
Abstract
Nerve injury-induced changes in pain-associated genes contribute to genesis of neuropathic pain and comorbid anxiety. Phosphorylated CTD interacting factor-1 (PCIF1)-triggered N6, 2′-O-dimethyladenosine (m6Am) mRNA modification represents an additional layer of gene regulation. However, the role of PCIF1 in these disorders is elusive. Here, we report PCIF1 is increased in glutamatergic neurons of the hindlimb region of the primary somatosensory cortex in mouse with neuropathic pain and anxiety, but not inflammatory pain or anxiety alone. Serpine-1 mRNA-binding protein-1 (SERBP1) is identified as a PCIF1 cofactor, their complex mediates m6Am deposition onto mRNA. Blocking SERBP1-PCIF1 upregulation in glutamatergic neurons of the hindlimb region of the primary somatosensory cortex abolishes m6Am gain on maf1 homolog, negative regulator of RNA polymerase III (Maf1), elevates MAF1 protein, and mitigates neuropathic pain and anxiety. Conversely, mimicking this increase adds m6Am onto Maf1, reduces MAF1, and induces comorbidity symptoms. These findings highlight the significance of m6Am in neuropathic pain-anxiety comorbidity and identify SERBP1–PCIF1 in glutamatergic neurons of the hindlimb region of the primary somatosensory cortex as a potential therapeutic target.
Similar content being viewed by others


Introduction
Nerve injury-induced neuropathic pain, a chronic and refractory disease, affects 7–10% of the world’s population1,2. What is worse is that approximately one-third of neuropathic pain patients have clinically relevant emotional symptoms such as anxiety. The latter further increases the difficulty of treating neuropathic pain and leads to a further decline in the patients’ quality of life3. Current treatments for neuropathic pain have the limited effectiveness and/or produce severe adverse effects, partially due to unclear and complex mechanisms underlying the comorbidity of pain and anxiety. Among many brain regions associated with nociceptive sensation and emotion processing4, primary somatosensory cortex (S1) is known to be crucial for pain and emotional modulation5. Clinical and preclinical studies have shown abnormal activation of S1 neurons in chronic neuropathic pain patients6,7,8 and in animal models of neuropathic pain9,10. Comorbid neuropathic pain and anxiety are triggered at least in part through abnormal hyperexcitability and ectopic discharges in S1 neurons11. Recently developed strategies have enabled the precise pinpointing of neuronal function in individual layers of S1 in the sensation and emotional progression of nociception12,13. Therefore, understanding the dysregulation of genes in specific S1 neurons may provide targeted approaches for the management of neuropathic pain and anxiety disorders.
mRNA modification plays an important role in the regulation of gene expression14. N6-methyl-2′-O-methyladenosine (m6Am) is one of the most prevalent mRNA modifications14,15,16. The m6Am site is located at the first transcribed nucleotide adjacent to the m7G cap in the 5′-UTR of mRNA17,18. Like m6A methylation, m6Am RNA modification represents an additional layer of reversible and dynamic gene regulation. Phosphorylated CTD interacting factor 1 (PCIF1) was recently identified as the sole known specific N6-methyltransferase for m6Am. RNA m6Am is written by PCIF1 and erased by fat-mass and obesity-associated protein (FTO) and is involved in mRNA stability, transcription, and translation19,20,21,22. PCIF1 interacts with C-terminal binding protein 2 (CTBP2) to facilitate m6Am deposition in mRNA23, suggesting the potentially important role of CTBP2 in PCIF1 control of m6Am. Therefore, PCIF1- or FTO-induced dysregulation of m6Am modification may produce physiological defects and participate in the pathological processes underlying various diseases24,25,26,27,28. However, it remains unknown whether and how m6Am has a critical role in neuropathic pain. Serpine1 mRNA-binding protein 1 (SERBP1) is recently discovered to have chaperone-like properties, and plays crucial role in mRNA stability29. Increasing evidence have linked SERBP1 to brain function and development, especially neurogenesis and synaptogenesis30,31,32. Yet, it is elusive whether SERBP1 is involved in neuropathic pain.
Here we demonstrate that peripheral nerve injury induces a selective upregulation of PCIF1 protein (but not FTO) within glutamatergic neurons of the hindlimb region of primary somatosensory cortex (S1HLGlu). This PCIF1 elevation is both necessary and sufficient for the induction and maintenance of neuropathic pain hypersensitivity. Crucially, we establish SERBP1 as an essential cofactor that collaborates with PCIF1 to mediate m6Am deposition on target mRNAs in S1HLGlu neurons under neuropathic pain-anxiety comorbidity conditions. The SERBP1-PCIF1 complex exerts its effects, at least partially, through adding m6Am modifications on maf1 homolog, negative regulator of RNA polymerase III (Maf1) mRNA. This epigenetic alteration leads to decreased MAF1 expression in S1HLGlu neurons. These findings suggest that the SERBP1-PCIF1 complex may serve as a promising therapeutic target for addressing the comorbidity of neuropathic pain and anxiety disorders.
Results
Peripheral nerve injury results in the increases in m6Am and PCIF1 in primary somatosensory cortex
To mimic clinical neuropathic pain in the patients, we conducted the spared nerve injury (SNI) model in mice. Consistent with previous studies33, after SNI surgery, mechanical allodynia as shown by an increase in paw withdrawal frequency (PWF) to 0.07 g and 0.4 g von Frey stimuli and heat hyperalgesia (Supplementary Fig. 1a–d). In addition, its comorbid anxiety-like behaviors occurred on day 21 after SNI demonstrated by spending less time in the central area in the open field (OFT) test (Supplementary Fig. 1e, f) and in the open arms with having fewer open-arm entries in the elevated plus maze (EPM) test (Supplementary Fig. 1h–j). As expected, locomotor function is intact in all SNI and sham mice (Supplementary Fig. 1g). These observations indicate that SNI successfully results in neuropathic pain and anxiety-like behaviors.
To determine the dynamics of mRNA modifications in the primary somatosensory cortex of the hind limb (S1HL) after peripheral nerve injury, we first examined the levels of m6A, m6Am, ac4C, m5C, and m7G in S1HL mRNA on day 7 (initial phase) and day 21 (maintenance phase) after SNI surgery. The LC–MS/MS assay showed that the levels of m6Am and m7G were significantly increased in S1HL on days 7 and 21 post-SNI, the level of m6A was also markedly decreased on day 21 after SNI (Fig. 1a). These observations suggest that m6Am and m6A in S1HL may be implicated in the genesis of neuropathic pain. To further examine how m6Am and m6A are changed in S1HL after SNI, we analyzed the expression of three RNA methyltransferases (PCIF1 for m6Am; METTL3 and METTL14 for m6A) and 2 RNA demethylases (FTO for m6Am; ALKBH5 and FTO for m6A) in the S1HL after surgery and found that only the level of PCIF1 was markedly increased in contralateral S1HL on day 7 or day 21 after SNI (Fig. 1b). This increase was also observed on days 3, 14, 28 and 35 after SNI (Fig. 1c), as well as was seen on day 14 in S1HL of SNI female mice (Supplementary Fig. 1k). However, levels of neither m6Am nor PCIF1 protein were altered in the ipsilateral S1HL (Supplementary Fig. 1l, m) or in the contralateral primary motor cortex (M1), which is adjacent to the S1HL (Supplementary Fig. 1n, o), post-SNI. Consistently, the amount of Pcif1 mRNA in the contralateral S1HL was increased from day 7 to 35 after SNI (Fig. 1d). Similar results were observed in another preclinical mouse model of chronic constriction injury (CCI)-induced neuropathic pain (Fig. 1e, f), as well as in the cisplatin injection-induced chemotherapy neuropathic pain model (Fig. 1g) and in the type 2 diabetes mellitus model caused by high-fat diet-induced obesity (Fig. 1h). Interestingly, the level of PCIF1 was not substantially changed in the contralateral S1HL in animal model of chronic inflammatory pain caused by plantar injection of complete Freund’s adjuvant (CFA) from 2 hour to day 7 post-CFA (Fig. 1i). No change in PCIF1 level in contralateral S1HL was also observed from 10 min to 24 h after formalin injection (Fig. 1j). In addition, the expression of S1HL PCIF1 was not significantly altered after the mice were subjected to 10 days of chronic restraint stress (CRS) (Fig. 1k), a well-established rodent model of anxiety34. Additionally, due to the concurrent elevation of both m7G and m6Am levels, we examined the expression of the m7G methyltransferase METTL1. The Western blot results revealed a significant increase in METTL1 expression in the contralateral S1HL following SNI (Supplementary Fig. 1p). Taken together, these data suggest that PCIF1 upregulation in S1HL is neuropathic pain-specific and that this upregulation may be responsible for m6Am increase in S1HL under neuropathic pain conditions.

a m5C, ac4C, m6Am, m7G, and m6A levels in contralateral primary somatosensory cortex (S1HL) on days 7 and 21 after surgery (n = 8 mice). b Expression level of METTL3, METTL14, PCIF1, ALKBH5, and FTO protein in the contralateral S1HL after surgery (n = 10 mice). c, e Expression of PCIF1 protein in contralateral S1HL at different times after spared nerve injury (SNI; n = 10 mice) or chronic constriction injury (CCI; n = 8 mice) or sham surgery. d, f Quantitative analysis of Pcif1 mRNA expression in contralateral S1HL by qRT-PCR at different times post-SNI or post-CCI or sham surgery (n = 8 mice). g–k Expression of PCIF1 protein in contralateral S1HL in a model of cisplatin-induced neuropathic pain (Cisp), diabetic neuropathic pain (DNP) or chronic restraint stress (CRS) (n = 8 mice). i, j Expression of PCIF1 protein in contralateral S1HL at different times after complete Freund’s adjuvant (CFA)injection (Saline group: n = 8 mice, CFA group: n = 10 mice) or formalin (FM) injection (Saline group: n = 12 mice, FM group: n = 10 mice). l, m Co-expression analysis of PCIF1 with neuron (NeuN), astrocyte (S100β) or microglia (Iba1) immunofluorescence staining in S1HL of naïve mice (n = 4 mice). Scale bar, 200 μm. n Schematic showing isolation of GABAergic neurons and glutamatergic neurons from the L2/3, L5, and L6 layers of S1HL of mice subjected to SNI surgery for quantitative analysis of Pcif1 mRNA expression. o, p Expression of Pcif1 mRNA in GABAergic or glutamatergic neurons of S1 layers 2-6. n = 12 single cells form mice. The data are represented as mean ± SEM. For RNA modifications, mRNA and protein, S1 samples from two mice were pooled to create one sample. One-way ANOVA followed with Dunnett’s post hoc test was used for panels (a, b). Two-way ANOVA followed with Fisher’s LSD post hoc test was used for panels (c–f, i, j, o and p). Student’s unpaired t-test (two-tailed) was used for panels (g, h and k).
Increased PCIF1 in S1 is restricted to layer 5 excitatory neurons
Next, we examined the distribution pattern of the upregulated PCIF1 in S1HL. Double immunofluorescent labeling assays showed that approximately 81.3% of PCIF1-labelled-cells were positive for NeuN (a marker specific to neurons) and 8.6% for S100β (a marker of astrocytes) (Fig. 1l, m). None of PCIF1-labelled cells were positive for Iba1 (a marker of microglia). It is evident that PCIF1 is expressed predominantly in neurons in S1HL. The neurons in layers 2/3, 5, and 6 of S1HL have different functions and different responses to nociceptive signals13,35,36,37. To determine which layers and neuronal types38 are involved in PCIF1 expression in neuropathic pain, we cross-bred the tdTomatofl/fl mice with CaMK2α-Cre mice or Vgat-Cre mice to label excitatory and inhibitory neurons, respectively, in the different layers of S1HL39,40 and then individually collected these neurons from specific layers for single-cell RT-PCR assay of Pcif1 mRNA 21 days after SNI or sham surgery (Fig. 1n; Supplementary Fig. 1q, r). Contralateral excitatory (glutamatergic) neurons from layer 5 of S1HL (S1L5Glu) exhibited a marked increase in the level of Pcif1 mRNA on day 21 after SNI surgery, whereases S1L2/3Glu and S1L6Glu neurons showed no change in Pcif1 mRNA expression after SNI surgery (Fig. 1o). The amount of Pcif1 mRNA was not altered in inhibitory neurons in layers 2/3, 5, or 6 (Fig. 1p). To further validate these findings, we employed laser capture microdissection41 and selectively isolated fluorescence-labeled excitatory and inhibitory neurons from layers 2/3, 5, and 6. Consistent with the observations from single-cell RT-PCR assay, Pcif1 mRNA was significantly increased only in S1L5Glu neurons (Supplementary Fig. 1t, u). These results indicate that SNI-induced increase of Pcif1 mRNA occurs specifically in the glutamatergic neurons of S1HL layer 5.
Blocking PCIF1 upregulation in S1HLGlu neurons attenuates the SNI-induced nociceptive hypersensitivity
Does the upregulated PCIF1 in S1HLGlu neurons participate in nerve injury-induced nociceptive hypersensitivity? To this end, we examined the effect of specifically blocking the PCIF1 upregulation in S1HLGlu neurons on the development and maintenance of the SNI-induced nociceptive hypersensitivity using CaMK2α promoter-driven AAV2/9-Pcif1-shRNA (shRNA) strategy (Fig. 2a). As expected, the SNI-induced increase of PCIF1 protein in S1HL from the control Scr-treated SNI mice was not seen in the shRNA-treated mice (Fig. 2b). Notably, SNI-induced mechanical allodynia and heat hyperalgesia were significantly blocked (Fig. 2c–e) or attenuated (Fig. 2f–h) in the shRNA-microinjected mice. Basal responses to mechanical and thermal stimuli on the contralateral side of SNI mice and on either side of the Sham mice were not affected by microinjection of the virus (Supplementary Fig. 2b–g).

