
Abstract
The scientific understanding and demonstration of the immobilization of heavy metals (notably lead and chromium) in food contact materials is crucial for the safety of the environment and humans. To tackle this issue lead crystal glasses doped between 50 and 2000 ppm of Cr2O3 were altered under severe aqueous alteration conditions (acetic acid 4%, pH 2.4, 70 °C, 330 days). The increase in Cr leads to a decrease in the Pb release into the solution. The leached Cr content was lower than Pb, itself much lower than alkalis. Structural characterization before and after alteration shows the presence of Cr(III) exclusively, with no redox changes in the course of alteration. Cr and Pb are partially retained during alteration near cationic domains, which are less accessible to protonated species. Cr favors the Si recondensation in the rich non-bridging oxygen (NBO) mixing sites, leading to an increase of glass polymerization within the altered layer.
Similar content being viewed by others

Chromium is used as a colorant in the glass industry rendering different colors as a function of its oxidation state, notably blue for Cr(II), green for Cr(III) and yellow for Cr(VI). The latter was introduced in the 1970s as a replacement for uranium in tinting wine bottles after uranium was banned due to well-documented human health risks. But in 2001, after working on Pb, Cd and Hg, a group of international experts gained an interest in hexavalent chromium following a report by the World Health Organization1 about the toxicity of Cr(VI) towards the environment and human health. These experts highlighted the scientific challenge of accurately distinguishing between Cr(III), considered harmless, and Cr(VI) in various media, including solids and liquids, as well as inhibiting the oxidation of Cr(III) to Cr(VI). The latter must be carefully controlled and immobilized, particularly in the case of glass containers intended for food consumption or packaging, where prolonged contact between the glass and the edible contents occurs. A method was then proposed for the detection of Cr(VI) in daily glass containers with a claimed sensitivity of 2 mg/kg of glass. The method comprises total dissolution of the glass through strong acidic attack and analysis of Cr(VI) by spectrophotometry2. About 20 years later, the European commission has lowered the authorized Cr(VI) content to 0.005 mg/kg for liquid or sticky toy material3.
Another comparison can be made with plastic materials, since the European Regulation on articles intended to come into contact with food was amended in 20204 taking into account the great difficulty to differentiate Cr(III) from Cr(VI). Therefore two different regimes were promulgated, (i) if it can be proved that the presence of hexavalent chromium in the plastic material can be excluded because it is not used or formed during the entire production process, a migrating limit of 3.6 mg/kg is applied but, (ii) if it is not the case, then the second regime is enforced: a migration limit of 0.01 mg/kg corresponding to the detection limit for total chromium according to current tests performed by the National Reference Laboratories.
The trend for lowering the authorized thresholds raises the question of the scientific limits for Cr(VI) routine detection and draws attention to the fundamental understanding of the structural role and behavior of chromium in glass. Addressing these challenges from a materials science perspective, using advanced analytical techniques, could prevent the complete banning of chromium in all oxidation states from industrial use, provided it can be definitively demonstrated that Cr(III) cannot transform into Cr(VI). Currently, Cr(VI) is subject to authorization in Europe under Appendix XIV of the Registration, Evaluation, Authorization, and Restriction of Chemicals (REACH) regulation, which lists substances of very high concern (SVHC)5. This regulation compels industrial manufacturers to reconsider their use of chromium, which remains essential in its Cr(III) form, particularly in the long-standing process of producing green-colored lead crystal glass.
The main goal of our study was to understand the effects of chromium on the structure and durability of lead crystal glass. The alteration of lead crystal glass has already been studied through various works in the literature, especially with acetic acid solution6,7 and at the same leaching temperature, 70 °C6,8. These works have already evidenced the rapid initial leaching of alkalis at the first stage of alteration which is then inhibited over time by the increase of the diffusion distance and the repolymerization of the glass silicate network with the formation of a diffusive barrier of a few hundreds of nanometers. The release of lead is significantly lower than that of alkalis, and its leaching rate becomes nearly negligible after the reorganization of the silicate network, indicating effective retention of lead in the altered layer over the course of several years8. But to date, the impact of the leaching of chromium on the other constitutive elements of lead crystal glass (Si, Pb, Na and K) and on the solution chemistry have not been reported. The structure of Cr-free lead crystal glass of the same composition has already been examined extensively by solid-state high-resolution nuclear magnetic resonance (NMR)6,8,9. Building on from this knowledge, further structural characterizations would allow to determine the structural role of chromium and its interactions with the constitutive elements in a lead crystal glass matrix.
To answer all these questions, 4 ingots of lead crystal glasses with roughly 50, 250, 500 and 2000 ppm of chromium oxide (Cr2O3) were prepared by adding the coloring oxide to the plain crystal glass creating a palette of green hues from light tender green to deep forest green. These glasses were then leached in acetic acid 4% (v/v), considered as the reference medium for food contact studies10, at 70 °C to accelerate alteration mechanisms, in the form of powder and monoliths up to 330 days. Monoliths were analyzed by micrometric X-ray fluorescence (µ-XRF) and X-ray absorption near edge spectroscopy (XANES) at the Cr K-edge. These techniques allowed us to determine the oxidation state of chromium in the pristine and altered glass, tracking potential disproportionation mechanisms of this element induced by the alteration process. These results were supported by optical absorption spectroscopy performed on the pristine glass slabs. Finally the structural changes of the pristine glass structure brought by the addition of chromium were investigated by solid-state 29Si and 207Pb NMR spectroscopy.
