
Abstract
We previously demonstrated that kynurenine pathway (KP) dysregulation associates with Parkinson’s disease (PD) and its symptoms. Here, we profiled 16 KP-related markers in a second, independent cohort; plasma: n = 202 (116 PD (“OFF”), 86 controls); CSF: n = 183 (108, 75). Consistent with previous findings, we detected significantly higher concentrations of neurotoxic 3-hydroxykynurenine in plasma and lower concentrations of neuroprotective kynurenic acid along with higher neurotoxic quinolinic acid/kynurenic acid ratios in CSF of PD patients. Additionally, 10 markers showed sex-based differences, with more pronounced dysregulation in females. These 10 markers loaded to a single principal component linked to higher UPDRS I and II scores. Together, this suggests a composite signature of KP dysregulation in PD that is associated with worse symptoms and more prevalent in women. This work shows that KP dysregulation in peripheral and central compartments is linked to symptom severity in PD and warrants further systematic studies unraveling sex-dependent metabolic differences.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is the second most common age-related neurodegenerative disorder worldwide and the fastest growing in prevalence1. Global case estimates more than tripled from ~2.5 million in 19902 to over 8.5 million in 20193 and continue to rise4 at rates not explained by population aging alone2,3,4. The disease places a tremendous burden on patients, caregivers, and health care systems due to its long duration of progressive disability, and despite ongoing efforts, there is a lack of disease-modifying treatments available that can slow or stop its course5.
Clinically, PD is characterized by motor symptoms (bradykinesia with rigidity and/or tremor)6 but also involves various non-motor symptoms (e.g., depression, pain, dementia, sleep problems, autonomic dysfunction) that often severely impact quality of life7. Neuropathologic hallmarks include a marked loss of dopaminergic neurons in the substantia nigra and the presence of intraneuronal inclusions containing misfolded α-synuclein8. The latter vary widely in severity and distribution throughout the brain, often correlating with symptom severity9. In addition to dopaminergic neuron loss, extra-nigral neuron loss (e.g., noradrenergic, serotonergic, cholinergic) also occurs to varying extents8. The mechanisms leading to neuronal loss in PD patients are multifactorial and incompletely understood. Mitochondrial dysfunction and oxidative stress10 are hypothesized to contribute along with iron dyshomeostasis11,12,13,14,15,16 and glutamate excitotoxicity17, to which dopaminergic neurons are particularly vulnerable13,18. Chronic neuroinflammation is another major component19, and growing evidence suggests a role for the kynurenine pathway (KP), a metabolic pathway with increased activity during inflammatory states that can exacerbate all of the above neurodegenerative factors20,21.
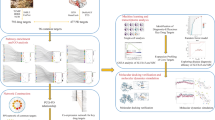
The KP (Fig. 1) catabolizes over 90%22 of dietary tryptophan (TRP) in the brain and periphery first into l-kynurenine (KYN), then into several branches of neuroactive and immunoregulatory metabolites (kynurenines) that can have neuroprotective or neurotoxic properties23. Two of the most well-studied kynurenines are the neurotoxic intermediates 3-hydroxykynurenine (3HK), produced directly from KYN, and quinolinic acid (QUIN), produced downstream of 3HK. In vitro studies and in vivo studies in mice have demonstrated that production along the 3HK-QUIN branch occurs primarily in macrophages and, in the brain, in microglia24,25 and neurons26. Both 3HK and QUIN can be beneficial, serving as intermediates in the synthesis of nicotinamide adenine dinucleotide (NAD+)27, a coenzyme essential to cellular energy production and redox homeostasis that gradually declines with age28. In excess, however, they also can be harmful via multiple mechanisms—3HK primarily via production of free radicals29,30 and QUIN mainly via selective agonist effects at NMDA receptors that promote glutamate excitotoxicity (but also via gliotoxic, pro-inflammatory, and pro-oxidant properties as well as blood-brain-barrier (BBB) permeability effects)31. Interestingly, these neurotoxic effects are normally balanced by neuroprotective effects of other kynurenines, including kynurenic acid (KYNA), an ionotropic glutamate receptor antagonist able to counteract the excitotoxic effects of QUIN32, and anthranilic acid (AA) and picolinic acid (PIC), both of which have iron-chelating properties33,34. This balance can be disrupted, however, by inflammation (i.e., in disease states), as pro-inflammatory cytokines increase KYN production (at the expense of serotonin (5-hydroxytryptamine (5HT)) production) and disproportionately increase 3HK-QUIN production relative to KYNA and other neuroprotective kynurenines. Consistent with this concept, studies report changes in the plasma and CSF of PD patients that indicate a dysregulated activation of the KP20,21, including our own work in which higher plasma concentrations of 3HK, lower CSF concentrations of KYNA, and a higher QUIN/KYNA ratio were associated with symptom severity35. The involvement of the KP in PD is further supported by genetic evidence; point mutations36 or deletions37 in the ACMSD gene (encoding the KP enzyme that generates PIC at the expense of QUIN) have been found in sporadic PD and a familial syndrome featuring tremor and parkinsonism, respectively.

Rectangles highlight the metabolites measured in the present study, with fill indicating neuroprotective (blue) or neurotoxic (red) properties. The KP begins with the catabolism of over 90% of the essential amino acid tryptophan (TRP) into L-kynurenine (KYN), with a relatively small amount converted to serotonin (5HT). KYN is then further catabolized along several branches into multiple neuroactive metabolites, known as kynurenines. The central branch is the 3-hydroxykynurenine-quinolinic acid (3HK-QUIN) branch, which leads to synthesis of NAD+, a coenzyme essential for cellular energy production and redox homeostasis that declines with age. Buildups of 3HK and QUIN can both contribute to oxidative stress, and QUIN is neurotoxic by multiple other mechanisms, including glutamate excitotoxicity primarily due to selective agonist effects on NMDA receptors. Along other branches and off-branches, neuroprotective kynurenines are produced that balance the effects of 3HK and QUIN. These include kynurenic acid (KYNA), a glutamate receptor antagonist, and anthranilic acid (AA) and picolinic acid (PIC), both metal chelators. Enzymes (in italics) along the pathway drive the relative production of kynurenines and are influenced by pro-inflammatory cytokines, such as IFN-γ. The ratios of 3HK and QUIN to neuroprotective kynurenines are altered in inflammation and many neurological and neuropsychiatric diseases, including Parkinson’s disease.
Sex-based differences in the KP have been observed in the general population, including lower KYNA, KYNA/3HK, and KYNA/QUIN ratios in females, similar to KP differences observed in depressed individuals38. These differences have been suggested to contribute to the ~two-fold higher risk of depression in females compared to males39,40,41. Sex also influences both PD risk (PD is ~1.4 times more common in males than females)2 and clinical phenotypes. For instance, several studies show that female PD patients are more likely to present the more benign tremor-predominant subtype at onset42,43 but report more severe depression, anxiety, and pain43,44,45,46, mirroring the differences seen in the general population39,40. Although sex could be an important factor in KP dysregulation of relevance to clinical manifestation, its role in KP dysregulation in PD remains relatively unexplored.
Here, we quantified KP metabolites in CSF and plasma from a set of clinically well-characterized PD and control individuals enrolled in the multisite BioFIND study47, to validate and expand our previous findings35 in an independent cohort. The blood and CSF samples were collected in OFF state, which allowed us to study the biological changes without the direct and/or acute influence of PD medications. In addition, we sought to understand whether sex-associated signatures of KP dysregulation occur in PD and how these associate with clinical features.
