
Abstract
Staphylococcus aureus is a major cause of bacterial infection-related deaths. Increasing antimicrobial resistance highlights the urgent need for effective preventative strategies. Antibody-mediated opsonophagocytosis, the key mechanism for protection against S. aureus, is disabled by critical virulence factors such as Staphylococcal protein A (SpA) and leukocidin AB (LukAB). In our study, we combined genetically detoxified vaccine candidates SpA* and LukAB RARPR-33 with a TH1 adjuvant aiming to restore host antibody functionality. To evaluate these vaccine candidates, we developed both surgical site infection (SSI) and superficial wound infection (SWI) models in minipigs. Our results showed a significant reduction in bacterial load and systemic dissemination in the SSI model, while skin infection severity was markedly decreased after intradermal immunization in the SWI model. This study introduces a novel S. aureus vaccine strategy by targeting immune evasion factors SpA and LukAB, utilizing potent TH1 adjuvants, and employing minipig challenge models.
Similar content being viewed by others


Introduction
Staphylococcus aureus is a major human pathogen and is included in the World Health Organization’s list of top priority pathogens (so-called “ESKAPE” pathogens) because of its public health importance as one of the most common causes of death due to bacterial infection globally, and because of its high level of resistance to antibiotics1,2,3,4. S. aureus causes community and nosocomial infections, and frequently infects the skin and soft tissue, surgical wounds and implants, and causes pneumonia, bacteremia and sepsis2. S. aureus bacteremia is associated with high ( >20%) mortality2,5. Methicillin-resistant S. aureus (MRSA) is one of the most common causes of death due to resistant bacteria globally, second only to resistant extra-intestinal pathogenic Escherichia coli6. Increasingly limited antibiotic choices and a clear need for preventative strategies have fueled numerous vaccine developments targeting S. aureus, of which none has been successful to date7. The lack of success after decades of effort can be partly attributed to four key issues: the use of poorly representative rodent infection models that have yielded encouraging results unable to be replicated in human clinical trials; prolonged focus on capsular polysaccharides and surface proteins as vaccine antigens; failure to induce CD4 TH1/ TH17 responses necessary for preventing skin infections, pneumonia, deep-seated surgical site or mucosal infections; and most importantly, lack of targeting the key immune evasion factors Staphylococcal protein A (SpA) and leukocidin AB (LukAB) in S. aureus vaccine efficacy studies7,8,9. These two proteins have been subject to evolutionary pressure to allow S. aureus to evade antibody-induced opsonophagocytosis.
In contrast to many bacterial pathogens that escape immunity by way of antigenic variation (such as Streptococcus pneumoniae, E. coli, Neisseria meningitidis, N. gonorrhoeae, and many others), S. aureus is relatively antigenically conserved. Instead, S. aureus affects immune escape by producing a myriad of potent virulence factors that inhibit neutrophil chemotaxis (for example chemotaxis inhibiting protein of S. aureus), complement evasion (such as Staphylococcal complement inhibitor), CD4 T-cell immunity (T cell superantigens), and leukocidins effectively lysing human and pig neutrophils to prevent opsonophagocytosis10,11,12. SpA and LukAB are critical virulence factors that effectively disable the host opsonophagocytic killing mechanism. SpA is a B cell superantigen and opsonophagocytic antibody blocker that is expressed on the bacterial cell surface and freely secreted by most S. aureus isolates13. SpA conveys dual immune escape mechanisms; it contains five domains that bind to immunoglobulin G (IgG) Fc gamma regions in a non-specific configuration (or ‘upside down’), effectively blocking opsonophagocytosis by host cells14. Additionally, SpA suppresses an effective immune response by binding to membrane-bound VH3 immunoglobulins on B cells. This triggers expansion and production of a VH3-biased non-specific antibody response, to the detriment of other S. aureus-specific antibody targets13,15. Astonishingly, SpA binds to ~30% of the B cell pool during an infection in humans, compared to around 0.01% for conventional antigens, effectively disabling the host immune response13. The criticality of SpA as a virulence factor was demonstrated by Falugi et al., 2013, who showed that blocking IgG-Fc and Fab-VH3 binding capacity of SpA restored immune functionality in mice, including phagocytosis and development of a wide antibody repertoire, respectively, and thus established SpA as an attractive vaccine antigen candidate16.