Results
Glass composition
The glasses shown in Fig. 1 were found to be homogeneous and fully vitreous under scanning electron microscopy (SEM) down to the micrometer scale. They were analyzed by electronic micro-probe analyzer (EMPA) and the results are available in Table 1 displaying Cr concentrations ranging from 0.002 mol% (BAC_Cr50) to 0.028 mol% (BAC_Cr2000) and minimal composition differences besides the Cr content.

As-cast glass ingots are on the top of the image (5 cm wide and thick each) and polished monoliths are at the bottom, each measuring 5 × 5 × 2 mm.
Leaching of chromium colored lead crystal glasses
Glass powders of BAC_Cr50, BAC_Cr250, BAC_Cr500, BAC_Cr2000 were altered for 330 days with a high SAgeo/V ratio (500 m−1 calculated from Eq. (1) combined with the elemental concentrations (Eq. (2)) and density (Eq. (3)) giving access to the Equivalent Thickness (ETh) of glass altered per element. The ETh for Si, Pb, Na and K, the main glass constituents, are represented for 112 days in Fig. 2a–d, respectively.

The alkalis are released through an ion-exchange mechanism (linear release as function of the square root of time), as expected from the literature6,7,8. The evolution of ETh(Na, K, Pb) have been represented as a function of the square root of time, as displayed in Supplementary Fig. 1a for Na, 1b for K and Fig. 4a for Pb. The trend for Na and K is fully linear confirming the diffusion-controlled mechanism of alteration.
Silicon showed the lowest ETh of all measured elements, about 100 times smaller than for alkalis, indicative of a slow hydrolysis reaction of the silicate network compared to the fast ion-exchange mechanism. This results in a linear release of Si as a function of time. There is no relation between Cr content in the glass and the ETh of Na, K, and Si. The increasing concentration of dissolved silica reduces the rate of Si‒O‒Si hydrolysis and favors the reorganization of the alteration layer11. Polymerization may contribute to the formation of a diffusive barrier, along with other phenomena, which could explain the sharp decrease in alkali release observed after 14 days12.
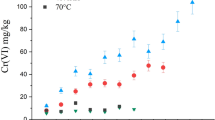
Unlike the other constitutive elements studied (Na, K and Si), the leaching of Pb showed a different behavior. The release of Pb under diffusive conditions is only observed over short timescales. The reorganization within the alteration layer subsequently leads to further slowing down the release of Pb. The final sampling at 330 days confirmed this behavior. (Fig. 3). Additionally, the ETh(Pb) showed a clear correlation with the Cr content in the glass, demonstrating a decrease in ETh(Pb) as the Cr content increased. Moreover, like for Si, the ETh(Cr) evolution is linear as a function of time (Fig. 3). At 330 days, the Cr and Pb leached layer is about 200 and 500 nm, respectively.

Evolution of the equivalent thicknesses (ETh) of chromium colored lead crystal glasses for Pb and Cr during alteration at 70 °C and pH = 2.4 in acetic acid 4% (v/v) with a SAgeo/V ratio of 500 m−1 (glass powder 63-125 μm).
For Pb, a domain of linearity between 1 and 14 days of alteration can be seen in Fig. 4a, from which apparent diffusion coefficients were calculated using Eq. 5 for each of the Cr-bearing glasses. These coefficients D(Pb) are presented as a function of the logarithm of the molar content in Cr of the glasses in Fig. 4b, exposing a linear correlation between the decrease of the diffusion coefficient of Pb and the logarithm of the molar Cr content in these glasses.

a Evolution of the equivalent thicknesses (ETh) of Pb as a function of square root of time from Cr lead crystal glasses. b Apparent diffusion coefficients of Pb after 14 days of alteration calculated from Eq. (5) as a function of the logarithm of the Cr content in the glass.
Pristine glass structure and properties
For many decades, various relationships between alteration behavior and glass structure have been highlighted and investigated by the glass community across a wide range of glass compositions, including lead crystal glass6. As the results of the present work revealed the specific role of Cr, investigations into the Cr redox state and Cr impact on the pristine glasses structure were conducted. The oxidation state of Cr was first determined by optical absorption spectroscopy to gain information on the configuration of Cr in the bulk pristine glasses. Then, the structure of glasses was addressed through 29Si and 207Pb solid-state NMR.
Optical absorption spectroscopy
For Cr, the most common oxidation states found in silicate glasses are Cr(III) and Cr(VI), which have close but distinct absorption bands in the visible and near ultraviolet regions of the electromagnetic spectrum13. The presence of both species in the Cr-bearing pristine glasses has been investigated by optical absorption spectroscopy and the results are shown in Fig. 5. The spectra obtained for the pure crystalline reference compounds of Cr(III) and Cr(VI) (extracted from Bamford (1977)14) are displayed in Fig. 5. The spectrum of Cr(III) is characterized by two large Gaussian shaped bands of absorption centered at 15,000 and 22,000 cm−1 which are responsible for the green color of Cr(III)-bearing materials. They account for the spin-allowed, parity forbidden d–d transitions of octahedral Cr3+: 4A2g → 4T2g and 4A2g → 4T1g. The most oxidized species of Cr, Cr(VI), is identified by an unique Gaussian shaped absorption band, centered at 27,500 cm−1 and yields yellowish colored glasses, especially when no other Cr species are present. The spectra obtained from pristine Cr-bearing glasses were collected over large areas of the glasses (about 7 mm2) and are presented above the reference spectra clearly showing the presence of Cr(III). The increasing intensity of the bands located at 15,000 and 22,000 cm−1 aligned with the increase of the total Cr-content in the glasses.

The position of the absorption bands characteristic of the two Cr oxidation states are indicated by the dotted experimental spectra obtained on reference oxide compounds by Bamford14. Spectra are normalized to their maximal intensity and to the thickness of the sample. Spectra have been shifted vertically for better readability.
