
Abstract
Well-compatible with current industrial manufacturing and diverse applications, interfacial polymerization (IP) and IP-based polyamide nanofiltration membranes have evoked extensive research. Wherein, aside from tailoring pore sizes, regulating charged features of membranes remains challenging. Re-visiting principles of interfacial amide condensation, HCl byproducts are found exerting disparate effects in interfacial diffusion and ensuing amidation reaction. Exemplified by classic aqueous monomers–piperazine, H+ byproducts or equivalents, backflowing from IP zone and associating with amine groups of up-migrating piperazine or surrounding oligomers to alleviate the amidation, unexpectedly escort more amines to terminate polyamide chains for enhanced positive moieties. Resulting NF membranes synergize steric and charge effects for high Li+/Mg2+ selectivity of ~68 concurrent with water permeance of ~9 L m−2 h−1 bar−1, superior in polyamide membranes. Hence, backflow H+ from the organic phase, typically cleared away in routine IP, is developed as the novel strategy to configure spatial charges of resulting membranes, further expanding IP’s versatility in fabricating functional membranes.
Similar content being viewed by others

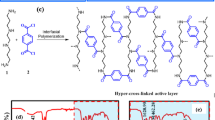

Introduction
Polyamide (PA) composite nanofiltration (NF) membranes have been extensively applied in water softening and purification, resource extraction and recovery, and also high-value chemical separations, owing to the inherently selective separation features1,2,3,4,5. Yet, further advancing the separation selectivity is dictated by the fine regulation of the PA selective layer, mostly comprising nanoporous and potentially charged structures, which, however, encountered numerous challenges6,7,8,9,10. Especially, on the basis of the currently well-established industrial manufacturing way, it is imperative and attractive for one-step reaping the desired NF performance.
Since interfacial polymerization (IP) has been invented to fabricate composite separation membranes, it serves as the most extensively exploited and classic method of manufacturing NF membranes. Consequently, great efforts have been devoted to delving deep into the IP process and, in turn optimizing the PA structure to directly and efficiently achieve desired membrane properties1,5,11,12. Wherein, unlike the relative ease of tailoring membrane pore sizes, precisely regulating the charge distribution of NF membranes remains knotty, both of which play key roles in separative characteristics. On the other hand, a great deal of research, based on experimental or mathematical means, has attempted to delineate details and figure out the complete landscape of processes of the IP as well as polymer growth11,12,13,14,15,16,17,18,19,20. Therein, all kinds of factors, such as concentration and proportion of monomers, properties of solvents, bulk/cross-interface diffusion of participants, polymer precipitation, and effects of affiliated by-products, have long been systematically pondered over, providing profound insights into the IP process of simple procedures but complex mechanisms. Nevertheless, a complete understanding of IP remains elusive, constrained by its inherent complexity and current undetectability. That said, some qualitatively known factors still hold the potential to inspire new avenues for regulating IP and optimizing membrane performance. For instance, HCl by-products of interfacial amide polycondensation, always mentioned but tentatively discussed only in the early days of studying IP, mostly backflowing from the reaction zone to the two-phase interface, have been roughly assessed to significantly or negligibly impose adverse side effects on the progress of IP, usually under various premises11,13,14,15. It is partially because the generated HCl by-products were finite, which challenged the real-time monitoring during the IP process, and likely resulted in limited effects on resultant membrane performance, as hardly manifested well in the filtration tests. However, revisiting principles of well-established IP in this study, backflow HCl byproducts or equivalents were first systematically investigated and viewed as the IP regulator to modulate charge distributions and yet pore structures of as-prepared NF membranes.
This research’s aims to use one regulator to place significantly disparate impacts on two sequentially occurring processes of IP made protons (H+), carried by HCl or equivalents, as the optimal selection to explore, referring to the piperazine (PIP)- trimesoyl chloride (TMC) system. Although well-acquainted with the generation and impact of protons during amidation reactions in homogeneous systems, the exploration of using relatively vast amounts of protons affecting the IP has not yet been implemented to date. In other words, by amplifying or intensifying the effects, the study of protons backflowing to the interface becomes tangible and achievable. It seems to disobey common sense to strengthen the role of the protons, and yet the resultant achievement of such lower rejection of Na2SO4 than MgCl2, characteristics of positive NF membranes, is astounding. In these designed experiments, PCl3 mimicked HCl to associate with diffusing PIP in n-hexane while eventually hydrolyzed by water to produce several-fold protons protonating PIP at the interface (Fig. 1A). The protonated PIP, plus protons from interfacial hydrolysis of PCl3 and byproducts of amidation reactions, markedly retarded the polycondensation of PIP and TMC, and in turn strived for plenty of time for PIP to diffuse across the interface. Notably, the further polycondensation of already-formed oligomers was also compromised as a result of possibly associating with protons. On the other hand, it might be controversial that protonated PIP slowly diffused21, however, they just accounted for a very small portion in comparison to the PIP pool and barely brought about a loss of PIP diffusion flux (detailed later). Hence, it is obtainable that impacts of HCl or equivalents were exerted significantly on the amidation reaction of PIP and TMC, markedly distinguishing from slightly adverse effects on PIP diffusion behavior. Thereafter, the separation performance of as-prepared NF membranes, completely different from that of conventional negative PIP-based NF membranes, ensures a niche of selectively rejecting muti-valent cations (e.g., Mg2+) versus multivalent anions (e.g., SO42-), or precise separation of Li+/Mg2+ frequently discussed recently. Briefly, backflowing protons or masses of protons in organic phase, relative to up-migrating PIP, were designed as the novel strategy to improve the positive charge density of PA membranes, by simultaneously and discrepantly affecting the interfacial diffusion and ensuing amidation reaction of IP, both of which determine PA structures. Meanwhile, another profound significance of this finding is to complement one piece of the jigsaw of classic IP, well-compatibly and directly applied in current industrial manufacturing lines for NF membranes of strengthened membrane positivity.

A The schematic illustration of H+ or its equivalents functioning as a “H+ fence” to increase positively-charged (blue) moieties by fading away negatively-charged (red) moieties of the polyamide chains. B Water permeance and xylose rejections, and C salt rejections of NF membranes fabricated with increasing PCl3 in n-hexane based on the formulation of 2.0 wt% PIP in water and 0.10 wt% TMC in n-hexane.
Results
Thin film composite PA membranes, usually fabricated via IP of PIP and TMC, are dominating various NF applications. While it is obtainable for tailoring specified pore sizes to adapt various practical separation scenarios, the alteration of conventional negatively charged features of PIP-based NF membranes via one-step routine IP is still full of challenges. Herein, one IP regulation strategy is offered by highlighting HCl byproducts (H+) or equivalents, which migrate downwards and meet with the upwardly-diffusing PIP first at the interface and subsequently in the organic phase (n-hexane). Specially, protons (H+), originally carried by PIP, entering into n-hexane and then moving towards the aqueous phase as part of byproducts, and their equivalents necessarily migrate downward to the interfacial zone to militate, both of which consequently are collectively called backflow H+. The differentiated effects of backflow H+, namely, slightly retarding the diffusion process and significantly alleviating the violent amide polycondensation of PIP, escort more PIP monomers crossing the interface of immiscible phases. It ultimately facilitates the formation of polyamide chains terminated with more amine groups replacing the conventionally terminal acyl chloride groups, which implies the limited negative carboxylic groups (red moieties in Fig. 1A) and increased positive amine groups (blue moieties in Fig. 1A) of polymers on the top surface. The novel PA NF membranes are gifted with the characteristics of positively charged NF membranes, which highly reject MgCl2 than Na2SO4, when concurrently equipped with proper membrane pore size, and potentially stand out in the selective separation of Li+/Mg2+ (Fig. 1B and C).