S. aureus releases an array of effective leukocidins that kill immune cells and evade cellular immunity17. LukAB is a human-specific leukocidin that can target and kill pig cells but not rodent cells. It causes extracellular or intracellular neutrophil killing by forming pores in the cell membrane, which allows engulfed bacteria to escape, preventing host clearance and increasing the bacterial load18,19. LukAB has also been shown to lyse or inhibit the function of dendritic cells, with downstream effects on the proliferation and activation of T cells, and is critical for the cytotoxicity of both MRSA and methicillin-sensitive S. aureus (MSSA) towards human polymorphonuclear cells (PMNs)19,20,21.
Antibody-mediated opsonophagocytosis is one of the main immune mechanisms used for S. aureus clearance (Fig. 1A), as demonstrated by increased susceptibility to S. aureus bacteremia in patients with neutrophil deficiencies, and increased susceptibility to S. aureus pneumonia and skin diseases in patients with interleukin (IL)-17 and IL-17 receptor deficiencies22,23,24,25. Consequently, an effective vaccine needs to disarm the immune evasion factors, SpA and LukAB, that target phagocytic cells like neutrophils and macrophages and restore antibody-mediated opsonophagocytosis (Fig. 1B). Moreover, while prevention of S. aureus bacteremia is a key goal of vaccination, prevention of skin and deep surgical site infections and pneumonia requires a CD4 TH1/TH17-biased (interferon-gamma [IFN-γ]/IL-17 cells) immune response, which is necessary to drive the relocation of circulating neutrophils and macrophages to the infection site, and optimize the humoral antibody response9,25,26 (Fig. 1C). This necessitates the use of a potent adjuvant that enables dendritic cells to produce the polarizing TH1/TH17 cytokines.

A Antibody-mediated opsonophagocytic killing is the primary immune mechanism against S. aureus; B In the unvaccinated state, S. aureus escapes clearance by SpA and LukAB toxin; C a TH1/ TH17 immune response is needed for protection against deep surgical site and skin infection. IFNγ interferon gamma, IgG Immunoglobulin G, SAUR S. aureus, TH1/ TH17 T helper type 1/17 cells, Trm tissue-resident memory T cells, IL17A Interleukin 17A, LukAB leucocidin AB SpA Staphylococcal protein A.
In the present study, we developed a vaccine candidate that combined genetically detoxified, biologically inactive forms of SpA and LukAB with the TH1 adjuvant AS01B, aiming to block two critical S. aureus immune evasion mechanisms to allow restoration of neutrophil function, opsonophagocytosis, and bacterial clearance (Fig. 1A). Here we discuss the rationale for the choice of vaccine antigens and the results of vaccination in preclinical minipig models of surgical site infection (SSI)8 (Fig. 2A) and superficial wound infection (SWI) (Fig. 2B).

A Modified schematic presentation of the surgical site minipig model as was described in ref. 8. B Schematic presentation of the superficial wound minipig model. A full thickness 2 cm long incision is made under anesthesia exposing the muscle layer. The challenge is released into the wound with a pipettor while moving the tip down the length of the wound. The incision remained open for the 8-days infection period. Eight days after infection the animal is euthanized and the skin from both sides of the incision is collected and homogenized. Samples were processed as described for the SSI model. Immunization schedule is shown below each picture. CFU colony-forming units, D day, IM intramuscularly, ID intradermal.
Results
SpA* protein, the genetically inactivated variant of SpA as a lead vaccine candidate
Wild-type SpA is not considered safe for use in humans and a genetically detoxified variant, SpAKKAA, was originally developed by the Schneewind and Missiakas group at the University of Chicago27. SpAKKAA successfully blocked the virulence of S. aureus and protected mice against MRSA challenge but was associated with safety concerns related to incomplete inactivation of the SpA virulence traits28. A new variant, SpAKKE (SpA*) contains three amino acid substitutions (Gln9,10Lys, Ser33Glu) in each of the five immunoglobulin-binding domains that constitute this antigen. These substitutions prevent interactions of SpA with Fc-IgG and VH3-Fab of all immunoglobulins28. SpA* has markedly reduced ability to crosslink IgE, addressing safety concerns associated with SpAKKAA28. In mice colonized with S. aureus, vaccination with SpA* induced SpA-specific antibodies, showed efficacy in promoting decolonization, and was linked to a broader immune response compared to unvaccinated animals28. As such, SpA* was selected for further preclinical investigation in our prototype S. aureus vaccine.
