
Abstract
Many drug failures in clinical trials are due to inadequate safety profiles. We developed an in-silico side effect genetic priority score (SE-GPS) that leverages human genetic evidence to inform side effect risk for a given drug target. We construct the SE-GPS in the Open Target dataset using post-marketing side effect data, externally test it in OnSIDES using side effects reported from drug labels and then generate a SE-GPS for 19,422 protein coding genes and 502 phecodes, of which 1.7% had a SE-GPS > 0. To consider drug mechanism, we incorporated the direction of genetic effect into a directional version of the score called the SE-GPS-DOE. We observe that restricting to at least two lines of genetic evidence conferred a 2.3- and 2.5-fold increased risk in side effects in Open Targets and OnSIDES respectively, with increased enrichments in severe drugs. We make all predictions publicly available in a web portal.
Similar content being viewed by others


Introduction
The development of a novel therapeutic from discovery to market is a rigorous and expensive process1, yet side effects often remain undetected until the later stages of clinical trials or even after drug approval2. A considerable proportion of these later-stage side effects are linked to the drug’s action at the primary biological target (‘on-target’) rather than secondary targets (‘off-target’)3, underscoring the inherent challenges in drug discovery, including the limited time frame and sample size of clinical trials and poor translation from animal to human studies4,5,6. These shortcomings can result in millions of dollars lost annually and account for approximately a quarter of all clinical trial safety failures3. Since such safety failures represent a significant bottleneck in drug discovery, optimizing prioritization frameworks of drug targets at the preclinical stage is essential to selecting safer targets.
Although numerous factors influence a drug’s safety profile7, understanding the biological role of the intended drug target is fundamental to selecting an effective and safe target. Human genetic data provides a valuable resource that can help predict the effect of lifelong modulation of a drug target by providing support for a causal relationship between the perturbed drug target and phenotypic outcomes4. Providing evidence of phenotypic risk, or lack of it, can provide support for or against the on-target safety of drug modulation. For example, the discontinuation of the SPR inhibitor (Quartet Medicine) due to the observation of concerning neurological effects in toxicology studies4, is supported by loss of function (LOF) variants in the SPR gene associated with neurological disorders8,9. Evidence showing that rare homozygotes for LOF variants have markedly lower low-density lipoprotein cholesterol but have no apparent deleterious health consequences supported the probable safety of inhibition of PCSK910,11.
With the continual expansion of human genetic data, numerous retrospective analyses have utilized Mendelian disease and genome-wide association data to demonstrate the predictive value of incorporating diverse types of genetic evidence, encompassing common and rare variants, to predict side effects7,12,13. These studies have shown that drug targets supported by human genetic evidence have an increased risk of side effect occurrence. However, despite the development of several prioritization scores that use multiple lines of genetic evidence to predict drug indications14,15, there is currently no framework that predict targets likely to elicit side effects. Furthermore, the directionality of the genetic effect has not been considered in these previous studies; this is required to determine if the genetic risk for the phenotypic outcome is in the same direction as the drug target modulation.
In this study, we have developed a side effect genetic priority score (SE-GPS) for 19,422 genes and 502 drug side effects, based on the presence of genetic evidence from four distinct genetic features using nine sources of genetic evidence. These features include: 1) clinical variant evidence from ClinVar16, HGMD17 and OMIM18, consolidated into a single feature quantified as the number of overlapping entries; 2) single coding variants encompassing pLOF and missense single variants curated from Genebass19 and RAVAR20; 3) Gene burden tests from Open Targets15 and RAVAR and 4) genome-wide association (GWA) loci, represented by two separate features: Locus2Gene21 and eQTL phenotype22. This approach extends from our prior work on the development of a genetic priority score (GPS) for drug indications14. We have utilized two sources of side effect data to consider side effects reported during clinical trials and those detected following market approval. Our method demonstrates that the SE-GPS can effectively highlight drug targets likely to elicit a drug side effect and provides a useful framework for incorporating direction of genetic effect with clinical trial safety data.
Results
Construction of the drug genetic dataset
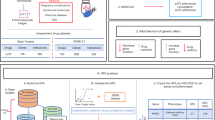
In this study, we calculated the SE-GPS to inform the likelihood of a side effect across 19,422 genes and 502 phecodes, using a similar approach to our previously published GPS14. We summarize this framework in Fig. 1.

A workflow of the data sources and steps to construct the SE-GPS and SE-GPS-DOE as outlined in this analysis. The SE-GPS and SE-GPS-DOE were created in the Open Target dataset (discovery), validated in OnSIDES and then generated for 19,422 genes and 502 phecodeX integers, for which 15,139 genes linked to 499 phenotypes had support from at least one genetic feature and directional evidence (n = 146,011). SE side effect, OT Open Targets, SE-GPS side effect genetic priority score, SE-GPS-DOE side effect genetic priority score with direction of effect.
We utilized two datasets that report side effect data: Open Targets15 as our discovery dataset, which compiles post-marketing surveillance data from the FDA Adverse Event Reporting System (FAERS)23, and OnSIDES as our validation dataset24, which extracts adverse drug reactions from drug labels reported during clinical trials. To measure the frequency of reported side effects and compare differences in side effect reporting across clinical trial data (OnSIDES) and post-marketing data (Open Targets), we plotted the ratio of reporting frequency (RRF), calculated as the normalized count of drugs associated with a given side effect from Paccanaro et al.5. Supplementary Fig. 1 shows the RRF of each side effect in Open Targets, correlated against the side effect data in OnSIDES. Both datasets indicate that most reported side effects are drug-specific, with similar reported frequency (r = 0.71). To construct the drug datasets, we mapped the side effects and drug indication data to phecodeX integer terms across 16 phecode categories, similar to the GPS, and outlined additional quality control steps in the Methods. We observe that a proportion of side effects in Open Targets and OnSIDES shared their phecode terms with the drug indication (9.18% and 11.41%, respectively). This overlap is likely due to several reasons, including side effects that result from an exaggerated pharmacological response directly related to the drug’s therapeutic effect, misclassification of disease symptoms as side effects, and issues with data reporting. For example, the drug levothyroxine sodium reports hypothyroidism as both an indication and side effect. To ensure this overlap did not drive our genetic enrichment analyses, we excluded those side effects where the drug was approved for an indication that shared the same phecode term. We retained this side effect filter, i.e. removal of phecode terms that matched a drug indication, for all subsequent analyses, as in previous studies7,12. Following quality control, the Open Target dataset comprised 1003 drugs, 752 genes, 360 unique drug indications and 445 unique side effects, whereas the OnSIDES dataset consisted of 777 drugs, 688 genes, 366 unique drug indications and 424 unique side effects.
Using gene–phecode pairs as the common identifier, we combined both side effect datasets with the nine human genetic data sources at the gene–phecode level, consolidating these into four genetic features to use for analysis: Clinical Variant, Single Variant, Gene Burden, and GWA Trait to reflect the different types of genetic support. These features were constructed as follows: the Clinical Variant feature was derived from genetic data from ClinVar16, HGMD17 and OMIM18, consolidated into a single feature recorded as the number of overlapping entries. The Single Variant feature comprised pLOF single variants curated from Genebass19 and RAVAR20, while the Gene Burden feature consisted of gene burden tests curated from Open Targets15 and RAVAR. Lastly, the GWA trait feature consisted of genes identified from genome-wide association significant variants identified using Locus2Gene21 and eQTL phenotype22. For Single Variant, Gene Burden and GWA trait, we binarized the features based on the presence or absence data from either source. A detailed description of each data source is provided in the Methods, resulting in an analyzable data matrix in which the Clinical Variant predictor is encoded as 0, 1, 2 or 3 and all other predictors are binary (0 or 1), across 9,749,844 gene–phecode pairs comprising 19,422 protein-coding genes, 502 phecode terms and 16 phecode categories. Across this matrix, 1.7% of gene–phecode pairs had a least one source of genetic evidence. An overview of these gene–phecode observations across is shown in Supplementary Fig. 2.
