
Abstract
The development of efficient catalysts for ethane dehydrogenation (EDH) to ethylene remains a challenge due to the lack of direct material property-performance relationships at the most elementary level. Here, we introduce the first application of ZrO2-based catalysts for EDH in a chemical looping mode. Their performance in the first 1 minute, primarily via the oxidative dehydrogenation, highlights their potential for large-scale ethylene production. LaZrOx achieves a space-time yield of 2.26 \({{\mbox{kg}}}_{{{\mbox{C}}}_{2}{{\mbox{H}}}_{4}}\cdot {{\mbox{kg}}}_{{\mbox{cat}}}^{-1}\cdot {{\mbox{h}}}^{-1}\) at about 80% ethylene selectivity and 50% ethane conversion at 700 °C. Mechanistic studies have identified the reactivity and availability of lattice oxygen as crucial descriptors for mitigating coke formation and suppressing combustion reactions. These properties can be tuned by exposing less stable ZrO2 crystal planes or incorporating metal-oxide promoters. Strongly adsorbed oxygen species can also participate in ethane oxidation. Thus, this study establishes a catalyst system for chemical looping EDH and provides insights for designing more efficient EDH catalysts.
Similar content being viewed by others

Introduction
Ethylene is a vital building block in the petrochemical industry for the production of a wide range of chemicals and polymers, including polyethylene, ethylene oxide, and vinyl acetate1,2. Global demand for this olefin continues to grow, with the United States and China leading production efforts, each with annual growth rates of approximately 3%3. However, traditional steam cracking methods for ethylene production are highly energy-intensive and contribute significantly to carbon dioxide emissions, raising substantial environmental concerns3.
The above issues have prompted the exploration of alternative production methods, with catalytic ethane dehydrogenation (EDH) emerging as a promising energy-efficient pathway. In contrast to the non-oxidative propane dehydrogenation (PDH), which has been successfully commercialized4,5,6,7,8,9, the chemical inertness of ethane and thermodynamic limitations of non-oxidative EDH10 are significant barriers to the commercialisation of such a process. Of particular interest is oxidative EDH, which utilizes oxygen to remove hydrogen from ethane as water, reducing operational temperatures and lowering carbon dioxide emissions11. Despite these advantages, oxidative EDH faces several challenges, including overoxidation of ethane/ethylene, explosion risks, limited industrial scalability, and the need to separate air to obtain pure oxygen1. To address the limitations of the oxidative approach, chemical looping oxidative dehydrogenation (CL-ODH) has been identified as a potential solution12,13,14,15. In this approach, ethane and air are fed alternately to the catalysts, allowing for greater control over the reaction conditions and minimizing side reactions as well as explosion risks.
While considerable progress has been made in the design of catalysts for both oxidative and non-oxidative EDH16,17,18,19,20,21, significant challenges remain. In oxidative EDH, easily reducible catalysts can enhance activity but also promote combustion reactions, reducing ethylene selectivity. In the CL-ODH approach, catalysts must have high oxygen storage capacity and rapid oxygen transfer rates to ensure long catalyst lifetimes22. Too fast oxygen transfer may, however, be detrimental for achieving high ethylene selectivity due to overoxidation of the latter. Furthermore, maintaining the physicochemical stability of oxygen carriers through multiple redox cycles is essential for ensuring durable operation23. Efficient ethylene desorption from the catalyst surface is crucial in both oxidative and non-oxidative processes, as it improves the selectivity to ethylene by inhibiting the formation of coke, which leads to catalyst deactivation24. The mechanism of coke formation in EDH is not well understood, in comparison to PDH25,26,27,28. Therefore, it is essential to elucidate the dehydrogenation and coke formation pathways and the factors influencing them to develop next-generation EDH catalysts with industrially relevant ethylene selectivity and ethane conversion as well as high durability/on-stream stability, and productivity.
In our previous studies on PDH29,30,31,32,33,34, we introduced an alternative approach for developing alkane-dehydrogenation catalysts based on ZrO2. In these catalysts, the active sites are coordinatively unsaturated Zr cations (Zrcus), which are created by the removal of lattice oxygen through treatment with a reducing agent at high temperatures. Experimental studies have confirmed that treating ZrO2-based materials with H2 at temperatures exceeding 550 °C results in the formation of anion vacancies due to the removal of lattice oxygen35. Notably, the excess electrons formed in this process are localized at the vacancies rather than leading to the formation of Zr3+ species36. Moreover, the stability of both stoichiometric and defective ZrO2 surfaces is strongly influenced by the reaction temperature and the partial pressures of reducing and oxidizing agents35.
Considering the aforementioned challenges and the findings of our previous PDH studies, the present study was aimed to elucidate the potential of ZrO2-based catalysts for CL-ODH and to provide the fundamentals for controlling product selectivity and catalyst productivity. To achieve this goal, we prepared a series of catalysts that differed in the type of metal (La, Y or Ce) oxide promoter for ZrO2, which is relevant to the ability of the latter to release its lattice oxygen. Their catalytic performance was assessed during different times on ethane stream to distinguish between CL-ODH or EDH and to evaluate these approaches in terms of their efficiency regarding the formation of ethylene. Material property-performance relationships were established by combining in situ and operando time-resolved UV-vis and DRIFT spectroscopic studies with temporal analysis of products and kinetic and mechanistic studies with time resolutions varying from sub-milliseconds to seconds. Density functional theory (DFT) calculations were instrumental to rationalize the findings.
Results
Ethane dehydrogenation
Using gas chromatography (GC) and mass spectrometry (MS) operating with resolutions of minutes and seconds, respectively, we analysed products formed over the developed catalysts under industrially relevant conditions using a feed with 56 vol% ethane (Supplementary Fig. 1). These techniques were complementary to identify different regimes (oxidative and non-oxidative) of ethane conversion to the target and side products and to elucidate the role of lattice oxygen and in situ formed defects in these processes (see section ‘Methods’ for details). Doping ZrO2 with Y, La, and Ce notably enhanced the initial (after the first 5 min on stream) C2H6 conversion (Fig. 1a and Supplementary Fig. 2). Furthermore, we identified two distinct stages in the catalytic process: (i) the first 15 minutes on stream, where the conversion over all catalysts decreased, while the selectivity to ethylene increased with increasing time on stream; (ii) after 15 min on stream, where both conversion and selectivity did not change further. Notably, the formation of side products, COx (CO and CO2) and CH4, after the first 5 min on stream (the first GC data point) was minimal. Coke formation was the dominant side product. With increasing time on stream, ethylene emerged as the main product but at lower ethane conversion. However, on-line monitoring via MS (Supplementary Fig. 3) revealed that combustion side reactions played a significant role during the first 5 min on stream, with CeZrOx showing the highest activity in this respect. These observations prompted further investigation of ZrO2-catalysed ethane conversion during different times on stream using MS, driven by (i) the potential to control combustion side reactions through doping ZrO2 with different metal oxides and (ii) the feasibility of periodically removing coke deposits via air or oxygen, i.e., employing a chemical looping strategy.

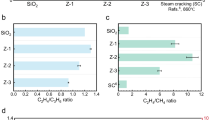
a Ethane conversion versus time on stream, measured using an on-line GC (Reaction conditions: T = 700 °C, WHSV = 6.09 h-1, C2H6/N2/He = 56/9/35). b Ethane conversion within the first 1, 5, and 10 min on reaction stream measured using an on-line MS (Reaction conditions: T = 700 °C, WHSV = 6.09 h-1, C2H6/Ar/He = 56/9/35 vol%). The error bars represent the standard deviation of five independent measurements. c Dependence of ethylene selectivity on ethane conversion determined at the 5th minute on ethane stream (GC results). Data for ZrO2 and LaZrOx within the first 1 min on ethane stream (MS results) and commercial ethane steam cracking (CESC) technology37 are also shown for comparison. d Assessment of space time yield of ethylene production (STY) over ZrO2 and LaZrOx within the first minute with previously reported non-oxidative and oxidative EDH catalysts. All referenced catalysts and citations are listed in Supplementary Table 1.