LukAB CC8Δ10C toxoid and newly designed LukAB RARPR-33 toxoid
Isogenic mutants to systematically dissect the roles of individual cytotoxins produced by S. aureus revealed that the bi-component leukotoxin LukAB, is predominantly responsible for death of human phagocytes29. Compared to the other S. aureus bicomponent leukocidins, LukAB displays the highest toxicity against primary human PMNs, as shown by the lowest LD50 values in Fig. 3A. S. aureus clonal complex (CC) 8 LukAB can be genetically detoxified by deleting the 10 C-terminal amino acid residues in LukA (LukAB CC8Δ10C) or more specifically, by mutating the glutamic acid at position 323 in the LukA C terminus to alanine (E323A)30. LukAB CC8Δ10C adjuvanted with AS01B was initially evaluated in the minipig SSI model8. In this model, detoxification of LukAB did not compromise immunogenicity, as immunization with LukAB CC8Δ10C boosted LukAB-neutralizing antibodies and reduced bacterial load following S. aureus challenge8. These promising initial findings supported further investigation of genetically detoxified LukAB as a candidate vaccine antigen.

A LukAB shows highest toxicity at the lowest concentrations among leukocidins produced by S. aureus.B Lower in vitro cytotoxicity of LukAB RARPR-33 than prototype LukAB CC8Δ10C. CC clonal complex, Hlg gamma-hemolysin locus, LukAB leukocidin AB, LukED leukocidin ED, PVL Panton-Valentine leukocidin, PMN polymorphonuclear cells, WT wild-type.
One limitation of the prototype LukAB CC8Δ10C vaccine antigen was that it did not address the sequence heterogeneity of LukA and LukB observed among different S. aureus clonal complexes31. CC8 LukAB and CC45/CC30 LukAB differ enough that they require different cellular receptors to maximize host cell lysis31. Thus, antibodies generated against CC8 LukAB may not sufficiently cross-react with LukAB variants from other clonal complexes such as CC45/CC30 to provide adequate protection. We therefore devised a strategy to construct a novel LukAB heterodimer antigen, LukAB RARPR-33, consisting of LukA from CC8 and LukB from CC45. The antigen harbored five amino acid substitutions in LukA, including the LukA CC8 E323A detoxifying mutation30, along with a single substitution in LukB, which were aimed at further inactivating and stabilizing the chimeric antigen.
Lack of cytotoxicity of the LukAB RARPR-33 toxoid on human PMNs
The cytotoxicity of LukAB RARPR-33 toxoid, as compared to LukAB CC8 and CC45 toxin and LukAB CC8Δ10C toxoid, was assessed at concentrations up to 40 µg/ml on human PMNs obtained from five donors. Based on CellTiter measurements, maximum cytotoxicity was observed for the LukAB CC8 and CC45 toxin at approximately 1.25 μg/ml toxin upon 1-hour exposure of PMNs to toxin (Fig. 3B). For LukAB RARPR-33, the percentage of dead cells measured with CellTiter was approximately 10% at a concentration of 40 µg/ml, which was less than 50% of the cytotoxicity observed with LukAB CC8Δ10C, illustrating the superior detoxification of LukAB RARPR-33.
Preclinical proof of concept with the prototype LukAB CC8Δ10C vaccine and synergy of SpA* + LukAB + AS01B
We constructed a pilot vaccine combining SpA* and LukAB CC8Δ10C with the TH1 adjuvant AS01B. Minipigs received three intramuscular (IM) immunizations, separated by three weeks. The minipigs were challenged with a S. aureus CC398 strain in the minipig SSI model8, and the bacterial burden was determined 8 days after challenge (Fig. 2A). SpA* + LukAB CC8Δ10C and the separate SpA* and LukAB CC8Δ10C components in combination with AS01B all induced statistically significant reductions in colony forming units (CFUs) in deep and mid-level muscle compared to the adjuvant only group (Fig. 4). The combined SpA* + LukAB CC8Δ10C toxoid vaccine was the only vaccine that significantly inhibited dissemination of S. aureus to the spleen, showing the benefit of the combination of antigens over each antigen administered alone.

