
Abstract
Although genome-wide association studies have provided valuable insights into the genetic basis of complex traits and diseases, translating these findings to causal genes and their downstream mechanisms remains challenging. We performed trans expression quantitative trait locus (trans-eQTL) meta-analysis in 3734 lymphoblastoid cell line samples, identifying four robust loci that replicated in an independent multi-ethnic dataset of 682 individuals. The trans-eQTL signal at the ubiquitin specific peptidase 18 (USP18) locus colocalised with a GWAS signal for systemic lupus erythematosus (SLE). USP18 is a known negative regulator of interferon signalling and the SLE risk allele increased the expression of 50 interferon-inducible genes, suggesting that the risk allele impairs USP18’s ability to effectively limit the interferon response. Intriguingly, the USP18 trans-eQTL signal would not have been discovered in a meta-analysis of up to 43,301 whole blood samples, reaffirming the importance of capturing context-specific genetic effects for GWAS interpretation.
Similar content being viewed by others


Introduction
Genome-wide association studies (GWAS) have provided valuable insights into the genetic basis of complex traits and diseases. However, translating GWAS findings to actionable drug targets has remained challenging, particularly when the functions of the associated genes are unknown. A promising technique to identify the effector genes of GWAS variants as well as their downstream regulatory consequences is trans gene expression and protein quantitative trait loci (trans-QTL) analysis. Trans-QTL studies test for association between genetic variants across the genome and expression levels of all measured genes or proteins1. In a prominent example, an erythrocyte-specific regulatory element first identified as a trans protein QTL (trans-pQTL) for foetal haemoglobin (HbF) was used to design the first ever gene editing therapy for sickle-cell disease2,3.
Trans-QTLs are especially promising, because 60%-90% of gene and protein expression heritability is located in trans4, most associations detected in large-scale pQTL studies are located in trans5, and cis-QTL discovery is starting to saturate after 10,000 samples5. Furthermore, most complex trait heritability has been proposed to be mediated by trans-QTL effects4. However, current large-scale trans-eQTL and trans-pQTL studies have been limited to easily accessible bulk tissues such as whole blood6,7 or plasma5,8,9,10. Bulk tissue studies are subject to cell type composition effects which can be difficult to distinguish from true intracellular trans-QTLs1,6. The whole blood and plasma studies are also likely to miss cell type and context specific regulatory effects. In contrast, trans-eQTL studies in other tissues and purified cell types have had limited statistical power due to small sample sizes (typically less than one thousand samples), enabling the discovery of only very large effects and potentially underestimating pleiotropic effects on multiple target genes11,12,13,14,15,16,17,18.
A key limitation in our understanding of how trans-eQTLs contribute to complex traits and how they interact with cis-eQTL is the lack of well-characterised disease-associated trans-eQTL signals4. Two most prominent examples include the adipose-specific KLF14 locus associated with type 2 diabetes16,19 and the IRX3/5 locus associated with obesity20,21. At the KLF14 locus, the lead variant (rs4731702) is a cis-eQTL for the KLF14 transcription factor and was associated with the expression of 385 target genes in trans, 18 of which also had independent cis associations for other metabolic traits19. The simultaneous regulation of multiple target genes in trans-eQTL regulatory networks seems to be a general property of many known trans-eQTL signals6,12,14. However, what proportion of trans-eQTL target genes directly mediate the disease or trait associations as opposed to being independent ‘bystanders’ with minimal direct causal effect has remained unclear.
We performed the largest trans-eQTL meta-analysis in a single cell type, comprising 3734 lymphoblastoid cell line (LCL) samples across nine cohorts (MetaLCL). LCLs are obtained by transforming primary B-cells with Epstein-Barr virus22. LCLs have been widely used as a resource for human genetics, from banking cells from rare genetic disorders, through control material in laboratories to prevent repetitive blood sampling, to the study of tumorigenesis, mechanisms of viral latency and immune evasion22. Furthermore, Epstein-Barr virus has been epidemiologically linked to several autoimmune diseases in which B cells are implicated to play a pathogenic role, such as multiple sclerosis (MS)23,24 and systemic lupus erythematosus (SLE)25 with recent studies starting to elucidate the potential molecular mechanisms underlying these associations26,27,28. Thus, trans-eQTLs discovered in LCLs might provide insights into the pathogenesis of these autoimmune diseases, especially in the context of chronic exposure to viral stimuli.
After stringent quality control, we identified four highly robust trans-eQTL associations that replicated in an independent cohort (n = 682) and were associated with multiple target genes. At the USP18 locus, the trans-eQTL signal colocalised with a GWAS association for SLE. The SLE risk allele was associated with increased activity of the type I interferon signalling pathway and increased expression of several classical interferon response genes. While there is robust evidence for the potential causal role of increased interferon signalling in SLE pathogenesis, we find that the expression of many individual interferon response genes is unlikely to have a direct causal effect on SLE. Our results caution against blindly using trans-QTL associations for target gene prioritisation without clear understanding of the trans-QTL mechanism and robust genetic evidence from cis-acting variants implicating the same gene. To support secondary use of our data, we have made the complete MetaLCL cis and trans-eQTL summary statistics for 18,792 genes publicly available via the eQTL Catalogue FTP server.
Results
Large-scale trans-eQTL meta-analysis in a single cell type
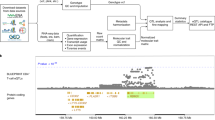
We performed a large-scale trans-eQTL study, utilising data from LCLs collected from 3734 donors (2238 female, 1496 male) across nine cohorts of European ancestries (Supplementary Table 1). To avoid confounding by technical factors, we performed association testing separately in each cohort and meta-analysed the results (Fig. 1A). After excluding cis associations located within 5 Mb of the target gene, we identified 79 suggestive independent trans-eQTL loci at p < 1 × 10−11 threshold (Fig. 1B). To identify robust signals associated with multiple target genes and reduce the risk of false positives caused by cross-mappability29, we further required each locus to be associated with at least five independent target genes (p < 5 × 10−8) with low cross-mappability scores (see Methods). This filtering reduced the number of candidate loci to six (Fig. 1B), four of which replicated in an independent multi-ethnic cohort of 682 individuals30. These four replicating trans-eQTL loci were located near the BATF3, MYBL2, USP18, HNF4G genes (Supplementary Data 1, Supplementary Fig. 2). While the strong trans-eQTL signal near the BATF3 transcription factor (2294 targets at FDR 5%) has been previously reported31, the other three seem to be novel. Remarkably, the trans-eQTL targets at the MYBL2 locus were consistent with direct activation by the MYBL2 transcription factor (Supplementary Note 1), indicating that our analysis is identifying biologically interpretable signals.

A Overview of the study design and participating cohorts. B Significant trans-eQTLs detected in the meta-analysis. The upper scatter plot shows the number of trans-eQTL target genes detected at each trans-eQTL locus with p < 5 x 10−8. Six trans-eQTL loci with the most target genes have been labelled with the name of the closest cis gene. The lower scatter plot shows all significant loci for each tested gene at the more stringent p < 1 × 10−11 threshold. Cis associations are located on the diagonal while putative trans associations are located off diagonal. The points represent two-sided p-values from inverse-variance weighted meta-analysis. Source data are provided as a Source Data file.