Association of genetic features with drug side effects
We performed univariate associations assessing the enrichment of the four genetic features with the drug side effects outcome in the Open Target and OnSIDES datasets. Given the variation in the number of unique drug-side effect pairs across the PhecodeX categories (Supplementary Fig. 3), and to account for disease heterogeneity, we adjusted for the 16 phecodeX categories as covariates. We observed significant associations of each feature in both datasets (Fig. 2). Furthermore, we examined the association between each genetic feature and drug side effects within each disease category, which revealed variability in the strength of enrichments across categories and between genetic features (Supplementary Figs. 4, 5). Notably, the single variant feature had a lower number of observations overall, and thus when stratified by category, this resulted in much wider confidence intervals.

Forest plot of ORs with 95% CI calculated for each genetic feature with drug side effects, adjusted for 16 phecode categories using a logistic regression model. Side effects were removed where the drug is approved for an indication that shares the same phenotype term. The genetic features are grouped by color according to their genetic evidence category. For each feature the proportion of unique genes with genetic evidence and an observed side effect over the total number of unique genes with genetic evidence is shown is recorded in red on the y-axis and the proportion of unique phenotypes with genetic evidence and an observed side effect over the total number of unique phenotypes with genetic evidence is recorded in blue. Panel a displays results for Open Targets (n = 1,254,900 independent drug–gene–phenotype combinations), whereas panel b displays results for OnSIDES (n = 1,158,368 independent drug–gene–phenotype combinations). The statistical test was two-sided and ORs with 95% CIs are defined in the forest plot as circles and error bars. The red dashed line represents the null odds ratio (OR = 1). CI confidence interval, OR odds ratio.
Construction of the SE-GPS
We next constructed the SE-GPS based on the cumulative effects of the four genetic features with drug side effects. Specifically, we used 80% of the Open Target dataset as the training set and applied a multivariable mixed-effect regression model of the association of the four genetic features with drug side effects as the outcome to obtain the effect sizes from the association of each feature to use as weights in the score (Eq. (1)). Similar to the univariate model, we included the 16 phecode categories as covariates in the model. We included each drug as the random-effect variable and included a crowdsourced severity score to weight the occurrence of the side effects by their severity25. With the remaining 20% of the data, we used the effect sizes of the genetic feature as weights to calculate the SE-GPS by summing the observations corresponding to each genetic feature, weighted by the effect size estimate (Eq. (2)). We repeated this process in a five-fold cross-validation framework and observed consistent effects of each genetic feature with drug side effects between the five cross-validated sets (Supplementary Fig. 6; Supplementary Table S1). We note that across the five-training test splits, only ~ 3% of all gene–phecode pairs had a SE-GPS greater than zero (Supplementary Figs. 7, 8). Within each cross-validated test set, we assessed the association between the SE-GPS and drug side effects using a logistic regression model, with drug side effect as the outcome, the SE-GPS as the predictor, and the 16 phecodeX categories included as covariates (Supplementary Table S2). We used cross-validated test 2, which had the highest OR and applied the coefficients from this mixed-effect model to further validate the SE-GPS in the OnSIDES dataset. In both Open Targets and OnSIDES we see that, similar to the GPS, multiple genetic features with varying effects, contribute to the highest SE-GPS (Supplementary Figs. 7, 9).
Association of the SE-GPS with drug side effects
In Open Targets and OnSIDES, 3.6% and 3.2% of gene–phecode pairs had a SE-GPS > 0. We evaluated the overall association of the SE-GPS with drug side effects. We observed a 1.8-fold increase risk in drug side effects (95% confidence interval (CI) = 1.7–1.9, P < 7.1 ×10−169) in Open Targets and 1.9-fold (95% CI = 1.8–2.0, P < 1.2 ×10−240) in OnSIDES.
We next explored the strength of enrichment of the SE-GPS by stratifying the drug dataset. First, we considered the number of gene targets per drug, the number of side effects per drug and the number of drugs per side effect. We found that drugs with one gene target and drugs with only a few side effects exhibited the strongest enrichments in both Open Targets (Fig. 3a) and OnSIDES (Fig. 3b). This is likely due to the reduced complexity of these drugs, enabling a clearer relationship between the target and phecode to be defined. Additionally, we found that drug-specific side effects had a stronger enrichment than side effects more commonly observed, recapitulating similar findings shown by Minikel et al.7. Second, we stratified the SE-GPS by the side effect phecode category (Fig. 3c, d). We observed significant variability in the odds ratio, highlighting that the impact of genetics is more pronounced in certain side effect categories than others. Infectious disease-related SEs had large odds ratios in both Open Targets and OnSIDES, whereas congenital-related SEs were not significant in either dataset. Furthermore, the degree of enrichment differs between categories when comparing side effects reported in Open Targets and OnSIDES, potentially reflecting differences in side effect reporting between clinical trials and post-marketing surveillance.

a Forest plot showing ORs with 95% CI for the association between the presence of a SE-GPS > 0 (binarized as 1) and drug side effects, adjusted for 16 phecode categories using logistic regression. This was performed across the full Open Target dataset (n = 1,254,900 independent drug–gene–phenotype combinations) with the OR colored in red and stratified by the number of gene targets per drug (1, 2–5, 5+; blue), the number of side effects per drug (1–4, 5–10, 10+; purple) and the number of drugs per side effect (1–10,11–30, 30+; green). For each feature, unique genes (red) and unique phenotypes (blue) are recorded on the y-axis. b Replication analysis of (a) using the OnSIDES dataset (n = 1,158,368 independent drug– gene–phenotype combinations). c Forest plot showing ORs with 95% CI for the association between the presence of a SE-GPS > 0 (binarized as 1) and drug side effects, stratified by phenotype category in the Open Target dataset. d Replication analysis of (c) using the OnSIDES dataset. The sample size of each stratified group is detailed in the Source Data. The statistical test was two-sided and ORs with 95% CIs are defined in the forest plot as circles and error bars. Filled circles indicate an OR with a significant P-value. The red dashed line represents the null odds ratio (OR = 1). CI confidence interval, N number, OR odds ratio, SE side effect.
The most serious side effects reported are denoted as boxed warnings, the highest safety-related warnings assigned by the FDA. Thus, these side effects are arguably the most important to predict. To investigate the utility of our score to predict severe side effects, we next restricted our dataset to drugs with a boxed warning, or drugs withdrawn due to the risk of toxicity. Within this restricted set of drugs, we observed an increase in OR of 2.0 (95% CI = 1.6–2.4, P < 1.6 × 10−12) in Open Targets and an OR of 3.7 in OnSIDES (95% CI = 2.8–4.9, P < 4.1 ×10−22). We further evaluated these enrichments stratified by drug grouping and disease category (Supplementary Fig. 10); however, we note much larger overlapping confidence intervals due to lower observations.
Association of the high SE-GPS extremes with drug side effect
We previously observed that at increased increments of the GPS there was an increased likelihood of a gene being a successful drug target14. Thus, by applying score thresholds, we next considered whether higher SE-GPS had a greater side effect risk. In the Open Targets dataset, using 0.3 increment cutoffs of the SE-GPS, we observed a similar relationship. The top 0.49%, 0.08% and 0.02% of the SE-GPS (equivalent to scores greater than 0.6, 1.2 and 1.8) conferred a 2.3-, 3.2- and 4.8-fold increased effect of having a drug side effect, respectively (Fig. 4a). We selected a cutoff greater than 0.6 as our initial threshold to define a high SE-GPS, corresponding to evidence from at least two genetic features (Supplementary Fig. 7), which reflects an OR > 2.3 and corresponds to 358 genes and 254 phecodes. We replicated these findings using the OnSIDES dataset, however the enrichment was less pronounced and lacked a clear linear progression. The top 0.49%, 0.06% and 0.01% of the SE-GPS (equivalent to scores greater than 0.6, 1.2 and 1.8) conferred 2.5-, 2.0- and 2.4-fold increased effect of having a drug side effect, respectively (Fig. 4b). Nonetheless, despite not observing increased enrichment across higher thresholds in OnSIDES, we observed in both datasets that incorporating evidence from at least two lines of genetic evidence can identify a subset of targets with a greater likelihood of side-effect risk.

