
Abstract
Longitudinal serological proteomic dynamics during antiviral therapy (AVT) in chronic hepatitis B (CHB) patients with liver fibrosis remain poorly characterized. Here, using four-dimensional data-independent acquisition mass spectrometry (4D-DIA-MS), paired liver biopsy (LBx)-proven serum samples from 130 CHB liver fibrosis patients undergoing short-term (78 weeks) or long-term (260 weeks) AVT are analyzed. Our findings show that prolonged AVT drives progressive serological proteomic remodeling in fibrosis regressors, characterized by a temporal inversion in the activation of the complement and coagulation cascades. Using machine learning algorithms trained on the 4D-DIA-MS discovery cohort, we develop a logistic regression model incorporating a seven-protein panel for short-term AVT and a three-protein panel for long-term AVT, respectively, both of which demonstrate moderate discriminatory capabilities for fibrosis regression. Subsequent external validation in an independent cohort (n = 54) with serial LBx assessments at baseline, 78 weeks, and 260 weeks, where serological proteins are quantified using parallel reaction monitoring mass spectrometry (PRM-MS), further confirms their generalizability. Furthermore, our longitudinal trajectory analysis highlights that the long-term proteomic signature exhibits greater stability compared to the short-term panel. This study proposes and validates duration-adapted serological proteomic panels as non-invasive tools for monitoring histological fibrosis regression in on-treatment CHB patients.
Similar content being viewed by others
Introduction
Chronic hepatitis B (CHB) infection remains a principal driver of liver fibrosis burden worldwide1. While liver fibrosis regression has been well-documented in CHB patients following successful suppression of hepatitis B virus (HBV), only 40%-60% of patients achieve regression by antiviral therapy (AVT)2,3. Recent studies indicate that histological non-regression of fibrosis is positively associated with increased risks of hepatocellular carcinoma and liver-related mortality4,5, which can be distressing for patients after prolonged treatment when their fibrosis fails to regress or even worsens. This clinical reality underscores the critical need for longitudinal monitoring strategies to assess liver fibrosis regression during AVT.
Although liver biopsy (LBx) remains the gold standard for assessing the histological status of HBV-related liver fibrosis, its inherent drawbacks (e.g., sampling variability and procedural risks) limit its use for serial monitoring of liver fibrosis6. In recent years, transient elastography (TE), a non-invasive test (NIT), has been developed and shown to be effective for evaluating liver fibrosis in treatment-naïve patients due to its reliability and reproducibility7. However, liver stiffness measured by TE is largely influenced by inflammation rather than fibrotic remodeling during AVT8,9, indicating that TE is not suitable for monitoring liver fibrosis regression in CHB patients on AVT. Similarly, established serological indices and emerging biomarkers, such as platelet counts, platelet-based models (aspartate aminotransferase to platelet ratio index [APRI] and fibrosis-4 index [FIB4]), Wisteria floribunda agglutinin-positive Mac-2-binding protein (M2BPGi), Chitinase-3-like protein 1 (CHI3L1), and Golgi protein 73 (GP73), show good diagnostic potential for treatment-naïve liver fibrosis10,11,12,13. However, they have proven inadequate or nonspecific for accurately evaluating liver fibrosis regression13.
Previous studies have revealed that baseline heterogeneity in host and viral factors—such as age, gender, alcohol consumption, comorbidities, and the natural course of HBV—significantly affects antiviral outcomes, including virologic suppression, biochemical remission, and fibrosis regression14. This variability complicates the effectiveness of routine NITs and the identification of novel biomarkers for monitoring liver fibrosis regression during AVT. Our prior clinical studies also demonstrated that the serological biochemical indices related to HBV infection and liver inflammation significantly improved following a period of AVT, regardless of changes in histological fibrosis2,4,5,15. Therefore, we posit that the changes in the serological proteomic profile may be also highly sensitive to the recovery of virology and inflammation, posing a critical barrier to the discovery of serological proteomic biomarkers for liver fibrosis regression in CHB patients.
To our knowledge, the dynamic serological proteomic profiling of CHB liver fibrosis patients during AVT remains largely unexplored, and there are currently no effective NITs that accurately reflect fibrosis regression in on-treatment CHB patients. However, recent advancements in omics methodologies and the capabilities of machine learning (ML) enable hypothesis-free screening of even thousands of potential biomarker candidates for disease diagnosis and treatment efficiency evaluation16. Consequently, this study employed four-dimensional data-independent acquisition mass spectrometry (4D-DIA-MS) to characterize the comprehensive serological proteomic dynamics during AVT in CHB patients with liver fibrosis, using paired LBx-proven serum samples (n = 130). Additionally, multiple ML models were developed to identify potential serological biomarkers that can distinguish between regressive and non-regressive CHB liver fibrosis patients following AVT. The performance of these models was validated internally through bootstrap resampling and calibration curves, as well as externally in an independent cohort (n = 54) where serological proteins were quantified using parallel reaction monitoring mass spectrometry (PRM-MS).
Results
Study design and patient characteristics
The study design workflow is illustrated in Fig. 1. We analyzed paired serum samples from 130 CHB patients with liver fibrosis receiving AVT, comprising two treatment duration groups: short-term (78 weeks, n = 94) and long-term (260 weeks, n = 36). Paired pre-/post-treatment LBx were obtained for all short-term patients and 15 long-term patients; the remaining 21 long-term patients with baseline clinical diagnosis of compensated cirrhosis provided only post-treatment LBx. Fibrosis regression occurred in 59.6% (56/94) of short-term and 69.4% (25/36) of long-term treated patients, as determined by standardized histological criteria (see “Methods”). Longitudinal serological proteomic profiling was performed using 4D-DIA-MS (discovery cohort), with data randomly divided into training (60%) and testing (40%) sets through stratified sampling on the regression status for ML development. We then employed bootstrap resampling (1000 iterations) and calibration curve analysis for internal validation, complemented by external validation in an independent PRM-MS cohort (n = 54) with serial LBx at baseline (Ishak score ≥ 2), 78 weeks, and 260 weeks. This validation cohort demonstrated fibrosis regression rates of 59.3% (32/54) and 77.8% (42/54) following short-term and long-term treatment, respectively. Furthermore, the model performance was assessed in subgroups of patients from both the 4D-DIA-MS discovery cohort (training and testing sets) and the PRM-MS validation cohort, specifically focusing on three clinically relevant populations: patients with significant baseline fibrosis (Ishak ≥ 3), patients without steatosis at baseline (non-steatosis), and patients with substantial histological improvement versus non-regression following AVT (designated as ΔIshak ≥ 1). Finally, model performance was compared against routine NITs, including liver stiffness measurement (LSM), APRI, and FIB4, across both the 4D-DIA-MS discovery and PRM-MS validation cohorts. The demographic and clinical characteristics of the enrolled patients in the 4D-DIA-MS discovery cohort (training and testing sets) and PRM-MS validation cohort are summarized in Table 1. Detailed subgroup sample sizes are provided in Supplementary Table 1.

Paired serological proteomes from CHB liver fibrosis patients undergoing either short-term or long-term AVT were profiled using 4D-DIA-MS technology. The resulting 4D-DIA-MS data were randomly divided into training and testing sets. ML models, incorporating five feature selection methods and four ML algorithms, were developed using the training set and subsequently validated in the testing set to identify optimal panels for differentiating fibrosis regression status during AVT. The performance of the prioritized models was internally validated through bootstrap resampling and calibration curves, and externally assessed in an independent cohort where serological proteins were quantified using PRM-MS. Additionally, the models were compared with routine NITs and evaluated in clinically relevant subgroups of patients. Abbreviations: 4D-DIA-MS four-dimensional data-independent acquisition mass spectrometry, APRI aspartate aminotransferase to platelet ratio index, ALT alanine aminotransferase; AST aspartate aminotransferase, AVT antiviral therapy, FIB4 fibrosis-4 index, LSM liver stiffness measurement, ML machine learning, NIT non-invasive test, PLS partial least squares, PLT platelet, PIR predominantly progressive, indeterminate, predominantly regressive, PRM-MS parallel reaction monitoring mass spectrometry, SVM support vector machine. Created in BioRender. Chen, W. (2025) https://BioRender.com/84noo7b.