Results
Demographic and clinical characteristics of cohort
All plasma (n = 202) and CSF (n = 183) samples were obtained from participants in the BioFIND study during the practically defined OFF medication state (early morning before PD medications, ~12 h since last dose)47. The collection of samples from this cohort and their demographic characteristics were previously described47. We here display cohort characteristics stratified by sex in Table 1.
Group differences in kynurenine metabolite concentrations
We first compared individual KP-related marker concentrations and ratios in PD vs Control groups using one-way ANCOVA, controlling for age and sex (Fig. 2 and Supplementary Tables 1 and 2)35. Significant group differences are highlighted in Fig. 2 and detailed below.

a, b Higher mean concentrations of 3HK and QUIN, two neurotoxic kynurenines, c, d lower mean concentrations of KYNA and PIC, two neuroprotective kynurenines, and e–g higher mean ratios of neurotoxic:neuroprotective kynurenines in the PD group compared to Control in plasma and/or CSF. Gray dots indicate individual, log-transformed concentrations or ratios. Bars and error bars reflect estimated mean and 95% CI from one-way ANCOVA, after controlling for age and sex as covariates. Asterisks indicate raw P value of group difference (****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05). Second generation P values are shown directly above asterisks when significant (Pδ = 0; i.e., the 95% CI of the difference in means did not overlap the specified null interval ( ± 10%)), indicating particularly robust effects. 3HK 3-hydroxykynurenine, KYNA kynurenic acid, KYN kynurenine, PIC picolinic acid, QUIN quinolinic acid, PD Parkinson’s disease.
Higher neurotoxic and lower neuroprotective kynurenines in PD. The PD group had significantly higher mean 3HK concentrations in both plasma (3.3 vs 2.9, P < 0.001) and CSF (1.2 vs 0.93, P = 0.039; Fig. 2a) and higher mean CSF QUIN concentrations (3.0 vs 2.8, P = 0.008; Fig. 2b), but lower mean KYNA concentrations in both plasma (3.1 vs 3.2, P = 0.026) and CSF (0.47 vs 0.68, P < 0.001; Fig. 2c) and lower mean plasma PIC concentrations (2.9 vs 3.1, P = 0.044; Fig. 2d). To account for multiple comparisons and increase the likelihood of detecting meaningful effects, we also generated second generation P values (SGPVs)48, based on a null interval of +/−10%. The most robust of the group differences, with SGPVs (pδ) at 0 (indicating significance)48, were the increased concentration of 3HK in plasma and decreased concentration of KYNA in CSF in PD (Fig. 2 and Supplementary Table 2).
Higher neurotoxic vs neuroprotective KP metabolite ratios in PD. The PD group also had significantly higher ratios of neurotoxic (e.g., 3HK and QUIN) to neuroprotective kynurenines (e.g., KYNA, PIC) in both plasma and CSF (Fig. 2e–g and Supplementary Table 2). These included higher 3HK/KYNA (plasma: 0.60 vs −0.39, P < 0.001; CSF: 0.75 vs 0.25, P < 0.001), QUIN/KYNA (plasma: 2.1 vs 1.9, P = 0.004; CSF: 2.6 vs 2.1, P < 0.001), and QUIN/PIC (plasma: 2.3 vs 2.0, P = 0.004; CSF: 0.77 vs 0.53, P = 0.035) ratios. Of these, SGPVs were significant for plasma and CSF 3HK/KYNA and CSF QUIN/KYNA (Fig. 2 and Supplementary Table 2).
Sex-based differences in KP metabolites
We next determined the influence of sex or sex*PD interactions on each KP-related marker concentration or ratio, controlling for age as covariate. Ten markers and 3 ratios showed evidence of sex-based differences or interactions (Fig. 3a–j; for complete set of two-way ANCOVA results and full distributions, please see Supp. Table 3 and Supplementary Figs. 1 and 2). In all cases, the sex-based differences involved females with PD (red in Fig. 3) showing higher concentrations of neurotoxic or lower concentrations of neuroprotective kynurenines, higher neurotoxic ratios, and/or a more pronounced difference in mean concentration or ratio in PD compared to Control. Effects of adjusting additionally for BMI or disease duration were minimal, thus only age was retained as covariate in the final models.

Interaction plots for a TRP, b 5HT, and c–g KP metabolites and h–j ratios show estimated mean concentrations and 95% CIs for males and females within PD and Control groups after controlling for age in two-way ANCOVA. Sex*PD interaction term was included in all models; significant interactions (P < 0.05) are shown at bottom of respective plots. Asterisks reflect Bonferroni-adjusted P values of post-hoc, pairwise comparisons for simple main effects, with females vs males within PD or Control groups in black and PD vs Control within males or females colored by sex. *P <0.05; **P <0.01, ***P <0.001, ****P <0.0001. Second generation P values also are shown directly above asterisks when significant (Pδ = 0; i.e., the 95% CI of the difference in means did not overlap the specified null interval ( ± 10%)), indicating particularly robust effects. TRP tryptophan, 3HK 3-hydroxykynurenine, QUIN quinolinic acid, KYNA kynurenic acid, KYN kynurenine, PIC picolinic acid, NTA nicotinamide, PD Parkinson’s disease.
Sex*PD Interactions. Interactions between sex and PD diagnosis were statistically significant for plasma 5HT (P = 0.037), plasma 3HK (P = 0.018), CSF 3HK (P = 0.020), and the plasma 3HK/KYNA ratio (P = 0.007; Fig. 3b, c, h and Supplementary Table 3). Post-hoc tests of simple main effects of PD within male and female subgroups showed that mean plasma 5HT concentrations were only significantly lower in PD compared to Control in females (females: 3.8 vs 4.6, P = 0.007; males: 4.8 vs 4.8, P = 0.962; Fig. 3b), mean plasma 3HK concentrations were significantly higher in PD in both sexes but to a greater extent in females (females: 3.5 vs 2.8, P <0.0001; males: 3.2 vs 2.9, P = 0.011; Fig. 3c), and mean CSF 3HK concentrations were only significantly higher in PD in females (females: 1.4 vs 0.90, P = 0.002; males: 0.99 vs 1.0, P = 0.933; Fig. 3c). Simple main effects of sex within PD and Control groups showed that mean plasma 5HT concentrations were significantly lower in females only in PD (PD: 3.8 vs 4.8, P < 0.001; Control: 4.6 vs 4.8, P = 0.505; Fig. 3b), and mean 3HK concentrations were significantly higher in females than males only in PD in both plasma (PD: 3.5 vs 3.2, P = 0.003; Control: 2.8 vs 2.9, P = 0.593; Fig. 3c) and CSF (PD: 1.4 vs 0.99, P = 0.005; Control: 0.90 vs 1.0, P = 0.513; Fig. 3c). All SGPVs for simple main effects of sex within the PD group and for PD within females for markers with sex*PD interactions (3HK, 5HT, and plasma 3HK/KYNA) were significant (Pδ = 0; Fig. 3b, c, h). Among the remaining sex-associated KP markers, for which the interaction term was non-significant, most indicated a significant main effect of sex and/or trend toward sex*PD interaction; these are detailed in Supplementary Table 3.
