
Abstract
Viral entry is a critical step in the infection process. Klebsiella spp. and other clinically relevant bacteria often express complex polysaccharide capsules that act as a barrier to phage entry. In turn, most lytic phages targeting Klebsiella encode depolymerases for capsule removal. This virus-host arms race leads to extensive genetic diversity in both capsules and depolymerases, complicating our ability to understand their interaction. This study exploits the genetic information encoded in Klebsiella prophages to model the interplay between the bacteria, the prophages, and their depolymerases, using a directed acyclic graph and a sequence clustering-based method. Both approaches show significant predictive ability for prophage capsular tropism and, importantly, are transferrable to lytic phages. In addition to creating a comprehensive database linking depolymerase sequences to their specific targets, this study demonstrates the predictability of phage-host interactions at the subspecies level, providing insights for improving the therapeutic and industrial applicability of phages.
Similar content being viewed by others


Introduction
Host recognition is considered a critical step for successful bacteriophage (or phage) infection1. This process is mediated by virus-encoded receptor-binding proteins (RBPs), which mostly comprise tail fiber and tail spike proteins. Tail fiber proteins are generally non-enzymatic and bind to a protein receptor on the host surface2. In contrast, tail spikes are characterized by the presence of a polysaccharide-degrading domain, also referred to as depolymerase3,4. The depolymerase domain is responsible for the specific recognition and degradation of complex sugars like exopolysaccharides (EPS), capsular polysaccharides (CPS) or lipopolysaccharide (LPS)5. This primary binding to the capsule by the depolymerase allows subsequent access to the secondary receptor located on the membrane surface before entry into the host6,7. Once inside, the phage continues its cycle by either replicating into the bacteria and propagating, or remaining dormant as a prophage in the host, referred to as a lysogen.
Phages are particularly relevant against ESKAPE pathogens, which are regarded as being among the most urgent threats to global health8. This group includes Klebsiella pneumoniae, a Gram-negative bacterium with a highly diverse capsule with 77 serotypes and over 180 K-loci (KL types) identified through analysis of the K-locus, the region responsible for capsule biosynthesis9,10. The KL type is the most determining factor of infectivity for most phages targeting Klebsiella11,12,13, but our ability to determine the KL tropism of a given phage based on depolymerase sequence alone remains limited. This challenge is further complicated by the rapid evolution of the capsule locus in bacteria and depolymerases in phages, frequently subjected to horizontal gene transfer (HGT)14,15,16,17,18,19.
Recent advances in the field of machine learning have paved the way for addressing important questions related to phages20. Many of these advancements have been grounded in the attention mechanism, which can be defined as the ability of a model to focus on specific parts of the input to enhance its performance21,22,23. This has led to the development of transformer architectures, including protein language models (PLM)24. PLMs, such as Evolutionary Scale Modeling 2 (ESM2), can capture complex patterns and relationships between amino acids in protein sequence into embedded representations25. These embeddings have been successfully applied to downstream tasks such as sequence or token classification, yielding remarkable results26,27. PLMs offer a promising approach for modeling phage-bacteria interactions, enabling the extraction of relevant sequence features and improving predictive accuracy28,29,30,31. However, in some specific settings, such as the therapeutic one, predictions at the genus and species levels are not satisfactory as phages typically infect a few strains within a species32,33.
Three aspects are crucial for attaining a higher level of resolution when predicting the outcome of phage-bacteria interactions: (i) extensive high-quality training data, (ii) strong biological premises and (iii) state-of-the-art machine learning techniques. Recent studies34,35 made great progress in this direction, but still reported limitations related to the lack of training data. While this is an inherent limitation when using experimentally validated phage-bacteria interactions, here this challenge is addressed by leveraging information contained in Klebsiella lysogens. Large numbers of prophage-encoded depolymerase domain sequences were retrieved alongside with the K-locus of the host to predict the capsular specificity of depolymerases based on their sequence. This approach introduced several technical challenges for modeling: phages often encode multiple depolymerases with varying specificities, individual depolymerases can exhibit multi-KL targeting (i.e., binding to multiple capsular types), and frequent HGT among depolymerases obscures direct sequence-KL correlations36,37,38.
To address this multi-label classification task and its inherent challenges, we conceptualized a hierarchical framework, akin to a binary relevance method39. First, KL-specific binary classifiers were designed to isolate depolymerase features associated with individual KL types. Second, an ensemble recommender system integrated predictions across classifiers to rank KL targets based on the output probabilities, explicitly accommodating the multi-label nature of depolymerase-KL interactions. Two complementary approaches were employed to model interactions between depolymerase domains, prophages, and bacterial hosts: a directed acyclic graph-based model (TropiGAT) and a sequence clustering-based model (TropiSEQ). Moreover, these approaches enabled the generation of a comprehensive database linking depolymerase domain sequences to their respective KL targets, offering valuable insights into phage-bacteria interactions. This resource holds promise for therapeutic applications, such as targeted phage therapy, and industrial uses, where accurate KL type specificity is crucial40,41.
Results
A comprehensive profiling of prophage-encoded depolymerases targeting Klebsiella
To gather a collection of prophages with labeled KL type, all Klebsiella genomes were first downloaded from the NCBI database (n = 14,601), retaining those with a confident assigned KL type (n = 12,003). The screening of these bacterial genomes identified 77,802 prophages. Clusters of prophage sequence showing high identity (99% over more than 80% of their genome) were identified to remove redundancy, resulting in a total of 16,077 prophage vOTUs. Then, for each prophage within a prophage vOTU, the KL of the last common ancestor of the infected bacteria was identified and used as a proxy of the most probable KL type of the host at the time of phage infection (see method section). Out of the 77,802 prophages, only 3500 presented an ambiguous target KL type, resulting in a collection of 74,302 KL-labeled prophages.
Next, the depolymerase domain sequences within these prophages were screened using three methods (Fig. 1A). Two methods relied on alignment against experimentally validated depolymerases (BLASTp) and HMM profiles of domains associated with polysaccharide-degrading enzymes. The third method employed DepoScope, a machine learning tool combining a fine-tuned ESM-2 protein language model with convolutional neural networks to detect structural folds characteristic of depolymerases42. Identification with one of the three methods was sufficient to include a depolymerase in our dataset. This identified 19,600 depolymerase domain sequences, with 3908 unique sequences after removing redundancies (100% coverage and 100% percentage identity). Interestingly, for 80% of the prophages, no depolymerase was detected, suggesting prophage degradation or alternative modes of infection33,43. For the remaining 15,230 prophages with at least one depolymerase, ~72% harbored a single depolymerase, ~20% encoded two depolymerases, with some encoding as many as 12. This diversity resulted in an average of 1.3 depolymerase sequences per prophage across the database. The depolymerase domain can take on different structural folds, as previously described in studies of carbohydrate-active enzymes42,44,45. In the present prophage analysis, these folds were found in varying frequencies: the most prevalent fold was the right-handed β-helix, representing 69% (2722) of the identified sequences, followed by the n-bladed β-propeller at 18% (714). The other folds represented a smaller fraction, among which the TIM β/α-barrel with 7.5% (294), the α/β hydrolase fold with 3% (114), and finally the triple-helix (32) and α/α toroid (29) represented less than 1% (Fig. 1B, C).

A Pipeline for the identification of depolymerase domain sequences from prophage proteomes. B Venn diagram of the depolymerase domain identified through the 3 methods employed. They involved the deep learning based method Deposcope, a method based on HMM profiles of proteins with depolymerase polysaccharides degrading activity using HHMscan and BLASTp to scan the proteins against a collection of characterized depolymerases from the literature. C Proportions of the depolymerase folds identified. D Bar plot of the number of prophages carrying at latest one depolymerase on the y-axis across each KL types represented on the x-axis. The KL types represented by less than 5 prophages were assigned to the “Other” category.