Comparable serological proteomic kinetics in fibrosis regressors versus non-regressors during AVT
Our 4D-DIA-MS analysis quantified a total of 816 serological proteins, which underwent rigorous quality filtering (Supplementary Fig. 1). This process involved the exclusion of immunoglobulins and their fractions, proteins with more than 50% missing values across all samples. As a result, approximately 36.5% of the initial identifications were removed, yielding 518 high-confidence proteins with median coefficients of variation of around 10% across baseline and post-treatment time points (Supplementary Fig. 2). Treatment-induced serological proteomic changes were quantified as post-/pre-treatment (post/pre) ratios for subsequent analyses. Based on the post/pre ratios of 518 robust proteins, hierarchical clustering (HCL) with average linkage and the Euclidean distance metric revealed that the variation trends in the serological proteome were generally comparable between fibrosis regressors and non-regressors in both short-term and long-term AVT groups (Fig. 2a). Principal component analysis (PCA) of the post/pre ratios confirmed this observation, showing no systematic separation between regression status groups or treatment duration cohorts (Fig. 2b). To further assess inter-patient proteomic divergence, we implemented the scale-invariant “1-r” similarity metric. However, the integration of “1-r” values into HCL algorithm failed to distinguish fibrosis regression status in either treatment duration group (Fig. 2c). These complementary analytical approaches demonstrate fundamental similarities in AVT-induced proteomic dynamics between patients with differing fibrosis outcomes. This conserved kinetic signature across regression status groups underscores the challenges of conventional hypothesis-driven approaches for identifying serological biomarkers of histological fibrosis improvement during AVT.

a The heatmap illustrates the overall dynamics of serological proteomes between fibrosis regressors and non-regressors following AVT, represented by the Log2-transformed post/pre ratios of protein abundance (n = 81 for R; n = 49 for NR; n = 94 for short-term; n = 36 for long-term). Darker shades of grey indicate lower ratios, while darker shades of red indicate higher ratios. Both the row post/pre ratios and the column patient samples were clustered using HCL with average linkage and Euclidean distance metrics. b PCA plots compare short-term versus long-term patients and fibrosis regressors versus non-regressors, based on the Log2-transformed post/pre ratios of protein abundance (n = 81 for R; n = 49 for NR; n = 94 for short-term; n = 36 for long-term). Grouped samples are distinguished by distinct colors. c The heatmap depicts the divergence of serological proteomic kinetics between any two patients undergoing AVT, based on the scale-invariant measure “1-Pearson’s correlation coefficient (r)”. Total patients: n = 94 for short-term; n = 36 for long-term; n = 81 for R; n = 49 for NR. Short-term patients: n = 56 for R; n = 38 for NR. Long-term patients: n = 25 for R; n = 11 for NR. Darker shades of grey signify lower divergence, while darker shades of red signify higher divergence. Both row and column “1-r” values were clustered using HCL with average linkage and Euclidean distance metrics. Sample groups are color-coded. Abbreviations: AVT antiviral therapy, NR non-regressor, R regressor.
Prolonged AVT induces progressive serological proteomic shifts in fibrosis regressors and non-regressors
Following short-term AVT, fibrosis regressors exhibited 25 uniquely decreased and 9 increased proteins, while non-regressors showed 38 exclusively decreased and 17 increased proteins, with 50 proteins commonly downregulated and 11 upregulated in both groups (Fig. 3a, b and Supplementary Table 2). However, post/pre ratios of these differentially abundant proteins (DAPs) demonstrated limited discriminatory capacity, with all area under the curve (AUC) values < 0.70 for distinguishing regression status (Fig. 3a, b and Supplementary Table 2). In contrast, fibrosis regressors following long-term AVT displayed pronounced serological proteomic remodeling, characterized by 90 uniquely decreased and 33 increased proteins, whereas non-regressors showed minimal specific changes (13 decreased and 14 increased proteins), alongside 22 commonly downregulated and 10 upregulated proteins (Fig. 3c, d and Supplementary Table 3). Notably, 33 DAPs in regressors following long-term AVT achieved clinically relevant discriminatory power (AUC > 0.70) using post/pre ratios, highlighting their potential as biomarkers for fibrosis regression (Fig. 3c, d and Supplementary Table 3). Comparatively, the extent of serological proteomic changes was more pronounced in fibrosis regressors than non-regressors after long-term AVT.

a, c Venn diagrams illustrate the specifically or commonly upregulated (↑) and downregulated (↓) DAPs between fibrosis regressors and non-regressors following short-term or long-term AVT. DAPs post-AVT were identified using a two-sided paired Student’s t-test or a two-sided Wilcoxon signed-rank test, in conjunction with a FC criterion. Proteins meeting both criteria (p-value < 0.05 and FC > 1.2) were defined as statistically significant. Short-term patients: n = 56 for R; n = 38 for NR. Long-term patients: n = 25 for R; n = 11 for NR. b, d Heatmaps depict the average abundance of DAPs in fibrosis regressors and non-regressors before and after AVT. The intensity of red indicates higher abundance, while darker shades of grey represent lower abundance. The AUC values, calculated based on the post/pre ratios of DAPs to differentiate fibrosis regressors from non-regressors, are visualized (Short-term patients: n = 56 for R; n = 38 for NR. Long-term patients: n = 25 for R; n = 11 for NR). Darker colors represent higher AUC values, and lighter colors correspond to lower AUC values. DAPs with AUC values å 0.7 are marked. DAP groups are color-coded. e, f Significantly enriched KEGG pathways of DAPs that were either unique to or shared between R and NR after short-term or long-term AVT. DAVID employs a modified Fisher’s exact test (one-sided) for KEGG pathway enrichment analysis, with Benjamini-Hochberg correction for multiple testing. An adj.p value < 0.05 was considered statistically significant. g Venn diagrams show the representative pathways that were specifically or commonly enriched (↓, decrease; ↑, increase) between fibrosis regressors and non-regressors in the short-term or long-term patient groups. Abbreviations: AUC area under the curve, DAP differentially abundant protein, Post post-treatment, Pre pre-treatment, Short-R regressor in the short-term group, Short-NR non-regressor in the short-term group, Long-R regressor in the long-term group, Long-NR non-regressor in the long-term group.
Subsequentially, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of DAPs stratified by fibrosis regression status and AVT duration revealed temporal pathway divergence. Specifically, decreased DAPs—whether regressor-specific, shared, or non-regressor-specific—triggered by short-term AVT were predominantly enriched in cell adhesion molecules; in contrast, increased DAPs that were specific to non-regressors or shared by both groups were significantly associated with complement-coagulation cascades (Fig. 3e). Following long-term AVT, decreased DAPs (regressor-specific, shared, or non-regressor-specific) converged on fibrogenesis pathways, including cell adhesion molecules and extracellular matrix (ECM)-receptor interactions, while increased DAPs specific to regressors or shared by both groups sustained complement and coagulation cascades activation (Fig. 3f). Notably, suppression of fibrogenesis persisted across both groups regardless of therapy duration; however, the complement-coagulation responses exhibited a temporal inversion: non-regressors dominated after short-term AVT, whereas regressors prevailed after long-term AVT (Fig. 3g). This divergence underscores the capacity of sustained AVT to drive progressive serological proteomic remodeling in fibrosis regressors, surpassing the changes observed in non-regressors.