PC2, a sex-associated signature of KP dysregulation in PD
We next used principal components analysis (PCA) to determine whether the ten sex-associated markers could be condensed onto a single principal component (PC), or summary index. If so, this PC could reflect a composite signature of KP dysregulation and facilitate downstream analysis of its clinical relevance. We performed PCA of all 16 markers in all subjects and then tested for sex-based differences and sex*PD interactions among the top PCs. The top 2 PCs explained 45.8% of all variance (PC1: 29.3%, PC2: 16.5%). Individual scores and factor loadings for PC1 and PC2 are summarized in Fig. 4 (Supplementary Table 4 shows the complete set of loadings).

PCA of all 16 KP-related markers was performed, pooling all subjects with complete plasma and CSF data (N = 183). a Individual PC1 scores (x axis) vs PC2 scores (y axis) in Control (left) and PD groups (right), with color indicating male and female subgroups, suggest higher PC2 scores in PD in females. Ellipses reflect 95% CIs. b Two-way ANCOVA confirms significantly higher PC2 scores in PD in females, with a strong sex*PD interaction. Interaction plots (left) show estimated mean PC scores (with 95% CI) for each subgroup after controlling for age, with full distributions shown at right. Asterisks indicate Bonferroni-adjusted P values of simple main effects from post-hoc, pairwise comparisons (****P < 0.0001). Effects of sex within PD and Control groups are in black font; effects of PD within males or females are colored by sex. c PC1 and PC2 loadings, i.e., associations between PC scores and individual marker concentrations, illustrate the pathway signature reflected by each PC. PCA principal component analysis, TRP tryptophan, KYN kynurenine, 5HT 5-hydroxytryptamine (serotonin), 3HK 3-hydroxykynurenine, QUIN quinolinic acid, KYNA kynurenic acid, PIC picolinic acid, NTA nicotinamide, AA anthranilic acid.
In Fig. 4a, individual PC1 scores plotted against PC2 scores show that PC2 scores (but not PC1 scores) appeared higher overall in female PD patients than other subgroups. In Fig. 4b, interaction plots from two-way ANCOVA confirm this observation, revealing a highly significant sex*PD interaction for PC2 (P < 0.0001) and lack of significant sex- or PD-related differences or interaction for PC1. Tests of simple main effects showed that mean PC2 scores were markedly higher in PD but only in females (females: 1.4 vs −0.15, P < 0.0001; males: −0.55 vs -0.59, P = 0.904; Fig. 4b) and markedly higher in females but only in PD (PD: 1.4 vs −0.55, P < 0.0001; Control: −0.15 vs −0.59, P = 0.189; Fig. 4b). Notably, even though PC1 did not show significant main effects of sex, PD, or sex*PD interaction, PC1 scores were strongly associated with age as a covariate (P < 0.001). In contrast, PC2 scores were not associated with age (P = 0.326). PC1 could therefore be interpreted as a general signature of aging in the KP, whereas PC2 could be interpreted as a signature of KP dysregulation in PD that occurs predominantly in females.
To determine the KP signatures reflected by PC1 and PC2, we examined factor loadings for each marker (Fig. 4c). Loadings for PC1 were uniformly positive and showed that higher PC1 scores (associated with higher age), reflected higher plasma and CSF KYN (loadings: 0.40, 0.37, respectively) and QUIN (0.38, 0.39) and to a lesser extent higher plasma and CSF KYNA (0.26, 0.28) and 3HK (0.27, 0.25), plasma AA (0.25), and CSF TRP (0.22). Loadings for PC2 scores were mostly negative and showed that higher PC2 scores (associated with female PD patients) reflected lower levels of plasma TRP (−0.34) and 5HT (−0.29), lower levels of neuroprotective kynurenines (PIC (−0.45, −0.37) and NTA (−0.36, −0.29) in plasma/CSF and KYNA (−0.25) in plasma), and higher levels of neurotoxic kynurenines (3HK (0.24, 0.21) in plasma/CSF, QUIN (0.13) in CSF). In sum, all markers with sex-based differences in Fig. 3 loaded to PC2. Notably, PIC, NTA and 5HT also loaded to PC3 (Supplementary Table 4 and Supplementary Fig. 3), but, in contrast to PC2, in PC3 plasma and CSF PIC (−0.48, −0.53) loaded inversely to plasma and CSF NTA (0.37, 0.26) and plasma 5HT (0.37). Two-way ANCOVA showed that PC3 scores, unlike PC2 scores, were not associated with PD or sex (Supplementary Fig. 3).
Associations between PC2 scores and UPDRS I and II
To establish the clinical relevance of the sex-associated signature of KP dysregulation summarized by high scores on PC2 while controlling for age-associated KP changes summarized by high scores on PC1, we used k-means clustering to classify PD patients according to PC1 and PC2 scores (Fig. 5). We identified 5 clusters (high PC1/high PC2, high PC2, low PC1, high PC1, and low PC2; Fig. 5a). We then compared UPDRS I, II, III, and IV sub-scores and total MoCA scores among the clusters. Notably, the proportion of female-to-male patients among clusters significantly differed (Chi-square=17.894, P < 0.001), with the highest proportion of female patients in the high PC2 cluster (Cluster 2: 20 females, 9 males) and the lowest in the low PC2 cluster (Cluster 5: 2 females, 11 males).

a K-means cluster plot showing individuals in the PD group (sexes pooled) clustered by PC1 and PC2 scores. Highlighted in yellow is Cluster 2, representing PD patients with purely high PC2 scores (n = 29, including 9 males and 20 females), reflecting lower levels of neuroprotective kynurenines and serotonin and higher levels of neurotoxic kynurenines. Highlighted in blue is Cluster 5, representing PD patients with purely low PC2 scores (n = 13, including 11 males and 2 females). b One-way ANCOVA results, showing estimated mean ( ± 95% CI) UPDRS I and II scores for each cluster after controlling for age, sex, and disease duration. Asterisks indicate P values of post-hoc pairwise comparisons with Bonferroni correction. *P <0.05; **P <0.01. Dashed line marks cutoff for mild/moderate symptom severity.
We observed significant differences among the clusters in mean UPDRS I (F(4, 106) = 5.1, P < 0.001) and II (F(4, 106) = 3.7, P = 0.008) scores after controlling for age, sex, and disease duration in one-way ANCOVA (Fig. 5b). Pairwise comparisons with Bonferroni correction showed that the high PC2 cluster (Cluster 2) had a significantly higher mean UPDRS I score (13.2 ± 2.0) compared to three other clusters (1,3 and 5) and a higher mean UPDRS II score than two other clusters (3 and 5; Fig. 5b). Moreover, the high PC2 cluster (Cluster 2) was the only cluster for which estimated mean UPDRS I and II sub-scores fell in the moderate range of symptom severity. There were no significant differences among clusters in UPDRS III “ON” or “OFF”, UPDRS IV, or MoCA scores (P > 0.268).
To determine whether the lack of association with UPDRS III scores applied only to PC2 or to any individual KP markers, we performed post-hoc testing to establish whether CSF concentrations of any individual markers and ratios that differed between PD and Control groups were associated with UPDRS III “ON” or “OFF” scores. Results showed that CSF KYNA (P = 0.023) and PIC (P = 0.003) each were negatively associated with UPDRS III “ON” scores in PD patients after controlling for age, sex, and disease duration in separate regression analyses for each marker (Supplementary Table 5). When all CSF markers were entered into the model, backward selection retained only PIC as a significant predictor (P < 0.002).