Consequently, the performance of the developed catalysts within the first 1, 5, and 10 min on stream was separately assessed in five ethane and air alternating cycles in each period (Supplementary Fig. 4). The conversion of C2H6 over ZrO2, YZrOx, and LaZrOx gradually decreased with increasing time on stream, whereas an increase was identified for CeZrOx (Fig. 1b). This difference can be attributed to the product selectivity (see Supplementary Fig. 5). Coke formation is the primary cause of the decrease in C2H6 conversion over ZrO2, YZrOx, and LaZrOx, because active sites are progressively covered by this product. Additionally, the extent of coke formation directly affects ethylene selectivity of these catalysts. In contrast, CeZrOx demonstrated a distinct behaviour. Combustion reactions played an important role within the first 1 min on ethane stream (Supplementary Fig. 3d, Supplementary Fig. 5c). As the reaction progressed further, the formation of carbon oxides decreased, while coke formation gradually became the dominant side reaction, affecting both C2H6 conversion and C2H4 selectivity. Notably, within the first 10 minutes, the impact of the combustion side reactions on ethane conversion was significantly stronger than that of coke formation. Furthermore, ethylene selectivity is closely tied to the extent of these two competing reactions. In addition, for all catalysts, cracking reactions of C2H6/C2H4 to CH4 also contribute to the loss of this selectivity. Their contribution increases with rising time on stream (Supplementary Fig. 5b). All catalysts exhibit good durability (Supplementary Fig. 4), which is further illustrated by the performance achieved by ZrO2 in a 10-cycle EDH/regeneration test (Supplementary Fig. 6).
Motivated by the above results, we selected ZrO2 and LaZrOx for further in-depth analysis, considering both C2H6 conversion and C2H4 selectivity of all ZrO2-based catalysts. First, we prepared the selectivity-conversion relationships for ethylene using the initial performance (after the first 5 min on stream) determined by GC and the integral performance determined by MS during the first 1 min on stream (Fig. 1c). We also compared these results with the corresponding data from a commercial ethane steam cracking (CESC) technology37. It is obvious that 1 min operation is advantageous. Although the C2H6 conversion over ZrO2 and LaZrOx is lower than that of the CESC technology, the C2H4 selectivity surpasses that of the latter. Furthermore, we compared the space-time yield (STY) of C2H4 formation within the first 1 minute over ZrO2 (2.14 \({{\mbox{kg}}}_{{{\mbox{C}}}_{2}{{\mbox{H}}}_{4}}\,{{\mbox{kg}}}_{{\mbox{cat}}}^{-1}\,{{\mbox{h}}}^{-1}\)) and LaZrOx (2.26 \({{\mbox{kg}}}_{{{\mbox{C}}}_{2}{{\mbox{H}}}_{4}}\,{{\mbox{kg}}}_{{\mbox{cat}}}^{-1}\,{{\mbox{h}}}^{-1}\)) with previously reported non-oxidative and oxidative EDH catalysts (Supplementary Table 1). Our catalysts outperform most of those reported in the literature (Fig. 1d).
It is important to note that the productivity reported in this study is based solely on the EDH cycle (excluding the catalyst regeneration cycle), which reflects the intrinsic catalyst activity. However, in a practical chemical looping process, at least two reactors will be used, operating alternately. This allows the EDH reaction and catalyst regeneration to take place simultaneously, but in different reactors. Such operation minimizes the impact of regeneration time on overall process productivity3. Therefore, although our STY values do not account for the regeneration step, our approach still reflects a realistic scenario where the regeneration time does not become a bottleneck. Overall, these results suggest that CL-ODH over ZrO2 and LaZrOx represents a promising approach to ethylene production with significant application potential.
Temporal analysis of products
Driven by the high time resolution and the low pulse size (0.01–10 nmol)38, we applied a temporal analysis of products reactor for providing deeper mechanistic insights into the formation of target and side gas-phase products in the absence of gas-phase O2. It is important to note that such small pulse sizes in TAP experiments are not meant to simulate practical operating conditions. Rather, they conform to established TAP protocols, where low-dosage operation in high vacuum is crucial to eliminate heat and mass transfer effects, thereby allowing intrinsic kinetic analysis of surface reactions. In contrast, our chemical looping experiments were performed at ambient pressure under continuous flow using an industrially relevant feed of 56 vol% C2H6 and a WHSV of 6.09 h⁻¹, providing realistic evaluation of catalyst performance under process-relevant conditions.
In a single pulse experiment at 700 °C using a C2H6/Ar = 1 mixture, C2H4 and CO2 were identified as the reaction products of C2H6 oxidation over the oxidized ZrO2, LaZrOx, and CeZrOx catalysts, indicating the concurrent occurrence of selective and combustion side reactions. The response of C2H4 appears directly after the response on unreacted C2H6, while the response of CO2 is shifted to higher times (Fig. 2a–c). As no gas-phase O2 was pulsed, these reaction products, CO2 completely and C2H4 at least partially, must be formed with the participation of lattice oxygen of the catalysts. Based on the appearance order (the time of the maximal concentration) of the responses of C2H4 and CO2, it can be suggested that the latter product should be mainly formed through oxidation of the target olefin. Notably, the CO2 response is broad with a pronounced tailing compared to C2H4, suggesting its stronger adsorption on the catalyst surface.

Normalized transient responses of C2H6, C2H4 and CO2 formed over a ZrO2, b LaZrOx, and c CeZrOx after pulsing a C2H6:Ar = 1:1 mixture at 700 °C. d The ratio of the amount of CO2 formed in a series of C2H6 pulses (C2H6:Ar = 1:1) over ZrO2-based at 700 °C to that in the first pulse. e The ratio of the total amount of CO2 formed in a series of C2H6 pulses (C2H6:Ar = 1:1) over different catalysts to that over CeZrOx. f The selectivity to C2H4 versus the reduction degree of the catalysts.
As the extent of combustion side reactions is closely linked to the availability and reactivity of lattice oxygen species in the catalysts, we performed multipulse experiments with a single C2H6 pulse size of about 1016 molecules. It should be mentioned that the total amount of C2H6 pulsed in these tests corresponds to about 2% of the amount of ethane passed through the reactor in the 1st-minute tests discussed in section ‘Ethane dehydrogenation’. CeZrOx showed the highest conversion in the first pulse followed by LaZrOx and ZrO2, which decreased with increasing number of C2H6 pulses, with the strength of the decrease depending on the kind of the promoter for ZrO2 (Supplementary Fig. 7). The changes in the conversion are caused by the changes in the amounts of CO2 and C2H4 formed (Supplementary Fig. 8). For all catalysts, the amount of CO2, reflecting the number of active surface oxygen species involved in the combustion reactions, in the last C2H6 pulse was about 10 times lower than in the first pulse (Fig. 2d). The cumulative CO2 production revealed that the total number of active surface oxygen species available for the combustion side reactions was highest on CeZrOx, followed by LaZrOx and ZrO2 (Fig. 2e). Using the amounts of CO2 and C2H4 in each individual pulse, we calculated the cumulative amount of lattice oxygen removed under assumption that the formation of one CO2 and one C2H4 needs 7 (C2H6 + 7 O = 2CO2 + 3H2O) and 1 (C2H6 + O = C2H4 + H2O) lattice oxygen, respectively. Considering this amount and the total amount of lattice oxygen available in the catalysts, we estimated how the degree of reduction of the catalysts changed with increasing number of C2H6 pulses. Figure 2f shows that ZrO2 and YZrOx selectively oxidize ethane to ethylene even at very low reduction degrees. In contrast to these catalysts, the selectivity to ethylene formed over fully oxidized (very low reduction degree) CeZrOx is close to zero but increases with increasing reduction degree. The difference should be related to the ability (number and/or easiness) of these materials to provide their lattice oxygen for ethane oxidation. Thus, these findings suggest that regulating both the quantity and type of active surface oxygen species of EDH catalysts may provide a strategy for controlling combustion side reactions, thereby enhancing the overall EDH performance.