Serological proteomic panels for monitoring liver fibrosis regression in CHB patients following short-term AVT
Based on post/pre ratios of the specific DAPs identified in fibrosis regressors and non-regressors in response to short-term AVT from the training cohort, we employed five feature selection methodologies: univariate logistic regression, backward stepwise regression, least absolute shrinkage and selection operator (Lasso) regression, partial least squares (PLS) regression, and support vector machine (SVM). The selected protein panels were subsequently evaluated through four ML algorithms (logistic regression, PLS regression, SVM, and random forest) using ten iterations of fivefold cross-validation. Performance was independently validated in the corresponding testing cohort. As shown in Fig. 4a, the logistic regression model incorporating DAPs selected through univariate feature selection demonstrated optimal performance, achieving training and testing AUCs of 0.85 (95% confidence interval [CI]: 0.74–0.95) and 0.66 (95% CI: 0.47–0.84), respectively, for distinguishing fibrosis regression status. The final panel comprised seven proteins (Supplementary Fig. 3a, b): cholesteryl ester transfer protein (CETP), contactin 1 (CNTN1), paraoxonase 3 (PON3), isocitrate dehydrogenase 1 (IDH1), prosaposin (PSAP), serpin family A member 5 (SERPINA5), and complement factor H related 4 (CFHR4).

a AUC values (95% CI) of ML models developed using five feature selection methods and four ML algorithms. These models were constructed from the training set and validated in the testing set derived from short-term patients in the 4D-DIA-MS discovery cohort. The AUC value (95% CI) of the optimal short-term panel is highlighted in dark red. Training set: n = 34 for R; n = 23 for NR. Testing set: n = 22 for R; n = 15 for NR. b Receiver operating characteristic (ROC) curve analysis of the selected short-term panel and ML model for monitoring fibrosis regression status in short-term patients from the PRM-MS validation cohort (IDH1 was not detected, which was imputed using the mean abundance of IDH1 in the training set). Data were presented as AUC values with 95% CI. PRM-MS validation cohort: n = 32 for R; n = 22 for NR. c AUC values (95% CI) of the selected optimal short-term panel and ML model across clinically relevant populations, including patients with baseline Ishak scores ≥ 3, non-steatosis patients at baseline, and those with ΔIshak ≥ 1 post-short-term AVT. These values are presented for both the 4D-DIA-MS discovery cohort (including the training set, testing set, and the total of both sets) and the PRM-MS validation cohort (IDH1 was not detected, which was imputed using the mean abundance of IDH1 in the training set). Detailed subgroup sample sizes are provided in Supplementary Table 1. Abbreviations: 4D-DIA-MS four-dimensional data-independent acquisition mass spectrometry, AUC area under the curve, PLS partial least squares, PRM-MS parallel reaction monitoring mass spectrometry, SVM support vector machine.
To address potential overfitting, we first performed internal validation using 1,000 bootstrap resamples in the training set (Supplementary Fig. 4a, b). The optimism-corrected AUC remained stable at 0.85 (medium), with calibration analysis showing strong agreement between predicted and observed outcomes (Brier score = 0.154). This performance substantially exceeded that of conventional NITs, including the post/pre ratios of LSM (AUC = 0.64, 95% CI: 0.56–0.72; Brier score = 0.231), APRI (AUC = 0.56, 95% CI: 0.50–0.61; Brier score = 0.238), and FIB4 (AUC = 0.56, 95% CI: 0.50–0.62; Brier score = 0.241) in predicting fibrosis regression. Then, external validation in an independent PRM-MS cohort confirmed the generalizability of the model, yielding an AUC of 0.79 (95% CI: 0.66-0.91) for fibrosis regression status discrimination (Fig. 4b). Moreover, subgroup analyses demonstrated consistent performance across clinically relevant populations: baseline Ishak ≥ 3, non-steatosis patients at baseline, and those with ΔIshak ≥ 1 post-AVT in both the 4D-DIA-MS discovery cohort (training set, testing set, and the total of both sets) and the PRM-MS validation cohort (Fig. 4c). Collectively, this optimized serological proteomic panel demonstrates robust performance across multiple validation frameworks and clinical subgroups, establishing its potential as a non-invasive biomarker for monitoring short-term AVT-induced fibrosis regression in CHB patients.
Serological proteomic panels for monitoring liver fibrosis regression in CHB patients after long-term AVT
To identify serological biomarkers of fibrosis regression following long-term AVT, we extended our analytical pipeline to evaluate post/pre ratios of specific DAPs in fibrosis regressors and non-regressors undergoing prolonged AVT. Using five feature selection methods and four ML algorithms with ten iterations of fivefold cross-validation, univariate logistic regression identified a minimal serological panel comprising three proteins: serpin family A member 7 (SERPINA7), CD163 molecule (CD163), and CFHR4 (Supplementary Fig. 3c, d). While the random forest model incorporating this three-protein panel showed peak training performance (AUC = 0.93, 95% CI: 0.84–1.00) and testing performance (AUC = 0.70, 95% CI: 0.40–1.00), it exhibited limited generalizability in the external PRM-MS validation cohort (AUC = 0.55, 95% CI: 0.47–0.63) (Fig. 5a, b). In contrast, the logistic regression model incorporating the same three-protein panel demonstrated consistent discriminative capacity across the training set (AUC = 0.87, 95% CI: 0.71–1.00), testing set (AUC = 0.68, 95% CI: 0.28–1.00), and external PRM-MS validation cohort (AUC = 0.75, 95% CI 0.63–0.88) (Fig. 5a, b). Internal validation via 1,000 bootstrap resamples confirmed the robustness of the model, with an optimism-adjusted AUC of 0.87 and excellent calibration (Brier score = 0.138), surpassing conventional NITs, including the post/pre ratios of LSM (AUC = 0.59, Brier = 0.214), APRI (AUC = 0.72, Brier = 0.184), and FIB-4 (AUC = 0.77, Brier = 0.195) (Supplementary Fig. 4c, d). Additionally, subgroup analyses further validated stable performance among clinically relevant strata (baseline Ishak ≥ 3, non-steatosis at baseline, and ΔIshak ≥ 1 post-AVT) across both discovery and validation cohorts (Fig. 5c), underscoring the stability and generalizability of the model for diverse patient populations.

a AUC values (95% CI) for ML models developed using five feature selection methods in conjunction with four ML algorithms. These models were constructed from the training set and subsequently validated in the testing set derived from long-term patients in the 4D-DIA-MS discovery cohort. The AUC values (95% CI) for the optimal long-term panels are highlighted in dark red. Training set: n = 15 for R; n = 7 for NR. Testing set: n = 10 for R; n = 4 for NR. b ROC curve analysis of the selected long-term panel, used to monitor fibrosis regression status in long-term patients from the PRM-MS validation cohort (CD163 was not detected, which was imputed using the mean abundance of IDH1 in the training set). Data were presented as AUC values with 95% CI. PRM-MS validation cohort: n = 42 for R; n = 12 for NR. c AUC values (95% CI) of the ML models across clinically relevant populations, including patients with baseline Ishak scores ≥ 3, non-steatosis patients at baseline, and those exhibiting a ΔIshak ≥ 1 post-long-term AVT. These values are presented for both the 4D-DIA-MS discovery cohort (comprising the training set, testing set, and the aggregate of both sets) and the PRM-MS validation cohort (CD163 was not detected, which was imputed using the mean abundance of IDH1 in the training set). Detailed subgroup sample sizes are provided in Supplementary Table 1. Abbreviations: 4D-DIA-MS four-dimensional data-independent acquisition mass spectrometry, AUC area under the curve, PLS partial least squares, PRM-MS parallel reaction monitoring mass spectrometry, SVM support vector machine.
