
Abstract
High-energy and resource-abundant anode-free sodium metal batteries suffer from limited lifespan owing to dendrite growth and rapid capacity fading at early stages, particularly at high rate and high capacity. Here we report a preliminary surface-passivation strategy by highly fluorinated electrolyte, instantly forming a dense inorganic-dominant primitive solid-electrolyte interphase. Driven by dipole-dipole interaction, the spontaneously formed solid-electrolyte interphase is sufficiently dense to resist solvent decomposition and moisture attack, meanwhile regulating Na plating/stripping at high current densities and areal capacities up to 8 mA cm-2 and 5 mAh cm-2, respectively. The fabricated anode-free batteries demonstrate long-term stability at high cathode loadings (10–15 mg cm-2) and high rates (1–3 C), even with moisture contained. Impressively, fast-charging Ah-level anode-free pouch cells deliver energy density up to 150 Wh kg-1 (0.5 C) based on total cell weight, achieving power density as high as 152 W kg-1 and long lifespan up to 700 cycles at 1 C.
Similar content being viewed by others


Introduction
Sodium metal batteries (SMBs) have been extensively studied as a cost-effective alternative to lithium-ion batteries due to raw materials abundance, high theoretical specific capacity (1166 mAh g−1), and low electrode potential (− 2.71 V vs. standard hydrogen electrode) of metallic Na1,2,3. Compared to sodium-ion batteries (SIBs) with insufficient energy density (~ 90–160 Wh kg−1), SMBs are expected to increase the energy density beyond 200 Wh kg−1, which rivals the commercial lithium-ion batteries (LIBs), manifesting great potential in achieving high-energy-density energy storage systems4,5. To further raise the full-cell energy density, meanwhile simplifying the manufacturing process that involves soft and highly reactive Na metal, anode-free sodium batteries (AFSBs) have been proposed6,7 (Supplementary Note 1). In AFSBs, metallic Na does not exist in the initially assembled cell at the negative electrode side, but electrochemically forms on the current collector during the first charging step, by the Na ions solely stored in the positive electrode8. Accordingly, the zero excess of Na inventory bears its limited consumption and requires higher reversibility of metallic Na. Recently, delightful progress has been made in demonstrating long-life Ah-level AFSBs, achieving high energy density at mild or moderate rates9,10,11, based on ether electrolytes because of their compatibility with Na metal negative electrode. However, the power density of rechargeable batteries is equally important12,13. Further improvements are still required to develop simultaneously energy-dense and power-dense AFSBs for practical application.
As the holy grail of advanced energy storage systems, realizing both high energy density and power density demands battery cycling stability at high current density, high capacity, and high depth of discharge (DoD), i.e., “three-high” principle, which determines the threshold of charging/discharging rate, the amount of energy stored, and the minimized inactive cell mass/volume, respectively. However, the practice of this “three high” principle raises tremendous challenges for SMBs, especially when these conditions are simultaneously involved14,15. At high current densities, dendrite growth readily occurs among the unevenly deposited Na and undesirable side reactions become aggravated with excessive formation of “dead Na”16,17,18,19. While sodium dendrites eventually grow to cause short circuit that results in sudden cell failure and dangerous thermal runaway, the accumulating “dead Na” leads to rapid capacity fading due to Na inventory loss20,21,22, rendering poor battery lifespan. Both dendrite growth and “dead Na” formation further deteriorate when areal capacity increases, because the large volume change during charging/discharging cycles induces repeated breakage of solid-electrolyte interphase (SEI), constantly degrading the morphology of Na deposits and depleting active Na. Notably, the unremitting sacrifice of active Na is fatal at high DoD, or low negative to positive capacity ratio (N/P ratio), where the amount of supplementary Na is limited23,24. In AFSBs (DoD = 100% or N/P = 0), without excess Na, the capacity decay further accelerates under inadequate conditions, ending up with drastically shortened cycle life. Therefore, high-energy-density and high-power-density AFSBs complied with the “three high” principle renders strict requirements to the stability and reversibility of metallic Na.
Establishing a desirable SEI is crucial to the stable cycling of Na metal negative electrode by inhibiting excessive interfacial reactions between electrolytes and highly reactive Na metal, guiding homogeneous Na deposition, enabling fast sodium ion transport, and accommodating large volume change. Recently, decent cell performances have been achieved at mild or moderate operating conditions by constructing inorganic-rich SEI through electrolyte engineering25,26,27,28,29,30,31,32, particularly those enriched with NaF, in virtue of its high interfacial energy, high mechanical strength and fast sodium ion diffusion33,34. However, the reported NaF-rich SEI are still regarded as heterogeneous and organic-inorganic mixed. Instead of the ideal model of a bilayer structure with an inner inorganic layer and an outer organic layer, the electrochemically formed SEI are more like a mosaic assemblies of randomly distributed inorganic and organic microphases due to the inevitable solvent decomposition or sluggish film-forming kinetics of fluorine-contained ingredients35,36,37. Such mosaic patches of inorganic and organic phases readily result in largely differentiated Na+ migration and uneven Na deposition, particularly at high current densities. So far, the realization of long-cycle-life AFSBs at “three high” conditions has been rarely reported by SEI modification, considering that a sufficiently dense and homogeneous inorganic-dominant SEI is still hardly acquired at an early stage. On the other hand, the significance of primitive SEI38, i.e., the passivation layer chemically formed upon immediate contact between sodium metal and electrolytes, is often overlooked. As the SEI is constituted by insoluble decomposition products of both chemical and electrochemical reactions between electrolyte and metal, a primitive SEI can form in prior to the electrochemical formation process, which plays a key role in passivating the metallic electrode beforehand and mitigate the unwanted solvent decomposition during electrochemical reactions.
In this article, by tuning electrolyte chemistry, we demonstrate a dense and homogeneous inorganic-dominant SEI in situ formed on sodium metal at an early stage, that withstands high current densities, high capacities, and high depth of discharge (low N/P ratio), as well as water corrosion and high-temperature operation (Fig. 1). Unlike the commonly reported ion-dipole and ion-ion interactions modulating Na+ solvation structures, a unique dipole-dipole interaction is induced by employing the non-coordinating diluent methyl perfluorobutyl ether (MPE) in diethylene glycol dimethyl ether (diglyme), rearranging the electron density distribution on solvent molecules and altering the preferential reduction order of solvated clusters. Without entering Na+ solvation structure to involve in the electrochemical formation of SEI, the highly fluorinated non-coordinating diluent directly contributes to forming NaF-dominant primitive SEI by a spontaneous chemical reaction. Interestingly, the chemically non-reactive-to-Na MPE is excited in the solvent-solvent complex due to the redistribution of electron density triggered by dipole-dipole interaction, becoming preferentially decomposed on the Na metal surface with facilitated C-F bond cleavage. The loosely bonded fluorine atoms release and chemically form a self-limiting passivation film in a short time frame, even in the absence of either sodium salts or applied current. Benefiting from the favorable decomposition thermodynamics and fast reaction kinetics, such a primitive SEI is dominated by the inorganic NaF phase in a compact manner, which isolates the fresh Na metal from the electrolyte and moisture, inhibiting interfacial side reactions and water corrosion, that minimizes the waste of active Na once after its formation. The homogeneous nature of NaF-rich SEI leads to evenly distributed Na+ influx and migration39, enabling uniform yet dense Na deposition and suppressing metal dendrite growth at elevated current densities and large areal capacities. Consequently, using carbon coated aluminum foil (C@Al) as negative electrode/current collector and sodium vanadium phosphate (NVP) as positive electrode, anode-free coin cells (N/P ratio = 0) exhibit stable cycling up to 480 cycles, at high cathode loadings (up to 15 mg cm−2) and high rates (up to 3 C), even using moisture-containing electrolytes (5000 ppm). Impressively, Ah-level anode-free C@Al| |NVP pouch cells achieve a long lifespan of 700 cycles at high rates and lean electrolyte conditions (~ 2 g Ah−1), realizing high energy densities (up to 150 Wh kg−1) and high power densities exceeding 152 W kg−1 based on total cell weight.

Dipole-dipole interaction between electron-rich O atom (δO-) in G2 and electron-deficient H atom (δH+) in MPE that alters electron density distribution in solvent-solvent complex and facilitates C-F bond cleavage for spontaneous and fast surface passivation. The dense NaF-dominant primitive SEI protects Na metal at an early stage and ensures the reversible cycling of Na metal under harsh conditons.
Results
Electrolyte design and properties
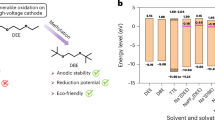
The electrolyte design commenced with a theoretical screening of fluorinated solvents candidates (Supplementary Table 1), by examining both their properties and capability of forming a dense and homogeneous NaF-rich electrode-electrolyte interphase. Hence, three critical descriptors were considered, that are molecular orbital (MO) energy levels, C-F Laplacian bond order (LBO), and F/C molar ratio. Considering the low solvation ability and salt solubility of solvents containing electron-withdrawing groups such as fluorine31, as shown in Supplementary Fig. 1, these fluorinated solvents are supposed to serve as a co-solvent to diglyme (G2), chosen for its moderate viscosity and good solvation capability. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of these solvent candidates were determined by density functional theory (DFT) calculations (Fig. 2a and Supplementary Data 1). The molecular orbital energy indicates the preferential decomposition between the fluorine-containing solvents and fluorine-free G2, thereby featuring their film-forming ability in terms of thermodynamics. LBO was calculated as a comparative and qualitative indicator to distinguish relative bond strength trends among different C–F bonds in fluorinated solvents (Supplementary Figs. 2–12), where the fluorine atoms with lower LBO value are more likely to be released, promoting solvent defluorination and film formation35. Furthermore, a high F/C molar ratio of solvent molecules is expected, which provides an abundant source of fluorine atoms for surface fluorination40. As shown in Fig. 2b, solvents that fall closer to the upper left corner are expected to be more effective in forming fluorine-containing SEI. Based on the above considerations, methyl perfluorobutyl ether (MPE) was chosen as the ideal cosolvent, that the film-forming electrolyte was composed of 1 M NaPF6 in a binary solvent of G2/MPE mixture, denoted by NGMPE (Supplementary Note 2). The conventional electrolyte of 1 M NaPF6 in G2 was chosen as the base electrolyte for comparison41, referred as NG2.

