
Abstract
Pharmacogenetics can enhance cardiovascular disease (CVD) treatment by tailoring drug therapy to genetic profiles and minimising trial-and-error approaches. Genetic variability influences responses to common CVD drugs, including antiplatelet drugs (clopidogrel and aspirin), anticoagulants (warfarin), statins, and antihypertensives (ACE inhibitors and β-blockers). Understanding genetic polymorphisms can improve efficacy and safety. Despite this progress, further research is needed to optimise pharmacogenomic applications and advance personalised medicine to improve CVD treatment outcomes.
Similar content being viewed by others


Introduction
Cardiovascular disease (CVD) is the leading cause of mortality globally, with the number of deaths increasing from 12.1 million in 1990 to 20.5 million in 20211. Pharmacogenetics is a relatively new area of pharmacology that focuses on pharmacogenetic factors that can significantly improve clinical outcomes by reducing side effects and increasing the efficacy of medications used to treat CVD. Therefore, understanding the impact of genetic polymorphisms on the pharmacokinetics and pharmacodynamics of therapeutic agents is imperative for healthcare practitioners2.
Efforts to improve the therapeutic efficacy of currently available drugs have been made through pharmacogenetic research, and the findings suggest the value of developing new drugs based on the association of variations in genes critical for drug action with variable outcomes of drug therapy. These variants often have smaller individual effect sizes and can determine the variability in drug responses in an individual or across a population. Thus, the primary objective of this review is to provide a narrative synthesis of the current knowledge on the pharmacogenetics of CVD drugs, with a dual focus on summarising key research findings and offering guidance on the clinical applicability of genetic markers. By exploring genetic polymorphisms that influence drug metabolism, transport, and therapeutic targets, this review aims to: (1) Highlight the most clinically actionable genetic polymorphisms of certain genes such as CYP2C19, VKORC1 and SLCO1B1, which have demonstrated significant impacts on drug efficacy and safety in the context of CVD therapy; (2) Discuss emerging genetic variants that remain within the research domain and evaluate their potential to transition into clinical practice; (3) Examine real-world challenges, including controversies, limitations, and barriers to implementing pharmacogenetic testing in routine CVD management; and (4) Provide actionable insights into how pharmacogenetic testing can be integrated into clinical workflows to improve personalised medical outcomes for patients with CVD.
The pharmacogenetics of antiplatelet therapy
The pharmacogenetics of purine receptor antagonists
Patients with acute coronary syndrome (ACS) or stroke and those undergoing percutaneous coronary intervention (PCI) are treated with oral antiplatelet therapy, such as ticagrelor, prasugrel and clopidogrel3,4.
The therapeutic target of thienopyridine and non-thienopyridine family drugs
The therapeutic target of these drugs is to act against platelets by blocking the purine receptor P2Y12 (P2RY12), which prevents adenosine diphosphate (ADP) from stimulating platelet aggregation5. Clopidogrel is the most frequently used antiplatelet P2Y12 inhibitor. Clopidogrel combined with aspirin is a well-recognised dual antiplatelet therapy to prevent ischaemic cardiovascular events3.
The metabolism of thienopyridine and non-thienopyridine family drugs
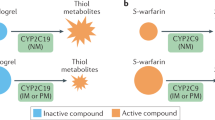
Clopidogrel is a second-generation prodrug of the thienopyridine family5. The transport of clopidogrel to the intestinal tract and its conversion into active components by various enzymes in the liver is necessary for its therapeutic effect. The absorption of clopidogrel in the small intestine is regulated by P-glycoprotein (P-gp) encoded by the ATP-binding cassette subfamily B member 1 (ABCB1) gene. Approximately 85% of the absorbed clopidogrel in the intestine is hydrolysed to inactive metabolites by carboxylesterase 1 (CES1) and then eliminated in the urine or faeces. Approximately 15% of clopidogrel is metabolised by cytochrome P450 in the liver, where it is first transformed into 2-oxo-clopidogrel, an intermediate metabolite, by CYP2C19, CYP1A2, and CYP2B6. Subsequently, 2-oxo-clopidogrel is transformed into an active thiol metabolite, which is catalysed by the CYP2C9, CYP3A4, CYP2B6 and CYP2C19 isoforms and the paraoxonase (PON)-1 enzyme. The active metabolites irreversibly bind to the P2RY12 on the platelet membrane to inhibit ADP, preventing the binding of fibrinogen to the GPIIb/IIIa receptor and ADP-mediated activation of the GPIIb/IIIa complex, thus preventing platelet activation and exerting antiplatelet effects (Fig. 1)6,7.

The intestinal absorption of clopidogrel is restricted by P-glycoprotein (P-gp), which is encoded by ABCB1. Carboxylesterase 1 (CES1) hydrolyses 85% of its uptake into inactive metabolites. Only 15% of inactive clopidogrel is oxidised by liver-resident cytochrome P450 (CYP450) enzymes to 2-oxo-clopidogrel, which is also inactive. Subsequently, CYP450 and paraoxonase enzymes convert 2-oxo-clopidogrel into active metabolites (thiols), which bind to the P2Y12 receptor on the platelet surface. This binding prevents the activation of the GPIIb/IIIa complex by ADP, which inhibits platelet activation (modified from refs. 6,7). Figure created with Bio Render.com.
Third-generation thienopyridine prasugrel exhibited better pharmacokinetic properties than clopidogrel. Following intestinal absorption of prasugrel, which is regulated by P-gp, prasugrel is metabolised by intestinal esterification to the intermediate metabolite thiolactone. This requires only one hepatic metabolic step to produce active metabolites, primarily by CYP3A4 and CYP2B6, with minor contributions from CYP2C19 and CYP2C94. Cyclopentyltriazolopyrimidine (CPTP) ticagrelor is a non-thienopyridine quickly absorbed in the intestines. CPTP acts directly on P2RY12 through reversible binding and does not require additional biotransformation for activation4.
Genetic polymorphisms affecting Clopidogrel response
Despite being one of the most widely used antiplatelet medications, clopidogrel has variable effects in certain patient populations. Genetic polymorphisms in CYP2C19 are among the main variables that influence an individual’s response to clopidogrel8. CYP2C19 plays a pivotal role in the formation of active metabolites, being involved in both metabolic steps as illustrated in Fig. 1. However, CYP2C19 gene polymorphisms are common, with more than 25 alleles identified, each conferring varying degrees of enzymatic activity and contributing to interpatient variability in antiplatelet effects9.
Among loss-of-function (LOF) alleles, CYP2C19*2 and *3 are the most prevalent and widely investigated. Homozygous or heterozygous patients are categorised as poor or intermediate metabolizers, respectively, because of their almost absent or reduced enzymatic activity9. Ellithi et al. reported that CYP2C19 LOF carriers exhibited a consistent reduction in active clopidogrel metabolite levels, increased platelet reactivity, and increased risk of ischaemic events10. Previous studies have suggested that the risk of clopidogrel resistance increases in the presence of the CYP2C19*2 and *3 polymorphic alleles11,12. Moreover, these alleles may be independent predictors of elevated platelet reactivity in patients treated with clopidogrel13. The presence of CYP2C19*2 and *3 alleles may be considered independent risk factors for stroke recurrence in patients with ischaemic stroke14. Furthermore, research findings indicate that patients with LOF of CYP2C19 exhibit a higher frequency of ischaemic events and an increased risk of stent thrombosis15. Two meta-analyses of clopidogrel-treated patients revealed that CYP2C19 LOF allele carriers were more likely to experience stroke and other combined vascular events than were noncarriers16,17. This finding validates the increased risk of clopidogrel failure in the prevention of cardiovascular events in patients with CYP2C19 LOF variants16,17.
The CYP2C19*17 allele has been linked to increased enzyme activity, and depending on the number of alleles present, it is associated with “rapid” metabolism (1 *17 allele) and 'ultra-rapid' metabolism (2 *17 alleles)9. CYP2C19*17 is strongly associated with an enhanced response to clopidogrel and a higher risk of bleeding18,19. Another study did not show a significant link between CYP2C19*17 and platelet inhibition or the risk of high platelet response13. In real-world genotype-guided antiplatelet therapy, the CYP2C19*17 allele was not significantly associated with post-PCI prescription decisions or clinical outcomes20. These findings suggest that the CYP2C19 *1/*17 and *17/*17 genotypes have limited clinical utility in guiding antiplatelet therapy after PCI20.
Numerous studies have identified significant interethnic variability in CYP2C19 polymorphisms. The most prevalent allele in Asian populations is CYP2C19*2, and its frequency varies by region. The frequency of the CYP2C19*2 allele in the Iranian population was 21.4%21, which is higher than that in Swedish (14.4%)22, German (15%)23, Ethiopian (13.6%)24, and Zimbabwean (13.1%)25 populations but lower than that in Japanese (23%)26 and Chinese-Taiwanese (32%)26 populations. The Filipino population had the highest frequency of the CYP2C19*2 allele (39%)26. However, the prevalence of the CYP2C19*2 allele has noticeably increased from Iran and Western Asia toward India, reaching its highest frequency, over 75%, in Melanesian populations27. Additionally, the CYP2C19*3 allele was detected with a prevalence of 1.7% in the Iranian population21 but was absent in the Canadian28 and Danish29 populations. However, the CYP2C19*3 allele was more frequent in other populations such as the Japanese (10.4%)26 and Korean (11.6%)30, whereas it was comparable to that reported in the Ethiopian population (1.8%)31. The prevalence of the CYP2C19*3 variant, commonly referred to as an Asian mutation32, has increased progressively from the western to eastern regions of Asia. Previous research has identified the highest frequency of the CYP2C19*3 allele in Southeast Asian populations, with 37% reported in Indonesians and 34% in the Iruna population of New Guinea33. Moreover, the frequency of the CYP2C19*17 allele in the Iranian population was 27.1%21 greater than that in other populations, including Denmark (20.1%)31, Norway (22%)34, African-Americans (21%)35, Ethiopia (17.9%)36, Korea (1.5%)37 and Japan (1.3%)38. This roughly resembles the frequency recorded for the Polish population (27.2%)39. Therefore, assessing the allele and genotype frequencies of CYP2C19 across various ethnic groups offers valuable insights for tailoring treatments to enhance therapeutic efficacy, minimise adverse effects, and support the progression of personalised medicine21.
Current guidelines and expert consensus recommend routine genetic testing in patients receiving clopidogrel20,40. According to evidence-based recommendations of the Clinical Pharmacogenetics Implementation Consortium (CPIC), the administration of clopidogrel to individuals with a poor or intermediate CYP2C19 metabolizer phenotype should be avoided. Instead, these patients should be administered an alternative P2Y12 inhibitor such as ticagrelor or prasugrel. However, clopidogrel can be considered for individuals with a normal, rapid, or ultra-rapid CYP2C19 metabolizer phenotype20,40. Recent meta-analyses have shown that patients undergoing PCI who benefit the most from alternative antiplatelet therapy may be identified using CYP2C19 genotype-guided therapy4,41. Clopidogrel is still commonly prescribed for ACS and non-ACS patients undergoing PCI, as well as for patients with other cardiovascular and neurovascular indications or who are at increased risk for bleeding20. Although newer and more potent P2Y12 inhibitors, such as prasugrel and ticagrelor, have demonstrated superior efficacy in reducing ischaemic events compared with clopidogrel in randomised trials involving patients with ACS, they are frequently associated with a higher risk of bleeding complications. Real-world studies involving patients with ACS have not consistently demonstrated that newer P2Y12 inhibitors outperform clopidogrel in terms of their clinical effectiveness. Clopidogrel remains a cornerstone of antiplatelet therapy, supported by robust evidence across various clinical contexts, underscoring its critical role in routine medical practice42.
A precision medicine strategy that used CYP2C19 genetic test results to deliver ticagrelor or prasugrel to LOF carriers and clopidogrel to non-carriers provided a more well-rounded therapeutic approach, as it lowered the risk of bleeding and ischaemic events compared to the universal use of ticagrelor or prasugrel43. Therefore, current evidence supports the use of CYP2C19 genetic testing before prescribing oral P2Y12 inhibitors in patients with ACS or those undergoing PCI43.