As shown in Fig. 1B and C, without the addition of PCl3, the NF membrane, interfacially formed using 2.0 wt% PIP in water and 0.10 wt% TMC in n-hexane, had the highest xylose rejection of 87.6 ± 2.3% and the lowest water permeance, which also had the smallest pore size of 0.26 nm (Supplementary Fig. 1 and Supplementary Table 1). Notably, the PIP concentration in the above formulation is relatively high compared to those reported in the literature. Following the increase of PCl3 in n-hexane, a series of NF membranes were fabricated with the performance of gradually decreased rejections of xylose as well as incremental water permeance. Meanwhile, the rejections of both NaCl and Na2SO4 almost monotonically decreased, while those of MgCl2 as well as MgSO4 had the consistent trend of ascending at first and then descending. Specifically, when PCl3 was more than 10 × 10-3 mol L-1 in n-hexane, it resulted in consistently greater rejection of NaCl relative to Na2SO4, as was similarly maintained for that of MgCl2 versus MgSO4. The phenomenon clearly evidenced the significant enhancement of membrane positivity. Nonetheless, at low concentration of PCl3 only gradually reduced Na2SO4 rejections were observed, which remained higher than that of NaCl, probably due to size sieving predominating over charge effects of as-fabricated membranes. Overall, during systematically varying PCl3 from 0 to 20 × 10−3 mol L-1 in n-hexane, MgCl2 rejection remained relatively stable while Na2SO4 rejection dropped sharply from ~92.4% to ~16.5%, producing a widening rejection difference that reached nearly ~50% at maximum. Therefore, adding PCl3 in n-hexane has remarkably improved the density of =NH (potentially positively-charged groups), which significantly benefited the permeation of SO42- and rejection of Mg2+ via enhanced charge effects.
Furthermore, the role of PIP concentration was also determined in experiments (Fig. 2). Generally, elevating PIP concentration favored the narrowing of membrane pores as proved by increased rejections of both xylose and NaCl, and decreased water permeance (Fig. 2A and B). However, utilizing 1.0 wt% PIP in water formed the loosest polyamide layer with only ~47.9% xylose rejection because of backflow H+ simultaneously restricting both PIP supply and subsequent interfacial amidation, which is detrimental to effectively rejecting MgCl2. It revealed that maintaining sufficiently high PIP concentrations is essential for optimal membrane performance. Beyond that, similar experimental results were also achieved by varying TMC in n-hexane, whose concentration was limited to the range generally used previously (Fig. 2C and D). Specifically, adopting 0.05 wt% (1.2 × 10−3 mol L−1) TMC, the xylose rejection of fabricated NF membrane was about 12%. These results have informed us of the paramount importance of simultaneously using concentrated PIP and proper PCl3 (backflow H+) to benefit the fabrication of positively charged membranes.

A Water permeance and xylose rejections, and B salt rejections by NF membranes fabricated with gradually increased PIP in water and fixed 0.10 wt% TMC in n-hexane. C Water permeance and xylose rejections, and D salt rejections by NF membranes fabricated with fixed 2.0 wt% PIP in water and gradually increased TMC in n-hexane.
As previously reported, the NF membrane, fabricated by the regular IP of 2.0 wt% PIP in water and 0.10 wt% TMC in n-hexane, had a granular membrane surface of low roughness, as well as a flat cross-section appearance with the typical thickness of ~57 nm (Fig. 3 and Supplementary Fig. 5A). Comparatively, with 1 × 10−3 mol L−1 PCl3 added in n-hexane, bigger granules spread all over the membrane surface, resulting in the significantly enhanced roughness of 21.1 ± 2.4 nm, and annular protuberances began to present especially according to the cross-sectional morphology. When PCl3 in n-hexane went up to 4 × 10−3 mol L−1, there were amounts of crater-like shapes occupying the membrane surface; and fascinatingly, nearly one-by-one void structures undulated and comprised the continuous PA nanofilms with the largest thickness of ~150 nm in whole despite the monolayer thickness of ~34 nm (Fig. 3, Supplementary Figs. 5B and 7). Further rising PCl3 to 20 × 10−3 mol L−1, although some ring-like structures (less violent than craters) scattered on the membrane surface, both morphologies of the surface and cross-section became smoother than that of the NF membrane fabricated without PCl3 in n-hexane. Furthermore, using 1.0 wt% PIP in water and 4 × 10−3 mol L−1 PCl3 in n-hexane was able to form similar membrane morphologies of that fabricated by 2.0 wt% PIP in water and 20 × 10−3 mol L−1 PCl3 in n-hexane, while the higher 3.0 wt% PIP led to craters crowded together, which appeared to represent more dramatic IP, producing PA layer ending up with ~160 nm entire thickness and ~42 nm monolayer thickness. In addition, the crater-like and uneven surface morphology induced by additional PCl3 was validated by AFM images of peeled-off polyamide layers (Supplementary Fig. 5), which also corresponded to plenty of pits and comparably significant roughness on the back surface from the bottom-up perspective (Supplementary Fig. 6). It suggested that IP regulation completely impacted the formed PA structure in three-dimensional pattern, not limited to the surface structure.

A AFM topographic images; B SEM surface morphology; C TEM cross-sectional appearance; D Superficial O/N ratios; and E Surface zeta potentials. The membranes were fabricated with a fixed 2.0 wt% PIP in water and the extra addition of 0, 1, 4, 20 × 10-3 mol L−1 PCl3 in n-hexane, respectively, or with 4 × 10−3 mol L−1 PCl3 in n-hexane and 1.0 wt% or 3.0 wt% PIP in water (in the order from left to right).
