary Fig. 2a, b). These monoclonal cells were then expanded, and the culture supernatants were harvested to collect CARmRNA@aCD206 sEVs containing anti-MSLN CAR mRNA and anti-CD206 scFv modifications.
We isolated CARmRNA@aCD206 sEVs by differential ultracentrifugation and characterized its morphology by transmission electron microscopy (TEM) after uranyl acetate staining. As shown in Fig. 1c, CARmRNA@aCD206 sEVs displayed oval morphology. The size of CARmRNA@aCD206 sEVs was measured by nanoparticle tracking analysis (NTA), with a main peak size of ~124 nm. To assess the stability of CARmRNA@aCD206 sEVs, they were resuspended in PBS or EVs-depleted serum, and their size was measured with NTA over time (Supplementary Fig. 3a–c). Additionally, we examined the morphological changes of the CARmRNA@aCD206 sEVs in EVs-depleted serum over time using TEM (Supplementary Fig. 3d). These assessments demonstrated that both the particle size and morphology of CARmRNA@aCD206 sEVs remained essentially stable over 5 days, indicating its excellent stability in serum. Furthermore, Western blot analysis was performed to characterize confirmed the expression of sEV markers (HSP70, CD63, TSG101, CD9) in CARmRNA@aCD206 sEVs, without cellular contamination (Calnexin) (Fig. 1d). To validate the successful genetic modification, we examined the expression of the engineered proteins in cell lysates and sEVs by Western blot. As expected, robust expression of both anti-CD206 scFv and L7Ae fusion proteins was detected in CARmRNA@aCD206 sEVs (Fig. 1e). Critically, flow cytometry confirmed the prominent surface display of anti-CD206 scFv on CARmRNA@aCD206 sEVs with proper outward-facing orientation of the antigen-binding domains (Fig. 1f and Supplementary Fig. 4).
Next, we quantified the CARmRNA@aCD206 sEVs to establish the relationship between sEV protein concentration and particle number, which is essential for accurate dosage calculations (Supplementary Fig. 5). Additionally, RT-qPCR was employed to assess the mRNA content within sEVs, normalizing mRNA copy numbers to the total EV particle count and expressing the results as copies per 1 × 106 sEVs. As illustrated in Fig. 1g, each 1 × 106 CARmRNA@aCD206 sEV, CARmRNA@sEV, and CARmRNA@aHA sEV contained ~693, 720, and 686 CAR mRNA copies, respectively. These findings demonstrate that CAR mRNA was successfully loaded into the engineered sEVs. Importantly, the minor variations in CAR mRNA copy numbers between these sEV types ensure consistency and comparability, facilitating subsequent functional validation experiments.
Given that effective delivery of CAR mRNA to target cells requires the co-expression of CAR mRNA and aCD206 within the same EV, it was essential to evaluate the co-localization of the scaffold proteins LAMP2 and CD6351. To enable detection, we engineered 293T cells to secrete LAMP2-mCherry sEVs, CD63-GFP sEVs, and a combination of LAMP2/mCherry-CD63/GFP sEVs. Following the purification of these sEVs by ultrafiltration size exclusion chromatography (UF-SEC), we examined the co-localization of the fluorescent signals using confocal microscopy. As shown in Supplementary Fig. 6, no double-positive vesicles were detected in the UF-SEC-purified mixed vesicles (composed of LAMP2-mCherry sEVs and CD63-GFP sEVs), indicating that the vesicles remained intact and well-dispersed after purification. In contrast, ~72% of sEVs from cells co-expressing LAMP2-mCherry and CD63-GFP displayed dual fluorescence, suggesting a high likelihood of co-localization of LAMP2 and CD63 within the same vesicle. This finding supports the feasibility of simultaneously loading CAR mRNA and CD206-scFv onto single sEVs, thereby enhancing the targeted delivery efficiency of CAR mRNA.
To investigate the mechanism underlying mRNA loading into sEVs, we developed several controls with specific modifications: (1) Free sEV: sEVs secreted by unmodified HEK293T cells; (2) L-sEV: sEVs derived from HEK293T cells expressing only CD63-L7Ae; (3) mRNACAR-sEV: sEVs from HEK293T cells expressing only anti-MSLN CAR; (4) mRNACAR-C/D box-sEV: sEVs from HEK293T cells expressing only anti-MSLN CAR with a C/D box; (5) mRNACAR-L-sEV: sEVs from HEK293T cells co-expressing CD63-L7Ae and anti-MSLN CAR; (6) mRNACAR-C/D box-L-sEV: sEVs from HEK293T cells co-expressing CD63-L7Ae and anti-MSLN CAR with a C/D box. RT-qPCR analysis of CAR mRNA content in sEVs revealed that CARmRNA@aCD206 sEVs contained significantly higher levels of CAR mRNA compared to other groups (Fig. 1h), indicating that the co-expression of L7Ae and C/D box is critical for effective mRNA loading into sEVs.
Previous studies have shown that the sEV membrane can protect internal mRNA from extracellular ribonuclease degradation32. We validated this protective effect by subjecting CARmRNA@aCD206 sEVs to RNase A digestion and analyzing CAR mRNA level. As shown in Fig. 1i, CAR mRNA abundance in CARmRNA@aCD206 sEVs was not affected by RNase A treatment alone. However, membrane disruption with Triton X-100 prior to RNase A incubation resulted in significant CAR mRNA degradation. Taken together, these results verified the successful generation of engineered sEVs surface-modified with anti-CD206 scFv and internally loaded with CAR mRNA.
CARmRNA@aCD206 sEVs enable in vitro CAR-M generation
We next evaluated the capacity of CARmRNA@aCD206 sEVs to induce CAR-M generation in vitro. Macrophages within the tumor microenvironment typically exhibit an immunosuppressive phenotype characterized by high CD206 expression52,53. Given that IL-4-polarized M2 bone marrow-derived macrophages (BMDM) exhibit an immunosuppressive phenotype with CD206 expressing, we utilized M2 BMDMs as target cells to model the targeted delivery of CAR mRNA via CARmRNA@aCD206 sEVs.
To assess the uptake of DiI-labeled sEVs by BMDMs, we co-incubated the sEVs with M2 BMDMs and assessed their internalization using confocal microscopy at 4 h and 24 h. In this experiment, a DiI-PBS control group was included, wherein PBS treated with DiI under the same conditions as the sEVs, to eliminate potential interference from residual DiI uptake by macrophages during the sEV labeling process. Additionally, we replaced the anti-CD206 scFv on CARmRNA@aCD206 sEVs with non-targeting scFv (anti-HA flag scFv) to create CARmRNA@aHA sEVs, to verify the targeting specificity of CARmRNA@aCD206 sEVs. We also tested the targeting ability of CARmRNA@aCD206 sEVs in M2 BMDMs pre-treated with anti-CD206 antibodies (CARmRNA@aCD206 sEV+aCD206 group). Our results demonstrated that the internalization of CARmRNA@aCD206 sEVs by M2 BMDMs was significantly higher than that of non-targeted CARmRNA@sEVs or CARmRNA@aHA sEVs at both 4 h and 24 h (Fig. 2a). Notably, the uptake of sEVs by macrophages increased from 4 h to 24 h, indicating a time-dependent pattern of sEV internalization. Moreover, the presence of anti-CD206 antibodies effectively inhibited the internalization of CARmRNA@aCD206 sEVs in macrophages, thereby confirming the critical role of the CD206 in mediating targeted delivery to M2 macrophages.

a Representative confocal images showing the targeting and internalization of indicated sEVs by M2 bone marrow-derived macrophages (BMDM) after co-incubation for 4 h and 24 h. sEVs are labeled with DiI (red), M2 BMDMs with DiO (green), and nuclei with DAPI (blue). To ensure consistent fluorescence intensity, sEVs staining with 2 μM DiI dye was strictly controlled for 10 min. Scale bar: 20 μm. b Representative flow cytometry images (left) and statistical analysis (right) depicting the internalization of different sEVs by M2 BMDMs at various time points (n = 5 independent experiments). c Representative confocal microscopy images showing the expression of EGFP in M2 BMDMs treated with the indicated sEVs. Scale bar: 50 μm. d, e Representative flow cytometry images and statistical analysis showing the CAR expression in macrophages after co-incubation with the indicated sEVs (n = 5 independent experiments). f The impact of Cx43-S368A and Lamp2-aCD206 modifications on EV-mediated mRNA delivery and subsequent translation was evaluated by assessing the Nanoluc protein signal in treated cells. EVs without Cx43-S368A and Lamp2-aCD206 modifications served as controls (n = 3 independent experiments). g, h Representative flow cytometry image and statistical analysis of CAR protein levels corresponding to different concentrations of CARmRNA@aCD206 sEVs (n = 5 independent experiments). i, j Representative flow cytometry image and statistical analysis showing CAR expression in macrophages over time after co-incubation with the indicated sEVs (n = 5 independent experiments). For (a–f) and (i, j), the dosage of sEVs used was 4 × 106 mRNA copies/mL. UTD: un-treated, Data are presented as mean ± SD. For (a, c), experiment was repeated three times independently with similar results. Data were analyzed using one-way ANOVA with Tukey’s test in (e, f). **** p ≤ 0.0001. Source data are provided as a Source Data file.
To further quantify the kinetics of sEV uptake, we performed flow cytometry analyses at multiple time points, measuring the fluorescence intensity of DiI-labeled sEVs internalized by macrophages54,55. Our findings revealed that the uptake of CARmRNA@aCD206 sEVs increased progressively over time, and the uptake efficiency of sEVs in the CARmRNA@aCD206 Exo group began to decline gradually after 10 h (Fig. 2b). Nevertheless, even at 24 h, the macrophage uptake of sEVs in the CARmRNA@Exo or CARmRNA@aHA sEVs group did not exceed that observed in the CARmRNA@aCD206 Exo group. The enhanced uptake of CARmRNA@aCD206 sEVs can be attributed to their specific binding to CD206 receptors on macrophages, facilitating more efficient internalization. Consequently, these results demonstrated that anti-CD206 scFv engineering successfully conferred macrophage-targeted delivery ability, enabling CAR mRNA to enter the cells for translation.
As a proof of concept, we engineered an EGFP mRNA reporter system (EGFPmRNA@aCD206 sEV) and demonstrated substantial EGFP expression in treated M2 BMDMs (Fig. 2c), confirming successful sEV mRNA delivery and translation. The anti-CD206 scFv engineered sEV containing non-functional CAR mRNA (lacking the anti-MSLN scFv construct) was used for comparison (MockmRNA@aCD206 sEV). In addition, CAR protein expression in CARmRNA@aCD206 sEV-treated BMDMs was assessed by flow cytometry, revealing CAR expression in an average of 73% of macrophages (Fig. 2d, e). Treatment with the translation inhibitor cycloheximide (CHX) resulted in a significant decrease in the proportion of CAR-expressing macrophages, indicating that the CAR protein was primarily translated from the CAR mRNA delivered by CARmRNA@aCD206 sEVs. However, it is noteworthy that BMDMs in the CARmRNA@aCD206 sEV+CHX treatment group still displayed detectable levels of CAR protein. While this residual CAR protein may partially result from incomplete inhibition of CAR mRNA translation, it is also possible that some CAR protein is directly transferred from the sEV membrane to the macrophage membrane.
To assess the contribution of anti-CD206 scFv and Cx43 S368A to the mRNA delivery efficiency, we prepared four types of sEVs loaded with Nanoluc mRNA. After transducing these sEVs into M2 BMDMs, functional Nanoluc protein levels were measured in cell lysates using a luciferase assay11. As shown in Fig. 2f, the fluorescence intensity of Nanoluc protein in M2 BMDMs treated with sEVs expressing Cx43 S368A, anti-CD206 scFv, or both components increased by 5.7, 4.2, and 18.4 times, respectively, compared to untreated sEVs. These results suggest that both Cx43 S368A and anti-CD206 scFv contribute to enhanced mRNA delivery efficiency, although the effect was less pronounced than that achieved with vesicular stomatitis virus glycoprotein (VSV-G), as reported in the literature56,57,58,59. We hypothesize that anti-CD206 scFv may enhance mRNA delivery efficiency by promoting sEV uptake and endosomal escape60. Given that the isoelectric point of most antibodies exceeds 8, anti-CD206 scFv would carry a positive charge in the acidic endosomal environment61,62. This positive charge could enhance the interaction between the sEV and the negatively charged phospholipids of the endosomal membrane, destabilizing the membrane and facilitating mRNA release into the cytoplasm63,64. However, this proposed mechanism requires further experimental validation.
Furthermore, the proportion of CARmRNA@aCD206 sEV-mediated CAR-M generation demonstrated a concentration-dependent increase as the concentration of sEVs increased (Fig. 2g, h). Encouragingly, CARmRNA@aCD206 sEVs instigated transient CAR expression peaking at 48 h and declining over ensuing days, consistent with the nature of the mRNA lacking genomic integration (Fig. 2I, j). Collectively, these results demonstrated that CARmRNA@aCD206 sEVs could efficiently deliver CAR mRNA into M2 BMDMs and induce transient expression of CAR protein.
hCARmRNA@aCD206 sEV-induced CAR-Ms exhibit potent antigen-specific anti-tumor effects in vitro
CAR-Ms have been reported to exert anti-tumor effects through antigen-specific phagocytosis, cytokine production, and immunomodulation15. Hence, we evaluated whether CARmRNA@aCD206 sEV-induced CAR-Ms possess similar functionality. To investigate CAR-mediated antigen-specific tumor phagocytosis, we constructed a B16 cell line stably overexpressing the CAR target mesothelin (B16-MSLN, Supplementary Fig. 7). M2 BMDMs pre-treated with PBS, MockmRNA@aCD206 sEVs, CARmRNA@sEVs, or CARmRNA@aCD206 sEVs were co-cultured with CFSE-labeled parental B16 cells (MSLN‒) or B16-MSLN cells (MSLN+), and phagocytic activity was measured by flow cytometry. CARmRNA@aCD206 sEV-treated M2 BMDMs displayed robust enhancement of antigen-specific phagocytosis against B16-MSLN tumor cells (Fig. 3a). Confocal microscopy demonstrated extensive co-localization of BMDMs with MSLN+ target cells in the CARmRNA@aCD206 sEV group, further validating the enhanced phagocytic uptake (Fig. 3b). Furthermore, we directly visualized the phagocytic process of CARmRNA@aCD206 sEV-treated M2 BMDMs on B16-MSLN cells by live cell imaging (Supplementary Movie 1).