USP18 is a negative regulator of interferon response at a lupus GWAS locus
To prioritise the four trans-eQTL loci for follow-up analysis, we performed GWAS lookup using the Open Targets Genetics Portal32 and found that only the USP18 locus lead variant (chr22_18166589_T_C, rs4819670) was in high LD (r2 > 0.9) with an annotated GWAS hit. Specifically, the USP18 lead variant was identical to a GWAS lead variant reported for SLE in East Asians33. Using the point estimation of colocalisation (POEMColoc) method, we confirmed that the two signals colocalised (PP4 = 0.97) (Fig. 2A)34. The colocalisation also replicated in an SLE GWAS meta-analysis across the UK Biobank (UKBB)35, FinnGen36 and Million Veterans Program (MVP)37 biobanks (Supplementary Fig. 3)38. At this locus, we identified 40 trans target genes at false discovery rate (FDR) 5% that were all strongly enriched for Reactome interferon signalling (R-HSA-913531, p = 1.1 × 10−26) and interferon alpha/beta signalling (R-HSA-909733, p = 1.7 × 10−21) pathways. The rs4819670-C allele was associated with decreased expression of multiple canonical type I interferon response genes (e.g., ISG15, IFI44, OAS1-3) (Fig. 2B). Reassuringly, we observed consistent effect sizes across nine sub-cohorts in our meta-analysis (I2 heterogeneity statistic = 0.46, Supplementary Fig. 2, Supplementary Data 2). The rs4819670-C was also associated with decreased risk of systemic lupus erythematosus (SLE) in East Asians33 as well as in the MVP-FinnGen-UKBB meta-analysis38.

A Regional association plot for the SLE GWAS with POEMColoc imputed summary statistics and regional association plot for the lead trans-eQTL gene (HERC5) at the USP18 locus. The trans-eQTL lead and GWAS lead variants (rs4819670) are identical and in high LD with a missense variant (rs3180408) in the USP18 gene. The points represent two-sided p-values from inverse-variance weighted meta-analysis. The original regional association plot for the SLE GWAS is shown on Supplementary Fig. 4. B Volcano plot of the trans-eQTL target genes. Genes with FDR < 5% are highlighted in red. C USP18 down-regulates type I interferon signalling by restricting the access of Janus-associated kinase 1 (JAK1) to the type I interferon receptor42. Source data are provided as a Source Data file.
We next sought to identify the most likely causal gene at the USP18 locus. Since the trans-eQTL effect was detected in monocultures of LCLs39, this implies that the causal cis effect mediating the trans associations must also be present in the same cell type (i.e., it cannot be mediated by a trans-acting factor such as a cytokine produced by some other cell type). Although the rs4819670 variant was located in an intron of USP18, we did not detect a significant cis-eQTL effect for USP18 or any other neighbouring gene neither in our meta-analysis nor the full eQTL Catalogue release 640. We also did not detect a splicing QTL for USP18 or other neighbouring genes in eQTL Catalogue release 6. However, the rs4819670 lead variant was in high linkage disequilibrium (LD) with a USP18 missense variant rs3180408 (chr22_18167915_C_T, ENSP00000215794.7:p.Thr169Met) in both European and East Asian populations (r = −1, r2 = 1 in EAS and r = −0.98, r2 = 0.96 in EUR 1000 Genomes superpopulation). Furthermore, the rs3180408 missense variant was the lead variant for SLE in the MVP-FinnGen-UKBB meta-analysis (Supplementary Fig. 3). Notably, USP18 is a known negative regulator of interferon signalling and a rare loss-of-function mutation in USP18 causes severe type I interferonopathy (Fig. 2C)41,42. Similarly, USP18 knock-out in human macrophages increased the expression of several canonical interferon response genes upon stimulation with interferon-beta43. Thus, these results indicate that USP18 is the most likely causal gene at this trans-eQTL locus.
However, identifying the exact causal variant remains challenging. While the absence of cis eQTL and splicing QTL evidence suggests that the likely causal mechanism is the rs3180408 missense variant, the missense variant was predicted to be benign by all tested variant effect prediction tools available from Ensembl VEP44. Alternatively, there could be other genetic variants in the region that are not captured by current genotype imputation reference panels (such as structural variants). One potential strategy to assess the functional impact of the rs3180408 missense variant on trans-eQTL target gene expression would be genome editing, but our power calculations (see Supplementary Note 2) suggest that this would be extremely challenging due to the small expected effect size of the variant.
Role of aberrant interferon signalling in lupus pathogenesis
Several studies have suggested that causal GWAS genes are enriched in shared pathways or biological processes45,46,47. To further characterise the potential role of USP18 target genes in lupus, we performed additional trans-eQTL meta-analysis across the nine discovery cohorts and one replication cohort (total n = 4416). This increased the number of significant USP18 target genes to 50 (FDR < 5%). Notably, 18/50 target genes overlapped the Reactome interferon alpha/beta signalling (R-HSA-909733) pathway (hypergeometric test, p = 4.14 × 10−24) and 26/50 genes overlapped a consensus set of interferon response genes (n = 124) identified by Mostafavi et al.48 (p = 1.44 × 10−39, Supplementary Data 3). Reassuringly, 40/50 genes were also more highly expressed in peripheral blood mononuclear cells from SLE cases compared to controls49 (Supplementary Data 3), consistent with the established role of increased interferon signalling in SLE50.
To better understand the role of the USP18 target genes in the interferon alpha/beta signalling pathway, we focussed on the 60 genes belonging to the Reactome R-HSA-909733 interferon alpha/beta signalling pathway and divided them into three categories - category I: proteins involved in signal transduction via IFNAR1/2 receptor (n = 13 genes, including the multi-gene interferon-alpha gene cluster, Fig. 3A); category II: downstream transcriptional targets of the interferon signalling (38 genes from the Reactome R-HSA-1015702 sub-pathway, Fig. 3B) and category III: other pathway genes (n = 9) not belonging to the first two categories (Supplementary Fig. 5). We found that 16/50 USP18 targets were shared with the 38 category II genes (transcriptional targets of interferon response) (p = 1.18 × 10−29, Fisher’s exact test). In contrast, only 2/50 USP18 trans-eQTL target genes (STAT1 and ISG15) were shared with the 13 category I genes (IFNAR1/2 receptor signal transduction proteins) and none were shared with the 9 category III genes. This suggests that the USP18 trans-eQTLs are primarily capturing the transcriptional targets of interferon response (category II), consistent with the established role of USP18 in regulating these genes (Fig. 2C)42.

A Upstream regulators of interferon response genes (IFNA* contains multi-gene interferon-alpha gene cluster). B Downstream transcriptional targets of the interferon signalling (HLA* marks the HLA region). The increased gene expression is marked in red, while reduced gene expression is marked in blue. The visualisation illustrates the effect on USP18 target genes in relation to the SLE risk allele. DE - differential gene expression in SLE cases versus controls49; GWAS - GWAS hits for SLE33, ChEMBL, phase III - SLE phase III clinical trials from ChEMBL56, PID - genes causing primary immunodeficiency from Genomics England. Source data are provided as a Source Data file.
Next, we assessed if there were additional lupus GWAS signals overlapping the three categories of interferon response genes defined above. We first used the Open Targets Genetics portal to extract the prioritised target genes for 108 lupus GWAS loci from Yin et al.33. This revealed that three prioritised lupus genes (USP18, STAT1, IFNA1-17) were shared with the 13 category I genes (IFNAR1/2 signal transduction proteins, Fig. 3A) and four prioritised genes (IRF1/5/8 and OAS1) were shared with the 38 category II genes (transcriptional targets of interferon response, Fig. 3B). Out of these, IRF1/5/8 are themselves transcription factors involved in the regulation of interferon production51,52, and stronger IRF1 binding across many GWAS loci has been associated with higher Crohn’s disease risk53. OAS1 represents a classical antiviral gene and here the SLE GWAS lead variant is in perfect LD with a fine-mapped splice QTL for OAS1 in the eQTL Catalogue (Supplementary Fig. 6)40. The consequences of this splice variant have been extensively studied in the context of Sjögren’s syndrome and COVID-19 hospitalisation54,55. Unfortunately, we were not able to check if there were additional sub-threshold hits for lupus near the interferon alpha/beta signalling pathway genes, because the genome-wide summary statistics from Yin et al.33 were not available.
We also overlapped interferon alpha/beta signalling pathway genes with ongoing or completed phase III clinical trials for SLE extracted from the ChEMBL database56. We identified three category I (interferon signal transduction) genes (IFNAR1, JAK1 and TYK2) that have been targeted by a clinical trial for SLE (Fig. 3A). While the trials targeting JAK1 and TYK2 are currently ongoing, a randomised control trial of anifrolumab, a human monoclonal antibody to type I interferon receptor subunit 1 (IFNAR1), found it to be an effective treatment for SLE57. None of the category II genes (transcriptional targets of interferon signalling, Fig. 3B) and category III genes (Supplementary Fig. 5) are currently in a phase III clinical trial for SLE (Fig. 3B).