Analysis of anion vacancy and coke formation
Following the results of the catalytic tests and the temporal analysis of products, we aimed to provide the fundamentals of (i) lattice oxygen removal and (ii) coke formation using near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) and operando UV-vis spectroscopy, which offers a distinct advantage in the analysis of coke formation in alkane dehydrogenation reactions31,39. Using H2 as a reducing agent, we performed in situ UV-vis tests with ZrO2, YZrOx and LaZrOx at 700 °C within 60 seconds. The obtained UV-vis spectra expressed in the form of the relative Kubelka-Munk function (F(Rrel), Eqs. 2 and 3) are characterized by two positive peaks with maxima at about 300 nm and about 460-500 nm (Supplementary Figs. 9a, 10a, and 11a). The first signal is due to the decrease in the intensity of charge-transfer transitions in ZrO2 caused by the removal of lattice oxygen as seen from the difference spectra (Supplementary Figs. 9b, 10b, 11b). The second peak can be due to electron transitions related to anion vacancies. Gionco et al.40 reported that when such defects are formed in ZrO2, several transition energy levels of 3.5-3.8 eV above the valence band appear. Dynamic information about the formation of anion vacancies in ZrO2, YZrOx and LaZrOx was derived by analysing the temporal evolution of the bands at about 460–500 nm (Supplementary Figs. 9c, 10c, and 11c). The intensity increased with increasing time on H2 stream confirming the continuous formation of anion vacancies. The H2-induced changes in the UV-vis spectrum of CeZrOx were similar to those described above but were overlapped by the absorption associated with the reduction of CeO2 (Supplementary Fig. 12).
Further confirmation of anion vacancy formation was provided through NAP-XPS measurements conducted on ZrO2 and LaZrOx during H2 reduction (Supplementary Fig. 13). Even after 100 min of H2 treatment, the Zr in both catalysts remains in the +4 oxidation state, and the La in LaZrOx consistently remains at +3. However, the binding energies of O 1 s and Zr 3 d in ZrO2, and those of O 1 s, Zr 3 d, and La 3 d in LaZrOx, are shifted by 0.8 and 1.0 eV, respectively, to higher values. These shifts are attributed to the modulation of the Fermi level position in oxides, resulting from the removal of lattice oxygen and the subsequent formation of anion vacancies41. Moreover, a comparative analysis of these shifts indicates that LaZrOx exhibits significantly larger displacements than ZrO2, implying that LaZrOx consumes a greater amount of lattice oxygen during H2 reduction.
To roughly quantify the amount of oxygen provided by the catalysts for H2 oxidation, we carried out temperature-programmed reduction tests (H2-TPR) using a feed with 5 vol%H2 in Ar. The obtained outlet profiles of H2 and H2O are shown in Supplementary Fig. 14 (also see the discussion below this figure). The intensity of the H2O signal starts to increase when the intensity of the H2 signal decreases as a result of H2 oxidation to H2O. Moreover, the higher the intensity of the H2O signal, the stronger the decrease in the intensity of the H2 signal. The highest amount of H2 oxidized to H2O related to the catalyst amount was determined for CeZrOx, while ZrO2 consumed the lowest amount of H2 (Supplementary Table 2). However, when considering the specific surface area of the catalysts, ZrO2 outperforms LaZrOx and YZrOx in terms of its ability to provide oxygen for H2 oxidation.
In contrast to H2, different changes in the operando UV-vis spectra were observed when ethane reacted with ZrO2-based catalysts at 700 °C (Fig. 3a–d). F(Rrel) increased in the whole range of wavelength between 250 and 1000 nm with increasing time on ethane stream. No obvious bands could be seen in the spectra. Based on a previous study39, low-condensed aromatic species and polyaromatic graphitic species absorb at low and high wavelength, respectively. To check if anion vacancies were also formed under EDH conditions, we analysed the UV-vis spectra within the first minute on ethane stream. As shown in Supplementary Figs. 15–17, ZrO2, YZrOx, and LaZrOx only exhibited the formation of anion vacancies within the first 7, 8, and 9 s, respectively. Beyond these times, coke formation commenced, with progressive accumulation evident over time. The absorption bands attributed to anion vacancies were gradually overlapped by those corresponding to coke deposits. In contrast, CeZrOx displayed a distinct behaviour (Supplementary Fig. 18). The concentration of anion vacancies continued to increase throughout the first minute, with no detectable absorption bands associated with carbon deposits. This lack of coke-related features indicates that coke formation was entirely suppressed during this period. Consistent with this observation, MS data in Supplementary Fig. 4 revealed no COx signals during the reoxidation stage following the first minute of reaction for CeZrOx. These results collectively confirm that CeZrOx effectively inhibits coke formation under EDH conditions, distinguishing it from other ZrO2-based catalysts.

a–d Operando UV-vis spectra and corresponding fitted results. e–h Temporal evolution of different normalized peak areas. i–l Temporal evolution of subband δ area during EDH (Inverted triangles: experimental data, solid line: simulated results). EDH was performed at 700 °C using a feed containing 56 vol% C2H6 in Ar.
To better visualize the temporal evolution of the UV-vis spectra with rising time on ethane stream, the spectra were simply fitted using Gaussian functions. For all catalysts, a minimal number of such functions was established to be 6 for getting a proper description of the experimental data (Fig. 3a–d). According to previous studies31,42, the absorption subbands from shorter to longer wavelengths (designated as α, β, γ, δ, λ, and φ) were attributed to various coke species, spanning from low-concentration aromatic intermediates to polyaromatic graphite-like deposits. The temporal evolution of the peak areas for these six subbands is shown in Fig. 3e–h, enabling a clearer understanding of the dynamic changes in coke formation. The data indicate distinct differences in coke formation kinetics, both among the subbands on the same catalyst and across different catalysts. For ZrO2 and LaZrOx, the formation of coke species mainly proceeded in two stages: (i) an initial rapid accumulation followed by a slowdown and (ii) a subsequent acceleration before decelerating again as the deposition rate approached zero. In contrast, YZrOx exhibited only the first stage, with no observable second stage of coke accumulation. CeZrOx, however, presented a more complex behaviour. During the initial reaction stage (up to ~1 min), no detectable coke deposits were observed, coinciding with the formation of new anion vacancies (Supplementary Fig. 18). Thus, changes in the absorption subband intensities within ~10 min cannot be solely ascribed to coke deposits but must also consider the evolving concentration of anion vacancies.
To quantify the observed trends in coke formation, apparent rate constants were determined by analysing the temporal evolution of the peak area corresponding to subband δ, which represents one of the dominant coke species across all ZrO2-based catalysts. A simplified kinetic model (Eq. 1) was applied:
Where f(t) represents the total amount of coke deposits associated with the subband δ, A1 and A2 denote the amounts of coke deposits formed during the first and second stages, respectively. k1 and k2 are the corresponding rate constants of coke formation for each stage. t is the time on ethane stream. t0 marks the transition time between the two stages (in minutes).
Figure 3i–l display the experimental data alongside the fitted curves derived using the model described in Eq. 1, with the corresponding parameters summarized in Supplementary Table 3. As shown, the model effectively captures the experimental trends for all catalysts. For ZrO2 and LaZrOx, the A1 and k1 values in the first stage are smaller than the corresponding A2 and k2 values in the second stage. This observation supports the definition of the first stage as the coke formation induction period, likely attributed to the gradual activation of the catalyst surface or suboptimal conditions for coke formation. The second stage, referred to as the coke formation acceleration period, exhibits a higher coke formation rate, potentially resulting from the reaction conditions approaching steady-state or structural changes on the catalyst surface that enhance the coke formation process. For YZrOx, the coke formation induction period appears to be very short, with the catalyst rapidly transition into the acceleration period. In contrast, the behavior of CeZrOx is more complex, making it difficult to assign specific physical meanings to the A1 and k1 values. This complexity arises from overlapping phenomena, including the continuous formation of anion vacancies and the delayed onset of detectable coke deposits.
Further, the total amount of coke deposits formed in the course of EDH during 35 minutes on ethane stream was determined by integrating the profiles of CO2 formed in temperature-programmed oxidation of spent catalysts (Supplementary Fig. 19). This follows the order YZrOx > LaZrOx > CeZrOx > ZrO2. Catalyst acidic sites are typically considered to play a key role in the formation of carbon deposits1. To evaluate whether such relationship holds for our catalysts, we correlated the amount of coke formed per square meter of the catalysts with the density of Brønsted or Lewis acid sites, as measured by IR spectroscopy of adsorbed pyridine (Supplementary Fig. 20 and Supplementary Table 4). Brønsted acid sites do not seem to be relevant for coke formation, while Lewis acid sites should play an important role in this side reaction of EDH over the ZrO2-based catalysts (Supplementary Fig. 21). Mechanistically, the formation of coke begins with the adsorption of ethylene. The adsorbed molecules first react with each other to form small olefinic and aromatic structures, which subsequently undergo further oligomerization and condensation reactions to produce larger graphitic structures1. On this basis, the spatial distribution of the initial coke precursors should be relevant for coke formation; the higher their density, the higher the probability for their transformation to coke.