a Phagocytosis of M2 BMDMs pre-treated with the indicated sEVs to CFSE-labeled B16 (MSLN−) cells or B16-MSLN (MSLN+) cells, as determined by flow cytometry (n = 5 independent experiments). b Representative confocal microscopy images showing the phagocytosis of B16-MSLN cells by M2 BMDMs pre-treated with the indicated sEVs. Macrophages were labeled with DiO (green) and B16-MSLN cells were labeled with DiI (red). Experiment was repeated three times independently with similar results. Scale bar: 20 μm. Cytotoxicity of M2 BMDMs pre-treated with the indicated sEVs against c B16 cells and d B16-MSLN cells at effector (pre-treated M2 BMDMs) to target (B16 or B16-MSLN cells) ratios of 10:1, 5:1, 2:1, 1:1 or 1:2 (n = 3 independent experiments). Cytokine concentrations of e IFN-γ, f TNF-α, and g IL-10 released by M2 BMDMs pre-treated with indicated sEVs after co-incubation with B16-MSLN cells (n = 6 independent experiments). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. For (a–g), the dosage of sEVs used was 4 × 106 mRNA copies/mL. Data are presented as mean ± SD. Data were analyzed using one-way ANOVA with Tukey’s test in (a) and (e–g). **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, ns: not significant. Source data are provided as a Source Data file.
Next, we evaluated the antigen-specific cytotoxicity of treated M2 BMDMs against tumor cells by luciferase-based cytotoxicity assay. The result showed that CARmRNA@aCD206 sEV-treated M2 BMDMs selectively enhanced cell death of B16-MSLN cells but not B16 cells in a dose-dependent manner, affirming their acquisition of antigen-specific anti-tumor cytolytic capacity (Fig. 3c, d). Consistent with the protein expression levels, CARmRNA@sEV-treated M2 BMDMs exhibited reduced killing due to lower CAR expression. In addition, we measured the levels of cytokines secreted in the different treatment groups after co-incubation with B16-MSLN or B16 cells. ELISA analyses demonstrated that treatment with CARmRNA@aCD206 sEVs could selectively augment the secretion of pro-inflammatory cytokines interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α), when co-incubated with B16-MSLN melanoma cells (Fig. 3e, f). In contrast, no significant increase in these cytokines was observed upon treatment of parental B16 cells (Supplementary Fig. 8a, b). Concurrently, this treatment effectively suppressed the production of the immunosuppressive cytokine interleukin-10 (IL-10) in the presence of B16-MSLN cells, while no such suppression was noted with parental B16 cells (Fig. 3g and Supplementary Fig. 8c). These results demonstrated that CARmRNA@aCD206 sEV-induced CAR-Ms could exert enhanced antigen-specific anti-tumor effects.
CARmRNA@aCD206 sEVs reprogram macrophages toward an immunostimulatory M1-like phenotype
Given the potent anti-tumor activities demonstrated thus far, we next explored whether CARmRNA@aCD206 sEV-mediated programming might also elicit phenotypic re-polarization of immunosuppressive M2-like macrophages toward immunostimulatory M1-like cells12,14. M2 BMDMs pre-treated with PBS, MockmRNA@aCD206 sEVs, CARmRNA@sEVs, or CARmRNA@aCD206 sEVs were co-cultured with B16-MSLN cells and analyzed for the expression of classic M1 (CD86, MHC II) and M2 (CD206) markers by flow cytometry. Strikingly, CARmRNA@aCD206 sEV treatment upregulated CD86 and MHC II while downregulating CD206, indicating phenotypic shift to an M1-like state (Fig. 4a–c). To clarify the process of BMDM re-polarization, we further measured the expression of key macrophage markers (CD80, MHC II, and CD206) following sEV internalization (Supplementary Fig. 9a–c) and CAR expression (Supplementary Fig. 9d–f) in BMDMs. Flow cytometry results showed that none of the three markers changed significantly in either phase. These findings suggest that the re-polarization of BMDMs to an M1-like phenotype was primarily driven by the interaction between CAR-Ms and tumor cells, rather than by the internalization of engineered sEVs or CAR expression alone. This observation supported the idea that CAR-M engagement with tumor cells was a critical step in guiding macrophages toward an anti-tumor M1-like state, thereby minimizing nonspecific cell killing.

(Representative flow cytometry plots (upper panel) and statistical analysis (lower panel) showing the expression of a CD86, b MHC II, and c CD206 on pre-treated M2 BMDMs after co-incubation with B16-MSLN cells (n = 5 independent experiments). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. d Heatmap depicting mRNA expression levels of cytokines and chemokines associated with macrophage phenotype and activation in the pre-treated M2 BMDMs after co-incubation with B16-MSLN cells (n = 3 independent experiments). Gene expression was normalized to GAPDH. G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. e Western blot showing phosphorylation of AKT, NF-κB P65 and ERK in the pre-treated M2 BMDMs after co-incubation with B16-MSLN cells. G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. Experiment was repeated two times independently with similar results. f T cell proliferation in a Transwell assay after co-incubation of pre-treated M2 BMDMs with B16-MSLN cells (n = 3 independent experiments). g Maturation of DCs (CD11c+CD80+CD86+) in a Transwell assay after co-incubation of pre-treated M2 BMDMs with B16-MSLN cells (n = 6 independent experiments). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. Data are presented as mean ± SD. Data were analyzed using one-way ANOVA with Tukey’s test in (a, b, c, g). *** p ≤ 0.001, **** p ≤ 0.0001. Source data are provided as a Source Data file.
To further define the immune profile of CARmRNA@aCD206 sEV-treated macrophages after co-incubated with B16-MSLN tumor cells, we performed RT-qPCR to assess the expression of cytokines and chemokines associated with macrophage polarized states. CARmRNA@aCD206 sEV treatment enhanced transcripts associated with M1 polarization (Fig. 4d). Moreover, Western blot analysis revealed upregulated phosphorylation of AKT, NF-κB p65, and ERK signaling pathways in CARmRNA@aCD206 sEV-programmed CAR-Ms upon antigen stimulation, indicating activation of pro-inflammatory signaling pathways (Fig. 4e). Finally, we validated the immunostimulatory effects of CARmRNA@aCD206 sEV-programmed CAR-Ms on other immune cell types in co-culture assays. CARmRNA@aCD206 sEV-treated M2 BMDMs promoted T cell proliferation (Fig. 4f) and DC maturation (Fig. 4g) when respectively co-cultured in Transwells, affirming their capacity to remodel the tumor microenvironment towards a pro-inflammatory state conducive to anti-tumor immunity. Collectively, these results indicated the capacity of this sEV-based platform to reprogram immunosuppressive macrophages into immunostimulatory CAR-Ms with the potential to reverse the immunosuppressive tumor microenvironment.
CARmRNA@aCD206 sEV mediated in vivo CAR-M manufacturing
Given the observation that CARmRNA@aCD206 sEVs are effective in CAR-M production in vitro, we further investigated whether CARmRNA@aCD206 sEVs could mediate effective CAR-M production in vivo using a murine lung metastatic tumor model. The lung metastatic tumor model was successfully established by intravenously injecting B16-MSLN cells into mice. Histological examination of the lung tissues confirmed the successful formation of lung metastatic tumors (Supplementary Fig. 10). Additionally, immunofluorescence staining revealed a substantial infiltration of macrophages within the lung tumor tissues, comprising both tumor-associated macrophages (TAM) and alveolar macrophages (AM) (Supplementary Fig. 11). These observations demonstrated that the established lung metastasis model effectively recapitulates the tumor microenvironment, which is characterized by a substantial presence of macrophages. This validation was crucial for evaluating the efficacy of our engineered CARmRNA@aCD206 sEVs in targeting and modulating macrophage populations within pulmonary melanoma metastases.
To improve the bioavailability of CARmRNA@aCD206 sEVs for the treatment of lung metastatic cancer, we compared the biodistribution after intravenous and inhalation administration. Consistent with previous work65, intravenous injection led to sEV accumulation predominantly in the liver and spleen (Fig. 5a and Supplementary Fig. 12). Given the limitations of intravenous administration, we developed an inhalation approach to better target lung tissue. Inhaled CARmRNA@aCD206 sEVs showed sustained lung accumulation over time, with detectable levels even at 48 h after inhalation (Fig. 5b and Supplementary Fig. 13). Furthermore, after DiI-labeled CARmRNA@aCD206 sEV inhalation, we performed lung tissue immunofluorescent staining and fluorescence microscopy to track the distribution of sEVs within the tumor. Melanoma cells were stained with anti-Melan-A to clearly identify the tumor area. The results show that sEVs could successfully localize within the tumor, indicating their ability to penetrate the tumor tissue (Supplementary Fig. 14).

a The biodistribution of CARmRNA@aCD206 sEVs after intravenous or inhalation administration in a lung metastatic tumor model (n = 3 mice). b The biodistribution of CARmRNA@aCD206 sEV over time after inhalation in a lung metastatic tumor model (n = 3 mice). c The expression of Fluc in the major organs 24 h after inhalation of PBS or FlucmRNA@aCD206 sEV (n = 3 mice). Fluc as a surrogate marker for CAR expression. d Representative flow cytometry images (left) and statistical analysis (right) showing CAR protein expression in macrophages 24 h after inhalation of the indicated agents (n = 6 mice). Gated in macrophages. G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aHA sEV, G5: CARmRNA@aCD206 sEV. e Representative flow cytometry images (left) and statistical analysis (right) showing CAR protein expression in macrophages in response to varying concentrations of CARmRNA@aCD206 sEVs (n = 6 mice). f Representative flow cytometry images (left) and statistical analysis (right) showing the percentage of various immune cells (macrophages, dendritic cells, T cells and B cells) among CAR+ immune cells in the tumor tissue (n = 6 mice). Gated in CAR+ immune cells. g Representative flow cytometry plots (left) and statistical analysis (right) showing CAR protein expression in macrophages over time after inhalation of CARmRNA@aCD206 sEV (n = 6 mice). h Representative immunofluorescence images exhibiting CAR (green) macrophages (F4/80, red) in tumor tissues harvested 24 h after CARmRNA@aCD206 sEV inhalation (n = 3 mice). Scale bar: 20 μm. For (a–d) and (f, g), the dosage of sEVs used was 2 × 109 mRNA copies/kg. Data are presented as mean ± SD. Data were analyzed using one-way ANOVA with Tukey’s test in (d). **** p ≤ 0.0001. Source data are provided as a Source Data file.
To ascertain whether CARmRNA@aCD206 sEVs could reprogram macrophages into CAR-Ms in vivo, we first utilized the firefly luciferase (Fluc) reporter as a surrogate to evaluate translational capacity after inhalation. B16-MLSN tumor-bearing mice were administered FlucmRNA@aCD206 sEVs by inhalation. At 24 h post-inhalation, bioluminescence was readily detectable in the lung (Fig. 5c and Supplementary Fig. 15), confirming successful lung delivery and macrophage-mediated protein production.
Strikingly, ~28% of macrophages, including both TAMs and AMs, expressed the CAR protein (Fig. 5d), and CARmRNA@aCD206 sEV-mediated CAR-M generation was concentration-dependent (Fig. 5e). Among these, ~30% of TAMs and 21% of AMs exhibited CAR protein expression. This differential distribution is likely influenced by the intrinsic CD206 expression levels of the respective macrophage subsets and their abundance within the tumor microenvironment (Supplementary Fig. 16). Furthermore, flow cytometric analysis of lung tissues revealed that within tumor tissues, CAR-M cells are predominantly CAR⁺ TAMs, whereas in normal lung tissues, CAR⁺ AMs are more prevalent (Supplementary Fig. 17).
Given the strong correlation among monocytes, AMs, and TAMs, we further investigated the expression levels of CAR protein on monocytes. Flow cytometry analysis revealed only minimal expression of CAR protein on infiltrating monocytes within the tumor tissue, with virtually no detectable CAR expression on peripheral blood monocytes (Supplementary Fig. 18). These findings suggest that CARmRNA@aCD206 sEVs lack targeting efficacy towards monocytes. In contrast, these results highlight the high specificity of CARmRNA@aCD206 sEVs in selectively targeting the macrophage population within the lung tumor microenvironment.
To investigate the protein translation of CAR mRNA mediated by CARmRNA@aCD206 sEVs within lung immune cells, we analyzed the composition of CAR protein-expressing immune cells following inhalation of CARmRNA@aCD206 sEVs. The results revealed that macrophages were the predominant immune cells expressing the CAR protein sEV (Fig. 5f and Supplementary Fig. 19). The predominance of CAR expression in macrophages compared to dendritic cells (DC) may be due to the abundance of macrophages in the tumor microenvironment. Notably, CARmRNA@aCD206 sEVs also induced the generation of a small population of CAR-T cells in vivo, with proportions comparable to those in the CARmRNA@sEV-treated group due to a lack of specific T cell targeting. (Supplementary Fig. 20). To assess their immunological activity, we evaluated the expression of CD69, an activation marker, on these CAR-T cells using flow cytometry. CAR-T cells from the CARmRNA@aCD206 sEV-treated group showed significantly higher CD69 expression compared to T cells from the PBS-treated group, indicating enhanced activation (Supplementary Fig. 21a, b). Additionally, the CAR-T cells isolated from CARmRNA@aCD206 sEV-treated group exhibited higher levels of anti-tumor cytokine secretion (Supplementary Fig. 21c) and improved tumor killing efficiency in a dose-dependent manner (Supplementary Fig. 21d). These results suggest that CAR-T cells generated in vivo may also play a role in tumor killing. However, despite the similar proportions of CAR-T cells between the groups, the T-cell response in the CARmRNA@aCD206 sEV-treated group was significantly stronger than that in the control group. This suggests that the enhanced T-cell response observed may be primarily attributable to CAR-M interactions, rather than to the CAR-T cells themselves.