There is an emerging consensus that rare mutations in genes prioritised for autoimmune diseases from GWAS studies can often also cause primary immunodeficiencies (PIDs)58,59,60. For example, loss-of-function mutations in USP18 cause rare type I interferonopathy41,42. At the same time, GWAS studies for SLE and other autoimmune diseases are still only powered to detect variants with large effects. Thus, knowing if a gene causes PID might be a useful (if noisy) indicator that the same gene might be discovered in a future larger autoimmune GWAS study. We obtained the list of genes causing either PID or monogenic inflammatory bowel disease from Genomics England61 and overlapped those with the three categories of interferon response genes defined above. We found that 10/13 category I genes (interferon signal transduction) have previously been implicated in causing PID, including USP18 and all three phase III drug candidates for SLE (Fig. 3A). In contrast, only 8 of the 38 category II genes (transcriptional targets of interferon response) have been implicated in PIDs (Fig. 3B), including OAS1 and IRF8 also detected by SLE GWAS. Finally, none of the category III genes have been implicated in PIDs (Supplementary Fig. 5).
Triangulation of evidence from prioritised lupus GWAS target genes, phase III clinical trial information and overlap with primary immunodeficiency genes reaffirms modulation of aberrant interferon alpha/beta signalling in B-cells as an emerging therapeutic opportunity for SLE (category I, Fig. 3A)60,62. This is further supported by recent studies demonstrating that depleting autoreactive B-cells via anti-CD19 CAR T cell therapy is an effective therapy for SLE and other autoimmune diseases63,64. In contrast, most trans-eQTL targets of USP18 are transcriptional targets of interferon response (category II, Fig. 3B) and it is far less clear what are the potential causal roles of these genes in SLE pathogenesis.
Replication of the USP18 trans-eQTL signal in whole blood
To understand the context-specificity of the USP18 trans-eQTL signal, we performed additional replication in the eQTLGen Phase 2 trans-eQTL meta-analysis of up to 43,301 whole blood samples. We observed that the USP18 missense variant rs3180408 was nominally associated (p < 0.05) with the expression of 7/50 USP18 target genes, including our lead target gene HERC5 (p = 0.037) as well as canonical interferon response genes IFI44 and ISG15 (Supplementary Data 4). For 6/7 nominally significant associations, the effect direction was concordant between the LCL and whole blood meta-analyses, but the effect size was an order of magnitude smaller in whole blood (Supplementary Data 4). We further tried to replicate the USP18 trans-eQTL association in naive B cells using single-cell RNA-seq data from 844 individuals from the OneK1K cohort18, but none of the tested genes were associated with the trans-eQTL lead variant (Supplementary Data 5). Thus, even at these large sample sizes, the USP18 trans-eQTL signal would not have been discovered in whole blood or naive B-cells.
To understand the potential reasons for the attenuated effect in whole blood, we compared the expression level of the USP18 gene across 49 GTEx tissues. We found that USP18 had the highest expression in LCLs (median transcripts per million (TPM) = 45.3) and one of the lowest in whole blood (median TPM = 0.46). Since USP18 is itself an interferon response gene and LCLs are characterised by a strong interferon signature driven by active infection with the Epstein-Barr virus, we characterised the expression of USP18 in naive B-cells as well as B-cells stimulated with interferon-alpha and TLR7/8 agonist R848 for 16, 40 and 64 h. We found that the expression level of USP18 in B-cells was upregulated by ~3.5-fold after 16 h of stimulation and stayed elevated for at least 64 h (Supplementary Fig. 7). This suggests that the very strong active interferon signalling and associated upregulation of USP18 transcription in LCLs is required for the trans-eQTL signal to be detected.
Discussion
We performed the largest trans-eQTL study in a single cell type where we profiled the expression of 18,792 genes in 3737 individuals from nine cohorts. We then replicated these findings in an independent multi-ancestry LCL cohort of 682 individuals. After careful quality control, we identified six independent loci that were associated with five or more target genes, and that were unlikely to be driven by cross-mappability artefacts. While we primarily focussed on the SLE-associated USP18 locus in our analysis, we have publicly released the complete genome-wide summary statistics from our MetaLCL project via the eQTL Catalogue FTP server. In addition to disease-specific colocalisation applications, we expect that our summary statistics will motivate the development and application of novel summary-based aggregative trans-eQTL mapping methods65,66,67.
While the GWAS signal for SLE at the USP18 locus was discovered in East Asian cohorts33, our trans-eQTL analysis was based on samples of predominantly European genetic ancestry. The shared GWAS and trans-eQTL lead variant (rs4819670) is common in both genetic ancestry groups (EAS MAF = 0.16; EUR MAF = 0.36), indicating that allele frequency differences alone cannot explain why the SLE GWAS association has not been detected in European ancestry individuals. However, prevalence, severity and age of onset of SLE varies considerably between ancestry groups68. Furthermore, heterogeneity of GWAS effect sizes between European and East Asian SLE GWAS studies has been reported68. Mechanisms of this genetic heterogeneity are unknown, but one plausible explanation is the presence of gene-environment interactions (e.g., diet or exposure to regional pathogens). In contrast, eQTL effect sizes in cultured LCLs are highly concordant between genetic ancestry groups30, indicating that our use of European-ancestry samples to interpret a GWAS hit discovered in East Asians is unlikely to systematically bias our results in this case. Furthermore, the USP18 trans-eQTL signal replicated in a multi-ancestry cohort30, indicating that the trans-eQTL effect is shared across ancestries. Finally, the USP18 SLE GWAS signal is also present in the MVP-FinnGen-UKBB meta-analysis (Supplementary Fig. 3).
Despite the strong evidence for the critical role of type I interferon response in SLE pathogenesis49,50 and three active clinical trials, we were surprised to see that of the 50 USP18 target genes, only OAS1 had an independent cis-association with SLE. Expanding the analysis to interferon response genes from Reactome further implicated IRF1/5/8 genes and the HLA region, but most interferon response genes were not detected in the SLE GWAS. One potential explanation for this could be the limited statistical power of the SLE GWAS that profiled 13,377 cases and 194,993 controls, identifying a total of 113 loci33. Furthermore, Liu et al. demonstrated that if multiple effector genes (‘core’ genes) are co-regulated by shared trans factors, with shared directions of effects (which seems to be the case for the interferon response genes), then nearly all heritability would be due to trans effects, further reducing the power to detect cis-acting signals at individual target genes4.
However, interferon response involves rapid upregulation of a broad transcriptional regulatory network of genes with diverse biological functions, only a subset of which might have a direct causal effect on SLE. This is supported by the fact that among the 38 interferon response genes (category II), only OAS1, ADAR, PSMB8, SAMHD1 and the IRF transcription factors have been implicated in causing primary immunodeficiencies (Fig. 3B). The remaining interferon response genes might thus be better thought of as biomarkers of the complex effect of interferon signalling on multiple parts of the immune system50,69. Similarly, it has been previously shown that variants in the IL6R region that are associated with circulating C-reactive protein (CRP) concentrations, are also associated with coronary artery disease (CAD) risk70, but variants in the CRP region are not71. Thus, plasma levels of CRP do not seem to have a direct causal effect on CAD risk, but can still act as a molecular readout (biomarker) of the IL6R-mediated inflammatory response that does seem to have a causal effect72. These observations suggest that widespread horizontal pleiotropy in gene regulatory networks could be a general property of trans-QTLs and could help explain why using trans-pQTL signals in Mendelian randomisation analysis has had low specificity for identifying known drug targets73,74. Instead, we propose that target genes identified from large-scale trans-QTL studies could be better thought of as drug response biomarkers for drugs targeting the cis gene responsible for the trans association8.