a Viral injection schematic and Pcif1-shRNA vector design. b S1HL PCIF1 levels significantly decreased 35 days post AAV-CaMK2α-shPcif1 in SNI mice (n = 16 mice). c–e Pre-injection of AAV-CaMK2α-shPcif1 into contralateral S1HL prevented SNI-induced mechanical allodynia (c, d) and thermal hyperalgesia (e) (n = 10 mice). f–h Post-injection of shPcif1 reversed established contralateral SNI-induced mechanical allodynia (f, g) and thermal hyperalgesia (h) (n = 8 mice). i Experimental timeline in Pcif1fl/fl mice. j Schematic and images showing reduced PCIF1 in contralateral S1HLGlu neurons after AAV-CaMK2α-Cre injection in Pcif1fl/fl mice. Scale bar, 20 µm. k, l Pre-injection of AAV-CaMK2α-Cre reduced ipsilateral S1HL PCIF1 (k, n = 12 mice) and m6Am (l, n = 8 mice) levels post-SNI. m–o Pre-injection of AAV-CaMK2α-Cre prevented SNI-induced mechanical hypersensitivity (m, n) and thermal hyperalgesia (o) (n = 10 mice). p Left: Fiber photometry setup for Ca2+ imaging in awake Pcif1fl/fl mice (drawn by figdraw.com). Right: image shows AAV-CaMK2α-GCaMP6s (Green)/AAV-CaMK2α-mCherry (Red). Scale bar, 500 µm. Heatmaps (q), average Ca2+ transients (r) and AUC quantification of the GCaMP6s signal (3-7 seconds, s) from S1HLGlu neurons of Pcif1fl/fl mice receiving 0.4 g von Frey stimulation (n = 24 trails from 8 mice). t Patch-clamp schematic in S1HLGlu neurons. u–x Sample traces (u), statistical data (v), and rheobase of the spike (w) for action potential firing and the membrane potential (x) recorded from S1HLGlu neurons (n = 13 cells from 4 mice). e PWF: paw withdrawal frequencies; PWL: Paw withdrawal latencies. OFT: open filed test; EPM: elevated plus maze; Gs: GCaMP6s. The data are represented as mean ± SEM. S1 samples from two mice were pooled to create one sample (b, l, k). One-way ANOVA followed with Tukey’s post hoc test was used for panels (b, k, l, s and v–x). Two-way ANOVA followed with Tukey’s post hoc test was used for panels (c–h and m–o).
Given that shRNA may have potential off-target effects, we generated the Pcif1fl/fl mice (Supplementary Fig. 2h, i), in which the endogenous Pcif1 mRNA in S1HLGlu neurons could be knocked down under the control of the CaMK2α promoter in the presence of Cre recombinase. We found that microinjection of AAV-CaMK2α-Cre (AAV-Cre), not control AAV-mCherry, into the contralateral S1HL 21 days before surgery dramatically reduced the expression of PCIF1 in contralateral (not ipsilateral) S1HLGlu neurons of Pcif1fl/fl mice 15 days post-surgery (Fig. 2l–k). Similarly, the SNI-induced elevation of m6Am was also attenuated in the S1HL on day 16 post-surgery in the Pcif1fl/fl mice pre-microinjected with AAV-Cre (Fig. 2l). This pre-microinjection also mitigated the SNI-induced pain and anxiety-like behaviors from days 3 to 14 after surgery (Fig. 2m–o; Supplementary Fig. 2j–r).
Given the broad expression of CaMK2α in cortical pyramidal neurons, we utilized Rbp4 (specifically localized to cortical layer 5 excitatory neurons)42 promoter -driven AAV2/9-Rbp4-Pcif1-shRNA (shRNA) to specifically knock down PCIF1 in the excitatory neurons of S1L5. As expected, the SNI-induced increase of PCIF1 in S1HL was reversed by microinjection of AAV2/9-Rbp4-Pcif1-shRNA on day 21 after treatment, this reversal resulted in the attenuation of nerve injury-induced mechanical allodynia and thermal hyperalgesia (Supplementary Fig. 3a–h). In contrast, microinjection of AAV2/9-Rbp4-Pcif1-shRNA in the motor cortex (M1) slightly decreased PCIF1 level on the contralateral side in both sham and SNI mice, but did not affect the paw withdrawal response to mechanical or thermal stimuli in these animals (Supplementary Fig. 3i–p).
Blocking PCIF1 upregulation in S1HLGlu neurons inhibits the SNI-induced increase in neuronal activity
To examine whether PCIF1 affects the activity of S1HLGlu neurons, we intracranially pre-injected AAV-CaMK2α-GCaMP6s with AAV-Cre or control AAV-eGFP into the S1HL of Pcif1fl/fl mice 3 weeks before SNI surgery. We then recorded the GCaMP6s signal, an indicator of Ca2+ signaling intensity, from S1HLGlu neurons upon application of a strong mechanical stimulus (0.4 g von Frey filament) to the surgery-side hind paw in the awake mice (Fig. 2p). Mechanical stimulation elicited a significant increase in GCaMP6s signal in S1HLGlu neurons of Pcif1fl/fl mice 7 days after SNI in the control eGFP-injected mice. This increase was ameliorated by injection of Cre in S1HLGlu neurons of Pcif1fl/fl mice (Fig. 2q–s). Given that the magnitude of the increase in firing rate upon stimulation represents the capacity of the neuronal population in response to input signals, we further examined the activity of S1HLGlu neurons with/without Pcif1 via whole-cell current clamp recordings in S1HL brain slices obtained 7 days after SNI surgery (Fig. 2t). Consistent with the GCaMP6s fiber photometry results, S1HLGlu neurons in the slices from SNI mice exhibited higher evoked firing rates than those in slices from Sham mice after injection of depolarizing currents (500 ms, 0–200 pA) (Fig. 2u, v). However, no differences were observed in the rheobase of spike and resting membrane potential of S1HLGlu neurons between Sham and SNI mice (Fig. 2w, x). Pcif1 depletion in S1HLGlu neurons via intracranial pre-injection of AAV-Cre in Pcif1fl/fl mice blocked the nerve-injury-induced increase in firing rate in the contralateral S1HL neurons, compared with control injection (Fig. 2v). These results suggest that Pcif1 is required for the activation of S1HLGlu neurons under neuropathic pain conditions.
Mimicking nerve injury-induced PCIF1 upregulation leads to nociceptive hypersensitivity
To explore whether PCIF1 upregulation in S1HLGlu is sufficient for the initiation of neuropathic pain, we intracranially injected AAV2/9-EF1a-DIO-Pcif1 (DIO-Pcif1) into unilateral S1HL of naïve CaMK2α-Cre mice (Fig. 3a, b; Supplementary Fig. 4a). As expected, on day 21 after DIO-Pcif1 injection, the levels of PCIF1 and m6Am were increased by 1.51-flod and 1.512-fold, respectively, compared those after DIO-mCherry (as a control) injection in S1HL (Fig. 3c, d). More importantly, the mice injected with DIO-Pcif1, but not control DIO-mCherry, exhibited the enhanced responses to both mechanical and heat stimuli on the side contralateral to the injection (Fig. 3e–g) and the production of anxiety-like behaviors (Supplementary Fig. 4b–g). Locomotor function is normal in all injected mice (Supplementary Fig. 4c). Spontaneous pain was also observed on day 21 after DIO-Pcif1 injection, as demonstrated by a preference for the chamber paired with lidocaine in the conditioned place preference task (Fig. 3h). These nociceptive-like responses were not seen on the ipsilateral side after injection of DIO-Pcif1 or DIO-mCherry (Supplementary Fig. 4h–j). To further validate this effect, we unilaterally injected AAV-Rbp4-Pcif1 to specifically upregulate PCIF1 in the S1L5Glu neurons of naïve mice (Supplementary Fig. 4k). Overexpression of PCIF1 in S1L5 Rbp4+ neurons significantly led to the development of nociception-like behavior (Supplementary Fig. 4m–r). In contrast, the upregulation of PCIF1 through the injection of AAV2/9-EF1α-DIO-Pcif1 (DIO-Pcif1) in the M1 of CaMK2α-Cre mice did not produce enhanced nociceptive responses to mechanical or thermal stimuli (Supplementary Fig. 4s–z). Taken together, these data suggest that upregulation of PCIF1 in S1HLGlu neurons induces neuropathic pain–like symptoms.

a Timeline for virus injection and behavior tests in CaMK2α-Cre mice. b Schematic of the experimental paradigm (left) and a representative image showing mCherry expression in S1HL after injection of AAV-DIO-Pcif1 or AAV-DIO-mCherry into CaMK2α-Cre mice (right). Scale bar, 500 µm (left). PCIF1 (c) and m6Am (d) levels significantly increased 28 days post AAV-DIO-Pcif1 (vs. mCherry) in CaMK2α-Cre mice (n = 8 mice). PCIF1 upregulation in S1HLGlu neurons induced contralateral mechanical allodynia (e, f), thermal hyperalgesia (g), as measured by conditioned place preference (h) (e–g: n = 8 mice, h: n = 7 mice). i Schematic (drawn by figdraw.com) and image of GCaMP6s recording setup in awake mice (scale bar: 500 µm). Scale bar, 500 µm. Heatmaps (j), average Ca2+ transients (k), and AUC quantification of the GCaMP6s signal (l) from S1HLGlu neurons of CaMK2α-Cre mice receiving 0.4 g von Frey stimulation (n = 24 trails from 8 mice). m Patch-clamp schematic in S1HLGlu neurons. Sample traces (n), statistical data (o), and rheobase of the spike (p) for action potential firings and the membrane potential (q) recorded from S1HLGlu neurons in mice with upregulated PCIF1 expression, as described for (c, d) above (DIO-mCherry group: n = 14 cells from 4 mice, DIO-Pcif1 group: n = 13 cells from 4 mice). The data are represented as mean ± SEM. S1 samples from two mice were pooled to create one sample (c, d). Student’s unpaired t-test (two-tailed) was used for (c, d, h, p and q). Two-way ANOVA followed with Bonferroni’s post hoc test was used for (e–g and o). One-way ANOVA followed with Bonferroni’s post hoc test was used for (l).
PCIF1 upregulation enhances the activity of S1HLGlu neuron
We next sought to determine if PCIF1 upregulation in S1HLGlu neurons could also increase S1HL neuronal activity through measuring GCaMP6s signal (Fig. 3i). We co-injected DIO-GCaMP6s and DIO-Pcif1 (or DIO-mCherry) AAV virus into S1HL of naïve CaMK2α-Cre mice 21 days before the experiment. Stimulation with a 0.4 g von Frey filament induced a significantly higher GCaMP6s signal in S1HL from the CaMK2α-Cre mice injected with DIO-Pcif1, compared with the CaMK2α-Cre mice injected with DIO-mCherry (Fig. 3j–l). In patch clamp studies, further research has revealed that S1HLGlu neurons in slices from DIO-Pcif1-treated mice displayed a significant higher evoked firing rate than S1HLGlu neurons from DIO-mCherry-injected mice (Fig. 3m–o). No differences in the rheobase of spike and resting membrane potential of S1HLGlu neurons were seen between these two groups (Fig. 3p–q). Thus, PCIF1 upregulation increases the activation of S1HLGlu neurons.
PCIF1 interacts with SERBP1 to catalyze m6Am modification
How does PCIF1 upregulation in S1HL contributes to neuropathic pain? Given that the catalytic activity of epigenetic enzymes often requires the contribution of binding proteins43, we searched the binding partners of PCIF1 through performing an absolute quantitative LC–MS/MS analysis of the proteins pulled down by endogenous PCIF1 protein, using its specific antibody in human HEK293T and mouse HT22 cells. The approximately 165 and 126 common candidate proteins, respectively, from HEK293T and HT22 cells, were achieved (Supplementary Data 2). Among the candidate proteins, SERBP1 displayed high enrichment in both cells (Fig. 4a; Supplementary Data 2). Although SERBP1 expression were not changed in S1HL days 7 after SNI surgery (Fig. 4b), SERBP1 binding to PCIF1 was increased by 1.49-fold. This increase could be blocked by Pcif1 knockdown using its shRNA. Immunofluorescent staining showed the predominant localization of SERBP1 in the neurons, but not in astrocytes or microglia, in S1HL (Supplementary Fig. 5a, b).

a The SERBP1 peptide identified by an MS analysis after an anti-PCIF1 co-immunoprecipitation assay in HEK293T cells. b Co-immunoprecipitation (IP) analysis of SERBP1 binding to PCIF1, using anti-PCIF1. Tissue was collected from the contralateral S1HL on day 21 after SNI or sham surgery in wild-type mouse premicroinjected with AAV-CaMK2α-shPcif1 (to knock down Pcif1 in glutamatergic cells) or a scrambled control. Input, the purified protein control. IB: immunoblotting (n = 10 mice). S1 samples from two mice were pooled to create one sample. c Construction schematic for the mammalian two-hybrid system; the luciferase reporter, with full-length Serbp1 and Gal4, and the VP16 transcription factor with a PCIF1 domain. The binding activity of SERBP1 and different PCIF1 domains (N-WW, d; helical, e; MTase, f; C-terminal, g) at 48 h after their co-transfection into HEK293T cells (n = 3 dishes of cells). h m6Am levels in HT22 cells after 48-hour transfection using: Vehicle (RNase-free water), SERBP1 plasmid with Control (empty vector), SERBP1 with Δ-MTase, SERBP1 with Δ-Helix, or SERBP1 with PCIF1 (n = 4 dishes of cells). i Schematic of CRY2/CIBN optogenetic regulation of the m6Am level through control of PCIF1 and SERBP1 binding activity. Serbp1 and Pcif1 were fused to the Cry2 and Cibn vectors, respectively. j, k Diagram showing Serbp1-Cry2 and Pcif1-Cibn co-transfection and activation of their interaction by blue light (j), and the quantitation of m6Am levels (k) (n = 3 dishes of cells). PBS, transfection of PBS control. The data are represented as mean ± SEM. One-way ANOVA followed with Tukey’s post hoc test was used for (b). One-way ANOVA followed with Dunnett’s post hoc test was used for (d–h and k).
To identify which domain of PCIF1 could interact with SERBP1, we first carried out an in vitro mammalian two-hybridization experiment via constructing report system (Fig. 4c). The reporter was significantly activated after co-transfection of pBIND-Serbp1 with the pACT plasmid containing PCIF1 helical domain or methyltransferase (MTase) domains, but not N terminal domains in HEK293T (Fig. 4d–g; Supplementary Fig. 5c). Although the reporter of the C-terminal domain is activated, the intensity is much smaller than that of the helical domain or MTase domain. This suggests that SERBP1 could interact with the helical and MTase domains of PCIF1.