To prepare the dataset for modeling, redundant prophages originating from the same bacterial clone were removed, yielding a final dataset of 8,871 prophages, each labeled with one of 128 distinct KL types. (Fig. 1D). The distribution of prophages in the database was not uniform across KL types. Indeed, ~44% of the prophages were assigned to 6 KL types: KL107 (1121), KL64 (897), KL47 (551), KL106 (488), KL17 (481) and KL2 (351). Among the remaining 122 KL types, half contained between 20–300 prophages, while the other half had fewer than 20 prophages.
Developing models to predict depolymerase tropism
To optimize individual KL classifiers, two different approaches were employed: a DAG-based approach, denoted TropiGAT, and a sequence clustering-based approach, referred to as TropiSEQ. In TropiGAT, depolymerases encoded by a given prophage were represented as embedding vectors computed using the ESM2 model. These vectors were aggregated using two strategies: an attention-based method and a baseline averaging method. The aggregated vector was then passed through a forward neural network for binary classification. Each model was trained on unique sets of depolymerases corresponding to a specific KL type. Model performance was evaluated using weighted MCC values, a robust metric for classifier quality that accounts for the number of training instances. The attention-based aggregation outperformed the baseline, achieving a weighted MCC of 0.547 compared to 0.528 for the averaging method (P = 0.0477). The attention-based aggregator generalized well for the most represented KL types, notably KL17 (n = 223), KL102 (n = 128) and KL3 (n = 147) with MCC scores > 0.8 (Pearson coefficient = 0.41; P-value < 1.10-5) (Supplementary Data 1). In contrast, KL types with limited training data, such as KL31, KL6, and KL9 (fewer than 25 prophages), performed poorly, with MCC scores near zero, suggesting limited predictive capacity for those KL types. Given its superior performance, the attention-based aggregator was retained for further analysis (Fig. 2A–C and Supplementary Data 2A).

The elements of the architectures are described using as an illustration the scenario of a lysogen with two prophages, one carrying two de polymerase sequences and the other carrying one 1. A Description of the architecture of TropiGAT. The input is modelized with graphs, where the bacteria, prophage and the encoded depolymerase are represented as nodes and their relationship by edges. The features of the depolymerase are aggregated by the encoder before being classified. B Description of the architecture of TropiSEQ. The depolymerase domain sequences are retrieved and then used to scan the collection of representative depolymerase domain clusters using BLASTp. The vector representing the presence-absence of each of the depolymerase domain cluster is fed into a Random Forest classifier. C TropiGAT’s (n = 85KL types) and D TropiSEQ’s (n = 108KL types) scatter plots representing the MCC scores on the y-axis, along with the number of infecting prophages on the x-axis, used for training. The green line in bold represents the regression line, and the colored area the deviation. r coefficient = Pearson correlation coefficient.
In TropiSEQ, prophages were represented as binary vectors indicating the presence or absence of depolymerase domain clusters. These vectors were classified using two classifiers: Random Forest and logistic regression. Among the tested configurations, the Random Forest classifier at a clustering threshold of 0.85 achieved the best performance, with a weighted MCC of 0.367 and was therefore retained for further analysis. Similar to TropiGAT, TropiSEQ performed best for KL types with abundant training data, though this trend was less pronounced (Pearson coefficient = 0.35; P-value < 5.10-4) (Fig. 2B–D and Supplementary Data 2B). Well-represented KL types such as KL17 and KL60 exhibited strong predictive performance. However, some KL types with comparable sample sizes, such as KL62 (n = 124) and KL2 (n = 151), showed more modest MCC scores of 0.35 and 0.30, respectively. Surprisingly, certain KL types with limited training data, such as KL7 (n = 39) and KL128 (n = 37), performed unexpectedly well, achieving MCC scores of 0.57 and 0.79, respectively. Despite these promising results, caution is warranted when interpreting the predictive power of underrepresented KL types due to their limited sample sizes. Further evaluation of the models is required to assess their efficiency.
Ensemble strategy for multi-label prediction
Both TropiGAT and TropiSEQ presented varying performances, with the strongest results observed for KL types supported by abundant training data. These findings motivated the development of an ensemble strategy to integrate predictions across KL types and enhance overall performance. Two strategies were compared to assess the impact of training data curation on model performance: the first utilized unique sets of depolymerases associated with each KL type, enabling the exclusion of identical training instances to promote generalization. The second strategy weighted training data by the natural distribution of depolymerase-KL pairs across lysogens, emphasizing evolutionarily prevalent interactions. Evaluation against a dataset of 25 experimentally validated right-handed β-helix depolymerases revealed complementary strengths. TropiGAT ranked 11 of 25 depolymerase-KL pairs within the top 10 predictions, whereas TropiSEQ achieved 14 correct rankings (Supplementary Data 3). When predictions of both models were combined, 16 targets (64%) were prioritized within the top 10, demonstrating synergistic value (Table 1). Distinct prediction patterns were observed across KL types: depolymerases targeting KL47 were consistently ranked first by both methods, while those targeting KL64 achieved ranks between 1 and 2. Conversely, KL1 and KL102 depolymerases were ranked outside the top 10 by both methods, aligning with reported divergence between prophage and lytic phage depolymerases12.
Performance was further quantified using the mean rank metric. TropiGAT performed optimally when trained on unique depolymerase sets (mean rank of the correct label: 24.09 vs. 29.33 for weighted data), whereas TropiSEQ worked best with infectious event-weighted training (mean rank of the correct label: 31.42 vs. 36.23 for unique sets). Consequently, the final ensemble combined TropiGAT (trained on unique depolymerases) and TropiSEQ (trained on weighted data).
Discrepancies in method-specific predictions highlighted their complementary roles. The depolymerase KP32gp37 (targeting KL3) was ranked third by TropiGAT but outside the top 15 by TropiSEQ. Conversely, the depolymerase depoKP36 (targeting KL63) was ranked first by TropiSEQ but 23rd by TropiGAT. A particularly interesting case was the depolymerase of Klebsiella phage vB_KpnP_KpV74, which TropiSEQ ranked first and second for interactions with KL2 and KL13, two KLs with known cross-reactivity46,47.
In conclusion, the ensemble approach, combining TropiGAT and TropiSEQ, included 64% of targets within the top 10 predictions, highlighting its potential for guiding phage therapy and host range prediction.
Benchmarking TropiGAT and TropiSEQ: a comparative evaluation of ranking systems for predicting phage-KL type interactions on lytic phage
Typically, phage host range is determined by incubating isolated phages with a collection of bacterial strains, a process that becomes increasingly tedious as the number of strains increases. To evaluate the applicability of the developed models as recommendation systems, a comprehensive dataset was generated from three publicly available infection matrices. These matrices were used to benchmark model performance. In the matrix by Ferriol-Gonzales et al, 202413, interactions between 71 phages and 77 bacteria covering as many different KL types were presented. The context of that study was the isolation of phages capable of infecting the full diversity of the 77 serotypes in Klebsiella. In the second matrix, Beamud et al, 202312 described the outcome of the interaction between 46 isolated phages and 138 bacterial strains covering 59 different KL types. There, the bacterial strains were sequenced in a clinical context. Finally, Townsend et al, 202148 presented 30 phages isolated and tested against 24 bacterial strains covering 18 different KL types. Two depolymerases were present in the training data and therefore discarded for further analysis. Overall, 89 different KL types were covered in the matrix. Each phage within these matrices was re-annotated for depolymerases using the pipeline described above. The re-annotation resulted in the identification of 249 depolymerase domain sequences in 126 phages targeting bacteria for which the KL type was predicted with a good level of confidence. The depolymerase domain folds consisted of 139 right-handed β-helix, 75 n-bladed β-propeller, 34 triple-helix and 1 α/α toroid (Supplementary Data 4).