Comparison of short-term and long-term serological proteomic panels for monitoring liver fibrosis regression in on-treatment CHB patients
A systematic comparison of short-term and long-term proteomic panels revealed distinct temporal dynamics in their ability to monitor AVT-induced fibrosis regression. As illustrated in Supplementary Fig. 5, the short-term panel lost its discriminative capacity for regression status after prolonged AVT, while the long-term panel similarly failed to stratify outcomes following short-term AVT (AUC < 0.60 across discovery and its subgroup cohorts). This finding confirms the duration-dependent remodeling of serological proteomes between fibrosis regressors and non-regressors. Subsequently, cross-panel analysis identified CFHR4 as the sole conserved biomarker shared between the long-term and short-term panels. CFHR4 demonstrated moderate diagnostic performance for long-term AVT response across the training set (AUC = 0.81, 95% CI: 0.60–1.00), testing set (AUC = 0.70, 95% CI: 0.39–1.00), and combined discovery cohort (AUC = 0.72, 95% CI: 0.53–0.90), but a poor diagnostic performance in the external PRM-MS validation cohort (AUC = 0.60, 95% CI: 0.43–0.77) (Supplementary Fig. 6).
Additionally, longitudinal trajectory analysis revealed critical divergences in protein behavior. Long-term panel proteins, including SERPINA7, CD163, and CFHR4, exhibited consistent directional changes across both the 4D-DIA-MS discovery and PRM-MS validation cohorts. Specifically, SERPINA7 (CD163 was not detected) was downregulated, while CFHR4 was upregulated in regressors after long-term AVT (Fig. 6a). Notably, these proteins displayed a trend of inverse trajectories during pre-treatment fibrogenesis—SERPINA7 and CD163 (p < 0.05) were elevated, whereas CFHR4 was suppressed in cases of progressive fibrosis (Supplementary Fig. 7)—establishing their dual relevance as biomarkers for both fibrogenesis and regression. In stark contrast, short-term panel proteins exhibited cohort-specific variability during AVT (Supplementary Fig. 8), with inconsistent inverse relationships to baseline fibrotic progression (Supplementary Fig. 7). This result underscores superior biological fidelity of long-term proteomic signatures, which consistently reflect bidirectional fibrosis dynamics compared to the transient and context-dependent fluctuations observed in short-term markers. Furthermore, functional characterization through gene set enrichment analysis (GSEA) linked the dynamics of the long-term panel (CFHR4 upregulation and CD163 downregulation) to the regulation of complement-coagulation cascades during sustained AVT (Fig. 6b). Collectively, these findings establish the temporal specificity of proteomic biomarkers in fibrosis monitoring and highlight the clinical necessity for duration-adapted serological panels. Additionally, long-term signatures offer robust mechanistic insights into AVT-induced fibrosis regression, potentially through the modulation of coagulation and complement pathways.

a Box plots with overlaid longitudinal trajectories illustrate the dynamic alterations in serological levels of long-term panel proteins (SERPINA7, CFHR4, CD163) between baseline (Week 0) and post-long-term AVT (Week 260) in fibrosis regressors versus non-regressors. Data are derived from the 4D-DIA-MS discovery cohort (training set: n = 15 pairs for R and n = 7 pairs for NR; testing set: n = 10 pairs for R and n = 4 pairs for NR) and the PRM-MS validation cohort (n = 42 pairs for R and n = 12 pairs for NR). All paired serum samples were biological replicates, with each pair originating from an individual patient. Normalized protein abundances were compared using a two-sided paired Student’s t-test (for normally distributed data) or a two-sided Wilcoxon signed-rank tests (for non-parametric data). A p-value < 0.05 was considered statistically significant. The box plots display the median (50th percentile; centre line), the 25th and 75th percentiles (box bounds), and the minima and maxima (whiskers) within 1.5 times the interquartile range (IQR) from the box bounds. b GSEA was performed on the complement and coagulation cascade pathway using proteins (CFHR4 and CD163) stratified into high- and low-expression groups based on median abundance thresholds in the 4D-DIA-MS discovery cohort. GSEA uses permutation-based significance testing to evaluate directional enrichment of gene sets in ranked gene lists, inherently employing a one-sided approach to detect coordinated over-representation at extremes of the expression profile. The enrichment plot displays the trajectory of the enrichment score across rank-ordered proteins, with the leading edge subset (indicated by the vertical red line) representing the proteins driving pathway enrichment. The p-value for the enrichment analysis is shown in the plot, highlighting the statistical significance of the enrichment. Abbreviations: 4D-DIA-MS four-dimensional data-independent acquisition mass spectrometry, NR non-regressor, PRM-MS parallel reaction monitoring mass spectrometry, R regressor.
Discussion
The high prevalence of HBV-related liver fibrosis remains a significant contributor to end-stage cirrhosis and hepatocellular carcinoma17, highlighting the growing need for reliable NITs to assess liver fibrosis. Current diagnostic approaches prioritize NITs such as APRI, FIB-4, and imaging elastography as first-line evaluations for antiviral-naïve patients with HBV-related liver fibrosis18. However, these methods present notable limitations, including indeterminate results and a stronger correlation with liver inflammatory activity than with histological fibrotic remodeling8,19,20,21, which diminishes their utility in reflecting LBx-verified fibrosis regression following AVT. Additionally, emerging evidence suggests that baseline heterogeneity in host-pathogen interactions may also influence AVT-induced liver fibrosis regression14, posing challenges for both the implementation of conventional NITs and the development of novel biomarkers to monitor fibrotic resolution during treatment.
Our prior investigation established the LBx-dependent “Beijing Classification” system as a validated framework for assessing post-AVT fibrosis regression2,4,5. While histological evaluation remains the gold standard, serum-based NITs may offer distinct advantages in clinical practicality and patient acceptability22. Leveraging advancements in high-throughput proteomics, we employed cutting-edge 4D-DIA-MS to analyze paired serum samples from CHB patients with histologically confirmed liver fibrosis undergoing AVT. The 4D-DIA-MS technique combines data-independent acquisition with ion mobility separation, enhancing the accuracy and depth of proteomic quantification23. Using this innovative platform, we identified over 500 reproducibly quantified proteins across longitudinal serum samples following rigorous quality control. However, unsupervised clustering approaches were unable to differentiate between fibrosis regressors and non-regressors after AVT, based on the post/pre ratios or scale-invariant measures of changes in these robust proteins. This convergence in serological proteome dynamics likely reflects near-universal virological suppression (undetectable HBV DNA) and inflammatory normalization (ALT normalization) across our cohort, even with short-term AVT. These findings further support the notion that serological proteomic fluctuations during AVT primarily reflect virological and inflammatory normalization, presenting substantial challenges in identifying serological protein biomarkers of histological fibrosis improvement.
While serological proteomic trajectories showed broad similarities between fibrosis regressors and non-regressors during AVT, longitudinal analysis revealed that prolonged AVT drives divergent proteomic remodeling in regressors, surpassing the changes observed in non-regressors. This aligns with our recent clinical and transcriptomic observations demonstrating the superior fibrosis regression rates following long-term versus short-term AVT (80.4% versus 65.2%, respectively)24, coupled with the enhanced restoration of ECM-related gene signatures specifically in regressors during prolonged therapy25. These temporal patterns suggest that the increased detection of reduced DAPs in regressors after long-term AVT likely reflects cumulative histological improvements achieved through sustained AVT. Intriguingly, proteomic profiling uncovered treatment-duration-dependent shifts in pathway activation. Although fibrogenesis suppression persisted in both groups irrespective of AVT duration, short-term AVT preferentially amplified complement-coagulation cascades in non-regressors—a pattern reversed with prolonged therapy, where regressors displayed dominant activation of these pathways. Studies have demonstrated that the complement and coagulation system plays a pivotal role in liver homeostasis and immune responses, with its activation contributing to beneficial effects on liver immune response and regeneration26. Therefore, the temporal switch in complement-coagulation activation implies that long-term AVT may synergistically enhance immunoregulatory competence and microenvironmental homeostasis, even without overt fibrosis regression.