Detailed reaction pathways in the course of EDH
From the above analysis, it is evident that ethane reacts with lattice oxygen of the ZrO2-based catalysts to facilitate oxidative dehydrogenation. To check if strongly adsorbed oxygen species formed during oxidative catalyst treatment can also participate in this reaction, we performed in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) measurements (Supplementary Fig. 22) and oxygen isotopic exchange experiments (Supplementary Fig. 23). The DRIFTS results show that diatomic oxygen species (O22-) can be formed from gas-phase O2 and are stable in Ar at 700 °C for at least 10 min, the time between air and ethane cycles in our CL-ODH tests. These species react readily with ethane (Supplementary Fig. 22). In addition to gas-phase O2, lattice oxygen of zirconia can also give O22- as indirectly suggested by the results of oxygen isotopic exchange experiments (Supplementary Fig. 23). These complementary findings underscore the dual origin and the nature of the active oxygen, which are critical for enhancing the oxidative dehydrogenation of ethane.
Subsequently, in situ DRIFTS was used to identify intermediate species on the catalyst surface and monitor their transformation under actual reaction conditions. The IR spectra obtained after exposure of the catalysts to C2H6 at 700 °C are characterized by absorption bands in four regions, i.e., 3800–3600, 3200–2700, 2450–2050, and 1650−1250 cm−1 (Fig. 4). They can be assigned to hydroxyl groups, gas-phase ethane/ethylene, CO/CO2 and various surface oxygenates/carbon-containing intermediaries, respectively. Their assignment was made based on previous studies and the relevant details are summarized in Supplementary Tables 5 and 6. It is important to note that the types of the IR bands are essentially identical for all ZrO2-based catalysts tested. However, the rate of increase in IR band intensity varied with each catalyst, suggesting differences in the kinetics of intermediate formation. These variations are likely to influence catalytic performance and efficiency in the ethane dehydrogenation process.

a IR bands in the range of 4000–3400 cm-1. b IR bands in the range of 4000–3400 cm-1. c IR bands in the range of 4000–3400 cm-1. d IR bands in the range of 4000–3400 cm-1. EDH was performed at 700 °C using a feed containing 56 vol% C2H6 in Ar.
By combining the in situ DRIFTS results with those of our temporal analysis of products with second (Supplementary Fig. 3) and submillisecond resolution (Fig. 2, Supplementary Figs. 6 and 7) as well as time-resolved UV-vis spectroscopic analysis (Fig. 3) and oxygen isotopic exchange results (Supplementary Fig. 23), we propose that EDH over ZrO2-based catalysts proceeds via the reaction pathways summarized in Fig. 5. This reaction over fully oxidized catalysts should be initiated by the formation of C2H5* groups (1493 and 1303 cm−1, Fig. 4d) through the cleavage of the C-H bond in C2H6 molecule by active oxygen species (diatomic oxygen and lattice oxygen) on the catalyst surface.

Primary pathways of ethane conversion are represented by routes I-III. Route I: combustion side reactions leading to COx and H2O, route II: oxidative ethane dehydrogenation with lattice oxygen, and route III: non-oxidative ethane dehydrogenation to ethylene and hydrogen. Consecutive transformations of ethylene to methane and coke are represented by routes IV and V, respectively.
Route I: Combustion side reactions
The generation of CO2 at the initial reaction stage confirms the ability of oxidized ZrO2-based materials to provide their active oxygen for ethane/ethylene oxidation. Our time-resolved data (Fig. 4c, Supplementary Fig. 22b, c, and Supplementary Fig. 3) indicate that the depletion of diatomic oxygen species during their reaction with ethane and the formation of COx occur almost simultaneously. Once the diatomic oxygen is exhausted, the COx production levels off, even as the oxidative dehydrogenation reaction proceeds. These observations suggest that C2H5* species, formed as ethoxide on the diatomic oxygen sites, are readily oxidized to formate (1570 and 1350 cm−1, Fig. 4d) or acetate (1535 and 1440 cm-1, Fig. 4d) intermediates. They subsequently undergo unselective oxidation to gaseous/adsorbed CO/CO2 (2450–2050 cm-1, Fig. 4c), simultaneously generating anion vacancies. CO can also consume surface OH groups, as evidenced by the appearance of negative bands at 3800–3600 cm-1 (Fig. 4a). In this process, two OH groups are consumed to produce CO2, H2O, and anion vacancies. It cannot, however, be completely excluded that diatomic oxygen also oxidize ethane to ethylene.
Route II: Oxidative ethane dehydrogenation
In parallel to the combustion reactions, C2H5* groups can undergo subsequent β-H elimination. This process results in the formation of ethylene (2984 cm-1, Fig. 4b), most of which desorbs from the surface of the catalysts. DFT calculations were performed to investigate the reaction pathways of oxidative ethane dehydrogenation over stoichiometric ZrO2(-111). The calculated Gibbs free energy profile at 973 K and ambient pressure and the optimized structures of intermediates and transition states are shown in Supplementary Fig. 24. The activation of C-H bond over ZrO2(-111) occurs on the Zr-O sites and leads to the formation of Zr-C2H5 and O-H intermediates. The second hydrogen abstraction leads to the formation of adsorbed C2H4 and Zr-H site. After ethylene desorption, one H atom located at Zr diffuses to the Zr atom bonded to the O-H site. Formation of H2O and anion vacancy has the highest barrier in the entire process (2.40 eV) and is highly endergonic by 2.23 eV. This step is the rate-determining one.
Moreover, our thermodynamic analysis (Supplementary Table 7) confirms that both oxidative and non-oxidative dehydrogenation of ethane are thermodynamically favorable at 700 °C, with the oxidative route proceeding in a presence of gaseous oxygen being more favorable. Although the reaction feed does not contain gaseous oxygen, oxidative pathway with the participation of the lattice oxygen of ZrO2 is more likely to dominate in the early stage of the reaction, prior to significant depletion of this oxygen.
Route III: Non-oxidative ethane dehydrogenation
With progressing removal of lattice oxygen of ZrO2 through the above-mentioned processes, coordinatively unsaturated zirconium cations are formed and can be involved in the non-oxidative dehydrogenation of ethane to ethylene31. According to the result of DFT calculations (Supplementary Fig. 25), the reaction occurs with participation of three Zr atoms, two of which surround an anion vacancy (Zrcus sites) and proceeds through stepwise first and second hydrogen abstraction. The sequential recombination of two H atoms with the formation of adsorbed H2 is highly endergonic by 1.08 eV and can be considered as the rate-determining step. For comparison, we also carried out DFT calculations on the same reaction pathway over a stoichiometric ZrO2(-111) surface. The corresponding free energy profile is presented in Supplementary Fig. 26, revealing an overall energy span of 2.75 eV, which is thermodynamically reasonable for a catalytic process at 700 °C. While this energy span is notably higher than that on the defective surface (1.21 eV), it still indicates that fully oxidized ZrO2 possesses intrinsic catalytic potential. These results highlight the beneficial role of surface defects in lowering the reaction barriers, but also demonstrate that both pristine and defective ZrO2 surfaces can contribute to ethane activation under high-temperature conditions.
Routes IV and V: Methane formation and coke deposits
The presence of Zrcus sites, which are Lewis acid sites, is also important for ethylene adsorption. Adsorbed ethylene can isomerize to form ethylidene, which subsequently dehydrogenates to form ethylidyne followed by the cleavage of the C-C bond in the latter to produce CHx (x = 1-3) species39, such as methyl groups (2874 and 1376 cm-1, Fig. 4b, d). In the presence of adsorbed hydrogen species, CHx species can be hydrogenated to form CH4 (Route IV). In addition, the dehydrogenation and oligomerization of adsorbed ethylene species contribute to the formation of coke precursors (Route V). The appearance of absorption IR bands at 1597 and 1510 cm-1 (Fig. 4d), attributed to the vibrations of the benzene ring skeleton, indicates the formation of aromatic hydrocarbon species, which may originate from aliphatic hydrocarbons43. These species are considered as coke precursors that eventually form coke through further dehydrogenation and polymerization44,45.