From the Beer–Lambert law, the absorbance of a glass depends on the molar extinction coefficient of its constituents, the length of the light path and the concentration of the optically active species, in our case Cr(III) and Cr(VI). It has been calculated that for Cr(III) in alkali silicate glasses, the molar extinction coefficient εCr(III) is around 18–20 L mol−1 cm−114,15,16, whereas for Cr(VI) the molar extinction coefficient associated with the absorption band at 27,500 cm−1 is around 4200 L mol−1 cm−1 in the same glass matrices13,17,18. Hence an absorbance more than 200 times greater for Cr(VI) compared to Cr(III) at equal concentrations and light path length. Considering our least concentrated sample, with a given light path and a given total Cr concentration of 0.002 mol%, the absorbance of equal concentrations of Cr(III) and Cr(VI), which depends directly on their respective molar extinction coefficients, is then 200 times more intense for Cr(VI) than Cr(III). Consequently, a 200 times lower concentration of Cr(VI) than Cr(III) can be detected. Since only two absorption bands are detected at 15,000 and 22,000 cm−1, corresponding to Cr(III) signal only, it can be assumed that the detection limit for Cr(VI) is at least 200 times lower than the total Cr amount which has been attributed to Cr(III), that is 1 × 10−5 mol%. Thus, the optical absorption spectroscopy demonstrated that the Cr-bearing pristine glasses contained Cr(III) only with a detection limit for Cr(VI) of 1 × 10−5 mol%.
29Si and 207Pb NMR spectroscopy
The distribution of Qn species in the glass is accessed via 29Si MAS NMR, which was performed on Cr-bearing glasses. The results are presented in Fig. 6 for the two extrema of Cr-bearing glasses (0.002 and 0.028 mol%) showing a distribution centered on Q3 and Q4 species. Tetracoordinated silicate units Q4 have four bridging oxygens (i.e. oxygen bonding two silicate units at a corner). A slight variation in the distribution of Q3 and Q4 species can be observed, with a slight decrease in Q3 and a corresponding increase in Q4 species as the Cr content increases. Surprisingly the increase of a paramagnetic compound like chromium oxide to the glass batch did not affect the intensity of the 29Si MAS NMR spectra obtained for Cr-bearing glasses, while a significant effect was observed in the 207Pb signal intensity as displayed in Fig. 7 where the signal intensity of Cr2000 (0.028 mol% Cr) is approximately twice that of Cr50 (0.002 mol%).

29Si MAS NMR spectra were performed at 11.72 T and normalized to the same sample mass, using identical experimental conditions.

Spectra are normalized to the same sample mass, using identical experimental conditions.
The spectra were normalized to the same sample mass, with identical experimental conditions allowing direct quantitative comparison of the obtained results. Additionally, for the glass with the highest Cr content, the lineshape of the 29Si MAS NMR spectrum did not vary with the recycle delay as shown in Supplementary Fig. 2. Considering the paramagnetic nature of Cr(III), and according to previous studies using contrast in the NMR signal induced by differential spin-lattice relaxation rates (see ref. 19 for example), this suggests that no phase segregation occurred, which would have led to a preferential relaxation of specific Si sites.
The growing intensity of the 207Pb NMR spectra observed in Fig. 7 can only be related to the increase of the Cr content in the glass samples. This could be explained by the shortening of 207Pb relaxation time T1 by paramagnetic Cr, leading to higher intensity signals. No detailed investigation of 207Pb T1 was carried out, mainly because of the very long time required to collect the data (several days). The NMR lineshape, acquired under static, i.e. non-spinning conditions, is close to that of an environment dominated by a large chemical shift anisotropy (CSA) tensor, as already measured in lead silicate glasses20,21. Then, NMR showed that the proximity between Pb and Cr atoms suggested by the increase in 207Pb spectra intensity upon to the addition of Cr.
Structure of altered glasses
XANES on bulk glasses
The behavior of Cr in lead crystal glasses exposed to harsh alteration conditions was investigated using X-ray absorption near edge spectroscopy (XANES) at the Cr K-edge on both pristine and altered glasses at the ID21 beamline of the European Synchrotron Radiation Facility (ESRF, France)22. While optical absorption spectroscopy established the absence of Cr(VI) in the pristine state, the potential changes in oxidation state caused by the local restructuring of the altered layer required the investigation of Cr speciation specifically in the surface region. Two approaches were followed. First, XANES was performed with a large, unfocused beam (100–350 µm) directly at the surface of pristine and altered glass monoliths. Then, µ-XANES analyses were obtained with a focused beam (0.26 × 0.8 µm2, v × h), on a cross-section of altered glass, both in the most external area and in a deeper region. In the first approach, photons are spread over a large area, decreasing the dose and consequently the risk of beam damage. However, it should be taken into account that at the Cr K-edge, the attenuation length of X-ray is 7 µm, larger than the alteration layer (6 µm). Therefore, a small contribution from deep, unaltered glass should not be neglected. In the second approach, the photons are laterally concentrated in a very small probe. Radiation tests were carried out by repeating XANES acquisitions at the same positions and revealed the formation of chromate, Cr(VI), under the beam. To slow down this oxidation reaction, different adaptations to the standard set-up were done. The beam intensity was reduced with a 50 µm Al attenuator; the energy step was increased from 0.4 eV (as used in unfocused mode) to 0.6 eV; the starting energy was increased from 5.950 to 5.985 keV, to avoid unnecessary exposure of the samples to photons. Finally, the beam fast shutter was opened only during data collection. Spectra were collected as single scans, over different points, from the surface to the depth of the glass. An initial µ-XRF map was systematically collected prior to µ-XANES spectra to locate the alteration layer. The first method yielded good quality spectra as shown in Fig. 8, albeit, with merged signals of the pristine and altered glass. The second approach allowed the targeting of alteration layers only but with a low X-ray dose to preserve the samples producing noisier spectra as shown in Fig. 9b.