There is conflicting information regarding polymorphisms in ABCB1, which encodes a P-gp transporter and is involved in the efflux process of several medications, including clopidogrel44. Approximately 50 single-nucleotide polymorphisms (SNPs) are found in ABCB1; the most studied is C3435T, which is linked to P-gp7. Variations in P-gp expression between individuals caused by polymorphisms in the ABCB1 gene could change clopidogrel absorption in vivo and the quantity of parent medication reaching the liver45. The 3435 T allele may be connected to clopidogrel hyporesponsiveness, as one study on the ABCB1 C3435T gene polymorphism revealed that patients with the TT and CT genotypes had decreased clopidogrel absorption and an increased risk of adverse cardiovascular events compared to patients with the CC genotype46. These findings are in line with those of a study on patients with coronary artery disease (CAD) and ACS, which revealed that patients with the TT genotype had a greater risk of major adverse cardiovascular events (MACEs) than those with the CC or CT genotype47. Nevertheless, some recent studies have failed to discover a relationship between the effectiveness of clopidogrel and ABCB1 gene polymorphisms48, suggesting that the ABCB1 genotype has no effect on platelet reactivity in patients receiving clopidogrel49. Furthermore, the risk of clopidogrel resistance is not strongly correlated with the presence of the ABCB1 mutant allele12,50. It is still highly debatable whether the ABCB1 C3435T gene polymorphism influences the antiplatelet effect of clopidogrel and whether this polymorphism can predict high platelet reactivity following clopidogrel treatment. In contrast, the TT genotype may or may not be a factor in modifying treatment plans for patients who do not respond well to clopidogrel51. There is currently no clinical role for testing these polymorphisms in patients treated with clopidogrel, as the test has not demonstrated clinical utility at this time.
CES1 plays an essential role in clopidogrel biotransformation. CES1 hydrolyses 85% of the prodrug clopidogrel to an inactive carboxylic acid derivative. Thus, cytochrome P450 enzymes ultimately convert a small amount of clopidogrel into its active metabolites. Changes in enzyme activity can result from mutations in CES1, which encodes members of the carboxylesterase family that are important hepatic enzymes for drug clearance52. Variations in the CES1 gene result in changes in the activity of the CES1 enzyme, which is negatively correlated with the generation of active metabolites of clopidogrel and impacts the antiplatelet effects of this drug7,19,53. According to Gao et al., the CES1 G143E (rs71647871) polymorphism is anticipated to be a predictor of platelet hyporesponsiveness to clopidogrel because this mutation results in reduced enzyme catalytic activity51. Mirzaev et al. reported that the CES1 rs2244613 polymorphism is associated with an increased risk of clopidogrel resistance, elevated platelet reactivity, and increased enzymatic activity54. Nevertheless, other studies have demonstrated that the CES1 rs2244613 SNP is not associated with the risk of MACEs and has no effect on the effectiveness of clopidogrel in inhibiting platelet aggregation55. However, further research is needed to determine the precise influence and correlation of CES1 rs2244613 SNP with high platelet reactivity, given the absence of related studies and inconsistent results51. At present, testing for these polymorphisms in patients receiving clopidogrel is not clinically indicated, as there is no established evidence supporting its clinical usefulness.
The key enzyme in the second step of clopidogrel metabolism is paraoxonase-1 (PON1). Based on in vitro findings, PON1 is the rate-limiting enzyme in the formation of the active thiol metabolite of clopidogrel56. A study conducted by Bouman et al. revealed an association between the presence of PON1 QQ192 and a significant decrease in PON1 activity, leading to decreased levels of the active metabolite clopidogrel and an increased risk of stent thrombosis56. Subsequently, another study on patients with or without ACS revealed that the PON1 Q-allele was strongly linked to poor cardiovascular outcomes but not to the antiplatelet effect of clopidogrel57. In contrast, the PON1 192R variants were found to reduce the impact of clopidogrel on platelet aggregation. The impact of the PON1 192Q allele on platelet function and clopidogrel efficacy is considered minimal, suggesting that the PON1 192R variant may independently increase the risk of high platelet reactivity13. A study examining the impact of PON1 polymorphisms on MACEs and clopidogrel efficacy revealed that patients with the RR genotype had a greater risk of clopidogrel resistance; however, no association with clinical outcomes was detected58. Furthermore, in patients with ACS, the RR genotype is a risk predictor for PCI followed by revascularization59. However, a recent study showed that the PON1 Q192R polymorphism had no effect on clopidogrel pharmacokinetics and antiplatelet activity60. Thus, there is still a discrepancy regarding which allele, Q or R, interferes with the effect of the PON1 Q192R polymorphism on the response to clopidogrel.
Previous research on P2RY12 has mainly focused on T744C, C34T, and G52T variants. The P2RY12 744T allele is associated with an increased risk of MACEs and high on-treatment platelet reactivity after PCI61. A meta-analysis did not reveal any impact of the P2RY12 T744C polymorphism on clopidogrel resistance or high on-treatment platelet reactivity62. Similar results have been reported by Nie et al. 63. Two recent observational studies of patients undergoing percutaneous neurointervention revealed no correlation between high platelet reactivity during treatment and the T744C allele64. A study of Chinese patients undergoing PCI revealed a correlation between genetic variations in C34T and G52T and reduced clopidogrel responsiveness and adverse clinical events65. Furthermore, a meta-analysis revealed that patients on clopidogrel may be at risk for increased platelet reactivity owing to the presence of the T alleles of C34T and G52T61. Interestingly, another meta-analysis revealed that G52T and C34T variations in the P2RY12 gene were associated with cardiovascular events in the Chinese population, but not in the Caucasian population66.
Research has indicated inconsistencies in the findings of ongoing investigations on the effect of P2RY12 T744C on clopidogrel effectiveness. Studies on C34T and G52T have produced encouraging results but are not direct enough to guide clinical care. P2RY12 gene correlation studies offer research ideas for predicting platelet reactivity following clopidogrel treatment, but there are insufficient data to justify the clinical implementation of these ideas. The effects of genetic polymorphisms on clopidogrel response are summarised in Table 1.
The pharmacogenetics of aspirin
Acetylsalicylic acid (ASA), also referred to as aspirin, is a vital medication that is frequently used as antiplatelet therapy to prevent recurrent ischaemic events in patients with ischaemic stroke67. Clopidogrel combined with aspirin is a well-established, commonly used dual antiplatelet therapy for individuals with ischaemic heart disease to reduce ischaemic risk68.
The therapeutic targets of aspirin
The therapeutic target of this drug is to inhibit the function of prostaglandin G/H synthases 1 and 2 (PTGS1 and PTGS2), which are also referred to as cyclooxygenases (COX)-1 and -2, respectively (Fig. 2). COX-2 is highly inducible, primarily by inflammation, whereas COX-1 is constitutively produced. Mature platelets express the COX-1 enzyme, which catalyses the transformation of arachidonic acid (AA) into prostaglandins G2 and H2, producing thromboxane A2 (TXA2). When TXA2 is released into the bloodstream, it binds to TXA2 receptors on nearby platelets and activates them. Furthermore, TXA2 collaborates with other chemicals released by activated platelets such as fibrinogen, adenosine diphosphate (ADP), and factor V to accelerate the process69.

Aspirin is an inhibitor of cyclooxygenase (COX) that catalyses the transformation of arachidonic acid (AA) into prostaglandins G2 and H2. Thromboxane A2 (TXA2) is formed by thromboxane synthase and binds to the TXA2 receptors of nearby platelets, thereby serving as a potent platelet activator (modified from ref. 69). Figure created with Bio Render.com.
Genetic polymorphisms affecting aspirin response
Genetic variations that alter drug metabolism, leading to aspirin resistance (AR), are frequently reported phenomena that can cause treatment failure in patients, even in those who experience the well-established benefits of ASA70. Two genes, PTGS1 and PTGS2, encode COX-1 and COX-2, which are enzymes that catalyse the conversion of AA to prostaglandins and thromboxane69. Both PTGS1 and PTGS2 have been studied extensively. The C allele and CC genotype of the rs1330344 variant of the PTGS1 gene encoding COX-1 are linked to AR and increased levels of AA-induced aggregation69. A similar finding was noted by Li et al. 71. In Chinese stroke patients receiving aspirin therapy, the CC genotype has been linked to poor functional outcomes72. The C allele has also been linked to a higher risk of ischaemic stroke in the Chinese population73. Nevertheless, the outcomes have not been consistent, and this polymorphism may have increased COX-1 activity and did not affect the platelet response to aspirin74,75. Additionally, rs10306114, another SNP in PTGS1, did not correlate with AR69. Although PTGS2 is a highly polymorphic gene, its expression and function are affected only by a few SNPs76,77. Recently, in patients with ischaemic stroke carrying the GC or CC genotype, the rs20417 C allele was found to be strongly correlated with AR. Importantly, not all studies corroborated or validated these findings78.
The specific TXA2 receptor (TBXA2R) is encoded by the TBXA2R gene, and variations in this gene can affect ADP-induced platelet aggregation. The TBXA2R polymorphism rs4523 has been linked to platelet aggregation in patients with CAD who receive aspirin. Patients with the TT and CT genotypes showed greater platelet aggregation than those with the CC genotype69. In previous investigations, TT homozygotes also demonstrated elevated platelet reactivity related to AR79. The other SNP (rs1131882) of the TBXA2R gene is not associated with AR69,80.
The platelet membrane receptors P2Y1 and P2Y12 are essential for platelet aggregation, thrombosis and antiplatelet medication pharmacology81. While the inhibition of P2RY12 inhibits ADP-induced platelet activation, ADP amplifies multiple signalling pathways to activate platelets through autocrine and paracrine mechanisms82. Goodman et al. reported no correlation between these polymorphisms and AR83.
Genetic variations in platelet endothelial aggregation receptor 1 (PEAR1) were previously thought to be linked to platelet aggregation and the response to aspirin84. However, the relationship between these findings and cardiovascular clinical outcomes remains unclear. PEAR1 encodes a type 1 membrane protein that is expressed in endothelial cells and platelets. Consequently, a clinical outcome analysis of aspirin and the PEAR1 rs12041331 G > A variant has recently been published19. The authors examined bleeding, cardiovascular incidents, and cerebrovascular events; however, none of these clinical outcomes was significantly associated with this PEAR1 variant. However, the effects of PEAR1 rs12041331 might vary depending on the type of stroke and whether aspirin is being administered for primary or secondary stroke prevention. According to a recently published pharmacogenetic analysis by Li et al., PEAR1 rs12041331 was significantly associated with functional outcomes only in patients with the small-artery occlusion subtype of stroke treated with aspirin alone85. Thus, the reason for the discrepancy in the results for the PEAR1 rs12041331 variant between the studies could be the difference in disease severity among patients. Thus, it is possible that PEAR1 rs12041331 interacts with aspirin only in patients with specific stroke subtypes. Further research is required to confirm these results.
Platelet adhesion and aggregation control depend on glycoprotein IIb/IIIa (GPIIb/IIIa)86. Different levels of aspirin sensitivity have been associated with variations in the integrin subunit beta 3 (ITGB3) gene, which encodes the fibrinogen glycoprotein GPIIb/IIIa. Szczeklik et al. found that PlA1/A2 allele carriers had higher AR than wild-type carriers87. According to a meta-analysis by Goodman et al., AR measured using platelet function assays was not associated with PlA1/A2 polymorphisms in patients with CVD83. Another meta-analysis confirmed the relationship between the single-nucleotide substitution rs5918 in the ITGB3 gene of GPIIIa and clinical outcomes in patients receiving aspirin, showing no significant association with MACEs88. Other studies have reported contradictory results, with no significant association between glycoprotein IIIa P1A1/A2 polymorphism and AR or stroke development89,90.
α-2A-adrenergic receptor (ADRA2A) gene polymorphism is associated with AA-induced aggregation. ADRA2A encodes α-2A-adrenergic receptor, which is involved in epinephrine-induced platelet aggregation. Greater levels of AA-induced aggregation were linked to the minor allele T (rs4311994) of ADRA2A. This allele has been linked to increased platelet reactivity to aspirin in individuals with type 2 diabetes91, but these outcomes are not always repeatable92.
