
Abstract
Hormone receptor-positive (HR + )/HER2−negative (HER2 − ) breast cancer is the most common subtype, with biomarker-driven therapies improving outcomes. Circulating tumor DNA (ctDNA) analysis enables minimally invasive assessment of somatic alterations to guide therapy. However, assay choice impacts clinical utility, and access remains inconsistent. This study compares single-gene and panel-based sequencing for assessing PIK3CA mutations and broader genomic profiling. We conducted a prospective, multicenter study analyzing 161 plasma samples from 146 patients before initiating a new line of palliative therapy using the SiMSen-Seq (SSS) assay for PIK3CA hotspot mutations, the AVENIO ctDNA Expanded assay (77 genes) and mFAST-SeqS for tumor fraction estimation. High concordance (92.6%) was observed between SSS and AVENIO for PIK3CA mutations. AVENIO identified additional actionable alterations, including ESR1 (17.5%) and PI3K pathway alterations (40.6%), and together with tumor fraction estimation, improved interpretation of negative liquid biopsy findings. These findings support broader ctDNA profiling in clinical practice while highlighting accessibility challenges.
Similar content being viewed by others


Introduction
Hormone receptor-positive (HR + )/HER2−negative (HER2−) breast cancer is the most common subtype, accounting for approximately 70% of all breast cancer cases1. Endocrine therapy remains the backbone of treatment concepts for this subtype, and the addition of cyclin-dependent kinase 4/6 inhibitors (CDK4/6i) has significantly improved patient outcomes2,3,4,5. Although treatment resistance inevitably arises, recent advances in next-generation biomarker-driven therapies offer new opportunities to address this clinical challenge. Key developments include drugs targeting PI3K-altered pathways, such as alpelisib6, inavolisib7, and capivasertib8, which target PIK3CA, AKT or PTEN alterations. Additionally, oral selective estrogen receptor degraders (SERDs), exemplified by elacestrant9, have improved efficacy in patients with ESR1 mutations. These emerging agents aim to overcome drug resistance, prolong disease control, and refine precision medicine strategies for HR + /HER2− breast cancer.
Liquid biopsy has transformed therapeutic guidance for metastatic breast cancer by facilitating minimally invasive, dynamic, and repeatable analysis of circulating tumor DNA (ctDNA)10,11,12,13. In the metastatic setting, ctDNA provides distinct advantages over tissue biopsies, as it more effectively captures spatial and temporal heterogeneity, detects emerging resistance mechanisms, and simultaneously reduces the burden of invasive tumor biopsies for patients. It may also be the only source of tumor material when tissue is inaccessible. In precision oncology, ctDNA testing guides treatment selection by identifying resistance mechanisms and informing therapeutic decisions14,15,16,17.
The choice of ctDNA assay remains critical, as assay characteristics such as sensitivity, specificity, and breadth can affect clinical utility. Our previous research demonstrated that targeted approaches, exemplified by SiMSen-Seq, effectively detected predefined mutations, including PIK3CA, with high sensitivity and specificity18. Although various commercial platforms are available, particularly within the United States, global access remains inconsistent, and there is no international consensus on the optimal selection of platforms. A position paper by the American Society of Clinical Oncology highlighted the importance of assay standardization, technical harmonization, and comparative studies to ensure robust and reliable ctDNA testing in clinical trials and routine practice19. The magnitude of the complexity of predictive genomic alterations is anticipated to grow as new treatments are developed and require ongoing evaluation and cross-comparison of testing methodologies, considering both analytical performance and practical implementation across diverse healthcare settings.
In this study, we conducted a cross-comparison of the highly sensitive SIMSen-Seq assay with the broader AVENIO ctDNA Expanded panel for detecting hotspot PIK3CA mutations in advanced HR + /HER2− breast cancer. We evaluated assay concordance and sensitivity using 161 prospectively collected ctDNA samples obtained before initiating first-line or subsequent palliative treatment. Furthermore, we investigated the added value of broader panel sequencing, particularly its ability to identify additional clinically relevant mutations and improve the interpretation of samples with low tumor fractions.
Results
Patient characteristics and plasma samples
One hundred forty-six patients with HR + /HER2− advanced breast cancer were included in this analysis. Figure 1 illustrates the sample selection process, and Table 1 summarizes the clinical and pathological characteristics of the total study cohort. For patients with multiple blood draws, data from the first blood draw were used for the description.

A total of 161 plasma samples from 146 patients were analyzed using three distinct ctDNA assays. Thirteen samples were excluded from the comparison analysis: Ten due to failure of the SSS assay, and three due to insufficient cfDNA availability for AVENIO testing.
The median age of patients at the time of blood sample collection was 64.9 years (25th–75th percentile: 55.3–74.8 years), and most patients were female (97.3%). The predominant histological subtype was breast cancer of no special type (NST, 75.3%), followed by invasive lobular carcinoma (ILC, 15.8%) and mixed types (5.5%). At the time of diagnosis of advanced disease, 41.1% of the patients presented with de novo metastatic disease, 57.5% had recurrent metastatic disease, and 1.4% had locally advanced disease. The most common metastatic sites at enrollment were non-liver visceral locations (56.2%), followed by liver metastases (24.7%) and bone-only disease (19.2%). Most patients (90.4%) had one to three metastatic sites.