a HOMO and LUMO energy levels of G2 and fluorinated solvents. b Screening of fluorinated solvents for design of film-forming electrolytes. c Raman spectra of neat solvents and electrolytes. MD snapshots of (d) NG2 and (e) NGMPE electrolytes, with an inset showing their representative solvation structures, where Na, C, H, O, P, and F is marked by blue, gray, white, red, violet, and cyan, respectively. f RDFs of the NG2 (upper) and NGMPE (lower) electrolytes. g The representative Na+ cation solvate species (SSIP, CIP and AGG) in NG2 and NGMPE electrolytes, as well as their schematic illustrations. h Temperature-dependent ionic conductivities of the electrolytes with the corresponding activation energies. i MSD of Na+ ions in the electrolytes. j Wettability of the electrolytes on Celgard 2325 separator demonstrated by their contact angles.
The Na+ solvation structures and coordination environments in NG2 and NGMPE electrolytes were examined by Raman spectroscopy (Fig. 2c). Compared to the pure G2 solvent, NG2 exhibited an additional peak at 741 cm−1 assigned to the P-F stretch peak of coordinated PF6-, while the characteristic peak of free G2 molecules for C-O stretching vibration showed a blue-shift from 850 cm−1 to 870 cm−1 after dissolving NaPF6, implying the strong Na+-G2 interactions42,43,44. However, in NGMPE, the characteristic peaks of Na+-G2 and Na+-PF6- pairs remained nearly unchanged upon the addition of MPE. Meanwhile, the peaks of free MPE molecules were inherited in NGMPE (780 cm−1 and 750 cm−1). Therefore, MPE is considered as a “non-coordinating” diluent, failing to engage in the salt dissociation nor participate in the Na+ solvation structure. The negligible Na+-MPE interaction arises from the non-polar nature of MPE, with |electrostatic potential minimum (ESPmin)| << electrostatic potential maximum (ESPmax) (Supplementary Fig. 13 and Supplementary Table 2), rendering limited Na+ affinity28,45. The non-coordinating attribute of MPE was further corroborated by classic molecular dynamics (MD) simulations of NG2 and NGMPE (Supplementary Data 1). As shown in the MD snapshots (Fig. 2d, e), the main solvation structure of Na+ was maintained through MPE incorporation, that Na+ is solely coordinated to PF6- anions and G2 molecules in the primary solvation sheath, with MPE molecules uniformly dispersed outside of the solvation structure. In the radial distribution function (RDF) analysis (Fig. 2f), the dominant occupation of primary solvation sheath by PF6- anions and G2 solvents was indicated by the prominent peaks of Na-FPF6−and Na-OG2 pairs appear around 2.4 Å in both electrolytes, while MPE molecules are absent within the radii of primary and secondary solvation sheaths, indicated by the peaks of Na-OMPE and Na-FMPE at the distance as far as 6.5 Å, consistent with previous inference. Noteworthily, without entering Na+ solvation structures, the non-coordinating diluent spatially compressed the solvation clusters (Supplementary Figs. 14, 15) and reduced solvent molecules interacting with Na+, thereby allowing more anions entering the solvation shells. The ratio of PF6- to G2 coordinated with Na+ increased from 0.460 in NG2 to 0.534 in NGMPE. Moreover, compared to NG2, NGMPE becomes predominantly constituted by contact ion pairs (CIP) and aggregated ions (AGG), with a reduced content of solvent-separated ion pairs (SSIP), which is widely acknowledged to mitigate excessive solvent decomposition and benefit the construction of inorganic-rich SEI (Fig. 2g). The proportion of representative solvation structures from MD simulations was also corroborated by the results of Raman spectroscopy (Supplementary Fig. 16).
The physicochemical properties of electrolytes are optimized by the incorporation of fluorinated diluent MPE. Their ionic conductivities were collected at various temperatures, as shown in Fig. 2h, where NGMPE exhibited higher ionic conductivity compared to NG2. The increased ion mobility in NGMPE is ascribed to its reduced viscosity with low-viscosity diluent MPE and the facile movement of the compressed solvating clusters46. The charge carrier mobility in these electrolytes were compared by calculating the mean-squared displacements (MSD) over a time interval for 9000 ps (Fig. 2i), that NGMPE exhibited a much higher Na+ diffusion coefficient of 4.3 × 10−8 cm2 s−1 than NG2 of 1.8 × 10−8 cm2 s−1. The oxidative stability of NG2 and NGMPE was further probed by linear sweep voltammetry (LSV) in Na| |Al half cells (Supplementary Fig. 17). As the base electrolyte, NG2 started decomposition at 4.2 V, the oxidation threshold voltage of the fluorinated electrolyte NGMPE was extended up to 4.6 V. The appearance of the small bump of oxidative current density at 4.8 V implied that the decomposition of NGMPE may passivate the electrode surface and inhibit the further oxidation of electrolyte at higher voltage47,48. Owing to the reduced viscosity, NGMPE also showed improved surface wettability by demonstrating smaller contact angles on the separators (Fig. 2j and Supplementary Note 3) and various current collectors, i.e., Cu and Al foil (Supplementary Figs. 18, 19), which endows it with uniform distribution on the electrode-electrolyte interface for homogeneous SEI formation and rapid electrolyte infiltration in large-size pouch cells even under lean electrolyte conditions49,50.
Facile formation of primitive SEI
Unlike most of the electrolyte-induced SEI that gradually establishes by the electrochemical reactions during cell cycling, a dense and homogeneous SEI can form by the chemical reactions with the incorporation of MPE, upon immediate contact between electrolyte and Na metal, referred to as “primitive SEI”38. The formation of primitive SEI in NGMPE is spontaneous and occurs in a very short time frame. The surface evolution was monitored through time after immersing Na metal in NG2 and NGMPE electrolytes, as well as their corresponding solvents (Fig. 3a and Supplementary Fig. 20). As expected, the surface of Na metal immersed in G2 and MPE solvents remained shiny throughout the test, indicating the non-reactivity of neat solvents to Na and absence of catalytic impurities, e.g., metal impurities or trace water. In contrast, surface change occurred on Na metal immersed in NG2 and NGMPE electrolytes, in which the metal chunk in NG2 ended up with a rusty and mottled surface, while that in NGMPE was fully covered by a uniform surface layer. The composition of the thus-formed surface layer in NGMPE was characterized by X-ray diffraction analysis (XRD), the characteristic peaks of NaF were identified in its diffraction pattern, indicating crystalline NaF in situ formed on metallic Na (Fig. 3b). By collecting the surface layer, intense and sharp diffraction peaks of NaF were solely observed, with eliminated interference of Na substrate (Supplementary Fig. 21 and Supplementary Note 4). The formation of such a crystalline NaF primitive SEI was further proved in actual batteries, through XRD characterization of sodium metal after infiltrating actual amount of electrolyte (80 μL) in coin cells and cell rest of 12 h (Supplementary Fig. 22). Interestingly, an identical surface layer was in situ formed on the Na metal immersed in G2-MPE solvent mixture without addition of sodium salts, albeit in a slightly slower rate. It implies that the spontaneous chemical reaction was not driven by the ion-ion interactions or ion-dipole interactions involving dissociated Na+ cations or PF6- anions, instead being triggered by a unique dipole-dipole interaction between solvent molecules. Although the dipole-dipole interactions are rarely reported in previous studies and considered extremely weak, which are usually several orders of magnitude weaker than the commonly discovered ion-ion interactions or ion-dipole interactions51, they can still greatly alter the electrolyte stability and properties. Based on DFT calculations, G2-MPE solvent clusters have a lower LUMO energy than individual solvents (Fig. 3c), implying its greater tendency for reduction at the Na metal surface52. Such an unusual system was also validated by additional DFT calculations based on different basis sets, which yielded similar trends and consistent results (Supplementary Table 3). The underlying dipole-dipole interactions were investigated by the 1H nuclear magnetic resonance (NMR) spectroscopy and 2D 1H–1H correlation spectroscopy (2D 1H–1H COSY). Considering the subtle interactions within the solvent-solvent pairs, the NMR experiments were performed using a coaxial internal insert tube to conserve the pristine microstructure of the solvents (Supplementary Fig. 23)53. The presence of intermolecular interaction is discerned by the diverse chemical shifts in the 1H spectra of the G2-MPE mixture compared to those of G2 and MPE single solvents. As shown in Fig. 3d, as the 1H-NMR of MPE in binary G2-MPE mixture exhibited an obvious downfield shift, the hydrogens in G2 molecules are found shifting upfield after the introduction of MPE, which is rather counterintuitive because the strongly electron-withdrawing F atom in MPE usually results in de-shielding effect of H atom54. It may be explained by the strong electron-donating capability of the methyl group in MPE that induces polarization of electrons and enhances electron cloud density toward H atoms in G2, as a result of the proximal solvent molecules due to dipole-dipole interaction. The dipole-dipole interaction induced by the attraction of electron-rich O (δO-) in G2 and electron-deficient H (δH+) in MPE, as supported by the ESP distribution (Supplementary Fig. 13), was further confirmed in both neat solvent mixture and electrolyte by the 2D 1H–1H COSY51,53,55,56. In addition to the interference signals of intramolecular 1H coupling, which manifest in high intensities due to their much closer proximity, the 1H–1H coupling signals (marked by circles) were detected for the intermolecular solvent-solvent interactions in the solvent mixture of G2-MPE (Fig. 3e). The 1H–1H coupling demonstrates the shortened distance between G2 and MPE solvent molecules driven by the δO-(G2)-δH+(MPE) interaction, which leads to their overlapped H electron clouds. As a result, the dipole-dipole interaction alters the electron cloud density and distribution among the solvent molecules, thereby reforming the bond strength and facilitating the C-F cleavage. As the weak dipole-dipole interaction is proved in the neat solvent mixture, such an intermolecular interaction is found further enhanced by dissolving sodium salts, owing to the polarizing effect of Na+ on the G2 molecules (Fig. 3f). It corresponds to the previous observation in the primitive SEI formation (Supplementary Fig. 20), that the unique dipole-dipole interactions between G2 and MPE induced the spontaneous defluorination of MPE and surface passivation in G2-MPE binary solvent, while the release of fluorine atoms was further facilitated by the enhanced dipole-dipole interactions, thereby the accelerated formation of primitive SEI. This often-overlooked dipole-dipole interaction, even though being rather weak, may commonly exist in the multinary electrolyte systems, and it should be considered as a critical design rule for primitive SEI formation. This is particularly important for AFSBs with limited Na inventory, that the rapid formation of a self-limiting primitive SEI minimizes the Na wastage in the repeated electrochemical formation cycles.