Generally, the IP of PIP and TMC is so instant and drastic, plus 2.0 wt% PIP being relatively high, as to rapidly form the dense PA network and in turn inevitably restrict PIP diffusion and thus the expansion of the reaction zone, which is usually responsible for the limited thickness, normally lower than 60 nm, and the smooth membrane morphology. However, proper addition of PCl3 would limit the polymerization prior to adversely affecting the PIP diffusion, allowing for further diffusion of PIP and hence resulting in the entire thickness of PA layer increasing from ~57 nm to ~150 nm. In light of above inferences, relevant processes of free IP in place of practical IP were viewed (Supplementary Fig. 8). After 2 min of free IP, 4 × 10−3 mol L−1 PCl3 in n-hexane caused the formed PA layer optically thicker and rougher, which became more pronounced when free IP extending to 48 h, standing by that the H+ regulation alleviated IP and enlarged the reaction zone in turn. Nonetheless, concentrating PCl3 up to 20 × 10−3 mol L-1 in n-hexane brought about the membrane thickness reducing to ~46 nm. The shifty membrane thickness and emergent voids, accompanied by the varying PCl3 concentration, indirectly reflected the significant discrepancy of the polycondensation process suppressed by PCl3 or backflow H+. In other words, the enriched backflow H+, associated with increased PIP, hindered the local ongoing polycondensation process, but hardly impaired PIP, further moving forward to extend the reaction zone where PIP/TMC polycondensation could start over. Or say, under circumstances of H+ effects being weakened, for example, the existing H+ away from the interface was absorbed by supplemented PIP from below, or the interfacial H+ made its way downward into the aqueous solution, the polycondensation proceeded again. Specifically, void structures might result from intermittent interruptions and uneven continuities of the interfacial polycondensation within local reaction domains. Besides, the loose assemblies or aggregates of PCl3 and PIP in n-hexane, which are easily overlooked and may be viewed as salts as a whole, having a slow-moving speed in theory, might play some role in the polycondensation process and contribute to forming the cavities. As is also evidenced, excess H+ was able to severely suppress the polycondensation so as to generate a smooth, thin, and loose polyamide layer if using 20 × 10-3 mol L−1 PCl3 in n-hexane. Apparently, the PIP concentration also mattered; for instance, 1.0 wt% PIP in water was not able to supply enough PIP diffusing into the organic phase, which led to the smooth and thin PA layer, while 3.0 wt% PIP in water enabled the fast delivery of more PIP, so that craters and cavities came into being in outer and inner sections of PA layers, respectively. In conclusion, the distinguishing impacts of backflow H+, simulated by the hydrolysis of PCl3 at the interface, were significantly exerted on the follow-up polycondensation in comparison to the diffusion behavior of aqueous monomers, which opened up a new strategy for regulating PA structure.
Further on, some explorations were conducted to answer for the higher rejection of MgCl2 than Na2SO4. According to the NF mechanism, positively charged NF membranes would facilitate the transmembrane transport of SO42- rather than Mg2+. Actually, the superficial O/N ratios of resultant membranes steeply decreased from ~1.42 to ~1.1 when PCl3 coexisted with TMC in n-hexane, evidencing the increased density of amine groups (Fig. 3D, Supplementary Tables 3, 4 and 5). Furthermore, by deconvoluting the high-resolution XPS scan spectra, the introduction of PCl3 in n-hexane indeed resulted in the evident decrease of O-C=O groups from 6.9% to about 5.0%, and remarkable increase of =NH/=NH2+ groups from 10.7% to more than 15.0% on the membrane surface (Supplementary Fig. 9 and Supplementary Table 2). Despite lower O/N ratios were readily recognized to indicate a higher crosslinking degree or smaller pore sizes of NF membranes, it contradicted the sequentially descending xylose rejections by respective NF membranes fabricated with the ascending addition of PCl3 in n-hexane (Fig. 1C). Noteworthily, the vertical distribution patterns of N elements were not the case that the closer it is to the top surface, the higher its density. On the contrary, when PCl3 was beyond 4 × 10−3 mol L−1 in n-hexane, the outer surface of resultant membranes had slightly enhanced O/N ratios according to the angle resolved XPS detection (Fig. 3D). In other words, if non-involved with errors of XPS characterization, the distinctions among the O/N ratios of the shallower top membrane surfaces, induced by adding PCl3 in n-hexane, were mainly related to the late stage of IP. More specifically, highly concentrated PIP in aqueous solutions definitely promised sufficient PIP supply for the entire amidation reaction, while, following the increasing cross-linking degree even affected by the IP regulation, the availability of PIP was bound to be restricted at the late stage of IP. Plus, the nearly identical concentration of TMC in organic phase, covering the entire IP process, would otherwise dominate in the end. As a consequence, the O/N ratios of the shallower top membrane surface slightly elevated, typically implying the polyamide structure gradually loosened outwardly, purfled with negative carboxylic groups on the outermost surface. Therefore, the decreased O/N ratios supported the increased moieties of PIP terminating PA chains or the enhanced amine density, which was partly because H+ from PCl3 slacked the amide polycondensation to benefit the continuous cross-interface diffusion and supplement of PIP monomers.
However, these NF membranes, characterized by the zeta potential analyzer, had negative potentials similar to conventional negative membrane surfaces (Fig. 3E). It somehow confronted with the common understanding for that such lower rejection of Na2SO4 than MgCl2, typical rejection characteristic of positively-charged NF membranes, which should have been proofed by positive values of measured potentials. In stark contrast, owing to forming the densest polyamide layer with no addition of PCl3 in n-hexane, the corresponding NF membrane exhibited the weakest negative potentials across all tested pH conditions (Fig. 3E). Beyond charged chemical groups detected, it is seemingly rational in light of the streaming potentials being also closely tied to sections of PA networks through which electrolyte solutions flow, thus necessitating to consider the crosslinking degree and thickness of tested NF membranes (Supplementary Note 1).
Nonetheless, surface potentials of various NF membranes at specified pH were parallelly compared. Following the gradual increment of PCl3 in n-hexane, surface potentials of corresponding NF membranes at pH =~3 stepped up because of the protonation of =NH, as is clear evidence of the enhanced =NH density; Beyond that, when the pH of background solutions ranged from 5 to 11, related potential values of serial NF membranes consistently kept the trend of first increasing and then decreasing, as conformed to the variation tendency of carboxyl density (Fig. 3E, Supplementary Figs. 9 and 10). Especially, the addition of 20 × 10−3 mol L−1 PCl3 in n-hexane brought about the lowest membrane potential of ~-66 mV despite of concurrence with ~16% Na2SO4 rejection and ~65% MgCl2 rejection, which, together with ~17% xylose rejection, indicated that such loosely cross-linked NF membrane still functioned as positively charged NF membranes. Apparently, zeta potentials measured for the top surface of corresponding membranes, usually viewed as the indirect proof of functional groups on the membrane surface, did not like the markedly decreased O/N ratios to act as powerful evidence of the improved positivity of NF membranes. When there was 4 × 10−3 mol L−1 PCl3 in n-hexane, only varying PIP in water from 1.0 wt% to 3.0 wt% formed the NF membranes of similar traits in both surface potentials and elemental compositions. Furthermore, referring to the quantification of amine groups using the ion-binding method and the speculation of potential chemical structural units by ToF-SIMS, it confirmed that more remarkably increased density of amine groups occured in resultant polyamide layers owing to PCl3 addition (Supplementary Fig. 11, Supplementary Note 1). In final, in combination with the rejection performance, the exploitation of backflow H+ is a feasible and facile way to improve the membrane surface positivity through increasing unreacted amine groups or the PIP-terminated moieties via one-step IP.