Furthermore, we observed that CAR expression in programmed macrophages declined over the following days (Fig. 5g), consistent with the transient nature of mRNA-mediated protein production. Additionally, immunofluorescence microscopy confirmed the co-localization of CAR and the macrophage marker F4/80, providing further validation of successful macrophage programming (Fig. 5h). In summary, inhaled CARmRNA@aCD206 sEVs successfully penetrated tumors in our murine lung metastatic tumor model, and efficiently programmed macrophages into transient CAR-expressing effectors.
Therapeutic efficacy and biocompatibility of CARmRNA@aCD206 sEVs against lung metastatic cancer in vivo
Having confirmed the feasibility of programming CAR expression in macrophages, we next evaluated whether this strategy could mediate meaningful anti-tumor effects in vivo. Lung metastatic cancer was established in C57BL/6 mice by intravenous injection of B16-F10 cells expressing luciferase (B16-MSLN-FLuc, Supplementary Fig. 22) to allow bioluminescent monitoring of tumor progression. Seven days after inoculation (Fig. 6a), bioluminescence imaging confirmed the successful establishment of the mouse model, and mice were randomized into four treatment groups: PBS, MockmRNA@aCD206 sEV, CARmRNA@sEV, and CARmRNA@aCD206 sEV. The former two groups served as negative controls to assess non-specific effects of the delivery vehicle, while CARmRNA@sEVs lacking macrophage targeting assessed the importance of cell-specificity for maximal therapeutic efficacy. Serial bioluminescence imaging revealed markedly delayed tumor progression in the CARmRNA@aCD206 sEV group compared to all other groups, indicating potent therapeutic efficacy (Fig. 6b, c). The lack of efficacy after administration of untargeted CARmRNA@sEVs confirms the critical role of the CD206-targeting strategy for efficient in vivo CAR mRNA delivery to macrophages. Assessment of tumor burden revealed a striking reduction in both the number and size of pulmonary tumor nodules, achieved by CARmRNA@aCD206 sEV treatment (Fig. 6d). Lung histopathology confirmed near-normal lung architecture following CARmRNA@aCD206 sEV therapy compared to effacement by invasive tumors in control cohorts. Importantly, PBS or MockmRNA@aCD206 sEV-treated mice succumbed to metastatic disease within 30 days, whereas 100% of CARmRNA@aCD206 sEV-treated mice survived beyond 60 days post-tumor inoculation (Fig. 6e). CARmRNA@sEVs lacking targeted delivery also showed inferior survival benefits, highlighting the improved therapeutic efficacy enabled by selective modification of immunosuppressive macrophages. Additionally, unlike PBS-treated animals, which showed a steady weight loss reflecting disease burden, body weights remained stable after CARmRNA@aCD206 sEV therapy, suggesting maintenance of overall health (Fig. 6f). Collectively, inhaled CARmRNA@aCD206 sEVs successfully programmed native lung macrophages to orchestrate robust anti-tumor responses, and dramatically improved survival in lung metastatic tumor model.

a Schematic illustration of treatment and immune analysis schedules in a murine lung metastatic tumor model. b Bioluminescence images of tumor-bearing mice treated with PBS, MockmRNA@aCD206 sEVs, CARmRNA@sEVs, or CARmRNA@aCD206 sEVs on days 0, 7, 14, and 21 (n = 3 mice). c Bioluminescence intensity analysis reflecting the tumor burden of mice in each group (n = 3 mice). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. d Representative lung images (upper panel) and H&E-stained sections (lower panel) from mice in each group after different sEV treatments for 14 days (n = 3 mice). Scale bar: 100 μm. e Survival analysis of B16-MSLN tumor-bearing mice in different groups (n = 10 mice). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. f Body weight curve of B16-MSLN tumor-bearing mice after different treatments (n = 3 mice). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. g Serum biochemical indices of healthy mice treated with PBS or CARmRNA@aCD206 sEV (n = 3 mice). h H&E staining of the major organs (heart, liver, spleen, lung, kidney) in the mice after PBS or CARmRNA@aCD206 sEV inhalation (n = 3 mice). Scale bar: 50 μm. i Body weight curve of mice after inhalation of PBS or CARmRNA@aCD206 sEVs (n = 3 mice). For (b–i), the dosage of sEVs used was 2 × 109 mRNA copies/kg. Data are presented as mean ± SD. Source data are provided as a Source Data file.
Next, we evaluated the biocompatibility of CARmRNA@aCD206 sEVs in C57BL/6 mice. Healthy C57BL/6 mice were randomly divided into two groups for inhalation of PBS or CARmRNA@aCD206 sEVs every 3 days. Blood samples were then collected for biochemical analysis. The liver function indicators alanine aminotransferase (ALT), aspartate aminotransferase (AST), alanine aminotransferase (ALP), and the kidney function indicators blood urea nitrogen (UREA) and creatinine (CR) were evaluated. Biochemical analysis showed that the levels of these indicators were within normal limits, and the mice treated with CARmRNA@aCD206 sEVs showed no significant difference compared to the mice in the PBS-treated group (Fig. 6g). The results demonstrated that CARmRNA@aCD206 sEVs exhibited negligible toxicity to the liver or kidney in mice. In addition, we further evaluated the safety of CARmRNA@aCD206 sEVs on major organs (heart, liver, spleen, lung, and kidney) by histopathological analysis. Hematoxylin and eosin (H&E) staining was used for pathological morphological analysis, and no obvious pathological morphological abnormalities were found in any group (Fig. 6h). Body weight was also monitored for 30 days and showed no significant difference between the two groups (Fig. 6i). These results indicated that CARmRNA@aCD206 sEVs had good biocompatibility and hold great potential for in vivo applications.
To assess the potential of CARmRNA@aCD206 sEV as an allogeneic off-the-shelf therapy, we prepared sEVs modified with anti-CD206 scFv (aCD206 sEV) without loading mRNA. Healthy C57BL/6 mice were administered aCD206 sEVs via aerosol inhalation, after which we measured the expression levels of the activation marker CD69 on T cells and the concentrations of key cytokines in the serum. Our results indicated that no significant immune activation was observed in the aCD206 sEV-treated group (Supplementary Fig. 23), supporting the suitability of CARmRNA@aCD206 sEV as a safe and effective allogeneic off-the-shelf therapeutic option.
Reprogramming of the tumor microenvironment by CARmRNA@aCD206 sEVs in vivo
To elucidate the immune mechanisms underlying the robust anti-tumor effects of CARmRNA@aCD206 sEVs, we performed further flow cytometric analysis of immune cells within the tumor microenvironment. Strikingly, we observed a profound increase in the proportion of M1-like macrophages along with a concomitant reduction in M2-like macrophages in CARmRNA@aCD206 sEV-treated mice (Fig. 7a, b). The increased abundance of M1 macrophages may be attributed to the phenotypic shift induced by CARmRNA@aCD206-mediated programming of macrophages. Given the critical role of M1 macrophages as professional phagocytes and antigen-presenting cells, we postulated that CARmRNA@aCD206 sEV-programmed macrophages could orchestrate downstream anti-tumor immune responses. Indeed, CARmRNA@aCD206 sEV treatment elicited a marked infiltration of cytotoxic CD8+ T cells in the tumor compared to controls, concomitant with heightened infiltration of granzyme B-expressing CD8+ T cells (Fig. 7c, d). Furthermore, CARmRNA@aCD206 sEVs significantly reduced immunosuppressive regulatory T cells (Treg) and myeloid-derived suppressor cells (MDSC) within the tumor (Fig. 7e, f), indicating that tumor-mediated immunosuppression was alleviated.

a, b Representative flow cytometry plots and statistical analysis of M1 macrophages (CD45+CD11b+F4/80+CD80+) and M2 macrophages (CD45+CD11b+F4/80+CD206+) in the tumor microenvironment (n = 6 mice). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. c, d Representative flow cytometry plots and statistical analysis of tumor-infiltrating CD8+ T cells (CD45+CD3+CD8+) and activated CD8+ T cells (CD45+CD3+CD8+GzmB+) in the tumor microenvironment (n = 6 mice). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. e, f Representative flow cytometry plots and statistical analysis of Treg cells (CD45+CD3+CD4+CD25+FOXP3+) and MDSCs (CD45+CD11b+Gr1+) infiltrating in the tumor microenvironment (n = 6 mice). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. Cytokine concentrations of g IFN-γ, h TNF-α, i IL-10 and j TGF-β in tumor tissues of each group, as quantified by ELISA kits (n = 6 independent experiments). G1: PBS, G2: MockmRNA@aCD206 sEV, G3: CARmRNA@sEV, G4: CARmRNA@aCD206 sEV. For (a–j), the dosage of sEVs used was 2 × 109 mRNA copies/kg. GzmB: Granzyme B. Data are presented as mean ± SD. Data were analyzed using one-way ANOVA with Tukey’s test in (b, d, f) and (g–j). **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. Source data are provided as a Source Data file.
To further investigate the immune-activating effects of CARmRNA@aCD206 sEVs, we analyzed the expression of cytokines in the tumor microenvironment. The pro-inflammatory cytokines interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α), crucial mediators of anti-tumor immunity, were markedly upregulated in CARmRNA@aCD206 sEV-treated mice. In contrast, the immunosuppressive cytokines interleukin 10 (IL-10) and transforming growth factor beta (TGF-β) were substantially decreased (Fig. 7g–j). Collectively, these results demonstrated that CARmRNA@aCD206 sEV-programmed macrophages profoundly reprogrammed the tumor microenvironment to a pro-inflammatory, immune-activated state conducive to tumor eradication.
Induction of durable anti-tumor immune memory by CARmRNA@aCD206 sEVs impede tumor recurrence
Tumor recurrence after initial clinical remission remains one of the most pressing challenges in cancer treatment66. Given the critical role of immune memory in providing long-term protection against recurrence, we investigated whether CARmRNA@aCD206 sEVs could establish durable immunological memory. Central memory T cells (TCM) and effector memory T cells (TEM) are key mediators of durable anti-tumor immunity67. TCM home to lymph nodes, proliferate rapidly upon re-exposure to cognate antigens, and secrete IL-2 to propagate the memory response. In contrast, TEM circulate systemically and rapidly produce inflammatory cytokines such as IFN-γ to mediate immediate cytotoxicity against target cells upon antigen re-encounter. By analyzing the immune memory cells in lymph nodes prior to tumor rechallenge, we found that both TCM (CD3+CD8+CD44+CD62L+) and TEM (CD3+CD8+CD44+CD62L‒) frequencies were profoundly increased in CARmRNA@aCD206 sEV-treated mice compared to controls (Fig. 8a, b).

a Representative flow cytometry plots and b statistical analysis of naïve T cells (CD45+CD3+CD8+CD44‒CD62L+), TCM cells (CD45+CD3+CD8+CD44+CD62L+), and TEM cells (CD45+CD3+CD8+CD44+CD62L‒) in the lymph nodes of B16-MSLN-bearing mice treated with PBS (G1), MockmRNA@aCD206 sEV (G2), CARmRNA@sEV (G3), or CARmRNA@aCD206 sEV (G4) (n = 6 mice). c Schematic illustration of the experiment to assess CARmRNA@aCD206 sEV-induced long-term immune memory to inhibit tumor recurrence. d Bioluminescence images showing the tumor burden of naïve or CARmRNA@aCD206 sEV-cured mice rechallenged with B16 or B16-MSLN cells (n = 3 mice). e Survival analysis of naïve or CARmRNA@aCD206 sEV-cured mice rechallenged with B16 or B16-MSLN cells (n = 10 mice). For (a, b) and (d, e), the dosage of sEVs used was 2 × 109 mRNA copies/kg. Data are presented as mean ± SD. Data were analyzed using one-way ANOVA with Tukey’s test in (b). **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. Source data are provided as a Source Data file.
To identify the specific macrophage population mediating the immune memory. We isolated the CAR-AM and CAR-TAM from the CARmRNA@aCD206 sEV-treated mice, and the AM and TAM from the PBS treated mice. T-cell immunophenotypic changes were then determined in vitro after co-incubation with spleen-isolated T cells. The results showed that both CAR+ AMs and CAR+ TAMs could promote the differentiation of naïve T cells into central memory T cells (TCM) and effector memory T cells (TEM) (Supplementary Fig. 24), indicating that CAR+ AMs and CAR+ TAMs induced by CARmRNA@aCD206 sEVs were all involved in the development of long-term immune memory against lung metastases.
Based on the observed memory T cell responses, we next explored whether CARmRNA@aCD206 sEVs could prevent tumor recurrence in a rechallenge model. As depicted in Fig. 8c, after eradication of pulmonary B16-MSLN tumors with CARmRNA@aCD206 sEVs, cured mice were rechallenged with B16-MSLN cells to model recurrence. As controls, tumor naïve mice were likewise challenged. Strikingly, CARmRNA@aCD206 sEV-cured mice exhibited profound resistance to B16-MSLN rechallenge, with no detectable tumor formation even by day 21 (Fig. 8d). Notably, CARmRNA@aCD206 sEV-cured mice were able to resist rechallenge with B16 tumor cells to some extent, despite the absence of the CAR-specific target antigen MSLN in B16 tumor cells. The results suggested that CARmRNA@aCD206 sEV-induced CAR-Ms might lead to epitope spreading by cross-presentation of intracellular tumor antigens ingested during phagocytosis of whole tumor cells. Moreover, CARmRNA@aCD206 sEV-cured mice demonstrated dramatically prolonged survival compared to naïve mice after B16-MSLN or B16 rechallenge (Fig. 8e). Collectively, these findings demonstrated the potent capacity of CARmRNA@aCD206 sEVs to establish durable anti-tumor immune memory that prevents recurrence after treatment cessation.