a Optical observation of the surface evolution of Na metal immersed in neat solvents, solvent mixtures, and electrolytes. b XRD patterns of surface layers formed in film-forming electrolyte NGMPE, by immersing Na foil in the fluorinated electrolyte for 2 h. c LUMO energy levels of neat solvents, solvent mixture, and electrolytes, where Na, C, H, O, P, and F is marked by purple, gray, white, red, brown, and cyan, respectively, with red and blue shades corresponding to low and high electron density regions. d NMR spectra of 1H of neat solvents and solvent mixture. COSY spectra of (e) solvent mixture G2 + MPE showing the dipole-dipole interactions in the absence of either cations or anions, and (f) binary electrolyte NGMPE showing the enhanced dipole-dipole interactions. ToF-SIMS 3D reconstructions of depth-profiling of representative secondary ion species (individuals and overlays) in the sodium metal cycled in (g) NG2 electrolyte and (h) NGMPE electrolyte for 10 cycles at 8 mA cm−2 and 4 mAh cm−2. i Low-magnification cryogenic transmission electron microscopy (cryo-TEM) of the in situ formed primitive SEI after exposure to NGMPE electrolyte for 2 h. j High-resolution TEM (HRTEM) of the primitive SEI, with lattice fringes of crystalline regions marked in different colors. k Fast Fourier transform (FFT) of cryo-HRTEM of the primitive SEI.
Dense and water-resistant robust SEI
Following the prior formation of a favorable primitive SEI, a dense and homogeneous NaF-dominant SEI was established on the sodium metal after its chemical and electrochemical formation in the NGMPE electrolyte. To reveal the compositional distribution and chemical nature of SEI formed at high current densities in different electrolytes, time-of-flight secondary ion mass spectrometry (ToF-SIMS) and X-ray photoelectron spectroscopy (XPS) were implemented on the negative electrode of Na metal cycled at 8 mA cm−2. The depth profiles of various representative secondary ion species, such as the organic fragments C2HO- and CHO2- and inorganic fragments NaF2- and F2-, were collected by the high-resolution ToF-SIMS. In Fig. 3g, the upper stratum of the SEI in NG2 electrolyte is dominated by the organic species C2HO- and CHO2-, mainly attributed to the reduction of G2 solvent57,58. Meanwhile, the inorganic moieties NaF2- and F2- from NaPF6 decomposition are interspersed among the organic phases, constituting a typical mosaic SEI structure with random assembly of organic and inorganic components. In sharp contrast, inorganic species NaF2- and F2- are uniformly and densely distributed throughout the SEI induced by NGMPE, along with very little organic species (Fig. 3h). The NaF-enriched SEI in NGMPE benefited from both the decomposition of PF6- salt anions during electrochemical formation cycles and the cleavage of the C-F bond of highly fluorinated MPE solvent during the chemical formation process. The fluorine-dominant SEI in NGMPE was also validated by in-depth XPS. In the F 1 s spectra (Supplementary Fig. 24), the peak intensity of Na-F signals near 684 eV attenuates through increasing etching depth in the NG2 because of the decreased amount of inorganic components. In NGMPE, the Na-F signals remain strong throughout different etching times, emphasizing the NaF-rich nature of SEI formed by the fluorinated electrolyte, which is in accord with the results from ToF-SIMS analysis. Furthermore, the in situ formed primitive SEI was investigated by cryo-TEM. The low-magnification cryo-TEM shows a dense and thin layer of spherical nanocrystals on Na metal surface after its exposure to NGMPE electrolyte for 2 h (Fig. 3i), of which the surface morphology remains unchanged by extending its exposure to 48 h (Supplementary Fig. 25), indicating the facile formation of such a dense primitive SEI and its self-limiting nature. The composition and crystallinity of the inorganic-dominant primitive SEI are obtained from cryo-HRTEM of the spherical regions via identification of lattice fringes and analysis of FFT. As shown in Fig. 3j, the primitive SEI is mainly composed of crystalline NaF, with its (200) planes predominantly exposing, which is also implied by a clear and almost continuous circle of the corresponding diffraction spots in the FFT of cryo-HRTEM (Fig. 3k) and complied with the XRD results59.
Impressively, the NaF-dominant SEI from the NGMPE electrolyte was sufficiently dense to suppress interfacial side reactions and dendrite growth, and even resisted water corrosion. The water-resistant SEI was testified by immersing Na metal in an ethanol solution of 25 vol% water and G2 solvent containing 20000 ppm of water, respectively, after the chemical formation of primitive SEI (Fig. 4a, b). In both solutions, volatile reactions manifested on bare Na metal, with excessive gas emission and heat release (Supplementary Movies 1, 2), while no obvious change was observed on Na metal from NGMPE, owing to the complete isolation of fresh Na by the dense primitive SEI. A comparative analysis of the gas evolution during battery cycling in different electrolytes, as an indicator of solvent decomposition and interfacial reactions, was conducted by in situ differential electrochemical mass spectrometry (DEMS) using Na metal symmetric cells (Fig. 4c). At the initial rest stage (0–2 h), strong signals of H2 production were detected in both electrolytes, indicating a chemical reaction was initiated on the Na metal electrode surface upon immediate contact with electrolytes. However, the H2 signal is much narrower and appears earlier in NGMPE, implying the fast kinetics of chemical reaction forming self-limiting primitive SEI at a very early stage, which is consistent with the previous optical observation. Then, the Na| |Na symmetric cells were cycled at a high current density of 5 mA cm−2 for 10 cycles. In the base electrolyte NG2, intensive peaks arise during the discharging and charging steps, which means volatile gas emission is induced by the excessive side reactions at high current density. The symmetric cell with NGMPE exhibited a flat and smooth baseline throughout the battery cycling, demonstrating the sufficiently compact SEI formed on sodium metal that effectively inhibits continuous electrolyte decomposition and interfacial reactions even at high current density. Such a dense inorganic-rich SEI also regulates the uniform Na+ influx and homogeneous Na deposition, suppressing dendrite growth at high current densities, which was affirmed by in situ optical microscopy (OM) (Fig. 4d, Supplementary Movies 3, 4 and Supplementary Note 5). At a high current density of 3 mA cm−2, the surface morphology of the Na metal electrode rapidly deteriorates, and dendritic and mossy Na readily appear in the NG2 electrolyte. After 10 min of Na plating, large Na dendrites caused a short-circuit of the cell. On the contrary, in NGMPE, Na surface remained even and flat during the Na plating process, where the thickness of Na electrode gradually increases with time, without any cracks or significant surface morphological change (SEI breakage). The integrity of SEI maintained during Na deposition is also contributed by its high mechanical strength. Thereby, an atomic force microscope (AFM) was further performed on the SEI formed in NG2 and NGMPE, to investigate their mechanical stability and surface morphology (Fig. 4e, f). The NaF-rich SEI in NGMPE features a high Young’s modulus of 921 MPa with an average surface roughness of 11.4 nm, rendering a robust and flat SEI compared to that from NG2 (587 MPa and 40.3 nm). Furthermore, the charge transfer kinetics of the thus-formed SEI was confirmed by the Tafel curves (Fig. 4g), that the NaF-dominant SEI formed by the NGMPE electrolyte exhibited a higher exchange current density than that of the NG2, suggesting the facilitated ion transportation kinetics.

Water corrosion test of surface-passivated sodium metal in (a) water-containing ethanol solution and (b) water-containing solvents. c Voltage profiles and DEMS signals for m/z = 2 (H2) emission of Na symmetric cells, using NG2 and NGMPE electrolytes, at a current density of 5 mA cm−2 and areal capacity of 2.5 mAh cm−2. d In situ observation of Na deposition and surface morphology evolution by optical microscopy at ca urrent density of 3 mA cm−2. AFM height images of SEI formed in (e) G2 electrolytes and (f) NGMPE electrolyte at 5 mA cm−2 and 5 mAh cm−2 after 10 cycles, with the average surface roughness and Young’s modulus indicated. g Tafel curves for Na plating/stripping in the electrolyte of NG2 and NGMPE.