The association of increasing SE-GPS with drug side effects was investigated by binning the drug dataset into 0.3 increments of the SE-GPS and comparing SE-GPSs greater or equal to each increment with SE-GPSs equal to zero. A logistic regression model was performed for each increment bin with drug side effect as the outcome variable and the SE-GPS bin as the predictor variable, adjusting for phecode categories as covariates. The statistical test was two-sided and ORs with 95% CIs are defined in the forest plot as circles and error bars, with filled circles indicating an OR with a significant P-value < 0.05 after correcting for multiple testing. Points are colored along a blue-to-red gradient, with blue representing lower OR values and red representing higher OR values. Panel a displays results for Open Targets (n = 1,254,900 independent drug–gene–phenotype combinations) whereas panel b displays results for OnSIDES (n = 1,158,368 independent drug–gene–phenotype combinations). The gray vertical line represents the null odds ratio (OR = 1). CI confidence interval, OR odds ratio, SE-GPS side-effect genetic priority score.
We further assessed the enrichment of high SE-GPS when using threshold cutoffs in the severe side effect dataset. We observed a marked enrichment of the SE-GPS with severe side effects for drugs with boxed warnings or withdrawn in Open Targets with the top 0.73% and 0.10% of the SE-GPS (equivalent to scores greater than 0.6 and 1.2) conferring 2.5 and 4.5-fold increased effect of having a drug side effect, respectively. A similar marked enrichment was also observed in OnSIDES, with the top 0.65% and 0.10% of the SE-GPS (equivalent to a score greater than 0.6 and 1.2) conferring 5.2- and 6.1-fold increased effect of having a drug side effect, respectively. However, much larger confidence intervals were also observed due to the lower number of observations (Fig. 5).

The Open Target and OnSIDES datasets were restricted to drugs with a boxed warning or drugs withdrawn due to toxicity risk and the side effect phecodes matching the toxicity class. The association of increasing SE-GPSs with these severe drug side effects was investigated by binning the boxed warning dataset into 0.3 increments of the SE-GPS and comparing SE-GPS greater or equal to each increment with SE-GPS equal to zero. A logistic regression model was performed for each increment bin with drug side effect as the outcome variable and the SE-GPS bin as the predictor variable, adjusting for phecode categories as covariates. The statistical test was two-sided and ORs with 95% CIs are defined in the forest plot as circles and error bars, with filled circles indicating an OR with a significant P-value < 0.05 after correcting for multiple testing. Points are colored along a blue-to-red gradient, with blue representing lower OR values and red representing higher OR values. Panel a displays results for Open Targets (n = 69,290 independent drug– gene–phenotype combinations) and panel b displays results for OnSIDES (n = 30,652 independent drug– gene–phenotype combinations). The gray vertical line represents the null odds ratio (OR = 1). CI confidence interval, OR odds ratio, SE-GPS side-effect genetic priority score.
Incorporating the direction of genetic effect
When determining whether a gene target is likely to elicit a side effect, it is crucial to consider the drug’s mechanism of action, as inhibition and activation can lead to distinct phenotypic outcomes. To consider this mechanism, we next adapted the directional version of the GPS to create a side effect genetic priority score with direction of effect (SE-GPS-DOE) that mimics the response of a drug. Using the Open Target dataset for each gene target-phecode pair, we incorporated loss-of-function (LOF) evidence for targets with an inhibitory mechanism and gain-of-function (GOF) evidence for targets with an activation mechanism across the four combined genetic features. We used LoGoFunc26, a machine-learning method for genome-wide prediction of pathogenic LOF, GOF and neutral genetic variants, and estimates of effect from quantitative trait loci (QTL) to infer the direction of the associated genetic effect14. We provide a summary of the number of genes and phecodes for each of the predicted LOF genetic features in Supplementary Fig. 11 and GOF features in Supplementary Fig. 12, noting a much larger number of LOF observations. We restricted the analysis to drugs with either an activator or inhibitor mechanism and observed similar effects of each genetic feature with drug side effects between the five cross-validated sets (Supplementary Fig. 13; Supplementary Table S3). We then used these effect sizes as weights to calculate the SE-GPS-DOE. We recorded the SE-GPS-DOE as either a positive score, if the weighted sum of effect sizes across the LOF predictions reflected drug inhibition, or negative, if the weighted sum of effect sizes across the GOF predictions reflected drug activation (Supplementary Figs. 14, 15). We evaluated the association of the positive SE-GPS-DOE with drug side effects restricting to inhibitor drugs and observed a 1.9-fold (CI = 1.8–2.0, P < 4.9 × 10−86) increase in drug side effects. Similarly, restricting the analysis to the negative SE-GPS DOE within activator drugs resulted in a 2.1-fold (CI = 1.8–2.5, P < 1.2 × 10−18) increase (Fig. 6). Similar to the SE-GPS, we evaluated the association of the SE-GPS-DOE within each cross-validated test set (Supplementary Table S4) to select the coefficients to calculate the SE-GPS-DOE in the OnSIDES dataset. In OnSIDES, we observed similar enrichments for the positive SE-GPS DOE and negative SE-GPS-DOE (Fig. 6). In Open Targets and OnSIDES, the proportion of gene–phecode pairs with an inhibitory drug mechanism was significantly greater (88.2% and 81.9%, respectively) than those with an activator mechanism.

Association between positive SE-GPS-DOE and drug side effects among inhibitor drugs in the Open Target (n = 853,065) and OnSIDES (n = 625,606) datasets, and between negative SE-GPS-DOE and drug side effects among activator drugs in the Open Target (n = 297,705) and OnSIDES (n = 385,215) datasets. In logistic regression models, non-zero SE-GPS-DOE values were binarized and compared to scores of 0, adjusting for 16 phecode categories. The statistical test was two-sided and ORs with 95% CIs are defined in the forest plot as circles and error bars. The red dashed line represents the null odds ratio (OR = 1). CI confidence interval, OR odds ratio, SE-GPS-DOE side-effect genetic priority score with direction of effect.
We next considered whether applying threshold cutoffs for SE-GPS-DOE, was associated with a greater side effect risk. In the Open Targets dataset, restricting to inhibitor drugs, we observed that the top 0.27% and 0.04% of the positive SE-GPS-DOE (equivalent to a SE-GPS-DOE greater than 0.6 and 1.2, respectively) conferred a 2.4- and 3.7 -fold increased effect of having a drug side effect, respectively (Supplementary Fig. 16). When restricting to activator drugs, however, we did not observe this increase enrichment. The top 0.08% and 0.02% of the negative SE-GPS-DOE (equivalent to a SE-GPS-DOE greater than 0.6 and 1.2, respectively) conferred a 3.3- and 2.5- fold increased effect of having a drug side effect, respectively (Supplementary Fig. 16). We did not observe the same increased enrichment in OnSIDES, but note that there were limited number of observations with directional evidence (Supplementary Fig. 17). Due to the fewer observations and the fact that LOF and GOF directional predictions are based on inference, we suggest using the SE-GPS-DOE as a complementary score to the SE-GPS.
Extending the SE-GPS to 19,422 genes and 502 phecodes
Finally, we extended both methods to 19,422 protein-coding genes and 502 phecodes, of which 18,436-genes and 46 phecodes were not included in either the Open Target or OnSIDE dataset. We identified 15,139 genes linked to 499 phecodes for a total of 146,011 observations with support from at least one genetic feature and directional evidence. Among these observations, 74.8% had a directional score for target inhibition. Furthermore, given that only a small fraction of protein-coding genes is currently considered druggable, we assessed the proportion of targets with genetic evidence classified as druggable genes. Out of 15,139 genes, 3818 genes were identified as druggable with significantly higher SE-GPS compared to non-druggable genes (Mann–Whitney test, P < 2.5 ×10−215).