In addition to the efficacy studies mentioned earlier, several other recent studies have assessed the correlation between genetic variants and side effects of aspirin on the gastrointestinal tract. Researchers have discovered a variant in the intron of EYA1 with a statistically significant association with the side effects of aspirin in the gastrointestinal tract, raising the possibility of aspirin-induced peptic ulcer disease. Lower EYA1 expression in the epithelium of the upper gastrointestinal tract may alter this risk; however, further research is needed to determine the functional underpinnings of this relationship93. According to recently published candidate gene association studies, PTGS1, NOS3, CHST2, and GSTP1 variants have been linked to gastrointestinal bleeding in aspirin-treated patients94,95,96,97.
Genetic testing holds significant value in personalised medicine, as tailoring treatment strategies for AR patients based on genetic polymorphisms may reduce treatment failures and improve clinical outcomes, although further studies are imperative98. The effect of genetic polymorphisms on aspirin response is summarised in Table 2.
The pharmacogenetics of anticoagulants
The pharmacogenetics of warfarin
Globally, warfarin is the most frequently prescribed oral anticoagulant for treating and preventing thromboembolic disorders and preventing stroke in patients with atrial fibrillation (AF)99. Warfarin is given as a racemic mixture of its R and S isomers. The S isomer is three to five times stronger than the R isomer and is metabolised via a different pathway. CYP2C9 is the main enzyme involved in S-warfarin metabolic clearance, whereas CYP1A1, CYP1A2 and CYP3A4 are among the cytochrome P450 enzymes that remove R-warfarin (Fig. 3)100.

Warfarin inhibits the formation of the active form of vitamin K. Warfarin—clinically available as an equal mixture of R and S enantiomers—inhibits vitamin K oxidoreductase complex 1 (VKORC1) and, therefore, prevents the reduction of vitamin K1 to vitamin KH2. KH2 is necessary for γ-glutamyl carboxylase (GGCX) expression and activation of clotting factors II, VII, IX, and X (modified from ref. 101). Figure created with Bio Render.com.
The mechanism of action of warfarin
The mechanism by which warfarin produces its anticoagulant therapeutic effect is by inhibiting the enzyme vitamin K oxidoreductase complex 1 (VKORC1). This enzyme converts vitamin K epoxide to vitamin K1, which is subsequently converted to vitamin KH2. Vitamin KH2 is an important cofactor for the enzyme γ-glutamyl carboxylase (GGCX), which activates clotting factors II, VII, IX, and X, which are essential for blood clotting. Vitamin K1 is metabolised by the CYP4F2 enzyme to hydroxyl-vitamin K1, an inactive metabolite, to reduce the amount of vitamin K1 that can be reduced to vitamin KH2101. A narrow therapeutic index and wide inter-individual variation in dose requirements and response to treatment pose challenges to warfarin treatment and increase the risk of treatment complications. As a result, regular monitoring of the international normalised ratio (INR) is necessary to guarantee optimal anticoagulation and lower the risk of either a low or high INR (which carries the risk of thrombosis or bleeding, respectively)101,102.
Genetic polymorphisms affecting warfarin response
Genetic polymorphisms significantly influence dose requirements, yet they are infrequently incorporated into clinical practice. Warfarin was the first medication to have established pharmacogenetic dosing guidelines101. According to the 2017 CPIC guidelines, genotype-guided warfarin therapy is based on variations in CYP2C9, VKORC1, CYP4F2 and rs12777823. These guidelines use validated pharmacogenetic algorithms to determine appropriate dosing.
Patients with CYP2C9 and VKORC1 polymorphisms may require adjusted starting doses to achieve the intended anticoagulant effects because these variations contribute to reduced warfarin metabolism and altered sensitivity101,103. The effectiveness of a genotype-guided warfarin dosing strategy compared to either a fixed (e.g. 5 mg/day) or clinically guided dosing strategy has been investigated in three large randomised controlled trials: COAG, EU-PACT, and GIFT. In the EU-PACT and GIFT clinical trials, over 90% of the patients in the COAG group were of European ancestry, whereas 27% were of European ancestry. EU-PACT and GIFT demonstrated improved outcomes, while COAG found no distinction between genotype-guided and clinically guided dosing approaches104.
The primary metabolizer of S-warfarin and several other commonly prescribed drugs is CYP2C9, an enzyme belonging to the CYP2C subfamily of cytochrome P450 enzymes105. Studies have shown that CYP2C9*2, *3, *5, *6, *8 and *11 are linked to decreased function of the CYP2C9 enzyme, which lowers the clearance of S-warfarin, the more active enantiomer, and consequently lowers patient dosage requirements. Individuals with these genotypes are more likely to experience severe bleeding complications and thus require lower doses. The most prevalent alleles in individuals of European ancestry are *2 and *3, whereas those in individuals of African ancestry are primarily the *5, *6, *8 and *11 alleles101,103,104.
The target enzyme of warfarin, vitamin K epoxide reductase protein, is encoded by the VKORC1 gene. The rate-limiting step in vitamin K recycling is the conversion of vitamin K epoxide to vitamin K, which is catalysed by VKORC1. SNPs in this gene contribute to warfarin resistance, and extremely high doses of medication are required for therapeutic anticoagulation. A common variant in the VKORC1 regulatory region, c.1639 G > A (rs9923231), is associated with decreased VKORC1 expression and patients with this variant require lower warfarin doses. Asians are more likely to carry the -1639 AA (susceptible) genotype than individuals of European descent, whereas African-Americans are more likely to carry the -1639 GG (reduced sensitivity) genotype. This difference explains the higher reported doses of warfarin required in Africans compared to Europeans and the frequently lower reported doses required in Asians101,103.
As a counterpart to VKORC1, CYP4F2 prevents vitamin K from building up by converting it to hydroxyl-vitamin K1 and eliminating it from the vitamin K cycle. Enzyme activity is affected by the presence of the CYP4F2*3 variant, which leads to increased availability of vitamin K1 for the reduction of vitamin K hydroquinone and activation of clotting factors. In the Asian and European cohorts, the *3 allele was associated with higher warfarin doses than the *1 allele. No association was found among the populations of African descent101,103. The Food and Drug Administration (FDA) has approved warfarin-labelling information, but does not currently list CYP4F2 as a biomarker. However, in addition to testing for CYP2C9 and VKORC1 variants, some clinical laboratories may test for the CYP4F2*3 allele. The 2017 CPIC warfarin dosing guidelines include recommendations based on the CYP4F2 genotype when results are available103. Another variant, rs12777823, located close to the CYP2C cluster on chromosome 10, is associated with reduced warfarin dosage requirements. Reduced S-warfarin clearance was also associated with the SNP rs12777823. This SNP is prevalent in the general population but appears to be specific to African-Americans regarding its relationship with warfarin response101.
Mal, T Cell Differentiation Protein Like (MALL) gene has recently been identified as a factor that could indirectly influence warfarin response through its interactions with caveolin-1106. The integrity of endothelial cells, which is responsible for controlling the haemostatic pathway107, is regulated by the caveolin-1 membrane protein108. As the MALL-encoded protein interacts with caveolin-1, the indirect influence of MALL on the anticoagulant action of warfarin is biologically plausible. MALL has been reported to influence warfarin dosage solely in patients of African descent, although direct evidence of biological plausibility is still required106. A recent study emphasised the necessity for larger pharmacogenomic studies of warfarin in patients of African descent to identify genetic variants specific to this group and ultimately enhance the quality of anticoagulant therapy for this under-researched population106. The use of genetic information to guide warfarin dosing in clinical practice has been influenced by ethnic group variations due to genetic differences that affect drug metabolism and response. Variations in CYP2C9 result in slower warfarin metabolism, which increases the risk of bleeding. CYP2C9*2 and *3 genetic variants were more common in European populations than in other ethnic groups, whereas the *5, *6, *8, and *11 alleles were more frequent among African populations. Individuals with these variations usually need lower dosages of warfarin101,103,104. VKORC1 polymorphisms are more prevalent in Asian and African ethnicities and are associated with a higher sensitivity to warfarin, making these individuals require lower doses101,103. Because of the impact of the CYP4F2* 3 variant on enzyme activity, clotting factors are activated, which increases the risk of clotting. The prevalence of CYP4F2*3 variation was higher in Asian and European cohorts than in African cohorts, and higher doses of warfarin were needed101,103. Another variant, rs12777823, which is close to CYP2C, has been linked to decreased S-warfarin clearance. This SNP is specific to African-Americans and is required to reduce the warfarin dosage101. The recently discovered MALL locus may have an indirect impact on warfarin response, particularly in African-American patients. Although it has been associated with warfarin dosage requirements, further research is needed to confirm its biological plausibility106.
The pharmacogenetics of lipid-lowering drugs
The pharmacogenetics of hydroxymethylglutaryl coenzyme A reductase inhibitors
Statins, also called 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, are the first-line therapy for lowering lipid levels and reducing CVD risk109,110,111.
The therapeutic target of statins
Statins exert their therapeutic effects by inhibiting the active site of HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis, resulting in lower levels of low-density lipoprotein cholesterol (LDL-C) in the blood. The conversion of HMG-CoA to mevalonic acid is inhibited by preventing substrate access to this site, which leads to reduced hepatic cholesterol synthesis and upregulation of LDL receptors (LDLRs). This further leads to a decrease in the amount of LDL-C in the blood and an increase in LDL-C removal from circulation112. However, not every patient responds well to statins, and some do not achieve their target cholesterol reduction levels. Moreover, a large proportion of patients experience side effects. Statin-induced myopathy (SIM) and statin-associated muscle symptoms (SAMs) are the most frequently reported side effects of statins, and these side effects result in poor adherence to or cessation of statin pharmacotherapy regimens113,114. Many genes have been studied to determine their potential influence on various interactions of statins. Genes involved in absorption, distribution, and metabolism are associated with fat metabolism, and these differences can be affected115.
Genetic polymorphisms affecting statin response
Liver cells contain a protein known as organic anion transporting polypeptide 1B1 (OATP1B1), encoded by the solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene. Toxins, medications, hormones, and bilirubin are all transported by OATP1B1 into the liver, where they are eliminated from the body. The OATP1B1 protein typically transports several medications, including statins (such as rosuvastatin and pitavastatin)109. Pitavastatin and rosuvastatin have been suggested by Prueksaritanont et al. as probe substrates for OATP1B, and they have demonstrated relative sensitivity and selectivity to OATP1B inhibitors. Pitavastatin was meant to be a more selective and sensitive clinical OATP1B probe than rosuvastatin116. The effect of the missense variant rs4149056 in SLCO1B1, which causes impaired hepatic clearance and elevated plasma statin concentrations, has been studied117. When simvastatin was used, a clear correlation was observed between increased exposure to simvastatin and a greater risk of developing myopathy118. The SLCO1B1 SNP has been shown to decrease the OATP1B1 transporter activity119. As a result, rosuvastatin concentration and plasma exposure were elevated, increasing the risk of SIM117.
The ATP-binding cassette G2 (ABCG2) protein, also referred to as breast cancer resistance protein (BCRP), functions as an intermediary for the cellular outflow of various exogenous substances, such as antibiotics, chemotherapeutic agents, and dietary toxins, as well as endogenous substances such as oestrogens120. In tumour cells, ABCG2 causes resistance to several anticancer medications; however, in normal cells, ABCG2 appears to protect cells against cytotoxic substances120. Rosuvastatin is actively taken up into hepatocytes from portal circulation, undergoes minimal metabolism, and is largely excreted unchanged into the bile, leading to faecal elimination. Research has demonstrated that an ABCG2 genetic variant increases the plasma concentration of simvastatin121 and is linked to a greater risk of SIM122. According to previous studies, SNPs in ABCG2 decrease ABCG2 expression123,124.