Aneuploidy-based estimation of tumor fraction using mFAST-SeqS in 161 samples from 146 patients revealed a median z-score of 2.47 (25–75th percentile: 1.27–5.53), indicating low overall tumor levels in our cohort, as z-scores below three fall outside the quantitative range and are not considered informative. Using our previously established cut-off of ≥ 3, 64/161 samples (39.8%) had elevated z-scores, indicating the presence of ctDNA20. Using a linear mixed-effects model, which accounted for repeated measurements, we observed significantly higher z-scores in 2 L samples compared to 1 L samples (adjusted mean increase of 10.1, 95%CI: 1.66–18.43, p = 0.019). Given the non-normal distribution of z-scores, we performed a sensitivity analysis using log10-transformed z-scores. The results remained significant, with z-scores in 2 L samples being 1.57 times higher than in 1 L samples (95% CI: 1.09–2.26; p = 0.015), corresponding to a 57% increase (Supplementary Fig. 1).
Concordance between AVENIO and SSS for PIK3CA mutation detection in liquid biopsy samples
161 blood samples were analyzed using both the SSS and AVENIO assays. Of these, 13 samples (8.1%) were excluded because of insufficient coverage in the SSS assay (n = 10) or insufficient amount of cfDNA for the AVENIO assay (n = 3). Consequently, 148 samples were included in the head-to-head comparison. Since our SSS assay was designed to target only 11 PIK3CA mutations from the SOLAR-1 trial, only the PIK3CA mutations covered by both assays were included in the concordance analysis. Using a mixed-effects logistic regression model to account for repeated measurements, the estimated probability of detecting PIK3CA variants was 38.4% (95% CI: 29.81–46.95%) with the SSS assay and 36.85% (05% CI: 26.88–46.81%) with the AVENIO assay. Overall, the SSS assay identified 64 PIK3CA variants, whereas the AVENIO assay identified 62 variants. Both assays found most alterations in three known hotspots, including H1047, E545, and E542.
The AVENIO assay demonstrated high concordance with SSS (Supplementary Table 1) in detecting PIK3CA mutations. The estimated adjusted overall agreement, calculated using a mixed-effects model to account for repeated measurements, was 92.6% (137/148; 95% CI, 88.3%–96.8%). The estimated adjusted positive percent agreement (PPA) was 88.7% (52/58; 95% CI, 79.4%–98.1%), while the adjusted negative percent agreement (NPA) was 94.3% (85/90; 95% CI, 89.4%–99.1%). The details of the discordant results are summarized in Table 2. PIK3CA mutations were exclusively identified using SSS in seven samples (4.7%), whereas AVENIO uniquely detected mutations in five samples (3.4%). Notably, in four of the five samples in which SSS did not detect a PIK3CA mutation, the VAF was below the limit of detection for SSS. These samples also had low tumor fractions, as indicated by a z-score <3 and an hVAF value < 1%. Conversely, of the seven samples in which AVENIO did not detect a PIK3CA mutation, five had high tumor fractions (z-scores ≥3 or hVAF ≥1%). Finally, Bland-Altman analysis demonstrated that the AVENIO assay is highly accurate in estimating PIK3CA VAFs with a mean difference of 0.62% (Fig. 2A). Linear mixed-effects modeling further confirmed a strong linear relationship between VAFs measured by the two assays (coefficient 0.94, 95% CI: 0.89–0.99; p < 0.001), highlighting a consistent agreement between the two assays in quantifying PIK3CA mutations (Fig. 2B).

A Bland-Altman plot showing the agreement between PIK3CA VAFs. B Linear regression analysis with a mixed-effects model demonstrates a strong correlation between the two methods (coefficient: 0.94, 95% CI: 0.89–0.99; p < 0.001). C Classification of PIK3CAwt liquid biopsy samples detected by SSS and AVENIO. Bar plots show the proportion of PIK3CAwt samples classified as true negative (green) and non-informative (grey) using different criteria. hVAF# includes only pathogenic or likely pathogenic variants, and hVAF* additionally includes VUS.
Validation of negative liquid biopsy results
Interpreting negative liquid biopsy results is a common challenge in clinical practice, especially when differentiating true negative results from samples lacking sufficient tumor-derived material for accurate analysis. To address this, we first used our previously established z-score ≥ 3, corresponding to a tumor fraction of approximately 5–10%, as a criterion to classify samples as informative due to elevated tumor content20,21. Among the 90 samples classified as PIK3CA wild-type by SSS, 34 (37.8%) had a z-score ≥ 3, indicating sufficient tumor content for reliable mutation detection.
However, mFAST-SeqS z-scores are only informative above 3 (corresponding to a 5% tumor fraction). Moreover, tumor fraction estimates may underestimate ctDNA levels in samples with minimal genomic imbalances or low tumor aneuploidy, potentially excluding cases with detectable but low tumor fractions. To address this limitation and fully leverage the genomic data from AVENIO, we utilized the hVAF of all selected somatic pathogenic or likely pathogenic variants as a more sensitive proxy for tumor fraction, enhancing detection sensitivity. A sample was classified as ctDNA informative if it harbored at least one variant with an hVAF of ≥1%. Among the 148 samples analyzed, 89 had a z-score below 3, and of these, 40 samples (44.9%) were classified as ctDNA informative based on the hVAF criterion. This finding highlights the added value of more comprehensive mutation analysis for detecting and quantifying ctDNA based on additional mutations.