Currently, while ML offers transformative potential for biomarker prioritization in omics discovery pipelines, translational implementation remains challenging due to frequent overfitting and lack of clinical validation16. To address these limitations, we implemented rigorous partitioning of our 4D-DIA-MS proteomic dataset into independent training and testing cohorts prior to feature selection, ensuring no data leakage during model development27. This strategy capitalized on the strength of 4D-DIA-MS in generating reproducible, untargeted proteomic profiles—ideal for unbiased discovery—while leveraging the capacity of ML to decode complex biomarker signatures. Notably, the optimized logistic regression models, incorporating a seven-protein panel for short-term AVT and a three-protein panel for long-term AVT, demonstrated superior discrimination of fibrosis regression compared to conventional NITs in internal validation. Critically, we bridged discovery to clinical applicability through PRM-MS-based external validation in an independent cohort, exploiting the precision of PRM for targeted quantification28, and confirmed the model robustness across platforms. Subgroup analyses further reinforced clinical utility, showing consistent performance across diverse populations (baseline Ishak ≥ 3, non-steatosis patients at baseline) and treatment responders (ΔIshak ≥ 1) in both 4D-DIA-MS discovery and PRM-MS validation phases. This multilayered validation framework—spanning technical reproducibility (4D-DIA-to-PRM transition), cohort independence (training-testing-external cohorts), and clinical heterogeneity (subgroup consistency)—establishes a paradigm for translating ML-derived proteomic signatures into clinically actionable tools. Moreover, we systematically evaluated the incremental value of clinical parameters alongside serological proteomic features during model training. Unfortunately, integrative models combining protein and clinical data failed to outperform serological protein-only classifiers in discriminating fibrosis regression (Supplementary Fig. 9), suggesting that proteomic signatures may capture latent biological signals beyond conventional clinical metrics—a hypothesis requiring further mechanistic investigation.
Our longitudinal proteomic analyses revealed notable differences in AVT-induced biological remodeling based on treatment duration. Long-term AVT resulted in more pronounced serological proteome reorganization and pathway activation in fibrosis regressors compared to short-term AVT, highlighting duration-dependent functional adaptations. This temporal divergence provides a mechanistic basis for the observed specificity of our diagnostic panels: the short-term model (7-protein panel) was unable to effectively discriminate fibrosis regression status at long-term endpoints, while the long-term panel (3-protein signature) demonstrated limited utility during the early treatment phases. Importantly, the proteins included in the long-term panel exhibited consistent directional changes across both the 4D-DIA discovery and PRM validation cohorts, showing inverse trajectories during pre-treatment fibrogenesis. In contrast, the constituents of the short-term panel displayed inter-cohort variability in expression patterns, showing inconsistent inverse relationships with baseline fibrotic progression. The modest proteomic perturbations observed during the early treatment phases, combined with the biological noise inherent to acute therapeutic responses, likely contribute to the inter-cohort inconsistency and temporal instability of the short-term signatures. In contrast, the robustness of the long-term panel—validated across orthogonal platforms and diverse populations—positions it as a clinically actionable tool for identifying non-regressors after prolonged AVT. Except for the superior stability, the long-term panel also exhibited mechanistic relevance to complement-coagulation cascades, aligning with sustained functional modulation following sustained AVT. This stratification capability has direct translational relevance, enabling intensified monitoring of high-risk patients to mitigate the incidence of decompensated cirrhosis and hepatocellular carcinoma through timely clinical interventions.
To our knowledge, this study represents the pioneering longitudinal proteomic investigation characterizing on-treatment serological dynamics and biomarker trajectories associated with histologically confirmed fibrosis regression in CHB patients. The study presents several notable advantages. First, we used paired LBx-validated serum samples from multicenter randomized controlled trials, ensuring clinical relevance through standardized enrollment criteria and antiviral regimens2,4,5,15. The histopathological rigor was reinforced by robust inter-observer agreement (κ = 0.65; ICC = 0.71, 95% CI: 0.65–0.76), exceeding the thresholds necessary for reproducible fibrosis staging29. Second, we employed a dual-phase discovery-validation framework: 4D-DIA-MS facilitated unbiased proteome-wide profiling, while PRM-MS provided targeted verification, leveraging their complementary strengths in discovery depth and quantification precision23,28. Third, our multilayered validation strategy, which included internal bootstrapping, external PRM-MS validation, and subgroup analyses across different populations, confirmed the generalizability of the models while mitigating the risk of overfitting. However, our study also has limitations. 1. There was cohort asymmetry, with the long-term group having fewer paired serum samples than the short-term group in the 4D-DIA-MS discovery cohort, which may limit the power to detect subtle proteomic shifts. 2. While 4D-DIA-MS demonstrated robust reproducibility, its detection threshold may have excluded low-abundance proteins that are critical to fibrotic resolution. 3. The failure in quantifying IDH1 in the short-term panel and CD163 in the long-term panel by PRM-MS technique, which may have led to an underestimation of diagnostic performance. 4. Although serological biomarkers offer non-invasive clinical utility, their expression levels and regulatory dynamics may differ from those in liver tissue due to compartmentalized pathophysiology. Future studies integrating paired tissue-serum proteomics are needed to resolve this critical knowledge gap.
In conclusion, our study addressed a critical unmet need in CHB-related liver fibrosis management by linking proteomic dynamics to histological outcomes, offering actionable tools for monitoring fibrosis regression; the association of the long-term panel with complement-coagulation activation offers valuable mechanistic insights into AVT-induced functional restoration (Fig. 7). This study advances non-invasive fibrosis monitoring by integrating cutting-edge proteomics with ML-driven biomarker discovery. Its strengths lie in technical rigor, validation depth, and biological plausibility of identified pathways. However, limitations in cohort size, diagnostic performance, and mechanistic elucidation highlight the need for multicenter validation and functional studies to translate these findings into clinical practice.

Prolonged AVT induced progressive serological proteomic and functional shifts in both fibrosis regressors and non-regressors. Notably, serological proteomic recovery was more pronounced in fibrosis regressors compared to non-regressors following long-term AVT. Additionally, a seven-protein panel was identified for monitoring fibrosis regression during short-term AVT, while a three-protein panel was established for long-term AVT. Abbreviations: AVT antiviral therapy, CHB chronic hepatitis B, DAP differentially abundant protein, NR non-regressor, R regressor.
Methods
Study population
CHB patients with liver fibrosis undergoing AVT were screened from our previous nationwide multicenter randomized controlled clinical trials and their extension studies (ClinicalTrials.gov identifiers: NCT01938781, NCT01938820, NCT03777969, and NCT02849132). Inclusion criteria: age 18 years or older; availability of paired LBx pre- and post-treatment (either 78 or 260 weeks of AVT) or clinically diagnosed compensated cirrhosis at baseline with post-treatment LBx; baseline fibrosis Ishak score ≥ 2 if the pre-treatment LBx was available; availability of serum samples at both baseline and the time of post-treatment LBx; hepatitis B surface antigen (HBsAg) positive for at least 6 months before AVT; HBV DNA levels > 20,000 IU/ml for hepatitis B e antigen (HBeAg)-positive patients or > 2000 IU/ml for HBeAg-negative patients; and length of the LBx core ≥ 0.5 cm with at least five portal tracts. Exclusion criteria: current or historical liver decompensation (ascites, variceal bleeding, or encephalopathy); elevated serological alpha-fetoprotein (> 100 ng/mL) or creatinine ( > 1.5 times the upper normal limit); coinfection with hepatitis C or human immunodeficiency virus; other severe liver conditions or serious organ disorders; and pregnancy or lactation. Patients who met the inclusion and exclusion criteria were divided into a discovery group and a validation group for subsequent serological biomarker discovery. To ensure internal consistency across all participating centers, we implemented standardized protocols that included the following aspects: all centers uniformly applied identical eligibility criteria for patient enrollment; all participants received a standardized AVT regimen with entecavir as the therapeutic backbone; systematic clinical evaluations were conducted at six-month intervals following established follow-up protocols; and the primary outcomes focused on histological fibrosis regression and the incidence of predefined clinical endpoints. All enrolled patients provided written informed consent. The study was approved by the Ethics Committee of Beijing Friendship Hospital, Capital Medical University (2016-P2-021-04), and conducted in accordance with the Declaration of Helsinki. Patients and/or the public were not involved in the design, conduct, reporting, or dissemination plans of our research.