Plasma-CSF correlations
Finally, to evaluate plasma-CSF markers’ concordance across the BBB, we performed Pearson’s correlations (Supplementary Fig. 4). Median concentrations of the markers in plasma ranged from ~2–40 times higher than those in CSF (Supplementary Table 1). The markers with strongest and most consistent correlation between plasma and CSF across the four subgroups were QUIN (r = 0.50–0.81, P < 0.001) and PIC (r = 0.68–0.89, P < 0.001). KYN also had consistently moderate to strong plasma-CSF correlations (r = 0.50-0.65, P < 0.003). 3HK showed plasma-CSF correlations that were either only significant or stronger in PD (r = 0.44-0.62, P < 0.004 in PD). KYNA showed positive plasma-CSF correlations in both sexes in PD (r = 0.40–0.52, P < 0.01) but only in females in the Control group (r = 0.15, P > 0.05 in males, r = 0.43, P = 0.006 in females). TRP showed only weak (r < 0.4) or non-significant (P ≥ 0.05) plasma-CSF correlations.
Discussion
This work validates our previous findings of disturbances in peripheral and central levels of KP metabolites in PD and their association with symptom severity, demonstrating reproducibility of these findings across cohorts, sites, analytical platforms, and PD medication status. Moreover, this study is the first to demonstrate significantly elevated levels of QUIN and 3HK in addition to reduced KYNA in the CSF of PD patients, strengthening our previous work with robust findings in the central compartment. Our analysis also reveals, for the first time, sex-based differences indicative of heterogeneity in KP dysregulation in PD. These findings might open a new horizon to understanding sex-based differences in PD etiology and progression, which could ultimately impact prevention, prognosis and treatments.
We first showed that, consistent with our previous findings35, the PD group had a net neurotoxic profile of kynurenines in plasma and CSF consistent with an inflammatory activation of the pathway, including higher concentrations of neurotoxic 3HK (plasma and CSF) and QUIN (CSF), lower concentrations of neuroprotective KYNA (plasma and CSF) and PIC (plasma), and higher ratios of neurotoxic:neuroprotective kynurenines (plasma and CSF) compared to Controls.
Next, we identified sex-based differences in 10 of the KP-related markers in PD, all consistent with more pronounced dysregulation in females. Using PCA, we showed that all 10 of these markers loaded to a single principal component, PC2, and that PC2 scores were markedly higher on average in female PD patients than any other subgroup and not associated with age. Using PC loadings, we showed that higher PC2 scores reflected a composite signature of KP dysregulation involving lower levels of neuroprotective kynurenines (PIC, NTA, KYNA) and serotonin and higher levels of neurotoxic kynurenines (QUIN and 3HK) in plasma and/or CSF. We then identified a cluster of PD patients with high PC2 scores and showed that this cluster had significantly higher mean UPDRS I and II scores than the low PC2 cluster and was the only cluster scoring in the moderate severity range on average. Thus, we revealed a sex-associated signature of KP dysregulation in PD linked to worse symptom severity.
Previous clinical studies have implicated dysregulation of the KP as a feature of PD, although the size of the studies, type of biofluid, and the markers targeted have varied, with some inconsistencies in findings for specific markers20,21,49,50. Thus, it is worth underscoring the validation of our previous findings of higher plasma 3HK, lower CSF KYNA, and a higher QUIN/KYNA ratio in PD patients enrolled at Penn State University (PSU)35 in an independent cohort from the multisite BioFIND study47. The reproducibility is particularly remarkable given stark differences in clinical characteristics of the BioFIND cohort compared to PSU (namely, a much narrower range of disease duration, age, and symptom severity in BioFIND)35,47. Also, the BioFIND cohort was OFF medication at the time that plasma and CSF were collected, whereas the PSU cohort was ON medication. Finally, in the current study we used a different analytical protocol. Taken together, these data strongly support the potential generalizability of these biomarkers.
For both plasma and CSF, the sample size of the present cohort is a strength, with over 200 plasma samples and 183 CSF samples in total. In comparison, several past studies were based on a more moderate size of 20–35 subjects per group49,51. In a recent meta-analysis of KP changes in neurodegenerative disease, nine of the ten PD studies included had N < 35 per group, and over half had N < 2050. Including over 100 PD subjects with both plasma and CSF in the present study not only increased power but, importantly, afforded stratification by sex. The number of CSF samples in the present cohort was particularly high compared to past studies given that CSF is relatively difficult to obtain. Thus, in addition to differences that replicated35, we detected several additional significant differences in CSF in the BioFIND cohort (higher CSF 3HK and QUIN in PD) that are consistent with net neurotoxicity.
Our findings related to 3HK align with several other recent studies. 3HK was elevated in post-mortem CSF of 48 pathologically-confirmed PD patients compared to 57 controls in a metabolomic analysis52 and also higher in CSF in a smaller study of 20 living PD patients and 13 controls51. Another recent metabolomic analysis of over 100 idiopathic PD patients found higher serum 3HK as one of six metabolites significantly differing from controls; 3HK also was significantly higher in PD than in AD patients, suggestive of disease-specificity53. 3HK production is regulated by kynurenine 3-monooxygenase (KMO), which has been shown to be activated in brain following systemic pro-inflammatory insult (lipopolysaccharide) in rats54. 3HK produces free radicals at endogenous concentrations, thus its detrimental effects have been suggested to be mediated by oxidative stress30. More recent evidence, however, suggests 3HK exerts its damage primarily via mitochondrial dysfunction55. Interestingly, genetic mutations predicted to compromise KMO activity and consequently reduce 3HK generation have been associated with a later onset of PD, suggesting that a 3HK reduction affects PD progression56.
Lower KYNA has also been previously demonstrated in PD patients, including lower KYN and KYNA concentrations in post-mortem substantia nigra, putamen, and frontal cortex57, and lower KYNA concentrations in CSF49. Peripheral KYN and KYNA levels have been inconsistent, with both higher and lower levels reported (reviewed in ref. 32 and in the aforementioned meta-analysis50). This is in line with our own findings of markedly lower CSF KYNA across the BioFIND and PSU cohorts but significantly lower plasma KYNA only in the BioFIND cohort, with a smaller effect size. Central KYNA levels and QUIN/KYNA ratios are hypothesized to be influenced by astrocytes, as IFN-γ-stimulated cultures of human astrocytes produce large amounts of KYN and KYNA but lack KMO needed to produce along the 3HK/QUIN branch58. KYNA deficiency is significant in that its antagonistic effects at NMDA receptors and other glutamate receptors are thought to counteract the agonistic effects of QUIN that lead to glutamate excitotoxicity32. It is worth noting that we observed significantly lower CSF KYNA in both males and females with no sex*PD interaction, suggesting a more universal phenomenon in PD patients.
Aside from our prior study35 and a relatively large metabolomics study showing higher plasma QUIN and QUIN/KYNA ratios in PD with more pronounced differences in later stages59, relatively few studies have assessed QUIN and PIC, although both have plausible roles in neurodegeneration, and higher QUIN/PIC ratios are linked to suicidality60. QUIN is a potent neurotoxin via multiple mechanisms, including excitotoxicity31, and 3HK potentiates its neurotoxicity, as demonstrated in rat striatum61. PIC is an iron chelator, which can be important since iron is increased in the substantia nigra pars compacta in PD14,16. Our previous work showed that in PD and Control subjects, PIC correlated negatively with nigral R2*, an MRI measure of tissue iron content35. Interestingly, in PD subjects from the same cohort, MRI measurements of iron also correlated negatively with plasma 5HT (which was lower in PD patients than controls)62. Since low PIC and low 5HT both loaded to PC2 in the present study, it would be worth investigating their association with nigral iron accumulation in future studies.