Discussion
Capitalizing on the innate tumor-infiltrating capacity and abundance of macrophages within the tumor microenvironment, CAR-Ms have emerged as one of the most promising immunotherapies against solid tumors14. To date, several clinical trials are ongoing, including CAR-Ms for the treatment of MSLN-expressing solid tumors6. However, realizing the full therapeutic potential of CAR-Ms has been hampered by complex ex vivo manufacturing protocols and suboptimal biodistribution upon intravenous administration, posing major challenges for cancers such as lung metastatic cancer. To address these challenges, we propose a strategy for in situ generation of CAR-Ms within the tumor microenvironment by inhalation of CARmRNA@aCD206 sEVs. Our results demonstrated that CARmRNA@aCD206 sEVs could target and deliver MSLN-specific CAR mRNA to macrophages to generate CAR-Ms in vivo, thereby achieving enhanced phagocytosis, immune activation, and tumor microenvironment reprogramming. In preclinical models of lung metastatic and recurrent cancer, inhaled CARmRNA@aCD206 sEVs induced profound tumor suppression. Therefore, this strategy bypasses the need for complex ex vivo cell manufacturing, dramatically reducing cost and lead time to therapy. Moreover, in situ generated CAR-Ms allow for optimal bioavailability while avoiding the potential side effects of systemic cell infusion therapies.
Although still in its infancy, the in vivo CAR immune cell generation strategy shows great promise compared to traditional ex vivo methods20. First, the in vivo CAR immune cell preparation strategy substantially streamlines the intricate and time-intensive process of cell preparation. Currently, typical ex vivo CAR T-cell generation requires a lengthy 2–4 weeks engineering process that includes peripheral blood isolation, T-cell purification, gene transduction and cell expansion68. In contrast, CAR-M production requires additional steps such as bone marrow mobilization and monocyte induction11. To shorten the production cycle of CAR-Ms, this study attempts to promote CAR-M generation by inhaling CARmRNA@aCD206 sEVs and detects tumor-infiltrating CAR-Ms in the lung within 24 h. This acceleration of therapy could prove critical for patients with aggressive cancers. Equally important, by bypassing extensive ex vivo processing, in vivo CAR generation promises to reduce manufacturing costs and facilitate broad availability. Second, the in vivo approach paves the way for scale-up production of CAR immune cell-related biopharmaceuticals. Conventional manufacturing requires individual ex vivo modification of each patient’s immune cells to avoid immunogenicity-mediated cell killing, which severely limits the production capacity of CAR immune cells20. In contrast, the in vivo strategy allows the use of a single biopharmaceutical to generate CAR-Ms in different patients21. In this study, the engineered sEVs used for CAR-M preparation have the advantages of easy preparation, long-term preservation, and easy transport, which are conducive to achieving large-scale preparation of CAR immune cell-related biopharmaceuticals and reducing preparation costs. Finally, direct in situ generation of CAR cells may augment their anti-tumor potency. It is well known that the insufficient ability of CAR immune cells to migrate and infiltrate solid tumors is one of the main reasons for their poor anti-tumor efficacy, and the strategy of generating CAR immune cells in situ is expected to overcome this challenge15. Owing to the superior tumor tissue penetration efficiency of sEVs, they serve as effective vectors for intratumoral drug delivery. As anticipated, our study demonstrated that the inhalation of CARmRNA@aCD206 sEVs proficiently transported CAR mRNA into the lung tumor tissue, facilitating in situ generation of CAR-Ms in substantial numbers, thereby enhancing their tumor penetration and anti-tumor capabilities. Hence, our strategy of generating CAR-Ms directly within tumor tissue via engineered sEVs holds great promise for solid tumor therapies.
For the in vivo generation of CAR-Ms, the choice of gene vector, delivery vehicle and route of administration are all critical for the ultimate anti-tumor efficacy67,69,70. In this study, mRNAs without the risk of insertional mutagenesis were selected as gene vectors for CAR expression. Given the transient expression profile of the mRNA, it holds promise for reducing the risk of a cytokine storm, a common complication associated with CAR immune cell therapies. Our study demonstrated that CAR expression on macrophages gradually declined within 1 week, suggesting that CAR mRNA has a favorable safety profile. To avoid enzymatic degradation of naked CAR mRNA, engineered sEVs were used to deliver CAR mRNA in this study. sEVs, as natural mRNA carriers, have distinct advantages, including low immunogenicity, modifiability, and better biological barrier crossing, making them a superior system for mRNA delivery. However, the incorporation of foreign mRNA into sEVs has been technically challenging71. Traditional methods such as co-incubation and electroporation have fallen short in encapsulating macromolecular mRNA. Drawing inspiration from the EXOtic device of Kojima et al.37, which facilitates the sorting of specific mRNA into sEVs, our study utilized a modified version of this device to encapsulate MSLN-specific CAR mRNA. To enhance the efficiency of mRNA delivery, we engineered sEVs to display anti-CD206 scFv on their surface. This modification resulted in improved targeting of macrophages expressing CD206 both in vitro and in vivo, and it significantly enhanced the internalization of CAR mRNA by macrophages, thereby reducing the adverse events associated with non-specific reprogramming. In addition to the gene vector and delivery vehicle, the route of administration is also critical for in vivo CAR-M generation. Typically, intravenously administered sEVs tend to accumulate predominantly in the liver and spleen, which is suboptimal for the treatment of extrahepatic tumors47,72. In contrast, our study demonstrated a preferential accumulation of sEVs in lung tissue after inhaled administration, which significantly enhanced targeted delivery to macrophages within the tumor microenvironment. This strategy is expected to increase the pulmonary bioavailability of the delivered drug and further improve biosafety.
A major limitation of conventional immunocytotherapy is tumor recurrence due to antigen loss mutations66,73. Excitingly, our approach to generate CAR-Ms in situ may provide durable protection against heterogeneous tumors. In a model of tumor recurrence, CAR-Ms generated via inhaled engineered sEVs potently resisted rechallenge, even when tumor cells no longer expressed the original CAR target antigen. This resistance likely stems from epitope spreading, whereby programmed CAR-Ms phagocytose cancer cells and present their full repertoire of neoantigens to prime polyclonal T cell responses. Consequently, tumor-specific T cells arise that extend beyond the specificity of the original CAR construct. These neoantigen-responsive T cells persist and continue to exert anti-tumor immunity against antigen-loss variants.
However, the CARmRNA@aCD206 sEV-mediated in situ CAR-M generation strategy has some limitations that need to be overcome to advance its clinical translation. First, although macrophages are the primary targets, our approach simultaneously induces CAR expression in a small subset of dendritic cells. Given the focus of this study on CAR-Ms, the potential modulatory effects of CAR-DCs on the tumor microenvironment have not been fully explored. Second, although we have investigated the biodistribution of these sEVs following pulmonary administration in mouse models, the extrapolation of these findings to larger mammals will require a comprehensive pharmacokinetic analysis, considering the interspecies variability of the respiratory system. Finally, while the initial assessments of the CARmRNA@aCD206 sEV-mediated CAR-M generation technique suggest a promising safety and efficacy profile, the long-term effects on host physiology require more extensive observation.
In conclusion, this study established a strategy for in situ generation of CAR-Ms in the lung tumor microenvironment by inhaled engineered sEVs. This strategy achieved localized reprogramming of macrophages into functional CAR-Ms, eliciting profound anti-tumor efficacy against lung metastasis and recurrence. Notably, this strategy bypasses the complex procedures associated with the ex vivo culture of CAR-Ms and significantly enhances CAR-M infiltration in tumor tissue, thereby mitigating the side effects associated with systemic therapeutic delivery. Thus, this work lays the foundation for unlocking the full potential of CAR-Ms and in situ cell reprogramming against lung metastasis and beyond.
Methods
Cell lines and plasmid construction
Human embryonic kidney 293T cells (HEK 293T, ATCC CRL-3216) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher) supplemented with 10% fetal bovine serum (FBS) under standard conditions (37 °C, 5% CO2). The B16-F10 murine melanoma cell line (B16, ATCC CRL-6475) was maintained in RPMI-1640 (Gibco) with 10% FBS. Primary T cells and DCs were cultured in serum-free cell culture medium (Zhuhai Baso Cell Science&Technology Co., Ltd) for expansion.
For macrophage-targeted sEV, an anti-CD206 single-chain variable fragment (scFv) was fused to the N-terminus of Lamp2, cloned and inserted into the lentiviral vector pCDH-CMV-mCherry-EF1α-Neo to yield the anti-CD206-Lamp2 plasmid. The construction of anti-HA flag-Lamp2 plasmid were similar to that of anti-CD206-Lamp2 plasmid, by replacing the anti-HA flag single chain variable fragment with anti-CD206 single chain variable fragment. For CAR expression, a second-generation anti-MSLN CAR cassette was synthesized with C/D box motifs inserted in the 3’ untranslated region (UTR), then subcloned and inserted into the pCDH lentiviral backbone, generating the anti-MSLN CAR-C/D box plasmid. In addition, L7Ae (an RNA binding protein) was fused to CD63 and inserted into pCDH-GFP-EF1α-Puro, generating the CD63-L7Ae plasmid. To further improve the efficiency of mRNA release into the cytoplasm of target cells, Cx43 S368A was linked to the CD63-L7Ae fusion construct via a P2A peptide, resulting in the generation of the CD63-L7Ae-P2A-Cx43 S368A construct. By subcloning the MSLN sequence into pCDH-CMV-P2A-FLuc-EF1α-Puro, pCDH-MSLN-FLuc-Puro was produced. Lentiviral packaging plasmids psPAX2 and pMD2.G were purchased from Addgene (Cambridge, USA). All nucleotide sequences were synthesized by BGI (Beijing, China) prior to cloning.
Animals and ethics approval
Female C57BL/6 mice (Stock No. GDMLAC-07) aged 5–8 weeks were obtained from Guangdong Medical Laboratory Animal Center and housed under specific pathogen-free (SPF) conditions in a barrier facility at the Fifth Affiliated Hospital of Sun Yat-sen University. The animals were maintained in a controlled environment with a temperature of 22 ± 2 °C, relative humidity of 50–60%, and a 12-h light/dark cycle. Experimental and control groups were co-housed in ventilated cages with sterilized food and water provided.
All animal procedures were performed in strict accordance with the guidelines of the Animal Care and Use Committee of Sun Yat-sen University and the NIH Guide for the Care and Use of Laboratory Animals. The study protocol was approved by the Animal Ethics Committee of the Fifth Affiliated Hospital of Sun Yat-sen University (Approval No. 00410). Sex was not included as a variable in the experimental design because the lung metastasis model used does not show known sex-specific differences in tumor progression or treatment response. Therefore, only female mice were used to reduce biological variability and the number of animals required.
The experimental endpoint was defined by either a predetermined time point based on study objectives or reaching humane endpoints, whichever occurred first. Humane endpoints included significant weight loss, severe distress, impaired mobility, or other signs of suffering, ensuring animal welfare was prioritized. Animals were euthanized by isoflurane inhalation until loss of reflexes was confirmed, followed by cervical dislocation to ensure death.
sEVs preparation for inhalation
To prepare inhalable sEVs, a monoclonal cell line capable of stably producing engineered sEVs was first generated. In brief, HEK 293T cells were sequentially transduced with lentiviral vectors encoding anti-CD206-Lamp2-Neo, CD63-L7Ae-P2A-Cx43 S368A-Hygro, and anti-MSLN CAR-C/D box-Puro, followed by selection with neomycin (Solarbio), hygromycin B (Solarbio), and puromycin (Solarbio), respectively. Each round of lentiviral transduction and selection was completed before proceeding to the next round. The process was as follows: (1) HEK 293T cells were transduced with lentivirus encoding anti-CD206-Lamp2-Neo. After 24 h, the transduced cells were seeded in 12-well plates and selected with 500 µg/mL G418. After 4 days of selection, single cells were sorted into individual wells of 96-well plates through limiting dilution and expanded under 500 µg/mL G418 for ~12 days. Using flow cytometry and western blot analysis, we confirmed that anti-CD206 scFv was successfully expressed on the secreted sEVs. A single-cell clone with good growth characteristics and high protein expression levels (designated as aCD206 293T) was chosen for the next round of transduction. (2) The aCD206 293T cells were transduced with lentivirus encoding CD63-L7Ae-P2A-Cx43 S368A-Hygro and selected with 300 µg/mL hygromycin, using a similar procedure. This resulted in a monoclonal cell line expressing both anti-CD206 scFv and L7Ae (designated as aCD206/L7Ae 293T). (3) Finally, the aCD206/L7Ae 293T cells were transduced with lentivirus encoding anti-MSLN CAR-C/D box-Puro and selected with puromycin to obtain a monoclonal cell line capable of producing anti-MSLN-CAR mRNA-containing sEVs. Control cell lines for producing corresponding engineered sEVs were established using the same process.
The obtained monoclonal cell lines were expanded in DMEM medium containing exosome-depleted FBS for 2 days. After this period, the cell culture supernatant was collected, and sEVs were isolated from the conditioned medium using differential ultracentrifugation. In brief, cellular debris was removed by centrifugation at 300 × g, 2000 × g, and 10,000 × g. Subsequently, the sEVs were precipitated through ultracentrifugation at 100,000 × g and resuspended in phosphate-buffered saline (PBS). The protein concentration of sEVs was quantified using the BCA assay (Thermo Fisher Scientific), while nanoparticle tracking analysis (NTA) was employed to determine the particle count per milligram of sEVs. Additionally, the mRNA content within the sEVs was quantified using RT-qPCR, targeting specific mRNA molecules per 1 × 106 sEVs as detailed in the subsequent sections. Based on the required mRNA copy number for the respective experiment, the sEVs containing the target mRNA were resuspended in PBS for in vitro or in vivo experiments.