Next, to investigate whether the interaction between SERBP1 and PCIF1 is essential for m6Am formation. First, we constructed two PCIF1 protein mutants — one lacking the MTase enzyme domain (pCMV-HA-Δ-MTase) and another lacking the Helical domain (pCMV-HA-Δ-Helix). Co-transfection of the pCMV-HA-Δ-MTase plasmid with the pCMV-Serbp1 plasmid resulted in a 3% increase in m6Am levels compared to the co-transfection of the control plasmid with the pCMV-Serbp1 in HT22 cells. In contrast, co-transfection of pCMV-HA-Δ-Helix with pCMV-Serbp1 resulted in a 33% increase (Fig. 4h). These findings indicate that the MTase enzyme domain is required for depositing m6Am in RNA. Moreover, Transfection with pCMV-PCIF1 (CDS full-length expression plasmid) and pCMV-Serbp1 increased m6Am levels by 49% compared to the same controls (Fig. 4h). Based on these findings, we conducted in vitro m6Am catalytic assays to validate the regulatory effects of the SERBP1-PCIF1 complex on RNA m6Am modification. Experimental data revealed that incubation of mRNA with purified SERBP1 protein alone did not alter m6Am levels. Strikingly, co-incubation of SERBP1 with purified MTase protein induced a 37% elevation in m6Am level compared to the MTase controls (Supplementary Fig. 5d). These findings suggest that, while SERBP1 is not an essential factor for PCIF1-mediated m6Am methylation catalysis, it significantly enhances the catalytic efficiency of PCIF1 in RNA m6Am modification. To obtain the desired mutant amino acids in SERBP1, we evaluated which amino acids within SERBP1 is crucial for the formation of the SERBP1-PCIF complex. Given that the structure of SERBP1 is unresolved, we combined AlphaFold3 (https://alphafoldserver.com) modeling44 and alanine-scanning mutagenesis45 to predict its interaction with PCIF1 and identify potential functional amino acids involved in binding. The prediction results revealed four SERBP1 binding sites on PCIF1, located at amino acid positions 15 (Asn, N), 16 (Arg, R), 21 (Phe, F), and 343 (Asp, D) (the N-terminal amino acid of SERBP1 designated as +1). (Supplementary Fig. 5e, f; Supplementary Table 1). Moreover, to assess the role of these candidate SERBP1 amino acids, we mutated each to alanine and generated individual mutant expression plasmids. LC-MS assay results showed that mutation at position 16 significantly decreased m6Am levels following co-transfection with a Pcif1 expression vector, compared to the wild-type Serbp1 plasmid (Supplementary Fig. 5g). In contrast, no significant changes in m6Am levels were observed after co-transfection with mutants at positions 15, 21, or 343 and the Pcif1 expression vector (Supplementary Fig. 5g). These findings indicate that the arginine (R) at position 16 of SERBP1 is essential for its interaction with PCIF1. Together, these findings indicate that the interaction between SERBP1 and PCIF1 is essential for m6Am formation.
To further confirm the role of interaction of SERBP1 with PCIF1 in catalyze m6Am, we employed the CRY2/CIBN optogenetic system, an excellent tool for detecting molecular interactions46. SERBP1 and PCIF1 were fused onto the photosensitive proteins CRY2 and CIBN, respectively (Fig. 4j). We hypothesized that the binding of SERBP1 to PCIF1 would be promoted when blue light activated the dimerization of CRY2 and CIBN. As expected, LC–MS showed that, after the application of blue light to HT22 cells co-transfected with Pcif1-Cibn and Serbp1-Cry2 for 48 h, the m6Am level was increased by 1.65-fold (Fig. 4k). Moreover, acute blue light increased the level of S1HL m6Am in naïve mice co-pre-injected with Pcif1-Cibn and Serbp1-Cry2 (Fig. 5a, b). These mice exhibited the enhanced responses to mechanical and heat stimuli on the contralateral side (Fig. 5c–e). Pre-treatment with their controls did not alter basal PWT or PWL on the ipsilateral side (Supplementary Fig. 5h–j). Together, our in vitro and in vivo findings suggest that the interaction between SERBP1 and PCIF1 facilitates m6Am deposition in RNA.

a Schematic of virus injections and optical fiber implantation for activation of the optogenetic CRY2/CIBN system. b m6Am levels at 10 min after 1-h administration of blue light in wild-type mice injected 5 days previously in S1HL with Lenti-Pcif1-Cibn (Pci-Cibn) and Lenti-Serbp1-Cry2 (Ser-Cry2) or their controls Lenti-Cibn (Ctrl-Cibn) and Lenti-Cry2 (Ctrl-Cry2) (n = 10 mice). Pre, Pci-Cibn/Ser-Cry2-injected mice without blue light; Post, Pci-Cibn/Ser-Cry2-injected mice after 1-h blue light. Paw-withdrawal frequencies (PWF) to 0.07 g (c) and 0.4 g (d) von Frey filaments and paw-withdrawal latencies (PWL) to heat stimuli (e) on the contralateral side of mice injected as described in B. Behavioral tests were conducted 10 min after the end of 1 h blue light administration (n = 11 mice). f, g The level of PCIF1 protein (f) and m6Am (g) on day 7 after microinjection of AAV-DIO-shSerbp1 (shSerbp1) or AAV-DIO-scrambled-shRNA (Scr) in S1HL of CaMK2α-Cre mice preinjected with AAV-DIO-Pcif1 (Pcif1) or AAV-DIO-mCherry (mCherry) (F: n = 12 mice, G: n = 10 mice). Effect of SERBP1 downregulation on the contralateral mechanical (h, i) and heat (j) hypersensitivities induced by PCIF1 upregulation by DIO-Pcif1 (n = 10 mice). Red arrows, DIO-Pcif1 or DIO-mCherry injection. Blue arrows, DIO-shSerbp1 or DIO-mCherry injection. The data are represented as mean ± SEM. S1 samples from two mice were pooled to create one sample (b, f and g). Student’s unpaired t-test (two-tailed) was used for (b). Student’s paired t-test (two-tailed) was used for (c–e). One-way ANOVA followed with Tukey’s post hoc test was used for (f and g). Two-way ANOVA followed with Tukey’s post hoc test was used for (h–j).
To address whether SERBP1 participates in neuropathic pain and anxiety, we used DIO-Pcif1 and DIO-shSerbp1 AAV virus to respectively upregulate the expression of PCIF1 and downregulate SERBP1 in CaMK2α-Cre mice (Fig. 5f). The LC–MS results revealed a strong decline in m6Am in S1HL mRNAs on day 21 after co-injection of DIO-Pcif1 and DIO-shSerbp1 (Fig. 5g), suggesting that knockdown of SERBP1 blocked the increase in m6Am even under the conditions of PCIF1 increase. SERBP1 knockdown in S1HL also ameliorated the contralateral heat and mechanical hypersensitivities induced by S1HL microinjection of DIO-Pcif1 (Fig. 5h–j). As expected, basal PWT and PWL responses were not changed on the ipsilateral side in these mice (Supplementary Fig. 5k–m). Additionally, SERBP1 knockdown in the L5 layer blocked anxiety-like behaviors induced by PCIF1 protein upregulation (Supplementary Fig. 5n–s). Our data strongly support the hypothesis that the contribution of PCIF1 in S1HL to neuropathic pain requires, at least in part, for its binding to SERBP1. Our findings suggest that SERBP1 regulates m6Am methyltransferase activity in vivo.
PCIF1 upregulation is responsible for gain of m6Am in Maf1 mRNA in S1HL after nerve injury
Next, we explored how PCIF1 upregulation in S1HLGlu neurons contributed to neuropathic pain and comorbid anxiety-like behavior. Considering that PCIF1 acts as a “writer” that adds m6Am to the cap site of mRNA, thereby negatively regulating protein translation through its interaction with SERBP1. Therefore, we conducted m6Am mRNA immunoprecipitation sequencing (m6Am-Exo-Seq, Supplementary Fig. 6a) in conjunction with ribosome-nascent chain complex-bound mRNA sequencing (RNC-Seq) and SERBP1 RNA immunoprecipitation sequencing (SERBP1-RIP-Seq) in the S1HL on day 21 post-SNI to identify the downstream targets of PCIF1 (Fig. 6a). About 16,068 genes were identified with m6Am modifications through m6Am-Exo-Seq in sham, SNI, and shPcif1 groups (Supplementary Fig. 6b). The unbiased m6Am-Exo-Seq profiling revealed that m6Am was significantly enriched in the 5’ UTR regions of mRNAs from sham, SNI, and shPcif1 mice (Fig. 6b). Additionally, the genomic sequences surrounding m6Am peaks exhibited a conserved XCA motif pattern (A = m6Am; X = C, G or T) (Fig. 6c), which is consistent with previous reports47. The SNI-induced increased m6Am within approximately 3,195 genes was blocked by PCIF1 knockdown (Supplementary Fig. 6c), among which, the translation levels of about 436 genes were reduced and their binding activity to SERBP1 increased after nerve injury (Fig. 6d). These 436 genes may be associated with synaptic organization processes (Fig. 6e). In particular, MAF1, a GABAAR-interacting protein involving in synapse organization, was ranked in top 10 of these 436 genes, and was one of dramatically decreased genes (Fig. 6f, g). Therefore, MAF1 was selected as a candidate target of PCIF1. The SERBP1-RIP-Seq results revealed that SERBP1 bound the mRNAs of about 18,316 genes in the neurons of the sham group, whereas it bound the mRNAs of about 17,773 genes in the SNI group. Further analysis indicated that there were 2238 differential genes between two groups, including Maf1 mRNA (Supplementary Fig. 6d). GO analysis indicates that these differentially genes, including 3195 m6Am-modified genes and 2238 genes bound by SERBP1, exhibit similar enrichment patterns (Supplementary Fig. 6e, f).

a Flowchart of the screening for downstream targets of PCIF1 by m6Am-Seq and RNC-Seq. SNI+shPcif1 indicates PCIF1 downregulation in S1HL of SNI mice via injection of AAV-shPcif1. b Distribution of m6Am peaks across mRNA segments of S1HL from three groups. c Motif analysis of m6Am peaks of mRNAs’ genomic sequences. The minus sign in X axis, upstream genomic nucleotides. d Venn diagram analysis revealed 436 genes consistently regulated by PCIF1 and SERBP1, based on integrated analysis of three complementary datasets: (i) genes with SNI-induced m6Am increases that were reversed by PCIF1 knockdown (m6Am-Exo-Seq), (ii) genes with increased SERBP1 binding post-SNI (SERBP1-RIP-Seq), and (iii) genes that exhibited reduced translation efficiency following SNI (RNC-Seq). e Gene ontology (GO) analysis of biological processes for the 436 genes. f Heatmap visualization of p-values and fold changes for the 436 genes. g Top 10 genes with most significance among 436 genes. h The representative image of m6Am peaks on Maf1 gene in S1HL. i Maf1 m6Am in S1HL on day 14 post-SNI measured via RNA immunoprecipitation (RIP)-PCR with four PCR primer pairs. The first two pairs include the RNA cap site (n = 8 mice). The forward F, and reverse R arrows represent paired PCR primers. j, k Maf1 m6Am (j) and the binding level of PCIF1 to Maf1 mRNA (k) in S1HL of SNI or Sham Pcif1fl/fl mice after preinjection of AAV-CaMK2α-Cre (Cre) or AAV-CaMK2α-mCherry (Ctrl) into S1HL (n = 10 mice). Maf1 m6Am level (l) and PCIF1 binding to Maf1 mRNA (m) on day 21 after injection of AAV-DIO-Pcif1 or AAV-DIO-mCherry into S1HL of naïve CaMK2α-Cre mice (n = 10 mice). For (i–m), data are represented as mean ± SEM. S1 samples from two mice were pooled to create one sample (j–m). Student’s unpaired t-test (two-tailed) was used for panels (i, j, k). One-way ANOVA followed with Tukey’s post hoc test was used for (l and m).
Consistent with the previous report21, the adenosine (A) at the start site of Maf1 mRNA was methylated with m6Am, and bioinformatics analysis confirmed that Maf1 mRNA contained a high confident, PCIF1-dependent m6Am site (Fig. 6h). To confirm the Maf1 m6Am level from m6Am-Exo-Seq, four pairs of primers were designed: the first two pairs (F1R1 and F1R2) covering the 5′UTR region including m6Am site, and the last two pairs (F3R3 and F4R4) covering the CDS and 3′UTR regions (Fig. 6i). We found that F1R1, F1R2, and F3R3 fragments were amplified from the immunoprecipitated m6Am complexes in S1HL tissue lysates. The levels of F1R1 and F1R2 fragments were increased on day 14 after SNI compared with the sham group, where the level of F3R3 fragment was unchanged (Fig. 6i). These observations are consistent with the sequencing results and demonstrates that nerve injury increases the m6Am level of Maf1 in S1HL. We then examined whether this m6Am gain in Maf1 mRNA after SNI is caused by PCIF1 upregulation in S1HL neurons. The SNI-induced m6Am increase could be blocked by Pcif1 knockout in S1HLGlu neurons (Fig. 6j). The PCIF1-RIP assay revealed that the basal binding between the Maf1 mRNA fragment and PCIF1 was rather weak in sham S1HL. In contrast, there was a striking elevation in the binding activity in S1HL on day 14 after SNI. This elevation was significantly prevented by specific Pcif1 knockout in S1HLGlu neurons (Fig. 6k). Moreover, PCIF1 overexpression in naïve mice produced an increase in m6Am sites in the Maf1 mRNA, as evidenced by a 2.09-fold increase in immunoprecipitative activity using the anti-m6Am antibody, compared with the control groups (Fig. 6l). Similarly, the binding of PCIF1 to Maf1 mRNA was markedly enhanced on day 28 after AAV injection (Fig. 6m). These findings suggest that the SNI-induced gain of m6Am in Maf1 mRNA can be attributed to the SNI-induced increase in PCIF1 in S1HLGlu neurons.
Furthermore, to assess the effect of SERBP1 on Maf1 m6Am levels, we assessed the changes in Maf1 m6Am following SERBP1 knockdown. Similar to PCIF1, the nerve injury-induced increase in m6Am levels of Maf1 was inhibited by SERBP1 knockdown using its shRNA in S1HLGlu neurons (Supplementary Fig. 6g). Conversely, overexpression of SERBP1 in S1HLGlu neurons of naïve mice resulted in an increase in Maf1 m6Am levels (Supplementary Fig. 6h). These results suggest that SERBP1 determines the level of m6Am in Maf1 mRNA.
Increased PCIF1 inhibits MAF1 expression in S1HL after SNI
To determine whether PCIF1 inhibits MAF1 expression in S1HL after SNI, we examined the effect of blocking the nerve-injury induced PCIF1 increase on MAF1 expression. Specific upregulation of Pcif1 in S1HLGlu neurons in naïve mice by injection of DIO-Pcif1 virus in CaMK2α-Cre mice decreased the level of MAF1 protein in the S1HL on day 21 after virus injection (Fig. 7a). Moreover, pre-injection of Cre, but not control mCherry AAV virus, prevented the SNI-induced decrease in MAF1 protein in the S1HL of Pcif1fl/fl mice on day 21 post-SNI (Fig. 7b). Double-labeled immunostaining revealed that approximately 83.5% of MAF1+ cells in S1HL were co-labeled by NeuN and 5.3% of MAF1+ cells were co-labeled with S100β (Fig. 7c). A subcellular analysis showed that most of the MAF1 staining located in the S1HL neurons (Fig. 7d). Our single-cell RT-PCR assay showed co-expression of Pcif1 mRNA, Maf1 mRNA, and Serbp1 mRNA in 7 of 8 S1HLGlu neurons (Fig. 7e).