CYP2C9 is responsible for the metabolism of certain statins, such as fluvastatin, pitavastatin, and rosuvastatin125. Genetic variations in CYP2C9 affect the ability of an individual to metabolise these drugs. The two most extensively studied variants, CYP2C9*2 and CYP2C9*3, significantly metabolise statins more slowly, leading to higher drug levels and an increased risk of side effects. CYP2C9 genetic variations have been shown to impact statin exposure and the risk of SAMS109.
Serum lipid levels and the response to statin therapy vary significantly among individuals126. While the data suggest that HMG-CoA reductase genotypes explain less than 2% of the variance in statin response, 6–15% of the variants in the gene encoding HMG-CoA reductase can be explained by alternative splicing127,128. An SNP located in intron 13 of the HMG-CoA reductase gene causes an alternate splicing event that involves exon 13 within the catalytic domain of the enzyme to inhibit its activity118. Alternative splicing polymorphisms of HMG-CoA reductase have a significant effect on the response of women to statin therapy, according to a study of a group of patients with familial hypercholesterolaemia129.
The type and quantity of LDLR mutations affect the patient’s response to lipid-lowering medications. Numerous randomised trials have shown that the risk of cardiovascular events is decreased by treatments that lower LDL-C levels by reducing LDL particles through upregulation of LDLR130,131. Recent data suggest that berberine-mediated LDL-C reduction depends on the 3′-untranslated region (3-UTR) of LDLR, which stabilises LDLR mRNA. SNPs in the 3-UTR are linked to both in vitro LDLR mRNA stability and in vivo LDL-C levels132. Additionally, Polisecki et al. reported that variations in this region might affect statin-mediated lipid reduction. Certain variations in LDLR have been linked to lipid-lowering responses following simvastatin administration133.
The main metabolic pathway of statins, including atorvastatin and simvastatin, is the CYP3A4 pathway134. Inducers or inhibitors of this enzyme can change the plasma concentration of statins, which may reduce the effectiveness of treatment or increase the risk of side effects such as myopathy135. It has been demonstrated that groups of affected individuals can differ in CYP3A4 activity by up to ten times, possibly due to differences in the genes encoding these enzymes. A study of patients with hypercholesterolaemia treated with atorvastatin revealed that the CYP3A4 promoter variant rs2740574 is associated with elevated LDL-C levels. Compared to carriers of the wild-type allele, carriers of the missense variant rs4986910 showed increased efficacy and decreased LDL-C levels136. Another study showed no correlation between the rs4986910 variant and atorvastatin response137. The rs2740574 variant has been reported to significantly reduce LDL-C levels in individuals with wild-type genotypes compared to those with the variant allele.
The CPIC guidelines offer recommendations for the use of pharmacogenetic information, including statins, in clinical practice. Dose adjustment based on pharmacogenomics involves altering the dosage of a medication according to an individual’s genetic profile to enhance the effectiveness of the drug and reduce the risk of adverse effects109. CPIC guidelines state how SLCO1B1, ABCG2, and CYP2C9 genotypes can guide statin therapy to reduce the risk of SAMS. Based on weak evidence and a lack of conclusive clinical applications, no recommendations have been provided for CYP3A4/5 and HMGCR109.
The pharmacogenetics of antihypertensive drugs
The pharmacogenetics of β-blockers
CVDs such as ischaemic heart disease, hypertension, cardiac arrhythmias, and heart failure are frequently treated with β-adrenergic receptor blockers or β-blockers138. The activation of the adrenergic nervous system influences the onset and progression of numerous cardiovascular and associated metabolic disorders139. β-blockers produce negative inotropic (reduced force of myocardial contraction), negative dromotropic (reduced conduction velocity through the atrioventricular node), and chronotropic (reduced heart rate [HR]) effects by modulating activation of the sympathetic nervous system138. Numerous investigations have verified a strong correlation between the HR-lowering effect of β-blockers and advantageous cardiovascular consequences140,141.
The therapeutic target of β-blockers
β-adrenergic receptors play a significant role in cardiac function via signal transduction controlled by G protein-coupled receptor kinase (GPCRK) phosphorylation interactions. The therapeutic goal of β-blockers is to prevent the binding of a ligand (catecholamines, either norepinephrine or epinephrine) to β-adrenergic receptors (β1AR and β2AR) by competing for the binding site. After the agonist binds to the βAR, it is linked to the Gs protein (Gαs) to activate adenyl cyclase (AC) and produce cyclic AMP (cAMP), which further phosphorylates GRK2 and then activates PKA, which in turn phosphorylates various intracellular substrates for practical function142. The heart is the primary target of β-adrenergic receptor stimulation. When this receptor is activated, the contractility (inotropy), HR, and conduction velocity (homotopy) of the heart increase. The cardiovascular system can be better regulated by taking advantage of β1AR and β2AR signalling. When catecholamines overstimulate the β-adrenergic receptors, they cause CVD, stroke, heart failure, and cardiac enlargement. Elevating the level of Gα causes βAR kinase (βARK) to become active, which in turn influences and accelerates the development of heart failure by stimulating the cardiomyocyte βARs. Furthermore, in a practical sense, an increase in GRKs limits heart failure pathogenesis via the desensitisation of βARs to control overstimulation142.
Genetic polymorphisms affecting β-blockers response
Although β-blockers are generally effective, their HR-lowering effects vary among individuals143. Genetic factors have been identified as the cause of this variability in the HR144. Approximately 25% of medications undergo biotransformation, significantly aided by cytochrome P450 2D6 (CYP2D6). Several β-blockers, including metoprolol, carvedilol, propranolol, labetalol, nebivolol, and timolol, are substrates of the CYP2D6 enzyme145. Metoprolol is a primary β-blocker whose pharmacokinetic variability, largely due to its dependence on CYP2D6 for metabolism, may lead to significant differences in pharmacodynamic responses among individuals146. Many studies have shown that CYP2D6 poor and intermediate metabolizers exhibit decreased apparent oral clearance of metoprolol. These pharmacokinetic differences primarily result in variations in heart rate (HR) response; however, other CYP2D6 phenotypes do not appear to significantly affect the response to β-blockers147,148,149. The CYP2D6 genotype is linked to a changed HR response to β-blocker therapy, and it has been demonstrated that the CYP2D6 phenotype is one of the most important indicators of variation in HR response to metoprolol150. Notably, the HR indicates the level of β1AR blockade when β-blocker therapy is administered. The correlations between HR and CYP2D6 genotype were consistent, but the correlations between blood pressure and other responses were weaker147. However, some studies have shown a positive correlation between the CYP2D6 genotype and blood pressure response147,151,152.
Sympathetic nervous system activation mediates chronotropic, dromotropic, and inotropic effects through β1AR, which are GPCRs encoded by adrenoceptor beta 1 (ADRB1). These receptors are mainly expressed in cardiac tissue153. The ADRB1 variants rs1801252 (ADRB1 145 A > G; Ser49Gly) and rs1801253 (ADRB1 1165 C > G; Arg389Gly) are the most prevalent and well-studied. The variant rs1801252 causes agonist-promoted downregulation, whereas the variant rs1801253 modifies G-protein coupling and G-protein-coupled cyclase activity and decreases cAMP production154,155. The strongest evidence points to a relationship between the ADRB1 genotype and reduced diastolic blood pressure observed in response to β-blockers. However, the ADRB1 genotype was not associated with changes in HR or systolic blood pressure. Additionally, the data indicated that patients with the ADRB1 haplotype would likely benefit the most from β-blocker therapy and have a lower chance of developing MACEs and chronic heart failure147.
β2AR is a G protein-coupled adrenergic receptor that is encoded by ADRB2. Vascular smooth muscle cells, bronchial smooth muscle cells, and cardiac myocytes express β2AR. The ADRB2 (46 A > G; Arg16Gly) SNP rs1042713 and ADRB2 (79 C > G; Gln27Glu) SNP rs1042714 are two common SNPs. The rs1042713 variant amplified the downregulation induced by agonists, which in turn led to a reduction in cAMP formation. Conversely, the variant rs1042714 exhibits resistance against agonist-induced downregulation, which ultimately leads to an increase in the formation of cAMP156,157. Recent data from individuals with CAD and chronic heart failure collectively indicated that the ADRB2 genotype is not associated with cardiovascular events147,158.
GPCRKs desensitise GPCRs that are occupied by ligands such as βARs, which eventually results in the downregulation of the receptor153. G protein-coupled receptor kinase 5 (GPCRK5) is highly expressed in cardiac tissue. The GPCRK5 Leu41 variant encodes a functional kinase with increased in vitro desensitisation of β1AR, which reduces receptor stimulation and cAMP generation159,160. For GPCRK5 variants, the homozygous genotype with the major allele (Gln41Gln) was linked to a better response to β-blockers during treatment. In contrast, carriers of the Leu41 allele do not benefit from β-blocker therapy147.
ADRA2C encodes α2C AR, a presynaptic GPCR that reduces the release of norepinephrine from sympathetic nerves upon activation. α2ARs inhibit adenylyl cyclase in response to endogenous catecholamine stimulation, which lowers cAMP levels and causes noradrenergic neurons to become hyperpolarized161. Individuals of African heritage are most likely to have the in-frame deletion rs61767072 (c.971_982del), which results in the loss of four amino acids (p.Gly324_Ala327del, sometimes called Del322-325), and is associated with decreased receptor function162. This leads to increased sympathetic nervous activity and an increased catecholamine response to the α2AR antagonist, yohimbine163.
Fewer studies are available related to individual variants of ADRA2C, GPCRK4, and GPCRK5 and their associations with β-blocker responses. Therefore, there is insufficient evidence to provide therapeutic recommendations for the individual variants of CYP2D6, ADRB2, ADRA2C, GPCRK4 and GPCRK5164.
The pharmacogenetics of angiotensin-converting enzyme inhibitors
Angiotensin-converting enzyme inhibitors (ACEIs) are frequently used to treat common conditions, such as heart failure, chronic renal disease, and hypertension.
The therapeutic targets of ACE inhibitors
The therapeutic effect of ACE inhibitors is to reduce the amount of angiotensin II, which increases sodium excretion in the urine, lowers blood pressure, and prevents the remodelling of smooth muscle and cardiac muscle cells165,166. The renin-angiotensin-aldosterone system (RAAS) interacts with ACEIs. The RAAS controls blood pressure by releasing angiotensinogen from the liver into the circulation, where renin converts angiotensinogen to angiotensin I. Angiotensin I (Ang I) is converted to angiotensin II (Ang II) by ACE. Ang II attachment to angiotensin receptors promotes blood vessel constriction, which increases blood pressure. Furthermore, Ang II stimulates aldosterone synthesis, which increases the ability of the blood to reabsorb salt and water and elevates blood pressure. ACEIs limit smooth muscle constriction in the vasculature and decrease aldosterone levels by blocking the ability of ACE to convert Ang I to Ang II and by lowering blood pressure. ACE is an essential enzyme involved in bradykinin degradation. Bradykinin builds up under conditions of ACE inhibition, and increased bradykinin levels may have side effects such as cough and angioedema167.
Genetic polymorphisms affecting ACE inhibitor response
Genes involved in the renin-angiotensin pathway are among those extensively studied in the pharmacogenetics of ACEIs. One of the most widely investigated genetic variants is the insertion/deletion (I/D) polymorphism in the ACE gene, characterised by the presence (insertion, I) or absence (deletion, D) of a 287-base pair Alu repeat sequence within intron 16. This polymorphism results in three genotypes: II, ID, and DD. The D allele is associated with elevated ACE enzyme activity and higher circulating ACE levels, whereas the I allele corresponds to lower ACE activity. These enzymatic differences can affect individual responses to ACEIs, which are commonly prescribed for hypertension and heart failure168,169,170. The distribution of the I and D alleles varies significantly across populations. In European populations, the alleles are relatively evenly distributed. In contrast, African populations tend to exhibit a higher frequency of the D allele, for example, ~70% in Morocco, with similar trends observed across the continent. Middle Eastern populations, including Saudi Arabia, also show a high prevalence of the D allele, estimated at around 72.5%. Conversely, East Asian populations such as those in China and Japan demonstrate a greater prevalence of the I allele, with frequencies ranging from 65 to 70%. These population-based differences underscore the potential role of genetic background in influencing both the efficacy and safety of ACEI therapy, reinforcing the importance of incorporating genetic factors into personalised treatment strategies171.