We demonstrate how this prediction tool could be used to supplement additional safety information during pre-clinical development or even inform the likelihood of a side effect for a novel gene target with no prior clinical trial evidence. First, we highlight several known examples of side effects where the direction of genetic effect supports the side effect observed in the Open Target or OnSIDES dataset (all with a SE-GPS > 0.6, Supplementary Data 1). Each example emphasizes the importance of fully understanding the mechanism of the gene targets involved in the therapeutic response. For example, it is unsurprising that inhibitors targeting SCN5A result in cardiac side effects as SCN5A encodes the main cardiac sodium channel Nav1.5. Furthermore, the identification of on-target side effects using this score can also reveal potential therapeutic targets by modulating the target in the opposite direction. Such an example in this table is the identification of alopecia as a side effect of daclizumab, an IL2RA inhibitor indicated to treat multiple sclerosis. The SE-GPS corroborated this observation, with Mendelian evidence from OMIM and genome-wide association evidence from L2G. IL2RA activation by rezpegaldesleukin is currently in phase IIb clinical trials to treat patients with severe to very severe alopecia areata (NCT06136741)27. Second, we evaluated the performance of the SE-GPS using two examples of well-known targets discussed by Carss et al.4, where genetic evidence has previously provided strong support for the observation of severe side effects for drugs that led to clinical trial failure (Supplementary Data 2). The SE-GPS provides strong support for gastrointestinal side effects following inhibition of DGAT1 and neurological disorders from inhibition of SPR. Despite the side effects associated with DGAT1, it remains an attractive target for many autoimmune, metabolic and oncological diseases28,29,30. Therefore, recognizing the possible gastrointestinal side effects of DGAT1 can enable appropriate monitoring, risk assessment and the development of more selective inhibitors31. Third, we highlight examples of current undrugged targets with high predicted druggability (DrugnomeAI >0.5)32, showcasing how the SE-GPS can be applied at target discovery to help identify potential on-target side effects for targets with no prior clinical trial evidence (Supplementary Data 3).
A large proportion of gene-phecodes prioritized by the SE-GPS are known therapeutic targets (10.8%, 17.9% and 42.9% with SE-GPS > 0, >0.6 and >1.2 in Open Targets, and 18.9%, 24.4% and 46.8% with SE-GPS > 0, >0.6 and >1.2 in OnSIDES, respectively). This is expected since we previously showed that targets with a high GPS are enriched for drug indications14 and as mentioned previously, a considerable proportion of side effects result from an exaggerated pharmacological response directly related to the drug’s therapeutic effect. Ultimately, weighing the value of the genetic evidence pointing to drug efficacy versus potential side effect risk is essential to integrating this data into the drug discovery pipeline. We have created a web portal sharing the results of the SE-GPS and SE-GPS-DOE for 15,139 protein-coding genes and 499 drug side effects (https://rstudio-connect.hpc.mssm.edu/sideeffect-geneticpriorityscore/).
Discussion
In this study, we introduce the SE-GPS to aid the prediction of side effects likely to be elicited by each target. By incorporating human genetic evidence to model the effect of target modulation, we establish a framework to identify potential on-target side effects related to the biology of the intended pharmacological target. We demonstrate how integrating multiple lines of genetic evidence can help inform the likelihood of an on-target side effect, providing evidence of known side effects while also suggesting potential side effect risk for targets with no clinical trial evidence. Furthermore, by incorporating the direction of genetic effect, we demonstrate the relevance of each genetic score to the direction of the therapeutic hypothesis, distinguishing between targets inhibited and targets activated. This distinction can separate a side effect from a drug repurposing opportunity, as illustrated by IL2RA.
We propose the SE-GPS as a tool for evaluating the safety of therapeutic targets, offering insights into potential side effects that could arise during clinical development. By predicting side effects at target discovery, we hope this can help reduce late-stage failures and prioritize drug targets with minimal adverse effects. Furthermore, using this framework to identify likely side effects can help inform preclinical and clinical trial design to ensure that these side effects are known and monitored. To facilitate the use of the SE-GPS, we have provided all scores for 15,139 genes linked to 499 phecodes. In addition, it may be preferrable to recalculate the SE-GPS in combination with additional sources of evidence or for phecodes not captured in our framework. We have thus provided the feature weights in Supplementary Table S5.
The overlap between targets with predicted side effect risk and known drug targets for similar drug indications emphasizes the importance of integrating all aspects of genetic evidence and disease biology when selecting a potential drug target to ensure that it is both effective and safe. Although we prioritize associations supported by multiple lines of genetic evidence by suggesting a cut-off of 0.6, this approach may overlook signals captured by a single line of evidence. For example, prior work by Walker et al.6 highlighted the association between HMGCR inhibition and increased diabetes risk, for which we observed a SE-GPS of 0.36, captured exclusively through GWA evidence. Thus, we provide this cut-off and framework as a starting point and recommend the addition of complementary genetic methods to further strengthen this evidence and capture genetic associations not currently included in the SE-GPS. One such example is the incorporation of somatic variant data from tumor tissues, included by Minikel et al., who similarly demonstrated that side effects with human genetic support are 2.0 times more likely to occur7. Additional methods includes Mendelian Randomization, which offers several advantages, particularly the ability to infer causality rather than associations6 and polygenic risk scores, which offer the opportunity to stratify patients in clinical trials according to disease risk33.
Although only 3.6% and 3.2% of gene–phecode pairs in Open Targets and OnSIDES have a SE-GPS > 0, this still reflects 11,620 and 9416 gene–phecode pairs associated with a 1.8- and 1.9-fold increased risk of side effects. This subset of gene-phecodes with SE-GPS > 0 highlights how genetic evidence can point to biologically relevant mechanisms underlying on-target adverse effects and provides a starting point for deeper phenotypic profiling. We expect this proportion to increase as GWA and rare variant evidence continues to expand. However, the absence of a SE-GPS should not be interpreted as evidence for the absence of a side effect, and additional functional and experimental validation is required when modulating a particular target.
There are several limitations to this study. First, there is a considerable difference in the side effects reported in clinical trials in the OnSIDES dataset compared to post-marketing data in the Open Target dataset. Clinical trials have a limited duration and a relatively small number of participants; therefore the side effects reported during this stage tend to be more common34. By contrast, post-marketing studies offer the advantage of capturing a broader range of side effects due to a larger, more diverse patient population and the extended period of exposure35. However, post-marketing data rely on spontaneous adverse event reporting systems, and hence there is less certainty that the reported side effect is solely attributable to the associated drug36. While Open Targets evaluates significant drug-ADR pairs using the likelihood ratio test (LRT) to control false discovery rates37, it is important to be aware of these potential misclassified side effects. In addition, there are also several limitations to the OnSIDES database, as described by the authors24. This database is constructed using computational extraction methods and therefore some side effects are incorrectly labeled or not extracted accurately. The use of MedDRA to record these events meant that not all side effects were included. Second, studies have indicated that clinical trials are biased toward White participants of European ancestry38. Since the likelihood of a drug resulting in a side effect can vary between people of different ancestry, a greater representation of clinical trial participants is necessary. This limitation is also reflected in the limited diversity of genetic data. Third, we do not distinguish between severe and moderate side effects when providing the SE-GPS. Therefore, the overall severity of each predicted side effect needs to be further explored to determine the potential impact on clinical trials. Fourth, while we make a significant effort to compile evidence across clinical variants, coding variants and GWAS variants, a significant limitation of this study is the use of binary case-control phenotypes. Expanding the breadth of phenotype data to incorporate predicted disease phenotypes that integrate a wealth of multi-modal data across diverse populations, will further expand the wealth of genetic associations available to elucidate the biological mechanisms of drug targets39,40. Furthermore, because we use a similar method and datasets as the GPS, many limitations discussed here14 also apply to this study. This includes discrepancies in the ascertainment of drug data between the Open Targets and OnSIDES datasets, potential misclassifications and exclusion due to the use of multiple ontologies, including ICD-10 and phecode terminology, the absence of a genetic feature that does not equate to evidence against a drug target and reliance on LoGoFunc inference for LOF and GOF predictions.