Building on these findings, we explored a combined approach that integrates both metrics to refine the classification of non-informative samples. Using a combined classification strategy of either a z-score ≥3 or an hVAF ≥1%, 49 of the 91 PIK3CA wild-type samples (52.7%) were classified as true negative results. Figure 2C illustrates the proportion of PIK3CA wild-type samples classified as informative under each criterion, highlighting the utility of combining the tumor fraction and hVAF metrics to refine the classification of true negative liquid biopsy results.
Collectively, our results demonstrate that combining tumor fraction estimates with hVAF of panel sequencing improves the reliability of negative liquid biopsy findings and provides critical support for guiding therapeutic decision-making.
Somatic and actionable genomic alterations using AVENIO assay
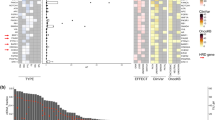
In this analysis, we included all somatic pathogenic and likely pathogenic mutations detected using the AVENIO assay in 143 patients. For the 15 patients with two blood draws, only the first time point of blood draw was considered for this analysis. Figure 3A shows an overview of all the somatic mutations in an oncoprint. Overall, pathogenic and/or likely pathogenic mutations were detected in 38/77 genes in 95/143 (66.4%) patients, with a median of 2 variants (range 1–11) per sample and a median VAF of 1.6% (range 0.1–74.4). The most frequently mutated genes in this cohort were PIK3CA (37.1%), TP53 (23.1%), ESR1 (17.4%), SMAD4 (5.6%), ERBB2 (4.2%), and RB1 (4.2%).

A Oncoprint depicting somatic pathogenic/likely pathogenic alterations detected in 95/143 HR + /HER2− advanced breast cancer patients. The upper bar graph illustrates the number of alterations per sample, while the right bar graph shows the frequency of alterations for each gene. B Comparison of mutation frequencies between 1 L (N = 62) and 2 L (N = 25) liquid biopsy samples. Bar plots display the percentage of cases with mutations in key genes, stratified by mutation type: missense mutations (green), multi-hit mutations (yellow), nonsense mutations (blue), and splice-site mutations (red). ESR1, AKT1, and RB1 mutations were numerically more frequent in 2 L samples, while the frequencies of PIK3CA, TP53, and SMAD4 mutations were similar across both groups.
Among the 53 samples harboring PIK3CA mutations, a total of 70 variants were detected using the AVENIO assay, with a median VAF of 4.3% (range 0.2-46.4). Of these, 62 (88.6%) variants are included among the mutations eligible in the INAVO120 trial7. Among the 95 patients in 1 L treatment, 35 had PIK3CA variants eligible for treatment with inavolisib. Notably, 13 variants (18.6%) were not covered by the SSS assay (Supplementary Fig. 2), but six were included in the INAVO-120 trial. Furthermore, out of the 53 patients with PIK3CA mutations eligible for the INAVO-120 trial, five (9.4%) would not have been identified using the SSS assay alone, highlighting the added clinical value of the AVENIO assay in capturing actionable mutations. Only 6/70 (8.6%) variants were subclonal (VAF less than 20% compared to the hVAF), aligning with previous reports that identified PIK3CA mutations as predominantly truncal events. When analyzing potential patients for inavolisib treatment based on the INAVO120 trial criteria, ctDNA profiling in our cohort revealed PIK3CA mutations in 8/95 (8.4%) 1 L patients who had either completed five years of adjuvant endocrine therapy or experienced disease progression within one year of treatment completion. Notably, 20.8% (11/53) of PIK3CA mutated samples were polyclonal, harboring multiple concomitant variants. In these samples, canonical SOLAR-1 PIK3CA mutations, including E541K, E545K, and H1047R, consistently demonstrated higher VAFs compared to other PIK3CA mutations, suggesting a convergent evolution of multiple clones (Supplementary Table 2). Among the 25 ESR1-mutated samples, a total of 37 variants were identified, with a median VAF of 1.1% (range 0.1-29.2). The most common variants were D538G (32.4%, 12/37) and Y537S (29.7%, 11/37). Notably, 32.0% (8/25) of ESR1-mutated samples exhibited polyclonality, harboring multiple concomitant ESR1 variants. Additionally, 32.4% (12/37) of ESR1 variants were subclonal, consistent with previous reports suggesting that ESR1 mutations emerge mainly as subdominant events during tumor evolution under treatment with aromatase inhibitors, contributing to endocrine resistance. Finally, we examined the frequency of mutations within the PI3K pathway, defined as the presence of one or more mutations in PIK3CA, AKT1, or PTEN genes, which were detected in 40.6% (58/143) of samples. As demonstrated in the previous section, using a combined approach of z-scores and hVAF, including variants of unknown significance (VUS), increased the number of informative results for both PIK3CA and ESR1 wild-type samples (Supplementary Fig. 3), highlighting the value of panel NGS ctDNA profiling.
Co-mutation analysis of all 143 samples revealed significant co-occurrence patterns, including PIK3CA and ESR1 in 14 samples (9.8%, unadjusted p-value = 0.040, Fisher’s exact test), PIK3CA and RB1 in 6 samples (4.2%, unadjusted p-value = 0.002, Fisher’s exact test), PIK3CA and ERBB2 in 4 samples (2.8%, unadjusted p-value = 0.026, Fisher’s exact test), and ESR1 and SMAD4 in 4 samples (2.8%, unadjusted p-value = 0.032, Fisher’s exact test).