Detection of mRNA-expression cassette integration by qPCR
After lentiviral transduction and clone selection, vector copy number (VCN) was assessed by real-time quantitative PCR (qPCR) to determine the number of integrated mRNA-expression cassettes per genome in individual monoclonal 293T cell lines. The detection method was adapted from previous reports74,75. Genomic DNA was extracted from CAR-expressing monoclonal 293T cell lines using the QIAamp DNA Mini Kit (QIAGEN), following the manufacturer’s instructions. DNA purity and concentration were measured with a NanoDrop UV spectrophotometer (Thermo Fisher Scientific), and samples were diluted to 10 ng/µL in nuclease-free water prior to qPCR.
CAR-specific primers and probe were custom synthesized by GenScript. The CAR-specific primers were: Forward, 5′-GCTGGACTTCGCCTGTGATA-3′; Reverse, 5′-GAGCCTGCTCCTCTTACTCC-3′; and Probe, 5′-FAM-CAAGTCCCGGCCAAGGGCGC-TAMRA-3′. The human single-copy gene RNase P (Applied Biosystems) served as the reference gene for normalization. Standard curves for both CAR and RNase P were generated using plasmids containing the respective sequences, with 10-fold serial dilutions ranging from 107 to 101 copies.
qPCR was performed on a CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories). Each 20 µL reaction contained 10 µL TaqMan Universal PCR Master Mix (Thermo Fisher Scientific), 0.5 µL each of forward and reverse primers, 0.5 µL TaqMan probe, 2 µL genomic DNA, and nuclease-free water. Thermal cycling comprised 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 60 s. No-template controls were included on each plate to monitor potential contamination. VCN was calculated using the formula: VCN = 2−ΔCt × 2, where ΔCt = Ct (CAR) – Ct (reference gene), and the factor of two accounts for the two alleles of the RNase P reference locus in the human diploid genome.
Detection of target gene VCN in cell lines by DNA FISH
DNA fluorescence in situ hybridization (FISH) was conducted to assess vector copy number (VCN) in stably transfected 293T cell clones, following established protocols76,77,78. Monoclonal 293T-derived stable clones were seeded onto coverslips in six-well plates, fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature, and washed three times with phosphate-buffered saline (PBS). Permeabilization was performed with 0.5% Triton X-100 for 5 min, followed by three PBS washes. Subsequently, the cells were dehydrated through a graded ethanol series in preparation for hybridization. FAM-labeled DNA FISH probes specific to the target gene were custom-designed and synthesized by BersinBio (Guangzhou, China). For denaturation, 30 µL of denaturation solution was applied to each slide, incubated at 75 °C for 5 min, immediately cooled on ice, and then dehydrated. Afterwards, the pretreated hybridization solution containing the probe was applied onto the slides, which were incubated overnight at 37 °C in a humidified chamber to allow hybridization. Following hybridization, coverslips were washed with saline-sodium citrate (SSC) buffers of increasing stringency to remove loosely bound probes. Nuclei were counterstained with DAPI. Slides were mounted using an anti-fade reagent and sealed. Fluorescence signals were visualized under a confocal laser scanning microscope, and the VCN was determined by counting the number of discrete fluorescent spots per nucleus.
Validation of integrated cassette and sEV component expression
To confirm the successful encapsulation of the target mRNA within the sEVs, we performed absolute quantification of target mRNA molecules in sEVs using a standard curve-based RT-PCR method. We generated a reference standard curve by: (1) PCR-amplifying the target gene from its plasmid template; (2) The PCR products were separated on a 1% agarose gel and purified using the QIAquick PCR Purification Kit (Qiagen) according to the manufacturer’s instructions; (3) Measuring the purified PCR product concentration by NanoDrop; (4) Serially diluting the product from 10−8 ng/µL to 100 ng/µL; (5) The diluted standards and RNA samples from engineered sEVs were analyzed by RT-PCR to obtain their respective Cq values. A standard curve was constructed based on the logarithm of known DNA concentrations of the standards versus their Cq values. From this curve, we estimated the amount of mRNA in each sEV sample. By combining the measured mRNA mass with dilution factors, total RNA content, and the molecular weight of the target gene, we calculated the total mRNA copy number. Finally, we normalized the mRNA copy number to the total sEV particle count, presenting it as copies per 1 × 106 sEVs.
Co-localization of CD63 and Lamp2
To ensure that co-localization observed was not an artifact of vesicle aggregation or fusion during purification and imaging, stable 293T cell lines were first established to separately secrete sEVs labeled with LAMP2-mCherry and CD63-GFP. These stable cell lines were generated by transducing 293T cells with lentiviruses encoding LAMP2-mCherry and CD63-GFP, respectively, followed by antibiotic selection to obtain resistant clones. After mixing the culture supernatants containing LAMP2-mCherry and CD63-GFP labeled vesicles, we purified the mixed vesicles using ultrafiltration size-exclusion chromatography (UF-SEC). Subsequently, we transduced the LAMP2-mCherry-expressing 293T stable cell line with a lentivirus encoding CD63-GFP and selected for co-expression of both fluorescent markers. The dual-expressing stable cell line was cultured, and sEVs were purified from the supernatant using the same UF-SEC method. The purified sEVs were diluted and placed on coverslips for confocal microscopy analysis.
Characterization of CARmRNA@aCD206 sEVs
Morphological characterization of CARmRNA@aCD206 sEVs was performed by transmission electron microscopy (TEM). sEVs were adsorbed onto copper grids and negatively stained with 2% uranyl acetate solution. Size distribution and particle concentration were measured by nanoparticle tracking analysis using a NanoSight NS300 system (Malvern Instruments Company, UK).
Expression of anti-CD206 scFv on CARmRNA@aCD206 sEVs
To confirm the presence of anti-CD206 scFv on the sEV surface, flow cytometric analysis was performed. sEVs were first coupled to 4 μm aldehyde/sulfate latex beads to enable detection. After incubation with biotinylated-CD206 (ACROBiosystems) to label surface scFvs, sEV-bound beads were stained with phycoerythrin (PE)-conjugated streptavidin (BioLegend, 1:200 dilution) and analyzed by flow cytometry using an Attune NxT cytometer (Thermo Fisher Scientific). Data were processed with FlowJo software (version 10.9). The gating strategy for flow cytometry is shown in Supplementary Fig. 25.
Nano flow cytometry analysis for EVs was performed as previously reported79,80,81,82. Specifically, Purified EVs were resuspended in 20 nm-filtered Dulbecco’s Phosphate Buffered Saline (DPBS, Thermo Fisher Scientific) to a final concentration of 1 × 10¹¹ particles/mL, as quantified by nanoparticle tracking analysis (NTA 3.0 analysis software, Malvern Panalytical). To minimize nonspecific binding, all protein reagents solutions were pre-cleared by centrifugation at 100,000 × g for 10 min at 4 °C, effectively reducing aggregate-induced background. Biotinylated CD206 (ACROBiosystems, 2 μg/mL) was titrated to an optimal working concentration of 10 µg/mL, determined by dose-response experiments. EVs were incubated with the biotinylated CD206 (10 µg/mL) for 1 h at 4 °C in the dark to preserve membrane integrity. After incubation, unbound antibodies were removed using qEV original/35 nm size exclusion chromatography (SEC) columns (Izon Science), ensuring the minimal retention of free antibody. PE-conjugated streptavidin (BioLegend) was added at 2.5 µg/mL, a concentration determined by saturation binding assays, and incubated for an additional hour under identical conditions. Consistent with recommendations to avoid signal artifacts, stained EVs were diluted 1,000-fold in filtered DPBS to a final concentration of about 1 × 10⁸ particles/mL. This approach reduced background fluorescence while maintaining Poisson-acceptable event rates. Data acquisition was performed on the CytoFLEX nano (Beckman Coulter) using the PE channel, and data were analyzed with FlowJo software (version 10.9). The gating strategy for flow cytometry is shown in Supplementary Fig. 25.
mRNA loading efficiency in sEVs
To investigate the mechanism of mRNA loading into sEVs, we developed several controls with specific modifications: (1) Free sEV: sEVs secreted by unmodified HEK293T cells; (2) L-sEV: sEVs derived from HEK293T cells expressing only CD63-L7Ae; (3) mRNACAR-sEV: sEVs from HEK293T cells expressing only anti-MSLN CAR; (4) mRNACAR-C/D box-sEV: sEVs from HEK293T cells expressing only anti-MSLN CAR with a C/D box; (5) mRNACAR-L-sEV: sEVs from HEK293T cells co-expressing CD63-L7Ae and anti-MSLN CAR; (6) mRNACAR-C/D box-L-sEV: sEVs from HEK293T cells co-expressing CD63-L7Ae and anti-MSLN CAR with a C/D box. Then, the mRNA content in these EVs was analyzed using RT-qPCR.
Western blot analysis
The expression of engineered proteins in sEVs and cells was verified by Western blot. Briefly, cells and sEVs were lysed with exosome protein specific lysis buffer (Umibio (Shanghai) Co., Ltd.) on ice for 20 min, and the supernatant was collected after centrifugation at 10,000 × g for 30 min. We then quantified the protein concentration using a BCA protein assay kit. Protein samples (20 μg/well) and protein marker (GenScript) were loaded and fractionated on 10% SDS-PAGE gels. After electrophoresis, the protein samples in the gels were transferred to PVDF membranes (Millipore). After blocking with 5% skim milk in TBST (Sigma-Aldrich), the membranes were incubated with specific primary antibodies overnight at 4 °C. The membranes were then washed three times with TBST and incubated with the appropriate HRP-conjugated secondary antibody (Cell Signaling Technology, 1:2000 dilution) for 2 h. After three washes with TBST, protein bands were visualized using ECL substrate (GE Healthcare) and chemiluminescence imaging.
Analysis of cellular gene expression by RT-qPCR
For analysis of gene expression changes in cells, RNA isolation, reverse transcription, and RT-qPCR were performed using primers specific for genes related to macrophage activation and polarization (CD40, CD80, CD86, iNOS, TNF-α, IFN-γ, CXCL-9, CXCL-10), immunosuppression (CD206, CD163, Arginase-1, TGF-β), anti-MSLN CAR, or the housekeeping gene GAPDH. Primers were designed using Primer-BLAST and synthesized by IDT. The DNA sequences used for primers in this study are provided (Supplementary Table 1).
Isolation and culture of primary murine macrophages
Bone marrow-derived macrophages (BMDM) were isolated from the femurs and tibias of female C57BL/6 mice. Bones were flushed with high-glucose DMEM containing 30% L929 cell conditioned medium to extrude bone marrow cells67. Erythrocytes were lysed using RBC lysis buffer (Sigma-Aldrich) per the manufacturer’s protocol. The remaining cells were cultured in high-glucose DMEM with 30% L929 conditioned medium for 7 days to differentiate into BMDMs. The medium was refreshed every 2 days. L929 conditioned medium provides a rich source of macrophage colony stimulating factor (M-CSF) to promote macrophage differentiation. It was generated by culturing L929 cells in high-glucose DMEM with 10% FBS for 10 days. To generate M2-polarized BMDMs, 20 ng/mL IL-4 (PeproTech) was added to the medium.
In vitro analysis of sEV internalization by BMDMs
To assess cellular uptake, M2-polarized bone marrow-derived macrophages (M2 BMDM) were seeded on glass bottom dishes (Nest Biotechnology, China) and labeled with the fluorescent dye DiO (green, Abmole, USA). The sEVs, including CARmRNA@sEVs, CARmRNA@aCD206 sEVs, and CARmRNA@aHA sEVs (where the anti-CD206 scFv was replaced with anti-HA scFv), were labeled with DiI (red, Abmole, USA). Additionally, a DiI-PBS control group was prepared by treating DiI in the same manner as the exosome labeling to exclude any effects from residual dye. For blocking experiments, M2 BMDMs were pre-treated with anti-CD206 antibodies to block CD206 before incubation with CARmRNA@aCD206 sEVs. Then, the DiI-labeled sEVs (20 μg/mL) were co-incubated with the DiO-labeled M2 BMDMs for either 4 h or 24 h at 37 °C, followed by thorough washing to remove unbound sEVs. After incubation, the cells were fixed with 4% paraformaldehyde for 5 min, nuclei were stained with DAPI (blue), and sEV internalization was visualized using confocal microscopy (Zeiss LSM 880). Data were analyzed with ZEN 2.3 blue edition. To achieve absolute quantification and analyze the kinetics of sEV uptake, BMDMs were harvested at multiple time points (4 h, 10 h, 16 h, and 24 h), washed thoroughly to remove any surface-bound sEVs, and analyzed using an Attune NxT cytometer (Thermo Fisher Scientific). Data were processed with FlowJo software (version 10.9). The gating strategy for flow cytometry is shown in Supplementary Fig. 25.
Engineered sEV-mediated mRNA translation efficiency in BMDMs
For visual evaluation of mRNA delivery, the CAR coding sequence was replaced with EGFP to generate EGFP mRNA-loaded sEVs. M2 BMDMs were incubated with EGFPmRNA@aCD206 sEVs (20 μg protein/mL, 4 × 106 mRNA copies/mL), EGFPmRNA@sEVs (20 μg protein/mL, 4 × 106 mRNA copies/mL), or PBS for 24 h. EGFP expression was visualized by confocal microscopy (Zeiss LSM 880), Data were analyzed with ZEN 2.3 blue edition.