Genetic variations at this locus correlate with hypertension172,173 and myocardial infarction174,175. As the presence of the ACE I/D gene polymorphisms explained more than half of the total phenotypic variance in ACE activity, it was demonstrated to have a strong correlation with plasma ACE levels. Studies have shown that individuals with genotype II have lower ACE concentrations than those with DD176. The DD genotype has been associated with increased ACE activity and levels, which increase the production of Ang II and ultimately elevate blood pressure177. In a study of male Malaysian hypertensive participants, those with the DD genotype showed a more robust response to ACEIs (lisinopril and enalapril)178.
Angiotensinogen (AGT), a primary structural gene in the RAAS pathway, is associated with hypertension179. Genetic variations in AGT lead to alterations in plasma AGT concentrations, which may contribute to the development of hypertension, CAD, and myocardial infarction180. The AGT SNPs M235T (rs699), M174T (rs4762) and A-6G (rs5051) are associated with serum AGT levels and hypotension in response to ACEIs181,182. Other studies have not found this association183. However, there was a strong correlation between hypertension and the T allele of M235T, which was linked to higher plasma AGT levels184. AGT SNPs (T174M and M235T) are among the most prevalent variants that cause hypertension in several populations180. There are other variants, such as G-217A, A-6G, A-20C and G-152A; however, their significance is not as great as that of variants T174M and M235T180.
The primary angiotensin II receptor AGTR1 mediates most of the physiological effects of AGT II, such as aldosterone secretion, pressure responses, renal tubular salt transport, and vascular contraction185. There is conflicting evidence in earlier research linking the (A1166C) AGTR1 polymorphism to cardiovascular disorders. According to a previous study, the AA genotype protects against CAD and myocardial infarction, whereas the (1166C) AGTR1 allele is a genetic marker that predisposes individuals to these conditions186. Other studies have not supported these conclusions187,188. According to a recent study published in 2022, AC heterozygosity may lower the risk of heart failure associated with AGTR1 polymorphism189. Previous research findings have indicated that there is no correlation between AGTR1 polymorphisms and response to ACEIs190,191. Frazier found that AGTR1 polymorphisms are linked to blood pressure response to antihypertensive drugs in black women192. Moreover, another study revealed that hypertensive Chinese individuals with AGTR1 polymorphism had lower blood pressure when treated with ACEIs193.
Common adverse effects of ACEIs, such as enalapril, have been associated with specific SNPs that influence their incidence. Enalapril, a prodrug targeting the RAAS, is rapidly converted to its active metabolite, enalaprilat. Among its side effects, a persistent dry cough is the most frequently reported. Genetic variations in the SLCO1B1 gene, which encodes the hepatic transporter OATP1B1, have been linked to reduced hepatic clearance of enalapril, resulting in elevated plasma concentrations. This accumulation increases bradykinin levels, thereby contributing to the incidence of cough. Furthermore, polymorphisms in the BDKRB2 gene, which encodes the bradykinin B2 receptor, have also been implicated in heightened susceptibility to ACEI-induced cough194.
According to a genome-wide association study (GWAS), mutations in potassium voltage-gated channel interacting protein 4 (KCNIP4) have also been linked to ACEI-induced cough195. The control of Kv4 potassium channels is one of the main functions of KCNIP4. Bradykinin buildup in the lungs can stimulate sensory nerve afferents when there is a mutation in the KCNIP4 gene, which causes problems in Kv4 channel regulation196. Therefore, patients with KCNIP4 polymorphisms that affect the regulation of Kv4 channels are more susceptible to the negative effects of ACEIs, because the resulting mutant proteins can stop bradykinin breakdown.
Angioedema is another potentially fatal side effect of ACEIs. Variants in immune-regulating genes (ETV6) and protein kinase C (PRKCQ) are risk factors for the side effects of ACEI, including angioedema. According to GWAS, PRKCQ variation is associated with a lower incidence of angioedema. In contrast, variations in ETV6 are linked to a greater risk of angioedema associated with ACEIs197. Angioedema is strongly linked to variations in the membrane metalloendopeptidase (MME), which encodes neprilysin. Neprilysin is a secondary enzyme that degrades vasoactive peptides such as bradykinin in conjunction with ACE. Mutations in MME may lead to decreased neprilysin function, which causes an increase in vasoactive peptides in the plasma under ACE-inhibition conditions. Vasoactive peptide build-up can lead to a greater frequency of coughing and angioedema. Thus, ACEI adverse events are more likely to occur in patients with reduced neprilysin levels197.
Discussion
Pharmacogenetics has become crucial for optimising CVD treatment, potentially reducing adverse drug reactions, and enhancing therapeutic efficacy. This review highlights significant advancements in our understanding of how genetic polymorphisms can influence CVD drug metabolism, transport, and therapeutic targets. The various prime-time genetic variants influencing cardiovascular drug responses discussed here include CYP2C19 polymorphisms affecting clopidogrel drug response, CYP2C9 and VKORC1 polymorphisms impacting warfarin dose, SLCO1B1 variants associated with statin-induced myopathy risk, and CYP2D6 polymorphisms influencing β-blocker metabolism, particularly for metoprolol. Additionally, ACE insertion/deletion (I/D) polymorphisms affect responsiveness to ACE inhibitors. Emerging or preliminary variants, on the other hand, refer to those recently identified or still under investigation, such as the MALL gene linked to warfarin response, EYA1 associated with aspirin-induced ulceration, and KCNIP4 linked to ACE inhibitor-induced cough. While these variants show promise, further research is needed to establish their clinical relevance. Healthcare professionals should recognise that while ‘prime-time’ variants can currently guide clinical decisions, the identification of emerging variants highlights the evolving nature of pharmacogenetics, which may hold significant clinical implications in the future.
Pharmacogenetic testing for drugs such as clopidogrel, warfarin, statins, and β-blockers enables more precise genotype-guided treatments. However, significant gaps remain in the integration of pharmacogenetic knowledge into routine clinical practice. For example, while CYP2C19 testing is well-documented in relation to clopidogrel response, its clinical adoption varies due to factors such as cost, limited access to genetic testing, and ongoing debates about its relevance across diverse populations. Additionally, research on non-European populations remains underrepresented, raising concerns about the broader applicability of pharmacogenetic findings across different ethnic groups. These disparities underscore the need for more inclusive studies to develop population-specific recommendations.
Innovative areas such as polygenic risk scores, machine-learning-based pharmacogenomic models, and next-generation sequencing offer promising avenues for identifying new biomarkers and treatment targets. To better understand genetic variability in drug responses, future research must integrate pharmacogenetic data with other omics approaches, including proteomics and metabolomics. The widespread implementation of personalised medicine in cardiovascular care depends on overcoming logistical and regulatory barriers, such as the affordability of genetic testing and the education of healthcare providers. While pharmacogenetics has significantly advanced our understanding of the variability in drug responses, fully incorporating it into clinical practice necessitates further investigation, infrastructure development, and collaboration among key stakeholders. Addressing these challenges will allow us to maximise the potential of pharmacogenetics in improving cardiovascular outcomes worldwide.
References
https://world-heart-federation.org/news/deaths-from-cardiovascular-disease-surged-60-globally-over-the-last-30-years-report/ (accessed on 29th April 2024).
Saunders, H., Harris, D. & Chirilă, R. M. Pharmacogenomics: introduction and use in clinical practice. Rom. J. Intern. Med. 58, 69–74 (2020).
Castrichini, M., Luzum, J. A. & Pereira, N. Pharmacogenetics of antiplatelet therapy. Annu. Rev. Pharmacol. Toxicol. 63, 211–229 (2023).
Galli, M. et al. Genetic testing in patients undergoing percutaneous coronary intervention: rationale, evidence and practical recommendations. Expert Rev. Clin. Pharmacol. 14, 963–978 (2021).
Falco, L. et al. Antioxidant properties of oral antithrombotic therapies in atherosclerotic disease and atrial fibrillation. Antioxidants 12, 1185 (2023).
Akkaif, M. A. et al. The role of genetic polymorphism and other factors on clopidogrel resistance (CR) in an Asian population with coronary heart disease (CHD). Molecules 26, 1987 (2021).
Saiz-Rodríguez, M. et al. Influence of CYP450 enzymes, CES1, PON1, ABCB1, and P2RY12 polymorphisms on clopidogrel response in patients subjected to a percutaneous neurointervention. Clin. Ther. 41, 1199–1212.e1192 (2019).
Lopez, J. et al. Role of genetic polymorphisms in clopidogrel response variability: a systematic review. Open Heart 10, e002436 (2023).
Lee, C. R. et al. Clinical pharmacogenetics implementation consortium guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin. Pharmacol. Ther.112, 959–967 (2022).
Ellithi, M., Baye, J. & Wilke, R. A. CYP2C19 genotype-guided antiplatelet therapy: promises and pitfalls. Pharmacogenomics 21, 889–897 (2020).
Su, Q. et al. Association of CYP2C19 polymorphism with clopidogrel resistance in patients with acute coronary syndrome in China. Med. Sci. Monit.25, 7138 (2019).
Zhuo, Z. -l. et al. Association between CYP2C19 and ABCB1 polymorphisms and clopidogrel resistance in clopidogrel-treated Chinese patients. Anatol. J. Cardiol. 19, 123 (2018).
Peng, W., Shi, X., Xu, X. & Lin, Y. Both CYP2C19 and PON1 Q192R genotypes influence platelet response to clopidogrel by thrombelastography in patients with acute coronary syndrome. Cardiovasc. Ther. 2019, 3470145 (2019).
Liu, G., Yang, S. & Chen, S. The correlation between recurrent risk and CYP2C19 gene polymorphisms in patients with ischemic stroke treated with clopidogrel for prevention. Medicine 99, e19143 (2020).
Li, Y.-J. et al. Association between CYP2C19 polymorphisms and clinical outcomes in patients undergoing stent procedure for cerebral artery stenosis. Sci. Rep. 11, 5974 (2021).
Biswas, M., Sukasem, C., Khatun Kali, M. S. & Ibrahim, B. Effects of the CYP2C19 LoF allele on major adverse cardiovascular events associated with clopidogrel in acute coronary syndrome patients undergoing percutaneous coronary intervention: a meta-analysis. Pharmacogenomics 23, 207–220 (2022).
Xi, Z. et al. CYP2C19 genotype and adverse cardiovascular outcomes after stent implantation in clopidogrel-treated Asian populations: a systematic review and meta-analysis. Platelets 30, 229–240 (2019).
Sibbing, D. et al. Cytochrome 2C19* 17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 121, 512–518 (2010).
Lewis, J. P. et al. Pharmacogenomic polygenic response score predicts ischaemic events and cardiovascular mortality in clopidogrel-treated patients. Eur. Heart J. Cardiovasc. Pharmacother. 6, 203–210 (2020).
Lee, C. R. et al. Impact of the CYP2C19* 17 allele on outcomes in patients receiving genotype-guided antiplatelet therapy after percutaneous coronary intervention. Clin. Pharmacol. Ther. 109, 705–715 (2021).
Dehbozorgi, M. et al. Prevalence of the CYP2C19* 2 (681 G> A),* 3 (636 G> A) and* 17 (‑806 C> T) alleles among an Iranian population of different ethnicities. Mol. Med. Rep. 17, 4195–4202 (2018).
Yamada, H. et al. CYP2D6 and CYP2C19 genotypes in an elderly Swedish population. Eur. J. Clin. Pharmacol. 54, 479–481 (1998).
Brockmöller, J. J., Rost, K., Gross, D., Schenkel, A. & Roots, I. Phenotyping of CYP2C19 with enantiospecific HPLCquantification of R-and S-mephenytoin and comparison with the intron4/exon5 G→ A-splice site mutation. Pharmacogenetics 5, 80–88 (1995).
Persson, I., Aklillu, E., Rodrigues, F., Bertilsson, L. & Ingelman-Sundberg, M. S-mephenytoin hydroxylation phenotype and CYP2C19 genotype among Ethiopians. Pharmacogenetics 6, 521–526 (1996).