Our findings of sex-based differences in the KP in PD are supported by evidence of sex-based differences in KP regulation in the general population38. Sex hormones also are linked to changes in KP metabolite levels (e.g., upon oral contraceptive use, during post-partum period) in younger cohorts38,63. Comparative studies of sex-based differences in the KP in PD are lacking, and our findings highlight the need for more studies in this direction. These findings could be relevant for the development of more specific therapeutic tools in PD as they raise the possibility that some of the observed biological differences could also be linked to sex-based differences in the course of PD.
Interestingly, even though we observed more pronounced KP dysregulation in females overall, and certain PD symptoms (e.g., tremor, depression, and pain) have been reported to be more severe or more common in females in past studies42,43,44,45,46, we did not detect more severe symptoms in PD females overall in terms of mean UPDRS subscores, and females performed slightly better on MoCA on average. One consideration in these clinical scores is that disease duration in the BioFIND cohort was restricted to 5–18 years from motor onset, and some sex differences could be stage dependent. Recently, in our analysis of AMP-PD data we showed that UPDRS I scores were more severe in females only in early-stage PD64.
Intriguingly, despite the lack of sex differences in UPDRS I and II overall, higher scores on both measures in PD were associated with higher scores on PC2, which strongly associated with female sex. A plausible explanation for this is that high PC2 scores were not universal among PD females or exclusive to females (the high PC2 cluster included only 25% of the PD cohort and had a 2:1 female-to-male ratio, so was sex-associated but not sex-specific). Thus, the overall proportion of female patients with the high PC2 KP signature may not have been enough to affect mean UPDRS I and II scores for females overall.
Another finding of interest concerning UPDRS was the association between PC2 and UPDRS parts I and II but not part III ON or OFF. UPDRS I and II are both self-report measures, with part I assessing non-motor aspects of daily living and part II assessing activities of daily living (ADLs). UPDRS III, on the other hand, is based on a one-time (5–10 min) clinician-rated assessment of motor severity that is prone to variation from both raters’ subjectivity and patients’ motor fluctuation65. Interestingly, a large, multicenter factor analysis of MDS-UPDRS parts I-IV showed that UPDRS I and II, but not III or IV, associated with quality of life (QoL) as assessed by Parkinson’s Disease Questionnaire-8 (PDQ8)66. Thus, the high PC2 KP signature may be more closely related to patient experience of non-motor symptoms, ADLs, and QoL than strictly motor severity in the clinical setting. Notably, in follow-up analyses that assessed whether any individual marker concentrations were associated with UPDRS III, we detected a significant association between lower CSF PIC concentrations and higher UPDRS III ON (but not OFF) scores, possibly linking PIC to motor severity. That PIC was not associated with UPDRS III OFF suggests PD medications may moderate this association.
Finally, we observed positive correlations across the BBB for several KP markers, supporting the use of plasma for biomarker purposes of pathway dysregulation when CSF cannot be obtained. These findings are mostly in line with a systematic review of concordance of peripheral and central kynurenines in humans67 showing that KYN and 3HK had moderate-strong concordance in psychiatric disorders and other pathophysiological conditions, whereas KYNA and TRP showed discordance. The highest plasma-CSF correlations we observed here were for QUIN and PIC. High plasma-CSF concordance for QUIN has been reported previously in a PD cohort, in individuals with Alzheimer’s disease, and in Control subjects49. This is interesting, given experimental evidence that TRP, KYN, 3HK, and AA readily cross the BBB but KYNA and QUIN have been shown to cross very poorly in rats67,68. Passive diffusion of QUIN across the BBB does not appear sufficient to explain the high plasma-CSF correlations we observed in PD (r > 0.75). It is unclear whether these correlations reflect passage of the kynurenines from peripheral to cerebral pools across the BBB, common influences on KP metabolism across these compartments, or a leaky BBB that has been compromised by chronic inflammation and/or aging24,67. A compromised BBB could allow infiltrating macrophages to become the major cerebral source of QUIN, given evidence that macrophage QUIN production is 20–32 times higher than that of microglia upon IFN-γ stimulation24,25. Even if macrophages are a major source, activated microglia could also contribute to central QUIN levels, since microgliosis can be a major aspect of PD pathology19 and microglia favor 3HK-QUIN production24,25.
It is important to note that the use of plasma biomarkers offers a relatively non-invasive measure of KP activity but may not reflect variation in regional concentrations in brain tissue. However, by adding CSF samples to the plasma samples in this study, we have greatly strengthened the data supporting a role for these metabolites within the central compartment. Other limitations include the cross-sectional design, precluding causal inference in the clinical associations, and strict selection criteria for the BioFIND study (e.g., only PD patients 5–18 years from symptom onset and exhibiting all three cardinal motor symptoms), yielding a more clinically homogeneous group than in typical clinical settings. Considering that despite these distinctions, we replicated findings in the KP from a more clinically diverse cohort at PSU, we can conclude that our findings extend to a moderate to advanced PD cohort. Future studies are needed to determine the underlying influences (e.g., cytokines, sex hormones, enzyme activity, chronic influence of anti-PD drugs) shaping the pathway activity, so that appropriate therapeutic targets can be determined.
In conclusion, we here demonstrate significantly elevated levels of the neurotoxins QUIN and 3HK in addition to reduced KYNA in the CSF of PD patients, together with similar alterations in plasma. We thus demonstrated the reproducibility of differences in KP-related markers in plasma and expanded and strengthened the findings in CSF. We also revealed a sex-associated composite signature of KP dysregulation in PD that suggests females with PD could be particularly prone to peripheral and central neurotoxicity and display a reduced amount of neuroprotective KP metabolites. PD subjects who showed this signature also had more severe symptoms. Our findings warrant systematic analysis of sex-based differences in KP dysregulation in future studies and further position the KP as a potential treatment target for PD.
Methods
Clinical cohort
The BioFIND cohort, described in detail in Kang et al.47, was recruited in response to a need for a cohort of well-characterized, clinically typical, moderate to advanced PD patients. All study protocols and recruitment strategies were approved by the institutional review boards for the University of Rochester Clinical Trials Coordination Center (CTCC) and individual sites47. All subjects provided written consent prior to participation. To maximize diagnostic accuracy, PD diagnosis was based on the United Kingdom PD Society Brain Bank clinical diagnostic criteria but modified to require all three classic motor signs of Parkinsonism (i.e., bradykinesia, rigidity, and resting tremor) instead of only two. An established medication response to a dopaminergic agent also was required, and patients were only included if they had disease duration of 5–18 years since symptom onset. For the current study, plasma samples were obtained from 116 PD patients and 87 control subjects who participated in the BioFIND study across 8 US sites from 2012 to 2015. CSF samples were obtained from 108 of the PD patients and 77 of the control subjects. One CSF sample had no detectable analyte, bringing the total to 76 control subjects. One Control subject had NTA concentrations in both plasma and CSF that were a magnitude of order higher than any other NTA concentrations in the study, so the subject was excluded, bringing the final totals to 86 plasma and 75 CSF samples for the Control group. The BioFIND study consisted of a baseline and follow-up visit. At the baseline visit (V1), PD patients were in the “ON” medication state (1–3 h after the last PD medication dose). At the follow-up visit (V2) within two weeks, PD patients were in the “OFF” state (early morning before PD medications, ~12 h after the last dose the night before). Clinical assessments, except for UPDRS III “OFF”, were performed at V1 (“ON” medication). All plasma and CSF samples in the current study were obtained from V2 (“OFF” medication). Demographic and clinical characteristics for the plasma cohort and CSF subcohort are provided following stratification by sex in Table 1.