Highly reversible Na metal at “three high” conditions
Due to the multifold merits of the compact and homogeneous NaF-dominant SEI, metallic Na demonstrated high reversibility at high current density, high areal capacity, and high depth of discharge. The Coulombic efficiency (CE) was measured in Na| |Cu and Na| |Al cells using untreated copper and aluminum foil. At 5 mA cm−2 and 5 mAh cm−2, the Na| |Cu cell using NGMPE electrolyte exhibited a remarkable CE of 99.95% averaged for 1000 cycles (Fig. 5a), significantly improved from the NG2 base electrolyte with fast short circuit. The electrochemical performance of its analogous with varying salt concentration and solvent ratio was also compared (Supplementary Table 4 and Supplementary Figs. 26, 27), validating the optimal recipe of NGMPE electrolyte. Further increasing the current density to 8 mA cm−2, average CE of 99.75% was still accomplished in Na| |Cu cell with NGMPE, with stable operation exceeding 800 cycles (Fig. 5b). The insets show the optical observation of the electrode surface after cycling, in which the Na metal remained shiny and smooth after cycling in NGMPE, while being rugged and rough in NG2. The surface degradation and unstable cycling in NG2 was corroborated by the electrochemical impedance spectroscopy (EIS) technique. The impedance of electrolyte/electrode interphase is large and erratic during the cell cycling in NG2, while that in NGMPE is stable and relatively small (Fig. 5c, d). For cost and weight reduction of current collectors regarding commercialization, the compatibility of electrolyte with aluminum (Al) foil was also investigated by Na| |Al cells. As a high CE of 99.90% in Na| |Al cell with NGMPE was realized for more than 1000 cycles at 3 mA cm−2 and 3 mAh cm−2 (Supplementary Fig. 28), more obvious improvements manifested in the cycle life at elevated current density and higher areal capacity conditions, which the NGMPE enabled high CE of 99.81% at 5 mA cm−2 and 5 mAh cm−2, or 99.61%, at 8 mA cm−2 and 4 mAh cm−2, on Al foil (Supplementary Figs. 29, 30). The stable Na plating/stripping at “three high” situations was further probed by Na| |Na symmetric cells. At moderate current density of 1 mA cm−2 and DoD of 50%, the long lifespan of symmetric cells was validated in NGMPE electrolyte, exceeding 7000 h, which was more than almost 20-fold longer compared to base electrolyte (Fig. 5e). At high current densities of 5 mA cm−2 and 8 mA cm−2, with a high DoD of 75%, the highly reversible Na metal and stable electrode-electrolyte interphase were maintained more than 1400 h and 1000 h, respectively, significantly improved from NG2 base electrolyte (Fig. 5f and Supplementary Fig. 31). The rate capability was also investigated in symmetric batteries, that the NGMPE reached high current density up to 15 mA cm−2, while the NG2 exhibited a fast short circuit at 5 mA cm−2 (Fig. 5g). As shown in Fig. 5h and Supplementary Table 5, the electrolyte engineered dense NaF-dominant SEI delivers promising electrochemical performance achieving high current density and high areal capacity, compared to the other studies on electrolyte modification.

Cyling performance of Na| |Cu cells at (a) 5 mA cm−2 and 5 mAh cm−2, and (b) 8 mA cm−2 and 4 mAh cm−2, with insets showing the surface morphology of Na metal after cycling. EIS of Na| |Cu cell cycled at 3 mA cm−2 and 3 mAh cm−2 in (c) NG2 and (d) NGMPE electrolytes, indicating the evolution of surface integrity. Symmetric Na| |Na cell at (e) 1 mA cm−2 and 1 mAh cm−2, DoD 50%, and (f) 5 mA cm−2 and 5 mAh cm−2, DoD 75%, (g) rate test from 0.1 mA cm−2 to 15 mA cm−2. h Comparison of half-cell performance in this work with those in other studies related to electrolyte engineering of SMBs (specification and corresponding reference are shown in Supplementary Table 5)28,36,41,57,58,78,79,80,81,82,83,84,85,86,87,88,89. Micro-CT 3D reconstruction of sodium metal after 50 cycles in (i) NG2 electrolyte and (j) NGMPE electrolyte at 8 mA cm−2 and 4 mAh cm−2, with the images of 2D horizontal slices at the same depth showing distribution and density of pores. k SEM images of surface morphology of sodium metal in NG2 and NGMPE electrolytes after 50 cycles at 5 mA cm−2 and 5 mAh cm−2.
As the prerequisites for the cycling stability and high reversibility, the dense Na deposition and suppressed side reactions in NGMPE were visualized by non-destructive micro-scale resolution X-ray computed tomography (micro-CT). As shown in Figs. 5i and j, after 50 cycles at 8 mA cm−2 and 4 mAh cm−2, abundant large-scale voids and cracks were detected throughout the volume of Na metal, due to uneven Na deposition and volatile gas evolution by the excessive interfacial reactions. The loosely deposited Na was revealed by the high porosity of 0.35%. On the contrary, Na metal cycled in the NGMPE electrolyte maintained dense and homogenous, benefited from the compact SEI that enabled uniform Na deposition and inhibited interfacial reaction even at high current density. The porosity of Na metal cycled in NGMPE was reckoned at merely 0.002%. The surface morphology of the Na metal negative electrode was also examined by scanning electron microscopy (SEM). After cycling at high current density and high areal capacity condition, the Na metal surface in NGMPE remained smooth and flat, while large cracks and voids emerged on Na metal surface in NG2 due to the uneven Na deposition and excessive interfacial reactions (Fig. 5k and Supplementary Fig. 32). Meanwhile, abundant residual Na were found on the aluminum foil current collector in NG2 after stripping to 0.3 V, accounting for the inferior CE of half-cells due to formation of “dead Na” (Supplementary Fig. 33).
Fast-charging and high-capacity AFSBs
The practicality of the designed fluorinated electrolyte and the effectiveness of dense NaF-dominant SEI were demonstrated in the full cells, rendering their fast charging, high capacity, low N/P ratio, and water tolerance. As shown in Fig. 6a, replacing the hard carbon negative electrode with Na metal and reducing its excess mass significantly escalates the gravimetric energy density of batteries. With very limited Na excess (N/P ratio = 1), the Na|NGMPE|NVP full cell maintained stable cycling and minimal capacity decay over 1200 cycles at high rate of 10 C and high cathode loading of 5 mg cm−2, while the base electrolyte full cell exhibited significantly fluctuating capacity and poor CE, which indicated the excessive soft short circuit and fast depletion of Na (Supplementary Fig. 34), manifesting in the poor surface morphology of post-cycling negative electrode of Na metal under SEM (Supplementary Fig. 35). Then, anode-free full cells (N/P ratio = 0) were fabricated by C@Al negative electrode/current collector and NVP positive electrode. At high cathode loading of 10 mg cm−2 and high rate of 3 C (1 C = 110 mA g−1), as well as lean electrolyte condition of 1 μL mg−1, the anode-free C@Al|NGMPE|NVP coin cell demonstrated long-term stability exceeding 400 cycles, with capacity retention of 80% (Fig. 6b and Supplementary Fig. 36). Further raising the active mass in positive electrode to 15 mg cm−2, the anode-free full cell still provided an initial discharging capacity of nearly 100 mAh g−1, with 80% of capacity retained after 480 cycles, under lean electrolyte and high rate of 1 C (Fig. 6c, d and Supplementary Fig. 37). The surface morphology of negative electrode has also been examined after cycling in anode-free batteries (Supplementary Fig. 38). According to total weight of active mass in positive and negative electrodes, the energy density of thus-fabricated anode-free coin-cell SMBs is calculated as high as 311 Wh kg−1, with power density up to 876 W kg−1 (Supplementary Tables 6, 7). On the contrary, the conventional electrolyte NG2 failed to establish a robust SEI that protects sodium metal negative electrode at “three high” conditions. Therefore, sodium dendrites readily formed on the host-less current collector and caused cell short-circuit (Fig. 6e) due to uneven Na deposition, while the limited Na inventory rapidly depleted during constant interfacial reactions, showing fast capacity decline. The superiority of the dense NaF-dominant SEI also ensures the stable cycling of sodium-metal batteries at harsh conditions, including moisture attack and high-temperature operation. To simulate the moisture attack, the prepared electrolytes was mixed with 5000 ppm water and employed in the anode-free C@Al| |NVP full cells60, with high cathode loading of 10 mg cm−2 and high rate of 1 C. Owing to the fast formation of compact primitive SEI formed by NGMPE, the anode-free C@Al| |NVP full cell maintained stable for 200 cycles in moisture-containing NGMPE electrolyte, with 83% of its initial capacity maintained (Fig. 6f and Supplementary Fig. 39). In contrast, the anode-free full cell using NG2 with 5000 ppm water showed rapid decay of capacity and unstable CE due to the water corrosion to loose SEI and sodium metal, ending up with nearly zero capacity after 200 cycles. Then, Na| |NVP full cells were fabricated by different electrolytes, with N/P ratio = 5 and cathode loading of 3 mg cm−2, to test their high-temperature performance. At 10 C and 60 °C, Na|NG2|NVP full cell exhibited low CE and fast capacity decay due to the aggravated interfacial reactions and depletion of Na, while the Na|NGMPE|NVP full cell showed great cycling stability with capacity retention of 81% after 1000 cycles (Fig. 6g).