Each spectrum resulted from the averaging of 2–8 spectra (for each sample the number of spectra averaged is indicated in the caption between brackets). a XANES spectra of the Cr-bearing samples before and after alteration. The spectra are shifted vertically for better readability. b XANES spectra obtained for BAC_Cr2000 before and after alteration and a spectrum from stichtite, a Cr(III) bearing mineral. c XANES spectra obtained for BAC_Cr2000 before and after alteration and spectra from crocoite and fornacite, Cr(VI) bearing minerals. The position of the main peak is indicated as well as the position of the pre-edge features.

a XRF mapping of K K-α with false colors from the edge of the altered coupon. The depleted zone in K is considered as indicative of the altered layer and the locations of the XANES spectra shown in b are marked on the map. b Cr K-edge experimental µ-XANES spectra from the altered zone (spectra altered_50 and altered_55) and the pristine zone (pristine_54) of the cross-section area presented in (a). XANES spectra obtained on bulk slabs of pristine and 330 days altered BAC_Cr2000 are also given for comparison.
Figure 8a displays the superposition of the mean spectrum acquired on bulk pristine and 330 days altered glass for each of the Cr-bearing glass. The same shape of spectrum was observed for all the glasses although the least concentrated glasses in Cr (BAC_Cr50 and BAC_Cr250) yield, evidently, noisier spectra. Excellent overlap of the pristine (solid lines) and altered (dashed lines) XANES spectra of each glass was observed with a slightly lower intensity of the main peak for the spectra collected on altered samples revealing small modification of Cr environment between the pristine and altered glasses. The same acquisitions were carried out on all the samples.
To confirm the oxidation state of Cr in our glasses, the experimental pristine and altered spectra of BAC_Cr2000 were compared to mineral references of Cr(III): stichtite (a magnesium and chromium carbonate), and Cr(VI): crocoite (PbCrO4) and fornacite (Pb2Cu(CrO4)(AsO4)(OH)), both of which contain Pb as in our samples. The results are shown in Fig. 8b, c, respectively. A good agreement can be noticed between the experimental spectra and the Cr(III) reference highlighting a key feature in the pre-edge region with a peak common to all spectra at 5.9913 keV. The features of this region of the spectrum, before the edge, are indicative of electronic transitions from orbital 1s to orbital 3d of transition metal cations like Cr. The intensity of the peak at 5.9913 keV is notably low for all spectra which is expected from the inversion of symmetry of p and d orbitals in the octahedral configuration imposed by Cr(III) valence. Regarding Cr(VI), which exhibits tetrahedral configuration, the geometry of orbitals p and d is favorable to their mixing resulting in an intense pre-edge at 5.9935 keV (Fig. 8c). The post-edge region echoes the Cr local environment which varies a lot among references compounds as awaited. Regarding the edge region in Fig. 8b, an offset of 3 eV between the white-line position of our samples and the stichtite Cr(III) reference is observed. Variation in the edge position is usually significant of different oxidation state.
In this case the shift towards lower energies could be interpreted as the presence of Cr(II) mixed with Cr(III) as described by Berry and O’Neill23. However, the presence of Cr(II), characterized by a large absorption band spreading from 10,000 to 20,000 cm−124, was not detected by optical absorption spectroscopy on the pristine glass (Fig. 5). Most likely, these differences are indicative of the different speciation of the Cr in the references and glasses of interest. Thus XANES spectra on the bulk pristine and altered Cr-bearing lead crystal glasses evidenced the presence of Cr(III), reinforcing the conclusions obtained by optical absorption spectroscopy in the pristine glasses.
µ-XRF and µ-XANES on cross-sections
XRF mapping of polished cross-sections of the altered monoliths enabled the identification of an alkali depleted layer outlining the pristine glass and corresponding to the alteration layer. An example of XRF mapping for potassium (K-K-α) obtained on the BAC_Cr2000 cross-section after 330 days of alteration is given in Fig. 9a. The layer appeared of constant thickness and perfectly parallel to the edge of the sample despite the inhomogeneity which arose from the sample preparation. The measured thickness of the potassium depleted altered layer was 6.00 ± 0.25 µm, corresponding to the green area at the top of Fig. 9a. The dark blue area represents the epoxy resin that embedded the sample, and the yellow region designates the pristine glass with the varying hues reflecting the local inhomogeneity created by the sample preparation and not actual concentration gradients of potassium.
By focusing the incident X-ray beam a resolution of 0.8 × 0.26 µm2 (h × v) was obtained allowing µ-XANES analyses in the pristine glass and in the altered layer distinctively. Three points of interest were selected, their location is indicated in Fig. 9a and the corresponding XANES spectra are presented in Fig. 9b alongside the spectra obtained on bulk slabs of BAC_Cr2000 with a defocused beam. As anticipated, due to the attenuated intensity of the beam used for focused XANES analyses on the cross-section, the spectra obtained display more background noise than those collected on the bulk glass using a defocused beam. Nonetheless, the comparison of spectra from the pristine glass, cross-section and bulk showed very similar shape and intensity, validating the presence of Cr(III) only in both cases. For the spectra obtained from the alteration layer of the cross-section (spectra ‘altered_50’ and ‘altered_55’), the edge position remained consistent with that observed in the bulk glass analyses (spectrum ‘BAC_Cr2000 (5) altered’) and in the pristine glass, located at 6.0075 keV. The pre-edge region (5.9800–5.9950 keV) of the cross-section spectra is not very well defined because of the low dose of X-ray used in the focused mode to avoid photoinduced oxidation. The presence of Cr(VI), indicated by a pre-edge at 5.9935 keV with greater intensity than that expected for Cr(III)13, should be distinguishable from the background noise. The absence of distinct features in this region can be interpreted as evidence for the lack of Cr(VI). Consequently, µ-XANES allowed the identification of Cr(III) only in pristine and altered Cr-bearing lead crystal glass samples with a Cr intake up to 0.028 mol%.