To benchmark the developed methods, their predictions were compared to SpikeHunter19, a sequence-cluster-based approach that associates depolymerases with KL types by analyzing prevalence in lysogen-derived clusters. Additionally, a Monte Carlo simulation (bootstrap = 1000) was implemented to model random predictions49. Predictions were evaluated to assess the extent to which they aligned with KL-type tropism reported in the matrices (Fig. 3 and Supplementary Data 5-6).

In the x-axis are represented KL types and in the y-axis their corresponding phages. In white: combination not tested; gray: no observable infection; dark gray: infection not predicted by neither of the predictors; green: infection predicted by TropiSEQ; golden: infection predicted by TropiGAT; red: infection predicted by both models.
Initial testing focused on helical depolymerases (i.e., right-handed β-helices and triple helices). When considering only the top-ranked prediction for each helical depolymerase, TropiSEQ correctly associated 48 of 164 depolymerases with a target KL type, outperforming SpikeHunter (44/164) and TropiGAT (25/164) (Fig. 4A). All three methods significantly surpassed the random approach (2/164). Among the top predictions, 13 depolymerase-KL pairs were consensus across all methods, while 29 were shared between TropiSEQ and SpikeHunter.

A Recall @N for TropiGAT (green), TropiSEQ (gold), the combination of TropiGAT and TropiSEQ (red) and the random approach (gray). Box plots of the MCC values reported for the KL types when only considering the helical depolymerases (right-handed β-helix & triple-helices) and n-bladed β-propeller when predicting with B TropiGAT and C TropiSEQ. *P = 0.011, **: P < 1.10e-6 (two-sided Welch t-test). The boxplots identify the mean values, interquartile ranges, and outliers for the different folds (n = 87 for the helical depolymerases, n = 67 for the n-bladed β-propeller).
Expanding the recommendation scope to the top five predictions significantly improved correct associations for TropiSEQ and TropiGAT: TropiSEQ achieved 76/164 correct associations, while TropiGAT identified 49, and the random approach reached 11. In contrast, SpikeHunter generated no additional predictions beyond the top-ranked KL type due to its cluster-based design, which restricts predictions to single KL associations per depolymerase19. This limitation arises from SpikeHunter’s reliance on sequence-cluster prevalence rather than probabilistic ranking, making it unsuitable for multi-label predictions. At higher rank thresholds, TropiGAT demonstrated progressive recall improvement, linking 62/164 depolymerases to KL targets within the top 15 predictions, compared to TropiSEQ’s 78/164. Collectively, the methods successfully associated 100/164 helical depolymerases with KL targets. TropiSEQ exhibited superior ranking precision, with a mean rank of the correct label of 1.8 versus TropiGAT’s 3.6 among the top 15 predictions. TropiSEQ also performed robustly on underrepresented KL types. For example, despite KL58 being represented by only six prophages in the training data, TropiSEQ ranked the right-handed β-helix of Klebsiella phage K58PH129C2 (CDS 47) first.
In contrast, TropiGAT demonstrated a unique ability to predict novel KL type associations, particularly for depolymerases with no homology to the training data. For instance, TropiGAT accurately predicted interactions between the triple-helix on CDS 25 of Klebsiella phage K29PH164C1 with KL24 (rank 4) and the right-handed β-helix on CDS 165 of phage NBNDMPCG with KL2 (rank 6), despite the absence of homology to the training data. These findings suggest TropiGAT leverages patterns beyond sequence similarity. By capturing folding patterns, the embeddings enable the model to infer structural relationships and make accurate predictions at a higher level. This observation draws attention to the top predictions that did not match the reported targets. Among these predictions, 54% (TropiGAT) and 37% (TropiSEQ) corresponded to KL types absent from the validation datasets, suggesting potential novel associations that have not yet been experimentally validated.
The depolymerase fold impacts the prediction of capsular specificity
The right-handed β-helix has been the most extensively characterized depolymerase fold50,51,52, while the characteristics and properties of other folds are less understood. The previous section has reinforced the idea that KL type tropism can be predicted for the right-handed β-helix and the triple-helix, but the predictability of other folds remains unclear. In order to explore these factors, the predictability of the different depolymerase folds was investigated for lytic phage dataset.
Significant differences in model performance were observed between folds. First, helical depolymerases showed higher correct association rates than n-bladed β-propellers in the top 15. TropiSEQ and TropiGAT correctly associated 48% (78/164) and 38% (62/164) of helical depolymerases, respectively. In contrast, for n-bladed β-propellers, correct associations were 22% (16/75) with TropiSEQ and 43% (32/75) with TropiGAT. Second, the mean rank of correct predictions for n-bladed β-propellers was significantly higher (poorer performance) at 4.21 (TropiSEQ) and 6.34 (TropiGAT), compared to 1.78 and 3.56 for helical depolymerases, respectively.
To further investigate these differences, MCC scores were computed for each model instance, corresponding to individual KL types, using a threshold of 0.5 for a positive prediction. For both TropiGAT and TropiSEQ, clear discrepancies in MCC scores were observed between helical depolymerases and n-bladed β-propellers. In TropiGAT, the average MCC score for helical depolymerases was 0.10, compared to 0.02 for n-bladed β-propellers (P = 0.011; Student t-test; Fig. 4B). For TropiSEQ, the difference was even more pronounced, with MCC scores of 0.20 and 0.0065 for helical depolymerases and n-bladed β-propellers, respectively (P < 0.0001; Welch t-test; Fig. 4C). These findings indicate that the models are more efficient at predicting KL-type tropism for helical depolymerases compared to n-bladed β-propellers.
Despite the overall lower predictability of n-bladed β-propellers, certain KL types exhibited strong model performance, suggesting some specificity in their predictive capabilities. For instance, KL21 achieved MCC scores of 0.28 and 0.56 for TropiGAT and TropiSEQ, respectively. TropiGAT also showed good performance for KL27 (0.42), KL52 (0.37), and KL57 (0.64). These findings indicate that KL-type tropism predictability for n-bladed β-propeller folds may be achievable for certain KL types. Notably, the underlying basis for this predictability appears to extend beyond simple protein sequence identity. Instead, it may depend on specific residues and structural features that are not readily captured by sequence clustering-based methods such as TropiSEQ. This observation highlights the need for further investigation to uncover the mechanisms driving these associations and to enhance the predictive performance of models across a broader range of depolymerase folds.
Analysis of depolymerase-KL association clusters
Associations between depolymerase sequences and KL types were analyzed using the learned features to uncover distinct patterns. For TropiSEQ, relationships between depolymerase domain clusters and KL types were explored using vector representations of clusters, which estimated interaction likelihoods. A probability threshold of 0.5 identified 550/989 clusters as associated with 96 KL types, with 110 clusters (20%) linked to ≥2 KL types. The most frequent associations included KL106-KL107, which shared seven depolymerase domain clusters, and KL47-KL64, which shared five. However, most clusters were small: 180 were singletons, 115 contained two sequences, and the largest clusters comprised 156 and 153 sequences.
TropiGAT employed an attentional aggregation layer to assign weights to depolymerase domains within prophages, reflecting their importance in KL-type predictions. Associations were deemed significant when attention weights exceeded 0.5 and predicted infection probabilities surpassed 0.8. This high-confidence threshold mapped 1627 depolymerase domains to 82 KL types.
Comparison of TropiGAT and TropiSEQ predictions revealed congruence in 1318/1761 (74.8%) depolymerase-KL associations. Cross-reactivity was observed in ~5% (79/1627) of depolymerases, with structural folds exhibiting distinct patterns. Notably, two n-bladed β-propellers were associated with 10 and 9 KL types, two TIM β/α-barrels were linked to 8 and 5 KL types, and three right-handed β-helices each targeted 4 KL types. These cross-reactive depolymerases may possess broad-spectrum activity, highlighting their potential for further functional and therapeutic uses. This observed cross-reactivity could also point towards conserved structural elements within the polysaccharides of the implicated KL types. Although the models do not directly learn correlations between KL types, these results could provide insights into similarities between such structures. These insights are especially valuable given the lack of known structural information for many KL types, thus guiding future research into these polysaccharide structures and their depolymerase interactions.