Aiming at lithium extraction from salt lakes rich in magnesium ions, it is widely accepted that the enhancement of Donnan exclusion, through enhancing the positive surface potentials of NF membranes, would increase the Li+/Mg2+ selectivity in theory. However, a significant challenge is posed to exploit the classical monomers and mainstream manufacturing ways with regard to improving the positivity of NF membranes. Only very few studies have adopted the combination of PIP/TMC monomers with ionic liquid or aromatic solvents to successfully fabricate the positive NF membranes8,22. Most of the research has turned to branched polyethyleneimine macromolecules or the newly synthetic monomers to prepare positively charged NF membranes3,23. To our knowledge, the H+ regulation of IP was first developed as a new strategy to enhance the positivity of the PA layer, which can perfectly match the current industrial production process.
The NF membrane was fabricated with 4 × 10−3 mol L−1 PCl3 addition in the IP system of 2.0 wt% PIP in water and 0.10 wt% TMC in n-hexane and denoted as NF4 membrane. The Li+/Mg2+ selectivity was assessed using synthetic brines for NF4 membrane and commercial DK membrane (Supplementary Fig. 4), demonstrating the pronounced Li+/Mg2+ separation performance under realistic conditions. All the tests maintained the pressure gauge reading of 5 bar with feeds of various components and concentrations. The rejection of MgCl2 by NF4 membrane was maintained above 96% while that of the DK membrane fluctuated around 93% (Fig. 4A and B). These relatively stable rejections were mainly determined by the steric effects. However, when LiCl solutions served as feeds, increased concentrations led to gradually decreasing rejections, with the NF4 membrane showing a more pronounced decline. Given that the membrane pore sizes of these two NF membranes (NF4: 0.30 nm; DK: 0.27 nm, Supplementary Table 1) were close to the hydrated radius of Li+/Cl− ions, steric effects would be partially responsible for the LiCl rejection. Thus, concentration polarization caused by high LiCl concentrations promoted the passage of LiCl through the membrane, manifested by a gradually reduced rejection as feed concentrations rose.

The water permeance, MgCl2 and LiCl rejections of A DK membrane and B NF4 membrane using single salt solutions, respectively; C The Mg2+ and Li+ rejections by DK membrane and NF4 membrane and D the Li+/Mg2+ selectivity of DK membrane and NF4 membrane using binary mixed solutions of diverse Mg2+/Li+ ratios based on constant LiCl concentration of 0.5 g L−1, respectively; −E The Li+/Mg2+ selectivity of DK membrane and NF4 membrane using binary mixed solutions of diverse Mg2+/Li+ ratios with MgCl2 background solutions fixed at 2 g L−1 or 10 g L−1, respectively; F The comparison of Li+/Mg2+ separation performance of DK membrane and NF4 membrane with that of PA NF membranes reported in recent years (Supplementary Table 7).
In addition to steric effects partially responsible for LiCl rejection, charge effects played a crucial role as well. Notably, Cl- has a smaller hydrated radius of 0.332 nm and a lower hydration free energy of −340 kJ mol−1 in comparison with Li+ of 0.382 nm and −475 kJ mol-13. As a result, Li+ ions are the dominant ions on NF membranes when rejecting LiCl. Although with larger pore sizes, the more positive NF4 membrane was unfavorable for the Li+ permeation through the membrane, while the DK membrane, featuring highly negatively charged, was conducive to the permeation of Li+. Namely, with the feeds consisting of the same concentrations of LiCl, the NF4 membrane had the higher rejection of LiCl. Consequently, following augmented concentrations of LiCl and thereby increasingly diminished charge effects, the reduction in LiCl rejection was markedly more substantial for the NF4 membrane compared to the DK membrane. Thus, the distinct charge effects of these two membranes underpinned the variations in the LiCl rejection as well as the discrepancy in the extent of decreasing LiCl rejection.
The selectivity was also evaluated using the binary mixture of fixing 0.5 g L−1 LiCl and increasing concentration of MgCl2 to achieve the Mg2+/Li+ ratios being 1, 5, 10, 20 and 40 (Fig. 4C and D). Both NF4 and DK membranes experienced slight variations in Mg2+ rejections (Fig. 4C). Specifically, a slight lift-up due to co-ions competition effects, reaching 98.6% and 96.7%, respectively, and then a decrease in Mg2+ rejections to 95.6% and 92.8%, respectively, resulting from the concentration polarization effects of elevated MgCl2 concentrations. Furthermore, relative to DK membrane, NF4 membrane showed more pronounced co-ions competition effects so as to more remarkably reduce the LiCl rejection (Fig. 4C). It was also found that synergistic effects of co-ions competition and concentration polarization caused by the gradually increased MgCl2 concentration led to the ascendant or steady rejection of Mg2+ and a decreased rejection of Li+ in mixed solutions within a certain range, contributing to improving the Li+/Mg2+ selectivity (Fig. 4D). For example, at a Mg2+/Li+ ratio of 20 in feeds, a negative Li+ rejection by NF4 membrane reached −118.0%, close to −137.6% of DK membrane. Noteworthily, excessively high Mg2+ concentrations observably reduced Mg2+ rejection. Moreover, osmotic pressure would be elevated to significantly impair the effective operation pressure, thereby adverse to water flux. When Mg2+/Li+ ratio was 40, the feed consisted of 0.5 g L−1 LiCl and 13.0 g L−1 MgCl2. The high osmotic pressure resulted in almost no water production for DK membrane and a reduction in the separation factor of NF4 membrane to 39.1. Therefore, combining the steric, charge, co-ions competition, and concentration polarization effects, the Li+/Mg2+ selectivity first increased and then decreased (Fig. 4D). The maximum separation factors were 67.7 for the NF4 membrane and 42.3 for the DK membrane, respectively.
The above-mentioned NF membranes were also briefly evaluated by fixing the ion concentration of Mg2+ while varying that of Li+. First, maintaining MgCl2 at 2 g L−1 in the binary mixed solutions, commonly adopted in the literature for assessment tests, ratios of Mg2+/Li+ were reduced from 80 to 10 by gradually adding LiCl (Fig. 4E and Supplementary Fig. 12). The Li+/Mg2+ selectivity for both NF4 and DK membranes fluctuated within narrow ranges of 56–59 and 33–43, respectively. However, when MgCl2 was increased to 10 g L−1 while maintaining the same Mg2+/ Li+ ratio range, the Li+/Mg2+ selectivity for both membranes showed a significant increase, mainly because of the slightly ascending rejection of MgCl2. Although the constant operation pressure rose from 5 to 10 bar, increasing MgCl2 concentration from 2 to 10 g L−1 remarkably raised the osmotic pressure, thereby decreasing the effective cross-membrane pressure difference and leading to a significant decrease in water flux. This also caused a slight increase in MgCl2 rejection while a more pronounced negative rejection of LiCl, ultimately pushing up the separation factor values. Subsequently, continuously increasing the LiCl in the binary mixed solution caused the negative rejection of Li+ to rebound, which in turn lowered the separation factor of Li+/Mg2+.