29Si NMR spectroscopy
After alteration, glass powder was sampled from each reactor. These powders were dried and analyzed by 29Si MAS NMR. Figure 6 displays the structural changes in the silicate network induced during the leaching time. These changes are characterized by a shift towards lower chemical shifts after alteration meaning a higher amount of Q4 species to the detriment of Q2 and Q3 species, all indicative of the increase of polymerization of the silicate network during alteration. It corresponds to the strengthening of the most superficial part of the altered layer that is acting as a diffusive barrier and is responsible for the drop of the alteration rate suggested in Fig. 2 through the ETh of Na, K and Pb after 14 days.
The extent of polymerization can be inferred from the increase of the area of the Q4 line. After alteration, Cr-rich glass BAC_Cr2000 had the largest proportion of Q4 species compared to BAC_Cr50. Consequently, it can be assumed that the presence of Cr in the glassy matrix favored the polymerization of the glass during alteration. Besides the slightly more polymerized silica network noticed for Cr-bearing glasses in the pristine state, this trend is then confirmed and amplified after alteration by the leaching mechanisms demonstrating the strong effect of Cr, even at low concentrations, on the structure of Cr lead crystal glasses.
Discussion
The presence of Cr(III) only in the investigated glasses has been established using XANES and optical absorption spectroscopy. Moreover, extensive studies of Cr(III) in various other Cr-containing glasses using the same analytical techniques arrived at the following conclusions. On one hand, XANES showed that Cr(III) was always found hexacoordinated with an octahedral geometry and low variations in its local environment15,23,25,26. On the other hand, optical absorption spectroscopy revealed the preferential localization of Cr atoms in cationic domains of the glass27 with a preference for alkalis over earth-alkalis15. Overall, Cr(III) was not uniformly distributed within the glass networks but was located in specific, somewhat disordered sites15.
Previous structural studies on lead crystal glass without Cr using 17O multi-quantum magic-angle spinning (MQMAS) NMR6 highlighted the presence of sites with mixed Pb and K, written: Si–O–(Pb, K), near non-bridging oxygens (NBOs). These sites exhibit the previously described characteristics for hosting Cr(III), namely a higher degree of chemical disorder in the cationic domains. 207Pb static NMR spectroscopy successfully demonstrated the proximity of Pb and Cr within the glass network. In lead crystal glasses, 10.6 mol% of PbO is too low to form a subnetwork of Pb units. Consequently, Pb is not considered as a network former, and the glass network is composed of silicate units, within whose interstices Pb and alkali metals are localized28,29. Thus, Cr cations could be positioned near these NBO domains. 29Si MAS NMR spectroscopy points to a slight increase in polymerization as the Cr content increases. These findings support the description of Cr(III) as a network hardener, reinforcing the network polymerization of the pristine glass.
From the structural and alteration data, the specific role of Cr can be further explored. Cr is partially retained near the cationic domains during alteration, also improving Pb retention though the role of network hardener. Pb and Cr entities are less accessible to protonated species, which reduces their mobility compared to alkali metals that are distributed throughout the entire glassy matrix. Consequently, their exchange and transport kinetics are slower compared to Na and K. The part where Cr and Pb are leached favors Si recondensation in the rich NBO mixing sites, leading in an increase of the glass polymerization within the altered layer, as observed by 29Si MAS NMR after leaching. The polymerization is clearly amplified when the Cr content in the glass increases.
The fact that the release kinetics of Pb after 14 days of alteration is lower than a purely diffusional behavior proportional to the square root of time may be linked to the increased polymerization in the alteration layer, which serves as an additional barrier. On the other hand, Cr is released similarly to Si, in agreement with the role of a network hardener in the silicate network.
The high sensitivity of lead crystal glass structure to very low contents of Cr can be outlined. In 2016, Liao et al.30 reported a double barrier effect leading to the immobilization of Cr under the form of Cr(III) in glass-ceramic material where Cr(III) was retained in crystallized spinel structure and in the glassy matrix. In this study, we extend this conclusion to a fully amorphous material, demonstrating how Cr used as a colorant is well retained while 50% of the alkalis are leached out of the glass developing micron-sized alteration layers.
The results are favorable from a toxicological and environmental standpoint, supporting the absence of Cr(VI) in both the pristine and altered glasses. Additionally, only a small amount of Cr was measured in the leachate after 330 days of alteration under extremely harsh conditions (70 °C, pH = 2.4, SAgeo/V = 500 m−1). Furthermore, the origin of the retention mechanism was clarified thanks to structural analysis tools like XANES at Cr K-edge and NMR of 29Si and 207Pb nuclei, overcoming the technical difficulties inherent to the paramagnetic and easily oxidized materials under study.
Thus, under very unfavorable food simulating conditions of alteration, the Cr contained in the original materials did not oxidize to Cr(VI) over the course of fabrication nor alteration of the glass and was well immobilized with Pb in the glass structure. Under normal conditions of use (significantly lower temperature and contact time), the expected releases of Pb and Cr should be well below the limits imposed by standardized tests (24 h at 25 °C)10.