a MAF1 protein levels on day 21 after injection of AAV-DIO-Pcif1 (AAV-Pcif1; to induce PCIF1 expression) or AAV-DIO-mCherry (DIO-mCherry) into S1HL of naïve CaMK2α-Cre mice (n = 10 mice). b The level of MAF1 protein in SNI or Sham Pcif1fl/fl mice preinjected with AAV-CaMK2α-Cre (CaMK2α-Cre; to knock out PCIF1) or AAV-CaMK2α-mCherry (CaMK2α-mCherry) into S1HL. Tissue was harvested 21 days after injection (n = 12 mice). c, d Co-expression analysis of MAF1 (red) with NeuN (a neuronal marker, cyan), S100β (an astrocyte marker, cyan), or Iba1 (a microglia marker, cyan) immunofluorescence staining in the S1HL of naïve mice. n = 4 mice. Scale bar, 200 μm. e Co-expression analysis of Maf1 with Serbp1 and Pcif1 in CaMK2α neurons by single-cell PCR. Numbers 1-8 represent eight individual neurons. M, DNA marker. f CRISPR-dCasRx/PCIF1 “writing” m6Am to the given site in Maf1 mRNA. gRNA, small guide RNA. g Identification of dCasRx-Pcif1 fusion protein expression on day 5 after microinjection of CRISPR-dCasRx-Pcif1 into S1HL. h, i The Maf1 m6Am (h) and MAF1 protein (i) levels on day 5 after co-microinjection of CRISPR-dCasRx-Pcif1 and gRNA-26 or gRNA-60 into S1HL in naïve mice (n = 10 mice). For (a, b and h, i), data are represented as mean ± SEM. S1 samples from two mice were pooled to create one sample (a, b, h and i). Student’s unpaired t-test (two-tailed) was used for (a). One-way ANOVA followed with Tukey’s post hoc test was used for (b, h and i).
Next, to further confirm the specific regulatory role of Maf1 mRNA m6Am in MAF1 protein expression by PCIF1, we carried out the CRISPR gene-editing system48. PCIF1 was fused with inactivated dCasRx protein (dCasRx/PCIF1 fusion protein) to specifically “write” m6Am to the start site “A” in Maf1 mRNA via guide RNA (gRNA; Fig. 7f). The dCasRx/PCIF1 fusion protein was detectable on day 5 after injection of lentivirus CRISPR-dCasRx-Pcif1 in naïve mice (Fig. 7g). Two gRNAs including gRNA-26 (26 to 44, the first nucleotide in the mRNA designated as +1) and gRNA-60 (60 to 79) located near the 5′UTR region were designed as previously described38,49. The m6Am level of Maf1 was increased by 7.03-fold and 4.87-fold compared to those in the control Scr group on day 5 after co-injection of CRISPR-dCasRx-Pcif1 and gRNA-26 or gRNA-60, respectively (Fig. 7h). Correspondingly, this co-injection significantly reduced the level of MAF1 expression in S1HL by 36% for gRNA-26 and 38% for gRNA-60 on day 5 after injection (Fig. 7i). To further confirm the specificity of CRISPR-dCasRx-Pcif1 with gRNA-26 for Maf1 mRNA, we tested its effects on Akt and Pik3r1 mRNA. These two genes, identified in our m6Am-Exo-Seq profiles as carrying m6Am modifications, also function as known regulators of Maf150,51. We found no changes in the m6Am levels of Akt or Pik3r1 mRNA, nor in their mRNA levels in S1, after the microinjection of CRISPR-dCasRx-Pcif1 and gRNA-26 (Supplementary Fig. 6i). In contrast, only Maf1 mRNA showed a significant increase in m6Am following the dCasRx-Pcif1/g26 treatment. Collectively, these results demonstrate that nerve-injury-induced MAF1 downregulation in S1HL results from m6Am elevation via PCIF1.
MAF1 mediates the PCIF1-induced increases in neuropathic pain
We then asked whether Pcif1-induced pain and anxiety-like behavior are due to Maf1 reduction. We first determined whether Maf1 in S1HLGlu neurons contributes to the neuropathic pain- and anxiety-like behavior. Intracranial injection of DIO-shMaf1 virus decreased the level of MAF1 protein in the S1HL of naïve CaMK2α-Cre mice day 28 post-injection (Fig. 8a). This shRNA-caused knockdown of Maf1 led to hypersensitivity to mechanical and thermal stimuli on the contralateral side (Fig. 8b–d), but not on the ipsilateral side (Supplementary Fig. 7a–c). Additionally, this knockdown also produced the anxiety-like behavior (Supplementary Fig. 7d-i). Moreover, m6Am increase-caused MAF1 downregulation via injection of CRISPR-dCasRx/Pcif1 and gRNA-26 (using same strategy described in Fig. 7f) led to the enhanced behavioral responses (Fig. 8e–g; Supplementary Fig. 7j–r), similar to shRNA effect described above. Injection of DIO-Maf1 virus rescued the SNI-induced decrease in MAF1 in S1HL (Fig. 8h). As expected, this specific rescue ameliorated the SNI-induced nociceptive hypersensitivity (Fig. 8i–k) and anxiety-like responses (Supplementary Fig. 7s–x) on the contralateral side, without affecting basal paw responses on the ipsilateral side of the SNI mice and on either side of the sham mice (Supplementary Fig. 7y–aa). These findings suggest that Maf1 is responsible for the genesis of neuropathic pain and anxiety behavior after SNI.

a Reduced S1HL MAF1 protein 21 days post AAV-DIO-shMaf1 injection in CaMK2α-Cre mice (n = 8 mice). Knockdown of S1HL Maf1 or Co-injection of CRISPR-dCasRx-Pcif1 + gRNA-26/60 induced contralateral mechanical allodynia (b, c, e, f) and thermal hyperalgesia (d, g) (n = 10 mice). h MAF1 levels increased after injection of AAV-DIO-MAF1 into CaMK2α-Cre mice (n = 12 mice). i–k Overexpressing MAF1 in contralateral S1HL attenuated SNI-induced mechanical allodynia (i, j) and thermal hyperalgesia (k) (n = 10 mice). l S1HL PCIF1/MAF1 levels after co-injection of AAV-DIO-Pcif1 ± AAV-DIO-Maf1 in naïve CaMK2α-Cre mice (n = 8 mice). m–o MAF1 overexpression blocked PCIF1-induced pain-like behaviors (n = 10 mice). p Schematic (left, drawn by figdraw.com) and GCaMP6s expression image (right) for fiber photometry. Heatmaps (q), average Ca2+ transients (r), and AUC quantification (s) of GCaMP6s signals recorded from S1HLGlu neurons as mice received 0.4 g von Frey stimulation (n = 24 trails from 8 mice). Scale bar, 200 µm. t Schematic of patch-clamp recordings from labeled S1HLGlu neurons in slices. Sample traces (u), statistical data (v), and rheobase of the spike (w) for action potential firing and the membrane potential (x) recorded from S1HLGlu neurons (n = 13 cells from 4 mice). Data are represented as mean ± SEM. S1 samples from two mice were pooled to create one sample (a, h and l). Student’s unpaired t-test (two-tailed) was used for (a) and (v). Two-way ANOVA followed with Bonferroni’s post hoc test was used for (b–d). Two-way ANOVA followed with Tukey’s post hoc test was used for (e–g). One-way ANOVA followed with Tukey’s post hoc test was used for (h, l, s, w and x). Two-way ANOVA followed with Tukey’s post hoc test was used for panels (i–k and m–o).
Next, we determined the role of Maf1 in Pcif1-induced nociceptive and anxiety-like behavior. Intracranial injection of DIO-Pcif1 AAV virus in S1HL of naïve CaMK2α-Cre mice not only increased the level of PCIF1 protein but also decreased the level of S1HL MAF1 expression. However, co-injecting DIO-Maf1 (but not control DIO-mCherry) blocked the PCIF1-overexpression-induced downregulation of MAF1, without affecting the PCIF1 upregulation caused by DIO-Pcif1 (Fig. 8l). Behaviorally, the enhanced nociceptive responses to mechanical and heat stimuli and anxiety-like behaviors in naïve CaMK2α-Cre mice with intracranial injection of DIO-Pcif1 in S1HL were attenuated by MAF1 overexpression caused by co-injecting DIO-Maf1 during the observation period (Fig. 8m–o; Supplementary Fig. 8a–f). Basal behavioral responses on the contralateral side were not affected (Supplementary Fig. 8g–i). Additionally, we explored whether the nociceptive response induced by Serbp1 upregulation could be blocked by Maf1 overexpression. As expected, intracranial injection of AAV-DIO-Serbp1 produced the augmented responses to mechanical and thermal stimuli in CaMK2α-Cre mice. However, these augmented responses were attenuated by the co-injection of AAV-DIO-Maf1 along with AAV-DIO-Serbp1 in these mice (Supplementary Fig. 8j–p). These results suggest that Serbp1 regulates pain behavior through the mediation of Maf1. Together, these findings indicate that MAF1 reduction is necessary for the PCIF1-SERBP1 complex to mediate PCIF1-induced nociceptive and comorbid anxiety-like behaviors.
Upregulation of MAF1 attenuates PCIF1-induced enhancement in Ca2+ signal intensity in S1HLGlu neurons
MAF1 is a key player in regulating calcium homeostasis and synaptic remodeling in neurons52,53. We finally examined whether upregulating MAF1 blocked the PCIF1-caused enhanced the Ca2+ signal intensity. Microinjection of DIO-GCaMP6s plus DIO-Pcif1 AAV virus (but not control DIO-mCherry) into S1HL of naïve CaMK2α-Cre mice led to a significant increase of GCaMP6s signal in response to 0.4 g von Frey filament stimulation, as measured on day 27 post-injection (Fig. 8p–s). This increase was markedly reduced by co-injection of DIO-Maf1. Notably, patch-clamp electrophysiological analyses revealed that Maf1 knockdown significantly increased action potential firing rates in pyramidal neurons (Supplementary Fig. 8s). Conversely, Maf1 overexpression completely abolished the PCIF1-mediated enhancement of neuronal excitability (Fig. 8v). These data suggest that MAF1 also participates in the process by which PCIF1 regulates neuronal activity in S1HL.
Discussion
Epitranscriptomic modifications have recently emerged as a focal point in chronic pain research due to their extensive regulatory effects on gene expression54. As one of the most prevalent RNA modifications, m6Am and its methyltransferase PCIF1 have been shown to involve in many biological processes and human diseases19,23,55,56,57. Bioinformatic analyses of m6Am profiling from 45 human and 16 mouse tissues have indicates that m6Am is brain tissue-specific58. Human and mouse m6Am modifications are often dysregulated in nervous-system-related diseases59, but it remains unclear exactly how m6Am is involved in brain-associated physiological and pathological functions. In the present study, we reported that an interaction between SERBP1 and PCIF1 after peripheral nerve injury contributes to deposition of m6Am on mRNA in the primary somatosensory cortex (S1). Furthermore, we demonstrated that the SERBP1–PCIF1 interacting complex in S1HL participates in the initiation and maintenance of neuropathic pain and anxiety-like behavior via targeting of m6Am to Maf1 mRNA. Our study provides the evidence to support a functional role for m6Am and its methyltransferase in the genesis of neuropathic pain and comorbid anxiety and indicates potential therapeutic strategies for the treatment of these disorders.
Several recent single-cell RNA sequencing reports showed that Pcif1 mRNA is expressed in the nervous system, including in S1 of mouse and macaque60,61. Immunofluorescence in vitro assays revealed that PCIF1 is localized predominantly in the nucleus in the cancer cells HCT116 and HT29 CRC26. Interestingly, an in vitro study reported that PCIF1 is enriched in both the nucleus and the cytoplasm in neck squamous cells23. Here, we found that PCIF1 was localized mainly in the nuclei of adult mouse S1 cortical neurons, consistent with the previous in vivo observations26. Neuronal distribution pattern of PCIF1 in S1 cortex suggests its potential role in nociceptive sensation and emotion processing.
Neuropathic pain is a common health problem, but its treatment is severely limited, partially because the molecular mechanisms underlying this disorder remain elusive. We here showed PCIF1 upregulation in S1 cortex in the preclinical mouse model of neuropathic pain with comorbid anxiety caused by peripheral nerve injury (SNI, CCI), diabetes, and chemotherapy, but not in the preclinical mouse model of chronic inflammatory pain or chronic restraint stress (to induce anxiety alone). These data strongly suggest that the upregulation of S1 PCIF1 is specific to neuropathic pain. Additionally, our findings support previous conclusions indicating that distinct mechanisms are involved in anxiety disorders compared to those induced by neuropathic pain62.
Importantly, the upregulation of PCIF1 occurred specifically in S1HLGlu neurons. A previous reports showed that peripheral nerve injury increased hyperactivity of S1HL pyramidal neuron35. We further showed that PCIF1 upregulation in S1HLGlu contributed to the development and maintenance of neuropathic pain and comorbid anxiety, demonstrated by the attenuation of nociception and anxiety-like behaviors and by the amelioration of enhanced Ca2+ signaling and S1HLGlu neuronal excitability after specific knock-down or conditional knock-out of PCIF1 in S1HLGlu in SNI mice. Thus, PCIF1 in S1HLGlu is likely a key initiator of neuropathic pain and comorbid anxiety.