Clinical examination
The clinical examination encompasses the collection of demographic data, routine blood tests, liver biochemistry analyses, and LSM. Detailed methodologies for these procedures are thoroughly documented in our prior publications2,4,15,30.
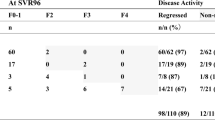
Histological assessment
Formalin-fixed paraffin-embedded blocks of LBx were sectioned into 5-μm-thick slices, stained with reticulin, and assessed by two pathologists who were blinded to the experimental conditions, timing of the LBx, and other clinical details. Inter-observer agreement for histopathological scoring was assessed using ICC and Cohen’s kappa (κ), with 95% confidence intervals calculated via bootstrapping (1000 iterations). Discordant cases were reviewed by a third senior pathologist to reach a consensus. Histological changes were evaluated using Ishak and/or PIR scores2. The PIR score represents a novel pathological classification designed to assess the dynamic changes in fibrosis quality2. It categorizes fibrosis into three types: predominantly progressive (characterized by thick, broad, loose, and pale septa with inflammation), predominantly regressive (marked by delicate, thin, dense, and splitting septa), and indeterminate (exhibiting a balance between progressive and regressive scarring). This classification underscores the significance of evaluating and recognizing the evolving nature of fibrosis, particularly in the context of AVT. For patients with paired LBx, a fibrosis regressor was defined as either a decrease in the Ishak score by ≥ 1 after treatment (ΔIshak ≥ 1) or a stable Ishak score accompanied by predominantly regressive changes according to the post-treatment PIR score. For patients clinically diagnosed with compensated cirrhosis before treatment, a fibrosis regressor was identified as having an Ishak stage ≤ 4 post-treatment (ΔIshak ≥ 1) or exhibiting predominantly regressive changes in those with Ishak fibrosis stages 5–6 after treatment. Conversely, a fibrosis non-regressor was defined as a patient who did not meet the criteria for fibrosis regression. Additionally, patients without liver steatosis at baseline (non-steatosis) were defined as having histological steatosis of less than 5% before treatment for those with paired LBx. For patients clinically diagnosed with compensated cirrhosis prior to treatment, non-steatosis was determined based on the absence of signs of fatty liver detected on ultrasound.
Clinical diagnosis of compensated cirrhosis
Patients diagnosed with compensated cirrhosis should meet at least one of the following criteria. (1) Presence of endoscopic examination: An endoscopic examination revealed esophageal and/or gastric varices, with the exclusion of non-cirrhotic portal hypertension. (2) Absence of endoscopic examination: If an endoscopic examination has not been performed, at least two of the following four criteria must be satisfied. i. Imaging examination: Ultrasound, computed tomography, or magnetic resonance imaging indicated typical changes associated with cirrhosis, including an irregular liver surface, granular or nodular liver parenchyma, with or without splenomegaly (defined as spleen thickness exceeding 4.0 cm or greater than 5 rib units). ii. Hematological indicators: Routine blood tests showed a platelet count of <100 × 109/L, with no alternative explanation. iii. Serum and coagulation abnormalities: Serum albumin levels <35 g/L or prothrombin time extended by more than 3 s (after discontinuation of thrombolytic or anticoagulant drugs for more than 7 days), or decreased cholinesterase levels (excluding the influence of anti-cholinesterase drugs). iv. Liver stiffness: LSM > 12.4 kPa.
Serum sample preparation
Blood samples were collected using standard venepuncture protocols into pro-coagulation vacuum tubes. Serum was obtained by centrifugation at 376 × g for 10 min at 4 °C and immediately frozen at −80 °C until use. Highly abundant serological proteins in each 40 μL sample were removed using the ProteoExtract® Albumin/IgG Removal Kit (MilliPore, MA, USA) following the vendor’s protocol, as albumin constitutes 70–90% of the serological proteome. The depleted serum was freeze-dried and reconstituted in SDS lysis buffer (Beyotime, Hangzhou, China). After centrifugation at 12,000 × g for 10 min at room temperature, the supernatant was collected, and protein concentration was detected using a bicinchoninic acid assay. Each sample containing 10 μg of protein was loaded onto a 12% SDS-PAGE gel for separation. The gel was stained with Coomassie Brilliant Blue using eStain LG (GenScript, Nanjing, China) and analyzed with an automatic digital gel imaging system (Tanon, Shanghai, China).
Equal amounts of protein (50 μg) were adjusted to a consistent concentration and volume based on the measured protein concentration. Samples were then treated with dithiothreitol (Thermo Fisher Scientific, MA, USA) at a final concentration of 5 mM and incubated at 55 °C for 30 min. Subsequently, 10 mM iodoacetamide (Thermo Fisher Scientific) was added and allowed to react in the dark for 15 min at room temperature. Protein precipitation was achieved by mixing samples with six volumes of pre-cooled acetone and incubating at −20 °C for overnight. After centrifugation at 8000 × g for 10 min at 4 °C, the resulting pellets were dissolved in 100 μL of 50 mM ammonium bicarbonate solution (Thermo Fisher Scientific) and digested with Trypsin (1 μg, Likuso, Shanghai, China) at 37 °C overnight. The enzymatic reaction was halted by adjusting pH to 3 with phosphoric acid (Sigma, MO, USA). Peptides were desalted using SOLA™ SPE (Thermo Fisher Scientific), freeze-dried in a vacuum freeze dryer, and then reconstituted with iRT peptides (1:10, Biognosys, Zug, Switzerland).
The peptide samples underwent fractionation using an Agilent 1100 HPLC System (Agilent, CA, USA). A gradient of mobile phases A (2% acetonitrile in HPLC-grade water) and B (90% acetonitrile in HPLC-grade water) was employed for reverse-phase chromatography. The gradient was programmed as follows: from 0 to 10 min, 2% B; from 10 to 10.01 min, 2% to 5% B; from 10.01 to 37 min, 5% to 20% B; from 37 to 48 min, 20% to 40% B; from 48 to 48.01 min, 40% to 90% B; from 48.01 to 58 min, 90% B; from 58 to 58.01 min, 90% to 2% B; from 58.01 to 63 min, 2% B. Separation of tryptic peptides occurred at a flow rate of 250 μL/min and was monitored at 210 nm and 280 nm. Fractions were collected every minute from 10 to 50 minutes into centrifuge tubes 1–10 sequentially, repeating this collection sequence until the gradient was complete. The separated peptides were subsequently lyophilized for mass spectrometry (MS) analysis.