Methods
Glass preparation
Four ingots of lead crystal glass with 50, 250, 500 and 2000 ppm of chromium oxide, as visible on the images in Fig. 1, were fabricated from a single batch of lead crystal glass. Out of each of these ingots, square shaped coupons measuring 5 × 5 × 2 mm were cut and all faces were polished to 1 µm (roughness of the order of a few nanometers) to obtain perfectly geometrical and smooth surfaces. The coupons were used for µ-XRF and XANES analyses. A few dozen grams of each glass were crushed, sieved and washed with acetone and absolute ethanol in an ultrasonic bath to obtain powders with particle size diameter ranging from 63 to 125 µm. The specific surface of each of these powders was measured with the BET method using Kr gas. The composition of the glasses was determined by electronic micro-probe analyzer (EMPA) and the results are displayed in Table 1. The standard deviation was calculated for each element of each glass based on 10 repetitions and the relative uncertainty is equal or below 5% of the given values for all elements, except Cr, for which the uncertainty rose up to 50% because of the very low quantities added.
Leaching experiments
Samples glasses were altered with two experiments following the same protocol of alteration, the first one using glass monoliths and the second one using glass powder, yielding different glass-surface-area-to-solution-volume-ratios (SA/V ratio). These were determined geometrically in both cases by precisely measuring the monolith dimensions and using Eq. (1) for powders. In both cases, long term alteration in static mode was performed in perfluoroalkoxy alkane reactors (PFA), at 70 ± 3 °C in 4% (v/v) acetic acid solution, which corresponds to a pH of 2.4 ± 0.1. Acetic acid solutions were prepared by diluting acetic acid (100%, Merck Suprapur) in 18 MΩ cm ultrapure water. For the alteration of glass coupons with chromium oxide content the initial SA/V ratio obtained for the alteration of 5 coupons (5 × 5 × 2 mm) of each glass in the same reactor was 2.45 ± 0.03 m−1. After 330 days of alteration, all the glass coupons were removed from the reactors and dried at 90 °C for 2 h before cooling down over night at room temperature and humidity. One altered coupons of each glass was embedded in epoxy resin and polished to obtain cross-sections that were then analyzed by µ-XRF and XANES at the ESRF synchrotron. As this experiment, with a very low SA/V ratio, did not allow to measure precisely of the leached-out element of the glasses, the second experiment with a high SA/V ratio was undertaken using about 10 g of 63–125 µm calibrated powder and 500 g of acetic acid 4% (v/v) solution to reach a SA/V ratio of 500 m−1, thus increasing the amount of leached elements in solution.
With SAgeo/V the glass-surface-area-to-solution-volume-ratio determined with the geometric approximation (m−1), Vsol the volume of solution in the experiment (m3), mglass the mass of glass in the experiment (g), ρ the density of the glass (g m−3) and Rmean the mean radius of glass particles in the experiment (m). The density of an equivalent glass without Cr was measured to be 3.03 g cm-3. Given the low Cr content, this density can be applied to all glasses in the series.
The alteration solution was regularly sampled from the reactors during the first 112 days of the experiment and the concentrations of glass constituents in the solution were analyzed by Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES) after acidification with nitric acid (1 mL HNO3 0.5 N, Suprapur grade). A final verification sampling was carried out at 330 days. The solution withdrawal generated by the samplings of 2 mL each is considered by recalculating the glass-surface-area-to-solution-volume ratio after each sampling.
For each solution sampling, the normalized mass loss of each element of each glass was calculated using Eq. (2).
With NLgeoi,t the normalized mass loss of element i after t days of alteration (g m−2), C(i)t the concentration of element i in solution after t days of alteration (g m−3), SAgeo/V the glass-surface-area-to-solution-volume-ratio determined with the geometric approximation after t days of alteration (m−1), xi the mass fraction of element i in the pristine glass.
The normalized loss per element can be translated to equivalent thicknesses of glass altered according to Eq. (3) and enable the calculation of alteration rates through partial derivation with respect to time given in Eq. (4).
With \({{\rm{E}}{\rm{T}}{\rm{h}}}_{{{\rm{g}}{\rm{e}}{\rm{o}}}_{i,t}}\) the equivalent thickness of glass altered for element i after t days of alteration (m), \({{\rm{N}}{\rm{L}}}_{{{\rm{g}}{\rm{e}}{\rm{o}}}_{i,t}}\) the normalized mass loss of element i in solution after t days of alteration (g m−2), ρ the pristine glass density (g m−3)
With ri(t) the release rate of an element i for a given time of alteration (g m−2 d−1) and NLgeoi,t the normalized losses of element i in solution after t days of alteration (g m−2).
The diffusion coefficients for species with interdiffusion behavior were calculated using Eq. (5).
With Di the diffusion coefficient of an element i for a given time of alteration (m2 s−1), ri(t) the partial derivative of normalized mass loss of element i with respect to time (g m−2 s−0.5) and ρ the pristine glass density (g m−3).
For this protocol, the overwhelming uncertainty in the calculation of ri(t), \({{\rm{ETh}}}_{{{\rm{geo}}}_{i,t}}\),), \({{\rm{N}}{\rm{L}}}_{{{\rm{g}}{\rm{e}}{\rm{o}}}_{i,t}}\) is the ICP-AES determination of elemental concentrations in solution. These come out with a 10% relative uncertainty, which is then reflected on the calculated data.