Discussion
The exploration of dominant factors in regulating the IP process using proper equivalents
In the IP regulation to elevate the membrane positivity, the added PCl3 in n-hexane does not react with TMC, nor does it interact with and thereby affect the bulk diffusion behavior of TMC (Supplementary Table 6). Exactly, when the interfacial diffusion was initiated, it is possible for PCl3 to weakly associate with PIP to potentially form aggregates. More importantly, PCl3 diffused downwards and contacted with water for instant hydrolysis, producing phosphorous acid (H3PO3) and hydrochloric acid (HCl). Because PCl3 concentration was at the level of 10−3 mol L−1, they tended to dissociate thoroughly on the water side of the interface, generating H+ (protons) that affected the IP. Therefore, it is essential to investigate the dominant factors of IP regulation, involving binding of organic additives with PIP that have diffused into n-hexane, or associations of PIP with HCl byproducts and H+ hydrated from PCl3.
For the optimal case of membrane fabrication conditions, with the extra addition of 4 × 10−3 mol L−1, PCl3 was completely hydrolyzed into 12 × 10−3 mol L−1 H+ belonging to HCl, and 8 × 10−3 mol L−1 H+ belonging to H3PO3. For deeply inquiring the fundamentals, some equivalents were selected mainly by referring to the common H+ (protons) effects and based on the pKa of HCl and H3PO3 at ambient temperature, although other physicochemical properties might potentially impact the IP. Specifically, the heat released by PCl3 hydrolysis is roughly comparable to that generated by the amidation reaction of PIP and TMC (Supplementary Figs. 17 and 18), as characterized by isothermal titration calorimetry and suggested unnecessary to be viewed as a contributory factor affecting IP. Therefore, 4 × 10−3 mol L−1 PCl3 in n-hexane was replaced with an equivalent amount of similar acyl chlorides, namely, acetyl chloride, phosphoryl chloride, ethylsulfonyl chloride, or thionyl chloride, with the prepared NF membranes named as NFAC, NFPC, NFSC, and NFTC, respectively; or with 12 × 10−3 mol L-1 acidic substances with various pKa values, namely, ethylsulfonic acid, 2-fluoroacrylic acid, pentafluorobenzoic acid, and trifluoroacetic acid, with the prepared NF membranes named as NFSA, NFFA, NFPA, and NFTA, respectively (Fig. 5). With above-mentioned replacements, the effects of IP regulation on membrane performance divided, which were confirmed and became more prominent by the further elevated concentration (Supplementary Fig. 13).

The general performance of NF membranes as fabricated by replacing the 4 × 10−3 mol L-1 PCl3 with 4 × 10−3 mol L-1 other acyl chlorides or 12 × 10−3 mol L-1 acids of various pKa as the additive in the organic phase.
According to the separation performance, the regulation effects of phosphoryl chloride and thionyl chloride during the IP process matched well with that of PCl3, but demanding a higher concentration for phosphoryl chloride and a lower concentration for thionyl chloride. However, acetyl chloride and ethylsulfonyl chloride did not generate similar effects. Conversely, acetyl chloride led to the formation of NF membranes with significantly lower rejection of MgCl2, while even further increasing the concentration of ethylsulfonyl chloride did not decrease the rejection of Na2SO4. It is well-known that acetyl chloride more preferably reacted with PIP, while ethylsulfonyl chloride carried on an extremely slow reaction with PIP as well as water in comparison with the IP of PIP and TMC. Moreover, ethylsulfonic acid, as the hydrolysis product of ethylsulfonyl chloride, has a higher pKa of ~1.8. The higher concentration of additional ethylsulfonyl chloride (12 × 10−3 mol L−1), simultaneously producing more ethylsulfonic acid and HCl in theory, failed to achieve the expected effects, possibly because of no enough protons supplied timely (Supplementary Fig. 13). Nonetheless, these results proved that possible associations of PIP and acyl chlorides in n-hexane were not dominant on the IP regulation.
Further, extra-added acids with various pKa in n-hexane were capable of regulating the IP differently. In particular, with pKa = 1.8 (25 °C) as a tipping point, which is also lower than pKa = 2.0 for H3PO3, acids of the lower pKa tended to decrease the Na2SO4 rejection of the fabricated membranes, while acids of the higher pKa achieved the lower MgCl2 rejection accordingly. It suggested that the proper content of HCl rather than H3PO3 hydrolyzed from PCl3 has taken dominant effects during the IP regulation. More specifically, it was found aggregates were generated in n-hexane as 4 × 10−3 mol L−1 acyl chlorides or 12 × 10−3 mol L−1 acids, selected reagents for mechanism exploration, were mixed with the saturated PIP n-hexane solutions, phenomenally confirming the potential interactions between PIP and above reagents that are easily perceived of. Nonetheless, during the actual IP process, PIP in n-hexane was so below the saturation state that there were no visible aggregates. The above-mentioned mixed processes were phenomenally similar with blending n-hexane solutions of PCl3 and PIP (Supplementary Fig. 15). Based on the general recognition of PIP/TMC IP occurring on the side of organic phase, it can be extrapolated that, on one hand, these acyl chlorides would react with PIP or might first associate with PIP weakly; on the other hand, these hydrolysates of acyl chlorides or additional acids directly combined with PIP strongly. These weak or strong aggregations between additives and PIP in the organic phase did not hinder the IP of PIP and TMC, but altered the proportion of PIP/TMC participating in the IP, thereby affecting the end groups of polymeric chains as well as the charged characteristics of PA layers. In other words, the strong associations with PIP, provided by the strong acids of pKa less than 1.8, played the dominating role in elevating the positivity of NF membranes. Hence, for PCl3, the generated HCl rather than H3PO3 was of vital importance. It is meaningful to define that backflow H+, including the case of only originating from the HCl byproducts, during the IP matters to configure the spatial charge of PA layers, because once adequate H+ is supplied to affect the IP, the resulting membranes are capable of reducing the rejection of Na2SO4. The corresponding strong associations with PIP did alleviate the instant and violent IP for the fast formation of dense PA layer and promise, plus a higher concentration of PIP as not usually adopted in the aqueous phase, more PIP transporting across the interface to finally terminate the PA chains.
The prediction of instant diffusion flux of PIP upon contacting the organic phase and the subsequent diffusion behavior in n-hexane
The PIP content in n-hexane, whether from direct dissolution or diffusion from aqueous PIP solutions, was investigated in detail. Direct dissolution of PIP flakes in pure n-hexane yielded ~8.0 × 10³ mg L−¹ PIP in n-hexane after sufficient dissolution over 24 h. In contrast, partition equilibrium between 2.0 wt% or 10.0 wt% PIP aqueous solutions and n-hexane resulted in about 8.5 and 14.5 mg L⁻¹ PIP in n-hexane, respectively, less than 1/500 of direct dissolution. Considering the detection limit of gas chromatography (GC), the diffusion dynamic of PIP from water into n-hexane was carried out using 15 wt% (150 × 103 mg L−1) PIP aqueous solutions and pure n-hexane solvent. At 10 min, about 9.0 mg L−1 PIP in n-hexane was achieved, and by 240 min, the diffusion process was close to equilibrium with about 15 mg L-1 PIP in n-hexane. Notably, the initial cross-interface transfer of PIP was so rapid that nearly 60% of the total diffusion occurring within the first 10 min. Driven by the concentration gradient, the PIP diffusion was positively correlated with the initial PIP concentration in aqueous solution. Convincingly and predictably, about 5.0 mg L−1 PIP promptly appeared at the interface when initiating IP using a 2.0 wt% PIP aqueous solution.