Dandara, C. et al. Genetic polymorphism of CYP2D6 and CYP2C19 in east-and southern African populations including psychiatric patients. Eur. J. Clin. Pharmacol. 57, 11–17 (2001).
Goldstein, J. A. et al. Frequencies of the defective CYP2C19 alleles responsible for the mephenytoin poor metabolizer phenotype in various Oriental, Caucasian, Saudi Arabian and American black populations. Pharmacogenetics 7, 59–64 (1997).
Sistonen, J. et al. Pharmacogenetic variation at CYP2C9, CYP2C19, and CYP2D6 at global and microgeographic scales. Pharmacogenet. Genomics 19, 170–179 (2009).
Jurima-Romet, M. et al. CYP2C19 genotyping and associated mephenytoin hydroxylation polymorphism in a Canadian Inuit population. Pharmacogenetics 6, 329–339 (1996).
Bathum, L., Andersen-Ranberg, K., Boldsen, J., Brøsen, K. & Jeune, B. Genotypes for the cytochrome P450 enzymes CYP2D6 and CYP2C19 in human longevity Role of CYP2D6 and CYP2C19 in longevity: role of CYP2D6 and CYP2C19 in longevity. Eur. J. Clin. Pharmacol. 54, 427–430 (1998).
Herrlin, K. et al. Bantu Tanzanians have a decreased capacity to metabolize omeprazole and mephenytoin in relation to their CYP2C19 genotype. Clin. Pharmacol. Ther. 64, 391–401 (1998).
Pedersen, R. S. et al. Linkage disequilibrium between the CYP2C19* 17 allele and wildtype CYP2C8 and CYP2C9 alleles: identification of CYP2C haplotypes in healthy Nordic populations. Eur. J. Clin. Pharmacol. 66, 1199–1205 (2010).
Hamdy, S. I. et al. Allele and genotype frequencies of polymorphic cytochromes P450 (CYP2C9, CYP2C19, CYP2E1) and dihydropyrimidine dehydrogenase (DPYD) in the Egyptian population. Br. J. Clin. Pharmacol. 53, 596–603 (2002).
Hsu, H.-L., Woad, K. J., Woodfield, D. G. & Helsby, N. A. A high incidence of polymorphic CYP2C19 variants in archival blood samples from Papua New Guinea. Hum. Genomics 3, 1–7 (2008).
Rudberg, I., Mohebi, B., Hermann, M., Refsum, H. & Molden, E. Impact of the ultrarapid CYP2C19* 17 allele on serum concentration of escitalopram in psychiatric patients. Clin. Pharmacol. Ther. 83, 322–327 (2008).
Kearns, G. L., Leeder, J. S. & Gaedigk, A. Impact of the CYP2C19* 17 allele on the pharmacokinetics of omeprazole and pantoprazole in children: evidence for a differential effect. Drug Metab. Dispos. 38, 894–897 (2010).
Sim, S. C. et al. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin. Pharmacol. Ther. 79, 103–113 (2006).
Kim, K. A., Song, W. K., Kim, K. R. & Park, J. Y. Assessment of CYP2C19 genetic polymorphisms in a Korean population using a simultaneous multiplex pyrosequencing method to simultaneously detect the CYP2C19* 2, CYP2C19* 3, and CYP2C19* 17 alleles. J. Clin. Pharm. Ther. 35, 697–703 (2010).
Sugimoto, K., Uno, T., Yamazaki, H. & Tateishi, T. Limited frequency of the CYP2C19* 17 allele and its minor role in a Japanese population. Br. J. Clin. Pharmacol. 65, 437–439 (2008).
Kurzawski, M. et al. Effect of CYP2C19* 17 gene variant on Helicobacter pylori eradication in peptic ulcer patients. Eur. J. Clin. Pharmacol. 62, 877–880 (2006).
Group, E. S. D. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: The Task Force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 42, 1289–1367 (2021).
Pereira, N. L. et al. Effect of CYP2C19 genotype on ischemic outcomes during oral P2Y12 inhibitor therapy: a meta analysis. JACC Cardiovasc. Interv.14, 739–750 (2021).
Patti, G., Micieli, G., Cimminiello, C. & Bolognese, L. The role of clopidogrel in 2020: a reappraisal. Cardiovasc. Ther. 2020, 8703627 (2020).
Pereira, N. L. et al. CYP2C19 genetic testing for oral P2Y12 inhibitor therapy: a scientific statement from the American Heart Association. Circulation 150, e129–e150 (2024).
Loo, T. W. & Clarke, D. M. Cysteines introduced into extracellular loops 1 and 4 of human P-glycoprotein that are close only in the open conformation spontaneously form a disulfide bond that inhibits drug efflux and ATPase activity. J. Biol. Chem. 289, 24749–24758 (2014).
Zhang, Y.-J., Li, M.-P., Tang, J. & Chen, X.-P. Pharmacokinetic and pharmacodynamic responses to clopidogrel: evidences and perspectives. Int. J. Environ. Res. Public Health 14, 301 (2017).
Pan, Y. et al. Outcomes associated with clopidogrel-aspirin use in minor stroke or transient ischemic attack: a pooled analysis of clopidogrel in high-risk patients with acute non-disabling cerebrovascular events (CHANCE) and platelet-oriented inhibition in new TIA and minor ischemic stroke (POINT) trials. JAMA Neurol. 76, 1466–1473 (2019).
Zhang, S. et al. A SNP involved in alternative splicing of ABCB1 is associated with clopidogrel resistance in coronary heart disease in Chinese population. Aging 12, 25684 (2020).
Samardzic, J. et al. Impact of continuous P2Y12 inhibition tailoring in acute coronary syndrome and genetically impaired clopidogrel absorption. J. Cardiovasc. Pharmacol. 75, 174–179 (2020).
Hou, X., Han, W., Gan, Q., Liu, Y. & Fang, W. CYP 2C19 and ABCB 1 genetic polymorphisms correlate with the recurrence of ischemic cardiovascular adverse events after clopidogrel treatment. J. Clin. Lab. Anal. 32, e22369 (2018).
Namazi, S. et al. Association of ABCB1 gene polymorphisms and clopidogrel responsiveness in Iranian patients undergoing percutaneous coronary intervention. Iran. J. Pharm. Res.19, 307 (2020).
Gao, H. et al. Advances and perspectives in methods for identifying high platelet reactivity. Heliyon 9, e22214 (2023).
Zhu, H.-J. et al. Carboxylesterase 1 as a determinant of clopidogrel metabolism and activation. J. Pharmacol. Exp. Ther. 344, 665–672 (2013).
Neuvonen, M. et al. Effects of genetic variants on carboxylesterase 1 gene expression, and clopidogrel pharmacokinetics and antiplatelet effects. Basic Clin. Pharmacol. Toxicol. 122, 341–345 (2018).
Mirzaev, K. B. et al. Effects of the rs2244613 polymorphism of the CES1 gene on the antiplatelet effect of the receptor P2Y12 blocker clopidogrel. Drug Metab. Pers. Ther. 34, 20180039 (2019).
Fathy, S. et al. Pharmacogenetic and clinical predictors of response to clopidogrel plus aspirin after acute coronary syndrome in Egyptians. Pharmacogenet. Genomics 28, 207–213 (2018).
Bouman, H. J. et al. Paraoxonase-1 is a major determinant of clopidogrel efficacy. Nat. Med. 17, 110–116 (2011).
Park, K. W. et al. Paraoxonase 1 gene polymorphism does not affect clopidogrel response variability but is associated with clinical outcome after PCI. PLoS ONE 8, e52779 (2013).
Zhang, Z., Chen, M., Zhang, L. & Zhao, Q. The impact of cytochrome 450 and Paraoxonase polymorphisms on clopidogrel resistance and major adverse cardiac events in coronary heart disease patients after percutaneous coronary intervention. BMC Pharmacol. Toxicol. 21, 1–9 (2020).
Ma, W. et al. Relationship of paraoxonase-1 Q192R genotypes and in-stent restenosis and re-stenting in Chinese patients after coronary stenting. Atherosclerosis 251, 305–310 (2016).
Mega, J. L. et al. PON1 Q192R genetic variant and response to clopidogrel and prasugrel: pharmacokinetics, pharmacodynamics, and a meta-analysis of clinical outcomes. J. Thromb. Thrombolysis 41, 374–383 (2016).
Wang, X. et al. Relationship between clopidogrel-related polymorphisms and variable platelet reactivity at 1 year: a cohort study from Han Chinese. J. Res. Med. Sci. 21, 111 (2016).
Cui, G., Zhang, S., Zou, J., Chen, Y. & Chen, H. P2Y 12 receptor gene polymorphism and the risk of resistance to clopidogrel: a meta-analysis and review of the literature. Adv, Clin. Exp. Med. 26, 343–349 (2017).
Nie, X. -y. et al. Haplotype of platelet receptor P2RY12 gene is associated with residual clopidogrel on-treatment platelet reactivity. J. Zhejiang Univ. Sci. B 18, 37 (2017).
Fu, H., Hu, P., Ma, C., Peng, F. & He, Z. Association of clopidogrel high on-treatment reactivity with clinical outcomes and gene polymorphism in acute ischemic stroke patients: an observational study. Medicine 99, e19472 (2020).
Li, M. et al. Associations between P2RY12 gene polymorphisms and risks of clopidogrel resistance and adverse cardiovascular events after PCI in patients with acute coronary syndrome. Medicine 96, e6553 (2017).
Zhao, K. et al. P2Y12 polymorphisms and the risk of adverse clinical events in patients treated with clopidogrel: a meta-analysis. Drug Res. 69, 23–31 (2019).
Rothwell, P. M. et al. Effects of aspirin on risk and severity of early recurrent stroke after transient ischaemic attack and ischaemic stroke: time-course analysis of randomised trials. Lancet 388, 365–375 (2016).
Levine, G. N. et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines: an update of the 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention, 2011 ACCF/AHA guideline for coronary artery bypass graft surgery, 2012 ACC/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease, 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction, 2014 AHA/ACC guideline for the management of patients with non–ST-elevation acute coronary syndromes, and 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery. Circulation 134, e123–e155 (2016).
Ikonnikova, A. et al. Genetic association study and machine learning to investigate differences in platelet reactivity in patients with acute ischemic stroke treated with aspirin. Biomedicines 10, 2564 (2022).
Ferreira, M. et al. The emergent phenomenon of aspirin resistance: insights from genetic association studies. Pharmacogenomics 21, 125–140 (2020).
Li, X. -l. et al. Genetic polymorphisms of HO-1 and COX-1 are associated with aspirin resistance defined by light transmittance aggregation in Chinese Han patients. Clin. Appl. Thromb. Hemost. 19, 513–521 (2013).
Cai, H. et al. Association between PTGS1 polymorphisms and functional outcomes in Chinese patients with stroke during aspirin therapy: interaction with smoking. J. Neurol. Sci. 376, 211–215 (2017).
Zhao, L. et al. Interaction between COX-1 and COX-2 increases susceptibility to ischemic stroke in a Chinese population. BMC Neurol. 19, 1–12 (2019).
Li, Q. et al. Frequency of genetic polymorphisms of COX1, GPIIIa and P2Y1 in a Chinese population and association with attenuated response to aspirin. Pharmacogenomics 8, 577–586 (2007).
Pettinella, C. et al. Cyclooxygenase-1 haplotype C50T/A-842G does not affect platelet response to aspirin. Thromb. Haemost. 101, 687–690 (2009).
Papafili, A. et al. Common promoter variant in cyclooxygenase-2 represses gene expression: evidence of role in acute-phase inflammatory response. Arterioscler. Thromb. Vasc. Biol. 22, 1631–1636 (2002).
Sharma, V. et al. Association of COX-2 rs20417 with aspirin resistance. J. Thromb. Thrombolysis 35, 95–99 (2013).
Wang, H. et al. Association of GPI a and COX-2 gene polymorphism with aspirin resistance. J. Clin. Lab. Anal. 32, e22331 (2018).