When comparing 1 L (N = 95) and 2 L samples (N = 37) (Fig. 3B), mutations in ESR1 were statistically more frequent in 2 L samples (Supplementary Fig. 4A, Fisher’s exact test, p-value = 0.002). Notably, among patients with ESR1, TP53, and PIK3CA mutations (PIK3CA in Supplementary Fig. 4B), multiple concomitant mutations were observed more frequently in 2 L samples. Mutations in AKT1 and RB1 were numerically but not statistically more frequent in 2 L samples (Fisher’s exact test for both, p = 0.134). Furthermore, TP53, PIK3CA, and SMAD4 mutation frequencies were similar between the two groups (Fisher’s exact test, TP53 p = 0.999, PIK3CA p = 0.999, SMAD p = 0.673). Finally, the frequencies of PI3K pathway mutations were similar between 1 L (40.0%) and 2 L (43.2%) samples (Supplementary Fig. 1C, Fisher’s exact test, p = 0.844).
To investigate longitudinal changes in ctDNA profiles, we analyzed matched plasma samples from 14 out of 15 patients, with 1 L samples collected at baseline and 2 L samples collected at the end of treatment (EOT) before starting 2 L therapy. These paired samples enabled the assessment of changes in somatic mutations and ctDNA dynamics throughout 1 L CDK4/6i treatment. Supplementary Fig. 5 illustrates the changes in VAFs and/or z-scores for 14 patients, revealing key patterns such as the emergence of new resistant mutations, the persistence of dominant clones, and clonal heterogeneity of molecular response. A representative clinical vignette is shown in Fig. 4, highlighting longitudinal molecular dynamics in the context of clinical

The patient started first-line therapy with fulvestrant and abemaciclib. Baseline plasma analysis (prior to treatment start) showed a z-score <3, with detectable mutations in IDH2 (3.5% VAF) and TERT (0.3% VAF). After approximately 44 months of therapy, a second follow-up sample revealed a marked increase in IDH2 VAF (from 3.5% to 24.5%), clearance of the TERT mutation, and emergence of a new mutation in AKT1 (8.0% VAF). The z-score at follow-up remained below 3. Concurrent CT imaging demonstrated disease progression, with stable bone metastases and newly identified liver metastases. Created in Biorender.com.
Discussion
Liquid biopsies have reshaped clinical practice for advanced breast cancer, enabling the detection of PIK3CA mutations that guide treatment targeting PI3K-AKT pathway6,7,8,22 and ESR1 mutations that inform SERD treatment9,23,24 with specific approval for the oral SERD elacestrant for liquid biopsy-based ESR1 mutation detection. This minimally invasive testing approach has advanced precision oncology in routine clinical practice by enabling real-time monitoring of tumor evolution, revealing resistance mechanisms, and facilitating biomarker-driven targeted treatment decisions12,25. Despite the growing utility of liquid biopsy, access to validated testing platforms remains a persistent global challenge. In the United States, FDA-approved assays are widely available and covered by insurance, facilitating their integration into clinical workflows. However, in many regions, including Europe, access is inconsistent due to limited cost coverage and availability of commercial platforms, necessitating centralized testing at academic centers.
To translate technical advances in liquid biopsy testing into benefits for patients, cross-comparing different testing strategies in real-world clinical settings is crucial. Previously, we demonstrated that SSS is a highly sensitive and specific method for detecting hotspot PIK3CA mutations18. With the advancements in next-generation sequencing (NGS) panel methods, testing capabilities have expanded to include broader genomic profiling, capturing additional actionable targets that influence decisions for approved treatments, such as ESR1, PTEN, and AKT1 as well as other clinically relevant mutations such as somatic BRCA1/2 or ERBB2, which may inform further targeted therapeutic strategies26. Recognizing the limitations of single-gene approaches like SSS in addressing these broader clinical needs, we sought to evaluate whether a panel-based assay, such as the AVENIO ctDNA Expanded Kit, could match the sensitivity of SSS for PIK3CA detection while providing additional insights into the evolving molecular landscape of HR + /HER2− breast cancer.
Our findings demonstrate a high concordance (92.6%) between SSS and the AVENIO assay for detecting PIK3CA mutations with strongly correlated VAFs. The broader genomic coverage of AVENIO assay uncovered additional PIK3CA mutations in 19% of samples. Moreover, the INAVO120 trial, a phase III randomized study evaluating inavolisib, a PI3K inhibitor, in combination with palbociclib and fulvestrant for a broader spectrum of PIK3CA mutations in higher risk 1 L populations, demonstrated improved PFS and OS in patients with HR + /HER2− advanced breast cancer who had progressed on endocrine therapy7. In our cohort, 37% of first-line patients harbored PIK3CA variants eligible for inavolisib treatment, highlighting the importance of expanded genomic profiling to identify actionable mutations and improve therapeutic opportunities for patients. Overall, only 8% of our first-line patients met the criteria for initiating inavolisib in the first-line setting. Although the sample size is small, it reflects a representative clinical population, with over 35% of patients having de novo metastatic disease.