Furthermore, the diffusion coefficients of species in two phases were measured using two-dimensional diffusion ordered spectra (2D DOSY) recorded on a nuclear magnetic resonance (NMR) spectrometer (Supplementary Fig. 16 and Supplementary Table 6). The diffusion coefficients of PIP in water were 0.7 × 10−9 m2 s−1 for 2.0 wt% and 0.8 × 10−9 m2 s−1 for 0.2 wt% solutions. Whilst PIP, from 2.0 wt% or even saturated PIP aqueous solution, could diffuse upwards into n-hexane to reach partition equilibrium, it was insufficient for NMR detection. The saturated n-hexane solution of PIP, prepared by directly dissolving flaky PIP in n-hexane, was exploited to determine the diffusion behavior of PIP in n-hexane, measured at 4.2 × 10-9 m2 s−1. Considering PIP in saturated n-hexane solution was almost 1000-fold that in low-concentrated PIP n-hexane solution prepared through partition equilibrium between 2.0 wt% PIP aqueous solution and n-hexane, the practical PIP diffusivity during IP was very prompt. Additionally, during the IP, additional PCl3 in n-hexane is theoretically inclined to associate with and thus impact the PIP moving; therefore some specified PCl3 was used to simulate the interference. Not leading to visible suspended species, the diffusion coefficient of PIP in n-hexane gradually increased from 4.2 to 4.3 × 10−9 m2 s−1 with increasing PCl3. Since the PIP self-diffusion detected by NMR belongs to the Brownian motion, the enhanced diffusivity often reflects the decreased numbers of PIP molecules in n-hexane, likely due to association with few PCl3. When an extra 4 × 10−3 mol L-1 PCl3 attending the IP process of 2.0 wt% PIP and 0.10 wt% TMC, the instant contact of two immiscible phases would introduce about 0.05 × 10-3 mol L−1 (5.0 mg L−1) PIP into the organic phase. Apart from polymerizing with TMC, PIP may either be captured by PCl3 or escape at a higher velocity because of stronger diffusivity in n-hexane. Further, as shown by NMR measurements, the migration of PIP in n-hexane was far faster than that of PIP in water, which was a benefit of the dispersal and replenishment of PIP in n-hexane.
The impacts of H+ on the interfacial diffusion and polymerization during the IP process
Focusing on the routine PIP-TMC IP, HCl byproducts were roughly estimated based on the resulting polymer weight, mostly flowing from the reaction zone to the interface and likely being adsorbed on amine groups of neighboring PIP or oligomers while fatefully protonating a negligible portion of PIP in the aqueous phase. Whereas very few TMC might contact with water at the interface, HCl production from acyl chloride hydrolysis was marginal as well. Hence, the total amount of above HCl is very unlikely led to appreciable variations of membrane performance, which was barely indicative of small variations of unreacted chemical groups. As a result, it may cause a lack of exploring the significant impact of H+ (HCl) byproduct as well as regarding it as a way of manipulating the fabrication of NF membranes. Actually, further polymerization of protonated PIP molecules or oligomers (e.g., associating with HCl) necessitated the foremost elimination of the associated H+, by generally assuming the latter occurred faster than the former in the overall IP system11. This implies that retarding H+ elimination, such as synchronously and amply introducing H+ when IP proceeds, will alleviate the IP. However, the introduced H+, if far less than the reservoir of PIP in aqueous phase, will hardly affect the PIP cross-interface diffusion to maintain a consistent supply for the IP. Consequently, the delicate experiment design reveals the differentiated effects of H+ regulator on two consecutive processes of IP, suggesting backflow H+ matters on the resulting polyamide networks, such as impacting the terminal groups.
A slight compromise on PIP diffusion behavior as a result of additional H+
At the moment the two immiscible phases were contacted to bring about the greatest concentration gradient of PIP between the aqueous phase and organic phase, the interfacial diffusion flux of PIP was maximized instantly. Notably, the self-diffusion coefficient of PIP in n-hexane is about 4.2 × 10−9 m2 s−1, and five-fold higher than 8.3 × 10−10 m2 s−1 of PIP in water. In other words, PIP, once partitioning into the organic phase, although primarily polymerized with TMC, would partly diffuse forward at a faster velocity. With an ample reservoir of PIP in the aqueous phase, PIP, once crossing the interface, would move faster and spread further, attributable to superior diffusion behavior in n-hexane (more than as previously assumed). If restricting the amide polycondensation, it would incline to protect more PIP moving forward from immediately participating in the polycondensation. PCl3 in n-hexane would diffuse downwards with a self-diffusion coefficient of about 5.2 × 10−9 m2 s−1 and undergo a violent hydrolysis with water at the interface, a process accelerated by the alkalinity of the PIP solution. This meant that plenty of H+, far exceeding the quantity of HCl byproducts from IP, was rapidly produced at the interface. For the addition of 4 × 10−3 mol L−1 PCl3 in the organic phase, the molarity of H+ was six-fold that of PCl3 but accounted for less than 10% of 2.0 wt% PIP (232 × 10−3 mol L−1) aqueous solution at most. In this case, the interfacial concentration of unprotonated PIP ranged from 212 to 222 × 10−3 mol L−1 (close to 1.9 wt%), in light of PIP having two secondary amine groups available. Wholly, PIP, protonated PIP at one end, and protonated PIP at both ends would diffuse into the organic phase, although less proportion for the latter two species was expected due to their suppressed diffusion abilities. Consequently, excessive PIP, namely 2.0 wt% PIP in water, maintained the persistent diffusion and guaranteed a sufficient supply of amine monomers for the IP process, which has not been refrained by the presence of H+ provided by PCl3 hydrolysis.
A dominant retardance in the PIP reaction induced by interfacial H+
During the IP of PIP and TMC, the lower solubility of TMC in water compared to that of PIP in n-hexane, along with water adversely affecting the amidation reaction, confined the practical reaction zone primarily within the organic phase11,16. Likewise, H+ or equivalents affecting the reaction process should mostly take place on the n-hexane side. If PIP partitioned into n-hexane according to the partition coefficient, though the actual diffusion was a dynamic process and also governed by reaction consumption, and fully underwent amidation reaction, HCl byproducts would double. Namely, 232 × 10−3 mol L−1 (2.0 wt%) PIP in water would lead to about 0.1 × 10−3 mol L−1 PIP partitioning into n-hexane and thus about 0.2 × 10−3 mol L−1 HCl indwelling in n-hexane at most as determined by diffusion equilibrium measurements. Very few of HCl byproducts would move upwards into the air while most of them, due to concentration gradient and the great density, would descend and collide with the oncoming PIP. Nevertheless, owing to the excess amount and strong alkalinity of PIP, adverse effects of HCl byproducts exerting on the IP often were neglected and rarely reflected on the formed membrane performance. In the case of adding 4 × 10−3 mol L−1 PCl3 in n-hexane, 20 × 10−3 mol L−1 H+, instantly produced in the formation of HCl or H3PO3 through interfacial hydrolysis, was over 100-fold H+ by-products of amide polycondensation. If 12 × 10−3 mol L−1 H+ (HCl) from PCl3 hydrolysis was considered crucial, it was 60 times greater than H+ by-products of amide polycondensation. Therefore, when IP was initiated, oligomers formed, and subsequently were inclined to adsorb H+ byproducts and introduced H+, thereby suppressing the quick extension of oligomer chains in the interfacial reaction zone. The HCl or H3PO3, although mostly distributed into the aqueous phase and capable of protonating a very small portion of PIP, inevitably encountered rising PIP to form assemblies of PIP-HCl or PIP-H3PO3 in theory, which managed to make a difference in the following polycondensation. Meanwhile, the dispersed PIP in the organic phase might weakly associate with PCl3 before polycondensation, which was insignificant and unstable but handicapped the proceeding of polycondensation to some extent. In short, the interfacial H+, belonging to strong acids of pKa less than 1.8, retarded the PIP approaching and polymerizing with TMC to markedly mitigate the formation of a dense polyamide layer (further discussed in Supplementary Note 2 based on the chemical equilibrium theory).