Discussion
This study presents an innovative framework for modeling phage-bacteria interactions at the subspecies level in Klebsiella spp., addressing a critical challenge in the field. By leveraging prophage data, the study developed predictive models to determine phage capsular tropism based on depolymerase domain sequences. The models demonstrated several key capabilities: resolving predictions at the individual depolymerase level, identifying cross-reactive depolymerases, and accurately predicting KL types for depolymerases lacking sequence homology with the training data. This approach provides an actionable framework for characterizing phage host range and is supported by a comprehensive database mapping depolymerase domain sequences to their target KL types. Compared to traditional associations through direct sequence homology, this strategy offers greater practicality and predictive accuracy, particularly for sequences with low similarity to known data. These advancements not only enhance our understanding of phage-host specificity but also offer innovative possibilities for applications in targeted phage therapy and industrial processes.
The developed methods, TropiGAT and TropiSEQ, function as binary classifiers designed to predict the KL type specificity of depolymerase sequences. They demonstrated high predictive performance for KL types with substantial training data, such as KL2, KL17, KL47, KL64, KL106, and KL107. These KL types are well-represented as they are associated with high-risk Klebsiella spp. clones53,54,55. Their frequent occurrence in clinical sequencing data, therefore, makes the models targeting them particularly relevant in this setting. Furthermore, the models showed encouraging results even for KL types underrepresented during training. For instance, despite having only 11 and 13 training examples, respectively, KL103 and KL157 achieved strong MCC scores of 0.697 and 1.0 when TropiGAT was evaluated on lytic phage sequences. While these underrepresented KL types performed well on the specific lytic phage dataset, their performance might not extend to sequences more distant from the training data, due to limited generalization ability. This challenge could be addressed in future work by enriching the training data with sequences from other bacteria employing the Wzy capsule synthesis system, such as Escherichia coli or Acinetobacter baumannii. These related systems, also targeted by phages with depolymerases56,57, could represent a valuable pool of new informative patterns for model improvement18,19.
Beyond direct prediction, these binary classifiers were leveraged to build a recommender system designed to prioritize candidate KL type specificities for uncharacterized depolymerases. This system proved effective, with TropiSEQ consistently ranking correct depolymerase-KL type pairs within the top five associations (lower mean rank of the correct target) and TropiGAT performing better at higher rank thresholds, indicating superior recall. However, certain models for low-data KL types exhibited poor classification performance by TropiGAT (MCC ≤ 0) yet were consistently assigned high ranks due to frequent, high-probability false positive predictions (e.g., involving KL4, KL34, KL26), introducing significant noise detrimental to the system's reliability. These unreliable classifiers should therefore be used with caution or filtered in practice. To capitalize on complementary strengths while mitigating such issues, we recommend a combined approach: utilizing TropiSEQ for high-precision initial candidate generation (top 5 predictions) followed by TropiGAT to refine results and capture less obvious specificities, including potential cross-reactivities which hold significant therapeutic value.
Evaluation of the models on the lytic phages revealed varying levels of predictive power across the different depolymerase domain folds, prompting questions on the origins of these discrepancies. The right-handed β-helix is the most described polysaccharides degrading depolymerase in phages due to its widespread presence and stable trimer structure58,59, typically targeting one or a few different KL types37. In contrast, while ongoing research explores the properties of the n-bladed β-propeller in bacteria and eukaryotes60, its characteristics in phages remain unclear. Several factors could account for the weak predictive power of this fold. Firstly, the scarcity of training data, as the detectable n-bladed β-propeller is less abundant than the right-handed β-helix in phages42. Secondly, recent studies demonstrated vast structural diversity within this fold. The β-propeller structure results from the duplication of four antiparallel β-strands, which can then act as a monomer or dimer, leading to variation in the number of blades61. This diversity could complicate the identification of patterns at the primary structure level. Finally, the n-bladed β-propeller can accommodate a wide range of substrates depending on environmental conditions62, exerting diverse functions that can go beyond catalytic activity63. This is exemplified by the n-bladed β-propeller encoded by Sugarlandvirus phages targeting Klebsiella spp., which exhibit the broadest host range in terms of KL types, supporting the idea that this depolymerase fold plays a crucial role in the breath of the infectivity spectrum64. These features align with the findings of this study, as phages encoding an n-bladed β-propeller tended to target more KL types than those lacking it, with an average of 3.1 versus 1.9 bacterial strain targets, respectively. Further research into the precise activity of this fold in phages is needed to gain deeper insights.
Focusing exclusively on the depolymerase domain sequence in modeling phage-bacteria interactions has limitations. The recognition process can be mediated by the tail fiber and occur independently of the depolymerase64, or in conjunction with it6, particularly in phages exhibiting depolymerase activity with specific KL types but not others. The complexity of the phage recognition system could lead to an overestimation of false negatives, to the extent that successful infections may not always involve depolymerase activity, despite the phage encoding one. Moreover, the ability of a phage to recognize its host does not necessarily lead to successful infection due to post-entry defense mechanisms. This scenario could lead to potential false positives in cases where a phage can exert depolymerase activity but fails to induce a successful infection. Another limitation arises from the dependence of the method on the initial bacterial strains adequately representing the diversity of the population. If the dataset lacks sufficient phylogenetic coverage, the approach may be prone to biases or artifacts, potentially failing to accurately infer the KL type of the ancestral bacteria.
To address limitations associated with the approach of this study, models could be expanded by integrating other factors involved in the recognition of the host. This could entail integrating tail fibers and the secondary bacterial receptors. Recent genomic foundation models capable of generating embedding representations for DNA sequence were developed with context windows spanning hundreds of thousands of kilobases65. These tools could be leveraged to encode the whole K-loci and allow the incorporation of more informative data about the capsule and its modifications. Such a unified model would represent an upgrade over the current set of binary classifiers for two main reasons. First, it would simplify the architecture and enable analysis of bacterial strains within the same KL type, thereby increasing sensitivity. Second, by allowing the model to learn depolymerase patterns across different KL types, it could facilitate knowledge transfer from well-represented KL types to those with fewer training examples, improving performance on underrepresented targets. Moreover, the model could be extended by integrating data on downstream processes such as phage replication, host defense systems (e.g, CRISPR), and agents involved in bacterial lysis (e.g, holin, endolysin)66. This would allow TropiGAT to predict more comprehensively the success of phage infection, making it a valuable tool for various applications, particularly in clinical settings.
In summary, this study demonstrates innovative approaches to modeling phage-bacteria interactions by combining advanced machine learning models and architectures with an evolutionary perspective. The significant role and versatility of the depolymerase sequence enables the scope of this work to go beyond the therapeutic applications, offering potential utility in industrial contexts as well.
Methods
Bacterial genomes and capsule typing
The genomes of Klebsiella strains were downloaded from the NCBI Reference Sequence Database through the PanACoTa package67. In order to retrieve all the strains from the Genus Klebsiella, the “prepare” command with the “-T 570” parameter was used. Genome assemblies with an L90 (the minimum number of contigs necessary to get at least 90% of the whole genome) value superior to 100 were discarded. The K-loci type (KL type) of the bacterial strains was then predicted using Kleborate (https://github.com/klebgenomics/kleborate)10. The strains that presented a level of confidence of “perfect”, “very high”, “high” and “good” were kept for the analysis. These stringent assembly quality criteria ensure high genomic data reliability for downstream analyses. Finally, the genomes were annotated with Prokka v.1.1368.