a Schematic illustration of battery configurations and estimated energy densities of sodium-ion batteries, sodium-metal batteries, anode-less sodium-metal batteries, anode-free sodium-metal batteries. b Full cell performance of anode-free C@Al| |NVP coin cells, using different electrolytes, at cathode loading of 10 mg cm−2 and 3 C, following three formation cycles at 0.1 C and seven formation cycles at 0.5 C. c Cycling stability of anode-free C@Al| |NVP coin cells, using different electrolytes, at cathode loading of 15 mg cm−2 and 1 C, following three formation cycles at 0.1 C and seven formation cycles at 0.5 C, with the corresponding voltage-capacity profiles of (d) NGMPE and (e) NG2 (Supplementary Note 6). Cycling performance of (f) anode-free C@Al| |NVP coin cells following three formation cycles at 0.1 C, using water-contained electrolytes (5000 ppm), at cathode loading of 10 mg cm−2 and 1 C, (g) anode-less Na| |NVP coin cells at 60 °C and 10 C, with cathode loading of 3 mg cm−2 and N/P ratio = 5.
High-energy-density and high-power-density pouch cells
Finally, Ah-level anode-free pouch cells were fabricated, achieving high energy density and high power density using the film-forming electrolyte NGMPE. As most of the battery research focuses on raising the energy density for large-sized pouch cells, the high power density of sodium-based batteries is seldom accomplished at high areal capacity and long cycling life13. In particular, regarding the increasing areal capacity and total capacity is already challenging for SMBs with host-less negative electrode, achieving high power density of fast-charging AFSMBs has rarely been reported, on account of the volatile interfacial degradation and zero Na excess. Considering the promising electrochemical performance of AFSMB coin cells ensured by the dense NaF-dominant SEI, Ah-level multi-layer anode-free pouch cells were fabricated by the fluorinated electrolyte NGMPE, based on high-loading double-sided NVP as positive electrode and thin C@Al as negative electrode/current collector (Fig. 7a, b and Supplementary Note 7). At a moderate rate of 0.5 C, the anode-free pouch cell exhibited a high initial capacity of 1.1 Ah and stable operation for 289 cycles (capacity retention > 80%), demonstrating a high energy density of 150 Wh kg−1 with lean electrolyte (2.1 g Ah−1), as shown in Fig. 7c, d, Supplementary Fig. 41 and Supplementary Table 8. Impressively, a long cycling life was obtained by the C@Al|NGMPE|NVP anode-free pouch cell at high rate of 1 C (0.82 Ah), with stable operation exceeding 700 cycles with 81% of capacity retained under lean electrolyte condition of 1.9 g Ah−1 (Fig. 7e, f and Supplementary Fig. 42). Calculated by the total weight of pouch cell (Fig. 7g and Supplementary Table 9), a high power density of 152 W kg−1 was achieved with high energy density of 107 Wh kg−1, demonstrating the state-of-the-art performance in power density and cycling life for Ah-level sodium-metal batteries (Fig. 7h and Supplementary Note 8 and Supplementary Table 10)

a Schematic illustration of battery configuration and components of the multi-layer Ah-level anode-free sodium metal batteries pouch cell. b Key parameters of the battery components and the size of the pouch cell. Cycling performance of the 1.2 Ah anode-free pouch cells at charging/discharging rate of (c) 0.5 C and (e) 1 C, following three formation cycles at 0.1 C. The voltage-capacity profiles of pouch cells cycled at (d) 0.5 C and (f) 1 C (Supplementary Note 9). g Total weight of the 1.2 Ah anode-free pouch cell. h Comparison of power density and energy density of Ah-level sodium-ion pouch cells and Ah-level sodium-metal pouch cells9,10,11,90,91,92,93,94,95,96,97,98,99,100,101,102,103.
Discussion
In summary, we have successfully demonstrated an in situ formation of dense and homogenous inorganic-dominant SEI on sodium metal, induced by a unique dipole-dipole interaction in a highly fluorinated electrolyte, which mitigated excessive loss of Na inventory from interfacial side reactions and water corrosion, meanwhile enabling uniform Na deposition at elevated current density and areal capacity. The weak dipole-dipole interaction between solvent molecules was discovered, through 2D 1H–1H COSY NMR, of great importance in rearranging electron density distribution on solvent molecules and driving the spontaneous defluorination of chemically inactive fluorine-containing solvents. An inorganic-rich primitive SEI chemically formed in a short time frame and at an early stage, in prior to the electrochemical formation process, minimizing Na loss from interfacial side reactions. The in situ formed SEI was dense that inhibited solvent decomposition and moisture attack to Na metal, but also regulated homogenous Na influx and uniform deposition even at high current density and large areal capacity. The limited cycle life of anode-free sodium metal batteries at “three high” conditions, i.e., high current density, high areal capacity, and high depth of discharge, was addressed by the well-designed electrolyte engineering and hardly-acquired dense SEI. The anode-free sodium-metal coin cells exhibited long cycling life up to 480 cycles at high cathode loadings (10 and 15 mg cm−2), fast charging/discharging rates (1 and 3 C) and zero N/P ratio, even using lean electrolyte (1 μL mg-1) or moisture-containing electrolyte (5000 ppm). The Ah-level (1.2 Ah) anode-free sodium-metal pouch cell achieved a long lifespan up to 700 cycles, with a high power density of 152 W kg−1 at a high rate of 1 C and lean electrolyte of 1.9 g Ah−1. A high energy density of 150 Wh kg−1 was also demonstrated at 0.5 C. Considering the long-standing bottlenecks of practical sodium metal batteries involve their inferior cycling stability with low energy density and low power density, this work offers a different perspective of electrolyte design towards robust primitive SEI construction for stable and high-performance sodium metal batteries. This work is expected to promote the development of energy-dense and power-dense energy storage systems and the large-scale application of anode-free sodium metal batteries in a feasible route.
Methods
Materials
1,1,2,2-tetrafluoroethyl 2,2,2-trifluoroethyl ether (99%), 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (98% + ), 1,2-bis(1,1,2,2-tetrafluoroethoxy)ethane (98%), perfluorocycloether (water ≤ 50 ppm), 3-Fluorophenetole (98%+), benzotrifluoride (98%+) were provided by Adamas-beta. Fluorobenzene (99%) and hexafluorobenzene (99%) were provided by Sigma-Aldrich. 1-Ethoxy-2,3-Difluorobenzene (98% GC), methyl perfluorobutyl ether (98% GC), and ethyl perfluorobutyl ether (98%) were provided by Aladdin. Diethylene glycol dimethyl ether (99.8%+), fluoroethylene carbonate (99.9%+), and sodium hexafluorophosphate (99.8%+) were acquired from DodoChem. All solvents were pre-dried using molecular sieves for a minimum of 72 h prior to their use and characterization. The base electrolyte NG2 was prepared by dissolving 1 M of NaPF6 in G2 solvent and the fluorinated electrolyte NGMPE was composed of 1 M of NaPF6 in G2 and MPE solvent mixture (volume ratio of 7:3). Metallic Na (99.5%+) and Na foil (99.8%+) were purchased from Sinopharm Chemical Reagent Co., Ltd. and Jiaxing Changgao New Materials Technology Co., Ltd. Commercial Na foil was approximately 1 mm in thickness, and metallic Na chunks were rolled and cut into Na foils with a thickness of approximately 0.5 mm for further use. NVP cathode were prepared by ball-milling sodium vanadium phosphate (NVP, 99.9%, MTI Corp) power (80%wt), Super P (MTI Corp), and PVDF (99.5%+, molecular weight: 1 million, Canrd) in 1-methyl-2-pyrrolidinone (NMP, Water ≤ 50 ppm, 99.5%, Adamas-beta) to form a sticky yet fluid slurry. The cathode slurry was casted on carbon-coated aluminum foil (thickness of 12 + 1 µm, Canrd) using an infrared drying flat coater (MSK-AFA-ES200, MTI Corp), then vacuum dried at 85 °C. The positive electrodes, with active mass loading of 3 mg cm−2, 5 mg cm−2, and 10 mg cm−2, were further punched into circular foils using a precision disc cutter (MSK-T10, MTI Corp) for coin-cell batteries assembly. The high-loading NVP positive electrode of active mass loading of 15 mg cm−2 (94 wt%) and commercial separator Celgard 2325 were supplied by Shenzhen Neware Technology Co. Ltd. The compacted density of the positive electrode was 2.1 g cm−3. All positive electrodes and separators were stored in an Ar-filled glove box and vacuum dried at 60 °C for 2 h before immediate usage in coin-cell batteries. The double-sided positive electrodes with high loading of 30 mg cm−2 (93 wt% active mass) and carbon-coated aluminum foil, for pouch cell fabrication, were provided by Foshan Ruinake Technology Co. Ltd. Other battery accessories, such as stainless-steel cell case, stainless-steel gasket, stainless-steel springs, aluminum laminated film, commercial ceramic-PVDF coated PE separators (12 + 4ccs + 2pcs) and cell tabs, were acquired from Guangdong Canrd New Energy Technology Co., Ltd.