In sum, H+ from PCl3 hydrolysis protonated such a small portion of PIP that insignificantly impacted the overall PIP diffusion flux. Nevertheless, the presence of H+ at the interface and protonated PIP in n-hexane remarkably slowed down the polycondensation rate. Comparatively, PCl3 additive spared abundant time for PIP diffusion during the IP process, which is the key to increase =NH density (potential positive groups) particularly close to membrane surface. Further, hydrolysates of additives or themselves gifted with the pKa less than 1.8 were found capable to produce membranes with high positive charge density. Plus, adequate addition, exemplified as the equivalence of 12 × 10−3 mol L−1 HCl, would lead to the conspicuous variations of membrane performance, such as having lower rejection of Na2SO4 than MgCl2. Hence, it testified that backflow H+ (HCl or equivalents) exerts disparate effects on two successive processes of IP, particularly braking the otherwise uncontrollable, violent amide polycondensation.
In this study, various affecting factors in the IP process were revisited and scrutinized. Backflow H+ or equivalents were leveraged to develop one novel IP regulation strategy for enhancing the positive charge density of NF membranes. Such membranes can comprise the key modules in accelerating lithium extraction from brines to satisfy the substantial demand in lithium-ion battery industry. Additionally, mechanisms were dissected through experimental and quantitative analyses, unveiling a theoretical foundation for the feasibility of IP regulation strategy—disparate effects of protons on interfacial diffusion and ensuing amidation reactions. Most importantly, based on the well-established PIP-TMC IP system that typically produced negatively-charged membranes, NF membranes of positive charge effects can now be easily prepared in one step. This advancement aligns seamlessly with existing industrial production lines, thereby expediting the scale-up manufacturing of NF membranes with superior Li⁺/Mg2+ selectivity. It also complement one piece of the jigsaw of classic IP and further expand IP’s versatility in fabricating functional membranes.
Methods
Materials and Chemicals
The polysulfone ultrafiltration membrane, supplied by Hunan Ovay Technology Co., Ltd. (China), was used as the support layer for the self-made nanofiltration (NF) membranes. Piperazine (PIP) (Sigma-Aldrich, USA, 99%), trimesoyl chloride (TMC) (J&K, China, 98%), and n-hexane (Fisher Scientific, UK, > 98%) were selected for IP. Phosphorus trichloride (PCl3), acetic chloride (CH3COCl, AC), phosphoryl chloride (POCl3, PC), ethane sulfochloride (CH3CH2SOCl, SC), thionyl chloride (SOCl2, TC), ethylsulfonic acid (CH2CH3SO3H, EA, pKa 1.8), 2-fluoroacrylic acid (CH2CHFCOOH, FA, pKa 2.5), pentafluorobenzoic acid (C6F5COOH, PA, pKa 1.5), and trifluoroacetic acid (CF3COOH, TA, pKa -0.3) were obtained to regulate the IP, whose pKa is determined according to the Bordwell pKa Table24. The neutral model molecules, including glycerol, D-(+)-xylose, D-(+)-glucose and sucrose, as well as inorganic salts, such as LiCl, NaCl, MgCl2, MgSO4, and Na2SO4, were used to assess the membrane performance. Above-mentioned reagents were of analytical grade and obtained from Sinopharm Chemical Reagent Co., Ltd. (China). All chemicals were used as received. The conductivity of deionized (DI) water was below 5 μS/cm.
Fabrication of NF membranes
For the fabrication of NF membranes, a lab-made module (15 × 15 cm2) was used to fix the polysulfone UF membrane as the support layer, where the PIP aqueous solution was poured in and then poured out after 2 min followed by removing the surplus solution with a rubber roller. Immediately, TMC in n-hexane was added in to cover the PIP-impregnated substrate for 1 min, forming the PA active layer via the IP. Subsequently, it was transferred into the oven of 75 °C for the curing of 3 min, and then rinsed with DI water. When regulating the IP, the above-described procedure was followed except that only the specified additive was added into the TMC n-hexane solution. With the addition of PCl3 from 0, 1, 2, 4, 20 × 10−3 mol L−1, the NF membranes, fabricated by 2.0 wt% PIP in water and 0.10 wt% TMC in n-hexane, were named as NF0, NF1, NF2, NF4 and NF20, respectively.
Characterization of NF membranes
The superficial and cross-sectional membrane morphologies of NF membranes were characterized by a field emission scanning electron microscope (FESEM) (Hitachi S5500, Japan) with an accelerating voltage of 5 kV and a transmission electron microscope (TEM, Hitachi H7650, Japan) with an accelerating voltage of 80 kV, respectively. The front and back surface morphologies of NF membranes were inspected using an atomic force microscope (AFM, Dimension ICON, Bruker, Germany) with the scanning area of 10 × 10 µm2. At least three AFM images were acquired for the average roughness. The element distribution of PA separation layer was detected by X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi, Thermo Fisher, USA) with the X-ray source of Al Kα. Three spots on each sample of 1 × 2 cm2 were used to analyze elemental compositions on the top membrane surface. Further, the shallower membrane surface was detected by changing the sample stage from 0o to 30o and 60o in sequence, and underneath the membrane surface was inspected by the mode of XPS depth-etching. The potential structural units constituting polyamide layer were analyzed and detected employing ToF-SIMS (Time of Flight Secondary Ion Mass Spectrometry, TOF-SIMS 5–100, IONTOF GmbH, Germany). The water contact angle (CA) was obtained with at least six different positions tested on each sample according to the standard sessile drop method using a goniometer (Contact Angle System OCA20, Data Physics Instruments GmbH, Germany). The zeta potentials of NF membranes were measured in triplicate for each membrane sample in the 0.1 mM KCl background electrolyte solution with pH adjusted from 2 to 11 using an electrokinetic analyzer (Anton Paar, Graz, Austria).