Prophage prediction
Prophages were predicted using the PhageBoost program based on a machine learning approach trained to distinguish the viral signal from the background signal of a bacterial genome, based on 1587 different features69. A threshold of 0.70 confidence was applied, allowing the number of called prophages to be in concordance with the reported mean number of prophages in Klebsiella spp. in previous studies11. In order to identify prophages belonging to the same viral operational taxonomic unit (vOTU), the pairwise average nucleotide identity (ANI) was computed between the prophages from the dataset using FastANI70. The ANI is defined as the mean nucleotide identity of orthologous gene pairs shared between two genomes. Two prophages were considered from the same vOTU if the calculated ANI was higher than 99% with a bi-directional coverage above 80%.
Phylogenetic tree
The PanACoTa pipeline was used in a similar manner as used by Haudiquet et al. 202114. Briefly, the set of genes present in the genomes of the dataset (or pan-genome) was determined with the “pangenome” command. The output was then used to compute the set of genes present in more than 99% of the genomes in the dataset (or core-genome) using the “corepers” command with the “-t 0.99” parameter. Subsequently, the genes from the core genome were aligned using the “align” command. The aligned nucleotide sequences of each gene were then concatenated, resulting in a 498,179 bp alignment. Phylogenetic inferences were executed with IQ-TREE (v.2.1.4-beta COVID-edition) using the ModelFinder Plus parameter “-m MFP”, with 1000 replicates for ultrafast bootstrap “-B 1000”71. The resulting best model was the (GTR+F+I) corresponding to the general time-reversible model with empirical base frequencies allowing a proportion of invariable sites. The tree was well supported with an average ultrafast bootstrap value of 99.85%.
Recovering the KL type of the infected ancestral bacteria
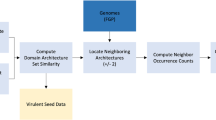
To limit the bias induced by the horizontal gene transfers of the K-locus, the KL type of the bacteria before phage infection, i.e., of the infected ancestor, was investigated for each prophage. First, the states of the ancestors of the bacteria from the dataset were inferred using the phylogenetic tree and the KL types computed for each strain with PastML (v1.9.34)72. The maximum likelihood algorithm MPPA with the recommended character evolution model F81 was used, allowing the attribution of multiple states when they had similar and high probabilities. Subsequently, a tailored algorithm was built with the Bio.Phylo package73 was used to identify the last common ancestor of the infected bacteria (Fig. 5A). This approach assumes a good phylogenetic sampling for which the ancestor likely represents the KL at the time of the infection. Of note, infection could have occurred earlier, and the KL-type of the last common ancestor might have already diverged from that of the infected ancestor. Overall, capsule swaps along the branches can obscure the reconstruction of the infected bacteria, with assumptions on longer branches being more susceptible to uncertainty (Fig. 5B–D). An overview of this process is provided in Fig. 5E.

A Outline of the algorithm used to determine the infected ancestor of each prophage to infer the KL type at the time of infection. Three scenarios are illustrated using phylogenetic trees to showcase how the methodology functions under different conditions, with KL types represented by blue and green colors. B In a straightforward scenario, the prophage is part of a vOTU identified in strains within the same clade (e.g., strains D3 and D4) but absent from other clades at the next hierarchical level (n + 1) connected to node B. Here, the KL type at the time of infection is inferred from the last common ancestor of these strains, corresponding to node C2. C In this scenario, the prophage's presence in strains D1, D3, and D4 suggests a single infection event. Consequently, the KL type at the time of infection aligns with that of the ancestor at node B. D In cases where the phylogenetic tree lacks adequate support (e.g., a missing branch connecting leaf D5 to node A), the inferred infected ancestor would be node B, despite the actual infection event occurring earlier. This would lead to an incorrect inference of the prophage's specificity as blue, due to the absence of additional information. E Schematic representation of the pipeline used to create a labeled collection of prophages from the Klebsiella genome dataset. Representations of phages and bacteria are from BioRender.
Identification of the depolymerase domain sequences
Phage sequences were annotated with Prodigal from Pyrodigal v.0.2.1 (https://github.com/althonos/pyrodigal), integrated in Phageboost. Sequences with a length less than 200 amino acids were discarded. Three different approaches were employed to identify the depolymerase domain sequences in the prophages. The first approach leveraged a database of Hidden Markov Models (HMM) profiles of domains associated with a polysaccharides-degrading (PD) activity42. Multiple sequence alignments (MSA) were built around the candidate protein sequences using MMseqs74 along the UniRef90 database. The resulting MSAs were then scanned against the HMM profiles of the PD domains using the HHsearch75. Hits with a score exceeding 20 and an alignment spanning at least 30 amino acids were considered candidate depolymerases.
In the second approach, prophage protein sequences were directly screened against a database of 333 depolymerase proteins from previously described phages12 using BLASTp76. Hits presenting a bitscore exceeding 75 were added to the set of candidate depolymerases. Subsequently, the candidate depolymerase set resulting from the two approaches above was pooled for downstream structural analysis. The 3D structures of the candidate depolymerases detected by the first and second approaches were predicted using ESMfold25. Then, the predicted structures were scanned using FoldSeek77 against a database of 3D domain folds involved in a PD activity. The folds represented in the database included the α/α toroid, the right-handed β-helix, the TIM β/α-barrel, the n-bladed β-propeller (with at least 4 antiparallel β-strands), the flavodoxin-like fold and the α/β hydrolase fold. The hits that presented a probability of being a true positive greater than 0.5, or 0.2 when the query was the right-handed β-helix, were considered depolymerase proteins. Next, the 3D structure of the depolymerase proteins was dissected into protein units using SWORD278. These protein units were scanned against the database of 3D structures of PD domains to infer the definitive depolymerase domain from the best match.
The third approach, independent of structural analysis, was based on the deep learning model DepoScope42. This model performs a binary classification of proteins for the identification of depolymerase proteins, as well as the delineation of the catalytic domain. The final depolymerase domain database was generated by sequences that were identified by any of the three methods. An overview of the subsequent data processing is given in Figs. 1A, 5E.
Directed acyclic graph-based approach for modeling prophage-CPS Interactions (TropiGAT)
Graphs represent complex relationships between objects using nodes to encode them and edges to encode their connections79. In this study, directed acyclic graphs (DAG) were designed with three node types: the infected bacteria (nodes A), the prophages (nodes B1) and the depolymerases (nodes B2). Two types of directed edges connected the nodes: (B2 → B1) linked depolymerases to the prophages where they were identified, and (B1 → A) linked prophages to their corresponding infected bacteria. Each node type was associated with specific features: nodes A were characterized by the KL type of the infected ancestor; nodes B1 had no initial specific feature; and nodes B2 were characterized by the 1280-dimension embedding representations of the depolymerase domain’s amino acid sequence, computed using the ESM2 model esm2_t33_650M_UR50D25.
Subsequently, a deep learning model was designed using an encoder-classifier-like architecture to process the input graph. For each node B1 (prophage), the encoder module aggregates features from the set of associated nodes B2 (depolymerases) using a graph attention layer (GATv2)80,81.
For that, a scoring function \(e:{{{\rm{R}}}}^{d}\times {{{\rm{R}}}}^{d}\,\to \,{{\rm{R}}}\) computes a score for every edge connecting a node B1 (annotated i) to its neighboring nodes B2 (annotated jwith and\(j\in B2\) represented by the features j), which indicates the importance of the features of the neighbor jto the node i
where \({{\bf{a}}},{{\bf{W}}}\in {{{\rm{R}}}}^{d^{\prime} }\) are learned. These attention scores are normalized across all neighbors using softmax, and the attention function is defined as:
Finally, GATv2 computes a weighted average of the features of the nodes B2 (followed by a non-linearity σ) as the representation hi with 1280 dimensions of the node i:
Through these attention coefficients, the model prioritize relevant neighbors based on the computed loss. This aggregation approach was compared to a baseline method that computes the average value across the set of feature vectors of the B2 nodes at each dimension82.