Options for optimizing ethylene selectivity and catalyst productivity
Based on the results of catalytic and characterisation tests, carbon oxides and coke deposits are two major side products lowering the selectivity to ethylene. Their impact depends, however, on the degree of reduction of ZrO2 and the ability to provide its lattice oxygen for ethane activation. The formation of carbon oxides and the oxidation of ethane to ethylene with the involvement of lattice oxygen occur in parallel at the beginning of EDH. As the former reaction needs higher number of lattice oxygen, its contribution with increasing time on ethane stream terminates faster in comparison with the selective reaction because the concentration of lattice oxygen decreases. Due to the higher oxygen storage capacity of CeZrOx in comparison with ZrO2, YZrOx, and LaZrOx, this catalyst operates unselectively and needs to reach higher reduction degree to hinder the formation of CO2 (Fig. 2, Supplementary Figs. 7 and 8). As the reaction progresses, the depletion of available oxygen species on the catalyst surface and within the bulk leads to the cessation of oxidative dehydrogenation in favour of non-oxidative dehydrogenation catalysed by Zrcus sites. However, the latter also participate in the formation of coke leading again to the loss of ethylene selectivity and catalyst deactivation (Fig. 1a, b). Remarkably, catalyst performance of all ZrO2-based catalysts was stabilized after 15 min on ethane stream, suggesting that certain Zrcus sites do not favour ethylene adsorption on them, thereby preventing coke formation (Fig. 3e–h). This observation highlights the need for further experimental and theoretical investigations to elucidate the distinct surface chemistries of these cation sites and their roles in sustaining catalyst activity over extended operation. Understanding these unique active sites could pave the way for improved catalyst design, offering enhanced stability and performance for ethane dehydrogenation processes.
Our findings also demonstrate that the kind of promoter is a critical descriptor for influencing the initial (mainly oxidative) EDH performance of ZrO2-based catalysts in view of reducing coke formation in favour of the desired reaction. These improvements are intrinsically linked to the availability and reactivity of lattice oxygen species of ZrO2. Therefore, the selection of a suitable promoter and its concentration requires careful optimization to maximize the dehydrogenation efficiency while minimizing side reactions.
Another promising modification strategy involves enhancing the availability of active oxygen species by regulating the exposed crystal planes of metal oxide catalysts. For instance, in the monoclinic phase of ZrO2 (m-ZrO2), the (-111) crystal plane is generally considered the predominant exposed surface due to its low surface energy31,46. However, using high-resolution transmission electron microscopy (HRTEM) for analysis of the present ZrO2, three additional crystal planes, (011), (002), and (100), were identified (Fig. 6a–c). Density functional theory (DFT) calculations suggest that for the extended m-ZrO2(-111) surface, the removal of lattice oxygen through the reaction with surface hydrogen species is highly endergonic (Supplementary Fig. 24). Additional calculations (Fig. 6d) demonstrated that for the less stable (011) crystal plane, the formation of anion vacancy is energetically more favourable. It is worth mentioning that according to several experimental and theoretical works31,32,47,48, ZrO2 nanocrystals possess high reactivity towards H2 and can undergo dehydration at milder conditions compared to the regular surfaces due to their structural flexibility and specific electronic structure. These findings imply that controlling exposure of specific crystal planes as well as nano structuring of ZrO2 can effectively enhance redox properties of ZrO2-based catalysts and accordingly their dehydrogenation performance.

a HRTEM image. b Fast Fourier transformed (FFT) image from Fig. 6a. c Inverse FFT images from Fig. 6b. d Gibbs free energy profiles as well as the optimized structures of intermediates and transition states obtained for the reaction of lattice oxygen with hydrogen on ZrO2(-111) and ZrO2(011) at 973 K and ambient pressure. Color scheme: Zr – light blue, O – red, C – grey, H – white.
Discussion
In summary, our study highlights the exceptional performance of bulk ZrO2-based catalysts for ethane dehydrogenation to ethylene in a chemical looping mode, driven largely by their abundance of highly active lattice oxygen species and strongly adsorbed oxygen species, which are ideally suited for alkane dehydrogenation. Using a combination of product analysis with resolution from sub-milliseconds to minutes and in situ/operando spectroscopic characterizations, we have demonstrated that appropriate elemental doping plays a crucial role in enhancing catalyst performance. Specifically, doping contributes to (i) reducing the number of Lewis acid sites and nonselective lattice oxygen species, and (ii) promoting lattice oxygen exchange and transport. These effects are instrumental in minimizing coke formation and mitigating side reactions such as overoxidation. Our study also reveals the dynamic nature of coke deposition and the existence of multiple reaction pathways, further underscoring the critical role that lattice active oxygen species play in determining EDH performance. The coexistence of multiple reaction pathways suggests a complex dehydrogenation mechanism that requires further investigation to optimize catalytic behaviour. Based on these findings, we propose targeted strategies for improving catalyst design, including the careful selection of doping elements and advanced surface engineering techniques to regulate the activity of lattice oxygen species. These insights not only deepen our understanding of ZrO2-based catalysts but also have broader implications for the development of next-generation bulk metal oxide catalysts.
Methods
Catalyst preparation
ZrO2 (Daiichi Kigenso Kagaku Kogyo Co. Ltd., Japan), YZrOx (14 wt% Y2O3, Daiichi Kigenso Kagaku Kogyo Co. Ltd., Japan), LaZrOx (9 wt% La2O3, Daiichi Kigenso Kagaku Kogyo Co. Ltd., Japan) and CeZrOx (17.4 wt% CeO2, Saint-Gobain NorPro, United States) were used as precursor materials. All of them were calcined at 700 °C in air for 4 h with a heating rate of 2 °C min-1 to obtain the final catalysts.
Catalyst characterization
Nitrogen physisorption was performed at 77 K using a Belsorp mini II setup (Bel Japan). The calcined samples were analysed after degassing at 300 °C. The specific surface area (SBET, m2⋅g-1) was obtained from the BET method.
X-ray diffraction (XRD) patterns were recorded on a Panalytical X’Pert Pro diffractometer with Cu Kα radiation (λ = 1.5406 Å, 40 kV, 40 mA) and an X’Celerator RTMS detector.
High-resolution transmission electron microscopy (HRTEM) images were taken on a FEI Tecnai G2 F20 operating at 200 kV.
Hydrogen temperature-programmed reduction (H2-TPR) and oxygen temperature-programmed oxidation (O2-TPO) experiments were conducted using an in-house-developed setup equipped with eight parallel fixed-bed reactors. The outlet gases were monitored by an online mass spectrometer (Pfeiffer Vacuum Omnistar GSD 320). For H2-TPR measurements, 100 mg of fresh catalyst was first pretreated in flowing air at 700 °C for 1 h, followed by purging with Ar for 10 min and then cooling to 50 °C under the same atmosphere. Subsequently, a gas mixture containing 5 vol% H2 in Ar (10 mL·min-1) was introduced into the reactor, and the sample was heated to 900 °C at a ramp rate of 10 °C·min-1. The signals for H2 (m/z = 2), H2O (m/z = 18), and Ar (m/z = 40) were recorded. For O2-TPO, 50 mg of spent catalyst collected after 35 min of time-on-stream testing was used. The sample was heated to 800 °C in a flow of 5 vol% O2 in Ar (20 mL·min-1) at a heating rate of 10 °C·min-1. The mass signals corresponding to Ar (m/z = 40) and CO2 (m/z = 44) were monitored.
Oxygen isotopic exchange experiments were carried out in the same setup used for the TPR and TPO tests. Each sample (100 mg) was calcined in 20 vol%16O2-N2 (12.5 mL·min-1) for 1 h, cooled down to room temperature under the same atmosphere and then purged with Ar (10 mL·min-1) at room temperature for 8 h to remove gas-phase 16O2. Hereafter, the samples were exposed to a flow of 5 vol % 18O2/Ar (10 mL min-1), and heated up to 900 °C with a ramp of 10 °C min-1. For detection of 16O2, 16O18O, and 18O2, the m/z signals of 32, 34, and 36 were, respectively, recorded. Ar was used as an internal standard.