Clinical assessments
Clinical assessments in the BioFIND study are described in detail in Kang et al.47. Clinical assessments used in the present study included the International Parkinson and Movement Disorder Society revision of the United Parkinson’s Disease Rating Scale (MDS-UPDRS) parts I, II, III (“ON” and “OFF”), and IV for PD patients (for control subjects, only part III was administered)20,69, and Montreal Cognitive Assessment (MoCA)70 for both PD patients and controls.
Biofluid collection
Methods for biofluid collection are detailed in Kang et al.47. Briefly, all participants were in a fasted state if possible or had a low fat meal the morning of the visit, and biofluids were drawn between 8 and 10 am. Biofluids were collected via standardized kits and according to standardized NINDS protocols. For plasma, blood was collected in 10 mL EDTA tubes. Within 30 min, blood samples were centrifuged at 4 °C for 15 min at 1500×g. Plasma was transferred in 1 mL aliquots into 2 mL microcentrifuge tubes, with care taken not to disturb the pellet. CSF was obtained via lumbar puncture. CSF was first collected in 50 mL conical tubes, then divided within 15 min into two 15 mL conical tubes for centrifugation at room temperature for 10 min at 2000×g. CSF was transferred in 1 mL aliquots into 2 mL microcentrifuge tubes. All samples were frozen within 30 min of collection at –80 °C, with exceptions in only 1.4% of those collected47, and stored at BioSEND. The experimental samples were later shipped to Van Andel Institute on dry ice.
Measurement of KP analytes
Metabolite analysis was conducted by the Van Andel Institute Mass Spectrometry Core (RRID: SCR_024903). Plasma samples were mixed with extraction solvent, which contained the internal standard mixture (90% acetonitrile with liquid chromatography-mass spectrometry (LC-MS) grade water at a 1:4 ratio), briefly vortexed, sonicated for 5 min, and incubated on wet ice for 10 min. After the incubation, the samples were centrifuged for 10 min at 17,000×g. The supernatant, which contains the metabolites of interest, was collected and dried under reduced pressure conditions for two hours in a GeneVac EZ-2 PLus speedvac (SP Scientific, Warminster, PA). Dried down extracts were then resuspended in 0.1% formic acid in LC-MS grade water via brief vortex. Once resuspended, samples were centrifuged and transferred to a glass autosampler vial containing a glass insert.
CSF samples were diluted twofold with solvent A (0.1% formic acid in LC-MS grade water) spiked with the ITSD mixture, added to SpinX tubes, and centrifuged for 5 min at 12,000×g at 4 °C. The filtrate was transferred to a glass autosampler vial containing a glass insert.
KP-related metabolites (TRP, 5HT, KYN, KYNA, 3HK, QUIN, PIC, NTA, AA, nicotinic acid (NIC)), were quantified using reverse phase ultra-high performance liquid chromatography (UPLC; 1290 Infinity II, Agilent Technologies, Santa Clara, CA) coupled to a triple quadrupole mass spectrometer (6470 Triple quadrupole, Agilent Technologies). In total, 5 µL of resuspended extracts were injected onto a Vanguard HSS T3 Pre-column that was connected to an Acquity HSS T3 analytical column. Elution conditions used a combination of Solvent A (0.1% formic acid in LC-MS grade water) and Solvent B (0.1% formic acid in 90% LC-MS grade acetonitrile) at a flow rate of 0.4 mL/min. Full LC-MS parameters, including LC gradient and MS transition list, are available as Supplementary Data 1. Skyline (v23.0) was used for peak picking and integration. For absolute quantitation, a stock mix containing each compound at the concentration indicated in Supplementary Data 1 was diluted 10-fold in water to form the top-level of and external calibration curve. This was then serially diluted into eight twofold dilution steps. Isotopically labeled internal standards (ISTD) were diluted 75 times from indicated stock concentrations (Supplementary Data 1) in extraction solvent and used to extract samples and standard curve. The peak area response ratio (analyte/ISTD) was used to calculate concentration of each analyte by linear or quadratic regression. Intra-assay coefficients of variability for plasma analytes: TRP 2.09%, 5-HT 1.71%, KYN 1.21%, KYNA 2.00%, 3-HK 3.40%, QUIN 2.45%, PIC 4.22%, NTA 1.01%, AA 6.70%, and NIC 6.22% and for CSF analytes: TRP 2.07%, KYN 1.58%, KYNA 8.98%, 3-HK 8.24%, QUIN 3.65%, PIC 4.02%, NTA 1.26%, NIC 6.89%. Lower limits of detection (LLOD) for each analyte were: TRP 36.6 nM, KYN 2.2 nM, KYNA 0.16 nM, 3-HK 0.29 nM, QUIN 4.15 nM, PIC 0.63 nM, NTA 0.98 nM, NIC 0.07 nM, AA 0.98 nM and 5-HT 0.73 nM. AA and 5HT were below LLOD in CSF, thus not included.
Statistical methods
Statistical analyses were performed using IBM’s Statistical Package for the Social Sciences (SPSS v.25) and RStudio (v2023.06.0). Comparisons of demographic variables were made using Student t tests for continuous variables and chi-square tests for categorical variables. Median concentrations and ratios were first calculated with interquartile ratios to summarize the data. Concentrations and ratios were then log-transformed prior to further statistical analyses, using the natural logarithm. All statistical tests were two-sided. Group comparisons of individual concentrations and ratios (PD vs Control) were performed using one-way ANCOVA with age and sex as covariates. The influence of sex and sex*PD interactions on individual concentrations and ratios was determined using two-way ANCOVA in SPSS, with models that included sex (male, female) and PD (PD, Control) as main factors, sex*PD interaction, and age as covariate. Main effects were calculated as the effect of sex overall averaged across PD and Control groups, or the effect of PD overall averaged across males and females. Simple main effects were calculated in R using the rstatix package71 by performing post-hoc pairwise comparisons with Bonferroni correction to determine the effects of sex within PD and Control groups and the effects of PD within males and females. As a sensitivity analysis, we also evaluated the effects of including BMI or disease duration as potential covariates. For BMI, we reran the two-way ANCOVAs with age and BMI as covariates. For disease duration, we ran one-way ANCOVAs (within PD) with age and disease duration as covariates. PCA was performed in R following standardization of all 16 marker concentrations in all subjects (PD and Control subjects pooled) using the factoextra package72. Following PCA, sex-based differences in PC1, PC2, and PC3 scores were determined using two-way ANCOVA controlling for age, using the same model as for the individual concentrations and ratios. For clinical relevance of PC1 and PC2, K-means clustering of PD patients based on PC1 and PC2 was performed in R using the factoextra package72. We explored 3-, 4-, and 5-cluster solutions, selecting the 5-cluster solution due to its ability to discriminate a small cluster of subjects who had both high PC2 scores and very high PC1 scores. Statistical testing of differences among clusters of PD patients in clinical UPDRS I, II, III “ON”, III “OFF”, and IV scores and MoCA scores was performed using one-way ANCOVA, with cluster as main factor and age, sex, and disease duration as covariates. Second generation P values (SGPVs) were used to account for multiple testing and simultaneously identify particularly robust associations48. Traditional P values only assess if there is evidence that two means or other point estimates differ, whereas SGPVs focus on whether or not the difference is large enough to be meaningful, by specifying a null interval. Statistically significant metabolite differences with a fold change near 1 are more likely to be false positives simply due to random noise or inherently high variability48. By setting a higher standard that differences must be greater than +/−10% (with 95% confidence), we enrich our results for associations that are both highly likely to replicate and have clinical meaning. Our null interval of +/−10% was chosen to match that of Blume et al.48. and also aligns with a concept in the metabolomics community that small differences in compound abundances can have important associated phenotypes. Because SGPVs are based on interval instead of point differences, their interpretation differs from that of traditional P values. SGPVs, indicated by Pδ, range from 0 to 1 and are significant only if equal to 0, indicating no overlap between the 95% CI and the null interval. SGPVs between 0 and 1 indicate partial overlap of these intervals and are considered inconclusive, and SGPVs equal to 1 indicate complete overlap, or equivalence48.