The updated features of B1 are then passed into the classifier. The classifier module consists of 3 linear layers, interspersed by batch normalization, LeakyRelu activation and dropout layers. The raw outputs of the final layer are passed through a sigmoid function, giving an appropriate estimate of the probability of node B1, given the nodes B2 it is connected to, to be able to infect the KL type of node A. The architecture was illustrated in Fig. 2A.
Sequence clustering-based approach for modeling prophage-CPS interactions (TropiSEQ)
The second approach for modeling phage-KL type interactions relied on a presence-absence matrix representing depolymerase domain repertoires within each prophage. To generate this matrix, depolymerase domain sequences identified in the prophages were clustered using CD-HIT, a rapid clustering tool for biological sequences83. A range of clustering thresholds (0.60, 0.65, 0.70, 0.75, 0.80, 0.85, 0.90, 0.95, 0.975) were assessed, each with a minimal alignment coverage of 80% set by the “-G 0 -aL 0.8” parameters. These thresholds were used to explore the effect of different levels of sequence similarity on the performance of the model. A presence-absence matrix was constructed to represent the depolymerase domain repertoire within each prophage. Each column of the matrix corresponded to a depolymerase domain sequence cluster generated by applying CD-HIT for a specific similarity threshold. Two models, a Random Forest and a logistic regression, were tested for classifying the matrix to predict phage-KL type interactions. The clustering threshold with the best predictive power was determined by computing the weighted Matthew Coefficient Correlation (MCC) for each KL type. The outline of this approach is detailed in Fig. 2B.
Training and evaluating the models
Both modeling approaches, TropiGAT and TropiSEQ, employed an ensemble method, generating distinct binary classifiers for each KL type (KL-classifier). Positive instances were defined as prophages encoding a depolymerase domain repertoire (or depolymerase set), linked to ancestral bacterial hosts carrying the corresponding KL type. To ensure high-quality training data, two curation strategies were applied to these positive instances. The first strategy consisted of keeping a unique representative of each set of depolymerases among the positive instances. The elimination of duplicate sets reduced redundancy and allowed for the best assessment of the model’s ability to generalize. The resulting dataset comprised 4271 training instances. The second strategy consisted of grouping the prophages of the positive instances based on vOTU, ancestral bacterial host, and depolymerase domain repertoire. Within each group, a single representative was retained to address the issue of highly similar genomes from bacterial strains sequenced during epidemic outbreaks. This grouping preserved the natural distribution of depolymerase-KL associations while reducing sampling bias. Although this approach risks inflating performance metrics by retaining phylogenetically related depolymerases, it may enhance confidence in predictions by emphasizing evolutionarily prevalent features. The strategy yielded 8871 training instances. Negative instances were randomly selected from prophages whose depolymerase domains did not overlap with positive instances, minimizing false negatives. The final dataset maintained a 1:5 ratio of positive to negative instances, ensuring a balanced training set for robust model evaluation.
For TropiGAT, each training instance consisted of a set of 1280-dimension tensors, corresponding to the embedding representation of the depolymerase domains from a prophage instance. Labels were assigned as 1 for positive instances and 0 for negative instances. For each KL type, a model was trained using fivefold cross-validation with a shuffle split method, where 70% of the data was used for training, 20% for validation, and 10% for testing. The final evaluation metrics (e.g., accuracy, MCC) were computed by averaging model performance across all folds during testing. Model instances were generated for KL types with at least 10 prophages, employing three forward layers in the classifier module. To prevent overfitting, early stopping was applied, halting training after 60 epochs if MCC performance showed no improvement. The hyperparameters were optimized using Optuna84.
For TropiSEQ, each training instance was encoded as a 989-dimensional binary vector, with each dimension indicating the presence or absence of a specific depolymerase domain cluster. The models also underwent fivefold cross-validation, using a stratified KFold method. Hyper-parameters were optimized via a Bayesian search using fivefold cross-validation through scikit-optimize. A summary of the training process is provided in Table 2.
The models developed for each KL type were structured as a recommender system to address a multilabel classification task. Binary classifiers generated prediction scores, which were ranked to prioritize KL types most likely to be targeted. Model success was determined by two key factors: the ability to generate high prediction scores and the capacity to rank target KL types higher than non-susceptible ones. Performance evaluation was conducted at two levels: specific KL-classifiers and recommender system-wide. At the KL-classifier level, metrics were computed focusing on binary classification for each KL type.
Predictions were classified as positive or negative using a threshold of 0.5. Standard performance metrics included: recall (\(\frac{{{\rm{TP}}}}{{{\rm{TP}}}+{{\rm{FN}}}}\)), accuracy (\(\frac{{{\rm{TP}}}+{{\rm{TN}}}}{{{\rm{TP}}}+{{\rm{TN}}}+{{\rm{FP}}}+{{\rm{FN}}}}\)), precision (\(\frac{{{\rm{TP}}}}{{{\rm{TP}}}+{{\rm{FP}}}}\)), F1 score (\(\frac{2\times {{\rm{precision}}}\times {{\rm{recall}}}}{{{\rm{precision}}}+{{\rm{recall}}}}\)), area under the ROC curve (AUC), and Matthews Correlation Coefficient (MCC; \(\frac{{{\rm{TP}}}\times {{\rm{TN}}}-{{\rm{FP}}}\times {{\rm{FN}}}}{\sqrt{({{\rm{TP}}}+{{\rm{FP}}})({{\rm{TP}}}+{{\rm{FN}}})({{\rm{TN}}}+{{\rm{FP}}})({{\rm{TN}}}+{{\rm{FN}}})}}\)), with TP, FN, TN, and FP representing true positives, false negatives, true negatives, and false positives, respectively. At the recommender system level, the mean rank of the correct label was used as the primary metric: ((1 / n) * Σ rank), where n is the total number of ranks. This metric reflects how well the system prioritizes the correct KL types among potential candidates.
Prediction on lytic phages
To assess the predictive capability of our models for lytic phages, two datasets were used. The first one consisted of lytic phage depolymerases whose association with KL types were experimentally validated. The second one comprised publicly available infection matrices of Klebsiella phages, which reported the phage-bacteria outcome for all the possible interactions12,13,48. To ensure the identification of all the depolymerase domain sequences in the lytic phages, the three identification methods described earlier were used, complemented with the information reported by the authors. The depolymerase domain sequences that appeared in the training data were discarded to avoid inflating the predictive power. A phage was considered effective against a given KL type if the authors reported an instance of the phage infecting a bacterium from the corresponding KL type. This criterion accounted for the fact that a depolymerase activity does not necessarily manifest as a visible halo in plaque assays3,85. For TropiGAT, predictions were made directly from the ESM2 representation of each depolymerase domain sequence. For TropiSEQ, the amino acid sequence of the depolymerase domains were first scanned against the representatives of the depolymerase domain clusters using BLASTp. Then, depolymerase domains were assigned to the cluster of the top hit only if the bitscore was exceeding 75 with a minimal alignment coverage of 80%. Finally, target KL types were inferred from the KL types associated with the corresponding depolymerase domain cluster.
Statistics & reproducibility
All statistical tests were performed using Python (version 3.11.4) with the scipy.stats module (version 1.11.1). The primary metric for evaluating individual KL-type classifier performance was the Matthews Correlation Coefficient (MCC). Pearson's correlation coefficient was used to assess the linear relationship between model performance (MCC scores) and the number of training instances. To test for significant differences between group means, such as comparing MCC scores across different depolymerase folds, two-sample, two-sided Student’s t-tests and Welch’s t-tests were utilized. The choice between these tests was determined by the equality of variances between the groups being compared, assessed using a two-tailed F-test with an alpha of 0.05. For each statistical test reported, the specific test, the corresponding p-value, and the sample size are provided in the Results section or figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Relevant data used related to the training of the models can be found at the Zenodo accession: 10.5281/zenodo.14065540. Those include: (i) the csv file containing the data used for the training and the evaluation of the models (ii) the weights and biases of the binary classifiers for each KL types trained with TropiGAT and TropiSEQ approaches. Source data are provided with this paper.