The SERBP1–PCIFI complex is required for the increased deposition of m6Am in mRNA in S1HLGlu neurons in SNI mice. Co-factors are known to contribute to process by which methyltransferases deposit modifications onto mRNA63,64. For instance, Wilms’ tumor 1–associating protein (WTAP) is a co-factor of METTL3, as their protein complex catalyzes the RNA m6A modification43. However, the mechanism underlying PCIFI deposition of m6Am onto mRNA is poorly understood. In this study, we demonstrate that SERBP1 interacts with PCIF1 to regulate m6Am deposition on mRNA, and that arginine residues at position 16 of SERBP1 are essential for this interaction. Indeed, SERBP1 is a well-known regulator of Serpine1 mRNA stability29, and was recently reported to have chaperone-like properties incell metabolism and histone methylation65,66,67. In addition, SERBP1 has a central role in brain function and development, especially neurogenesis and synaptogenesis30,31,32. SERBP1 is frequently localized in the cytoplasm and the nucleus because of the level of arginine methylation in the C-terminal region68,69. Consistently, the present study showed that SERBP1 was localized in both nucleus and the cytoplasm of S1HL neurons, where it plays a crucial role in the activity of PCIF1 and catalyzation of m6Am formation. We used a CRY2/CIBN optogenetic system to enhance SERBP1 binding to PCIF1. This binding resulted in an increase in the total level of m6Am in S1, as well as in nociceptive and anxiety-like behaviors. Notably, we successfully constructed the lentiviral vector Lenti-PCIF1-Cibn to overexpress PCIF1. However, due to the limitations of viral expression efficiency and vector capacity, this system resulted in only a modest 29% increase in m6Am levels. Even this slight increase seems sufficient to trigger mild pain-like behavioral manifestations in dark conditions. The identification of SERBP1 as a cofactor of PCIF1 represents a significant finding. This discovery is particularly important considering the ongoing challenges in understanding the mechanisms by which epigenetic modification proteins, including DNMTs, METTL3, NAT10, and PCIF1 deposit various epigenetic modifications, particularly the recently discovered m6A, m6Am, and ac4C modifications in DNA or RNA.
PCIF1 catalyzes m6Am methylation only at 5’ terminal of capped mRNAs, not within internal m6A methylation. Our m6Am-Seq assay revealed that many transcripts gained m6Am in contralateral S1 after nerve injury, and these gains correlate with the SNI-induced elevation in S1 PCIF1. SNI led to the changes in m6Am modification in many mRNAs, including both increases and decreases of m6Am levels in certain mRNAs. Unlike PCIF1, the expression of FTO, a known demethyltransferase of m6Am, was not altered in S1 after SNI. Which proteins are responsible for erasing the m6Am modification in mRNAs? Future studies will be necessary to address these questions. Peripheral nerve injury increased the level of m6Am level, but decreased the level of m6A, in S1. The abundance and alteration between m6Am and m6A do not appear to be correlated, consistent with previous findings58. As m6Am is adjacent to m7G in mRNA19,20, here, we found m7G was also increased in S1 under conditions of neuropathic pain. Whether their adjacent location is linked to this consistency remains to be determined.
MAF1 may participate in the role of PCIF1 in neuropathic pain and associated anxiety occurs. Blocking PCIF1 expression by microinjection of AAV-Cre virus into SNI Pcif1fl/fl mice or AAV-Pcif1-shRNA in SNI mice blocked the increases in Maf1 m6Am, rescued the decreased expression of MAF1 protein in S1 cortex, and blocked the development of neuropathic pain and associated anxiety. Mimicking nerve-injury-induced PCIF1 upregulation through S1HL injection of DIO-Pcif1 AAV virus in naïve CaMK2α-Cre mice increased Maf1 m6Am, reduced MAF1 protein expression in S1, and augmented the animals’ nociceptive and anxiety-like responses. Furthermore, we used the CRISPR-dCasRx system to show that PCIF1 has a direct effect on MAF1 expression in a Maf1 m6Am-dependent manner38,70,71,72,73. It is well documented that MAF1 is an endogenous regulator of neuronal excitability in optic neuropathy, learning and memory disorders, Alzheimer’s disease, and ischemic stroke52,74,75,76. MAF1 enhances GABAAR receptor activity, and thus has an inhibitory effect on neural activity53. The anti-nociceptive effect of reversing PCIF1 upregulation in SNI mice is likely due to rescue of the reduction in MAF1 expression. In line with this, PCIF1 upregulation enhanced stimulus-evoked calcium activity in S1HLGlu neurons, and this enhancement was blocked by overexpression of MAF1 in S1HLGlu neurons. However, we cannot rule out other mechanisms through which PCIF1 may be involved in neuropathic pain and associated anxiety. Our RNC- and m6Am-sequencing analyses revealed that, in addition to Maf1, changes in other genes such as Aacs and Zcchc8 were also identified following SNI, which could potentially contribute to m6Am-mediated modulation of neuropathic pain via distinct pathways. Whether these genes also mediate the role of PCIF1 in neuropathic pain and associated anxiety remains to be further investigated. In addition, sex dimorphism in pain and anxiety is a well-recognized clinical phenomenon77,78. As this study primarily used male mice, whether the above mechanisms are male-specific remains unclear. We will examine these mechanisms in mice in future studies.
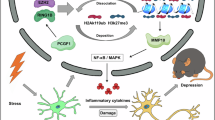
In summary, we have demonstrated that SERBP1 acts as a co-factor of PCIF1, and that the SERBP1–PCIF1 complex is essential for catalyzing m6Am modification. Blocking the nerve-injury-induced increase in S1HLGlu SERBP1–PCIF1 mitigated neuropathic pain and alleviated anxiety-like behaviors, without impairing of locomotor function. The effects of the SERBP1–PCIF1 complex on neuropathic pain occur, at least in part, through the m6Am modification of Maf1 in S1, leading to a reduced expression of MAF1 and an increase in neuronal excitability in the S1 cortex. (Fig. 9). This study establishes the initial link between RNA m6Am and neuropathic pain, along with associated comorbid anxiety. Our study suggests that the SERBP1–PCIF1 complex may be a potential clinical target for the management of this disorder.

Peripheral nerve injury induces an increase in the SERBP1-PCIF1 complex, which in turn regulates m6Am-controlled MAF1 expression in S1HL, leading to reduced inhibition and enhanced neuronal excitability (drawn by figdraw.com).
Methods
Animals
Healthy male and female BALB/c and C57BL/6 mice were obtained from Xuzhou Medical University. CaMK2α-Cre, Pcif1fl/fl and tdTomatofl/fl mice were purchased from Cyagen Biosciences. vGat-Cre mice were purchased from Shanghai Model Organisms Center, Inc. Mice were housed in standard conditions (Specific Pathogen-Free facility, 12 h light/dark cycle, temperatures of 23 ± 3 °C and humidity of 30–60%), with ad libitum access to food and water. All animal procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Xuzhou Medical University (the protocol number: 202207S038). BALB/c mice were used for preliminary validation of the viral construct. C57BL/6 mice were employed in all experiments except the preliminary viral construct validation. Female mice were used exclusively to examine the effect of sex on PCIF1 expression in the S1HL after SNI, and other experiments employed male mice. For each experiment, the animals were randomized to either the control or the experimental group. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Mouse genotyping
CaMK2α-Cre, vGat-Cre, tdTomatofl/fl and Pcif1fl/fl mice were identified by genotyping. A small mouse tail sample was collected, and DNA was extracted using the phenol-chloroform method. PCR analysis was performed to identify wild-type and mutant mice. PCR amplification was conducted using 500 ng of DNA in a 20 µL reaction volume, containing 10 μL of 2× Taq PCR MasterMix (Vazyme, China) and 1 µL of 10 µM primers. The primer sequences used in these reactions can be found in Supplementary Data 1. DNA and primers were denatured at 94 °C for 3 min, followed by 35 cycles of 94 °C for 30 s, 60 °C for 35 s, and 72 °C for 55 s, with a final extension at 72 °C for 5 min. Amplicons were separated using a 1.5% agarose gel and imaged with the Uvitec System (Q9 Alliance, Uvitec).
Animal models
Four peripheral nerve injury-induced neuropathic pain models—spared nerve injury (SNI)79, chronic constriction injury (CCI)80, diabetic neuropathic pain (DNP)81, and chemotherapy-induced neuropathic pain82—were prepared for experiments. For the SNI model, mice were anesthetized with 2% isoflurane. The left sciatic nerve was exposed and the common peroneal and tibial nerves were loosely ligated with nonabsorbent 5-0 chromic gut sutures. Subsequently, 1 mm sections distal to the sutures were removed, while the sural nerve was left intact. The pre-treatment of CCI model was similar to that in SNI model. After exposing the left sciatic nerve, three loose straps of 4-0 silk thread were placed around the sciatic nerve at intervals of about 1 mm at the proximal end of the trigeminal nerve.
For DNP model, mice were randomly assigned to either a normal chow diet (ND) group or a high-fat diet (HFD) group. After 4 weeks, citrate buffer solution was injected intraperitoneally into ND mice once a day for 3 days, while streptozotocin (STZ, 30 mg/kg, Beyotime) was injected into HFD-fed mice. Mice were evaluated according to the following criteria1: fasting glucose levels exceeding 16.7 mM2; elevated food and water consumption, increased urine output, and weight loss.
For chemotherapy-induced neuropathic pain model, cisplatin (2.3 mg/kg/day; MedChemExpress) or phosphate buffered saline (PBS) was injected intraperitoneally for two cycles of five daily injections followed by 5 days of rest. Mice were observed carefully for any abnormal behavioral changes every day after treatment.
Chronic restraint stress (CRS)34 is a common mouse model of anxiety/depression. Mice were placed in a well-ventilated 50 mL centrifuge tube for 2 h daily (10:00 a.m. to 12:00 p.m.) over 7 consecutive days. The control group received no treatment. Anxiety-like behavior was evaluated on the 8th day using the elevated plus maze and open field test.
Tissue collection
To ensure the reproducibility of the experiments, we designed rigorous methods to collect S1HL. Mice were anesthetized, euthanized, and immediately perfused with PBS through the heart left ventricle before the brains were removed. The brain was then placed in a rodent brain matrix (ST-1175, TOW-INT TECH). Coronal sections (500 µm) were cut using razor blades, and the slices were transferred to ice-cold PBS. According to the mouse brain atlas (second edition), the S1HL tissue was dissected using a razor blade and curved forceps under a stereomicroscope. The specific location of S1HL varies across different brain slices. The corresponding S1HL location was confirmed based on the morphology of the corpus callosum, the commissure, the lateral ventricle, and the hippocampal fimbria. The S1HL tissues were collected from Bregma: 0.38 to −0.22 mm.
Stereotaxic viral injections
Mice were anesthetized with isoflurane (2%) and immobilized on a stereotaxic device. Virus was injected into the brain (infusion speed 0.1 µl/min) with a Hamilton syringe containing a 33-gauge stainless steel needle connected to a stereotaxic infusion pump. The coordinates used to target the S1HL/S1L5 area was as follows: AP: −0.5 mm, ML: −1.7 mm, DV: −1.3 mm; that to motor cortex (M1) area was as follows: AP: −0.22 mm, ML: -1.0 mm, DV: −1.3 mm. The needle was left in place for 5 minutes after the injection to prevent backflow. After injection, the animals were placed in a clean cage, and the cage was kept warm until full recovery from anesthesia.
The AAV-CaMKIIα-shPcif1-WRPE (AAV2/9, 5.3 × 1012 vg/mL), AAV-CaMKIIα-Scr-WRPE (AAV2/9, 5.1 × 1012 vg/mL), AAV-Ef1α-DIO-Pcif1-mCherry (AAV2/9, 7.3 × 1012 vg/mL), AAV-Ef1α-DIO-mCherry (AAV2/9, 4.9 × 1012 vg/mL), AAV-RBP4-Pcif1-eGFP (AAV2/9, 8.6 × 1012 vg/mL), AAV-RBP4-shPcif1-eGFP (AAV2/9, 3.8 × 1012 vg/mL), AAV-RBP4-eGFP (AAV2/9, 6.5 × 1012 vg/mL), AAV-RBP4-Scr-eGFP (AAV2/9, 5.7 × 1012 vg/mL), Lenti-Pcif1-Cibn (8.0 × 109 VP/mL), Lenti-Serbp1-Cry2 (1.3 × 109 VP/mL), AAV-Ef1α-DIO-Serbp1-mCherry (AAV2/9, 3.3 × 1013 vg/mL), AAV-CaMKIIα-shSerbp1-WRPE (AAV2/9, 6.9 × 1012 vg/mL), AAV-Ef1α-DIO-Maf1-mCherry (AAV2/9, 5.2 × 1012 vg/mL), AAV-CaMKIIα-shMaf1-WRPE (AAV2/9, 1.6 × 1013 vg/mL) were generated through a systematic process involving vector construction and viral packaging. AAV-CaMKIIα-Cre (AAV2/9, 5.7 × 1012 vg/mL), AAV-CaMKIIα-GCaMP6s (AAV2/9, 6.1 × 1012 vg/mL) and AAV-hSyn-DIO-GCaMP6s (AAV2/9, 6.7 × 1012 vg/mL) were purchased from Obiosh Biotechnology (Shanghai, China).
Behavioral tests
Paw withdrawal frequency (PWF) and paw withdrawal latency (PWL) were measured to assess mechanical and thermal pain sensitivity38. Paw withdrawal frequencies (PWFs) in response to mechanical stimuli were measured using two calibrated von Frey filaments (0.07 g and 0.4 g, Stoelting Co.). In brief, the tested mouse was placed in a Plexiglas chamber to habituate for 30 min. The 0.07 g and 0.4 g von Frey filaments were applied to the plantar surface of each hind paw for 1 second, repeated 10 times at 5-minute intervals. The PWF was calculated as the percentage of positive responses among the 10 applications, using the formula: (number of paw withdrawals/10 trials) × 100. Paw withdrawal latencies (PWLs) were evaluated by recording the time taken to withdraw the hind paw in response to heat stimulation using the Model 336 Analgesia Meter, Series 8, IITC Life Science. Briefly, the tested mouse was placed in an individual Plexiglas cage on a glass plate. A beam of noxious light was directed at the center of each hind paw’s plantar surface. The PWL was measured as the interval from the initiation of the light beam to the hind paw withdrawal. The test was conducted five times at 5 min intervals per side. A 20 s cutoff time was implemented to prevent tissue injury.
Modified conditioned place preference (CPP) paradigm was employed to evaluate spontaneous pain behaviors in mice83. Mice were briefly placed in an apparatus consisting of two Plexiglas compartments connected by a tunnel. Compartment 1 featured a rough floor and walls adorned with horizontal black-and-white stripes. Compartment 2 featured a smooth floor and walls adorned with vertical black-and-white stripes. The time spent in each compartment was automatically recorded by the MED-PCIV CPP software. Initially, mice were preconditioned for 30 minutes with access to both compartments to familiarize them with the environment. Post-preconditioning phase, data analysis within 15 minutes assessed preexisting compartment bias in mice. Further testing was not performed on mice that spent over 80% or less than 20% of the total time in any one compartment. The connecting tunnel was closed for the next 3 days during the conditioning protocol. Mice were intrathecally injected with saline (5 µl) and then allowed to habituate for 15 min in the saline-paired compartment. Six hours later, 5 µl of 0.8% lidocaine in saline was injected intrathecally and paired with the opposite conditioning compartment. On the test day, the mice were placed in the connecting tunnel and explored both compartments freely for 15 min. The CPP score was defined as the test time spent in the lidocaine chamber divided by the preconditioning time spent in the lidocaine chamber.