Beyond PIK3CA, the AVENIO assay identified additional clinically actionable alterations, including ESR1 mutations in 17% of patients. Our findings are consistent with a prior large-scale study, which reported that 38–55% of patients harbored PI3K/AKT pathway alterations at the start of first-line therapy and observed an increase of ESR1 mutations over successive treatment lines27. Furthermore, we found that polyclonal mutations in both ESR1 and PIK3CA were more frequent in 2 L compared to 1 L samples, consistent with therapy-driven clonal selection and evolution15,28. Other resistance-associated alterations were observed, including SMAD4 (6%), RB1 mutations (4%), and ERBB2 (4%), albeit at lower frequencies. These alterations have been previously described as contributors to therapy resistance in HR + /HER2− breast cancer29,30,31,32. Together, our data emphasize the value of broader sequencing approaches such as AVENIO assay, not only for detecting actionable mutations but also for identifying resistance mechanisms and capturing tumor heterogeneity.
Our study highlights the pressing need for expanded multitarget testing. Our evaluation of the AVENIO platform focused on its clinical utility in first- and second-line settings for approved therapies in HR + /HER2− metastatic breast cancer. However, with the rapid advancement of clinical trials and the increasing number of approved targeted therapies, the platform may already face challenges in maintaining its relevance and clinical applicability. Recent trials, such as the EvoPAR-BR01 study (clinicaltrials.gov/study/NCT06380751), are now incorporating additional hotspot mutations and expanding their scope to include genes like BRCA1/2 and PALB2 to identify homologous recombination repair deficiency, evaluated for first-line treatment.
Negative liquid biopsy results present a persistent challenge, reflecting either the true absence of alterations or insufficient ctDNA for reliable analysis. Our previous work18 used aneuploidy-based tumor fraction estimates combined with SSS to improve confidence in interpreting negative results. Building on this, we demonstrate here that comprehensive genomic profiling with the AVENIO assay further enhances the reliability of negative liquid biopsy classifications. Our approach aligns with recent findings33, where samples with a ctDNA tumor fraction ≥1% were shown to have significantly improved concordance between liquid and tissue-based profiling, reducing the likelihood of false negatives. Together, these results emphasize the importance of integrating tumor fraction metrics with broader sequencing approaches to distinguish true negatives from cases requiring follow-up testing, ultimately improving clinical decision-making.
Our study has several limitations that should be considered when interpreting the findings. First, the AVENIO assay includes only 77 genes and excludes several key genes or gene regions that are potential therapeutic targets or implicated in therapy resistance. For example, the AVENIO assay focuses on detecting activating mutations in oncogenes and does not enable Indel calling in key tumor-suppressor genes BRCA1/2. Additionally, amplification of key genes associated with resistance to targeted therapies, such as CCND1, NF1, or FGFR114,34, are not captured, potentially underestimating the complexity of resistance mechanisms in HR + /HER2− breast cancer25,31. Second, the AVENIO assay only calls for copy number alterations (CNAs) in EGFR, ERBB2, and MET but lacks calling for other important genes that are increasingly recognized as important drivers of resistance and progression in breast cancer35. Third, our analysis did not consider clonal hematopoiesis of indeterminate potential (CHIP) mutations, which could introduce a bias, as CHIP mutations can mimic tumor-derived alterations in cfDNA36. Finally, the majority of 1 L and 2 L samples in our study were not matched from the same patients, limiting our ability to comprehensively evaluate longitudinal changes in ctDNA profiles and therapy-induced clonal evolution32. Addressing these challenges is crucial to ensure timely testing of metastatic HR + /HER2− breast cancer patients early in the evolution of their metastatic disease.
In conclusion, our study demonstrates high concordance between SSS and AVENIO for PIK3CA mutation detection while highlighting the added value of broader genomic coverage in identifying actionable and resistance-associated mutations. By integrating tumor fraction metrics with panel sequencing, we improved the interpretation of negative liquid biopsy results, contributing to more reliable clinical decision-making. These findings emphasize the importance of comprehensive ctDNA profiling for guiding therapy and monitoring tumor evolution in advanced HR + /HER2− breast cancer. Future research should prioritize expanded genomic targets, address assay limitations, and improve accessibility to translate these advances into benefits for patients globally.
Methods
Study design
This prospective, observational, multicenter study enrolled patients with HR + /HER2− advanced breast cancer before initiating first-line or subsequent palliative treatment. Hormone receptor-positive status was defined as estrogen-receptor expression with or without progesterone-receptor expression. HER2−negative status was defined as either 0 or 1+ intensity on immunohistochemical (IHC) testing and 2+ intensity on IHC testing with no amplification confirmed by in situ hybridization. All hormone receptor and HER2 assessments were performed at the local pathologies, and the most recently collected tissue from primary or recurrent cancer was used. Pathological and clinical information for this cohort was collected and managed using the secure REDCap electronic data capture tool hosted at the Medical University of Graz37,38.
Blood collection and cfDNA extraction
Blood samples were collected from patients before starting first- to fourth-line of palliative treatment, including 96 samples before first-line therapy (1 L), 52 samples before second-line therapy (2 L), 12 samples before third-line therapy (3 L), and 1 sample before fourth-line therapy (4 L). Thus, 161 plasma samples were collected from 146 patients (Fig. 1).