Code availability
The complete workflow, from data collection to model training and evaluation, is available for access and reuse at https://github.com/conchaeloko/DpoTropiSearch.
References
Silva, J. B., Storms, Z. & Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 363, fnw002 (2016).
Taslem Mourosi, J. et al. Understanding bacteriophage tail fiber interaction with host surface receptor: the key “Blueprint” for reprogramming phage host range. Int. J. Mol. Sci. 23, 12146 (2022).
Pires, D. P., Oliveira, H., Melo, L. D. R., Sillankorva, S. & Azeredo, J. Bacteriophage-encoded depolymerases: their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 100, 2141–2151 (2016).
Latka, A., Leiman, P. G., Drulis-Kawa, Z. & Briers, Y. Modeling the architecture of depolymerase-containing receptor binding proteins in klebsiella phages. Front. Microbiol. 10, 2649 (2019).
Topka-Bielecka, G. et al. Bacteriophage-derived depolymerases against bacterial biofilm. Antibiotics 10, 175 (2021).
Dunstan, R. A. et al. Mechanistic insights into the capsule-targeting depolymerase from a klebsiella pneumoniae bacteriophage. Microbiol. Spectr. 9, e01023–21 (2021).
Dunstan, R. A. et al. Epitopes in the capsular polysaccharide and the porin OmpK36 receptors are required for bacteriophage infection of Klebsiella pneumoniae. Cell Rep. 42, 112551 (2023).
Naghavi, M. et al. Global burden of bacterial antimicrobial resistance 1990–2021: a systematic analysis with forecasts to 2050. Lancet 404, 1199–1226 (2024).
Yang, Y. et al. The molecular basis of the regulation of bacterial capsule assembly by Wzc. Nat. Commun. 12, 4349 (2021).
Lam, M. M. C., Wick, R. R., Judd, L. M., Holt, K. E. & Wyres, K. L. Kaptive 2.0: updated capsule and lipopolysaccharide locus typing for the Klebsiella pneumoniae species complex. Microb. Genom. 8, 000800 (2022).
de Sousa, J. A. M., Buffet, A., Haudiquet, M., Rocha, E. P. C. & Rendueles, O. Modular prophage interactions driven by capsule serotype select for capsule loss under phage predation. ISME J. 14, 2980–2996 (2020).
Beamud, B. et al. Genetic determinants of host tropism in Klebsiella phages. Cell Rep. 42, 112048 (2023).
Ferriol-Gonz lez, C. et al. Targeted phage hunting to specific Klebsiella pneumoniae clinical isolates is an efficient antibiotic resistance and infection control strategy. Microbiol. Spectr. 12, e0025424 (2024).
Haudiquet, M., Buffet, A., Rendueles, O. & Rocha, E. P. C. Interplay between the cell envelope and mobile genetic elements shapes gene flow in populations of the nosocomial pathogen Klebsiella pneumoniae. PLOS Biol. 19, e3001276 (2021).
Fang, Q., Feng, Y., McNally, A. & Zong, Z. Characterization of phage resistance and phages capable of intestinal decolonization of carbapenem-resistant Klebsiella pneumoniae in mice. Commun. Biol. 5, 48 (2022).
Rendueles, O., De Sousa, J. A. & Rocha, E. P. Competition between lysogenic and sensitive bacteria is determined by the fitness costs of the different emerging phage-resistance strategies. eLife 12, e83479 (2023).
Tang, M. et al. Phage resistance formation and fitness costs of hypervirulent Klebsiella pneumoniae mediated by K2 capsule-specific phage and the corresponding mechanisms. Front. Microbiol. 14, 1156292 (2023).
Pas, C., Latka, A., Fieseler, L. & Briers, Y. Phage tailspike modularity and horizontal gene transfer reveals specificity towards E. coli O-antigen serogroups. Virol. J. 20, 174 (2023).
Yang, Y. Large-scale genomic survey with deep learning-based method reveals strain-level phage specificity determinants. GigaScience 13, giae017 (2024).
Nami, Y., Imeni, N. & Panahi, B. Application of machine learning in bacteriophage research. BMC Microbiol. 21, 193 (2021).
Graves, A., Wayne, G. & Danihelka, I. Neural turing machines. Preprint at arXiv https://doi.org/10.48550/arXiv.1410.5401 (2014).
Bahdanau, D., Cho, K. & Bengio, Y. Neural machine translation by jointly learning to align and translate. Preprint at arXiv https://doi.org/10.48550/arXiv.1409.0473 (2016).
Vaswani, A. et al. Attention is all you need. Adv. Neural Inf. Process. Syst. 30, 5998–6008 (2017).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, vol. 1 (Long and Short Papers) 1, 4171–4186 (Association for Computational Linguistics, 2019).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Kang, H. et al. Fine-tuning of the BERT model to accurately predict drug–target interactions. Pharmaceutics 14, 1710 (2022).
Sledzieski, S. et al. Democratizing protein language models with parameter-efficient fine-tuning. https://doi.org/10.1101/2023.11.09.566187 (2023).
Shang, J. & Sun, Y. CHERRY: a Computational metHod for accuratE pRediction of virus–pRokarYotic interactions using a graph encoder–decoder model. Brief. Bioinform. 23, bbac182 (2022).
Pan, J. et al. PTBGRP: predicting phage–bacteria interactions with graph representation learning on microbial heterogeneous information network. Brief. Bioinform. 24, bbad328 (2023).
Roux, S. et al. iPHoP: An integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria. PLOS Biol. 21, e3002083 (2023).
Gonzales, M. E. M., Ureta, J. C. & Shrestha, A. M. S. Protein embeddings improve phage-host interaction prediction. PLOS ONE 18, e0289030 (2023).
Domingo-Calap, P., Georgel, P. & Bahram, S. Back to the future: bacteriophages as promising therapeutic tools: bacteriophage-based therapy. HLA 87, 133–140 (2016).
de Jonge, P. A., Nobrega, F. L., Brouns, S. J. J. & Dutilh, B. E. Molecular and evolutionary determinants of bacteriophage host range. Trends Microbiol. 27, 51–63 (2019).
Gaborieau, B. et al. Prediction of strain level phage–host interactions across the Escherichia genus using only genomic information. Nat. Microbiol. 9, 2847–2861 (2024).
Boeckaerts, D. et al. Prediction of Klebsiella phage-host specificity at the strain level. Nat. Commun. 15, 4355 (2024).
Pieroni, P., Renniet, R. P., Ziola, B. & Deneer, H. G. The use of bacteriophages to differentiate serologically cross-reactive isolates of Klebsiella pneumoniae. J. Med. Microbiol. 41, 423–429 (1994).
Pan, Y.-J. et al. Klebsiella phage ΦK64-1 encodes multiple depolymerases for multiple host capsular types. J. Virol. 91, e02457–16 (2017).
Hsieh, P.-F., Lin, H.-H., Lin, T.-L., Chen, Y.-Y. & Wang, J.-T. Two T7-like bacteriophages, K5-2 and K5-4, each encode two capsule depolymerases: isolation and functional characterization. Sci. Rep. 7, 4624 (2017).
Zhang, M.-L., Li, Y.-K., Liu, X.-Y. & Geng, X. Binary relevance for multi-label learning: an overview. Front. Comput. Sci. 12, 191–202 (2018).
Ferriol-González, C. & Domingo-Calap, P. Phages for biofilm removal. Antibiotics 9, 268 (2020).
Wang, H., Liu, Y., Bai, C. & Leung, S. S. Y. Translating bacteriophage-derived depolymerases into antibacterial therapeutics: challenges and prospects. Acta Pharm. Sin. B 14, 155–169 (2024).