The elevated plus maze (EPM) and open field test (OFT) were employed to assess anxiety-like behaviors in mice84. The EPM consisted of four 30 cm × 6 cm arms (two closed arms and two open arms) and a central 6 cm × 6 cm platform. Mice were placed in the center of the apparatus and allowed to move freely. The number of entries and time spent in each arm was recorded by ANY-maze software (Stoelting Co). OFT was performed in a square arena (50 × 50 × 30 cm). The arena was evenly divided into 25 small squares (10 cm × 10 cm), with the central 9 squares defined as the central zone and the outer 16 squares defined as the peripheral zone. The mice were placed into the central zone of the apparatus and allowed to freely explore the arena for 10 min. The time spent in the center and the total movement distance were recorded by ANY-maze software.
RNA extraction and quantitative real-time polymerase chain reaction
Quantitative real-time PCR (qRT-PCR) was performed to determine mRNA expression levels85. To reverse transcribe into cDNA, 400 ng RNA was extracted from S1HLGlu neurons with RNAiso reagent (9109, Takara). RT‒qPCR was performed with 2× SYBR Premix ExTaqII (RR820A, Takara) and run on a LightCycler480. PCR cycling conditions were performed at 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 30 s. All RT‒qPCR primer sequences are listed in Supplementary Data 1.
Single-cell RT-PCR
CaMK2α-Cre and vGat-Cre mice were crossed with tdTomatofl/fl mice to label CaMK2α neurons and GABA neurons, respectively, in the nervous system. After genotyping the offspring, CaMK2α-td and vGat-td mice at 8 weeks of age were subjected to SNI or Sham surgery. On 21 days post-surgery, pain hypersensitivity was validated, and S1HL brain slices were prepared according to the brain slice electrophysiology protocols. Glass pipettes with appropriate tip diameters were pulled and used to approach the S1HL region under an inverted phase-contrast microscope. Fluorescently labeled neurons were aspirated into the pipette via gentle suction. The pipette was quickly removed from the solution, and its contents including the cell and surrounding fluid were expelled into a nuclease-free PCR tube containing 10 μL of cDNA lysis buffer. The samples were stored at −80 °C.
Nested RT-PCR was performed to amplify the Pcif1 mRNA fragment86. Two pairs of Pcif1 primers were designed: the first pair was identical to standard PCR primers, while the second pair bound to an internal region of the first PCR product (Supplementary Data 1), resulting in a shorter amplicon during the second RT-qPCR amplification. To ensure data reliability, we implemented rigorous quality control measures, including the use of reverse transcriptase-negative controls, post-collection imaging to confirm single-cell isolation, and PCR amplification of key cellular markers—such as Rbp4 for S1L5 excitatory neurons, Drd3 for S1L2/3 excitatory neurons, Ntsr1 for S1L6 excitatory neurons, Gad1 for inhibitory neurons, NeuN as a pan-neuronal marker, Gfap for astrocytes, and Gapdh as a loading control. These steps were followed by gel electrophoresis to exclude potential contamination from adjacent cortical layers.
Laser capture microdissection
Fluorescently labeled neurons in the S1HL region were isolated using laser capture microdissection87. The mouse brain was removed and then rapidly frozen in OCT compound using dry liquid nitrogen. The S1HL slices were sectioned (15 mm) using a cryostat (Leica Microsystems Wetzlar, Germany) and placed on an RNase-free glass slide. A single S1HL neuron was obtained using a laser microdissection system (LMD7000; Leica Microsystems). 20 neurons were pooled into one same sample for qPCR analysis.
Analysis of SERBP1-PCIF1 protein interaction
Protein sequences were submitted to AlphaFold Server (https://alphafoldserver.com) with default parameters88. The predicted models were validated by assessing the pLDDT (predicted lDDT-Cα) confidence scores, retaining the model with the highest pLDDT score for further analysis. The predicted structures were refined using the “Repair PDB” function in the FoldX (http://foldxsuite.crg.eu/) plugin within YASARA (YASARA Biosciences: www.yasara.org). PyMOL (http://www.pymol.org/pymol) was employed for structural visualization and measurement of hydrogen bond distances of amino acids that may be involved in binding. To evaluate residue-specific binding contributions, alanine scanning mutagenesis was systematically performed using FoldX’s position scan protocol89. Key candidate residues were substituted with alanine to calculate binding free energy changes (ΔΔG) between SERBP1 and PCIF1. Hydrogen bonds with donor-acceptor distances <3.5 Å were considered stable, ensuring optimal orbital overlap and electrostatic interactions90. Residues with ΔΔG > 2 kcal/mol upon alanine substitution were defined as energetically critical for the interaction89.
m6Am-Exo-seq
11 μg of mRNAs were mixed with NEBNext® Magnesium RNA Fragmentation buffer (E6150, NEB) on ice and incubated at 94 °C for 2 min to generate fragments of 200–300 bp, and purified using the RNA Cleanup & Concentration Kit (T2030, NEB) and eluted in 20 μL elution buffer. The purified mRNAs were treated with 20U T4 PNK (T0201, NEB) in T4 ligase buffer at 37 °C to phosphorylate uncapped and fragmented transcripts. Subsequently, XRN-1 (M0338, NEB), a 5’-phosphate-dependent exonuclease, was added to specifically digest the phosphorylated (uncapped and sheared) transcripts. Thus, 5’-capped transcript-enriched RNAs were left and purified using the RNA Cleanup & Concentration Kit. The mRNA decapping Enzyme (M0608, NEB) was added to remove m7G caps at 37 °C for 30 min, and then repurified with the RNA Cleanup & Concentration Kit and eluted in 20 μL buffer. As a result, approximately 1 μg of decapped 5’ RNA fragments were obtained for subsequent analysis. 1 μL (10%) of processed RNAs were used as input control, while the remainders were diluted with reaction buffer (1× DEPC-PBS + 0.5% Tween-20) to a final volume of 400 μL. Protein A/G Magnetic Beads (HY-K0202, MCE) were prepared by washing four times with 1 mL binding/wash buffer, then resuspended in 400 μL reaction buffer containing 4 μg anti-m6A antibody (A22411, ABclonal). After 2 h rotational incubation at 4 °C, the bead-antibody complexes were washed twice with 1 mL ice-cold binding/wash buffer. Then, the complexes were combined with RNA in 400 μL reaction buffer and incubated overnight at 4 °C with rotation. Bead-bound mRNAs were subsequently eluted using phenol-chloroform extraction and purified by RNA Cleanup & Concentration Kit.
Sequencing library preparation were conducted by Majorbio Co., Ltd. (Shanghai, China) using the SMART-Seq_V4 protocol (Clontech; San Diego, CA). Polyadenylated mRNA was reverse-transcribed from 10 ng total RNA using Oligo(dT) primers and MMLV reverse transcriptase, and generated single-stranded cDNA with 3’-terminal CCC overhangs, and amplified to nanogram-scale cDNA quantities using PCR. Subsequent fragmentation and adapter ligation were achieved through Tn5 transposase-mediated enzymatic cleavage coupled with end repair. Final library validation included quantification via Qubit 4.0 fluorometry prior to paired-end sequencing (2 × 150 bp) on the NovaSeq X Plus platform. Raw sequencing paired-end reads first underwent adapter trimming and quality assessment using fastp91 with default parameters.
The m6Am-Exo-seq data analyses were performed using protocols adapted from established methods21,25,55, with modifications as described below. Briefly, paired-end sequencing reads were first processed with Trimmomatic (v0.39)92 to remove adapters and low-quality bases. The cleaned reads were then aligned to the mouse reference genome (mm10) using HISAT2 (v2.1.0)93. To identify putative m6Am peaks, MACS2 (v2.1.1)94 was used with the following parameters: an effective genome size of 2.7 × 10⁹, the -nomodel option enabled, and a q-value cutoff of 0.05. Before peak calling, input samples were normalized to ensure equal sequencing depth. For motif analysis, consensus peaks from three biological replicates were identified through reciprocal overlap. Sequences spanning ±50 bp from peak centers were extracted from the sense strand. De novo motif discovery was then performed using MEME (v5.5.7)95. For metagene analysis, the GenomicRanges (v1.60.0) and Guitar (v2.24.0) packages in R were utilized, employing gene annotations derived from TxDb.Mmusculus.UCSC.mm10.knownGene.
To identify differentially expressed genes (DEGs) between two samples, the expression level of each transcript was calculated using the gene counts method, where single gene counts equals RIP-gene counts minus the corresponding input gene counts. Additionally, Gene Ontology (GO) enrichment analysis was performed to determine which DEGs were significantly enriched in specific GO terms, applying a Bonferroni-corrected P-value threshold of <0.05 compared to the whole transcriptome background. GO functional enrichment analyses were conducted using tools available at https://hiplot.cn and https://www.webgestalt.org.
SERBP1-RIP-seq
S1HL tissues were harvested from mice anesthetized with isoflurane. Tissues from four mice were pooled to create a single sample. Sample was homogenized in RIP lysis buffer (150 mM KCl, 25 mM Tris pH 7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% NP4, RNAse inhibitor, protease inhibitor), and incubated on ice for 30 min. Following centrifugation, the supernatant was collected and incubated with Anti-SERBP1 antibody overnight at 4 °C with gentle agitation. Subsequently, 100 μL of pre-washed protein A/G magnetic beads (HY-K0202, MCE) were added, and the mixture was incubated at room temperature for 2 h with rotation. RNAs bound to the beads were then eluted using phenol-chloroform extraction. RNA sequencing library was prepared using the SMART-Seq_V4 Ultra Low Input RNA Kit for Sequencing from Clontech (San Diego, CA). RNA sequencing, quality control, and data analysis were performed as described for m6Am-Exo-Seq.
RNC sequencing
Mouse tissue was removed rapidly and placed in 0.5 mL RB buffer (20 mM HEPES-KOH (pH 7.4), 15 mM MgCl2, 200 mM KCl, 100 µg/mL cycloheximide, 2 mM dithiothreitol) with 1% Triton, 5 µl PMSF (100 mM), and 200 U RNase inhibitor for each sample. The lysate was then incubated on ice for 30 min. Then it was centrifuged (11,000 × g, 10 min at 4 °C) and discard the precipitate. Take 10% supernatant liquid as the Input control, and slowly add 90% supernatant liquid to the upper layer of 9.5 mL sucrose buffer (30% sucrose in RB buffer). Polyribosome precipitate was collected after centrifuge (127,000 × g, 5 h at 4 °C). RNA was isolated from Input and polyribosome precipitation, and then submitted to Majorbio Co., Ltd. (Majorbio, China) for transcriptome sequencing. FPKM (fragments per kilobase per million) was used to normalize gene expression levels. RNA sequencing, quality control, and data analysis were performed according to the m6Am-Exo-Seq protocol described above. Translation efficiency (TE) was determined by the ratio of FPKM in polyribosome-seq to FPKM in input RNA-seq.
LC‒MS/MS
We quantified m6Am levels in the mRNA of S1HL (somatosensory cortex of the hind limb) using an LC–MS/MS assay25. Total RNA was extracted by RNAiso reagent (9108, Taraka). Extracting mRNA from 30ug total RNA by BeyoMag™ mRNA purification kit with magnetic beads (R0071L, Beyotime). The mRNA decapping Enzyme (M0608, NEB) was added to remove m7G caps, and then enzymatically hydrolyzed to nucleosides by Nucleoside Digestion Mix (M06459S, NEB), and extracted with phenol–chloroform. The upper clear aqueous layer was collected and filtered through 0.22 μm Millex-GV filters (EMD Millipore) into vials. Agilent 6410 QQQ triple-quadrupole LC mass spectrometer was used for mass spectrometry detection. Chromatographic separation was conducted on an Agilent 1290 Infinity II UHPLC platform equipped with a ZORBAX RRHD Eclipse Plus C18 analytical column (150 × 2.1 mm ID, 1.8 μm particle size) coupled to a matching guard column (5 × 3 mm ID, 1.8 μm; Agilent). The mobile phase comprised water and methanol (each containing 0.1% formic acid) delivered at 0.23 mL/min. The gradient program initiated with 5% methanol (0.5 min), followed by sequential steps: 5% → 15% methanol over 2.5 min, 15% → 95% methanol over 3 min, and column re-equilibration at 5% methanol for 4 min. For unmodified ribonucleoside analysis, samples were diluted and analyzed under isocratic conditions with 20% methanol. Mass detection employed an Agilent 6495 Triple Quadrupole mass spectrometer with positive electrospray ionization. RNA modification ratios (m6Am/A, m6A/A, ac4C/C, m5C/C, and m7G/G) were quantified using Agilent MassHunter software by integrating AUC values from mass spectrometry chromatograms25.
AAV plasmid construction and virus production
Full-length Pcif1, Serbp1, and Maf1 cDNA was amplified from mouse cortex cDNA, and the corresponding AAV virus with CaMK2α promoter were constructed. Primer sequences are listed in Supplementary Data 1. All constructed plasmids were confirmed by DNA sequencing. Viruses were generated in AAV293 cells, purified using an AAVpro Purification Kit (6232, Takara), and titrated with an AAVpro Titration Kit (6666, Takara). BALB/c mice were specifically employed for the initial validation of viral constructs and pilot experiments because of their increased susceptibility to viral transduction.
Cell line culture and transfection
HEK293T(H4-1401) and HT22(M8-0201) cells were purchased from Oricell, and authenticated by STR profiling. HT22 and HEK293T cells were cultured in DMEM with 10% fetal bovine serum at 37 °C in a humidified incubator. Cells were transfected with plasmid using ExFect Transfection Reagent (T101, Vazyme) following the manufacturer’s instructions.
CRISPR vector specifically targeting the m6Am site in Maf1
Full-length Pcif1 was cloned into BamHI-digested EF1a-dCasRx-2A-EGFP using the ClonExpress II one-step cloning kit (C112, Vazyme). Lentivirus gRNAs, including gRNA-26 (targeting +26 to +64 of Maf1 mRNA) and gRNA-60 (targeting +60 to +79), along with a negative control gRNA, were constructed by ligating to pLH-sgRNA1 using the primers listed in Supplementary Data 1. Lentivirus was collected by transfecting HEK293T cells with a lentiviral expression construct and identified by DNA sequencing.