Data availability
The datasets used and analyzed during the current study can be requested from BIOFIND at https://biofind.loni.usc.edu.
References
GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 16, 877–897 (2017).
GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 17, 939–953 (2018).
Ou, Z. et al. Global trends in the incidence, prevalence, and years lived with disability of Parkinson’s disease in 204 countries/territories from 1990 to 2019. Front. Public Health 9, 776847 (2021).
Zhu, J. et al. Temporal trends in the prevalence of Parkinson’s disease from 1980 to 2023: a systematic review and meta-analysis. Lancet Healthy Longev. 5, e464–e479 (2024).
Vijiaratnam, N., Simuni, T., Bandmann, O., Morris, H. R. & Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 20, 559–572 (2021).
Postuma, R. B. et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601 (2015).
Bloem, B. R., Okun, M. S. & Klein, C. Parkinson’s disease. Lancet 397, 2284–2303 (2021).
Jellinger, K. New developments in the pathology of Parkinson’s disease. Adv. Neurol. 53, 1–16 (1990).
Adler, C. H. et al. Unified staging system for Lewy body disorders: clinicopathologic correlations and comparison to Braak staging. J. Neuropathol. Exp. Neurol. 78, 891–899 (2019).
Lin, M. T. & Beal, M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 (2006).
Foley, P. B., Hare, D. J. & Double, K. L. A brief history of brain iron accumulation in Parkinson disease and related disorders. J. Neural Transm. 129, 505–520 (2022).
Lingor, P., Carboni, E. & Koch, J. C. Alpha-synuclein and iron: two keys unlocking Parkinson’s disease. J. Neural Transm. 124, 973–981 (2017).
Hare, D. J. & Double, K. L. Iron and dopamine: a toxic couple. Brain 139, 1026–1035 (2016).
Du, G. et al. Quantitative susceptibility mapping of the midbrain in Parkinson’s disease. Mov. Disord. 31, 317–324 (2016).
Oakley, A. E. et al. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 68, 1820–1825 (2007).
Sian-Hülsmann, J., Mandel, S., Youdim, M. B. & Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem 118, 939–957 (2011).
Caudle, W. M. & Zhang, J. Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp. Neurol. 220, 230–233 (2009).
Zádori, D., Klivényi, P., Toldi, J., Fülöp, F. & Vécsei, L. Kynurenines in Parkinson’s disease: therapeutic perspectives. J. Neural Transm. 119, 275–283 (2012).
Badanjak, K., Fixemer, S., Smajić, S., Skupin, A. & Grünewald, A. The contribution of microglia to neuroinflammation in Parkinson’s disease. Int. J. Mol. Sci. 22, 4676 (2021).
Venkatesan, D., Iyer, M., Narayanasamy, A., Siva, K. & Vellingiri, B. Kynurenine pathway in Parkinson’s disease—an update. eNeurologicalSci 21, 100270 (2020).
Chen, P. & Geng, X. Research progress on the kynurenine pathway in the prevention and treatment of Parkinson’s disease. J. Enzym. Inhib. Med. Chem. 38, 2225800 (2023).
Leklem, J. E. Quantitative aspects of tryptophan metabolism in humans and other species: a review. Am. J. Clin. Nutr. 24, 659–672 (1971).
Schwarcz, R., Bruno, J. P., Muchowski, P. J. & Wu, H. Q. Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477 (2012).
Espey, M. G., Chernyshev, O. N., Reinhard, J. F. Jr., Namboodiri, M. A. & Colton, C. A. Activated human microglia produce the excitotoxin quinolinic acid. Neuroreport 8, 431–434 (1997).
Guillemin, G. J., Smith, D. G., Smythe, G. A., Armati, P. J. & Brew, B. J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 527, 105–112 (2003).
Sathyasaikumar, K. V. et al. Cellular localization of kynurenine 3-monooxygenase in the brain: challenging the dogma. Antioxidants 11, 315 (2022).
Savitz, J. The kynurenine pathway: a finger in every pie. Mol. Psychiatry 25, 131–147 (2020).
Covarrubias, A. J., Perrone, R., Grozio, A. & Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 22, 119–141 (2021).
Eastman, C. L. & Guilarte, T. R. The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynurenine. Neurochem. Res. 15, 1101–1107 (1990).
Okuda, S., Nishiyama, N., Saito, H. & Katsuki, H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc. Natl. Acad. Sci. USA 93, 12553–12558 (1996).
Guillemin, G. J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 279, 1356–1365 (2012).
Ostapiuk, A. & Urbanska, E. M. Kynurenic acid in neurodegenerative disorders-unique neuroprotection or double-edged sword?. CNS Neurosci. Ther. 28, 19–35 (2022).
Chobot, V., Hadacek, F., Weckwerth, W. & Kubicova, L. Iron chelation and redox chemistry of anthranilic acid and 3-hydroxyanthranilic acid: a comparison of two structurally related kynurenine pathway metabolites to obtain improved insights into their potential role in neurological disease development. J. Organomet. Chem. 782, 103–110 (2015).
Grant, R. S., Coggan, S. E. & Smythe, G. A. The physiological action of picolinic acid in the human brain. Int. J. Tryptophan Res. 2, 71–79 (2009).
Heilman, P. L. et al. Tryptophan metabolites are associated with symptoms and nigral pathology in Parkinson’s disease. Mov. Disord. 35, 2028–2037 (2020).
Vilas, D. et al. A Novel p.Glu298Lys mutation in the ACMSD gene in sporadic Parkinson’s disease. J. Parkinsons Dis. 7, 459–463 (2017).
Martí-Massó, J. F. et al. The ACMSD gene, involved in tryptophan metabolism, is mutated in a family with cortical myoclonus, epilepsy, and Parkinsonism. J. Mol. Med. 91, 1399–1406 (2013).
Meier, T. B. et al. Kynurenic acid is reduced in females and oral contraceptive users: Implications for depression. Brain Behav. Immun. 67, 59–64 (2018).
Correia, A. S. & Vale, N. Tryptophan metabolism in depression: a narrative review with a focus on serotonin and kynurenine pathways. Int. J. Mol. Sci. 23, 8493 (2022).