In conclusion, we have developed the SE-GPS to aid the prediction of side effects in drug discovery. Our findings demonstrate that integrating multiple lines of genetic evidence can inform a range of side effects, mirroring our findings using the GPS to identify therapeutic targets. Further, the SE-GPS has increased predictive ability when considering severe side effects indicated by boxed warnings. We believe that this score will provide a valuable resource for assessing potential side effects, which should then be further explored to assess their risk compared to the overall therapeutic benefit.
Methods
Generation of discovery dataset
We collected and processed the drug, gene target, drug indication, drug side effect, drug mechanism of action and drug warnings data from the Open Targets Platform (version 25.03)15 as previously described14. We removed commonly observed side effects that were observed in at least 5% of drugs (corresponding to side effects observed in >187 drugs), similar to previous studies, as these side effects are less likely to reflect target-mediated mechanisms, and instead a likely consequence of off-target or systemic effects12. We excluded 62 common side effects (Supplementary Data 4) after which 10,208 side effects remained. We removed oncology drugs using the Anatomical Therapeutic Chemical (ATC) classification (version 2022AA) with an ATC code L01 and L02 (‘Antineoplastic agents’ and Endocrine Therapy) following the reasoning that oncology drugs have a different acceptable side effect profile. We mapped drug indications, recorded as EFO terms and drug side effects, recorded using MedDRA, (mappings outlined below) to phecodeX terms41, restricting the phecode terms to the integer part to aggregate similar codes. We excluded the phecode categories Neonatal and Pregnancy, resulting in 16 remaining PhecodeX categories. Finally, we removed any phecodes that lacked genetic evidence, excluding 46 phecode integer terms (Supplementary Data 5). We removed any drugs from our Open Target dataset that were also present in the replication dataset OnSIDES, however we note that 18.2% of gene-side effect pairs are still found in both datasets (Supplementary Fig. 18). In total, we extracted 1003 drugs, 752 genes, 360 unique drug indications and 445 unique side effects mapped to phecode integers. We list the side effects and drug indications mapped to phecode integers in Supplementary Data 6 and Supplementary Data 7.
Generation of validation dataset
We used the OnSIDES database (version 2.0_20231113)24 as our drug validation set and extracted drugs, side effect and drug warning data. Drug side effect and drug indication data were both recorded using MedDRA vocabulary terms. We extracted the drug indication data from ChEMBL42,43 and identified the gene targets of these drugs using the same target sources as described previously14: DrugBank (filtered to targets with known pharmacological action)44, ChEMBL (filtered to targets with a single protein)42,43, and a published list of FDA-approved therapeutic efficacy targets45. We repeated the same filtering steps as in Open Targets, removing common side effects (>101 drugs, 278 side effects removed), after which 4302 side effects remained and removed any phecodes that lacked genetic evidence, excluding 52 phecode integer terms (Supplementary Data 8; Supplementary Data 9). In total, we extracted 777 drugs, 688 genes, 366 unique drug indications and 424 unique side effects mapping to phecode integer terms. We list the side effects and drug indications mapped to phecode integers in Supplementary Data 10 and Supplementary Data 11.
Generation of boxed warnings datasets
We subsetted both drug datasets to drugs that either had a box warning or had been withdrawn due to toxicity risk. In Open Targets, these side effects are annotated as toxicity classes, which we then mapped to phecode categories as detailed in Supplementary Table S6. For each drug in the Open Targets warning set, we included only phecodes annotated with a side effect where the phecode category matched the toxicity class. In total, we extracted 135 drugs, 220 genes and 169 side effects mapped to phecodes and binarized each side effect as 1/0 across the 169 side effect phecode integers. In OnSIDES, the actual side effect terms were reported for the boxed drugs, hence we restricted the drugs in the OnSIDES warning set to these phecode side effects. Thus, this set represented a more accurate set of serious side effects. In total, we extracted 115 drugs, 225 genes and 79 side effects mapped to phecodes.
Genetic databases
We compiled genetic evidence from three types of genetic data (clinical variants, coding variants and GWAS traits) to explore the association between human genetic variation in drug target genes and drug side effects by utilizing multiple publicly available data sources. We mapped all genetic phenotypes to phecodeX terms, removed any phecodes mapping to Neonatal and Pregnancy categories, and restricted these codes to the phecodeX integer terms. We use ‘phecodes’ to reflect phecodeX integer codes throughout. We restricted our analysis to protein-coding genes, for which we obtained a list of 19,422 protein-coding genes from Ensembl (release 110)46. Each of these genetic features is described below.
Clinical variants
We collected clinical variant genetic evidence from three sources as previously described. EVA-ClinVar (sourced from Open Targets Platform version 25.03)16,47, HGMD Professional (version 2023.3)17 and OMIM (accessed August 28, 2023)18. We applied a more stringent filtering approach than previously described14, which resulted in a smaller number of gene–phenotype pairs. First, we extracted ClinVar evidence based on two steps. This evidence was filtered on clinical significance terms: likely pathogenic and pathogenic, and based on the confidence of the submission: criteria provided, multiple submitters, no conflicts, reviewed by expert panel and practice guidelines. We mapped these phenotype terms, recorded as EFO terms, to phecodes. In total, 1238 genes were associated with 1240 phenotypes that mapped to 192 phecodes. Second, we extracted disease-causing genes and likely disease-causing genes with at least three variant observations from HGMD, removing any variants classified as of unknown significance. We mapped these phenotype terms, recorded as HPO terms, to phecodes. In total, there were 13,486 genes associated with 14,705 phenotypes that mapped to 343 phecodeX integers. Third, we used Mendelian evidence from OMIM and used the same filtering steps performed by Nelson et al.48, restricting our selection, to entries with phenotype mapping code 3 (‘the molecular basis for the disorder is known; a mutation has been found in the gene’) and filtering these phenotypes by removing any terms containing ‘susceptibility’, ‘somatic’ or ‘response’ (drug response associations), as well as any flagged as questionable (‘?’) or representing non-disease phenotypes (‘[’)48. In total, 4009 genes were associated with 6622 phenotypes that mapped to 426 phecodeX integers.
Coding variants
Single variants
We used two different datasets to collate single variant genetic evidence. We used single variant tests from Genebass19 with P < 4.3 × 10−7 and confined our study to variants labeled either pLOF or missense. We restricted the analysis to traits labeled ‘ICD first occurrence’. We extracted 1021 genes associated with 464 phenotypes that mapped to 246 phecodeX integer codes. Secondly, we used the Rare Variant Association Repository (RAVAR)20, an open database that compiles rare variant associations obtained via a literature search using P < 1.0 × 10−6 and a minor allele frequency (MAF) less than 0.02. We mapped these phenotype terms, recorded as EFO terms, to phecodes. In total, 552 genes were associated with 297 phenotypes that mapped to 198 phecode integers.
Gene burden
We used two different datasets to combine gene burden genetic evidence. We used gene burden data sourced from Open Targets15 with, P < 1.0 × 10−7, and gene burden tests from RAVAR20 with P < 1.0 × 10−4. We mapped these phenotype terms, recorded as EFO terms, to phecodes. In total, 387 genes were associated with 148 phenotypes that mapped to 163 phecodeX integers from Open Targets, and 8694 genes associated with 1206 phenotypes that mapped to 390 phecodeX from RAVAR.