Protein mass spectrometry
We constructed a PCIF1-HA overexpression construct and fully transfected it into human HEK293T and murine HT22 cells (n = 2). The PCIF1 protein was pulled down using an HA-tag (AE105) antibody and subjected to proteomic analysis by GeneChem (Shanghai, China). LC-MS/MS was performed using a Q Exactive mass spectrometer coupled with an Easy nLC system (Thermo Fisher Scientific, MA, USA). Raw data were processed in Proteome Discoverer 2.2 (Thermo Fisher Scientific) for protein identification, followed by database searching using the embedded Mascot 2.6 engine. Protein identification was conducted against a custom database (detailed in the Chinese “Methods” section). Search parameters included: trypsin as the digestion enzyme with ≤2 missed cleavages; precursor mass tolerance of 10 ppm; fragment mass tolerance of 0.05 Da; carbamidomethylation (C) as a fixed modification; and oxidation (M) and acetylation (protein N-terminus) as variable modifications. Proteins were considered positively identified if at least one unique peptide achieved a significance threshold of FDR ≤ 0.01.
PCIF1-MTase protein expression in vitro and purification
The coding sequence of mouse PCIF1-MTase domain was cloned as an in-frame fusion to the His tagged vector PLou3. The PLou3-MTase expression vector was transformed into BL21(DE3) competent cells for protein expression. Induction of protein expression was carried out with 0.5 mM IPTG at 18 °C overnight. Bacteria were harvested at 1252 × g, 4 °C and the cell pellet was resuspended in protein purification lysis buffer (50 mM Tris-HCl pH 7.5, 0.25 M NaCl, 0.1% Triton-X, 1 mM PMSF, 1 mM DTT, and protease inhibitors) and lysed using an ultrasonic cell disruptor under low-temperature conditions with the following parameters: 30% amplitude (600 W), 3 s pulse duration, 5 s interval, and 2 min operation cycles followed by 2 min cooling periods until the solution became clear. The lysate was collected and centrifuged at 11,000 × g for 15 min at 4 °C to separate the supernatant from the pellet. For protein purification, the pre-packed Ni-NTA affinity column was equilibrated with 5 column volumes of lysis buffer. The clarified supernatant was then loaded onto the equilibrated column and allowed to incubate for at least 2 min to ensure sufficient interaction between the target protein and the resin. The flow-through was collected, and the column was washed with 10–15 column volumes of wash buffer to remove non-specifically bound proteins. Finally, the target protein was eluted using 5–10 column volumes of elution buffer with fraction collection. The purification efficiency was assessed by SDS-PAGE analysis.
In vitro methyltransferase assays
The 5’UTR mRNA was isolated following the m6Am-Exo-Seq protocol. In vitro methylation reactions were performed in a 50 μL system containing 50 nM MTase, SERBP1 protein (purchased from AtaGenix, China), and 4 μM 5’UTR mRNA in methylation buffer (50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM DTT, 5% glycerol) supplemented with 160 μM S-adenosylmethionine (B9003, NEB). The reaction mixture was incubated at 37 °C for 10 min, followed by heat inactivation at 65 °C for 20 min. RNA purification was carried out using the RNA Cleanup & Concentration Kit (T2030, NEB) according to the manufacturer’s instructions. The purified RNA samples were filtered through a 0.22 μm Millex syringe filter (Millipore) and subsequently subjected to LC-MS analysis for m6Am quantification.
Western blotting
After anesthetizing mice with isoflurane and perfusing them with physiological saline, S1HL tissue was taken from the mice. The collected tissue was washed twice with precooled PBS and lysed in RIPA buffer (P0013B, Beyotime). Following centrifugation at 11,000 × g for 15 min at 4 °C, the supernatant was collected and its concentration quantified using a BCA kit (P0012S, Beyotime). Following a 2 h block with 5% skimmed milk, the membranes were incubated overnight at 4 °C with primary antibodies (PCIF1, A21596, ABclonal; SERBP1, 10729-1-AP, Proteintech; MAF1, A15531, ABclonal). Following three 5 min rinses, the membranes were incubated with the appropriate secondary antibody (Beyotime) for 2 h at room temperature. The membranes were rinsed three times for 5 min each with TBS-Tween. Detection was conducted using ECL reagent (P0018FS, Beyotime) and a UVITEC System (Q9 Alliance, UVITEC). Protein bands were quantified using ImageJ software and normalized to H3 or β-Actin bands.
Co-immunoprecipitation (Co-IP)
Co-IP assay was conducted using an immunoprecipitation kit with protein A + G magnetic beads (P2179S, Beyotime) following the manufacturer’s protocol. The antibodies PCIF1 and SERBP1 were utilized in co-IP assays, with murine IgG and rabbit IgG serving as negative controls.
Immunohistochemistry
Animals were anesthetized with isoflurane and perfused with 4% paraformaldehyde. Then, the brains were taken out and postfixed for 6 h at 4 °C. Brains were dehydrated using 30% sucrose and sectioned at 30 µm with a freezing sledge microtome. The sections were washed thrice for 10 min in PBS, then blocked with 10% donkey serum and 0.4% Triton X-100 in PBS for 2 h. After three PBS rinses, the sections were incubated with the following primary antibodies at 4 °C for one or two nights: PCIF1 (1:500, A21596, ABclonal), NeuN (1:500, ab104224, Abcam), Iba1 (1:500, ab3554, Abcam), S-100β (1:500, S2532, Sigma), SERBP1 (1:500, 10729-1-AP, Proteintech), and MAF1 (1:500, A15531, ABclonal). After washing three times (10 min each) with PBS, the sections were incubated for 2 h at room temperature with the fluorescein-conjugated secondary antibody. Finally, the sections were stained with DAPI and examined using Olympus FV1000 laser confocal microscope.
In vivo calcium imaging
Localized calcium signals were recorded by fiber photometry of GCaMP6s fluorescence. Mice were injected with AAV-DIO-GCaMP6s (PT-0071, BrainVTA) and implanted with optical fibers prior to behavioral tests and fiber photometry recordings in S1HL. The fiber photometry system (Inper) has been described previously. The laser power at the tip of the optical fiber was adjusted to 20–40 μW. The photomultiplier tube current output was amplified and converted into a voltage signal by an amplifier, then digitized at 50 Hz and recorded by Inper-Studio software (Inper). Heatmaps and averaged Ca2+ traces were plotted with custom-written functions in InperDataProcess and GraphPad Prism. The raw photometry signal (X) was converted to Z-Scores: Z-Score = (X − mean)/standard deviation. The sample mean and standard deviation refer to values calculated from the 3 s before the stimulus. This approach was applied to the analysis of data collected before and after von Frey stimulation. We checked and verified that the photometry signals were free of movement artifacts: the eGFP signal originating from control AAV2/9-DIO-eGFP (PT-0086, BrainVTA) expression in S1HLGlu neurons did not change during epochs of pain behavior.
Brain slice electrophysiology
Mice were anesthetized with 2% isoflurane and intracardially perfused with 20 mL oxygenated ice-cold sACSF containing 85 mM NaCl, 24 mM NaHCO3, 1.25 mM NaH2PO4, 4.0 mM MgCl2, 2.5 mM KCl, 0.5 mM CaCl2, 75 mM sucrose, and 25 mM glucose. The brain was then sliced into 300 μm coronl sections containing S1HL using a vibrating microtome (VT1200s, Leica). Brain slices were incubated in ACSF (125 mM NaCl, 26 mM NaHCO3, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 2.5 mM KCl, 2.4 mM CaCl2, 11 mM glucose) for a minimum of 1 h at 32 °C. The brain slices were transferred to recording chamber and were perfused with ACSF at 3 ± 0.5 mL/min.
For electrophysiological recordings, an infrared (IR) differential interference contrast (DIC) microscope (BX51WI, Olympus) equipped with for fluorescence detection was used to visualize neurons in S1 slices. Whole-cell patch-clamp recordings were performed using a MultiClamp 700B amplifier, a Digidata 1440 A analog-to-digital converter, and pClamp 10.7 software (Axon Instruments/Molecular Devices). Pipettes (4–10 megaohms) made of borosilicate glass (VitalSense Scientific Instruments) were pulled using a horizontal puller (P1000, Sutter Instruments). Current-evoked action potential firing was recorded in current-clamp mode with internal solution containing 135mM K-gluconate, 5 mM KCl, 0.2 mM EGTA, 0.5 mM CaCl2, 10 mM HEPES, 2 Mg-ATP, 0.1 mM GTP. (osmolarity: 300 mOsm/kg, pH: 7.2). The above records were sampled at 10 kHz and filtered at 2 kHz. All experiments were conducted at room temperature (22–24 °C).
RNA immunoprecipitation
S1HL tissue was removed from mice and homogenized in RIP lysis buffer with a glass homogenizer. After centrifugation, the supernatant was sonicated and divided into three aliquots: Input, IgG, and IP. Anti-m6A antibody or IgG was added, respectively to the IP and IgG samples, and the mixture was incubated overnight at 4 °C. Then, magnetic beads were added and the mixture was incubated for another 2 h. The RNA adsorbed by the beads was eluted and extracted by TRIzol. Four pairs of primers were designed: the first two pairs were located in the 5′UTR region, covering the m6Am site (1st [F1R1]: +1 – +80; 2nd [F1R2]: +1 – +94, with the first nucleotide of mRNA designated as +1); the last two pairs were in the CDS (3rd [F3R3]: +516 – +669) and the 3′UTR (4th [F4R4]: +955 ~ +1109) regions. RT-qPCR was used to detect the m6Am levels in different regions of Maf1 mRNA. The PCR primers are shown in Supplementary Data 1.
Mammalian two-hybrid assay
To identify the domains of SERBP1 that interact with PCIF1, we carried out an in vitro mammalian two-hybridization experiment using a three-plasmid system: the pACT plasmid (encoding the herpes simplex virus VP16 activation domain upstream of an MCR and expressing the Renilla reniformis luciferase), the pBIND plasmid (encoding the yeast GAL4 DNA binding domain upstream of a multiple cloning region, MCR), and the reporter plasmid pG5luc (encoding firefly luciferase) were purchased from Promega (CheckMate Mammalian Two-Hybrid System). Four domains of Pcif1— the N terminal WW domain, helical, methyltransferase (MTase), and C-terminal domains, respectively inserted into the pACT vector to form five pACT-Pcif1-X construer (X represents the individual domain) through amplifing from mouse cDNA using primers (Supplementary Data 1). The full-length Serbp1 CDS was cloned into the pBIND vector (pBIND-Serbp1) to generate a Gal4-Serbp1 chimeric fusion protein (Fig. 4c). The determination of the interaction of SERBP1 with the specified PCIF1 domain was measured through the pG5luc luciferase level after cotransfection of the vectors in HEK293T cells. All constructs were verified by PCR and DNA sequencing. Detection of the reporter was carried out according to the manufacturer’s instructions.
Optogenetic manipulation
We used CIBN and CRY296 optogenetic approach to evaluate the effect of the interaction of SERBP1 and PCIF1 on deposition of m6Am level in RNA. Full-length Pcif1 and Serbp1 were inserted into Lenti-EF1α-Pcif1-Cibn and Lenti-EF1α-Serbp1-Cry2. For the in vitro experiment, both constructed plasmids were transfected into HT22 cells and 12 h treatment with 470 nm light (3-W energy, 4 h full light) was used to activate binding of CIBN to CRY2. Total RNA was extracted at 40 h after light treatment, and the m6Am level was analyzed via LC–MS/MS. For the in vivo experiment, Lenti-EF1α-Pcif1-Cibn and Lenti-EF1α-Serbp1-Cry2 were co-microinjected into S1HL of naïve mice, followed by implantation of optical fibers. 7 days after injection, mice were subjected to 465 nm light stimulation with the following laser parameters: 5 mW energy, 5 Hz pulse frequency, 100 ms pulse width, 5 s stimulation duration, 1 s interstimulus interval, 30 min total duration. Paw withdrawal frequencies to a mechanical stimulus (von Frey hairs) were recorded. Control group mice were injected with an empty virus.
Statistics and reproducibility
Statistical analysis was performed in GraphPad 8.0 software85. All experiments are repeated at least three times. Sample sizes were determined empirically according to literature in the field and our previous experience38. Animals were randomly assigned to experimental groups in all experiments. All behavioral experiments employed double-blind testing. To rule out the influence of locomotor impairments on behavior tests, we evaluated locomotor function by measuring the distance traveled in the open field test. Data are expressed as mean ± SEM. The study assumed normal data distributions without formal testing. Statistical analysis was performed using one-way or two-way ANOVAs, as well as paired or unpaired Student’s t tests. p value below 0.05 was deemed statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository97,98 with the dataset identifier PXD065634. The RNA-seq raw data generated in this study have been deposited in the National Center for Biotechnology Information SRA database under the reference PRJNA1282680, PRJNA1282880, PRJNA1282644. Source data are provided as a Source Data file. Source data are provided with this paper.
References
van Hecke, O., Austin, S. K., Khan, R. A., Smith, B. H. & Torrance, N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 155, 654–662 (2014).
Attal, N., Bouhassira, D. & Colvin, L. Advances and challenges in neuropathic pain: a narrative review and future directions. Br. J. Anaesth. 131, 79–92 (2023).
Mills, S. E. E., Nicolson, K. P. & Smith, B. H. Chronic pain: a review of its epidemiology and associated factors in population-based studies. Brit J. Anaesth. 123, e273–e283 (2019).
Li, X. Y. et al. Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science 330, 1400–1404 (2010).
Jin, Y. et al. A somatosensory cortex input to the caudal dorsolateral striatum controls comorbid anxiety in persistent pain. Pain 161, 416–428 (2020).
Damasio, A. R. et al. Subcortical and cortical brain activity during the feeling of self-generated emotions. Nat. Neurosci. 3, 1049–1056 (2000).
Talbot, J. D. et al. Multiple representations of pain in human cerebral cortex. Science 251, 1355–1358 (1991).
De Ridder, D., De Mulder, G., Menovsky, T., Sunaert, S. & Kovacs, S. Electrical stimulation of auditory and somatosensory cortices for treatment of tinnitus and pain. Prog. Brain Res. 166, 377–388 (2007).
Fitzcharles, M. A. et al. Nociplastic pain: towards an understanding of prevalent pain conditions. Lancet 397, 2098–2110 (2021).
Liu, Y. et al. Touch and tactile neuropathic pain sensitivity are set by corticospinal projections. Nature 561, 547–550 (2018).
Ishikawa, T. et al. Pain-related neuronal ensembles in the primary somatosensory cortex contribute to hyperalgesia and anxiety. iScience 26, 106332 (2023).
Pancholi, R., Ryan, L. & Peron, S. Learning in a sensory cortical microstimulation task is associated with elevated representational stability. Nat. Commun. 14, 3860 (2023).