To quantify CAR expression, M2 BMDMs were treated with various sEV formulations and analyzed by flow cytometry, following previously established protocols. Specifically, M2 BMDMs were incubated with PBS, MockmRNA@aCD206 sEVs, CARmRNA@sEVs, CARmRNA@aHA sEVs, or CARmRNA@aCD206 sEVs at a concentration of 4 × 106 mRNA copies/mL (20 μg protein/mL) for 24 h. In the CARmRNA@aCD206 sEV+CHX group, BMDMs were pre-treated with the translation inhibitor cycloheximide (CHX) prior to incubation with CARmRNA@aCD206 sEVs for 24 h. Following incubation, cells were thoroughly washed to remove unbound sEVs. BMDMs were then harvested and blocked with anti-mouse CD16/CD32 (Fc block, 1:200 dilution) to prevent non-specific antibody binding. Subsequently, cells were stained with anti-CD11b-Pacific Blue (BioLegend, Clone: M1/70, 1:500 dilution), anti-F4/80-APC (BioLegend, Clone: BM8, 1:500 dilution), and PE-labeled MSLN antibodies (ACROBiosystems, 1:50 dilution) for 45 min. After washing to eliminate residual dye, anti-MSLN CAR protein levels were quantified using flow cytometry.
To investigate the dose-dependent expression of CAR protein on BMDMs, M2 BMDMs were incubated with varying concentrations (0 mRNA copies/mL, 1 × 106 mRNA copies/mL, 2 × 106 mRNA copies/mL, 3 × 106 mRNA copies/mL, 4 × 106 mRNA copies/mL, 5 × 106 mRNA copies/mL) of CARmRNA@aCD206 sEVs for 24 h. Following incubation, cells were stained and washed with PBS containing 1% BSA, and CAR expression levels were analyzed by flow cytometry. Additionally, to assess the kinetics of CAR expression, M2 BMDMs treated with CARmRNA@aCD206 sEVs were evaluated for CAR levels at multiple time points post-treatment, specifically on days 1, 2, 4, and 7. Data were processed with FlowJo software (version 10.9). The gating strategy for flow cytometry is shown in Supplementary Fig. 25.
Analysis of tumor cell phagocytosis
To evaluate the effects of CAR expression on macrophage phagocytic activity, M2 BMDMs were treated with PBS, MockmRNA@aCD206 sEVs, CARmRNA@sEV, or CARmRNA@aCD206 sEVs. After 24 h, CFSE-labeled B16-MSLN or B16 cells were added at a 1:1 ratio and co-cultured for 8 h. Cells were stained with anti-CD11b-PE (BioLegend, Clone: M1/70, 1:500 dilution) and anti-F4/80-APC (BioLegend, Clone: BM8, 1:500 dilution) and analyzed by flow cytometry to quantify phagocytosis based on the CFSE+ signal within CD11b+F4/80+ macrophages. The gating strategy for flow cytometry is shown in Supplementary Fig. 25.
CARmRNA@aCD206 sEV-treated M2 BMDMs were stained with DiO (green) and co-incubated with DiI-labeled B16-MSLN cells (red) at a 1:1 ratio for 8 h. Phagocytosis was visualized by confocal microscopy (Zeiss LSM 880). Data were analyzed with ZEN 2.3 blue edition.
To dynamically assess phagocytosis, CARmRNA@aCD206 sEV-treated M2 BMDMs were co-cultured with B16-MSLN cells at a 1:1 ratio and imaged by live cell imaging (Axio Observer 7) at 1 min intervals.
In vitro cytotoxicity assays
To evaluate the specificity and cytotoxic efficacy of CAR-Ms against B16-MSLN tumor cells, a luciferase-based assay was employed. B16-MSLN cells were genetically engineered to overexpress firefly luciferase (B16-MSLN-Fluc), enabling the quantitative assessment of tumor cell viability through luminescence measurements. CAR-Ms were generated by treating macrophages with CARmRNA@aCD206 sEVs. Prior to the assay, B16-MSLN-Luc cells were seeded into 96-well plates at a density of 5000 cells per well and allowed to adhere overnight to ensure optimal growth conditions. Subsequently, CAR-Ms were co-cultured with B16-MSLN-Fluc cells for 24 h at varying effector-to-target (E: T) ratios (1:2, 1:1, 2:1, 5:1, and 10:1) to assess the dose-dependent cytotoxic effects. To ensure exposure of the luciferase enzyme to the substrate, 2% Triton X-100 lysis buffer was added before the luciferase substrate (D-luciferin potassium salt, Beyotime) was added to facilitate cell lysis. The bioluminescence signal was then measured with an IVIS Spectrum (PerkinElmer). The cytotoxicity of CAR-Ms generated by CARmRNA@aCD206 sEVs was calculated based on the total flux of the luciferase signal relative to tumor cells alone, using the following formula.
Cytotoxicity = [1 − (Sample signal−Background signal)/(Tumor alone signal−Background signal)] × 100%
Immunophenotypic changes in treated macrophages
To explore the polarization process of CAR-Ms induced by CARmRNA@aCD206 sEV-treated BMDMs, we designed an in vitro experiment to assess macrophage immunophenotypic changes at three key time: after internalization of engineered sEVs, following CAR expression, and upon engagement with B16-MSLN cells. Briefly, M2 BMDMs were treated with PBS, MockmRNA@aCD206 sEVs, CARmRNA@sEV, or CARmRNA@aCD206 sEVs for 4 h to facilitate sEV internalization and then cultured for an additional 24 h to induce CAR expression. Subsequently, the treated BMDMs were co-cultured with B16-MSLN melanoma cells at a 1:1 ratio for an additional 24 h. At the end of each experimental stage (after sEV internalization, CAR expression, and CAR-Ms/tumor cell engagement), the cells were harvested, stained with anti-CD11b-Pacific Blue (BioLegend, Clone: M1/70, 1:500 dilution), anti-F4/80-APC (BioLegend, Clone: BM8, 1:500 dilution), anti-CD86-PE (BioLegend, Clone: GL-1, 1:500 dilution), anti-CD86-PE/Cyanine7 (BioLegend, Clone: GL-1, 1:500 dilution), anti-CD206-FITC (BioLegend, Clone: C068C2, 1:500 dilution), and anti-MHC II-PE/Cyanine5 (BioLegend, Clone: M5/114.15.2, 1:500 dilution), PE-labeled MSLN antibodies (ACROBiosystems, 1:50 dilution). Samples were then analyzed by flow cytometry, and the data were processed using FlowJo software (version 10.9). The gating strategy for flow cytometry is shown in Supplementary Fig. 25.
Immunomodulatory effects of CARmRNA@aCD206 sEV-induced CAR-Ms on immune cells
To evaluate the potential enhanced immune-stimulatory effects of programmed CAR-Ms, Transwell assays were performed by seeding PBS, MockmRNA@aCD206 sEV, CARmRNA@sEV or CARmRNA@aCD206 sEV-treated M2 BMDMs in the bottom chamber and co-culturing them with B16-MSLN cells. Murine T cells were isolated from spleens of healthy C57BL/6 mice and labeled with CFSE (5 μM, Thermo Fisher) to track proliferation. CFSE-labeled T cells were seeded in the upper Transwell chamber (NEST Biotechnology) and co-cultured with BMDMs for 3 days before harvesting and immunostaining with anti-CD3-APC antibody (BioLegend, Clone: 17A2, 1:500 dilution) for flow cytometry. We also investigated the interactions of CAR-Ms and dendritic cells (DC) using Transwell assays. Bone marrow-derived DCs were generated by culturing bone marrow cells in RPMI 1640 complete medium with 20 ng/mL GM-CSF (PeproTech) for 7 days. The CFSE-labeled DCs were then co-cultured in the upper Transwell chamber with CAR-Ms in the bottom chamber for 24 h. DCs were harvested and stained for the dendritic cell markers anti-CD11c-FITC (BioLegend, Clone: N418, 1:500 dilution), anti-CD80-APC (BioLegend, Clone: 16-10A1, 1:500 dilution), anti-CD86-PE (BioLegend, Clone: GL-1, 1:500 dilution) to evaluate DC maturation (CD11c+CD80+CD86+) by flow cytometry. The gating strategy for flow cytometry is shown in Supplementary Fig. 25.
In vivo biodistribution of CARmRNA@aCD206 sEVs
To investigate the in vivo biodistribution of CARmRNA@aCD206 sEVs, a B16-MSLN lung metastatic tumor model was first established by intravenous injection of 5 × 105 B16-MSLN cells per mouse via the tail vein.
The CARmRNA@aCD206 sEVs were then labeled with the fluorescent dye DiR according to the manufacturer’s protocol. Unbound dye was removed by ultracentrifugation at 100,000 × g for 70 min, followed by washing three times with phosphate-buffered saline (PBS). The purified DiR-labeled CARmRNA@aCD206 sEVs were resuspended in PBS for further use.
For the biodistribution study, B16-MSLN tumor-bearing mice were randomly divided into two groups and administered DiR-labeled CARmRNA@aCD206 sEVs (10 mg protein/kg, 2 × 109 mRNA copies/kg) via intravenous injection or inhalation. At 12 h post-administration, the mice were euthanized and the major organs (heart, liver, spleen, lung, kidney, stomach, and intestine) were harvested. Ex vivo fluorescence imaging was performed on the excised organs using the Xenogen IVIS Spectrum system (Caliper Life Sciences, Hopkinton, MA, USA) to detect the presence of DiR-labeled CARmRNA@aCD206 sEVs. Data were analyzed with Living image software (Perkin Elmer, V4.3.1). Additionally, immunofluorescence staining of lung tumor sections was performed to visualize sEV penetration in tumor.
To investigate the time-dependent biodistribution profiles following pulmonary delivery, an additional cohort of B16-MSLN tumor-bearing mice received DiR-labeled CARmRNA@aCD206 sEVs (10 mg protein/kg, 2 × 109 mRNA copies/kg) via inhalation. The mice were euthanized at 6, 12, 24 or 48 h post-inhalation and the major organs were collected. Ex vivo fluorescence imaging was similarly conducted on the excised organs using the IVIS Spectrum system.
In vivo generation of CAR-Ms via CARmRNA@aCD206 sEVs
To evaluate the ability of our engineered sEVs in delivery mRNA in vivo, we constructed the reporter FlucmRNA@aCD206 sEVs containing firefly luciferase (Fluc) mRNA instead of the CAR construct. B16-MSLN tumor-bearing mice were administered FlucmRNA@aCD206 sEVs (10 mg protein/kg, 2 × 109 mRNA copies/kg) or PBS by inhalation. After 24 h, mice received an intraperitoneal injection of the luciferase substrate D-luciferin (150 mg/kg, Beyotime) and underwent bioluminescence imaging using the IVIS Spectrum system to quantify luminescence intensity in excised organs as a measure of sEV-mediated Fluc expression.
To further investigate whether CARmRNA@aCD206 sEVs could selectively deliver CAR mRNA and mediate protein expression in macrophages including TAMs and AMs, B16-MSLN tumor-bearing mice were randomized into five groups and inhaled with PBS, MockmRNA@aCD206 sEVs, CARmRNA@sEVs, CARmRNA@aHA sEVs or CARmRNA@aCD206 sEVs (10 mg protein/kg, 2 × 109 mRNA copies/kg). After 24 h, the mice were euthanized, and the lungs were extracted and digested using collagenase IV (1 mg/mL, Sigma-Aldrich) to obtain single cell suspensions. Erythrocytes were lysed with RBC lysis buffer (BioLegend) and Fc receptors were blocked with anti-CD16/32 antibodies (BioLegend, 1:200 dilution) before staining with fluorescently labeled antibodies. The cells were then stained with anti-CD45-PE/Cyanine7 (BioLegend, Clone: 30-F11, 1:500 dilution), anti-CD11b-Pacific Blue (BioLegend, Clone: M1/70, 1:500 dilution), anti-F4/80-APC (BioLegend, Clone: BM8, 1:500 dilution), anti-CD11c- APC/Cyanine7 (BioLegend, Clone: N418, 1:500 dilution), anti-ly6c-FITC (BioLegend, Clone: HK1.4, 1:500 dilution), anti-Siglec-F-PerCP/Cyanine5.5 (BioLegend, Clone: S17007L, 1:500 dilution), and PE-labeled MSLN antibodies (ACROBiosystems, 1:50 dilution) at 4 °C for 30 min. After three washes with PBS containing 1% BSA, CAR expression on macrophages (CD45+CD11b+F4/80+, including AMs (CD45+CD11c+F4/80+Siglec-F+) and TAMs (CD45+CD11b+F4/80+Siglec-F-)) and monocytes (CD45+CD11b+F4/80-ly6c+) were quantified by flow cytometry. We also evaluated CAR expression on other leukocytes (CD11c+dendritic cells, CD3+ T cells, CD19+ B cells) to verify targeted delivery and gene-specific expression in macrophages. All samples were stained with corresponding IgG antibodies as isotype controls. The gating strategy for flow cytometry is shown in Supplementary Fig. 25. Additionally, immunofluorescence staining of lung tumor sections was performed to visualize CAR protein expression and localization.
To further investigate the dose-dependent effects of CARmRNA@aCD206 sEV-mediated CAR-M generation, B16-MSLN tumor-bearing mice were inhaled with CARmRNA@aCD206 sEVs at various concentrations (0 copies/kg, 6 × 108 mRNA copies/kg, 1.2 × 109 mRNA copies/kg, 2 × 109 mRNA copies/kg, 2.4 × 109 mRNA copies/kg). After 24 h, the lungs were processed to obtain single cell suspensions, and CAR protein expression levels on macrophages were quantified by flow cytometry.
Immune memory response induced by in vivo generated CAR-Ms
To evaluate the immune memory response induced by mediated by CARmRNA@aCD206 sEVs, we isolated CAR-TAMs and CAR-AMs from the tumor tissues of mice inhaled with CARmRNA@aCD206 sEVs. Concurrently, T cells were isolated from the spleens of B16-MSLN bearing mice. The isolated CAR-AMs or CAR-TAMs were then co-cultured with the T cells at a ratio of 1:5 for 72 h. For negative controls, AMs (CD45+CD11c+F4/80+Siglec-F+) or TAMs (CD45+CD11b+F4/80+Siglec-F-) were isolated from the tumor tissue of tumor-bearing mice inhaled with PBS and co-cultured with T cells. A blank control group comprised T cells isolated from the spleen without the presence of macrophages. Following the co-culture period, T cells were harvested and stained with the following antibodies for flow cytometric analysis: anti-CD45-PE/Cyanine7 (BioLegend, Clone: 30-F11, 1:500 dilution), anti-CD3-Pacific Blue (BioLegend, Clone: 17A2, 1:500 dilution), anti-CD8-PE (BioLegend, Clone: 53-6.7, 1:500 dilution), anti-CD44-FITC (BioLegend, Clone: IM7, 1:500 dilution), and anti-CD62L-APC (BioLegend, Clone: MEL-14, 1:500 dilution). The proportions of TCM (CD45+CD3+CD8+CD44+CD62L+), TEM (CD45+CD3+CD8+CD44+CD62L‒) and naïve T cells (CD45+CD3+CD8+CD44‒CD62L+) were analyzed. The gating strategy for flow cytometry is shown in Supplementary Fig. 26.