Concha-Eloko, R. et al. DepoScope: accurate phage depolymerase annotation and domain delineation using large language models. PLOS Comput. Biol. 20, e1011831 (2024).
Fortier, L.-C. & Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365 (2013).
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 (2014).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577 (2022).
Solovieva, E. V. et al. Comparative genome analysis of novel Podoviruses lytic for hypermucoviscous Klebsiella pneumoniae of K1, K2, and K57 capsular types. Virus Res. 243, 10–18 (2018).
Domingo-Calap, P., Beamud, B., Vienne, J., González-Candelas, F. & Sanjuán, R. Isolation of four lytic phages infecting klebsiella pneumoniae k22 clinical isolates from Spain. Int. J. Mol. Sci. 21, 425 (2020).
Townsend, E. M. et al. Isolation and characterization of Klebsiella phages for phage therapy. PHAGE 2, 26–42 (2021).
Harrison, R. L., Granja, C. & Leroy, C. Introduction to Monte Carlo Simulation. in 17–21 https://doi.org/10.1063/1.3295638 (Bratislava (Slovakia), 2010).
Squeglia, F. et al. Structural and functional studies of a klebsiella phage capsule depolymerase tailspike: mechanistic insights into capsular degradation. Structure 28, 613–624.e4 (2020).
Knecht, L. E., Veljkovic, M. & Fieseler, L. Diversity and function of phage encoded depolymerases. Front. Microbiol. 10, 2949 (2020).
Klumpp, J., Dunne, M. & Loessner, M. J. A perfect fit: bacteriophage receptor-binding proteins for diagnostic and therapeutic applications. Curr. Opin. Microbiol. 71, 102240 (2023).
Wyres, K. L., Lam, M. M. C. & Holt, K. E. Population genomics of Klebsiella pneumoniae. Nat. Rev. Microbiol. 18, 344–359 (2020).
Cai, Z. et al. Clinical and molecular analysis of ST11-K47 carbapenem-resistant hypervirulent klebsiella pneumoniae: a strain causing liver abscess. Pathogens 11, 657 (2022).
Wang, J., Feng, Y. & Zong, Z. The origins of ST11 KL64 klebsiella pneumoniae: a genome-based study. Microbiol. Spectr. 11, e04165–22 (2023).
Abdelkader, K. et al. The specific capsule depolymerase of phage PMK34 sensitizes acinetobacter baumannii to serum killing. Antibiotics 11, 677 (2022).
Blasco, L. et al. Development of an anti-acinetobacter baumannii biofilm phage cocktail: genomic adaptation to the host. Antimicrob. Agents Chemother. 66, e01923–21 (2022).
Shneider, M. M. et al. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500, 350–353 (2013).
Buth, S. et al. Structure and biophysical properties of a triple-stranded beta-helix comprising the central spike of bacteriophage T4. Viruses 7, 4676–4706 (2015).
Saccuzzo, E. G. et al. Competition between inside-out unfolding and pathogenic aggregation in an amyloid-forming β-propeller. Nat. Commun. 15, 155 (2024).
Pereira, J. & Lupas, A. N. New β-propellers are continuously amplified from single blades in all major lineages of the β-propeller superfamily. Front. Mol. Biosci. 9, 895496 (2022).
Pandey, S., Mahanta, P., Berger, B. W. & Acharya, R. Structural insights into the mechanism of pH-selective substrate specificity of the polysaccharide lyase Smlt1473. J. Biol. Chem. 297, 101014 (2021).
Chen, C. K.-M., Chan, N.-L. & Wang, A. H.-J. The many blades of the β-propeller proteins: conserved but versatile. Trends Biochem. Sci. 36, 553–561 (2011).
Concha-Eloko, R., Barberán-Martínez, P., Sanjuán, R. & Domingo-Calap, P. Broad-range capsule-dependent lytic Sugarlandvirus against Klebsiella sp. Microbiol. Spectr. 11, e04298–22 (2023).
Nguyen, E. et al. Sequence modeling and design from molecular to genome scale with evo. https://doi.org/10.1101/2024.02.27.582234 (2024).
Lood, C. et al. Digital phagograms: predicting phage infectivity through a multilayer machine learning approach. Curr. Opin. Virol. 52, 174–181 (2022).
Perrin, A. & Rocha, E. P. C. PanACoTA: a modular tool for massive microbial comparative genomics. https://doi.org/10.1101/2020.09.11.293472 (2020).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Siren, K. et al. Rapid discovery of novel prophages using biological feature engineering and machine learning. NAR Genom. Bioinform. 3, lqaa109 (2021).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Ishikawa, S. A., Zhukova, A., Iwasaki, W. & Gascuel, O. A fast likelihood method to reconstruct and visualize ancestral scenarios. Mol. Biol. Evol. 36, 2069–2085 (2019).
Talevich, E., Invergo, B. M., Cock, P. J. & Chapman, B. A. Bio.Phylo: a unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinform. 13, 209 (2012).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Steinegger, M. et al. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 20, 473 (2019).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–245 (2023).
Cretin, G. et al. SWORD2: hierarchical analysis of protein 3D structures. Nucleic Acids Res. 50, W732–W738 (2022).
Veličković, P. Everything is connected: graph neural networks. Curr. Opin. Struct. Biol. 79, 102538 (2023).
Zaheer, M. et al. Deep sets. Adv. Neural Inf. Process. Syst. 30, 3391–3401 (2017).
Brody, S., Alon, U. & Yahav, E. How attentive are graph attention networks? Preprint at arXiv https://doi.org/10.48550/arXiv.2105.14491 (2022).
Hamilton, W., Ying, Z. & Leskovec, J. Inductive representation learning on large graphs. Adv. Neural Inf. Process. Syst. 30, 1024–1034 (2017).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Akiba, T., Sano, S., Yanase, T., Ohta, T. & Koyama, M. Optuna: A Next-generation Hyperparameter Optimization Framework. in Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining 2623–2631 (ACM, Anchorage, AK, USA, 2019).
Sillankorva, S., Neubauer, P. & Azeredo, J. Pseudomonas fluorescens biofilms subjected to phage phiIBB-PF7A. BMC Biotechnol. 8, 79 (2008).
Acknowledgments
This work was financially supported by Advanced Grant 101019724 - EVADER from the European Research Council (ERC) to R.S, and fellowship GRISOLIAP/2020/158 from the Conselleria d’Innovació, Universitats, Ciència i Societat Digital (Generalitat Valenciana) to R.S. and R.C.-E. P.D.-C was funded by ESCMID Research Grant 20200063, project PID2020-112835RA-I00 from MCIN/AEI/10.13039/501100011033, and project SEJIGENT/2021/014 from the Conselleria d’Innovació, Universitats, Ciència i Societat Digital (Generalitat Valenciana); B.B. was funded by a PhD fellowship from the Spanish MCIU (FPU16/02139). We acknowledge Flaticon.com and its contributing designers (Smashicons, Freepik, Muhammad Waqas Khan) for the use of various icons in figures and schematics.
Author information
Authors and Affiliations
Contributions
Conceptualization & Methodology: R.C.-E., B.B., P.D.-C., and R.S.; Data Curation, Software, Formal Analysis, Validation & Original Draft preparation: R.C.-E.; Manuscript Review & Editing: R.C.-E., B.B., P.D.-C., and R.S.; Supervision: B.B., P.D.-C., and R.S. All authors read and approved the final manuscript. The authors declare no competing interests.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks João Setubal, who co-reviewed with Guillermo Uceda-Campos, Petar Veličković, Xiaofang Jiang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Concha-Eloko, R., Beamud, B., Domingo-Calap, P. et al. Unlocking data in Klebsiella lysogens to predict capsular type-specificity of phage depolymerases. Nat Commun 16, 8798 (2025). https://doi.org/10.1038/s41467-025-63861-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63861-w