Ziegler, K. et al. Primary somatosensory cortex bidirectionally modulates sensory gain and nociceptive behavior in a layer-specific manner. Nat. Commun. 14, 2999 (2023).
Barbieri, I. & Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 20, 303–322 (2020).
Wei, C., Gershowitz, A. & Moss, B. N6, O2’-dimethyladenosine a novel methylated ribonucleoside next to the 5’ terminal of animal cell and virus mRNAs. Nature 257, 251–253 (1975).
Grozhik, A. V. & Jaffrey, S. R. Distinguishing RNA modifications from noise in epitranscriptome maps. Nat. Chem. Biol. 14, 215–225 (2018).
Keith, J. M., Ensinger, M. J. & Moss, B. HeLa cell RNA (2’-O-methyladenosine-N6-)-methyltransferase specific for the capped 5’-end of messenger RNA. J. Biol. Chem. 253, 5033–5039 (1978).
Liu, Z., Lan, P., Liu, T., Liu, X. & Liu, T. m6Aminer: predicting the m6Am sites on mRNA by fusing multiple sequence-derived features into a CatBoost-Based Classifier. Int. J. Mol. Sci. 24, 7878 (2023).
Akichika, S. et al. Cap-specific terminal N 6-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science 363, eaav0080 (2019).
Boulias, K. et al. Identification of the m6Am methyltransferase PCIF1 reveals the location and functions of m6Am in the transcriptome. Mol. Cell 75, 631–643.e638 (2019).
Sendinc, E. et al. PCIF1 catalyzes m6Am mRNA methylation to regulate gene expression. Mol. Cell 75, 620–630.e629 (2019).
Sun, H., Zhang, M., Li, K., Bai, D. & Yi, C. Cap-specific, terminal N6-methylation by a mammalian m6Am methyltransferase. Cell Res. 29, 80–82 (2019).
Li, K. et al. The CTBP2-PCIF1 complex regulates m6Am modification of mRNA in head and neck squamous cell carcinoma. J. Clin. Investig. 133, e170173 (2023).
Relier, S. et al. FTO-mediated cytoplasmic m6Am demethylation adjusts stem-like properties in colorectal cancer cell. Nat. Commun. 12, 1716 (2021).
Zhuo, W. et al. m6Am methyltransferase PCIF1 is essential for aggressiveness of gastric cancer cells by inhibiting TM9SF1 mRNA translation. Cell Discov. 8, 48 (2022).
Wang, L. et al. Role of PCIF1-mediated 5’-cap N6-methyladeonsine mRNA methylation in colorectal cancer and anti-PD-1 immunotherapy. EMBO J. 42, e111673 (2023).
Zhang, Q. et al. HIV reprograms host m6Am RNA methylome by viral Vpr protein-mediated degradation of PCIF1. Nat. Commun. 12, 5543 (2021).
Cong, S. et al. Integrative proteomic and lipidomic analysis of Kaili Sour Soup-mediated attenuation of high-fat diet-induced nonalcoholic fatty liver disease in a rat model. Nutr. Metab.18, 26 (2021).
Heaton, J. H., Dlakic, W. M., Dlakic, M. & Gelehrter, T. D. Identification and cDNA cloning of a novel RNA-binding protein that interacts with the cyclic nucleotide-responsive sequence in the Type-1 plasminogen activator inhibitor mRNA. J. Biol. Chem. 276, 3341–3347 (2001).
Go, C. D. et al. A proximity-dependent biotinylation map of a human cell. Nature 595, 120–124 (2021).
Shilenok, I. et al. SERPINE1 mRNA binding protein 1 is associated with ischemic stroke risk: a comprehensive molecular-genetic and bioinformatics analysis of SERBP1 SNPs. Int. J. Mol. Sci. 24, 8716 (2023).
Youn, J. Y. et al. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell 69, 517–532.e511 (2018).
Ma, L. et al. ZNF382 controls mouse neuropathic pain via silencer-based epigenetic inhibition of Cxcl13 in DRG neurons. J. Exp. Med. 218, e20210920 (2021).
Wang, D. et al. Lateral septum-lateral hypothalamus circuit dysfunction in comorbid pain and anxiety. Mol. Psychiatr. 28, 1090–1100 (2023).
Cichon, J., Blanck, T. J. J., Gan, W. B. & Yang, G. Activation of cortical somatostatin interneurons prevents the development of neuropathic pain. Nat. Neurosci. 20, 1122–1132 (2017).
Kim, W., Kim, S. K. & Nabekura, J. Functional and structural plasticity in the primary somatosensory cortex associated with chronic pain. J. Neurochem. 141, 499–506 (2017).
Okada, T. et al. Pain induces stable, active microcircuits in the somatosensory cortex that provide a therapeutic target. Sci. Adv. 7, eabd8261 (2021).
Zhang, M. et al. The Cytidine N-Acetyltransferase NAT10 Participates in Peripheral Nerve Injury-Induced Neuropathic Pain by Stabilizing SYT9 Expression in Primary Sensory Neurons. J. Neurosci. 43, 3009–3027 (2023).
Gradinaru, V., Mogri, M., Thompson, K. R., Henderson, J. M. & Deisseroth, K. Optical deconstruction of Parkinsonian neural circuitry. Science 324, 354–359 (2009).
Cassidy, R. M. et al. A lateral hypothalamus to basal forebrain neurocircuit promotes feeding by suppressing responses to anxiogenic environmental cues. Sci. Adv. 5, eaav1640 (2019).
Bordi, M. et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12, 2467–2483 (2016).
Onodera, K. & Kato, H. K. Translaminar recurrence from layer 5 suppresses superficial cortical layers. Nat. Commun. 13, 2585 (2022).
Ping, X. L. et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24, 177–189 (2014).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Nantasenamat, C., Prachayasittikul, V. & Bulow, L. Molecular modeling of the human hemoglobin-haptoglobin complex sheds light on the protective mechanisms of haptoglobin. PLoS ONE 8, e62996 (2013).
Kennedy, M. J. et al. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 7, 973–975 (2010).
Linder, B. et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772 (2015).
Lorsch, Z. S. et al. Stress resilience is promoted by a Zfp189-driven transcriptional network in prefrontal cortex. Nat. Neurosci. 22, 1413–1423 (2019).
Hofacker, D. et al. Engineering of effector domains for targeted DNA methylation with reduced off-target effects. Int. J. Mol. Sci. 21, 502 (2020).
Lai, C. et al. Maf1 suppression of ATF5-dependent mitochondrial unfolded protein response contributes to rapamycin-induced radio-sensitivity in lung cancer cell line A549. Aging 13, 7300–7313 (2021).
Palian, B. M. et al. Maf1 is a novel target of PTEN and PI3K signaling that negatively regulates oncogenesis and lipid metabolism. PLoS Genet 10, e1004789 (2014).
Han, Y. et al. Maf1 loss regulates spinogenesis and attenuates cognitive impairment in Alzheimer’s disease. Brain 16, awae015, (2024).
Smith, K. R. et al. Identification and characterisation of a Maf1/Macoco protein complex that interacts with GABAA receptors in neurons. Mol. Cell. Neurosci. 44, 330–341 (2010).
Sun, H., Li, K., Liu, C. & Yi, C. Regulation and functions of non-m6A mRNA modifications. Nat. Rev. Mol. Cell Bio 24, 714–731 (2023).
Wang, L. et al. PCIF1-mediated deposition of 5’-cap N6,2’-O-dimethyladenosine in ACE2 and TMPRSS2 mRNA regulates susceptibility to SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 120, e2210361120 (2023).
Mauer, J. et al. Reversible methylation of m6Am in the 5’ cap controls mRNA stability. Nature 541, 371–375 (2017).
Sikorski, P. J. et al. The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5’ cap modulates protein expression in living cells. Nucleic Acids Res. 48, 1607–1626 (2020).
Liu, J. et al. Landscape and regulation of m6A and m6Am methylome across human and mouse tissues. Mol. Cell 77, 426–440.e426 (2020).
Engel, M. et al. The role of m6A/m-RNA methylation in stress response regulation. Neuron 99, 389–403.e389 (2018).
Chen, A. et al. Single-cell spatial transcriptome reveals cell-type organization in the macaque cortex. Cell 186, 3726–3743.e3724 (2023).
Codeluppi, S. et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 15, 932–935 (2018).
Li, Z. Z. et al. Extracellular matrix protein laminin β1 regulates pain sensitivity and anxiodepression-like behaviors in mice. J. Clin. Investig. 131, e146323 (2021).
Schöller, E. et al. Interactions, localization, and phosphorylation of the m6A generating METTL3-METTL14-WTAP complex. RNA 24, 499–512 (2018).
Shen, L. Functional interdependence of N6-methyladenosine methyltransferase complex subunits in Arabidopsis. Plant Cell 35, 1901–1916 (2023).
Kosti, A. et al. The RNA-binding protein SERBP1 functions as a novel oncogenic factor in glioblastoma by bridging cancer metabolism and epigenetic regulation. Genome Biol. 21, 195 (2020).
Tsuboyama, K. et al. A widespread family of heat-resistant obscure (Hero) proteins protect against protein instability and aggregation. PLoS Biol. 18, e3000632 (2020).
Bolger, G. B. The RNA-binding protein SERBP1 interacts selectively with the signaling protein RACK1. Cell. Signal. 35, 256–263 (2017).
Lee, Y. J., Hsieh, W. Y., Chen, L. Y. & Li, C. Protein arginine methylation of SERBP1 by protein arginine methyltransferase 1 affects cytoplasmic/nuclear distribution. J. Cell. Biochem. 113, 2721–2728 (2012).
Lee, Y. J., Wei, H. M., Chen, L. Y. & Li, C. Localization of SERBP1 in stress granules and nucleoli. FEBS J. 281, 352–364 (2014).
Shao, J. et al. Synthetic far-red light-mediated CRISPR-dCas9 device for inducing functional neuronal differentiation. Proc. Natl. Acad. Sci. USA 115, E6722–e6730 (2018).
Zhou, H. et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat. Neurosci. 21, 440–446 (2018).
Ma, H. et al. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 34, 528–530 (2016).
Zhou, H. M. et al. DNA N6-methyladenine methylase N6AMT1 controls neuropathic pain through epigenetically modifying Kcnj16 in dorsal horn neurons. Pain 165, 75–91 (2024).
Chen, D. et al. Maf1 regulates axonal regeneration of retinal ganglion cells after injury. Exp. Neurol. 348, 113948 (2022).
Chen, K., Zhu, L., Guo, L., Pan, Y. B. & Feng, D. F. Maf1 regulates dendritic morphogenesis and influences learning and memory. Cell Death Dis. 11, 606 (2020).
Tsang, C. K. et al. Maf1 is an intrinsic suppressor against spontaneous neural repair and functional recovery after ischemic stroke. J. Adv. Res. 51, 73–90 (2023).
Luo, X. et al. IL-23/IL-17A/TRPV1 axis produces mechanical pain via macrophage-sensory neuron crosstalk in female mice. Neuron 109, 2691–2706.e2695 (2021).
Mogil, J. S. Qualitative sex differences in pain processing: emerging evidence of a biased literature. Nat. Rev. Neurosci. 21, 353–365 (2020).
Decosterd, I. & Woolf, C. J. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 (2000).
Bennett, G. J. & Xie, Y. K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107 (1988).
Furman, B. L. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. 1, e78 (2021).
Zhang, M., Du, W., Acklin, S., Jin, S. & Xia, F. SIRT2 protects peripheral neurons from cisplatin-induced injury by enhancing nucleotide excision repair. J. Clin. Investig. 130, 2953–2965 (2020).
Pan, Z. et al. Methyltransferase-like 3 contributes to inflammatory pain by targeting TET1 in YTHDF2-dependent manner. Pain 162, 1960–1976 (2021).
Yang, L. et al. DExH-box helicase 9 modulates hippocampal synapses and regulates neuropathic pain. iScience 27, 109016 (2024).
Pan, Z. et al. Downregulation of a Dorsal Root Ganglion-Specifically Enriched Long Noncoding RNA is Required for Neuropathic Pain by Negatively Regulating RALY-Triggered Ehmt2 Expression. Adv. Sci.8, e2004515 (2021).
Yin, H. et al. Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Mol. Ther. 18, 819–827 (2010).
Sakai, A. et al. MicroRNA cluster miR-17-92 regulates multiple functionally related voltage-gated potassium channels in chronic neuropathic pain. Nat. Commun. 8, 16079 (2017).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic Acids Res 33, W382–W388 (2005).
Jeffrey G. A., W. Saenger, definitions and concepts, Hydrogen Bonding in Biological Structures, 15–48 (1991)
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Feng, J., Liu, T., Qin, B., Zhang, Y. & Liu, X. S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7, 1728–1740 (2012).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009).
Valon L., A. MarÃn-Llauradó, T. Wyatt, G. Charras, & X Trepat. Optogenetic control of cellular forces and mechanotransduction. Nat. Commun. 8, 14396 (2017)
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–d1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–d1527 (2022).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (82371243 and 82171234 to Z-Q Pan, 82201391 to Q-H Wang, 32200818 to L Yang, 82171233 to H-J W), the Jiang Su-Specially Appointed Professor Project and the Natural Science Foundation of Jiangsu Education Department Key Project (22KJA320008 to Z-Q Pan), the Starting Grants of Excellent Talents of Xuzhou Medical University (L. Yang and Q. Wang), and the Postgraduate Research and Practice Innovation Program of Jiangsu Province Grant (KYC22-2929 to Y Huang). Mouse images in Figs. 2p, 3i, 5a, 8p, and 9 were generated by Figdraw.
Author information
Authors and Affiliations
Contributions
Z.-Q.P. and J.-L.C. conceptualized and supervised this study. Z.-Q.P. and Y.H. wrote the manuscript, Z.-Q.P., S.-S.-C.W., and Y.-X.T. revised the manuscript. Y.H., G.M., S.X., R.W., Y.L., Y.Z.(Zeng), Y.Z.(Zhao), Q.W., L.Y., H.H., and L.H. performed experiments and analyzed data, X.Z., H.W. and W.S. analyzed data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Y., Ma, G., Xie, S. et al. SERBP1-PCIF1 complex-controlled m6Am modification in glutamatergic neurons of the primary somatosensory cortex is required for neuropathic pain in mice. Nat Commun 16, 7225 (2025). https://doi.org/10.1038/s41467-025-62565-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62565-5
This article is cited by
-
When Heroes Fall: Reduced Expression of Heat-Resistant Obscure Proteins in Ischemic Stroke
NeuroMolecular Medicine (2025)