Zhao, J. et al. Association of thromboxane A2 receptor gene polymorphisms with cerebral infarction in a Chinese population. Neurol. Sci. 34, 1791–1796 (2013).
Peng, L. -l. et al. Associations of MDR1, TBXA2R, PLA2G7, and PEAR1 genetic polymorphisms with the platelet activity in Chinese ischemic stroke patients receiving aspirin therapy. Acta Pharmacol. Sin. 37, 1442–1448 (2016).
Storey, R. F. Biology and pharmacology of the platelet P2Y12 receptor. Curr. Pharm. Des. 12, 1255–1259 (2006).
Dorsam, R. T. & Kunapuli, S. P. Central role of the P2Y 12 receptor in platelet activation. J. Clin. Investig. 113, 340–345 (2004).
Goodman, T., Ferro, A. & Sharma, P. Pharmacogenetics of aspirin resistance: a comprehensive systematic review. Br. J. Clin. Pharmacol. 66, 222–232 (2008).
Herrera-Galeano, J. E. et al. A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler. Thrombos. Vasc. Biol. 28, 1484–1490 (2008).
Li, Z. et al. Platelet endothelial aggregation receptor 1 polymorphism is associated with functional outcome in small-artery occlusion stroke patients treated with aspirin. Front. Cardiovasc. Med. 8, 664012 (2021).
Fullard, J. F. The role of the platelet glycoprotein IIb/IIIa in thrombosis and haemostasis. Curr. Pharm. Des. 10, 1567–1576 (2004).
Szczeklik, A., Undas, A., Sanak, M., Frołow, M. & Węgrzyn, W. Relationship between bleeding time, aspirin and the PlA1/A2 polymorphism of platelet glycoprotein IIIa. Br. J. Haematol. 110, 965–967 (2000).
Wang, J. et al. Association among PlA1/A2 gene polymorphism, laboratory aspirin resistance and clinical outcomes in patients with coronary artery disease: An updated meta-analysis. Sci. Rep. 9, 13177 (2019).
Derle, E. et al. Aspirin resistance in cerebrovascular disease and the role of glycoprotein IIIa polymorphism in Turkish stroke patients. Blood Coagul. Fibrinolysis 27, 169–175 (2016).
Abderrazek, F. et al. The GPIIIa PlA polymorphism and the platelet hyperactivity in Tunisian patients with stable coronary artery disease treated with aspirin. Thromb. Res. 125, e265–e268 (2010).
Postula, M. et al. Genetic determinants of platelet reactivity during acetylsalicylic acid therapy in diabetic patients: evaluation of 27 polymorphisms within candidate genes. J. Thromb. Haemost. 9, 2291–2301 (2011).
Johnson, A. D. et al. Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat. Genet. 42, 608–613 (2010).
Bourgeois, S. et al. Genome-wide association between EYA1 and aspirin-induced peptic ulceration. EBioMedicine 74,103728 (2021).
Mallah, N. et al. A multicenter case–control study of the effect of e-nos VNTR polymorphism on upper gastrointestinal hemorrhage in NSAID users. Sci. Rep. 11, 19923 (2021).
Mallah, N. et al. Synergism interaction between genetic polymorphisms in drug metabolizing enzymes and NSAIDs on upper gastrointestinal haemorrhage: a multicenter case-control study. Ann. Med. 54, 379–392 (2022).
Forgerini, M. et al. Genetic variants in PTGS1 and NOS3 genes increase the risk of upper gastrointestinal bleeding: A case–control study. Front. Pharmacol. 12, 671835 (2021).
Handa, Y. et al. A novel gene associated with small bowel bleeding in patients taking low-dose aspirin. Dig. Liver Dis. 53, 841–845 (2021).
Zeng, W. et al. Genetic factors related to aspirin resistance using the Multiplate® device in Hong Kong Chinese patients with stable coronary heart disease. Heliyon 10, e34552 (2024).
Humbert, X. et al. Non-vitamin K oral anticoagulant treatment in elderly patients with atrial fibrillation and coronary heart disease. Int. J. Cardiol. 222, 1079–1083 (2016).
Kim, S.-Y. et al. Metabolism of R-and S-warfarin by CYP2C19 into four hydroxywarfarins. Drug Metab. Lett. 6, 157–164 (2012).
Duarte, J. D. & Cavallari, L. H. Pharmacogenetics to guide cardiovascular drug therapy. Nat. Rev. Cardiol. 18, 649–665 (2021).
Arwood, M. J. et al. Anticoagulation endpoints with clinical implementation of warfarin pharmacogenetic dosing in a real-world setting: a proposal for a new pharmacogenetic dosing approach. Clin. Pharmacol. Ther. 101, 675–683 (2017).
Johnson, J. A. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for pharmacogenetics-guided warfarin dosing: 2017 update. Clin. Pharmacol. Ther. 102, 397–404 (2017).
Oni-Orisan, A. et al. An introductory tutorial on cardiovascular pharmacogenetics for healthcare providers. Clin. Pharmacol. Ther. 114, 275–287 (2023).
Pratt, V. M. et al. Recommendations for clinical warfarin genotyping allele selection: a report of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn. 22, 847–859 (2020).
Asiimwe, I. G. et al. Meta-analysis of genome-wide association studies of stable warfarin dose in patients of African ancestry. Blood Adv. 8, 5248–5261 (2024).
Verhamme, P. & Hoylaerts, M. F. The pivotal role of the endothelium in haemostasis and thrombosis.Acta Clinica Belgica 61, 213–219 (2006).
Xu, L. et al. Caveolae: molecular insights and therapeutic targets for stroke. Expert Opin. Ther. Targets 19, 633–650 (2015).
Cooper-DeHoff, R. M. et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin. Pharmacol. Ther. 111, 1007–1021 (2022).
Ahangari, N. et al. Personalised medicine in hypercholesterolaemia: the role of pharmacogenetics in statin therapy. Ann. Med. 52, 462–470 (2020).
Alfonsi, J. E., Hegele, R. A. & Gryn, S. E. Pharmacogenetics of lipid-lowering agents: precision or indecision medicine?. Curr. Atheroscler. Rep. 18, 1–10 (2016).
Buhaescu, I. & Izzedine, H. Mevalonate pathway: a review of clinical and therapeutical implications. Clin. Biochem. 40, 575–584 (2007).
Mitchell, D., Guertin, J. R., Iliza, A. C., Fanton-Aita, F. & LeLorier, J. Economic evaluation of a pharmacogenomics test for statin-induced myopathy in cardiovascular high-risk patients initiating a statin. Mol. Diagn. Ther. 21, 95–105 (2017).
Leusink, M., Onland-Moret, N. C., de Bakker, P. I., de Boer, A. & Maitland-van der Zee, A. H. Seventeen years of statin pharmacogenetics: a systematic review. Pharmacogenomics 17, 163–180 (2016).
Schmitz, G. & Langmann, T. Pharmacogenomics of cholesterol-lowering therapy. Vasc. Pharmacol. 44, 75–89 (2006).
Prueksaritanont, T. et al. Pitavastatin is a more sensitive and selective organic anion-transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 78, 587–598 (2014).
Tornio, A. et al. SLCO1B1 polymorphism markedly affects the pharmacokinetics of lovastatin acid. Pharmacogenet. Genomics 25, 382–387 (2015).
Group, S. C. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N. Engl. J. Med. 359, 789–799 (2008).
Tirona, R. G., Leake, B. F., Merino, G. & Kim, R. B. Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European-and African-Americans. J. Biol. Chem. 276, 35669–35675 (2001).
Sparreboom, A. et al. Effect of ABCG2 genotype on the oral vioavailability of topotecan. Cancer Biol. Ther. 4, 650–653 (2005).
Keskitalo, J. et al. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 86, 197–203 (2009).
DeGorter, M. K. et al. Clinical and pharmacogenetic predictors of circulating atorvastatin and rosuvastatin concentrations in routine clinical care. Circ. Cardiovasc. Genet. 6, 400–408 (2013).
Kobayashi, D. et al. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab. Dispos.33, 94–101 (2005).
Imai, Y. et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol. Cancer Ther.1, 611–616 (2002).
Cid-Conde, L. & López-Castro, J. In Cardiovascular Risk Factors in Pathology (IntechOpen, 2020).
Jermendy, G. et al. Effect of genetic and environmental influences on cardiometabolic risk factors: a twin study. Cardiovasc. Diabetol. 10, 1–8 (2011).
Medina, M. W. & Krauss, R. M. Alternative splicing in the regulation of cholesterol homeostasis. Curr. Opin. Lipidol. 24, 147–152 (2013).
Medina, M. W. & Krauss, R. M. The role of HMGCR alternative splicing in statin efficacy. Trends Cardiovasc. Med. 19, 173–177 (2009).
Leduc, V., Bourque, L., Poirier, J. & Dufour, R. Role of rs3846662 and HMGCR alternative splicing in statin efficacy and baseline lipid levels in familial hypercholesterolemia. Pharmacogenet. Genomics 26, 1–11 (2016).
Catapano, A. L. et al. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Pol. Heart J. 74, 1234–1318 (2016).
Silverman, M. G. et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA 316, 1289–1297 (2016).
Muallem, H. et al. Quantitative effects of common genetic variations in the 3′ UTR of the human LDL-receptor gene and their associations with plasma lipid levels in the Atherosclerosis Risk in Communities study. Hum. Genet. 121, 421–431 (2007).
Polisecki, E. et al. Genetic variation at the LDL receptor and HMG-CoA reductase gene loci, lipid levels, statin response, and cardiovascular disease incidence in PROSPER. Atherosclerosis 200, 109–114 (2008).
Adams, C. D. Stockley’s drug interactions. Stockley’s Drug Interactions Pocket Companion 2013. J. Med. Libr. Assoc.102, 221 (2014).
Voora, D. et al. Pharmacogenetic predictors of statin-mediated low-density lipoprotein cholesterol reduction and dose response. Circulation: Cardiovasc. Genet. 1, 100–106 (2008).
Kajinami, K., Takekoshi, N., Brousseau, M. E. & Schaefer, E. J. Pharmacogenetics of HMG-CoA reductase inhibitors: exploring the potential for genotype-based individualization of coronary heart disease management. Atherosclerosis 177, 219–234 (2004).
Kadam, P., Ashavaid, T., Ponde, C. & Rajani, R. Genetic determinants of lipid-lowering response to atorvastatin therapy in an Indian population. J. Clin. Pharm. Ther. 41, 329–333 (2016).
Ibanez, B. et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 39, 119–177 (2018).
Ciccarelli, M., Santulli, G., Pascale, V., Trimarco, B. & Iaccarino, G. Adrenergic receptors and metabolism: role in development of cardiovascular disease. Front. Physiol. 4, 265 (2013).
McAlister, F. A., Wiebe, N., Ezekowitz, J. A., Leung, A. A. & Armstrong, P. W. Meta-analysis: β-blocker dose, heart rate reduction, and death in patients with heart failure. Ann. Intern. Med. 150, 784–794 (2009).
Kolloch, R. et al. Impact of resting heart rate on outcomes in hypertensive patients with coronary artery disease: findings from the INternational VErapamil-SR/trandolapril STudy (INVEST). Eur. Heart J. 29, 1327–1334 (2008).
Ali, D. C. et al. β-Adrenergic receptor, an essential target in cardiovascular diseases. Heart Fail. Rev. 25, 343–354 (2020).
Cotarlan, V., Brofferio, A., Gerhard, G. S., Chu, X. & Shirani, J. Impact of β1-and β2-adrenergic receptor gene single nucleotide polymorphisms on heart rate response to metoprolol prior to coronary computed tomographic angiography. Am. J. Cardiol. 111, 661–666 (2013).
Petrashevskaya, N. N., Koch, S. E., Bodi, I. & Schwartz, A. Calcium cycling, historic overview and perspectives. Role for autonomic nervous system regulation. J. Mol. Cell. Cardiol. 34, 885–896 (2002).
Shin, J. & Johnson, J. A. Pharmacogenetics of β-blockers. Pharmacotherapy 27, 874–887 (2007).