GWAS traits
We used two sources of GWAS evidence to combine GWAS trait genetic evidence. First, the eQTL phenotype was defined as genes with a shared association between a GWA phenotype from the Pan-UK Biobank22 and an expression quantitative trait loci (eQTL) using eQTL summary statistics from the Genotype-Tissue Expression (GTEx, v8) Portal across 49 tissues49, following the same filtering process as previously described14. Briefly, Pan-UK Biobank variants were lifted over from hg19 to hg38 using liftOverPlink50, genome-wide significant single-nucleotide variants (P < 5 × 10⁻⁸) were extracted, and these were intersected with eQTL significant variants that, for each tissue, had a nominal P-value less than the gene level threshold. We identified 5278 genes associated with 619 phenotypes that mapped to 188 phecodeX integers, collapsing across tissues. Second, we extracted GWAS association evidence with a Locus2gene21 score >0.5 from Open Targets15,21. To infer directionality, we incorporated the corresponding colocalization results for the GWAS credible sets with the overlapping molecular credible sets from expression QTL, protein QTL and splice QTL. The Open target Platform uses two methods, coloc, and eCAVIAR, and we restricted to coloc results with an H4 > 0.8, and eCAVIAR results with a colocalization posterior probability (CLPP) > 0.01. We identified 9,079 genes associated with 2364 phenotypes that mapped to 408 phecodeX integers.
Phenotype mapping
To integrate our drug and genetic data, we used phecodeX terms which represent clinically meaningful terms across the medical phenome41. We used several different ontologies and mapping methods to map each data source to phecode terms. These data sources are listed as follows:
Disease/Phenotype file from the Open Targets Platform (version 25.03)15.
Unified Medical Language System (UMLS) MRCONSO.RRF (version 2023AA)51.
HPO to PhecodeX restricting to ‘StrongEvidenceSpecific’52.
HPO to phecode map - Supplementary Table 1253.
EMBL-EBI Ontology Lookup Service (EBISPOT OLS) file that maps UK Biobank traits to EFO terms54.
MedDRA to ICD10 map, https://www.meddra.org/news-and-events/news/icd-10-meddra-mapping-now-available.
CUI to ICD codes, https://bioportal.bioontology.org/ontologies/ICD10CM/?p=summary (version 2023AA).
PhecodeX (Extended), version 1.0 Map to ICD-10, Phecode definitions and Phecode 1.2 to PhecodeX mapping Supplementary Table 741.
In addition, we manually mapped 1443 MedDRA codes to ICD10. All phenotype ontology terms were mapped first to ICD10 codes and then to phecodes, except for HPO terms which we were able to map directly to phecodeX terms.
Generation of the integrated drug-genetic dataset
At the gene–phecode integer level, we integrated the drug datasets with the nine data sources described above. We binarized each feature, drug indication and side effect across the 445 side effect phecode integers. Clinical variant features were consolidated into one feature, recorded as the number of overlapping entries. The two single variant features, two gene burden features and two GWA trait features were each combined to reflect the presence of either data source. In cases where drugs had multiple gene targets, these were repeated as multiple rows in the dataset to allow comparison between the drug side effects and genetic features at the gene level. Each drug-gene (n = 2820) pair is repeated for 445 side effect phecode integers giving a total of 1,254,900 rows. We formatted the OnSIDES validation dataset similarly where each drug-gene (n = 2732) pair is repeated for 424 side effect phecode integers giving a total of n = 1,158,368 rows.
Generation of the integrated gene–phecodeX integer dataset across 19,422 protein-coding genes and 502 phecodeX integer pairs
Similar to generating the drug-genetic datasets, we integrated the nine data sources described above for all 19,422 protein-coding genes for 502 unique phecodes. This resulted in a matrix of 9,749,844 gene–phecode pairs, for which 17,214 genes and 502 phecodes had support from at least one genetic feature. We integrated the drugnomeAI probability score32 and druggable genes were defined using the following sources: drugbank44, chembl55 and two published supplementary tables which list druggable genes45,56.
SE-GPS
We adapted the GPS method to construct the SE-GPS, which informs the likelihood of a drug side effect using genetic evidence from clinical variants, coding variants and GWA traits. We applied a mixed-effect regression model (using the lme4 R package, version 1.1-35.1)57 within a fivefold cross-validation framework, and extracted the association coefficients as weights for each genetic feature contributing to the score as detailed in Eq. (1) (Supplementary Table S1). In the mixed-effect model, the outcome variable was drug side effect, modeled against four phenotype-specific features (where clinical variant was coded as the number of overlapping features with values 0,1,2 or 3 while the remaining three features were binarized as 1 or 0) and 16 covariate categories, with each drug included as a random-effect variable.
where P(SE) represents the probability of the outcome, and βi are the fixed effect coefficients for the covariates: Clinical Variant (CV), Gene Burden (GB), GWA trait (GW) and Single Variant (SV), and the 16 PhecodeX categories included as covariates. A random intercept was included for drugs.
The occurrence of side effects was weighted by severity using a crowdsourced severity score across 2929 MedDRA terms25. We mapped these terms to phecodeX terms as described above. This model was carried out using the Open Target dataset, which was randomly split into five non-overlapping groups of unique gene–phenotype pairs. For each of the five folds, one-fold was used as the test set, while the remaining four folds were used as the training set in the mixed-effect regression model. In each 20% test set, scores were calculated as the weighted sum of the phenotype-specific features across each gene–phenotype as defined in Eq. (2):
where i = gene–phenotype, j = genetic feature, n = number of features, βj is the estimated association coefficient for the jth feature in the Open Target train dataset, and XOTji is the value for the feature column (0/1) from the Open Target test dataset for each gene–phenotype pair. We combine the five test folds for downstream analyses.
To validate the scores using OnSIDES, we first determined which beta estimates to use as weights from the five cross-validated folds. We used a logistic regression model to assess the association between the SE-GPS and drug side effects. In this model, drug side effect was the outcome variable, the SE-GPS was the predictor, and the 16 phecodeX categories were included as covariates. We selected the cross-validated test set that yielded the highest OR (Supplementary Table S2) and used the corresponding beta coefficients from the 80% training set to construct the SE-GPS in the OnSIDES dataset (Supplementary Table S5). In addition, we also applied these weights to the entire integrated gene–phecodeX integer dataset for each genetic feature across 19,422 protein-coding genes and 502 phecodeX integer pairs, of which 17,214 genes and 502 phecodes had support from at least one genetic feature.
Association analysis of the SE-GPS with drug side effects at 0.3 increments
The enrichment of drugs with side effects in relation to increasing SE-GPS was evaluated by binning the drug data into 0.3 increments, ranging from 0 to 2.1. At each 0.3 score increase, using a logistic regression model, the proportion of gene–phenotypes associated with drug side effects was determined by comparing gene–phenotypes with a SE-GPS greater than each bin threshold to drug data with no genetic evidence. The response variable was the presence of drug side effects, with the 16 phecode categories included as covariates.
SE-GPS-DOE
We developed a complementary directional version of the score, termed the SE-GPS-DOE, which integrates LOF evidence for gene–phenotype pairs associated with an inhibitory drug mechanism and GOF evidence for gene–phenotype pairs with an activator drug mechanism. This differs from our initial implementation of the GPS-DOE, where we used the strongest prediction of direction of effect to determine if the gene–phenotype pair suited an inhibitory or activation mechanism. We used the same methods to identify the direction of effect for each predictor as described for the GPS14. We used LoGoFunc26 to predict pathogenic LOF and GOF variants for ClinVar, HGMD, OMIM, Genebass single variant and RAVAR single variant. We restricted to predictions with a predicted probability >0.5; for genes with multiple variants, we used the prediction with the highest predicted probability for both LOF and GOF. For GWA traits, eQTL and Locus2gene, we used DOE from the QTL with the estimate of effect from the GWAS study. Similar to the LoGoFunc method, we used the strongest effect for LOF and GOF for genes with multiple variants.