A limitation of our trans-eQTL analysis is its susceptibility to cross-mappability artefacts (Supplementary Data 6). While heuristic approaches have been developed to filter such artefacts post hoc, these approaches are not guaranteed to remove all cross-mappability effects and might be too conservative at other loci29. Cross-mappability artefacts also tend to replicate well in independent cohorts29. Furthermore, as the sample size of trans-eQTL studies increases, the power to detect subtle cross-mappability effects as putative trans-eQTLs also increases. To avoid these false positives, we used a very conservative strategy of requiring each trans-eQTL locus to have at least five independent target genes that all pass the cross-mappability filter. As a result, we likely missed many true trans-eQTLs regulating single or few target genes (e.g., trans-eQTL effect near the CIITA transcription factor on multiple HLA genes that has been replicated in several independent studies6,31,75,76,77, Supplementary Data 1, Supplementary Fig. 8). Future large-scale trans-eQTL studies will likely require the development of novel methods to properly adjust for cross-mappability, such as explicit modelling of transcript compatibility read counts between cis and trans target genes78.
While large-scale trans-eQTL studies using both bulk and single-cell measurements are likely to continue for easily accessible tissues such as whole blood (e.g., eQTLGen Phase 279), it seems unlikely that we will be able to perform trans-eQTL studies comprising tens of thousands of individuals for all disease-relevant cell types and contexts. A promising alternative is to use arrayed CRISPR screens or single-cell approaches to identify downstream gene-regulatory effects of disease-associated genes or individual genetic variants46,80,81.
Methods
Datasets, samples and ethics
We used genotype and gene expression data from ALSPAC31,82,83, TwinsUK84, CoLaus85,86, GEUVADIS87, MRCA88, MRCE88, GENCORD89, GTEx v817 and CAP90 studies. For replication, we used data from the MAGE cohort30. The RNA sequencing and genotype data from the GEUVADIS and MAGE studies was publicly available as part of the 1000 Genomes project. For the other studies, we applied for access to individual-level data via relevant data access committees (DACs), explaining the aim of our project and the intent to publicly share meta-analysis summary statistics. Informed consent was obtained when research participants joined the ten studies listed above. The use of the CAP data for this project was approved by the National Heart, Lung and Blood Institute DAC. The use of the GTEx data for this project was approved by the National Human Genome Research Institute DAC. The use of the GENCORD data for this project was approved by the GENCORD DAC. The use of the MRCA and MRCE data for this project was approved by the Gabriel Consortium DAC. The use of TwinsUK data for this project was approved by the TwinsUK Resource Executive Committee. The use of the ALSPAC data for this project was approved by the ALSPAC Executive Committee. For the ALSPAC cohort, ethical approval for the study was obtained from the ALSPAC Ethics and Law Committee and the Local Research Ethics Committees. Consent for biological samples has been collected in accordance with the Human Tissue Act (2004). The CoLaus study was approved by the Institutional Ethics Committee of the University of Lausanne. Single-cell RNA-seq samples were sourced ethically, and their research use was in accord with the terms of informed consent under an institutional review board/ethics committee-approved protocol (UK Regional Ethics Committee approval granted to work at Wellcome Sanger Institute, protocol reference number 15/NW/0282; project was approved by the Ethics on Research Committee of the Institute of Neurobiology at Universidad Nacional Autonoma de Mexico (UNAM), with the approval number 110.H.).
Statistics and reproducibility
We performed genome-wide trans-eQTL meta-analysis across 3734 samples (2238 female, 1496 male) from 9 cohorts (Supplementary Table 1). Sex was assigned based on X and Y chromosome gene expression and genotype data as described previously91. We included all samples available to us into the discovery meta-analysis. We excluded a small number of samples due to issues with gene expression or genotype data quality (see below). To avoid potential confounding by population stratification, we exclude the 87 YRI ancestry individuals from the GEUVADIS dataset and 34 diverse ancestry samples from the GTEx dataset (defined based on genotype principal components). No statistical method was used to pre-determine sample size. However, evidence from several previous trans-eQTL studies with comparable or smaller sample sizes suggest that we should have sufficient power to detect trans-eQTLs with large effects7,9,11,12,13,14,15,16,18,19. Due to limited sample size of our study and significant computational cost involved, we did not perform sex-stratified trans-eQTL analysis. Biological sex was included as a covariate in association testing. We used power calculations to estimate if we had sufficient power to replicate the USP18 trans-eQTL effect in the MAGE cohort (Supplementary Note 2). The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Genotype data quality control and imputation
Pre-imputation quality control
Genotype imputation was performed as described previously40. Briefly, we lifted coordinates of the genotyped variants to the GRCh38 build with CrossMap v0.4.192. We aligned the strands of the genotyped variants to the 1000 Genomes 30x on GRCh38 reference panel93 using Genotype Harmonizer94. We excluded genetic variants with Hardy–Weinberg p < 10−6, missingness > 0.05 and minor allele frequency <0.01 from further analysis. We also excluded samples with more than 5% of their genotypes missing.
Genotype imputation and quality control
Most of the datasets were imputed using the 1000 Genomes reference panel based on the GRCh38 genome version. CoLaus dataset was imputed using the TOPMed Imputation Server95,96,97, while still aligning with the same reference genome version. Additionally, GEUVADIS, GTEx and MAGE cohorts utilised whole genome sequencing data aligned to the GRCh38 reference genome.
We pre-phased and imputed the microarray genotypes to the 1000 Genomes 30x on GRCh38 reference panel93 using Eagle v2.4.198 and Minimac496. We used bcftools v1.9.0 to exclude variants with minor allele frequency (MAF) < 0.01 and imputation quality score R2 < 0.4 from downstream analysis. The genotype imputation and quality control steps are implemented in eQTL-Catalogue/genimpute (v22.01.1) workflow available from GitHub. Subsequently, we used QCTOOL v2.2.0 to convert imputed genotypes from VCF format to bgen format for trans-eQTL analysis with regenie.
Gene expression data
Studies
We used gene expression data from seven RNA-seq studies (TwinsUK84, CoLaus85,86, GEUVADIS87, GENCORD89, GTEx v817, CAP90, MAGE30) and three microarray studies (ALSPAC31,82,83, MRCA88 and MRCE88).
RNA-seq quantification and normalisation
RNA-seq data were pre-processed as described previously91. Briefly, quantification of the RNA-seq data was performed using the eQTL-Catalogue/rnaseq workflow (v22.05.1) implemented in Nextflow. Before quantification, we used Trim Galore v0.5.0 to remove sequencing adapters from the fastq files. For gene expression quantification, we used HISAT299 v2.2.1 to align reads to the GRCh38 reference genome (Homo_sapiens.GRCh38.dna.primary_assembly.fa file downloaded from Ensembl). We counted the number of reads overlapping the genes in the GENCODE V30 reference transcriptome annotations with featureCounts v1.6.4.
We excluded all samples that failed the quality control steps as described previously91. We normalised the gene counts using the conditional quantile normalisation (cqn) R package v1.30.0 with gene GC nucleotide content as a covariate. We downloaded the gene GC content estimates from Ensembl biomaRt and calculated the exon-level GC content using bedtools v2.19.0100. We also excluded lowly expressed genes, where 95 per cent of the samples within a dataset had transcripts per million (TPM)-normalised expression less than 1. Subsequently, we used the inverse normal transformation to standardise quantification estimates. Normalisation scripts together with containerised software are publicly available at https://github.com/eQTL-Catalogue/qcnorm.
Microarray data processing
Gene expression from 877 individuals in the ALSPAC cohort was profiled using Illumina Human HT-12 V3 BeadChips microarray. We used the normalised gene expression matrix from the original publication31. In the MRCA cohort, gene expression from 327 individuals was profiled using the Human Genome U133 Plus 2.0 microarray. We downloaded the raw CEL files from ArrayExpress (E-MTAB-1425) and normalised the data using the Robust Multi-Array Average (RMA) method from the affy Bioconductor package101. In the MRCE cohort, gene expression from 484 individuals was profiled using the Illumina Human-6 v1 Expression BeadChip. As raw data was unavailable, we downloaded the processed gene expression matrix from ArrayExpress (E-MTAB-1428). In all three microarray datasets, we applied inverse normal transformation to each probe before performing trans-eQTL analysis. If there were multiple probes mapping to the same gene, the probe with the highest average expression was used.