Material characterizations
Raman spectra of electrolytes and solvents were performed on a Horiba HR Evolution spectrometer using a 532 nm laser with 0.32 cm−1 resolution. The electrolyte wettability was determined by their contact angle on PP separator (Celgard 2325), untreated copper foil, and untreated aluminum foil, respectively, using the pendent drop method by Zhijia ZJ-6900 measuring instrument. The X-ray diffraction (XRD) characterization of the surface layer formed on Na metal was conducted on Bruker D8 and Haoyuan DX-27mini over a diffraction angle range of 5°-90°, at a speed size of 5° min−1. The Na metal with in situ formed surface passivation was rinsed by neat G2 solvent and sealed by polyimide film (Kapton tape) on a quartz slide in an Ar-filled glove box (O2 < 0.01 ppm, H2O < 0.01 ppm), then directly transferred to the XRD instrument to avoid air exposure. The passivation layer was also solely collected after rinsing with neat G2 solvent and carefully scraped off using a razor blade in a glove box, followed by vacuum drying at 60 °C for 30 min before XRD test. The 1H nuclear magnetic resonance (NMR) spectra of solvents and electrolytes were recorded on a 400 MHz Bruker AVANCE NEO, while the 1H-1H correlation spectroscopy (COSY) spectra were acquired by a 500 MHz Bruker AVANCE III HD. To avoid disturbance from mixing the sample with the NMR solvent (deuterium oxide, D2O), a coaxial internal insert containing NMR solvent was put into the NMR tube with the sample solvents. Chemical shifts of 1H NMR and COSY spectra of all samples were quoted to the D2O residual proton signal at 4.79 ppm. Time-of-flight secondary ion mass spectroscopy (ToF-SIMS) analysis was performed on PHI nanoTOF II Time-of-Flight SIMS (ULVAC-PHI Inc., Japan), using a Bi3++ beam (30 keV, 2 nA) for detection and an Ar+ beam (3 kV, 100 nA) for depth profiling. The sputter area was 400 × 400 μm, and the area of analysis was 80 × 80 μm. The X-ray photoelectron spectroscopy (XPS) analysis was conducted by PHI Versaprobe 4 (ULVAC-PHI Inc., Japan) using Al Kα radiation (1486.6 eV), with depth profile acquired through Ar+ sputtering at 2 kV. To preserve the chemical composition of SEI on highly reactive Na metal, sample preparation and cell disassembly for ToF-SIMS and XPS was carried out immediately before characterizations in an Ar-filled glovebox, and a sealing transfer vessel was used for sample transfer without air exposure. The electrodes acquired from disassembled cells were rinsed several times using neat G2 solvent and completely dried in a glove box. In situ differential electrochemical mass spectrometry (DEMS) measurements (QAS-100, LingLu Instruments, Shanghai) was coupled with a battery test system (CT-8002Q-5V100mA-124, NEWARE, Shenzhen), using a customized stainless-steel battery case with a three-way valve that allows cell assembly in a glove box and transfer to test equipment without air exposure. The symmetric Na| |Na batteries for DEMS measurements were assembled by Na foils (⌀ 10 mm), Celgard 2325 membrane (⌀ 19 mm), with electrolyte usage of 50 μL. High-purity Ar (> 99.999%) purging was used to eliminate the residual air in the three-way valve before testing, and high-purity Ar was utilized as carrier gas with a flow rate of 2 mL min−1. The in situ observation of Na deposition in Na metal symmetric cells was enabled by optical microscopy (YD650, Yuescope), where the tested symmetric cells were assembled in a glove box using hand-rolled Na foils and sealed in situ battery cell with quartz windows. Atomic force microscopy (AFM) analysis was carried out in an Ar-filled glove box, using a Bruker Dimension Ico instrument. The non-destructive micro-computed tomography (Micro-CT) analysis of cycled Na metal was performed on Carl Zeiss, Xradia 515 Versa, with image processing and porosity calculated via DragonFly software. The Na metal was obtained after cell disassembly in a glove box and vacuum sealed in an aluminum-plastic bag to avoid sample oxidation. The morphology and microstructure of the sodium metal negative electrode were investigated by a field emission scanning electron microscopy (FE-SEM, JEOL, JSM-7610F), using a vacuum vessel to transfer the sample from the glove box to the SEM chamber. The Na metal foils for cryo-TEM imaging were dismantled from coin cells and immediately frozen in a modified Ar-filled glove box and then cryo-transferred to the cryo-TEM chamber. The cryo-TEM images are obtained with an electron dosage of 100 e Å−2.
Electrochemical measurements
All coin cells (CR2032) were assembled in an Ar-filled glove box (Mikarouna) with both O2 and H2O content lower than 0.01 ppm. Na| |Cu and Na| |Al half cells were fabricated using Na foil (⌀ 12 mm and a thickness of 1 mm), untreated Cu or Al foil (⌀ 10 mm), and Celgard 2325 membrane (⌀ 16 mm). The designated current density was calculated based on the area of the current collectors. A small amount of as-prepared electrolytes was injected (15 μL per cell). The half cells were precycled from 0–0.3 V for 5 cycles to eliminate electrode surface contaminants before electrochemical testing. Na| |Na symmetric cells were assembled by Cu foil (⌀ 10 mm) with pre-deposited Na and Celgard 2325 separator (⌀ 16 mm), with 30 μL electrolytes per cell. To control the depth of discharge (DoD) in symmetric cells and negative-to-positive (N/P) electrode capacity ratio, a certain amount of Na was galvanostatically plated on the corresponding current collectors in half-cells, i.e., Cu and C@Al foils, denoted by Na@Cu and Na@C@Al, respectively. The half- cells were disassembled in a glove box, and the pre-sodiated current collectors were immediately used in DoD and N/P ratio-controlled cells. Na@C@Al| |NVP full cells, with different N/P ratios, was assembled using as-prepared NVP positive electrodes (⌀ 12 mm), pre-sodiated C@Al current collectors (⌀ 12 mm), ceramic-PVDF PE separators (⌀ 16 mm), and lean electrolytes (1 μL mg−1). The PE separators with ceramics and PVDF coating were used for better bonding of positive electrodes with separators and reducing thermal shrinkage at high temperature test. The Na metal negative electrode was pre-activated at low specific currents of 0.2 C for 3 cycles to ensure fluorinated SEI formation before high-rate cycling. Anode-free C@Al| |NVP were assembled using as-prepared NVP positive electrodes (⌀ 12 mm), pristine C@Al current collectors (⌀ 12 mm), and lean electrolytes (1 μL mg−1). Note that the cell sealing procedure should be done in a short time frame after electrolyte injection to avoid excessive volatilization of lean electrolytes. Following the formation process of anode-free full cells by cycling at 0.1 C for 3 cycles and 0.5 C for 7 cycles, the charging-discharging cycling of coin cells was performed with a cut-off voltage of 2.7–3.8 V, using a NEWARE battery testing system (CT-4008Tn-5V20mA) within a climatic chamber at 25 °C. The fabrication of pouch cells was conducted in a dry room, including cathode slurry mixing, coating, roll pressing, stacking, welding, and sealing. The pouch cells, consisting of NVP positive electrodes, Celgard 2325 separators and C@Al current collectors, were vacuum dried at 85 °C for 24 h in a glove box, with three times of gas (Ar) replacement, before for electrolyte injection and vacuum sealing in an Ar-filled glove box. The pouch cells experienced high temperature standing at 45 °C and formation cycles before testing cycling performance. The charging-discharging cycling of the pouch cell was performed using a NEWARE battery testing system (CT-4008Tn-5V6A) at 25 °C. All electrochemical performance tests of batteries were conducted in the climatic chambers (NEWARE WHW-200L-0C-110V-5V100mA-160CH and WGDW-200L-20C-380V-H) with controlled temperatures.
The electrochemical impedance spectroscopy (EIS) of Na| |Cu half cells was conducted on Gamry Interface 1010E after different cycle numbers, using a potentiostatic signal within the frequency range of 1 MHz to 0.1 Hz with an amplitude of 10 mV and 10 points per decade. The temperature-dependent ionic conductivity of electrolytes was also evaluated by the same instrument within the frequency range of 1 MHz to 1 Hz, in a symmetric cell configuration with stainless steel (SS) electrodes and glass fiber (Whatman GF/D) separator. At each set point of temperature, the SS| |SS coin cells were left standing in the oven for at least 10 min before EIS measurements. The ionic conductivity was calculated based on the following equation: \(\sigma \,=\,L\,/\,(R\times A)\), where L is the thickness of the separator, R is the bulk resistance, S is the area of the SS electrode. Linear sweep voltammetry (LSV) was carried out on Corrtest CS2350M electrochemical workstation in Na| |Al cells by sweeping from the open circuit voltage to 6 V vs. Na+/Na, at a scan rate of 0.1 mV s−1. Tafel plots and exchange current density were acquired by Corrtest CS2350M electrochemical workstation in Na symmetric cells (after three formation cycles allowing complete SEI formation) at a scan rate of 1 mV s−1 in range of − 0.1 V to 0.1 V.
Computations and calculations
Molecular dynamics (MD) simulations were carried out using the GROMACS software package (version 2021.5)61, employing the Generalized Amber Force Field (GAFF) for parameterization62. A cubic simulation box with initial dimensions of 50 × 50 × 50 ų was constructed, followed by a full NPT equilibration process. In the box, electrolyte salt and solvent molecules were introduced using the Packmol software (version 18.169/Windows 64)63, with a number of 65 for Na⁺ and PF₆⁻ ions and 455 for G2 molecules in NG2 electrolyte system; with a number of 65 for Na⁺ and PF₆⁻ ions, 320 for G2 molecules, and 119 for MPE molecules in NGMPE electrolyte system. The tolerance parameter was set to 2 Å to ensure reasonable molecular packing. Initial relaxation of the system was achieved through energy minimization, followed by an annealing process conducted over a temperature range of 0 to 298.15 K. This annealing was performed with a time step of 1 fs until equilibrium was reached, spanning a total of 10,000 fs for the annealing phase to accelerate system stabilization. The temperature coupling time constant was set to 0.2, and the velocity-rescaling (V-rescale) thermostat was employed64. Parrinello-Rahman algorithms was applied to control the pressure at 1.01325 × 105 Pa with an isothermal compressibility constant of 4.5 × 10−5 and coupling time constant of 2.0 ps65. The simulations were run in the NVT ensemble, utilizing the Verlet integrator to maintain a stable temperature of 298.15 K66. A production MD simulation was subsequently conducted for 10 ns at the same temperature, with trajectory data processed and analyzed using GROMACS utilities for statistical evaluation. The classification of contact ion pairs (CIP), aggregate (AGG), and solvent-separated ion pairs (SSIP) was performed through manual selection, that determine as SSIP if no anion is present within 3 Å of Na⁺, and only solvent molecules are found in this range; CIP if both anions and solvent molecules are present within 3 Å of Na⁺, and the solvent molecules and anions within this range do not interact with other Na⁺ within their own 3 Å radius; AGG if the anions and solvent molecules within 3 Å of Na⁺ also appear within 3 Å of another Na⁺, forming a networked structure.