Göttingen minipigs (n = 3/group) received three vaccinations with 50 µg SpA*, 50 µg SpA* + 100 µg LukAB CC8Δ10C or 100 µg LukAB CC8Δ10C all adjuvanted with AS01B (25 µg MPL and 25 µg QS 21) or adjuvant control at 21-day intervals. Three weeks post the third immunization, animals were challenged with 106 CFU S. aureus CC398 in the minipig SSI model as previously described8, and the bacterial burden was determined 8 days later. Each point represents a single animal. Open symbols indicate censored values that are at the limit of detection of the plate reader. The mean of each group is indicated. Statistical significance was determined using Tobit model with Bonferroni correction; *P < 0.05. AS01B Adjuvant System AS01B CC clonal complex, LukAB leukocidin AB, SpA Staphylococcal protein A.
Bridging LukAB CC8Δ10C and LukAB RARPR-33 toxoids (+AS01B) in the minipig SSI model
To compare the efficacy of the improved LukAB RARPR-33 toxoid with LukAB CC8Δ10C, we determined the number of CFUs in skin, muscle and spleen after three immunizations with these LukAB variants combined with AS01B and challenged with S. aureus CC398 in the minipig SSI model. A significant decrease in CFUs was observed in the mid and deep muscle of groups immunized with both LukAB CC8Δ10C and LukAB RARPR-33 variants ( + AS01B) compared to the adjuvant only group in which high CFUs were observed in all tissues tested (Fig. 5). A small (non-significant) decrease in CFUs was also observed in top muscle after vaccination with either of the LukAB variants, whereas there was no significant decrease in CFU observed in skin compared to the adjuvant only group (Fig. 5). CFUs were also observed in the spleen of the control group immunized with adjuvant alone, while there was a significant decrease in splenic CFUs in animals vaccinated with LukAB CC8Δ10C or LukAB RARPR-33 ( + AS01B) (Fig. 5). The results suggest that LukAB CC8Δ10C and LukAB RARPR-33 induce similar efficacy in mid and deep muscle, and in reducing organ bacterial dissemination.

Göttingen minipigs (n = 3/group) were immunized with 100 µg LukAB CC8Δ10C or 100 µg LukAB RARPR-33, both adjuvanted with 250 µL AS01B (25 µg MPL and 25 µg QS 21). The control group received only adjuvant. At 21 days post the third immunization, animals were challenged with 106 CFU S. aureus CC398 in the SSI model. Eight days post challenge, the bacterial burden (log10 CFU per g of tissue) was determined at the surgical site and in the spleen. Each point represents a single animal. Open symbols indicate censored values that are at the limit of detection of the plate reader. The mean of each group is indicated. Statistical significance was determined using Tobit model with Bonferroni post hoc test to correct for multiple comparisons; *P < 0.05. AS01B Adjuvant System AS01B, CC clonal complex, LukAB leukocidin AB, SpA Staphylococcal protein A.
Cross-neutralization of LukAB variants by antibodies induced by vaccination of minipigs
To evaluate the functionality and breadth of vaccine-induced IgG against the LukAB toxin, we measured the ability of serum samples collected from minipigs on day -63 (pre-vaccination) and day 0 (3 weeks post the third immunization, prior to challenge) to neutralize killing of THP-1 cells caused by LukAB variants from nine clonal complexes. Serum samples were obtained from the bridging efficacy study described above. The fold increase in IC50 titers between day -63 (before immunization) and day 0 (after three immunizations and before challenge), was determined. Both vaccination with LukAB CC8Δ10C and LukAB RARPR-33 increased toxin neutralization titers at day 0 versus day -63, and the fold increase for each toxin was significantly higher compared to the adjuvant only group (except for LukAB CC8Δ10C for CC5 neutralization). The largest fold increases in toxin neutralization titers for all nine clonal complexes of LukAB toxins tested were observed in the group vaccinated with LukAB RARPR-33 compared to LukAB CC8Δ10C (Fig. 6) and this was statistically significantly higher for the LukAB toxins CC5, CC8, CC10, CC22b and CC398. No increase in toxin neutralization titers was observed in animals vaccinated with adjuvant alone. These results indicate that the CC8/CC45 chimeric nature of LukAB RARPR-33 did enhance the breadth of the elicited anti-LukAB antibodies as compared to a CC8-based toxoid.