4D-DIA-MS analysis
Serological proteome of the discovery cohort was quantified using 4D-DIA-MS by Shanghai OE Biological Technology Co., Ltd. (Shanghai, China), following established protocols. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was performed using an Evosep One system (Evosep Biosystems, Odense, Denmark) coupled to a timsTOF Pro 2 mass spectrometer (Bruker Daltonics, MA, USA). Samples were loaded onto Evotip C18 traps at 1 μL/min and separated on a 15-cm analytical column using the standardized 30SPD method (15-min effective gradient). Mobile phases consisted of 0.1% (v/v) formic acid in water (Solvent A) and 0.1% (v/v) formic acid in acetonitrile (Solvent B). Mass spectrometry operated in PASEF mode31 with shared parameters: capillary voltage at 1.5 kV, dry gas flow of 3.0 L/min at 180 °C, m/z range of 100–1700, and ramped collision energy (20–59 eV). For data-dependent acquisition (DDA), precursors were fragmented across ion mobility (1/K0) values of 0.85–1.3 Vs/cm2 using intensity-triggered MS/MS (isolation width: ±1 m/z; topN=12). Data-independent acquisition (DIA) employed extended ion mobility (0.6–1.6 Vs/cm2) with variable isolation windows covering the full m/z range, synchronizing cycle time with chromatographic peak width. DDA established spectral libraries, while DIA enabled quantitative profiling.
MS/MS spectra preprocessing was performed using Spectronaut Pulsar™ (v15.3, Biognosys) under the following parameters: Trypsin was specified as the proteolytic enzyme with allowance for up to two missed cleavages; methionine oxidation and N-terminal carbamidomethylation were set as variable and fixed modifications, respectively; a 1% false discovery rate (FDR) threshold was applied at three hierarchical levels (peptide-spectrum match, peptide, and protein) using the Target-Decoy strategy with mProphet algorithm implementation32. DDA-derived spectral libraries were used for DIA data processing, with precursor, peptide, and protein FDR thresholds set to 1%. Protein identification was conducted against the UniProt-reviewed human proteome database (uniprot-reviewed_yes+taxonomy_9606.fasta). Quantification was executed at the MS2 level using an intensity-based local normalization strategy, where peptide quantities were derived from the mean intensity of the top 1–3 ranked precursors, and protein quantities were subsequently calculated as the mean of 1–3 highest-abundance peptides33. To ensure data quality, the abundance matrix of serological proteins was cleaned by removing immunoglobulin or its fractions and proteins with over 50% missing data. Missing data imputation and proteomic data normalization were performed using the missForest and vsn packages, respectively. Additionally, the coefficient of variation was employed to evaluate the abundance variability of each serological protein among individuals.
Expression and functional analyses of serological proteome
Changes in serological proteome post-AVT were analyzed by calculating the Post/Pre ratios of MS signal intensity and visualized using the ComplexHeatmap package. The divergence in serological protein abundance among individuals was assessed using the one minus Pearson’s Correlation Coefficient (1-r) with the corrplot package. HCL with average linkage and Euclidean distance metric was used to visualize similarities and differences in serological protein abundance among individuals using the pheatmap package. PCA was performed to identify categorical variables (principal components, PCs) in the data based on observed variables, serving to verify the results of the HCL. PCA was implemented using the PCA function from the FactoMineR package, with confidence ellipse drawn using the factoextra package with default parameters. DAPs post-AVT were identified using a two-sided paired Student’s t-test or a two-sided Wilcoxon signed-rank test, combined with the fold change (FC) criterion. Proteins with a p-value < 0.05 and FC > 1.2 were considered statistically significant. Venn analysis was performed to identify specific and common DAPs between or among groups using the VENNY 2.1 tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html). Functional interpretation of DAPs was performed using KEGG pathways through the DAVID web server (https://davidbioinformatics.nih.gov)34,35, with an adjusted p-value < 0.05 considered statistically significant. GSEA was performed to interpret the molecular functions of proteins of interest based on Sangerbox 3.035. Samples were categorized into low and high subgroups based on the median Post/Pre ratio of protein of interest, with a p-value < 0.05 considered statistically significant. Visualization of DAPs of interest was implemented using ggplot2 package.
ML models
The ML analysis was conducted with the aim to identify potential serological proteins that could distinguish histological fibrosis regression in CHB patients with liver fibrosis following short-term or long-term AVT. Initially, the short-term or long-term data from 4D-DIA-MS discovery cohort were randomly divided into training and testing sets through stratified sampling on the regression status, comprising 60% and 40% of the patients, respectively, using the caret package. The training set was employed for model development, while the testing set was reserved for subsequent validation. Feature selection was then performed using five methods: univariate logistic regression, backward stepwise regression, Lasso regression, PLS regression, and SVM applied to specific DAPs in regressors or non-regressors from the training set. This process employed packages including stats, MASS, glmnet, ropls, and e1071. Following feature selection, ML models, including regularized Logistic regression with Ridge penalization, PLS regression, SVM, and random forest, were established based on the five selected features using the glmnet, kernlab, e1071, randomForest, caret, MLmetrics, and pROC packages. Models that incorporated relatively fewer serological proteins but demonstrated good performance in both training and testing sets were prioritized for further internal and external validations. Internal validation was conducted in the training set using bootstrap resampling (drawing datasets of equal size to the original training cohort with replacement over 1,000 iterations) and calibration curves by the boot and calibrate packages.
PRM-MS analysis
To validate candidate short-term and long-term biomarker panels in the independent validation cohort, a customized PRM-MS assay was performed by Shanghai OE Biological Technology Co., Ltd. (Shanghai, China). This targeted approach prioritized maximal coverage of candidate proteins. Serum protein preparation and DDA-based spectral library construction followed identical protocols to those described for 4D-DIA-MS experiments, ensuring methodological consistency. Targeted proteomics via PRM-MS was performed on a timsTOF Pro 2 instrument (Bruker Daltonics) coupled to a nanoflow liquid chromatography system. Tryptic peptides were separated on a reversed-phase C18 column (75 μm × 25 cm, 1.6 μm, 120 Å; IonOpticks, Melbourne, Australia) at 300 nL/min using mobile phase A (0.1% formic acid in H2O) and B (0.1% formic acid in acetonitrile) with a 60 min nonlinear gradient: 2 → 22% B (0–45 min), 22 → 37% B (45–50 min), 37 → 80% B (50–55 min), and 80% B (55–60 min). Mass spectrometry operated in positive-ion PRM-PASEF mode with precursor lists configured in timsControl PRM module. Key parameters included: 1.4 kV capillary voltage, 100–1700 m/z scan range, ion mobility window (1/K0) of 0.6–1.6 Vs/cm2, 100 ms ion accumulation/release time, and 10 min scheduled retention time windows, enabling high-efficiency targeted quantification.
Raw PRM data were analyzed with SpectroDive software (v11.8, Biognosys) through a two-step workflow: (1) automated peak extraction with subsequent manual validation and retention time alignment to ensure peptide identification accuracy and cross-sample consistency, followed by (2) protein quantification using the mean peptide quantity algorithm (arithmetic mean of normalized peptide intensities) embedded in the software. Sparse data points were imputed non-parametrically via the missForest package, and global normalization was applied using the vsn package.
Statistical analysis
Statistical analysis was performed using R 4.4.3 (https://www.r-project.org/) or GraphPad Prism 9 (GraphPad Software Inc., CA, USA). Continuous variables were presented as mean ± standard deviation or median (interquartile range), while categorical variables were summarized with counts and percentages. For comparisons between paired groups, continuous variables were analyzed using a two-sided paired Student’s t-test or a two-sided Wilcoxon signed-rank test, depending on the normality of data distribution. Continuous variables among three independent groups were compared using one-way ANOVA (for normally distributed data with equal variances) or the Kruskal-Wallis H test (for non-parametric comparisons). Categorical variables among three independent groups were analyzed using the Chi-square test (when expected frequencies ≥ 5) or the Fisher-Freeman-Halton exact test (when expected frequencies <5). A p-value of <0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw 4D-DIA-MS data, PRM-MS data, and their processed peptide and protein abundance data generated in this study have been deposited in the Proteomics Identifications Database (PRIDE, https://www.ebi.ac.uk/pride/) under accession codes PXD059174 and PXD063833.