We restricted the datasets to drugs classified as inhibitors or activators to match the direction of genetic effect with the direction of therapeutic modulation. Inhibitor drugs included drug mechanisms labeled as ‘inhibitor’, ‘blocker,’ ‘antagonist, ‘sequestering agent,’ ‘negative allosteric modulator,’ ‘inverse agonist,’ ‘allosteric antagonist,’ ‘antisense inhibitor,’ and ‘RNAi inhibitor’. Activator drugs encompassed ‘agonist,’ ‘positive modulator,’ ‘opener,’ ‘activator’ and ‘positive allosteric modulator’. We removed gene–phenotype pairs which had both an activator and an inhibitor mechanism from the dataset. The Open Target dataset was subsetted to 913 drugs, 723 genes, 356 drug indications and 445 side effects mapped to phecode integers. The OnSIDES dataset was subsetted to 698 drugs, 591 genes, 359 drug indications and 421 drug side effects mapped to phecode integers. To construct the SE-GPS DOE, we implemented a mixed-effect regression with fivefold cross-validation as described for the SE-GPS. In this model we included mechanism of action, categorized as either inhibitor or activator as an additional covariate. We then applied these weights to the remaining 20% test set, with GOF annotated as −1, LOF annotated as 1, and no genetic evidence or estimates of neutral annotated as 0. The GPS-DOE was calculated for each gene–phenotype observation, with positive scores reflecting LOF evidence for gene targets with an inhibitor mechanism and negative scores reflecting GOF evidence for gene targets with an activator mechanism. We validated these scores using the OnSIDES dataset, applying the association coefficients from the cross-validated sample that yielded the highest OR in a logistic regression model (Supplementary Table S4). In addition, we also applied these weights to the 19,422 protein-coding genes and 502 phenotype pairs.
Statistical analysis
We calculated the side effect ratio of reporting frequency (RRF) as detailed in equation 6 from Paccanaro et al.5. Specifically, for each side effect, the side effect ratio of reporting frequency (RRF) represents a normalized count of the number of associated drugs. This equation is as follows:
where Xij represents the entry in row i, column j of the matrix X, n represents the total number of drugs and Z is the maximum number of associations for the side effects.
We tested the association of each genetic feature, the SE-GPS and the SE-GPS-DOE with drug side effects in a univariate logistic regression model with drug side effects as the outcome using the glm function, adjusting for the 16 phecode categories as covariates. This equation is as follows:
where P(SE) represents the probability of the side effect outcome, β1Feature is the effect of the genetic feature of interest and Categoryj represent the k disease categories included as covariates to account for confounding across side effect classes.
Upset plots were generated using the UpSetR package (version 1.4.0) and violin plots were generated using the geom_violin function and specifying the scale = width from the R Package ggplot2 (version 3.3.6). All analyses were performed within the Mount Sinai High Performance Computing resource (Minerva) using R58.
Ethics statement
This study complies with all relevant ethical regulations, and no specific approval was needed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The processed Open Target and OnSIDE drug-genetic datasets are available at https://zenodo.org/records/1533413659. We provide the weights used to create the SE-GPS and the SE-GPS-DOE in Table S5. Furthermore, the SE-GPS and SE-GPS-DOE for 15,139 genes and 499 drug side effects are publicly available at https://rstudio-connect.hpc.mssm.edu/sideeffect-geneticpriorityscore/. Public data used in this study mentioned in the methods are available via the listed URLS: Open Target genetic evidence and clinical trial data (v25.03), https://platform.opentargets.org/downloads. OnSIDE drug data (version 2.0_20231113), https://github.com/tatonetti-lab/onsides/releases/tag/v2.0.0-20231113, Gene target and drug indication and mechanism of action data from ChEMBL (release 33), https://ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLdb/releases/chembl_33/, Drugbank (5.1.11), https://go.drugbank.com/releases/5-1-11. Supplementary table 2 from Santos et al.45.ADR severity score reported from Gottlieb et al. (Supplementary table 2)25. Ensembl (release 110), http://ftp.ensembl.org/pub/release-110/gtf/homo_sapiens/, Online Mendelian Inheritance in Man (OMIM) (accessed August 28, 2023), https://www.omim.org/downloads. Human Gene Mutation Database (HGMD) Professional (version 2023.3), https://www.hgmd.cf.ac.uk/ac/index.php. Single Variant association results from Genebass, gs://ukbb-exome-public/500k/results/variant_results.mt. Genebass (500 K), gs://ukbb-exome-public/300k/results/variant_results.mt. RAVAR gene level associations, http://www.ravar.bio/api/download/static/gene_fulltable.txt. RAVAR variant level associations, http://www.ravar.bio/api/download/static/snp_fulltable.txt. GTEx Analysis V8, https://www.gtexportal.org/home/datasets. Pan-UK Biobank, https://pan.ukbb.broadinstitute.org/downloads/index.html. UCSC liftover chain file, https://hgdownload.cse.ucsc.edu/goldenpath/hg19/liftOver/. ATC classification (Version 2022AA, uploaded 08/09/2022), Disease/Phenotype file from the Open Targets Platform (version 25.03)15. Unified Medical Language System (UMLS) MRCONSO.RRF (version 2023AA)51, https://www.nlm.nih.gov/research/umls/licensedcontent/umlsknowledgesources.html. PhecodeX map, https://phewascatalog.org/phewas/#phex. HPO to PhecodeX, https://github.com/emcarthur/phecode-HPO-map/blob/main/data/finalHPO-phecodeLinks/mapsFilteredByEvidenceType/hpo-phecode1.2_linkswithHPOchildrenExpansion_StrongEvidenceSpecific.tsv52. HPO to phecode map - Supplementary Table 1253. EMBL-EBI Ontology Lookup Service (EBISPOT OLS) file that maps UK Biobank traits to EFO, https://github.com/EBISPOT/EFO-UKB mappings/blob/master/ISMB_Mapping_UK_Biobank_to_EFO.pdf54. MedDRA to ICD10 map, https://www.meddra.org/news-and-events/news/icd-10-meddra-mapping-now-available CUI to ICD codes, https://bioportal.bioontology.org/ontologies/ICD10CM/?p=summary (version 2023AA) Source data are provided with this paper.
Code availability
Analytic code to create the SE-GPS and SE-GPS-DOE is available at https://github.com/rondolab/SE-GPS60.
References
Sun, D., Gao, W., Hu, H. & Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B. 12, 3049–3062 (2022).
Galeano, D., Li, S., Gerstein, M. & Paccanaro, A. Predicting the frequencies of drug side effects. Nat. Commun. 11, 4575 (2020).
Cook, D. et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat. Rev. Drug Discov. 13, 419–431 (2014).
Carss, K. J. et al. Using human genetics to improve safety assessment of therapeutics. Nat. Rev. Drug Discov. 22, 145–162 (2023).
Galeano, D. & Paccanaro, A. Machine learning prediction of side effects for drugs in clinical trials. Cell Rep. Methods 2, 100358 (2022).
Walker, V. M., Davey Smith, G., Davies, N. M. & Martin, R. M. Mendelian randomization: a novel approach for the prediction of adverse drug events and drug repurposing opportunities. Int. J. Epidemiol. 46, 2078–2089 (2017).
Minikel, E. V. & Nelson, M. R. Human genetic evidence enriched for side effects of approved drugs. PLoS Genet. 21, e1011638 (2025).
Bonafe, L., Thony, B., Penzien, J. M., Czarnecki, B. & Blau, N. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am. J. Hum. Genet. 69, 269–277 (2001).
Thony, B. & Blau, N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum. Mutat 27, 870–878 (2006).
Cohen, J. et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165 (2005).
Hooper, A. J., Marais, A. D., Tanyanyiwa, D. M. & Burnett, J. R. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 193, 445–448 (2007).
Nguyen, P. A., Born, D. A., Deaton, A. M., Nioi, P. & Ward, L. D. Phenotypes associated with genes encoding drug targets are predictive of clinical trial side effects. Nat. Commun. 10, 1579 (2019).