Trans-eQTL mapping and meta-analysis
We performed independent quality control and normalisation on all datasets and only included 18,792 protein coding genes in the analysis. Trans-eQTL analysis was conducted separately on each dataset with regenie102. For studies containing related samples (TwinsUK, MRCA and MRCE) and ALSPAC, both step 1 and step 2 commands were employed, while for other datasets with a smaller number of unrelated samples (Supplementary Table 1), regenie was run in the linear regression mode (step 2 only). We used sex and six principal components of the normalised gene expression matrix and six principal components of genotype data as covariates in the trans-eQTL analysis. All scripts used to run trans-eQTL are publicly available at https://github.com/freimannk/regenie_analysis. Subsequently, we performed an inverse-variance weighted meta-analysis across studies. Meta-analysis workflow is available at https://github.com/freimannk/regenie_metaanalyse.
We used a cis window of ± 5 Mb to assign identified eQTLs into cis and trans eQTLs. To determine significant loci, we excluded variants proximal ( ± 1.5 Mb) to the most highly associated variant per gene. This approach allowed us to identify distinct and robust signals while mitigating potential confounding effects from nearby variants. By applying these filters, we found 79 trans-eQTLs loci at a suggestive p-value threshold of 1 × 10−11.
Accounting for cross-mappability
A major source of false positives in trans-eQTL analysis is cross-mappability, whereby RNA-seq reads from gene A erroneously align to gene B, leading to very strong apparent trans-eQTL signals11,29. To exclude potential cross-mappability artefacts, we excluded all trans-eQTLs where there was high cross-mappability (cross-mappability score from Saha et al.29 > 1) between the trans-eQTL target gene and at least one protein coding gene in the cis region ( ± 1.5 Mb) of the trans-eQTL lead variant. Since some of the strongest cross-mappability artefacts affected one or few target genes (Supplementary Data 6), we further restricted our analysis to trans-eQTL loci that had five or more target genes with p < 5 × 10−8 and cross-mappability score <1.
Random-effect meta-analysis
To further assess the robustness of our meta-analysis results, we performed a random-effects meta-analysis on the ten lead variants identified by our primary analysis. We used the DerSimonian-Laird method implemented in PyMARE. We estimated the between-study variance (τ²) and assessed statistical significance using a z-score and a two-tailed p-value. All of the associations remained significant using the random-effect model (Supplementary Data 1).
Replication of trans-eQTL associations
MAGE
Since we used somewhat arbitrary thresholds to define the initial set of 10 loci (lead p < 1 × 10−11, five or more targets with p < 5 × 10−8), we sought to replicate our findings in an independent Multi-ancestry Analysis of Gene Expression (MAGE)30 cohort. MAGE consisted of data from 731 lymphoblastoid cell lines from the 1000 Genomes project, 682 of which also had whole genome sequencing data available. We used two strategies to assess replication. First, we assessed if the lead variant-gene pair was nominally significant (p < 0.05) in the replication dataset with concordant direction of effect. Based on this criterion, 7/10 loci replicated (Supplementary Data 1). Secondly, since all of our loci had multiple target genes, we used the pi1 statistic to estimate the proportion of FDR < 5% target gene at each locus that had a non-null p-value in the replication dataset103. We used the qvalue R package104 to calculate pi1 = 1-qvalue(5% FDR trans gene p-values)$pi0. For 3/10 loci, the proportion of non-null p-values was > 0.5 (Supplementary Data 1). Note that replication in an independent cohort does not help to reduce false positives due to cross-mappability, as cross-mappability artefacts tend to be highly replicable29.
eQTLGen consortium
The eQTLGen Consortium is an initiative to investigate the genetic architecture of blood gene expression and to understand the genetic basis of complex traits. We used interim summary statistics from eQTLGen phase 2, wherein a genome-wide eQTL analysis has been performed in 52 cohorts, representing 43,301 individuals.
All 52 cohorts performed cohort-specific analyses as outlined in the eQTLGen analysis cookbook (https://eqtlgen.github.io/eqtlgen-web-site/eQTLGen-p2-cookbook.html). Genotype quality control was performed according to standard bioinformatics practices and included quality metric-based variant and sample filtering, removing related samples, ethnic outliers and population outliers. Genotype data was converted to genome build GRCh38 if not done so already and the autosomes were imputed using the 1000 Genomes 30x on GRCh38 reference panel93 (all ancestries) using the eQTLGen imputation pipeline (eQTLGen/eQTLGenImpute).
Like the genotype data, gene expression data was processed using the eQTLGen data QC pipeline (eQTLGen/DataQC). For array-based datasets, we used the results from the empirical probe mapping approach from our previous study6 to connect the most suitable probe to each gene which has previously been to show expression in the combined BIOS whole blood expression dataset. Raw expression data was further normalized in accordance with the expression platform used (quantile normalization for Illumina expression arrays and TMM105 for RNA-seq) and inverse normal transformation was performed. Gene expression outlier samples were removed and gene summary information was collected for filtering at the central site. Samples for whom there were mismatches in genetically inferred sex, reported sex, or the expression of genes encoded from sex chromosomes, were removed. Similarly, samples with unclear sex, based on genetics or gene expression were removed.
An adaptation of the HASE framework106 was used to perform genome-wide meta-analysis. For genome-wide eQTLs analysis, this limits the data transfer size while ensuring participant privacy. At each of the cohorts, the quality controlled and imputed data was processed and encoded so that the individual level data can no longer be extracted, but while still allowing effect sizes to be calculated for the linear relationship between variants and gene expression (eQTLGen/ConvertVcf2Hdf5 and eQTLGen/PerCohortDataPreparations).
Centrally, the meta-analysis pipeline was run on the 52 cohorts. The pipeline which performs per cohort calculations of effect sizes and standard errors and the inverse variance meta-analysis is available at eQTLGen/MetaAnalysis. We included 4 genetic principal components, 20 gene expression principal components and other technical covariates (e.g., RNA integrity number) where available. Per every dataset, genes were included if the fraction of unique expression values was equal or greater than 0.8, variants were included based on imputation quality, Hardy–Weinberg equilibrium (HWE) and minor allele frequency (MAF) (Mach R2 ≥ 0.4, HWE p ≥ 1 × 10−6 and MAF ≥ 0.01). In an additional step, genes were filtered to include only those genes that were available in at least 50% of the cohorts and 50% of the samples.
OneK1K
The OneK1K dataset consisted of single-cell RNA sequencing (scRNA-seq) data from naïve B cell samples collected from 844 donors. We relied on the original cell type annotations provided by the authors. Following the same approach as in the primary analysis, the data were processed using regenie in linear regression mode (step 2 only). For the trans-eQTL analysis, we included sex, six principal components of the normalized, rank-based inverse normal-transformed gene expression matrix, and six principal components of genotype data as covariates.
Differential gene expression in SLE cases versus controls
We re-analysed the microarray gene expression data from Banchereau et al.49 to explore differential gene expression between SLE cases and controls. After downloading the processed data from GEO (GSE65391), we selected one sample from each individual for our analysis based on their earliest recorded visits. The filtered dataset comprised a total of 204 samples, including 46 samples from healthy individuals and 158 samples from individuals diagnosed with SLE. We also applied the inverse normal transformation to standardise the gene expression values. Subsequently, we used the Python statsmodels107 module to fit a linear model to identify genes that were differentially expressed between SLE cases and controls. We included gender, age and batch as covariates in all models.
Overlap between USP18 target genes and GWAS hits for SLE
We downloaded the list of prioritised target genes for the Yin et al. GWAS study (GCST011956) from the Open Targets Genetics Portal. We combined the list of genes prioritised by either the L2G or the closest gene approach, yielding n = 109 target genes. We then overlapped these target lists with the list of 50 trans-eQTL targets for the USP18 locus (FDR < 5%).