Plasma DNA was isolated, as described previously18,39. Briefly, 20 mL (range: 10-30 mL) of whole blood was collected into PAXgene Blood ccfDNA Tubes (Qiagen, Hilden, Germany) to preserve cell-free DNA (cfDNA). Plasma was separated by centrifugation of the blood samples at 1900 × g for 10 min. The resulting supernatant was carefully transferred and centrifuged at 1900 × g for 10 min to remove any residual cellular debris. Plasma was aliquoted into 2 mL tubes and stored at −80 °C until further processing. cfDNA was extracted from 1 to 4 mL plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany) or the QIASymphony PAXgene Blood ccfDNA Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Plasma DNA was quantified using the Qubit dsDNA HS Assay Kit (Life Technologies, Vienna, Austria).
mFAST-SeqS
As previously described, the mFAST-SeqS (modified Fast Aneuploidy Screening Test-Sequencing System) assay was used to estimate the tumor fraction in plasma cfDNA. The mFAST-SeqS (modified Fast Aneuploidy Screening Test-Sequencing System) assay detects chromosomal aneuploidy in cfDNA by using a z-score statistical approach to quantify LINE-1 count deviations from a normal chromosomal representation of control samples. A genome-wide z-score (z-score), calculated by summing the squared z-scores of individual chromosome arms, was used as a surrogate marker for tumor fraction. Genome-wide z-scores exceeding ±3 were considered indicative of chromosomal gains or losses21.
SiMSen-Seq assay forPIK3CA mutation detection
The SiMSen-Seq (SSS) assay was performed as previously described by our group18. Briefly, this technology utilizes molecular barcoding of individual DNA template strands, allowing all sequencing reads to be traced back to their original DNA template and correcting for PCR errors introduced during library preparation. For plasma samples, an average of 38.2 ng (range 8.8-103.8 ng) of cfDNA per sample was used for library preparation. Sequencing was performed on an Illumina NextSeq platform using 150 bp single-end reads, aiming for 10 million reads per sample. Across all interrogated positions, the average unique coverage was 2660× (range: 42–17,227×). Variant allele frequencies (VAFs) are reported as the percentage of alternate reads relative to the total reads at a given position. The detection limit was previously established as a VAF of 0.25%, with at least five consensus reads supporting the mutation18.
Molecular profiling of plasma samples
Molecular profiling was performed using the AVENIO ctDNA Expanded Kit (Roche, Basel, Switzerland), which targets 77 cancer-associated genes. We validated the assay in-house and confirmed the manufacturer’s specifications40. Library preparation was performed using an average of 40 ng of cfDNA (range: 10–50 ng) following the manufacturer’s protocol. Libraries were quantified using the Qubit dsDNA HS Assay Kit and pooled equimolarly. The pooled libraries were further quantified via qPCR and sequenced in batches of four to 16 libraries using 150 bp paired-end reads on an Illumina NextSeq (Mid or High Output kit) or NovaSeq6000 platform (SP flowcell, XP workflow). Sequencing generated an average of 35 million read pairs per sample (range: 12–81 million). After generating consensus reads, the data achieved an average read depth of 3845× (range: 107–28302×), with a median fragment length of 173 bp (range: 159–317 bp). Data analysis was performed using the AVENIO Oncology Analysis Software (version 2.0, Roche) with customized filtration settings for somatic variants. Variants with a minor allele frequency (MAF) ≥ 1% in GnomAD v2.1 or listed as common in dbSNP155 were excluded. Additionally, variants with less than 10 mutated reads, variants with VAF below 0.1%, likely germline variants (VAF ∼ 50% and z-score <3), copy number variants, and sequencing artifacts were manually excluded. Filtered variants were annotated using VarSeq (Golden Helix) and classified according to their pathogenicity using OncoKB and/or Franklin (GennoX)41. ctDNA levels were assessed as VAF (percentage of sequence reads observed matching a specific DNA variant divided by the overall coverage at that locus). For each sample, we identified the mutation with the highest VAF (hVAF).
Statistical analysis
All statistical analyses were performed using R version 4.4.2, Stata (version 18.5), or GraphPad Prism version 10.4.1. Descriptive analyses were conducted to describe patient baseline characteristics. Continuous variables were summarized as medians and interquartile ranges, and categorical variables were reported as counts and percentages. Descriptive statistics were used to summarize the detection rates and performance metrics of the SSS and AVENIO assays for PIK3CA mutations. Logistic and linear mixed-effects models with random intercepts at the patient level were applied to account for repeated measurements in some patients. Logistic mixed-effects models with patient ID as a random effect were used to evaluate assay concordance, including overall agreement, positive percent agreement (PPA), and negative percent agreement (NPA). Marginal probabilities were estimated, and 95% confidence intervals (CI) were calculated. For continuous outcomes, a linear mixed-effects model was applied. Model parameters were estimated using restricted maximum likelihood (REML), and the significance of fixed effects was assessed using Wald chi-square tests. All statistical tests were two-sided, and p-values below 0.05 were considered statistically significant.
Declaration of Generative AI and AI-assisted technologies in the writing process
During the preparation of this work, the author(s) used ChatGPT v4 for English editing. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Data Availability
The BAM files or FASTQ files for the cfDNA samples have been deposited in the EGA archive under accession number EGAS00001008200.
References
Burstein, H. J. Systemic Therapy for Estrogen Receptor–Positive, HER2−Negative Breast Cancer. N. Engl. J. Med. 383, 2557–2570 (2020).
Finn, R. S. et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 375, 1925–1936 (2016).
Johnston, S. et al. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 5, 5 (2019).
Hortobagyi, G. N. et al. Overall Survival with Ribociclib plus Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 386, 942–950 (2022).
Morrison, L., Loibl, S. & Turner, N. C. The CDK4/6 inhibitor revolution — a game-changing era for breast cancer treatment. Nat. Rev. Clin. Oncol. 21, 89–105 (2024).