Immune analysis of in vivo generated CAR-T cells mediated by CARmRNA@aCD206 sEVs
To investigate the immune activation of CAR-T cells generated in vivo mediated by CARmRNA@aCD206 sEVs, B16-MSLN bearing lung metastasis mice were inhaled with PBS or CARmRNA@aCD206 sEVs (10 mg protein/kg, 2 × 109 mRNA copies/kg). After 24 h, the mice were euthanized, and the lungs were extracted and digested using collagenase IV (1 mg/mL, Sigma-Aldrich) to obtain single cell suspensions. Erythrocytes were lysed with RBC lysis buffer (BioLegend) and Fc receptors were blocked with anti-CD16/32 antibodies (BioLegend, 1:200 dilution) before staining with fluorescently labeled antibodies. The cells were then stained with anti-CD45-PE/Cyanine7 (BioLegend, Clone: 30-F11, 1:500 dilution), anti-CD3-APC (BioLegend, Clone: 17A2, 1:500 dilution), anti-CD8-Pacific Blue (BioLegend, Clone: 53-6.7, 1:500 dilution), PE-labeled MSLN antibodies (ACROBiosystems, 1:50 dilution) and anti-CD69-PerCP/Cyanine5.5 (BioLegend, Clone: H1.2F3, 1:500 dilution) at 4 °C for 30 min. After three washes with PBS containing 1% BSA, the expression levels of anti-MSLN CAR on T cells and CD69 on CAR-T or T cells were quantified by flow cytometry. The gating strategy for flow cytometry is shown in Supplementary Fig. 26.
To further evaluate the antitumor efficacy of in vivo-generated CAR-T cells, we employed flow cytometry to isolate CAR-T cells from mice treated with CARmRNA@aCD206 sEVs. Concurrently, T cells were extracted from the lung tumor tissues of PBS-treated control mice and subsequently cultured in vitro. The isolated CAR-T cells or control T cells were then co-cultured with B16-MSLN-Fluc melanoma cells at varying effector-to-target (E: T) ratios (5:1, 2:1, 1:1, 1:2, 1:5). Following a 24-h incubation period, 2% Triton X-100 lysis buffer was added, and subsequently, the luciferase substrate (D-luciferin potassium salt, Beyotime) was added to facilitate cell lysis and expose the luciferase enzyme. Bioluminescence signals were quantified using an IVIS Spectrum imaging system (PerkinElmer). The cytotoxicity of CAR-T cells generated via CARmRNA@aCD206 sEVs was then calculated.
Immunofluorescence staining
Lung metastatic cancer tissues fixed with 4% paraformaldehyde were embedded in paraffin and sectioned at 4 μm for immunofluorescence staining. After standard dewaxing, hydration, and heat-induced epitope retrieval steps, sections were incubated with 10% goat serum for 1 h at room temperature to block non-specific binding sites.
To assess the macrophage population, the sections were incubated with anti-mouse F4/80 antibody (Thermo Fisher, Clone: BM8, 10 μg/mL), anti-mouse-Melan-A (Thermo Fisher, 1:100 dilution), anti-mouse-siglec-F (Thermo Fisher, clone: 1RNM44N, 20 μg/mL) primary antibodies overnight at 4 °C; For the detection of CAR expression, the sections were incubated with anti-mouse F4/80 antibody (Thermo Fisher, Clone: BM8, 10 μg/mL) and anti-flag antibody (Abcam, clone: EPR20018-251, 1:500 dilution) primary antibodies overnight at 4 °C; To evaluate the penetration of sEVs, the sections were incubated with anti-mouse-siglec-F (Thermo Fisher, clone: 1RNM44N, 20 μg/mL) primary antibodies overnight at 4 °C. After primary antibody incubation, the sections were washed three times with PBST and subsequently incubated with the appropriate secondary antibodies for 2 h at room temperature. Following three additional washes with PBST, the sections were counterstained with DAPI (Abmole, 1: 5000 dilution) for 15 min to visualize nuclei. Finally, the stained sections were analyzed using a digital slide scanner (P250 FLASH) to obtain high-resolution immunofluorescence images. Data were analyzed with CaseViewer 2.4 software.
Anti-tumor effects of CARmRNA@aCD206 sEVs in vivo
To evaluate the therapeutic efficacy of CARmRNA@aCD206 sEVs against lung metastatic cancer in vivo, we first generated a luciferase-expressing B16-MSLN-FLuc cell line using a lentiviral construct. C57BL/6 mice were injected via the tail vein with 5 × 105 B16-MSLN-FLuc cells to establish a lung metastatic tumor model, which was confirmed by bioluminescence imaging on day 7. Mice were then randomized into four treatment groups and administered PBS (G1), MockmRNA@aCD206 sEVs (G2, 10 mg protein/kg, 2 × 109 mRNA copies/kg), CARmRNA@sEVs (G3, 10 mg protein/kg, 2 × 109 mRNA copies/kg) or CARmRNA@aCD206 sEVs (G4, 10 mg protein/kg, 2 × 109 mRNA copies/kg) by inhalation every 3 days. Tumor burden was monitored weekly by injecting mice intraperitoneally with D-luciferin (150 mg/kg) and performing bioluminescence imaging with an IVIS Spectrum system. Body weight and survival were also monitored throughout the experimental period. Additionally, the lungs were extracted for flow cytometry and histological analysis, and tumor lysates were analyzed by ELISA for IFN-γ, TNF-α, IL-10 and TGF-β levels (Dakewe) per the manufacturer’s instructions.
Inhibition of tumor recurrence mediated by CARmRNA@aCD206 sEVs
We also examined whether CARmRNA@aCD206 sEV treatment could confer long-term protection against lung metastatic cancer. C57BL/6 mice were inoculated with B16-MSLN-Luc cells to establish lung metastasis. Beginning on day 7, mice received CARmRNA@aCD206 sEVs (10 mg protein/kg, 2 × 109 mRNA copies/kg) by inhalation every 3 days until complete tumor regression by bioluminescence imaging. Mice were then rechallenged with 5 × 105 B16-MSLN-Luc or B16-Luc cells by tail vein injection along with untreated control mice. Tumor burden and overall survival were monitored. In parallel studies, before tumor cells rechallenge, lymph nodes from mice treated with 100 μL PBS (G1), MockmRNA@aCD206 sEVs (G2, 10 mg protein/kg, 2 × 109 mRNA copies/kg), CARmRNA@sEVs (G3, 10 mg protein/kg, 2 × 109 mRNA copies/kg) or CARmRNA@aCD206 sEVs (G4, 10 mg protein/kg, 2 × 109 mRNA copies/kg) were harvested and immune memory cells in the lymph nodes were analyzed by flow cytometry.
Flow cytometric analysis of the tumor microenvironment
For analysis of immune cells in tumors and lymph nodes, the tissues were mechanically fragmented and digested with collagenase type IV (1 mg/mL, Sigma-Aldrich) at 37 °C for 30 min to obtain single cell suspensions. Before flow cytometry staining, the erythrocytes were lysed with RBC lysis buffer (BioLegend), and the nonspecific binding sites of the cells were blocked with anti-mouse CD16/CD32 (Fc block, 1:200 dilution).
Cells isolated from tumor tissues were stained with the following antibodies for flow cytometric analysis: anti-CD45-PE/Cyanine7 (BioLegend, Clone: 30-F11, 1:500 dilution), anti-CD11b-Pacific Blue (BioLegend, Clone: M1/70, 1:500 dilution), anti-F4/80-PerCP/Cyanine5.5 (BioLegend, Clone: BM8, 1:500 dilution), anti-CD80-APC (BioLegend, Clone: 16-10A1, 1:500 dilution), anti-CD206-FITC (BioLegend, Clone: C068C2, 1:500 dilution), anti-CD3-APC (BioLegend, Clone: 17A2, 1:500 dilution), anti-CD4-FITC (BioLegend, Clone: GK1.5, 1:500 dilution), anti-CD8-PE (BioLegend, Clone: 53-6.7, 1:500 dilution), anti-CD8- Pacific Blue (BioLegend, Clone: 53-6.7, 1:500 dilution), anti-Granzyme B-PE (BioLegend, Clone: QA16A02, 1:200 dilution), anti-CD25-PerCP/Cyanine5.5 (BioLegend, Clone: PC61, 1:500 dilution), and anti-Foxp3-PE (BioLegend, Clone: MF14, 1:200 dilution), and anti-Gr1-APC/Cyanine 7 (BioLegend, Clone: RB6-8C5, 1:500 dilution). The proportions of M1 macrophages (CD45+CD11b+F4/80+CD80+), M2 macrophages (CD45+CD11b+F4/80+CD206+), CD4+T cells (CD45+CD3+CD4+), CD8+T cells (CD45+CD3+CD8+), activated CD8+T cells (CD45+CD3+CD8+Granzyme B+), Treg cells (CD45+CD3+CD4+CD25+Foxp3+), and MDSCs (CD45+CD11b+Gr1+) in the tumor microenvironment were analyzed. The gating strategy for flow cytometry is shown in Supplementary Fig. 26.
Cells isolated from lymph nodes were stained with the following antibodies for flow cytometric analysis: anti-CD45-PE/Cyanine7 (BioLegend, Clone: 30-F11, 1:500 dilution), anti-CD3-Pacific Blue (BioLegend, Clone: 17A2, 1:500 dilution), anti-CD8-PE (BioLegend, Clone: 53-6.7, 1:500 dilution), anti-CD44-FITC (BioLegend, Clone: IM7, 1:500 dilution), and anti-CD62L-APC (BioLegend, Clone: MEL-14, 1:500 dilution). The proportions of TCM (CD45+CD3+CD8+CD44+CD62L+), TEM (CD45+CD3+CD8+CD44+CD62L‒) and naïve T cells (CD45+CD3+CD8+CD44‒CD62L+) were analyzed. The gating strategy for flow cytometry is shown in Supplementary Fig. 26.
Safety assessment of CARmRNA@aCD206 sEVs
To evaluate the biocompatibility of CARmRNA@aCD206 sEVs in vivo, we randomly divided female C57BL/6 mice into two groups and treated them with CARmRNA@aCD206 sEVs (10 mg protein/kg, 2 × 109 mRNA copies/kg) or the same volume of PBS by inhalation every 3 days. The body weight of the mice was monitored for 30 days. Mice were then euthanized, blood was collected for serum biochemistry analysis, and major organs were extracted for histological analysis. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), blood urea nitrogen (BUN) and creatinine (CREA) were quantified as indicators of liver and kidney function using commercial assay kits. The major organs were harvested and fixed in 4% paraformaldehyde. Hematoxylin and eosin (H&E) staining was performed to determine whether CARmRNA@aCD206 sEVs caused any tissue damage.
Immunogenicity evaluation of sEVs
To evaluated the potential of aCD206-engineered small extracellular vesicles (aCD206 sEVs) without CAR mRNA as an allogeneic, off-the-shelf therapeutic vehicle, we assessed the adaptive immune response of mice after aCD206 sEVs inhalation. To this end, health C57BL/6 mice were randomly divided into two groups and inhaled with PBS or aCD206 sEVs every 3 days. After 30 days treatment period, we analyzed the immune response in the mice using flow cytometry. Spleen tissues were harvested, mechanically dissociated, and enzymatically digested to obtain single-cell suspensions. After red blood cell lysis, the cells were stained with the following antibodies for flow cytometric analysis: anti-CD45-PE/Cyanine7 (BioLegend, Clone: 30-F11, 1:500 dilution), anti-CD3-Pacific Blue (BioLegend, Clone: 17A2, 1:500 dilution), anti-CD8-PE (BioLegend, Clone: 53-6.7, 1:500 dilution), anti-CD69- PerCP/Cyanine5.5 (BioLegend, Clone: H1.2F3, 1:500 dilution). Flow cytometric analysis was performed to quantify the proportion of CD3⁺ T cells and assess the expression levels of CD69, indicating T cell activation status. The gating strategy for flow cytometry is shown in Supplementary Fig. 26.
In parallel, serum samples were collected to measure the concentrations of key cytokines involved in immune modulation and tumor immunotherapy, including Granzyme B, interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-2 (IL-2). These cytokine levels were quantified using commercially available ELISA kits (Dakewe), following the manufacturers’ protocols.
Statistical analysis
GraphPad Prism 8.0 software was used for all statistical analyses. Data represent the mean ± standard deviation (SD) from at least three independent experiments. Differences between groups were analyzed by Student’s t test or one-way ANOVA. Kaplan-Meier analysis was used for survival curves. p ≤ 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All relevant data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper and are also available in Figshare: https://doi.org/10.6084/m9.figshare.29063477. Source data are provided with this paper.
References
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48 (2023).
Castaneda, M., den Hollander, P., Kuburich, N. A., Rosen, J. M. & Mani, S. A. Mechanisms of cancer metastasis. Semin. Cancer Biol. 87, 17–31 (2022).
Ganesh, K. & Massague, J. Targeting metastatic cancer. Nat. Med. 27, 34–44 (2021).
Hu, Q. et al. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat. Biomed. Eng. 5, 1038–1047 (2021).
Gerstberger, S., Jiang, Q. & Ganesh, K. Metastasis. Cell 186, 1564–1579 (2023).
Maalej, K. M. et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol. Cancer 22, 20 (2023).