Fourier transform infrared (FTIR) spectroscopy of pyridine adsorption (Py-IR) was conducted to differentiate the types of acid sites on the catalyst samples using a Nicolet 6700 spectrometer. Self-supporting wafers (10 mg) were prepared from the samples and placed in an in situ cell. The wafers were activated by heating at 300 °C for 1 h under vacuum (10-4 mmHg) to remove adsorbed species. After activation, the wafers were exposed to pure pyridine vapor at 30 °C for 30 min. Following pyridine adsorption, the samples were evacuated at 100 °C for 60 min to remove weakly adsorbed pyridine. All spectra were normalized to the weight of the wafer to ensure consistent comparison. The Brønsted and Lewis acid site densities were estimated from the absorption bands in the 1350–1600 cm-1 range of the IR spectra.
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were conducted using a Bruker Invenio-S FT-IR spectrometer, equipped with an MCT detector and a home-made cell with ZnSe windows. Approximately 80 mg of catalyst powder was loosely packed to form a smooth, flat surface above the copper grid of the sample holder within the sample cell. Mixed gases were introduced from top to bottom through the catalyst bed to ensure thorough contact. For the ethane dehydrogenation experiments, the sample was first pretreated at 700 °C with 20% O2/N2 (27 mL/min) for 60 min to activate the surface and remove any adsorbed contaminants. Next, a gas mixture containing 56 vol% C2H6 (balanced with Ar, 27 mL min-1) was directed into the sample cell at 700 °C for 35 min. For the atmosphere influence experiments, the sample was first pretreated at 700 °C with Ar (27 mL min-1) for 10 min to remove any adsorbed contaminants. Subsequently, a gas mixture containing 20 vol% O2 (balanced with Ar, 27 mL min-1) was fed into the cell at 700 °C for 60 min to activate the surface. Subsequently, the catalysts were purged with pure Ar for 10 min before switching to the reaction gas mixture (C2H6/Ar = 56/44 vol%, 27 mL⋅min-1). All IR spectra were collected every 30 s, with each spectrum generated by accumulating 100 scans at a resolution of 4 cm-1.
In situ X-ray photoelectron spectroscopy (XPS) measurements were performed in a laboratory Near Ambient Pressure X-ray photoelectron spectroscopy system (NAP-XPS, SPECS Surface Nano Analysis GmbH, Germany). The setup is equipped with a differentially pumped Phoibos 150 electron energy analyser with a nozzle of 500 µm, a monochromated Al Kα radiation source (E = 1486.6 eV) and a laser heating system for sample heating. The analysis chamber can be filled by five mass flow controllers (Brooks, GF40) with different gases and reaction mixtures up to a total pressure of 10 mbar. Reaction gases and formed products are monitored by a quadrupole mass spectrometer (QMS, MKS e-vision 2) attached to the lens system of the spectrometer (lens 1). The powder samples are pressed on a stainless-steel sample plate using a laboratory press with 5 mm diameter and a load of about 1 t. Temperature is monitored by a thermocouple on the sample plate pressed to the sample surface. The electron binding energies are reported without further referencing. The samples are heated to 700 °C with 10 K/min in 20%O2-80%Ar. The oxidized (exposed to 20%O2-80%Ar at 700 °C for 45 min), purged (exposed to pure Ar at 700 °C for 10 min), spent (exposed to 56%H2-44%Ar at 700 °C for 10, 50, and 100 min), and repurged (exposed to pure Ar at 700 °C for 105 min) catalysts were used for measurements. Each test includes survey, Zr 3 d, O 1 s, La 3 d and C 1 s spectra and takes about 30 min. The measurements are started after the given times in the atmosphere at the same conditions (gas mixture and temperature).
In situ/operando ultraviolet visible (UV-vis) measurements were performed on an in-house developed setup (Supplementary Fig. 1), equipped with an Avantes spectrometer, including an Ava-Light-DH-S-BAL deuterium-halogen light source, a high-temperature reflection UV-vis probe, and a CCD array detector. Specially, the UV-vis probe consisting of six radiating optical fibres and one reading fibre was threaded through the furnace to face the wall of the quartz tube reactor at the position where the catalyst (200 mg) was located. Before collecting UV-vis spectra, the sample was pretreated in a flow of 20 vol% O2 (balance Ar, 27 mL·min-1) at 700 °C for 1 h. After the oxidative treatment, the catalyst was purged with Ar for 15 min followed by treatment in 56 vol% H2 (balance Ar, 27 mL·min-1) or under reaction conditions (56 vol% C2H6 in Ar, 27 mL min-1) for 1 h. Meanwhile, UV-vis spectra were collected in the range from 225 to 1000 nm every 1 min. Note that BaSO4 (99.998%, Aldrich) was used as a white reference.
For analysing the reduction process or coke formation, we defined a relative reflectance (Rrel) as the ratio of the reflectance of sample treated in H2 (Rred) or with coke deposits (Rcoke) to the fully oxidized one (Roxi), as given in Eq. 2. From this relative reflectance, we then calculated the relative Kubelka-Munk function, F(Rrel), according to Eq. 3.
Temporal analysis of products
Ethane dehydrogenation tests were conducted using a temporal analysis of products (TAP-2) reactor, a pulse technique with a time resolution of approximately 100 μs36. Fresh catalyst (80 mg, particle size of 315–710 μm) was packed between two layers of quartz particles (250–355 μm) in the isothermal zone of a custom-made quartz reactor (inner diameter: 6 mm; length: 40 mm). Before the experiments, the catalyst was pre-treated by heating to 700 °C under an Ar flow (4 mL·min-1), followed by oxidation in an O2/Ar mixture (O2: 2 mL·min-1; Ar: 4 mL·min-1) for 1 h. After oxidation, the reactor was flushed with Ar (4 mL·min-1) for 15 min and subsequently evacuated to approximately 10-5 Pa. Single-pulse and sequential-pulse experiments were performed using a C2H6/Ar mixture (1:1 ratio) with a 0.8 s time delay between pulses. The feed mixture was prepared with C2H6 (Linde, 3.5) and Ar (Air Liquide, 5.0) without further purification. The pulse size was set to approximately 1016 molecules per pulse during tests with C2H6.
An online quadrupole mass spectrometer (HAL RC 301, Hiden Analytics) was used to analyse the feed components and reaction products. The following atomic mass units (AMUs) were monitored for mass spectrometric analysis: 44.0 (CO2), 30.0 (C2H6), 29.0 (C2H6), 28.0 (C2H6, C2H4, CO), 27,0 (C2H4), 16.0 (CH4), 2.0 (H2), and 40.0 (Ar). To improve the signal-to-noise ratio, each pulse was repeated 10 times, and the resulting data were averaged. The concentration of feed components and reaction products was determined using the signals from the respective m/z values, normalized to helium or argon, and calculated based on standard fragmentation patterns and sensitivity factors, which account for the different ionization probabilities of individual compounds.
Reaction evaluation
Ethane dehydrogenation tests were conducted at 700 °C and 1.1 bar using a custom-built setup with a continuous-flow fixed-bed quartz tubular reactor (Supplementary Fig. 1), capable of operating in both continuous flow and transient pulse modes. For both modes, 200 mg of the redox catalyst (particle size of 315-710 μm) was used. Prior to the reaction, the catalysts were pretreated by heating to 700 °C in a flow of 20 vol% O2 (balanced with He, 27 mL⋅min-1) for 1 h. Following this, the catalysts were purged with pure He for 10 min before switching to the reaction gas mixture (C2H6/Ar/He = 56/9/35 vol%, 27 mL⋅min-1). The reaction products and feed components were analysed using gas chromatography (GC) and mass spectrometry (MS) methods as detailed in the following sections.
GC method
In continuous flow mode, the feed components and reaction products were quantitatively analysed using an online GC (Agilent 6890) equipped with AL/S (for hydrocarbons), PLOT/Q (for CO2), and Molsieve 5 (for H2, Ar, and CO) columns as well as flame ionization and thermal conductivity detectors. The feed components and reaction products were analysed every 290 s.