Membrane performance test
The performance of NF membranes was evaluated using a lab-scale cross-flow filtration system containing three parallel filtration cells (CF016, Sterlitech, USA). Each cell had the effective filtration area of 20.6 cm2 and the flow channel height of 2 mm. Three separate coupons were mounted into the three cells to perform the triplicate tests. During the filtration process, both the permeate and the retenate were circulated into the feed bank until some samples were taken for characterization when the operation state became stable. These tests were conducted with the pressure gauge reading of 5 bar and the cross-flow velocity of 0.2 m s−1 at 20 ( ± 2) °C, with a pre-compaction of membrane coupons by filtering DI water at 8 bar. The performance of NF membranes was mainly determined by rejecting inorganic salts and neutral organic solutes. The inorganic salts including LiCl, NaCl, MgCl2, Na2SO4 and MgSO4 were separately dissolved in the DI water, each achieving a total ionic strength of 10 mmol L−1. The salt concentrations in the permeate and the feed were obtained according to electrical conductivity by a conductivity meter (Mettler Toledo, FE38, Switzerland). Glycerol, D-(+)-xylose, D-(+)-glucose, and sucrose were separately dissolved in the DI water for the feed concentration of 50 mg L−1. The rejection of neutral organic solute was calculated according to total organic carbon (TOC) in samples measured by a TOC analyzer (Shimadzu TOC-VCPH, Japan). The Mg2+ and Li+ in the binary mixed solutions were listed in Supplementary Table 8 and determined by inductively coupled plasma optical emission spectrometry (ICP-OES, iCAP RQ, Thermofisher, Germany).
The water permeance and water flux of NF membranes were separately calculated by Eq. (1) and Eq. (2):
where A is the water permeance (L m−2 h−1 bar−1), Jw is the water flux (L m−2 h−1), ΔP is the applied pressure (bar), S is the effective membrane area (m2), and V is the volume of permeate (L) collected within the specified time interval t (h).
The rejection of solutes was calculated by Eq. (3):
where R is the solute rejection (%), and cp and cf are the concentrations in the permeate and the feed, respectively.
The selectivity of Li+/Mg2+ of NF membranes was analyzed by Eq. (4):
where CLi+, p, and CMg2+, p, CLi+, f, and CMg2+, f are the concentrations of Li+ and Mg2+ in the permeate and the feed, respectively.
Data availability
All data are provided in the main text and Supplementary Information. All data are accessible from the corresponding author upon request.
References
Werber, J. R., Osuji, C. O. & Elimelech, M. Materials for next-generation desalination and water purification membranes. Nat. Rev. Mater. 1, 1–16 (2016).
Razmjou, A. et al. Design principles of ion selective nanostructured membranes for the extraction of lithium ions. Nat. Commun. 10, 5793 (2019).
Zhang, T., Zheng, W., Wang, Q., Wu, Z. & Wang, Z. Designed strategies of nanofiltration technology for Mg2+/Li⁺ separation from salt-lake brine: A comprehensive review. Desalination 546, 116205 (2023).
Jiang, Z. et al. Aligned macrocycle pores in ultrathin films for accurate molecular sieving. Nature 609, 58–64 (2022).
Wang, K. et al. Tailored design of nanofiltration membranes for water treatment based on synthesis-property-performance relationships. Chem. Soc. Rev. 51, 672–719 (2022).
Ritt, C. L. et al. Machine learning reveals key ion selectivity mechanisms in polymeric membranes with subnanometer pores. Sci. Adv. 8, 5771 (2022).
Epsztein, R., DuChanois, R. M., Ritt, C. L., Noy, A. & Elimelech, M. Towards single-species selectivity of membranes with subnanometre pores. Nat. Nanotechnol. 15, 426–436 (2020).
Guo, B. B. et al. Double charge flips of polyamide membrane by ionic liquid-decoupled bulk and interfacial diffusion for on-demand nanofiltration. Nat. Commun. 15, 2282 (2024).
Liang, Y. et al. Polyamide nanofiltration membrane with highly uniform sub-nanometre pores for sub-1 Å precision separation. Nat. Commun. 11, 2015 (2020).
Zhao, Y. et al. Differentiating solutes with precise nanofiltration for next generation environmental separations: A review. Environ. Sci.Technol. 55, 1359–1376 (2021).
Morgan, P. W. & Kwolek, S. L. Interfacial polycondensation. II. Fundamentals of polymer formation at liquid interfaces. J. Polym. Sci. 6, 299–327 (1959).
Freger, V. & Ramon, G. Z. Polyamide desalination membranes: Formation, structure, and properties. Prog. Polym. Sci. 122, 101451 (2021).
Bradbury, J. H., Crawford, P. J. & Hambly, A. N. Kinetics of an interfacial polycondensation reaction. J. Polym. Sci. 5, 1337–1347 (1967).
Enkelmann, V. & Wegner, G. Mechanism of interfacial polycondensation and the direct synthesis of stable polyamide membranes. Makromol. Chem. 177, 3177–3189 (1976).
Behera, S. & Suresh, A. K. Kinetics of interfacial polycondensation reactions – Development of a new method and its validation. Polymer 127, 28–44 (2017).
Freger, V. Kinetics of film formation by interfacial polycondensation. Langmuir 21, 1884 (2005).
Berezkin, A. V. & Khokhlov, A. R. Mathematical modeling of interfacial polycondensation. J. Polym. Sci. Part B: Polym. Phys. 44, 2698–2724 (2006).
Kuchanov, S. Development of a quantitative theory of polycondensation. Prog. Polym. Sci. 29, 563–633 (2004).
Nadler, R. & Srebnik, S. Molecular simulation of polyamide synthesis by interfacial polymerization. J. Membr. Sci. 315, 100–105 (2008).
Oizerovich-Honig, R., Raim, V. & Srebnik, S. Simulation of thin film membranes formed by interfacial polymerization. Langmuir 26, 299–306 (2010).
Gao, Y., Wang, X. & Huang, X. The veiled impacts of H⁺ on interfacial polymerization and its effects on nanofiltration performance. Environ. Sci. Technol. Lett. 10, 274–279 (2023).
Wang, H., Zhang, Q. & Zhang, S. Positively charged nanofiltration membrane formed by interfacial polymerization of 3,3′,5,5′-biphenyl tetraacyl chloride and piperazine on a poly(acrylonitrile) (PAN) support. J. Membr. Sci. 378, 243–249 (2011).
Peng, H. et al. Designing Gemini-electrolytes for scalable Mg2+/Li⁺ separation membranes and modules. Adv. Funct. Mater. 33, 2305815 (2023).
Acknowledgements
We acknowledge the financial support by the National Natural Science Foundation of China, grant No. 52300095 (Y. G.) and 52270007 (X.-m. W.).
Author information
Authors and Affiliations
Contributions
Y. G. conducted the study and drafted the manuscript. X.-M. W. supervised the research and revised the manuscript. Y. G., S. L. and X.-M. W. engaged in the data curation and discussion related to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Qilin Li, Amir Razmjou, Zhe Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, Y., Wang, Xm. & Liu, S. Backflow H+ during interfacial polymerization matters to configure spatial charges of polyamide membranes. Nat Commun 16, 8764 (2025). https://doi.org/10.1038/s41467-025-63851-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63851-y