André, F. et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2–negative advanced breast cancer: final overall survival results from SOLAR-1. Ann. Oncol. 32, 208–217 (2021).
Turner, N. C. et al. Inavolisib-Based Therapy in PIK3CA-Mutated Advanced Breast Cancer. N. Engl. J. Med. 391, 1584–1596 (2024).
Turner, N. C. et al. Capivasertib in Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 388, 2058–2070 (2023).
Bidard, F.-C. et al. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: Results From the Randomized Phase III EMERALD Trial. J. Clin. Oncol. 40, 3246–3256 (2022).
Klocker, E. V. et al. Clinical applications of next-generation sequencing-based ctDNA analyses in breast cancer: defining treatment targets and dynamic changes during disease progression. Mol. Oncol. https://doi.org/10.1002/1878-0261.13671. (2024).
Ignatiadis, M., Sledge, G. W. & Jeffrey, S. S. Liquid biopsy enters the clinic — implementation issues and future challenges. Nat. Rev. Clin. Oncol. 18, 297–312 (2021).
Amato, O., Giannopoulou, N. & Ignatiadis, M. Circulating tumor DNA validity and potential uses in metastatic breast cancer. Npj Breast Cancer 10, 21 (2024).
Heitzer, E., Haque, I. S., Roberts, C. E. S. & Speicher, M. R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet 20, 71–88 (2019).
Turner, N. et al. FGFR1 Amplification Drives Endocrine Therapy Resistance and Is a Therapeutic Target in Breast Cancer. Cancer Res. 70, 2085–2094 (2010).
Condorelli, R. et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 29, 640–645 (2018).
Costa, C. et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kα Inhibitors in Breast Cancer. Cancer Discov. 10, 72–85 (2020).
Turner, N. C. et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. https://doi.org/10.1016/s1470-2045(20)30444-7. (2020).
Suppan, C. et al. Sensitive and robust liquid biopsy-based detection of PIK3CA mutations in hormone-receptor-positive metastatic breast cancer patients. Brit. J. Cancer 126, 456–463 (2022).
Merker, J. D. et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 36, 1631–1641 (2018).
Suppan, C. et al. Untargeted Assessment of Tumor Fractions in Plasma for Monitoring and Prognostication from Metastatic Breast Cancer Patients Undergoing Systemic Treatment. Cancers 11, 1171 (2019).
Belic, J. et al. Rapid Identification of Plasma DNA Samples with Increased ctDNA Levels by a Modified FAST-SeqS Approach. Clin. Chem. 61, 838–849 (2015).
Browne, I. M., André, F., Chandarlapaty, S., Carey, L. A. & Turner, N. C. Optimal targeting of PI3K-AKT and mTOR in advanced oestrogen receptor-positive breast cancer. Lancet Oncol. 25, e139–e151 (2024).
Oliveira, M. et al. Dynamics of ESR1 mutation (ESR1m) circulating tumour DNA (ctDNA) in patients (pts) with estrogen receptor (ER)+ HER2− metastatic breast cancer (mBC) receiving camizestrant or fulvestrant: Exploratory analyses from the SERENA-2 trial. ESMO Open 9, 103012 (2024).
Jhaveri, K. L. et al. Imlunestrant with or without Abemaciclib in Advanced Breast Cancer. N. Engl. J. Med. https://doi.org/10.1056/nejmoa2410858. (2024).
Lloyd, M. R., Jhaveri, K., Kalinsky, K., Bardia, A. & Wander, S. A. Precision therapeutics and emerging strategies for HR-positive metastatic breast cancer. Nat. Rev. Clin. Oncol. 21, 743–761 (2024).
Andre, F. et al. Genomics to select treatment for patients with metastatic breast cancer. Nature 610, 343–348 (2022).
Bhave, M. A. et al. Comprehensive genomic profiling of ESR1, PIK3CA, AKT1, and PTEN in HR(+)HER2(-) metastatic breast cancer: prevalence along treatment course and predictive value for endocrine therapy resistance in real-world practice. Breast Cancer Res. Treat. 207, 599–609 (2024).
Sivakumar, S. et al. Tissue and liquid biopsy profiling reveal convergent tumor evolution and therapy evasion in breast cancer. Nat. Commun. 13, 7495 (2022).
Kindt, C. K. et al. Genomic alterations associated with resistance and circulating tumor DNA dynamics for early detection of progression on CDK4/6 inhibitor in advanced breast cancer. Int. J. Cancer. https://doi.org/10.1002/ijc.35126. (2024).
Yang, C. et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36, 2255–2264 (2017).
Davis, A. A. et al. Genomic complexity predicts resistance to endocrine therapy and CDK4/6 inhibition in hormone receptor-positive (HR+)/HER2−negative metastatic breast cancer. Clin. Cancer Res. 29, 1719–1729 (2023).
André, F. et al. Acquired gene alterations in patients treated with ribociclib plus endocrine therapy or endocrine therapy alone using baseline and end-of-treatment circulating tumor DNA samples in the MONALEESA-2, -3, and -7 trials. Ann. Oncol. 36, 54–64 (2025).
Rolfo, C. D. et al. Measurement of ctDNA Tumor Fraction Identifies Informative Negative Liquid Biopsy Results and Informs Value of Tissue Confirmation. Clin. Cancer Res. 30, 2452–2460 (2024).
Sokol, E. S. et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann. Oncol. 30, 115–123 (2019).