Single-cell differential gene expression in resting and stimulated B-cells
Sample collection, cell isolation and cryopreservation
Blood samples were collected from five healthy Mexican individuals (three males and two females). Peripheral blood mononuclear cells (PBMCs) were isolated using Vacutainer CPT tubes, according to manufacturer instructions. Samples were cryopreserved in RPMI 1640 culture media (Sigma), Fetal Bovine Serum (FBS) and Dimethyl sulfoxide (DMSO) and stored at −80 °C for 24 h, before being transferred to liquid nitrogen.
Thawing and stimulation
Cryopreserved PBMCs were thawed quickly and washed in 14 mL of room temperature complete RPMI 1640 media (10% FBS, 1% Penicillin-Streptomycin, 1% L-Glutamine). Cells were incubated at 37 °C, 5% CO2 for 2 h. Cells were then stimulated with interferon alpha (IFN-ɑ, Bio-techne) and R-848 (Resiquimod, Cambridge Bioscience) at a working concentration of 1000 U/mL and 2 µg/ml, respectively. Cells were incubated at 37 °C, 5% CO2 and harvested after 16 h, 40 h and 64 h of stimulation. Unstimulated cells were kept in culture without any stimuli for 16 h (i.e., 0 h of activation).
Multiplexing, CITE-seq staining & scRNA-seq
Upon harvesting, cells were resuspended in a cell staining buffer (Biolegend) and cell hashing and genotype-based multiplexing was performed. Donors of the same stimulation condition were mixed at equal ratios (each pool corresponded to a mix of cells from four to five different individuals). These pools were stained with the TotalSeq-C Human Universal Cocktail, V1.0 (137 cell surface proteins (CSP), Biolegend), in addition to a unique hashtag antibody oligonucleotide (HTO, Biolegend) which corresponds to the stimulation condition pool. After staining and washing, all stimuli condition pools were pooled together at equal ratios. This pool was then stained with live/dead dye 4,6-diamidino-2-phenylindole (DAPI, Biolegend) and dead cells were removed using fluorescence-activated cell sorting.
Cells were next processed using the 10X Genomics Immune Profiling 5’ high-throughput (HT) v2 kit, as specified by the manufacturer’s instructions. 1.15 × 105 cells were loaded into each inlet of a 10X Chromium X to create Gel Bead-in-emulsions (GEMs). Two 10X HT reactions were loaded per time point of sample processing (targeted recovery was 40,000 cells per 10X reaction). Reverse transcription was performed on the emulsion, after which cDNA and CITEseq supernatant were purified, amplified and used to construct RNA-sequencing and CSP sequencing libraries, respectively. These RNA and CSP libraries were sequenced at a 5:1 ratio, respectively, using the Illumina NoveSeq 6000 S4, with 100-bp paired-end reads and all 10X reactions were mixed at equal ratios and sequenced across two lanes.
Deconvolution of single cells by genotype
Each 10X reaction comprised a mix of cells from unrelated individuals. Thus, natural genetic variation was used to assign cells to their respective individuals. First, a list of common exonic variants was compiled from the 1000 Genomes Project phase 3 exome-sequencing data (MAF > 0.05). Next, cellSNP (v1.2.1) was used to generate pileups at the genomic location of these variants. These pileups, in combination with the variants called from genotyping in each individual, were used as an input for Vireo108 (v0.5.7). If any cell had less than 0.9 posterior probability of belonging to any individual or were of mixed genotypes they were labelled as ‘unassigned’ and ‘doublets’, respectively, and removed from downstream analysis.
Data processing and quality controls
Raw scRNA-seq and CITE-seq data were processed using the Cell Ranger Multi pipeline (v7.0.0, 10x Genomics). In brief, RNA and CSP library reads were first assigned to cells. RNA reads were then aligned to the GRCh38 human reference genome and CSP antibody reads were matched to the provided list of known barcodes. Ensembl version 93 was used as a reference for gene annotation, and gene expression was quantified using reads assigned to cells and confidently mapped to the genome. Additionally, Cell Ranger multi was used to deconvolute samples based on HTOs. It uses an algorithm which employs a latent variable model over a state space composed of each HTO used in the experiment to assign each cell to a stimulation condition or as a doublet.
Results from RNA and CSP quantification in Cell Ranger were imported into RStudio (v4.3.1) and analysed using Seurat (v5.0.1). Any cell identified as doublet or unassigned by Vireo and or antibody hashtag deconvolution method were excluded. 10X reactions were split by time point and stimuli condition. Cells with 1.5–2.5 median absolute deviations below the median of genes and counts detected were discarded. Additionally, cells with 3–4 median absolute deviation above the median for the percentage of mitochondrial reads detected were discarded. The resulting cells were then annotated by Azimuth109 (v0.5.0), using the Azimuth PBMC reference dataset that was generated as part of the Hao and Hao et al., 2021 paper109.
Pseudobulk and normalisation
Raw counts were pseudobulked by Azimuth annotated level 1 cell types (CD4, CD8, B, Mono, DC, NK, Other and Other T) per donor, per time point and per stimulation condition, via edgeR110 (v4.0.16). Pseudobulked raw counts were then counts per million (CPM) normalised and log2 transformed with edgeR.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The whole genome sequencing data for the GEUVADIS and MAGE studies was downloaded from the 1000 Genomes website (https://www.internationalgenome.org/data-portal/data-collection/30x-grch38). The GEUVADIS RNA-seq data was downloaded from the European Nucleotide Archive (ENA) under accession PRJEB3366. The MAGE RNA-seq data was downloaded from the ENA (accession PRJNA851328). The genotype and RNA-seq data from the GENCORD study was downloaded from European Genotype-phenotype Archive (EGA) under accessions EGAD00001000425 and EGAD00001000428. The microarray gene expression data from the MRCA and MRCE studies was downloaded from ArrayExpress (accessions E-MTAB-1425 and E-MTAB-1428) and the genotype data was downloaded from EGA (accession EGAS00000000137). The gene expression and genotype data from GTEx and CAP studies was downloaded from dbGaP (accessions phs000424.v8.p2 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000424.v8.p2] and phs000481.v3.p2 [https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000481.v3.p2]). The RNA-seq data from the TwinsUK study was downloaded from EGA (accession EGAD00001001086) and genotype data was obtained from TwinsUK (https://twinsuk.ac.uk/researchers/access-data-and-samples/request-access/). The gene expression data from SLE cases and controls is available from GEO under accession code GSE65391. The informed consent obtained from ALSPAC participants does not allow the microarray and genotype data to be made freely available through any third party maintained public repository. However, data used for this study can be made available on request to the ALSPAC Executive. The ALSPAC data management plan describes in detail the policy regarding data sharing, which is through a system of managed open access. Full instructions for applying for data access can be found here: http://www.bristol.ac.uk/alspac/researchers/access/. The ALSPAC study website contains details of all the data that are available (http://www.bristol.ac.uk/alspac/researchers/our-data/). The RNA-seq and genotype data from the CoLaus cohort can be accessed by directly contacting the cohort (https://www.colaus-psycolaus.ch/professionals/how-to-collaborate/). The MetaLCL full trans-eQTL meta-analysis summary statistics are available from the eQTL Catalogue FTP server (https://www.ebi.ac.uk/eqtl/Data_access/) and additional documentation is available on the project website (https://github.com/AlasooLab/MetaLCL). Source data are provided with this paper.
Code availability
The MetaLCL trans-eQTL analysis workflows are available from https://github.com/freimannk/regenie_analysis, the MetaLCL meta-analysis workflow is available from https://github.com/freimannk/regenie_metaanalyse. Additional documentation and code used to generate figures in the paper is available from https://github.com/AlasooLab/MetaLCL. The eQTL Catalogue genotype imputation, RNA-seq processing and data normalisation workflows are available from https://github.com/eQTL-Catalogue/genimpute, https://github.com/eQTL-Catalogue/rnaseq and https://github.com/eQTL-Catalogue/qcnorm. The eQTLGen genotype imputation, data quality control, data preparation and meta-analysis workflows are available from https://github.com/eQTLGen/eQTLGenImpute, https://github.com/eQTLGen/DataQC, https://github.com/eQTLGen/ConvertVcf2Hdf5, https://github.com/eQTLGen/PerCohortDataPreparations and https://github.com/eQTLGen/MetaAnalysis.