Ethane conversion (X(C2H6)) and selectivity to gas-phase products (S(i)) were calculated using the following equations:
where \(\dot{n}\) with “in” and “out” stand for the molar flows of gas phase components at the reactor inlet and outlet, respectively. \({\nu }_{i}\) is the stoichiometric coefficient for product i. Ar was used as internal standard to consider reaction-induced changes in the number of molecules.
MS method
In transient pulse mode, feed gas was injected repeatedly for 1, 5, and 10 min to study the catalytic performance of the catalysts over different time periods (Supplementary Fig. 4). After each pulse, the catalysts were purged with pure He for 10 mins. Following the purge, the catalysts were reoxidized using a pulse of an oxygen mixture (O2/Ar/He = 20/9/71 vol%, 27 mL⋅min-1) for 10 min, and then purged again with pure He for 5 min. This completed one redox cycle.
Reaction products from each pulse were identified and quantified using an online MS (Pfeiffer Vacuum Omnistar, GSD 350). Initially, the following m/z signals were monitored: H2 (2), He (4), CH4 (16, 15), C2H4 (28, 27, 26, 15), CO (28), C2H6 (30, 29, 28, 27, 15), Ar (40, 20), CO2 (44, 28). Subsequently, quantitative analysis was performed by integrating the characteristic peak areas of all reaction products. Notably, the C2H4 signal was corrected by subtracting the contribution of C2H6 from the m/z = 27 peak area, based on the established m/z = 30 to m/z = 27 ratio. Likewise, the CO signal was adjusted by deducting the contributions of C2H6, C2H4, and CO2 from the m/z = 28 peak, and the CH4 signal was corrected by removing the contributions of C2H6 and C2H4 from the m/z = 15 peak. Correction factors were applied to account for fluctuations in the total mole number during the reaction. Calibration factors for each reaction product relative to Ar were determined using calibration mixtures with known concentrations: C2H6 (C2H6/Ar/He = 56/9/35 vol%, 27 mL min-1), C2H4 (C2H4/Ar/He = 25/9/66 vol%, 27 mL min-1), CO (CO/Ar/He = 5/9/86 vol%, 27 mL min-1), CO2 (CO2/Ar/He = 5/9/86 vol%, 27 mL min-1), CO (O2/Ar/He = 20/9/71 vol%, 27 mL min-1) or CH4 (CH4/Ar/He = 5/9/86 vol%, 27 mL min-1). Finally, the concentration of all reaction products at the reactor outlet was calculated by applying these calibration factors to the integrated m/z signal areas. Additionally, coke formation was quantified by integrating the CO2 signal detected during the catalyst regeneration step.
Ethane conversion (X(C2H6)), product selectivity (S(i)), and space-time yield of ethylene (STY, \({{\mbox{Kg}}}_{{{\mbox{C}}}_{2}{{\mbox{H}}}_{4}}\,{{\mbox{Kg}}}_{{\mbox{cat}}}^{-1}\,{{\mbox{h}}}^{-1}\)) calculated using the following equations:
Where \({x}_{{C}_{2}{H}_{6}}\) is the concentration of C2H6 in the outlet gas mixture. xi and νi are the concentrations and the stoichiometric coefficients of reaction product i in the outlet gas mixture. \({\dot{n}}_{{C}_{2}{H}_{4}}\) stands for the molar flow of C2H4 at the reactor outlet, and \({M}_{{{\mbox{C}}}_{2}{{\mbox{H}}}_{6}}\) is the molecule weight of ethylene (28 g mol-1). The carbon balance for each pulse was calculated and found to be approximately 96–99 %.
To ensure measurement accuracy, each pulse was repeated 5 times for the same time period, and the data were averaged.
Computational details
All periodic DFT calculations49,50 were performed by using the Vienna ab initio simulation package (VASP, version 5.4.1) with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional51 and the projector-augmented wave (PAW) method52,53. The DFT-D3 method of Grimme54 was employed in all calculations to treat van der Waals interactions. An energy cutoff of 400 eV was applied. Geometry optimization was converged until the forces acting on the atoms were lower than 0.02 eV/Å. The energy threshold defining self-consistency of the electron density was set to 10-4 eV. Gaussian smearing with a width of 0.05 eV was used. The Climbing Image Nudged Elastic Band method55 with eight images and the Improved Dimer method56 were applied for finding transition states. Harmonic frequencies of adsorbates and transition states were calculated, and zero-point energy (ZPE) corrections resulted from the frequency analysis were included in the calculated energies. In the case of artificial (small) imaginary frequencies that persisted in some calculations, they were rounded to 50 cm-1 (real).
Since XRD analysis (Supplementary Fig. 27) proved the existence of monoclinic ZrO2, we created a corresponding ZrO2 model. A 7 × 7 × 7 Monkhorst-Pack k point grid was used for sampling the Brillouin zone57. The calculated lattice parameters of the unit cell are a = 5.141 Å, b = 5.254 Å, c = 5.288 Å, α = γ = 90°, β = 99.36°.
The stoichiometric p (2 × 2) supercells of the ZrO2 (-111) and ZrO2 (011) surfaces were created from the optimized primitive cell of ZrO2 using Materials Studio software. The slab model contained three layers with the bottom layer fixed at its bulk positions. A vacuum space of 15 Å was inserted between the slab and its periodic replicas. The Brillouin zone was sampled with a 2 × 2 × 1 Monkhorst-Pack mesh. Zero-point energies and the Gibbs free energies for the reaction temperature of 973 K and ambient pressure were calculated using the VASPKIT program58.
Data availability
All the data generated in this study are provided in the Supplementary Information and Source Data file. Data are available from the corresponding authors upon request. Source data are provided with this paper.
References
Sattler, J. J. H. B., Ruiz-Martinez, J., Santillan-Jimenez, E. & Weckhuysen, B. M. Catalytic dehydrogenation of light alkanes on metals and metal oxides. Chem. Rev. 114, 10613–10653 (2014).
Li, X., Pei, C. & Gong, J. Shale gas revolution: catalytic conversion of C1-C3 light alkanes to value-added chemicals. Chem 133, 1755–1801 (2021).
Najari, S. et al. Oxidative dehydrogenation of ethane: catalytic and mechanistic aspects and future trends. Chem. Soc. Rev. 50, 4564–4605 (2021).
Ryoo, R. et al. Rare-earth-platinum alloy nanoparticles in mesoporous zeolite for catalysis. Nature 585, 221–224 (2020).
Motagamwala, A. H. et al. Stable and selective catalysts for propane dehydrogenation operating at thermodynamic limit. Science 373, 217–222 (2021).
Zhao, D. et al. In situ formation of ZnOx species for efficient propane dehydrogenation. Nature 599, 234–238 (2021).
Hannagan, R. T. et al. First-principles design of a single-atom-alloy propane dehydrogenation catalyst. Science 372, 1444–1447 (2021).
Chen, S. et al. Defective TiOx overlayers catalyze propane dehydrogenation promoted by base metals. Science 385, 295–300 (2024).
Zeng, L. et al. Stable anchoring of single rhodium atoms by indium in zeolite alkane dehydrogenation catalysts. Science 383, 998–1004 (2024).
Liu, L., Wang, L. & Xiao, F. Zeolite catalysts for non-oxidative ethane dehydrogenation to ethylene. EES Catal. 2, 923–931 (2024).
Gao, Y. et al. A molten carbonate shell modified perovskite redox catalyst for anaerobic oxidative dehydrogenation of ethane. Sci. Adv. 6, eaaz9339 (2020).
Zeng, L. et al. Metal oxide redox chemistry for chemical looping processes. Nat. Rev. Chem. 2, 349–364 (2018).
Neal, L. M., Yusuf, S., Sofranko, J. A. & Li, F. Oxidative dehydrogenation of ethane: a chemical looping approach. Energy Technol. 4, 1200–1208 (2016).
Gao, Y., Neal, L. M. & Li, F. Li-promoted LaxSr2-xFeO4-δ core-shell redox catalysts for oxidative dehydrogenation of ethane under a cyclic redox scheme. ACS Catal. 6, 7293–7302 (2016).