Serum obtained from the study described in Fig. 5 was used in toxin neutralization assays. The fold increase in IC50 toxin neutralization titers between day 0 (three weeks post the third immunization, before challenge) and day -63 (pre-immunization) is shown as the geometric mean for each group against each LukAB variant. The different experimental groups are color coded. Statistically significant differences between LukAB CC8Δ10C and LukAB RARPR-33 are indicated. The differences between LukAB CC8Δ10C and LukAB RARPR-33 compared to the AS01B group are not shown in the Figure but were statistically significant P < 0.05, (except for LukAB CC8Δ10C, CC5 neutralization). Statistical differences were determined using a Tobit model with a Bonferroni post-hoc test to correct for multiple comparisons. TNA, toxin neutralization assay (in vitro). AS01B Adjuvant System AS01B, CC clonal complex, LukAB leukocidin AB, SpA Staphylococcal protein A.
Efficacy of combined SpA* + LukAB RARPR-33 + AS01B/GLA-SE in the minipig SSI model
To test the efficacy of the combined vaccine containing optimized LukAB RARPR-33, we determined the number of CFUs in three muscle sites (top, middle and deep), the skin and the spleen after three immunizations with SpA*, LukAB RARPR-33 and AS01B and challenge with S. aureus CC398 in minipigs. A significant reduction in CFU ( ~ 5 logs) was observed in the mid and deep muscle compared to the negative control group (Fig. 7A). In the spleen a significant reduction in CFU was also observed (Fig. 7A). No significant reduction in CFUs was observed in the skin or top muscle (Fig. 7A). These results indicate that the vaccine candidate reduces bacterial load in deep-seated infections in the minipig SSI model and reduces bacterial dissemination to the spleen.

Göttingen minipigs (n = 3) were immunized with 100 µg SpA* and 100 µg LukAB RARPR-33, adjuvanted with AS01B (25 µg MPL and 25 µg QS-21) or with GLA-SE (10 µg GLA, 2% SE). The control group received antigen-formulation buffer. Three weeks post the third immunization, animals were challenged with A 106 CFU S. aureus CC398, B 106 CFU S. aureus USA300, in the SSI model. Eight days post challenge the bacterial burden (Log10 CFU/gram of deep muscle) was determined at the surgical site and the spleen. C Göttingen minipigs (n = 3) were immunized with a formulation representing the SA4Ag vaccine composition (SA4Ag-like), composed of capsular polysaccharide conjugates of serotypes 5 and 8 (30 µg each), recombinant surface protein clumping Factor A (60 µg), and recombinant manganese transporter Protein C (200 µg) without an adjuvant, as described above. An additional group was immunized with 100 µg LukAB RARPR-33 adjuvanted with AS01B (25 µg MPL and 25 µg QS-21) and the negative control group received only AS01B. Each point represents a single animal. Open symbols indicate censored values that are at the limit of detection of the plate reader. The mean of each group is indicated. Statistical significance was determined using Tobit model with Bonferroni post hoc test to correct for multiple comparisons (circles: CC398; triangles: USA300). AS01b Adjuvant System AS01B, GLA-SE synthetic toll-like receptor 4 agonist in squalene oil-in-water emulsion, CFU colony-forming units, LukAB leukocidin AB, SA4Ag-like capsular polysaccharide conjugates of serotypes 5 and 8 recombinant surface protein clumping Factor A, and recombinant manganese transporter Protein C without adjuvant, SpA Staphylococcal protein A.