Bae, S. H., Lee, J. K., Cho, D. Y. & Bae, S. K. Simultaneous determination of metoprolol and its metabolites, α-hydroxymetoprolol and O-desmethylmetoprolol, in human plasma by liquid chromatography with tandem mass spectrometry: A pplication to the pharmacokinetics of metoprolol associated with CYP 2 D 6 genotypes. J. Sep. Sci. 37, 1256–1264 (2014).
Thomas, C. D. & Johnson, J. A. Pharmacogenetic factors affecting β-blocker metabolism and response. Expert Opin. Drug Metab. Toxicol. 16, 953–964 (2020).
Blake, C. M., Kharasch, E., Schwab, M. & Nagele, P. A meta-analysis of CYP2D6 metabolizer phenotype and metoprolol pharmacokinetics. Clin. Pharmacol. Ther. 94, 394–399 (2013).
Sehrt, D., Meineke, I., Tzvetkov, M., Gültepe, S. & Brockmöller, J. Carvedilol pharmacokinetics and pharmacodynamics in relation to CYP2D6 and ADRB pharmacogenetics. Pharmacogenomics 12, 783–795 (2011).
Hamadeh, I. S. et al. Impact of CYP2D6 polymorphisms on clinical efficacy and tolerability of metoprolol tartrate. Clin. Pharmacol. Ther. 96, 175–181 (2014).
Batty, J. et al. An investigation of CYP2D6 genotype and response to metoprolol CR/XL during dose titration in patients with heart failure: a MERIT-HF substudy. Clin. Pharmacol. Ther.95, 321–330 (2014).
Yuan, H. et al. Effects of polymorphism of the β1 adrenoreceptor and CYP2D6 on the therapeutic effects of metoprolol. J. Int. Med. Res. 36, 1354–1362 (2008).
Lymperopoulos, A. & French, F. Pharmacogenomics of heart failure. Methods Mol. Biol. 1175, 245–257 (2014).
Rau, T. et al. Impact of the β1-adrenoceptor Arg389Gly polymorphism on heart-rate responses to Bisoprolol and carvedilol in heart-failure patients. Clin. Pharmacol. Ther. 92, 21–28 (2012).
Joseph, S., Lynham, J., Grace, A., Colledge, W. & Kaumann, A. Markedly reduced effects of (−)-isoprenaline but not of (−)-CGP12177 and unchanged affinity of β-blockers at Gly389-β1-adrenoceptors compared to Arg389-β1-adrenoceptors. Br. J. Pharmacol. 142, 51–56 (2004).
Dishy, V. et al. The effect of common polymorphisms of the β2-adrenergic receptor on agonist-mediated vascular desensitization. N. Engl. J. Med. 345, 1030–1035 (2001).
Green, S. A., Turki, J., Bejarano, P., Hall, I. P. & Liggett, S. B. Influence of beta 2-adrenergic receptor genotypes on signal transduction in human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 13, 25–33 (1995).
Huang, J. et al. ADRB2 polymorphism Arg16Gly modifies the natural outcome of heart failure and dictates therapeutic response to β-blockers in patients with heart failure. Cell Discov. 4, 57 (2018).
Kurnik, D. et al. GRK5 Gln41Leu polymorphism is not associated with sensitivity to β1-adrenergic blockade in humans. Pharmacogenomics 10, 1581–1587 (2009).
Liggett, S. B. et al. A GRK5 polymorphism that inhibits β-adrenergic receptor signaling is protective in heart failure. Nat. Med. 14, 510–517 (2008).
Hein, L., Altman, J. D. & Kobilka, B. K. Two functionally distinct α2-adrenergic receptors regulate sympathetic neurotransmission. Nature 402, 181–184 (1999).
Small, K. M., Forbes, S. L., Rahman, F. F., Bridges, K. M. & Liggett, S. B. A four amino acid deletion polymorphism in the third intracellular loop of the human α2C-adrenergic receptor confers impaired coupling to multiple effectors. J. Biol. Chem. 275, 23059–23064 (2000).
Neumeister, A. et al. Sympathoneural and adrenomedullary functional effects of α2C-adrenoreceptor gene polymorphism in healthy humans. Pharmacogenet. Genomics 15, 143–149 (2005).
Duarte, J. D. et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6, ADRB1, ADRB2, ADRA2C, GRK4, and GRK5 genotypes and beta-blocker therapy. Clin. Pharmacol. Ther. 116, 939–947 (2024).
Ali, H. A. et al. Genetic susceptibility to angiotensin-converting enzyme-inhibitor induced angioedema: a systematic review and evaluation of methodological approaches. PLoS ONE 14, e0224858 (2019).
Liau, Y., Chua, I., Kennedy, M. A. & Maggo, S. Pharmacogenetics of angiotensin-converting enzyme inhibitor-induced angioedema. Clin. Exp. Allergy 49, 142–154 (2019).
Flaten, H. K. & Monte, A. A. The pharmacogenomic and metabolomic predictors of ACE inhibitor and angiotensin II receptor blocker effectiveness and safety. Cardiovasc. Drugs Ther. 31, 471–482 (2017).
Ollomo, B. et al. D Allele and DD genotype of i/d polymorphism in the ace gene in patients with hypertension, stroke and cancer prostate in libreville: a concern given the high frequencies of these signatures in gabonese population I/D polymorphism in the ACE gene in patients with hypertension, stroke and cancer prostate in Libreville: a concern given the high frequencies of these signatures in Gabonese population. J. Proteom. Genomics Res. 2, 3 (2019).
Cambien, F. et al. Plasma level and gene polymorphism of angiotensin-converting enzyme in relation to myocardial infarction. Circulation 90, 669–676 (1994).
Dzimiri, N., Basco, C., Moorji, A. & Meyer, B. F. Angiotensin-converting enzyme polymorphism and the risk of coronary heart disease in the Saudi male population. Arch. Pathol. Lab. Med. 124, 531–534 (2000).
Sarangarajan, R. et al. Ethnic prevalence of angiotensin-converting enzyme deletion (D) polymorphism and COVID-19 risk: rationale for use of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers. J. Racial Ethn. Health Disparities 8, 973–980 (2021).
Tchelougou, D. et al. Renin-angiotensin system genes polymorphisms and essential hypertension in Burkina Faso, West Africa. Int. J. Hypertens. 2015, 979631 (2015).
Martinez, E. et al. Angiotensin-converting enzyme (ACE) gene polymorphisms, serum ACE activity and blood pressure in a Spanish-Mediterranean population. J. Hum. Hypertens. 14, 131–135 (2000).
Baruah, S. et al. Insertion/insertion genotype of angiotensin i-converting-enzyme gene predicts risk of myocardial infarction in North East India. Biochem. Genet. 54, 134–146 (2016).
Zhao, W., Ma, S. & Cui, L. Meta-analysis of angiotensin-converting enzyme insertion/deletion polymorphism and myocardial infarction in Han Chinese. Genet. Mol. Res. 14, 8068–8076 (2015).
Pjević, M. D. et al. Analysis of the association between polymorphisms within PAI-1 and ACE genes and ischemic stroke outcome after rt-PA therapy. J. Pharm. Pharm. Sci. 22, 142–149 (2019).
Malueka, R. G. et al. The D allele of the angiotensin-converting enzyme (ACE) insertion/deletion (I/D) polymorphism is associated with worse functional outcome of ischaemic stroke. Int. J. Neurosci. 128, 697–704 (2018).
Vijayan, M. et al. ACE-II genotype and I allele predicts ischemic stroke among males in south India. Meta Gene 2, 661–669 (2014).
Jeunemaitre, X. et al. Molecular basis of human hypertension: role of angiotensinogen. Cell 71, 169–180 (1992).
Shahid, M. et al. Genetic polymorphism in angiotensinogen and its association with cardiometabolic diseases. Metabolites 12, 1291 (2022).
Jeng, J.-R. Left ventricular mass, carotid wall thickness, and angiotensinogen gene polymorphism in patients with hypertension. Am. J. Hypertens.12, 443–450 (1999).
Schmidt, R. et al. Angiotensinogen polymorphism M235T, carotid atherosclerosis, and small-vessel disease-related cerebral abnormalities. Hypertension 38, 110–115 (2001).
Mellen, P. B. & Herrington, D. M. Pharmacogenomics of blood pressure response to antihypertensive treatment. J. Hypertens. 23, 1311–1325 (2005).
Tran, T. T. et al. Association between AGT M235T and left ventricular mass in Vietnamese patients diagnosed with essential hypertension. Front. Cardiovasc. Med. 8, 608948 (2021).
Helin, K., Stoll, M., Meffert, S., Stroth, U. & Linger, T. The role of angiotensin receptors in cardiovascular diseases. Ann. Med. 29, 23–29 (1997).
Kontou, P. et al. Identification of gene expression profiles in myocardial infarction: a systematic review and meta-analysis. BMC Med. Genomics 11, 1–11 (2018).
Andrikopoulos, G. K. et al. The paradoxical association of common polymorphisms of the renin-angiotensin system genes with risk of myocardial infarction. Eur. J. Cardiovasc. Prev. Rehabil. 11, 477–483 (2004).
de Araújo, M. A. et al. The A1166C polymorphism of the angiotensin II type-1 receptor in acute myocardial infarction. Arq. Bras. Cardiol. 83, 404–408 (2004).
Gorący, I. et al. The genetic variants in the renin-angiotensin system and the risk of heart failure in polish patients. Genes 13, 1257 (2022).
Liu, Y. et al. A1166C polymorphism of the angiotensin II type 1 receptor gene and essential hypertension in Han, Tibetan and Yi populations. Hypertens. Res. 25, 515–521 (2002).
Staessen, J. A. et al. Genetic variability in the renin-angiotensin system: prevalence of alleles and genotypes. J. Cardiovasc. Risk4, 401–422 (1997).
Frazier, L., Turner, S., Schwartz, G., Chapman, A. & Boerwinkle, E. Multilocus effects of the renin–angiotensin–aldosterone system genes on blood pressure response to a thiazide diuretic. Pharmacogenomics J. 4, 17–23 (2004).
Su, X. et al. Association between angiotensinogen, angiotensin II receptor genes, and blood pressure response to an angiotensin-converting enzyme inhibitor. Circulation 115, 725–732 (2007).
Mas, S. et al. Pharmacogenetic predictors of angiotensin-converting enzyme inhibitor-induced cough: the role of: ACE:: ABO:, and: BDKRB2: genes. Pharmacogenet. Genomics 21, 531–538 (2011).
Mosley, J. D. et al. A genome-wide association study identifies variants in KCNIP4 associated with ACE inhibitor-induced cough. Pharmacogenomics J. 16, 231–237 (2016).
Strunk, R. C. Integration of mouse and human genome-wide association data identifies KCNIP4 as an asthma gene. PLoS ONE 8, e56179 (2013).
Pare, G. et al. Genetic variants associated with angiotensin-converting enzyme inhibitor-associated angioedema. Pharmacogenet. Genomics 23, 470–478 (2013).
Acknowledgements
This research work was funded by the Institutional Fund Projects under grant number (IFPIP: 990-247-1443). The authors gratefully acknowledge the technical and financial support provided by the Ministry of Education and King Abdulaziz University, Deanship of Scientific Research, Jeddah, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
A.S. performed the literature search and wrote the first draft. P.N.P. and M.R. critically revised the content and proofread the manuscript. A.B.A. contributed to manuscript editing and refinement. L.S.M. and M.A.B. literature review. M.I.N. critically revised the content. F.A. and M.A.-E. supervision. S.B. supervised the study and provided final approval of the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no potential conflicts of interest. Although Prof. Muhammad Abu-Elmagd serves as an editor at npj Genomic Medicine, he was not involved in any stage of the peer review or editorial process related to this manuscript.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shorbaji, A., Pushparaj, P.N., Al-Ghafari, A.B. et al. A narrative review of research advancements in pharmacogenetics of cardiovascular disease and impact on clinical implications. npj Genom. Med. 10, 54 (2025). https://doi.org/10.1038/s41525-025-00511-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-025-00511-6