Zhu, X. et al. Chemical looping beyond combustion - a perspective. Energy Environ. Sci. 13, 772–804 (2020).
Fung, V., Tao, F. & Jiang, D. Understanding oxidative dehydrogenation of ethane on Co3O4 nanorods from density functional theory. Catal. Sci. Technol. 6, 6861–6869 (2016).
Dai, G. L. et al. Periodic DFT study of the deep oxidation in the oxidative dehydrogenation of ethane over V2O5(001). Chin. J. Catal. 34, 906–910 (2013).
Xiong, C. Y. et al. Structure-performance relationships for propane dehydrogenation over aluminum supported vanadium oxide. ACS Catal. 9, 5816–5827 (2019).
Zhou, Y. L. et al. Sulfate-modified NiAl mixed oxides as effective C-H bond-breaking agents for the sole production of ethylene from ethane. ACS Catal. 10, 7619–7629 (2020).
Wang, J., Song, Y. H., Liu, Z. T. & Liu, Z. W. Active and selective nature of supported CrOx for the oxidative dehydrogenation of propane with carbon dioxide. Appl. Catal., B 297, 120400 (2021).
Liu, L. et al. Rivet of cobalt in siliceous zeolite for catalytic ethane dehydrogenation. Chem 9, 637–649 (2023).
Chen, S. et al. Coverage-dependent behaviors of vanadium oxides for chemical looping oxidative dehydrogenation. Angew. Chem. Int. Ed. 59, 22072–22079 (2020).
Chen, S. et al. Modulating lattice oxygen in dual-functional Mo-V-O mixed oxides for chemical looping oxidative dehydrogenation. J. Am. Chem. Soc. 141, 18653–18657 (2019).
Qin, X. et al. Domino catalysis for selective dehydrogenation of ethane with shifted thermodynamic equilibrium. Joule 7, 753–764 (2023).
Sattler, J. J. H. B., Mens, A. M. & Weckhuysen, B. M. Real-time quantitative operando Raman spectroscopy of a CrOx/Al2O3 propane dehydrogenation catalyst in a pilot-scale reactor. ChemCatChem 6, 3139–3145 (2014).
Bare, S. R. et al. Characterization of coke on a Pt-Re/γ-Al2O3 Re-forming catalyst: experimental and theoretical study. ACS Catal. 7, 1452–1461 (2017).
Pham, H. N., Sattler, J. J. H. B., Weckhuysen, B. M. & Datye, A. K. Role of Sn in the regeneration of Pt/γ-Al2O3 light alkane dehydrogenation catalysts. ACS Catal. 6, 2257–2264 (2016).
Wang, H. Z. et al. Coke formation on Pt-Sn/Al2O3 catalyst for propane dehydrogenation. Ind. Eng. Chem. Res. 57, 8647–8654 (2018).
Otroshchenko, T. et al. ZrO2-based alternatives to conventional propane dehydrogenation catalysts: active sites, design and performance. Angew. Chem. Int. Ed. 54, 15880–15883 (2015).
Otroshchenko, T. et al. ZrO2-based unconventional catalysts for non-oxidative propane dehydrogenation: factors determining catalytic activity. J. Catal. 348, 282–290 (2017).
Zhang, Y. et al. Control of coordinatively unsaturated Zr sites in ZrO2 for efficient C-H bond activation. Nat. Commun. 9, 3794 (2018).
Zhang, Y. et al. The effect of phase composition and crystallite size on activity and selectivity of ZrO2 in non-oxidative propane dehydrogenation. J. Catal. 371, 313–324 (2019).
Han, S. et al. Elucidating the nature of active sites and fundamentals for their creation in Zn-containing ZrO2-based catalysts for nonoxidative propane dehydrogenation. ACS Catal. 10, 8933–8949 (2020).
Zhang, Q. et al. The enhancing effect of Co2+ on propane non-oxidative dehydrogenation over supported Co/ZrO2 catalysts. J. Catal. 432, 115440 (2024).
Eder, D. & Kramer, R. The stoichiometry of hydrogen reduced zirconia and its influence on catalytic activity, Part 1: volumetric and conductivity studies. Phys. Chem. Chem. Phys. 4, 795–801 (2002).
Imparato, C. et al. Unraveling the charge state of oxygen vacancies in ZrO2-x on the basis of synergistic computational and experimental evidence. J. Phys. Chem. C 123, 11581–11590 (2019).
Zimmermann, H. & Walzlheinz, R. Ethylene. In Ullmann’s Encyclopedia of Industrial Chemistry 7th edn 465–529 (Wiley, 2009).
Morgan, K. et al. Forty years of temporal analysis of products. Catal. Sci. Technol. 7, 2416–2439 (2017).
Nijhuis, T. A., Tinnemans, S. J., Visser, T. & Weckhuysen, B. M. Towards real-time spectroscopic process control for the dehydrogenation of propane over supported chromium oxide catalysts. Chem. Eng. Sci. 59, 5487–5492 (2004).
Gionco, M. C. et al. Paramagnetic defects in polycrystalline zirconia: an EPR and DFT study. Chem. Mater. 25, 2243–2253 (2013).
Nenning, A. et al. Ambient pressure XPS study of mixed conducting perovskite-type SOFC cathode and anode materials under well-defined electrochemical polarization. J. Phys. Chem. C 120, 1461–1471 (2016).
Airaksinen, S. M. K., Bañares, M. A. & Krause, A. O. I. In situ characterisation of carbon-containing species formed on chromia/alumina during propane dehydrogenation. J. Catal. 230, 507–513 (2005).
Watanabe, K. et al. Catalytic selective ethane dehydrogenation at low-temperature with low coke formation. RSC Adv. 12, 24465–24470 (2022).
Carlos, L. et al. Characterization of residual coke during burning. Ind. Eng. Chem. Res. 31, 1017–1021 (1992).
Srihiranpullop, S. & Praserthdam, P. A new approach of coke characterization on metal and support for Pt/Al2O3 by combination of Al2O3 and Pt/SiO2. Catal. Today 93-95, 723–727 (2004).
Jaegers, N. R. et al. Heterolytic C-H activation routes in catalytic dehydrogenation of light alkanes on Lewis acid-base pairs at ZrO2 surfaces. J. Am. Chem. Soc. 146, 25710–25726 (2024).
Puigdollers, A. R., Illas, F. & Pacchioni, G. Reduction of hydrogenated ZrO2 nanoparticles by water desorption. ACS Omega 2, 3878–3885 (2017).
Pacchioni, G. Role of nanostructuring on the properties of oxide materials: the case of zirconia nanoparticles. Eur. J. Inorg. Chem. 2019, 751–761 (2019).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Heyden, A., Bell, A. T. & Keil, F. J. Efficient methods for finding transition states in chemical reactions: comparison of improved dimer method and partitioned rational function optimization method. J. Chem. Phys. 123, 224101 (2005).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Wang, V. et al. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Acknowledgements
Financial support by the State of Mecklenburg-Vorpommern is gratefully acknowledged. This work was also financially supported by the China Scholarship Council (202106365006) and the National Natural Science Foundation of China (22176216). We thank Dr. Henrik Lund for performing XRD analysis. The authors thank Daiichi Kigenso Kagaku Kogyo Co., Ltd. for providing ZrO2, YZrOx and LaZrOx and Saint-Gobain NorPro for providing CeZrOx.
Author information
Authors and Affiliations
Contributions
E.V.K. initiated and led the whole project. T.Z. prepared all catalysts and carried out characterization and catalytic tests as well as analysed them. T.O. performed the DFT calculations. V.A.K. carried out the TAP tests and analysed them. S.B. performed NAP-XPS tests and analysed them. T.Z. and E.V.K. wrote the first draft of this article with the input from all other co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, T., Otroshchenko, T., Kondratenko, V.A. et al. Regulation of lattice oxygen reactivity of ZrO2 to promote efficient chemical looping oxidative dehydrogenation of ethane. Nat Commun 16, 8556 (2025). https://doi.org/10.1038/s41467-025-64246-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64246-9
This article is cited by
-
Photothermal coupling effect boosts solar-to-fuel conversion of CO2 via ZnO/CeZrO2 heterostructure catalysts in a concentrated solar reactor
Research on Chemical Intermediates (2026)