Duffy, Á. et al. Tissue-specific genetic features inform prediction of drug side effects in clinical trials. Sci. Adv 6, eabb6242 (2020).
Duffy, Á. et al. Development of a human genetics-guided priority score for 19,365 genes and 399 drug indications. Nat. Genet. 56, 51–59 (2024).
Buniello, A. et al. Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucl. Acids Res. 53, D1467–D1475 (2024).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2017).
Stenson, P. D. et al. The Human Gene Mutation Database (HGMD(®)): optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 139, 1197–1207 (2020).
Hamosh, A. et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 30, 52–55 (2002).
Karczewski, K. J. et al. Systematic single-variant and gene-based association testing of thousands of phenotypes in 394,841 UK Biobank exomes. Cell Genom 2, 100168 (2022).
Cao, C. et al. RAVAR: a curated repository for rare variant-trait associations. Nucleic Acids Res. 52, D990–D997 (2024).
Mountjoy, E. et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 53, 1527–1533 (2021).
Karczewski, K. J. et al. Pan-UK Biobank GWAS improves discovery, analysis of genetic architecture, and resolution into ancestry-enriched effects. medRxiv 03, 24303864 (2024).
Khaleel, M. A., Khan, A. H., Ghadzi, S. M. S., Adnan, A. S. & Abdallah, Q. M. A Standardized dataset of a spontaneous adverse event reporting system. Healthcare 10, 420 (2022).
Tanaka, Y. et al. OnSIDES (ON-label SIDE effectS resource) database: extracting adverse drug events from drug labels using natural language processing models. medRxiv 03, 24304724 (2024).
Gottlieb, A., Hoehndorf, R., Dumontier, M. & Altman, R. B. Ranking adverse drug reactions with crowdsourcing. J. Med. Internet Res. 17, e80 (2015).
Stein, D. et al. Genome-wide prediction of pathogenic gain- and loss-of-function variants from ensemble learning of a diverse feature set. bioRxiv 06, 495288 (2022).
A phase 2b study to evaluate Rezpegaldesleukin (Rezpeg) in the treatment of adult patients with moderate-to-severe atopic dermatitis (REZOLVE-AD). ClinicalTrials.gov Identifier: NCT06136741. https://clinicaltrials.gov/study/NCT06136741 (2023).
Graham, K. L. et al. DGAT1 inhibits retinol-dependent regulatory T cell formation and mediates autoimmune encephalomyelitis. Proc. Natl Acad. Sci. USA 116, 3126–3135 (2019).
Oleszycka, E., Kwiecień, K., Grygier, B., Cichy, J. & Kwiecińska, P. The many faces of DGAT1. Life Sci. 362, 123322 (2025).
Deng, B. et al. The role of DGAT1 and DGAT2 in regulating tumor cell growth and their potential clinical implications. J. Transl. Med. 22, 290 (2024).
Zhang, H. et al. Discovery of novel DGAT1 inhibitors by combination of machine learning methods, pharmacophore model and 3D-QSAR model. Mol. Diversity 25, 1481–1495 (2021).
Raies, A. et al. DrugnomeAI is an ensemble machine-learning framework for predicting druggability of candidate drug targets. Commun. Biol. 5, 1291 (2022).
Fahed, A. C., Philippakis, A. A. & Khera, A. V. The potential of polygenic scores to improve cost and efficiency of clinical trials. Nat. Commun. 13, 2922 (2022).
Fukuto, K., Takagi, T. & Tian, Y. S. Predicting the side effects of drugs using matrix factorization on spontaneous reporting database. Sci. Rep. 11, 23942 (2021).
Klein, E. & Bourdette, D. Postmarketing adverse drug reactions: a duty to report? Neurol. Clin. Pract. 3, 288–294 (2013).
Palleria, C. et al. Limitations and obstacles of the spontaneous adverse drugs reactions reporting: Two “challenging” case reports. J. Pharmacol. Pharmacother. 4, S66–S72 (2013).
Huang, L., Zalkikar, J. & Tiwari, R. C. Likelihood ratio test-based method for signal detection in drug classes using FDA’s AERS database. J. Biopharmaceutical Stat. 23, 178–200 (2013).
Knepper, T. C. & McLeod, H. L. When will clinical trials finally reflect diversity? Nature 557, 157–159 (2018).
Chen, R. et al. Expanding drug targets for 112 chronic diseases using a machine learning-assisted genetic priority score. Nat. Commun. 15, 8891 (2024).
Garg, M. et al. Disease prediction with multi-omics and biomarkers empowers case–control genetic discoveries in the UK Biobank. Nat. Genet. 56, 1821–1831 (2024).
Shuey, M. M. et al. Next-generation phenotyping: introducing phecodeX for enhanced discovery research in medical phenomics. Bioinformatics 39, btad655 (2023).
Davies, M. et al. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 43, W612–W620 (2015).
Gaulton, A. et al. The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954 (2016).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018).
Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017).
Martin, F. J. et al. Ensembl. Nucleic Acids Res. 51, D933–D941 (2023).
Cook, C. E. et al. The European Bioinformatics Institute in 2016: Data growth and integration. Nucleic Acids Res. 44, D20–D26 (2015).
Minikel, E. V., Painter, J. L., Dong, C. C. & Nelson, M. R. Refining the impact of genetic evidence on clinical success. Nature 629, 624–629 (2024).
Aguet, F. et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Kuhn, R. M., Haussler, D. & Kent, W. J. The UCSC genome browser and associated tools. Brief Bioinform 14, 144–161 (2012).
Bodenreider, O. The Unified Medical Language System (UMLS): integrating biomedical terminology. Nucleic Acids Res. 32, D267–D270 (2004).
McArthur, E., Bastarache, L. & Capra, J. A. Linking rare and common disease vocabularies by mapping between the human phenotype ontology and phecodes. JAMIA Open 6, ooad007 (2023).
Bastarache, L. et al. Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science 359, 1233–1239 (2018).
Pendlington, Z. M. Mapping UK biobank to the experimental factor ontology (EFO) https://github.com/EBISPOT/EFO-UKB-mappings/blob/master/ISMB_Mapping_UK_Biobank_to_EFO.pdf (2019).
Zdrazil, B. et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 52, D1180–D1192 (2024).
Finan, C. et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 9, eaag1166 (2017).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J Stat Softw 67, 1–48 (2015).
R. Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2022).
Duffy, Á. & Do, R. Data repository for development of a genetic priority score to predict drug side effects using human genetic evidence (Zenodo, 2025).
Duffy, Á. & Do, R. Development of a genetic priority score to predict drug side effects using human genetic evidence (Zenodo, 2025).
Acknowledgements
R.D. is supported by the National Institute of General Medical Sciences of the NIH (R35-GM124836). M.V. is supported by the French National Research Agency (ANR) (ANR-21-CE45-0023-01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
A.D. and R.D. conceived and designed the study. A.D. performed the statistical analyses. A.D., R.C., D.S., J.K.P., M.V., A.S., Y.I., M.M., D.N.C., G.R., D.M.J. and R.D. provided administrative, technical and material support. A.D. and R.D. drafted the manuscript. R.D. supervised the study. All authors aided in the acquisition and interpretation of data and/or critical revision of the manuscript. A.D. and R.D. had access to and verified all of the data in the study.
Corresponding author
Ethics declarations
Competing interests
R.D. reported being a scientific co-founder, consultant and equity holder for Pensieve Health (pending) and being a consultant for Variant Bio and Character Bio. A.D. is a current full-time employee of GlaxoSmithKline. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Duffy, Á., Chen, R., Stein, D. et al. Development of a genetic priority score to predict drug side effects using human genetic evidence. Nat Commun 16, 8713 (2025). https://doi.org/10.1038/s41467-025-63762-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63762-y
This article is cited by
-
Development of a genetic priority score to predict drug side effects using human genetic evidence
Nature Communications (2025)