References
Aguet, F. et al. Molecular quantitative trait loci. Nat. Rev. Methods Prim. 3, 1–22 (2023).
Bauer, D. E. et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 342, 253–257 (2013).
Frangoul, H. et al. CRISPR-Cas9 gene editing for sickle cell disease and β-Thalassemia. N. Engl. J. Med. 384, 252–260 (2021).
Liu, X., Li, Y. I. & Pritchard, J. K. Trans effects on gene expression can drive omnigenic inheritance. Cell 177, 1022–1034.e6 (2019).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 1, 10 (2023).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Westra, H.-J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53, 1712–1721 (2021).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Pietzner, M. et al. Mapping the proteo-genomic convergence of human diseases. Science 374, eabj1541 (2021).
Bonder, M. J. et al. Identification of rare and common regulatory variants in pluripotent cells using population-scale transcriptomics. Nat. Genet. 53, 313–321 (2021).
Kolberg, L., Kerimov, N., Peterson, H. & Alasoo, K. Co-expression analysis reveals interpretable gene modules controlled by trans-acting genetic variants. Elife 9, e58705 (2020).
Kasela, S. et al. Pathogenic implications for autoimmune mechanisms derived by comparative eQTL analysis of CD4+ versus CD8+ T cells. PLoS Genet 13, e1006643 (2017).
Fairfax, B. P. et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014).
Brandt, M. et al. An autoimmune disease risk variant: A trans master regulatory effect mediated by IRF1 under immune stimulation?. PLoS Genet 17, e1009684 (2021).
Small, K. S. et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 43, 561–564 (2011).
Consortium, T. G. et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Yazar, S. et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 376, eabf3041 (2022).
Small, K. S. et al. Regulatory variants at KLF14 influence type 2 diabetes risk via a female-specific effect on adipocyte size and body composition. Nat. Genet. 50, 572–580 (2018).
Claussnitzer, M. et al. FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med. 373, 895–907 (2015).
Smemo, S. et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 (2014).
Young, L. S. & Rickinson, A. B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4, 757–768 (2004).
Bjornevik, K. et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 375, 296–301 (2022).
Bar-Or, A. et al. Epstein-Barr virus in multiple sclerosis: theory and emerging immunotherapies. Trends Mol. Med. 26, 296–310 (2020).
James, J. A. et al. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J. Clin. Invest. 100, 3019–3026 (1997).
Harley, J. B. et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 50, 699–707 (2018).
Lanz, T. V. et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 603, 321–327 (2022).
Thomas, O. G. et al. Heightened Epstein-Barr virus immunity and potential cross-reactivities in multiple sclerosis. PLoS Pathog. 20, e1012177 (2024).
Saha, A. & Battle, A. False positives in trans-eQTL and co-expression analyses arising from RNA-sequencing alignment errors. F1000Res 7, 1860 (2018).
Taylor, D. J. et al. Sources of gene expression variation in a globally diverse human cohort. Nature 632, 122–130 (2024).
Bryois, J. et al. Cis and trans effects of human genomic variants on gene expression. PLoS Genet 10, e1004461 (2014).
Mountjoy, E. et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 53, 1527–1533 (2021).
Yin, X. et al. Meta-analysis of 208370 East Asians identifies 113 susceptibility loci for systemic lupus erythematosus. Ann. Rheum. Dis. 80, 632–640 (2021).
King, E. A., Dunbar, F., Davis, J. W. & Degner, J. F. Estimating colocalization probability from limited summary statistics. BMC Bioinforma. 22, 254 (2021).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Verma, A. et al. Diversity and scale: genetic architecture of 2068 traits in the VA Million Veteran Program. Science 385, eadj1182 (2024).
MVP-Finngen-UKBB meta-analysis. https://mvp-ukbb.finngen.fi/about.
Ryan, J. L. et al. Clonal evolution of lymphoblastoid cell lines. Lab. Invest. 86, 1193–1200 (2006).
Kerimov, N. et al. eQTL catalogue 2023: new datasets, X chromosome QTLs, and improved detection and visualisation of transcript-level QTLs. PLoS Genet 19, e1010932 (2023).
Meuwissen, M. E. C. et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 213, 1163–1174 (2016).
Alsohime, F. et al. JAK inhibitor therapy in a child with inherited USP18 deficiency. N. Engl. J. Med. 382, 256–265 (2020).
Taylor, J. P. et al. CRISPR/Cas9 knockout of USP18 enhances type I IFN responsiveness and restricts HIV-1 infection in macrophages. J. Leukoc. Biol. 103, 1225–1240 (2018).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261 (2017).
Schnitzler, G. R. et al. Convergence of coronary artery disease genes onto endothelial cell programs. Nature 626, 799–807 (2024).
Weeks, E. M. et al. Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases. Nat. Genet. 55, 1267–1276 (2023).
Mostafavi, S. et al. Parsing the interferon transcriptional network and its disease associations. Cell 164, 564–578 (2016).
Banchereau, R. et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 165, 551–565 (2016).
Baechler, E. C. et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 100, 2610–2615 (2003).
Jefferies, C. A. Regulating IRFs in IFN driven disease. Front. Immunol. 10, 325 (2019).
Honda, K., Takaoka, A. & Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360 (2006).
Reshef, Y. A. et al. Detecting genome-wide directional effects of transcription factor binding on polygenic disease risk. Nat. Genet. 50, 1483–1493 (2018).
Li, H. et al. Identification of a Sjögren’s syndrome susceptibility locus at OAS1 that influences isoform switching, protein expression, and responsiveness to type I interferons. PLoS Genet 13, e1006820 (2017).
Banday, A. R. et al. Genetic regulation of OAS1 nonsense-mediated decay underlies association with COVID-19 hospitalization in patients of European and African ancestries. Nat. Genet. 54, 1103–1116 (2022).
Zdrazil, B. et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 52, D1180–D1192 (2024).
Morand, E. F. et al. Trial of anifrolumab in active systemic lupus erythematosus. N. Engl. J. Med. 382, 211–221 (2020).
Amaya-Uribe, L., Rojas, M., Azizi, G., Anaya, J.-M. & Gershwin, M. E. Primary immunodeficiency and autoimmunity: a comprehensive review. J. Autoimmun. 99, 52–72 (2019).
Schmidt, R. E., Grimbacher, B. & Witte, T. Autoimmunity and primary immunodeficiency: two sides of the same coin?. Nat. Rev. Rheumatol. 14, 7–18 (2017).
Ghodke-Puranik, Y., Olferiev, M. & Crow, M. K. Systemic lupus erythematosus genetics: insights into pathogenesis and implications for therapy. Nat. Rev. Rheumatol. 20, 635–648 (2024).
Primary immunodeficiency or monogenic inflammatory bowel disease (Version 4.191). https://panelapp.genomicsengland.co.uk/panels/398/.
Crow, M. K. Advances in lupus therapeutics: achieving sustained control of the type I interferon pathway. Curr. Opin. Pharmacol. 67, 102291 (2022).
Mackensen, A. et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 28, 2124–2132 (2022).
Müller, F. abian et al. CD19 CAR T-cell therapy in autoimmune disease — a case series with follow-up. N. Engl. J. Med. 390, 687–700 (2024).
Dutta, D. et al. Aggregative trans-eQTL analysis detects trait-specific target gene sets in whole blood. Nat. Commun. 13, 1–14 (2022).
Wang, L., Babushkin, N., Liu, Z. & Liu, X. Trans-eQTL mapping in gene sets identifies network effects of genetic variants. Cell Genom. 4, 100538 (2024).
Akinbiyi, T., McPeek, M. S. & Abney, M. ADELLE: A global testing method for trans-eQTL mapping. PLoS Genet 21, e1011563 (2025).