Sablin, M.-P. et al. Copy number alterations in metastatic and early breast tumours: prognostic and acquired biomarkers of resistance to CDK4/6 inhibitors. Br. J. Cancer 131, 1060–1067 (2024).
Razavi, P. et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 25, 1928–1937 (2019).
Harris, P. A. et al. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inf. 42, 377–381 (2009).
Harris, P. A. et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inf. 95, 103208 (2019).
Heitzer, E. et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 5, 30 (2013).
Weber, S. et al. Technical Evaluation of Commercial Mutation Analysis Platforms and Reference Materials for Liquid Biopsy Profiling. Cancers 12, 1588 (2020).
Chakravarty, D. et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017, 1–16 (2017).
Acknowledgements
This work was supported by Novartis, AstraZeneca, Daiichi Sankyo and Pfizer (no grant numbers). and by the Austrian Federal Ministry for Digital and Economic Affairs (Christian Doppler Research Fund for Liquid Biopsies for Early Detection of Cancer).
Author information
Authors and Affiliations
Contributions
N.Da and M.B. designed and supervised the study. N.Do, R.G., C.B., and L.P. performed plasma preparation and sequencing experiments. E.V.K., C.S., D.E., C.A., R.B., A.M.S., P.J.J., and G.R. contributed to patient care and data acquisition. N.Da and D.H. performed the statistical analysis, N.Da, M.B., and E.H. performed interpretation of results. N.Da, M.B., and E.H. drafted and revised the manuscript. All authors reviewed the manuscript and approved its final version.
Corresponding authors
Ethics declarations
Competing interests
M.B. received honoraria from Amgen, AstraZeneca, Daiichi Sankyo, Eli Lilly, Gilead, Menarini, MSD, Novartis, Pierre Fabre, Pfizer, Roche, Seagen, Stemline, travel funding from BMS, Daiichi Sankyo, MSD, Roche, Pierre Fabre, and research funding from Astra Zeneca, Daiichi Sankyo, Novartis, and Pfizer. P.J.J. has had a consulting or advisory role, received honoraria, research funding, and/or travel/accommodation expenses from Astra Zeneca, Bayer, Boehringer Ingelheim, Novartis, Pfizer, Servier, Roche, BMS and Celgene, Pierre Fabre, Janssen/Johnson&Johnson, MSD, Merck, Sanofi/Aventis, Ipsen, Amgen, Cycuria Therapeutics, Vessel. G.R. received honoraria from Amgen, AstraZeneca, Daiichi Sankyo, Eli Lilly, Gilead, MSD, Novartis, Roche, Seagen, Stemline, BMS, Consulting or Advisory Role from AstraZeneca, Daiichi Sankyo, Eli Lilly, Gilead, MSD, Novartis, Pfizer, Roche, Stemline, and travel support from Amgen, Daiichi Sankyo, Gilead, Roche. R.B. received honoraria from Amgen, Astra-Zeneca, BMS, Daichi, Eisai, Eli-Lilly, Gilead, Gruenenthal, MSD, MedMedia, Novartis, Pfizer, Pierre-Fabre, Roche, Seagen, Stemline, travel support from Astra-Zeneca, Daiichi, Eli-Lilly, MSD, Novartis, Stemline and Research Support from Daiichi. N.D. received travel/accommodation expenses from Daiichi Sankyo. A.M.S. received honoraria for lectures from AstraZeneca and travel and congress registration support from PharmaMar, MSD, Lilly, AstraZeneca, and Stemline Menarini. D.E. received honoraria from AstraZeneca, Gilead Sciences, Daiichi Sankyo, Pfizer, Roche/Genentech, Novartis, Lilly, Sirius Medical, Menarini Group, Consulting or Advisory Role from AstraZeneca, Daiichi Sankyo, Gilead Sciences, Novartis, Lilly, Menarini Group, MSD, Roche/Genentech, research funding from Sirius Medical, travel/accommodation expenses from Daiichi Sankyo, Pfizer, and Menarini Group. E.V.K. received honoraria/travel support from AstraZeneca, Daiichi Sankyo, Lilly, Gilead, Novartis, Roche, Stemline, and Pierre Fabre. E.H. received unrelated funding from Illumina, Roche, Servier, and PreAnalytiX, and received honoraria from Roche, Astra Zeneca and Incyte for advisory boards, not related to our study. C.S. has received honoraria from Astra-Zeneca, Daichi Sankyo, Eli-Lilly, Gilead, Novartis, Pfizer, Pierre-Fabre, Roche, Stemline-Menarini, travel support from Daiichi Sanyo, Eli-Lilly, MSD, Novartis, Roche. All other authors have declared no conflict of interest.
Ethical approval
This study was approved by the Austrian ethics committees (approval number 32–415 ex 19/20 for the Medical University of Graz, 1495/2023 for the Medical University of Vienna, and 1164/2022 for the Medical University of Innsbruck). Written informed consent was obtained from all patients. All procedures adhered to the ethical standards of the Declaration of Helsinki and the guidelines for good scientific practice required by the Medical University of Graz.
Consent to publication
All contributing authors read and approved the manuscript.
Informed consent
All patients provided written informed consent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Klocker, E.V., Dobrić, N., Graf, R. et al. Clinical impact of single-gene vs. panel sequencing in advanced HR + /HER2− breast cancer: insights and implications. npj Breast Cancer 11, 86 (2025). https://doi.org/10.1038/s41523-025-00805-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-025-00805-z