NMR spectroscopy
NMR data on 29Si were collected on a Bruker AVANCE II 500WB spectrometer operating at a magnetic field of 11.7 T, using a Bruker 4 mm (outer diameter of the rotor) cross-polarization MAS HX probe at a spinning frequency of 12.5 kHz. 29Si MAS NMR spectra were acquired using a saturation-recovery pulse sequence with a recovery delay of 20 s (increasing the recycling delay did not alter the lineshape). The 29Si NMR shifts were referenced against tetrakis(trimethylsilyl)silane powder (two lines at 9.9 and 135.3 ppm with respect to tetramethylsilane).
207Pb NMR spectra were acquired at a magnetic field of 7 T on a Bruker AVANCE II Spectrometer. Because of the very large width of the spectra (mainly due to a very large chemical shift anisotropy (CSA)20), the variable-offset cumulative spectroscopy (referred to as VOCS)20,31 has been employed under static conditions (i.e. non-spinning sample), using carrier frequency jump of 50 kHz (typically from −150 to +150 kHz with the origin taken at the center of the spectrum). Typically, 48,000 scans have been accumulated with a recycle delay of 2 s. For some offset frequency values, spectra at recycle delays of 0.5, 2, 8 and 16 s have been compared to quantitatively estimate the relaxation of the signal and the impact of the Cr content (which was found to shorten significantly the relaxation time of 207Pb).
µ-XRF and XANES at ESRF
Measurements were performed at the scanning X-ray microspectroscopy end station of the beamline ID21 of the European synchrotron radiation facility (ESRF, Grenoble)22. A Si(220) double-crystal monochromator was used to scan the X-ray beam energy across Cr K-edge (5.9760 keV). For the calibration, the first inflection point of the first-order derivative Cr K-edge XANES spectrum of a Cr metallic foil was set at 5.9900 keV. Photoinduced oxidation was evidenced by repeating XANES spectra on the same spot and observing growing intensity of pre-edge at 5.9935 keV indicative of oxidation into Cr(VI) occurring under beam exposition. To avoid these beam damage artefacts, especially in the targeted altered layer, two different modes of analyses were adopted: (i) defocused beam, to sense large areas with a minimal dose of X-rays and (ii) focused beam, µ-XANES, to target alteration layers specifically. In defocused mode, the beam size ranged from 100 to 350 µm, and acquisitions were carried out from 5.95 to 6.15 keV, with 500 steps of 0.4 eV. For XANES investigations, the incident beam was focused with a Kirkpatrick–Baez (KB) mirror system down to a spot size of 0.8 × 0.26 µm2 (h × v) and the beam was attenuated with a 50 µm Al foil. The energy range was reduced to 5.985–6.15 keV, with steps of 0.6 eV. All data were collected under a vacuum.
Optical absorption spectroscopy
UV–visible–near IR transmission spectra were recorded on Cr-bearing glass samples at room temperature on thin coupons with both faces polished with a double beam Perkin-Elmer Lambda-1050© spectrometer in the spectral range between 3030 and 40,000 cm-1 with a wavelength (λ) step and spectral resolution of 0.92 cm−1 (1 nm). After correction for the signal jump at 11,600 cm−1 due to detector change, the absorption spectra were normalized to sample thickness. The area analyzed was about 2 × 3.5 mm2 sensing a large portion of the glass slabs and allowing a highly representative measurement of the optical absorbance.
Data availability
The data that supports the findings of this study are available from the corresponding author upon reasonable request.
References
Andersen, et al. Chromium and chromium compounds. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 49: Chromium, Nickel and Welding p 256-447 (World Health Organization, Geneva, 1990), IARC, Lyon, France.
Guadagnino, E., Sundberg, P. & Corumluoglu, O. A collaborative study on the determination of hexavalent chromium in container glasses—a report of ICG/TC2 ‘Chemical Durability and Analysis’. Glass Technol. 42, 148–152 (2001).
E.U. Commission. E.U. Commission Directive (EU) 2018/725—of 16 May 2018—amending, for the purpose of adaptation to technical and scientific developments, point 13 of part III of Annex II to Directive 2009/48/EC of the European Parliament and of the Council on the safety of toys, as regards chromium VI (E.U. Commission, 2018).
E.U. Commission. E.U. Commission Regulation (EU) 2020/1245 of 2 September 2020 Amending and Correcting Regulation (EU) No. 10/2011 on Plastic Materials and Articles Intended to Come into Contact with Food (E.U. Commission, 2020).
E.U. Commission. E.U. Commission Regulation (EU) No. 125/2012 of 14 February 2012 amending Annex XIV to Regulation (EC) No. 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) (E.U. Commission, 2012).
Angeli, F., Jollivet, P., Charpentier, T., Fournier, M. & Gin, S. Structure and chemical durability of lead crystal glass. Environ. Sci. Technol. 50, 11549–11558 (2016).
Lecanuet, G., Rocca, E., Hee, P., Skaper, M.-A. & Rapin, C. Mechanism of alteration of the surface of lead crystal glass in contact with food: a chemical study of the surface layer. Appl. Surf. Sci. 580, 152281 (2022).
Brunswic, L. et al. Comparative study of the structure and durability of commercial silicate glasses for food consumption and cosmetic packaging. NPJ Mater. Degrad. 8, 82 (2024).
Collin, M. et al. Chemical durability of lead crystal glass: Comparison of short-term aqueous and atmospheric alteration at 90 °C. Int. J. Appl. Glass Sci. 12, 158–174 (2021).
E.U. ISO 7086:2019. Glass Hollowware in Contact with Food—Lead and Cadmium Emission (E.U. ISO, 2019).
Bunker, B. Molecular mechanisms for corrosion of silica and silicate-glasses. J. Non-Cryst. Solids 179, 300–308 (1994).
Gin, S. et al. Insights into the mechanisms controlling the residual corrosion rate of borosilicate glasses. NPJ Mater. Degrad. 4, 41 (2020).