Density functional theory (DFT) calculations were conducted with the Gaussian 09 W software suite67. Structural optimization and frequency analysis were performed using the B3LYP functional in combination with the 6–311 G* basis set68,69. The supplementary DFT calculations were based on basis sets b3lyp/6–311 g(d,p) em = gd3bj and b3lyp/def2svp, as well as functionals M052X and M062X in combination with 6–311 g(d,p)70,71. The initial structures of individual solvent molecules were generated using the “convert name to structure” function in ChemDraw. The initial solvation configurations were derived from the final equilibrated frames of MD simulations, which the representative solvation structures were selected based on the RDFs and coordination numbers obtained from the MD trajectories. Subsequently, these extracted structures were subjected to geometry optimizations at the DFT level using the B3LYP functional and 6–311 G(d,p) basis set, with Grimme’s D3 dispersion correction and the Solvation Model Density (SMD) to account for implicit solvation effects72,73,74. The static dielectric constant and the dynamic dielectric constant of the solvent were set to 7.18 and 1.9, respectively75. Key properties, including the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), Laplacian bond order (LBO), and electrostatic surface potential (ESP), were computed and visualized using GaussView 6.076 and Multiwfn77.
Data availability
The data that support the findings of this study are available from the corresponding author upon request. The raw data generated in this study and Supplementary Data 1 have been deposited in the Figshare database under accession code https://doi.org/10.6084/m9.figshare.28853579. Source data are provided in this paper.
References
Lee, B., Paek, E., Mitlin, D. & Lee, S. W. Sodium metal anodes: emerging solutions to dendrite growth. Chem. Rev. 119, 5416–5460 (2019).
Huang, J. et al. Recent progress and strategic perspectives of inorganic solid electrolytes: fundamentals, modifications, and applications in sodium metal batteries. Chem. Soc. Rev. 52, 4933–4995 (2023).
Vaalma, C., Buchholz, D., Weil, M. & Passerini, S. A cost and resource analysis of sodium-ion batteries. Nat. Rev. Mater. 3, 18013 (2018).
Yang, T., Luo, D., Liu, Y., Yu, A. & Chen, Z. Anode-free sodium metal batteries as rising stars for lithium-ion alternatives. iScience 26, 105982 (2023).
Usiskin, R. et al. Fundamentals, status and promise of sodium-based batteries. Nat. Rev. Mater. 6, 1020–1035 (2021).
Qian, J. et al. Anode-free rechargeable lithium metal batteries. Adv. Funct. Mater. 26, 7094–7102 (2016).
Nanda, S., Gupta, A. & Manthiram, A. Anode-free full cells: a pathway to high-energy density lithium-metal batteries. Adv. Energy Mater. 11, 2000804 (2021).
Deysher, G. et al. Design principles for enabling an anode-free sodium all-solid-state battery. Nat. Energy 9, 1161–1172 (2024).
Li, Y. et al. Interfacial engineering to achieve an energy density of over 200 Wh kg−1 in sodium batteries. Nat. Energy 7, 511–519 (2022).
Zhu, Q. et al. A 110 Wh kg−1 Ah-level anode-free sodium battery at −40 °C. Joule 8, 482–495 (2024).
Guo, D. et al. Low-temperature sodium–sulfur batteries enabled by ionic liquid in localized high-concentration electrolytes. Adv. Funct. Mater. 34, 2409494 (2024).
Baek, M., Kim, J., Jin, J. & Choi, J. W. Photochemically driven solid electrolyte interphase for extremely fast-charging lithium-ion batteries. Nat. Commun. 12, 6807 (2021).
Li, Y. et al. Origin of fast charging in hard carbon anodes. Nat. Energy 9, 134–142 (2024).
Cheng, X.-B., Zhang, R., Zhao, C.-Z. & Zhang, Q. Toward safe lithium metal anode in rechargeable batteries: a review. Chem. Rev. 117, 10403–10473 (2017).
Wang, X. et al. Building stable anodes for high-rate Na-metal batteries. Adv. Mater. 36, 2311256 (2024).
Jiao, S. et al. Behavior of lithium metal anodes under various capacity utilization and high current density in lithium metal batteries. Joule 2, 110–124 (2018).
Wang, H., Matios, E., Luo, J. & Li, W. Combining theories and experiments to understand the sodium nucleation behavior towards safe sodium metal batteries. Chem. Soc. Rev. 49, 3783–3805 (2020).
Huang, J. et al. 3D sodiophilic mixed-ion-electron-conducting framework of hierarchical nanowire arrays for ultra-stable sodium-metal batteries. Energy Storage Mater. 70, 103420 (2024).
Wu, M. et al. Weakly solvating electrolytes for safe and fast-charging sodium metal batteries. J. Am. Chem. Soc. 146, 35229–35241 (2024).
Gauthier, R. et al. How do depth of discharge, C-rate and calendar age affect capacity retention, impedance growth, the electrodes, and the electrolyte in Li-ion cells?. J. Electrochem. Soc. 169, 020518 (2022).
Choudhury, S. et al. Designing solid-liquid interphases for sodium batteries. Nat. Commun. 8, 898 (2017).
Pan, H., Hu, Y.-S. & Chen, L. Room-temperature stationary sodium-ion batteries for large-scale electric energy storage. Energy Environ. Sci. 6, 2338–2360 (2013).
Wang, Y. et al. A sodium–antimony–telluride intermetallic allows sodium-metal cycling at 100% depth of discharge and as an anode-free metal battery. Adv. Mater. 34, 2106005 (2022).
Huang, Z. et al. Long cycle life and high-rate sodium metal batteries enabled by an active/inactive Co-Sn alloy interface. Adv. Funct. Mater. 34, 2302062 (2024).
Yu, Z. et al. Molecular design for electrolyte solvents enabling energy-dense and long-cycling lithium metal batteries. Nat. Energy 5, 526–533 (2020).
Zheng, X. et al. Critical effects of electrolyte recipes for Li and Na metal batteries. Chem 7, 2312–2346 (2021).
Sun, B. et al. Design strategies to enable the efficient use of sodium metal anodes in high-energy batteries. Adv. Mater. 32, 1903891 (2020).
Wang, C. et al. A weakly coordinating-intervention strategy for modulating Na+ solvation sheathes and constructing robust interphase in sodium-metal batteries. Nat. Commun. 15, 6292 (2024).
Lee, K. et al. Fluorinated cyclic ether diluent for high-voltage lithium metal batteries. ACS Energy Lett. 9, 2201–2211 (2024).
Zhao, Y. et al. Fluorinated ether electrolyte with controlled solvation structure for high voltage lithium metal batteries. Nat. Commun. 13, 2575 (2022).
Zhao, Y., Zhou, T., Mensi, M., Choi, J. W. & Coskun, A. Electrolyte engineering via ether solvent fluorination for developing stable non-aqueous lithium metal batteries. Nat. Commun. 14, 299 (2023).
Rahman, M. M. et al. An inorganic-rich but LiF-free interphase for fast charging and long cycle life lithium metal batteries. Nat. Commun. 14, 8414 (2023).
Wang, Y. et al. Fluorine chemistry in rechargeable batteries: Challenges, progress, and perspectives. Chem. Rev. 124, 3494–3589 (2024).
Wan, H., Xu, J. & Wang, C. Designing electrolytes and interphases for high-energy lithium batteries. Nat. Rev. Chem. 8, 30–44 (2024).
Chen, S. et al. Tuning reaction kinetics of fluorinated molecules to construct robust solid electrolyte interphases on SiOx anode. Angew. Chem. 64, e202413927 (2025).
Wan, S. et al. Reductive competition effect-derived solid electrolyte interphase with evenly scattered inorganics enabling ultrahigh rate and long-life span sodium metal batteries. J. Am. Chem. Soc. 145, 21661–21671 (2023).
Zhang, Q.-K. et al. Homogeneous and mechanically stable solid–electrolyte interphase enabled by trioxane-modulated electrolytes for lithium metal batteries. Nat. Energy 8, 725–735 (2023).
Wu, H., Jia, H., Wang, C., Zhang, J. G. & Xu, W. Recent progress in understanding solid electrolyte interphase on lithium metal anodes. Adv. Energy Mater. 11, 2003092 (2021).
Zhang, Y. et al. Synergetic regulation of SEI mechanics and crystallographic orientation for stable lithium metal pouch cells. Nat. Commun. 15, 4454 (2024).
Liu, J. et al. Screening of F-containing electrolyte additives and clarifying their decomposition routes for stable Li metal anodes. Nat. Commun. 15, 9356 (2024).
Seh, Z. W., Sun, J., Sun, Y. & Cui, Y. A highly reversible room-temperature sodium metal anode. ACS Cent. Sci. 1, 449–455 (2015).
Hu, L. et al. Restructuring electrolyte solvation by a versatile diluent toward beyond 99.9% coulombic efficiency of sodium plating/stripping at ultralow temperatures. Adv. Mater. 36, 2312161 (2024).
Liang, H.-J. et al. Ether-based electrolyte chemistry towards high-voltage and long-life Na-Ion full batteries. Angew. Chem. Int. Ed. 60, 26837–26846 (2021).