Müller, N. & Schwarz, M. J. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol. Psychiatry 12, 988–1000 (2007).
Kessler, R. C., McGonagle, K. A., Swartz, M., Blazer, D. G. & Nelson, C. B. Sex and depression in the National Comorbidity Survey. I: lifetime prevalence, chronicity and recurrence. J. Affect Disord. 29, 85–96 (1993).
Haaxma, C. A. et al. Gender differences in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 78, 819–824 (2007).
Solla, P. et al. Gender differences in motor and non-motor symptoms among Sardinian patients with Parkinson’s disease. J. Neurol. Sci. 323, 33–39 (2012).
Kovács, M. et al. Impact of sex on the nonmotor symptoms and the health-related quality of life in Parkinson’s disease. Parkinsons Dis. 2016, 7951840 (2016).
Guo, X. et al. Gender and onset age-related features of non-motor symptoms of patients with Parkinson’s disease—a study from Southwest China. Parkinsonism Relat. Disord. 19, 961–965 (2013).
Georgiev, D., Hamberg, K., Hariz, M., Forsgren, L. & Hariz, G.-M. Gender differences in Parkinson’s disease: a clinical perspective. Acta Neurologica Scandinavica 136, 570–584 (2017).
Kang, U. J. et al. The BioFIND study: characteristics of a clinically typical Parkinson’s disease biomarker cohort. Mov. Disord. 31, 924–932 (2016).
Blume, J. D., D’Agostino McGowan, L., Dupont, W. D. & Greevy, R. A. Jr. Second-generation p-values: improved rigor, reproducibility, & transparency in statistical analyses. PLoS ONE 13, e0188299 (2018).
Sorgdrager, F. J. H. et al. Age- and disease-specific changes of the kynurenine pathway in Parkinson’s and Alzheimer’s disease. J. Neurochem. 151, 656–668 (2019).
Fathi, M. et al. Dynamic changes in metabolites of the kynurenine pathway in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: a systematic Review and meta-analysis. Front. Immunol. 13, 997240 (2022).
Iwaoka, K. et al. Impaired metabolism of kynurenine and its metabolites in CSF of parkinson’s disease. Neurosci. Lett. 714, 134576 (2020).
Lewitt, P. A. et al. 3-hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov. Disord. 28, 1653–1660 (2013).
Klatt, S. et al. A six-metabolite panel as potential blood-based biomarkers for Parkinson’s disease. NPJ Parkinsons Dis. 7, 94 (2021).
Connor, T. J., Starr, N., O’Sullivan, J. B. & Harkin, A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: a role for IFN-gamma?. Neurosci. Lett. 441, 29–34 (2008).
Reyes-Ocampo, J. et al. Mitochondrial dysfunction related to cell damage induced by 3-hydroxykynurenine and 3-hydroxyanthranilic acid: non-dependent-effect of early reactive oxygen species production. Neurotoxicology 50, 81–91 (2015).
Babu, H. W. S. et al. Association study between kynurenine 3-monooxygenase (KMO) gene and Parkinson’s disease patients. Mol. Neurobiol. 61, 3867–3881 (2024).
Ogawa, T. et al. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 42, 1702–1706 (1992).
Guillemin, G. J. et al. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J. Neurochem. 78, 842–853 (2001).
Chang, K. H. et al. Alternations of Metabolic Profile and Kynurenine Metabolism in the Plasma of Parkinson’s Disease. Mol. Neurobiol. 55, 6319–6328 (2018).
Brundin, L. et al. An enzyme in the kynurenine pathway that governs vulnerability to suicidal behavior by regulating excitotoxicity and neuroinflammation. Transl. Psychiatry 6, e865 (2016).
Guidetti, P. & Schwarcz, R. 3-Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur. J. Neurosci. 11, 3857–3863 (1999).
Jellen, L. C. et al. Low plasma serotonin linked to higher nigral iron in Parkinson’s disease. Sci. Rep. 11, 24384 (2021).
Sha, Q. et al. Associations between estrogen and progesterone, the kynurenine pathway, and inflammation in the post-partum. J. Affect Disord. 281, 9–12 (2021).
Lewis, M. M. et al. Clinical progression of Parkinson’s disease in the early 21st century: Insights from the accelerating medicine partnership (AMP-PD) data. Parkinsonism Relat. Disord. 130, 107186 (2025).
Lewis, M. M. et al. Clinical progression of Parkinson’s disease: insights from the NINDS common data elements. J. Parkinsons Dis. 10, 1075–1085 (2020).
Skorvanek, M. et al. Relationship between the MDS-UPDRS and quality of life: a large multicenter study of 3206 patients. Parkinsonism Relat. Disord. 52, 83–89 (2018).
Skorobogatov, K. et al. Brain versus blood: a systematic review on the concordance between peripheral and central kynurenine pathway measures in psychiatric disorders. Front. Immunol. 12, 716980 (2021).
Fukui, S., Schwarcz, R., Rapoport, S. I., Takada, Y. & Smith, Q. R. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J. Neurochem. 56, 2007–2017 (1991).
Goetz, C. G. et al. Movement disorder society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov. Disord. 23, 2129–2170 (2008).
Nasreddine, Z. S. et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J. Am. Geriatr. Soc. 53, 695–699 (2005).
Kassambara, A. rstatix: pipe-friendly framework for basic statistical tests. R package version 0.7.2, https://rpkgs.datanovia.com/rstatix/ (2023).
Kassambra, A. & Mundt, F. factoextra: extract and visualize the results of multivariate data analyses. R package version 1.0.7, https://github.com/kassambara/factoextra (2020).
Acknowledgements
This work was funded by the Michael J. Fox Foundation grant MJFF-010296 (PIs Lena Brundin and Xuemei Huang). We thank all the participants in this study for volunteering to provide biofluid samples and clinical assessments. Data and biospecimens used in the preparation of this article were obtained from the Fox Investigation for New Discovery of Biomarkers (‘BioFIND’) database (http://biofind.loni.usc.edu/). For up-to-date information on the study, visit https://www.michaeljfox.org/biospecimens. BioFIND is sponsored by the Michael J. Fox Foundation for Parkinson’s Research (MJFF) with support from the National Institute for Neurological Disorders and Stroke (NINDS). We thank the Van Andel Institute Bioinformatics and Biostatistics Core (RRID:SCR_024762) for their help with the statistical analysis.
Author information
Authors and Affiliations
Contributions
L.B. and X.H. provided study concept, experimental design, oversight and obtained funding. M.L.E.G. and M.M.L. provided support with study concept, experimental design and manuscript writing. R.D.S. provided oversight and development of metabolite measurements and methodological details. A.J. and C.I. performed metabolite extraction and LC-MS acquisition. Q.S. performed study logistics. L.K. and Z.M. provided statistical methodology and advice. L.C.J. statistically analyzed the data, prepared the figures and tables, and wrote the initial manuscript. All authors critically reviewed the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
M.M.L. is now employed by National Institute of General Medical Sciences. This article was prepared while the author was employed at Penn State University. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, Department of Health and Human Services, or United States government. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jellen, L.C., Escobar Galvis, M.L., Sha, Q. et al. Sex differences in peripheral and central dysregulation of the kynurenine pathway in Parkinson’s disease. npj Parkinsons Dis. 11, 116 (2025). https://doi.org/10.1038/s41531-025-00949-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-025-00949-6