Villain, O., Calas, G., Galoisy, L. & Cormier, L. XANES determination of chromium oxidation states in glasses: comparison with optical absorption spectroscopy. J. Am. Ceram. Soc. 90, 3578–3581 (2007).
Bamford, C. R. Colour Generation and Control in Glass Vol. 2, Glass Science and Technology (Elsevier, North-Holland, Amsterdam, New York, 1977).
Villain, O., Galoisy, L. & Calas, G. Spectroscopic and structural properties of Cr3+ in silicate glasses Cr3+ does not probe the average glass structure. J. Non-Cryst. Solids 356, 2228–2234 (2010).
Calas, G., Galoisy, L., & Cormier, L. The color of glass. In Encyclopedia of Glass Science, Technology, History, and Culture 1st edn (eds Richet, P., Conradt, R., Takada, A. & Yon, J.) 677–691 (Wiley, 2021).
Stålhandske, C. The impact of refining agents on glass colour. Glasteknisk Tidskrift 55, 65–71 (2000).
Nath, P., Paul, A. & Douglas, R. Physical and chemical estimation of trivalent and hexavalent chromium in glasses. Phys. Chem. Glasses 6, 203 (1965).
Stebbins, J. F., Kim, N., Andrejcak, M. J., Boymel, P. M. & Zoitos, B. K. Characterization of phase separation and thermal history effects in magnesium silicate glass fibers by nuclear magnetic resonance spectroscopy. J. Am. Ceram. Soc. 92, 68–74 (2009).
Fayon, F., Bessada, C., Massiot, D., Farnan, I. & Coutures, J. P. 29Si and 207Pb NMR study of local order in lead silicate glasses. J. Non-Cryst. Solids 232, 403–408 (1998).
Fayon, F., Landron, C., Sakurai, K., Bessada, C. & Massiot, D. Pb2+ environment in lead silicate glasses probed by Pb-LIII edge XAFS and 207Pb NMR. J. Non-Cryst. Solids 243, 39–44 (1999).
Cotte, M. et al. The ID21 X-ray and infrared microscopy beamline at the ESRF: status and recent applications to artistic materials. J. Anal. Spectrom. 32, 477–493 (2017).
Berry, A. J. & O’Neill, H. S. C. A XANES determination of the oxidation state of chromium in silicate glasses. Am. Mineral. 89, 790–798 (2004).
Iwamoto, N. & Makino, Y. State of the chromium ion in soda silicate-glasses under various oxygen pressures. J. Non-Cryst. Solids 41, 257–266 (1980).
Pantelouris, A., Modrow, H., Pantelouris, M., Hormes, J. & Reinen, D. The influence of coordination geometry and valency on the K-edge absorption near edge spectra of selected chromium compounds. Chem. Phys. 300, 13–22 (2004).
Farges, F. Chromium speciation in oxide-type compounds: application to minerals, gems, aqueous solutions and silicate glasses. Phys. Chem. Miner. 36, 463–481 (2009).
Calas, G., Majérus, O., Galoisy, L. & Cormier, L. Crystal field spectroscopy of Cr3+ in glasses:: compositional dependence and thermal site expansion. Chem. Geol. 229, 218–226 (2006).
Angeli, F., Brunswic, L., Charpentier, T. & Gin, S. Lead leaching in industrial crystal glasses: role of chemical composition, structure and surface treatments. In Lead in Glassy Materials in Cultural Heritage 297 (2024).
Kacem, I. B., Gautron, L., Coillot, D. & Neuville, D. R. Structure and properties of lead silicate glasses and melts. Chem. Geol. 461, 104–114 (2017).
Liao, C., Tang, Y., Liu, C., Shih, K. & Li, F. Double-barrier mechanism for chromium immobilization: a quantitative study of crystallization and leachability. J. Hazard. Mater. 311, 246–253 (2016).
Massiot, D. et al. Ga-71 and Ga-69 nuclear-magnetic-resonance study of beta-Ga2O3-resolution of 4-fold and 6-fold coordinated Ga sites in static conditions. Solid State Nucl. Magn. Reson. 4, 241–248 (1995).
Acknowledgements
This study was supported by the French Agency for Research (ANR, PRCE program, grant 18-CE08-0025). We acknowledge the European Synchrotron Radiation Facility for the provision of beam time on ID21 (proposal MA-4648) and we would especially like to thank the ESRF instrumentation service and development division for their efforts in instrumental developments for EBS and ID21 refurbishment. µXRF and XANES datasets acquired in ESRF can be found under DOI 10.15151/ESRF-ES-579825745. Théo Caroff from IMPMC is acknowledged for his script for data processing optical absorption spectra and Déa Jaïs (IMPMC) for the transcription of the Cr(III) and Cr(VI) spectra from Bamford.
Author information
Authors and Affiliations
Contributions
L.B. performed the leaching experiments. XANES and XRF measurements were carried out by M.T. and L.B. under the supervision of M.C. and data was curated by L.B. T.C. performed the NMR studies. L.G. ran optical absorption spectroscopy and processed data. F.A., S.G. and T.C. supervised the study and worked on the conceptualization, methodology and original draft. L.B., F.A., S.G., R.L., L.C., E.D.H., and D.N. helped with the paper editing.
Corresponding author
Ethics declarations
Competing interests
Stephane Gin is co-editor of npj-MD.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Brunswic, L., Angeli, F., Charpentier, T. et al. Insight into the structure and chemical durability of Cr-bearing lead crystal glass. npj Mater Degrad 9, 40 (2025). https://doi.org/10.1038/s41529-025-00586-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-025-00586-7