Yang, J. et al. Improved low-temperature performance of rocking-chair sodium-ion hybrid capacitor by mitigating the de-solvation energy and interphase resistance. Adv. Funct. Mater. 32, 2200566 (2022).
Fan, X. et al. All-temperature batteries enabled by fluorinated electrolytes with non-polar solvents. Nat. Energy 4, 882–890 (2019).
Lu, D. et al. Ligand-channel-enabled ultrafast Li-ion conduction. Nature 627, 101–107 (2024).
Kalhoff, J. et al. Enabling LiTFSI-based electrolytes for safer lithium-ion batteries by using linear fluorinated carbonates as (co) solvent. ChemSusChem 7, 2939–2946 (2014).
Bolloli, M. et al. Effect of carbonates fluorination on the properties of LiTFSI-based electrolytes for Li-ion batteries. Electrochim. Acta 161, 159–170 (2015).
Gao, Y. et al. Polymer–inorganic solid–electrolyte interphase for stable lithium metal batteries under lean electrolyte conditions. Nat. Mater. 18, 384–389 (2019).
Kwon, H. et al. Borate–pyran lean electrolyte-based Li-metal batteries with minimal Li corrosion. Nat. Energy 9, 57–69 (2024).
Wang, Y. et al. Weak solvent–solvent interaction enables high stability of battery electrolyte. ACS Energy Lett. 8, 1477–1484 (2023).
Park, S. et al. Replacing conventional battery electrolyte additives with dioxolone derivatives for high-energy-density lithium-ion batteries. Nat. Commun. 12, 838 (2021).
Wahyudi, W. et al. Hitherto unknown solvent and anion pairs in solvation structures reveal new insights into high-performance lithium-ion batteries. Adv. Sci. 9, 2202405 (2022).
Liang, H. et al. Solvent–solvent interaction mediated lithium-Ion (De) intercalation chemistry in propylene carbonate based electrolytes for lithium–sulfur batteries. ACS Nano 17, 18062–18073 (2023).
Xie, H. et al. Thermodynamic and kinetic behaviors of electrolytes mediated by intermolecular interactions enabling High-Performance Lithium-Ion Batteries. ACS Nano 18, 22503–22517 (2024).
Wang, M. et al. Temperature-responsive solvation enabled by dipole-dipole interactions towards wide-temperature sodium-ion batteries. Nat. Commun. 15, 8866 (2024).
Vaidyula, R. R. et al. Binary solvent induced stable interphase layer for ultra-long life sodium metal batteries. Adv. Mater. 36, 2312508 (2024).
He, J. et al. Tuning the solvation structure with salts for stable sodium-metal batteries. Nat. Energy 9, 446–456 (2024).
Han, B. et al. Probing the Na metal solid electrolyte interphase via cryo-transmission electron microscopy. Nat. Commun. 12, 3066 (2021).
Zhang, N. et al. 4.6 V Moisture-tlerant Electrolytes for Lithium-Ion Batteries. Adv. Mater. 36, 2408039 (2024).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Martínez, L., Andrade, R., Birgin, E. G. & Martínez, J. M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 (2009).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 159, 98–103 (1967).
Frisch, M. J. T. et al. Gaussian 09, Revision D.01 (Gaussian Inc., Wallingford CT, 2009).
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785 (1988).
Zhao, Y., Schultz, N. E. & Truhlar, D. G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2, 364–382 (2006).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Zheng, C. et al. Electrostatic shielding boosts electrochemical performance of alloy-type anode materials of sodium-ion batteries. Angew. Chem. Int. Ed. 62, e202214258 (2023).
Dennington, R., Keith, T. A. & Millam, J. M. GaussView, version 6.0.16 (Semichem Inc., Shawnee Mission, KS, 2016).
Lu, T. & Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Zheng, J. et al. Extremely stable sodium metal batteries enabled by localized high-concentration electrolytes. ACS Energy Lett. 3, 315–321 (2018).
Tomich, A. W. et al. A carboranyl electrolyte enabling highly reversible sodium metal anodes via a “fluorine-free” SEI. Angew. Chem. Int. Ed. 61, e202208158 (2022).
Li, J. et al. Weakly coordinating diluent modulated solvation chemistry for high-performance sodium metal batteries. Angew. Chem. Int. Ed. 63, e202400406 (2024).
Chen, J. et al. High energy density Na-metal batteries enabled by a tailored carbonate-based electrolyte. Energy Environ. Sci. 15, 3360–3368 (2022).
Wang, S. et al. Stable cycling of Na metal anodes in a carbonate electrolyte. Chem. Commun. 55, 14375–14378 (2019).
Li, P. et al. High-rate sodium metal batteries enabled by trifluormethylfullerene additive. Nano Res. 15, 7172–7179 (2022).
Zheng, X. et al. Knocking down the kinetic barriers towards fast-charging and low-temperature sodium metal batteries. Energy Environ. Sci. 14, 4936–4947 (2021).
Soni, C. B. et al. Novel organic molecule enabling a highly-stable and reversible sodium metal anode for room-temperature sodium-metal batteries. J. Energy Storage 71, 108132 (2023).
Sun, H. et al. A safe and non-flammable sodium metal battery based on an ionic liquid electrolyte. Nat. Commun. 10, 3302 (2019).
Yu, Q. et al. All-fluorinated low-solvation electrolytes for high-voltage sodium metal batteries with appealing stability. Adv. Funct. Mater. 34, 2401868 (2024).
Li, E. et al. Hierarchical doping electrolyte solvation engineering to achieve high-performance sodium-ion batteries in wide temperature. Energy Storage Mater. 73, 103805 (2024).
Lu, Z. et al. Electrolyte sieving chemistry in cuppressing gas evolution of sodium-metal batteries. Angew. Chem. Int. Ed. 61, e202206340 (2022).
Li, Y. et al. Sole-solvent high-entropy electrolyte realizes wide-temperature and high-voltage practical anode-free sodium pouch cells. Adv. Mater. 37, 2419764 (2025).
Yang, B. et al. High modulus Na2SiO3-rich solid electrolyte interphase enable long-cycle and energy-dense sodium metal battery. Adv. Funct. Mater. 34, 2407783 (2024).
Ruan, J. et al. Current collector interphase design for high-energy and stable anode-less sodium batteries. Nat. Sustain. 8, 530–541 (2025).
Liao, Y. et al. Pentafluoro (phenoxy) cyclotriphosphazene stabilizes electrode/electrolyte interfaces for sodium-ion pouch cells of 145 Wh Kg− 1. Adv. Mater. 36, 2312287 (2024).
Xue, L. et al. A flame-retardant and weakly solvated gel electrolyte for high-performance and high-safety Ah class sodium-ion batteries. Chem. Eng. J. 504, 158828 (2025).
Chen, H. et al. Designing advanced electrolytes for high-safety and long-lifetime sodium-ion batteries via anion–cation interaction modulation. J. Am. Chem. Soc. 146, 15751–15760 (2024).
Guo, Z. et al. Inorganic-enriched solid electrolyte interphases: A key to enhance sodium-ion battery cycle stability?. Small 20, 2407425 (2024).
Zhang, T. et al. Converting residual alkali into sodium compensation additive for high-energy Na-ion batteries. ACS Energy Lett. 8, 4753–4761 (2023).
Wang, X. et al. Achieving a high-performance sodium-ion pouch cell by regulating intergrowth structures in a layered oxide cathode with anionic redox. Nat. Energy 9, 184–196 (2024).
Zhang, T. et al. Negative lattice expansion in an O3-type transition-metal oxide cathode for highly stable sodium-ion batteries. Angew. Chem. 136, e202316949 (2024).
Ye, W. et al. Water-soluble multifunctional binder with extraordinary bonding strength for high-voltage sodium-ion batteries. Adv. Energy Mater. 14, 2400101 (2024).
Broux, T. et al. High rate performance for carbon-coated Na3V2 (PO4) 2F3 in Na-Ion Batteries. Small Methods 3, 1800215 (2019).
Chen, Y. et al. One stone two birds” design for hollow spherical Na4Fe3 (PO4) 2P2O7/C cathode enabled high-performance sodium-ion batteries from iron rust. EcoMat 5, e12393 (2023).
Zou, Z. et al. Integrated polyanion-layered oxide cathodes enabling 100,000 cycle life for sodium-ion batteries. Energy Environ. Sci. 18, 2216–2230 (2025).
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No.22179079 (C.W.) and No. 52350710795 (S.X.D.)). The authors express great appreciation to Bing Xiao from Carl Zeiss (Shanghai, China) for supporting micro-CT analysis. The authors would like to thank Zhenwei Wu from SCl-GO (www.sci-go.com) for contributing to the 2D 1H–1H COSY NMR analysis. The authors extend their gratitude to Yibo Su from Shiyanjia Lab (www.shiyanjia.com) for providing assistance with ToF-SIMS analysis. The authors are deeply grateful for the great support from Siqi Ma.
Author information
Authors and Affiliations
Contributions
J.H. conceived the research and designed the experiments. J.H. and X.A. conducted the main experiments and contributed to the data analysis. Z.C. contributed assistantly to the data visualization and figure drawing. J.H. and L.L. drafted the manuscript with support from C.W., H.K.L., and S.X.D. All authors participated in the result discussion and manuscript preparation. The work was supervised by C.W. and S.X.D.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, J., An, X., Cheng, Z. et al. Dipole-dipole interaction-induced dense primitive solid-electrolyte interphase for high-power Ah-level anode-free sodium metal batteries. Nat Commun 16, 8586 (2025). https://doi.org/10.1038/s41467-025-63593-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63593-x