Zhang, P., Zhang, G. & Wan, X. Challenges and new technologies in adoptive cell therapy. J. Hematol. Oncol. 16, 97 (2023).
Sly, L. M. & McKay, D. M. Macrophage immunotherapy: overcoming impediments to realize promise. Trends Immunol. 43, 959–968 (2022).
Li, W., Wang, F., Guo, R., Bian, Z. & Song, Y. Targeting macrophages in hematological malignancies: recent advances and future directions. J. Hematol. Oncol. 15, 110 (2022).
Lei, A. et al. A second-generation M1-polarized CAR macrophage with antitumor efficacy. Nat. Immunol. 25, 102–116 (2024).
Klichinsky, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38, 947–953 (2020).
Zhang, L. et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 13, 153 (2020).
Wang, X. et al. Metabolic Reprogramming via ACOD1 depletion enhances function of human induced pluripotent stem cell-derived CAR-macrophages in solid tumors. Nat. Commun. 14, 5778 (2023).
Liu, Q. et al. Adoptive cellular immunotherapy for solid neoplasms beyond CAR-T. Mol. Cancer 22, 28 (2023).
Pan, K. et al. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 41, 119 (2022).
Chen, Y. et al. CAR-macrophage: a new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 139, 111605 (2021).
Raguram, A., Banskota, S. & Liu, D. R. Therapeutic in vivo delivery of gene editing agents. Cell 185, 2806–2827 (2022).
Hamilton, J. R. et al. In vivo human T cell engineering with enveloped delivery vehicles. Nat. Biotechnol. 42, 1684–1692 (2024).
Nie, S., Qin, Y., Ou, L., Chen, X. & Li, L. In situ reprogramming of immune cells using synthetic nanomaterials. Adv. Mater. 36, e2310168 (2024).
Michels, A., Ho, N. & Buchholz, C. J. Precision medicine: in vivo CAR therapy as a showcase for receptor-targeted vector platforms. Mol. Ther. 30, 2401–2415 (2022).
Qin, Y. T. et al. Biomaterials promote in vivo generation and immunotherapy of CAR-T cells. Front. Immunol. 14, 1165576 (2023).
Nawaz, W. et al. AAV-mediated in vivo CAR gene therapy for targeting human T-cell leukemia. Blood Cancer J. 11, 119 (2021).
Labanieh, L. & Mackall, C. L. CAR immune cells: design principles, resistance and the next generation. Nature 614, 635–648 (2023).
Parayath, N. N. & Stephan, M. T. In situ programming of CAR T cells. Annu. Rev. Biomed. Eng. 23, 385–405 (2021).
Smith, T. T. et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 12, 813–820 (2017).
Rurik, J. G. et al. CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96 (2022).
Liu, C. et al. mRNA-based cancer therapeutics. Nat. Rev. Cancer 23, 526–543 (2023).
Wu, J., Wu, W., Zhou, B. & Li, B. Chimeric antigen receptor therapy meets mRNA technology. Trends Biotechnol. 42, 228–240 (2023).
Huang, P., Deng, H., Wang, C., Zhou, Y. & Chen, X. Cellular trafficking of nanotechnology-mediated mRNA delivery. Adv. Mater. 36, e2307822 (2024).
Zong, Y., Lin, Y., Wei, T., & Cheng, Q. Lipid nanoparticle (LNP) enables mRNA delivery for cancer therapy. Adv. Mater. 35, e2303261 (2023).
Dai, J. et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 5, 145 (2020).
Zhang, Y. et al. Recent advances in exosome-mediated nucleic acid delivery for cancer therapy. J. Nanobiotechnology 20, 279 (2022).
Cheng, Z., Li, M., Dey, R. & Chen, Y. Nanomaterials for cancer therapy: current progress and perspectives. J. Hematol. Oncol. 14, 85 (2021).
Mager, S. E. L. A., Breakefield, I. & Wood, X. O. MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 12, 347–357 (2013).
Welsh, J. A. et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 13, e12404 (2024).
Valadi, H. et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659 (2007).
Kojima, R. et al. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 9, 1305 (2018).
Zhang, M. et al. Engineered exosomes from different sources for cancer-targeted therapy. Signal Transduct. Target Ther. 8, 124 (2023).
Zhao, C. et al. Vesicular antibodies: shedding light on antibody therapeutics with cell membrane nanotechnology. Adv. Mater. 35, e2207875 (2023).
Radler, J., Gupta, D., Zickler, A. & Andaloussi, S. E. Exploiting the biogenesis of extracellular vesicles for bioengineering and therapeutic cargo loading. Mol. Ther. 31, 1231–1250 (2023).
Zhu, T., Chen, Z., Jiang, G. & Huang, X. Sequential targeting hybrid nanovesicles composed of chimeric antigen receptor T-Cell-derived exosomes and liposomes for enhanced cancer immunochemotherapy. ACS Nano 17, 16770–16786 (2023).
Xiao, Y. et al. Functionalized biomimetic nanoparticles combining programmed death-1/programmed death-ligand 1 blockade with photothermal ablation for enhanced colorectal cancer immunotherapy. Acta Biomater. 157, 451–466 (2023).
Zhu, T. et al. Nanovesicles derived from bispecific CAR-T cells targeting the spike protein of SARS-CoV-2 for treating COVID-19. J. Nanobiotechnology 19, 391 (2021).
Li, X. et al. Genetically programmable vesicles for enhancing CAR-T therapy against solid tumors. Adv. Mater. 35, e2211138 (2023).
Xiang, X., Wang, J., Lu, D. & Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target Ther. 6, 75 (2021).
Elsharkasy, O. M. et al. Extracellular vesicles as drug delivery systems: why and how?. Adv. Drug Deliv. Rev. 159, 332–343 (2020).
Dinh, P. C. et al. Inhalation of lung spheroid cell secretome and exosomes promotes lung repair in pulmonary fibrosis. Nat. Commun. 11, 1064 (2020).
Shi, M. M. et al. Preclinical efficacy and clinical safety of clinical-grade nebulized allogenic adipose mesenchymal stromal cells-derived extracellular vesicles. J. Extracell. Vesicles 10, e12134 (2021).
Abdelaziz, H. M. et al. Inhalable particulate drug delivery systems for lung cancer therapy: Nanoparticles, microparticles, nanocomposites and nanoaggregates. J. Control. Release 269, 374–392 (2018).
Liu, M., Hu, S., Yan, N., Popowski, K. D., & Cheng, K. Inhalable extracellular vesicle delivery of IL-12 mRNA to treat lung cancer and promote systemic immunity. Nat. Nanotechnol. 19, 565–575 (2024).
Corso, G. et al. Systematic characterization of extracellular vesicle sorting domains and quantification at the single molecule—single vesicle level by fluorescence correlation spectroscopy and single particle imaging. J. Extracell. Vesicles 8, 1663043 (2019).
Lin, Y., Xu, J. & Lan, H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J. Hematol. Oncol. 12, 76 (2019).
Gao, L. et al. Convection-enhanced delivery of nanoencapsulated gene locoregionally yielding ErbB2/Her2-specific CAR-macrophages for brainstem glioma immunotherapy. J. Nanobiotechnology 21, 56 (2023).
Xu, L. et al. Design of experiment (DoE)-driven in vitro and in vivo uptake studies of exosomes for pancreatic cancer delivery enabled by copper-free click chemistry-based labelling. J. Extracell. Vesicles 9, 1779458 (2020).
Franzen, C. A. et al. Characterization of uptake and internalization of exosomes by bladder cancer cells. Biomed. Res. Int. 2014, 619829 (2014).
Zickler, A. M. et al. Novel endogenous engineering platform for robust loading and delivery of functional mRNA by extracellular vesicles. Adv. Sci. 11, e2407619 (2024.
Hirose, H., Hirai, Y., Sasaki, M., Sawa, H. & Futaki, S. Quantitative analysis of extracellular vesicle uptake and fusion with recipient cells. Bioconjug. Chem. 33, 1852–1859 (2022).
Bonsergent, E. et al. Quantitative characterization of extracellular vesicle uptake and content delivery within mammalian cells. Nat. Commun. 12, 1864 (2021).
Albanese, M. et al. MicroRNAs are minor constituents of extracellular vesicles that are rarely delivered to target cells. PLoS Genet. 17, e1009951 (2021).
Chiang, C. L. et al. Dual targeted extracellular vesicles regulate oncogenic genes in advanced pancreatic cancer. Nat. Commun. 14, 6692 (2023).
Liu, S., Verma, A., Kettenberger, H., Richter, W. F. & Shah, D. K. Effect of variable domain charge on in vitro and in vivo disposition of monoclonal antibodies. MAbs 13, 1993769 (2021).
Gupta, P. et al. Antibodies with weakly basic isoelectric points minimize trade-offs between formulation and physiological colloidal properties. Mol. Pharm. 19, 775–787 (2022).
Ribovski, L., Joshi, B., Gao, J. & Zuhorn, I. Breaking free: endocytosis and endosomal escape of extracellular vesicles. Extracell. Vesicles Circ. Nucl. Acids 4, 283–305 (2023).
Hagedorn, L., Jurgens, D. C., Merkel, O. M. & Winkeljann, B. Endosomal escape mechanisms of extracellular vesicle-based drug carriers: lessons for lipid nanoparticle design. Extracell. Vesicles Circ. Nucl. Acids 5, 344–357 (2024).
Kang, M., Jordan, V., Blenkiron, C. & Chamley, L. W. Biodistribution of extracellular vesicles following administration into animals: a systematic review. J. Extracell. Vesicles 10, e12085 (2021).
Lahiri, A. et al. Lung cancer immunotherapy: progress, pitfalls, and promises. Mol. Cancer 22, 40 (2023).
Kang, M. et al. Nanocomplex-mediated in vivo programming to chimeric antigen receptor-M1 macrophages for cancer therapy. Adv. Mater. 33, e2103258 (2021).
Hovhannisyan, L., Riether, C., Aebersold, D. M., Medova, M. & Zimmer, Y. CAR T cell-based immunotherapy and radiation therapy: potential, promises and risks. Mol. Cancer 22, 82 (2023).
Chen, C. et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci. Transl. Med. 14, eabn1128 (2022).
Yang, Z. et al. Dual mRNA co-delivery for in situ generation of phagocytosis-enhanced CAR macrophages augments hepatocellular carcinoma immunotherapy. J. Control. Release 360, 718–733 (2023).
Shrivastava, S. et al. Exosome-mediated stable epigenetic repression of HIV-1. Nat. Commun. 12, 5541 (2021).
O’Brien, K., Breyne, K., Ughetto, S., Laurent, L. C. & Breakefield, X. O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 21, 585–606 (2020).
Xia, Y. et al. Engineering macrophages for cancer immunotherapy and drug delivery. Adv. Mater. 32, e2002054 (2020).
Kunz, A. et al. Optimized Assessment of qPCR-based vector copy numbers as a safety parameter for GMP-grade CAR T cells and monitoring of frequency in patients. Mol. Ther. Methods Clin. Dev. 17, 448–454 (2020).
Kandell, J., Milian, S., Snyder, R. & Lakshmipathy, U. Universal ddPCR-based assay for the determination of lentivirus infectious titer and lenti-modified cell vector copy number. Mol. Ther. Methods Clin. Dev. 31, 101120 (2023).
Alamri, A., Nam, J. Y. & Blancato, J. K. Fluorescence in situ hybridization of cells, chromosomes, and formalin-fixed paraffin-embedded tissues. Methods Mol. Biol. 1606, 265–279 (2017).
Hu, L. et al. Fluorescence in situ hybridization (FISH): an increasingly demanded tool for biomarker research and personalized medicine. Biomark. Res. 2, 3 (2014).
Zhong, L. et al. Augmenting L3MBTL2-induced condensates suppresses tumor growth in osteosarcoma. Sci. Adv. 9, eadi0889 (2023).
Welsh, J. A. et al. MIFlowCyt-EV: a framework for standardized reporting of extracellular vesicle flow cytometry experiments. J. Extracell. Vesicles 9, 1713526 (2020).
Welsh, J. A. et al. A compendium of single extracellular vesicle flow cytometry. J. Extracell. Vesicles 12, e12299 (2023).
Morales-Kastresana, A., Welsh, J. A. & Jones, J. C. Detection and sorting of extracellular vesicles and viruses using nanoFACS. Curr. Protoc. Cytom. 95, e81 (2020).
Kobayashi, H. et al. Precise analysis of single small extracellular vesicles using flow cytometry. Sci. Rep. 14, 7465 (2024).
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (82303799 (T.Z.) and 82072062 (X.H.)), the National Science and Technology Key Projects for Major Infectious Diseases (2017ZX10302301-002 (X.H.)), the Development Project of Foshan Fourth People’s Hospital (FSSYKF-2020003 (X.H.) and FSSYKF-2020017 (X.H.)).
Author information
Authors and Affiliations
Contributions
Conceptualization: T.Z., Y.X., Methodology: Y.X., T.Z., Z.C., Investigation: Y.X., T.Z., Funding acquisition: X.H., T.Z., Project administration: X.H., Supervision: X.H., T.Z., Writing—original draft: Y.X., T.Z., Writing—review & editing: X.H., T.Z., Y.X.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Samir El Andaloussi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiao, Y., Zhu, T., Chen, Z. et al. Lung metastasis and recurrence is mitigated by CAR macrophages, in-situ-generated from mRNA delivered by small extracellular vesicles. Nat Commun 16, 7166 (2025). https://doi.org/10.1038/s41467-025-62506-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62506-2
This article is cited by
-
Bioengineering of extracellular vesicles with scaffold proteins for drug delivery
Journal of Nanobiotechnology (2026)
-
The evolution of cellular-based immunotherapy in the treatment of gastric cancer: an overview of clinical trials
Clinical and Translational Oncology (2026)
-
Convergence of mRNA technology and chimeric antigen receptor therapy: targeted technology optimizing targeted therapy
Journal of Translational Medicine (2025)