Code availability
The R codes generated in this study are available in the Figshare database (https://doi.org/10.6084/m9.figshare.27087253). All R packages used in this study are publicly available through the Comprehensive R Archive Network (CRAN) or Bioconductor, with specific version numbers and persistent digital object identifiers (DOIs) provided in Supplementary Table 4.
References
Polaris Observatory, C Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol. Hepatol. 3, 383–403 (2018).
Sun, Y. et al. New classification of liver biopsy assessment for fibrosis in chronic hepatitis B patients before and after treatment. Hepatology 65, 1438–1450 (2017).
Chang, T. T. et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology 52, 886–893 (2010).
Sun, Y. et al. Regression of liver fibrosis in patients on hepatitis B therapy is associated with decreased liver-related events. Clin. Gastroenterol. Hepatol. 22, 591–601 e593 (2024).
Chang, X. et al. The utility of P-I-R classification in predicting the on-treatment histological and clinical outcomes of patients with hepatitis B and advanced liver fibrosis. Hepatology 79, 425–437 (2024).
Schmoyer, C. J., Kumar, D., Gupta, G. & Sterling, R. K. Diagnostic accuracy of noninvasive tests to detect advanced hepatic fibrosis in patients with hepatitis C and end-stage renal disease. Clin. Gastroenterol. Hepatol. 18, 2332–2339 e2331 (2020).
Martinez, S. M., Crespo, G., Navasa, M. & Forns, X. Noninvasive assessment of liver fibrosis. Hepatology 53, 325–335 (2011).
Dong, X. Q. et al. Declining in liver stiffness cannot indicate fibrosis regression in patients with chronic hepatitis B: a 78-week prospective study. J. Gastroenterol. Hepatol. 34, 755–763 (2019).
Wong, G. L. et al. On-treatment monitoring of liver fibrosis with transient elastography in chronic hepatitis B patients. Antivir. Ther. 16, 165–172 (2011).
Lian, M. J., Chen, Z. Q., Wang, Q. M., Zheng, G. S. & Hong, G. L. Diagnostic accuracy of mac-2-binding protein glycosylation isomer for diagnosing hepatitis B-related fibrosis: a meta-analysis. J. Dig. Dis. 23, 550–560 (2022).
Das, A. et al. Evaluation of Chitinase 3-like 1 (CHI3L1) as a noninvasive biomarker of hepatic fibrosis in patients with Hepatitis B virus-related compensated chronic liver disease. J. Fam. Med Prim. Care 10, 1694–1698 (2021).
Cao, Z. et al. Assessment of serum Golgi protein 73 as a biomarker for the diagnosis of significant fibrosis in patients with chronic HBV infection. J. Viral Hepat. 24, 57–65 (2017).
Sun, Y. M., Chen, S. Y. & You, H. Regression of liver fibrosis: evidence and challenges. Chin. Med. J. 133, 1696–1702 (2020).
Darmadi, D. et al. Factors affecting HBV DNA suppression in chronic hepatitis B patients treated with tenofovir disoproxil fumarate. F1000Res 11, 1521 (2022).
Sun, Y. et al. Persistent low level of hepatitis B virus promotes fibrosis progression during therapy. Clin. Gastroenterol. Hepatol. 18, 2582–2591 e2586 (2020).
Thiele, M. et al. Opportunities and barriers in omics-based biomarker discovery for steatotic liver diseases. J. Hepatol. 81, 345–359 (2024).
Shen, C., Jiang, X., Li, M. & Luo, Y. Hepatitis virus and hepatocellular carcinoma: recent advances. Cancers 15, https://doi.org/10.3390/cancers15020533 (2023).
Sterling, R. K. et al. AASLD Practice Guideline on imaging-based noninvasive liver disease assessment of hepatic fibrosis and steatosis. Hepatology, https://doi.org/10.1097/HEP.0000000000000843 (2024).
Huang, L. L. et al. Liver stiffness measurement can predict liver inflammation in chronic hepatitis B patients with normal alanine transaminase. J. Clin. Transl. Hepatol. 11, 817–826 (2023).
Abu-Freha, N. et al. Liver fibrosis regression and associated factors in HCV patients treated with direct-acting antiviral agents. Life 13, https://doi.org/10.3390/life13091872 (2023).
Kim, W. R. et al. Evaluation of APRI and FIB-4 scoring systems for non-invasive assessment of hepatic fibrosis in chronic hepatitis B patients. J. Hepatol. 64, 773–780 (2016).
Wong, V. W., Adams, L. A., de Ledinghen, V., Wong, G. L. & Sookoian, S. Noninvasive biomarkers in NAFLD and NASH—current progress and future promise. Nat. Rev. Gastroenterol. Hepatol. 15, 461–478 (2018).
Li, J., Smith, L. S. & Zhu, H. J. Data-independent acquisition (DIA): An emerging proteomics technology for analysis of drug-metabolizing enzymes and transporters. Drug Discov. Today Technol. 39, 49–56 (2021).
Chen, S. et al. Liver fibrosis showed a two-phase regression rate during long-term anti-HBV therapy by three-time biopsies assessments. Hepatol. Int. 18, 904–916 (2024).
Chen, W. et al. Matrisome gene-based subclassification of patients with liver fibrosis identifies clinical and molecular heterogeneities. Hepatology 78, 1118–1132 (2023).
Thorgersen, E. B. et al. The role of complement in liver injury, regeneration, and transplantation. Hepatology 70, 725–736 (2019).
Desaire, H. How (Not) to generate a highly predictive biomarker panel using machine learning. J. Proteome Res. 21, 2071–2074 (2022).
Guergues, J. et al. A semi-automated workflow for DIA-based global discovery to pathway-driven PRM analysis. Proteomics 25, e2400129 (2025).
Pavlides, M. et al. Interobserver variability in histologic evaluation of liver fibrosis using categorical and quantitative scores. Am. J. Clin. Pathol. 147, 364–369 (2017).
Zhang, M. et al. Persistent steatosis correlates with decreased fibrosis regression during anti-HBV treatment in patients with chronic HBV infection. J. Med. Virol. 95, e29156 (2023).
Meier, F. et al. Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell Proteom. 17, 2534–2545 (2018).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Zhang, H. & Bensaddek, D. Narrow precursor mass range for DIA-MS enhances protein identification and quantification in Arabidopsis. Life 11, https://doi.org/10.3390/life11090982 (2021).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Shen, W. et al. Sangerbox: A comprehensive, interaction-friendly clinical bioinformatics analysis platform. Imeta 1, e36 (2022).
Acknowledgements
This work was supported by National Key Research and Development Program (2023YFC2306900 [to H.Y.]), National Natural Science Foundation of China (82430023 [to H.Y.], 82130018 [to HY], 82470646 [to W.C.], 82170613 [to W.C.], 82300696 [to S.C.]), Beijing Municipal Science & Technology Commission (Z221100007422115 [to Y.S.]), and Young Talent Funds of Chinese Institutes for Medical Research (CX23YQB08 [to W.C.], CX23YQB12 [to Y.S.]).
Author information
Authors and Affiliations
Contributions
Conception and design: H.Y., W.C.; project supervision: H.Y., W.C.; sample collection: S.Y.C., Y.M.S., X.N.W., J.L.Z., B.Q.W., T.T.M., R.X.H.; data acquisition: M.Y.Z., W.C., H.Y., S.Y.C., Y.M.S.; data analysis: M.Y.Z., W.C.; data interpretation: M.Y.Z., W.C., H.Y., Y.M.S., S.Y.C.; manuscript drafting: W.C., M.Y.Z.; critical revision of the manuscript: W.C., M.Y.Z., H.Y.; all authors reviewed and edited this manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jordan J. Feld and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, M., Chen, S., Wu, X. et al. Serological proteomic characterization for monitoring liver fibrosis regression in chronic hepatitis B patients on treatment. Nat Commun 16, 7714 (2025). https://doi.org/10.1038/s41467-025-63006-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63006-z