Wang, Y.-F. et al. Identification of 38 novel loci for systemic lupus erythematosus and genetic heterogeneity between ancestral groups. Nat. Commun. 12, 772 (2021).
McNab, F., Mayer-Barber, K., Sher, A., Wack, A. & O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103 (2015).
Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet 379, 1214–1224 (2012).
C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC) et al. Association between C reactive protein and coronary heart disease: Mendelian randomisation analysis based on individual participant data. BMJ 342, d548 (2011).
Burgess, S. et al. Using genetic association data to guide drug discovery and development: review of methods and applications. Am. J. Hum. Genet. 110, 195–214 (2023).
Zheng, J. et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 52, 1122–1131 (2020).
Karim, M. A. et al. Systematic disease-agnostic identification of therapeutically actionable targets using the genetics of human plasma proteins. medRxiv https://doi.org/10.1101/2023.06.01.23290252 (2023).
Wong, D. et al. Genomic mapping of the MHC transactivator CIITA using an integrated ChIP-seq and genetical genomics approach. Genome Biol. 15, 494 (2014).
Civelek, M. et al. Genetic regulation of adipose gene expression and cardio-metabolic traits. Am. J. Hum. Genet. 100, 428–443 (2017).
Hore, V. et al. Tensor decomposition for multiple-tissue gene expression experiments. Nat. Genet. 48, 1094–1100 (2016).
Ntranos, V., Kamath, G. M., Zhang, J. M., Pachter, L. & Tse, D. N. Fast and accurate single-cell RNA-seq analysis by clustering of transcript-compatibility counts. Genome Biol. 17, 112 (2016).
EQTLGen consortium. https://www.eqtlgen.org/.
Morris, J. A. et al. Discovery of target genes and pathways at GWAS loci by pooled single-cell CRISPR screens. Science 380, eadh7699 (2023).
Weinstock, J. S. et al. Gene regulatory network inference from CRISPR perturbations in primary CD4+ T cells elucidates the genomic basis of immune disease. bioRxiv 2023.09.17.557749 https://doi.org/10.1101/2023.09.17.557749 (2023).
Boyd, A. et al. Cohort Profile: the ‘children of the 90s’-the index offspring of the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 42, 111–127 (2013).
Fraser, A. et al. Cohort profile: the Avon longitudinal study of parents and children: ALSPAC mothers cohort. Int. J. Epidemiol. 42, 97–110 (2013).
Buil, A. et al. Gene-gene and gene-environment interactions detected by transcriptome sequence analysis in twins. Nat. Genet. 47, 88–91 (2015).
Firmann, M. et al. The CoLaus study: a population-based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc. Disord. 8, 6 (2008).
Sönmez Flitman, R. et al. Untargeted metabolome- and transcriptome-wide association study suggests causal genes modulating metabolite concentrations in urine. J. Proteome Res. 20, 5103–5114 (2021).
Lappalainen, T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013).
Liang, L. et al. A cross-platform analysis of 14,177 expression quantitative trait loci derived from lymphoblastoid cell lines. Genome Res. 23, 716–726 (2013).
Gutierrez-Arcelus, M. et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife 2, e00523 (2013).
Theusch, E., Chen, Y.-D. I., Rotter, J. I., Krauss, R. M. & Medina, M. W. Genetic variants modulate gene expression statin response in human lymphoblastoid cell lines. BMC Genom. 21, 555 (2020).
Kerimov, N. et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 53, 1290–1299 (2021).
Zhao, H. et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007 (2014).
Byrska-Bishop, M. et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440.e19 (2022).
Deelen, P. et al. Genotype harmonizer: automatic strand alignment and format conversion for genotype data integration. BMC Res. Notes 7, 901 (2014).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Fuchsberger, C., Abecasis, G. R. & Hinds, D. A. minimac2: faster genotype imputation. Bioinformatics 31, 782–784 (2015).
Loh, P.-R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. A. affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Nica, A. C. et al. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 7, e1002003 (2011).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 100, 9440–9445 (2003).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Roshchupkin, G. V. et al. HASE: framework for efficient high-dimensional association analyses. Sci. Rep. 6, 36076 (2016).
Seabold, S. & Perktold, J. Statsmodels: econometric and statistical modeling with Python. in Proceedings of the 9th Python in Science Conference (SciPy, 2010). https://doi.org/10.25080/majora-92bf1922-011.
Huang, Y., McCarthy, D. J. & Stegle, O. Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 20, 273 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Acknowledgements
Most of the analysis presented in the paper were performed at the High Performance Computing Center, University of Tartu. We are extremely grateful to all the families who took part in the ALSPAC study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. This work received support from Christian Molina-Aguilar, Carina Uribe Díaz, and Alejandra Castillo Carbajal. We thank the FACS facility and sequencing pipelines at Wellcome Sanger Institute for their assistance in data generation, and the Human Genetics Informatics for their support in sequence data processing. We thank Bess L. Chau for assistance in the experiments involving generation of single-cell RNA-seq data from stimulated PBMCs. We thank Karatug Ozan Bircan, Abayomi Mosaku, Laura Harris and Helen Parkinson for assistance with hosting the full trans-eQTL summary statistics on the eQTL Catalogue FTP server. We thank Peep Kolberg for providing access to processed data from the OneK1K cohort. The cell membrane icon on Figs. 2C and 3A by Servier (https://smart.servier.com/) is licensed under Creative Commons Attribution 4.0. K.F. and L.S. were supported by a grant from Open Targets (grant no. OTAR2069). K.A. was supported by the Estonian Research Council (grant nos. PSG415 and MOB3ERC115). K.A. also received funding from the European Union’s Horizon 2020 research and innovation program (grant no. 825775). G.T., C.P.J., and T.S.R. were supported by Open Targets (grant no. OTAR2064) and the Wellcome Trust (ref. 220540/Z/20/A, ‘Wellcome Sanger Institute Quinquennial Review 2021-2026). S.B. was supported by the Swiss National Science Foundation (grant no. 310030_152724/1). K.K. was supported by the Estonian Research Council (grant no. PRG1117). A.M.-R. was supported by CONACYT-FORDECYT-PRONACES (grant no. 11311) and Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica–Universidad Nacional Autónoma de México (PAPIIT-UNAM) IN218023. A.L.H.-L is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM), and she receives fellowship 790972 from Consejo Nacional de Humanidades, Ciencias y Tecnologías CONAHCYT, México. The UK Medical Research Council and Wellcome (Grant no. 217065/Z/19/Z) and the University of Bristol provide core support for ALSPAC. ALSPAC GWAS data was generated by Sample Logistics and Genotyping Facilities at Wellcome Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe. This publication is the work of the authors and they will serve as guarantors for the contents of this paper.
Author information
Authors and Affiliations
Consortia
Contributions
K.F. performed trans-eQTL analysis on nine cohorts and also performed the meta-analysis across cohorts. A.B. performed trans-eQTL analysis on the CoLaus cohort. K.F., J.C., E.R.H., J.C.M., N.N., H.O., L.S., M.C.T., G.T., K.K., and K.A. interpreted the results and prioritised follow-up analyses. T.S.R. generated and analysed single-cell RNA-seq data under C.P.J. and G.T. supervision. A.L.H.-L. collected blood samples for single-cell RNA-seq under A.M.-R. supervision. R.W. performed replications in the eQTLGen Consortium under U.V. and L.F. supervision. S.B. and K.A. supervised the research. K.F. and K.A. wrote the manuscript with feedback from all authors.
Corresponding author
Ethics declarations
Competing interests
J.C. is an employee of Pfizer. E.R.H., M.C.T., and J.C.M. are employees of Bristol Myers Squibb. H.O. is an employee of GSK. N.N. was an employee of GSK while this work was conducted. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yong-Fei Wang, who co-reviewed with Yao Lei, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Freimann, K., Brümmer, A., Warmerdam, R. et al. Trans-eQTL mapping prioritises USP18 as a negative regulator of interferon response at a lupus risk locus. Nat Commun 16, 8795 (2025). https://doi.org/10.1038/s41467-025-63856-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63856-7