Similar results were obtained with the TH1 adjuvant GLA-SE (synthetic toll-like receptor [TLR]4 agonist in squalene oil-in-water emulsion) used instead of AS01B, in combination with the combined vaccine antigens (Fig. 7A).
We repeated the experiment using S. aureus USA300 as the challenge strain. Immunization with SpA* + LukAB RARPR-33 + AS01B resulted in a significant decrease in CFUs in deep muscle compared to control (Fig. 7B). CFU data for the other muscle layers were comparable to the results obtained with the CC398 strain (Fig. 7B).
To test our minipig surgical site infection model further, we also evaluated the efficacy of a formulation resembling SA4Ag (SA4Ag-like, Fig.7C) in the SSI minipig model. The SA4Ag-like vaccine composition is a formulation composed of capsular polysaccharide conjugates of serotypes 5 and 8 (CP5 and CP8), recombinant surface protein clumping Factor A (ClfA), and recombinant manganese transporter Protein C (MntC), resembling a vaccine tested in clinical trials that failed to show efficacy (SA4Ag)32. The SA4Ag-like formulation did not have any impact on CFU counts at the surgical site in the minipig SSI model, while the positive control LukAB RARPR-33 + AS01B vaccine composition again reduced the bacterial burden by ~5 logs (Fig. 7C).
Immunogenicity of SpA* and LukAB RARPR-33 in minipigs
Humoral immunity
To test immunogenicity of the vaccine in minipigs, blood samples were collected prior to each immunization and before challenge and were analyzed for antibody responses against vaccine antigen SpA* and LukAB WT variants CC8 and CC45 by enzyme-linked immunosorbent assay (ELISA).
In animals immunized with buffer alone, no measurable antibodies against SpA* and only low levels of anti-LukAB CC8 and CC45 IgG antibodies were measurable throughout the study, indicating the absence of pre-existing antibodies to SpA* (Fig. 8A) and the presence of pre-existing antibodies to LukAB (Fig. 8B). Immunization with SpA* + LukAB RARPR-33 + AS01B induced anti-SpA*, anti-LukAB CC8 and anti-LukAB CC45 antibodies that increased with consecutive doses (Fig. 8). These results indicate an induction of specific antibodies to the target antigens by the combined SpA* + LukAB RARPR-33 + AS01B vaccine candidate. The antibody responses are from the same study as the efficacy data shown in Fig. 7A. Similar antibody responses against SpA* and LukAB RARPR-33 were observed when the antigens were combined with adjuvant GLA-SE (data not shown).

Göttingen minipigs (n = 3) were immunized with 100 µg SpA* + 100 µg LukAB RARPR-33 + AS01B (25 µg MPL and 25 µg QS-21). The control group received antigen formulation buffer. Sera was collected before each immunization and 3 weeks post the third immunization, before challenge. The study is the same as shown in Fig. 7A. Specificity towards A SpA*, B LukAB CC8 or LukAB CC45 was determined by ELISA. IgG EC50 titers are shown. Each point represents a single animal. The geometric mean ± geometric standard deviation of each group is shown. Dotted line indicates limit of detection and is set at 30. Samples below this value are censored to 30. Statistical significance was determined after three immunizations between the animals immunized with SpA* + LukAB RARPR-33 combined with AS01B to the buffer control group and between pre-and post-three immunizations in the vaccinated animals, using a Tobit model, *P < 0.05. AS01B Adjuvant System AS01B, CC clonal complex, EC50 half maximal effective concentration, LukAB leukocidin AB, SpA Staphylococcal protein A.
Cellular response towards LukAB RARPR 33 and SpA*
Cellular responses to SpA* + LukAB RARPR-33 + AS01B were determined using peripheral blood mononuclear cells (PBMCs) harvested from minipigs prior to and after three immunizations. T cell proliferation was tracked by carboxifluorescein diacetate succinimidyl ester (CFSE) dilution upon ex vivo re-stimulation with SpA* or LukAB RARPR‑33.
The proliferation index (representing the fold increase in CFSE dilution between stimulated and unstimulated cells) of total CD3+ T cells increased in animals immunized with vaccines containing LukAB RARPR‑33 upon in vitro restimulation with LukAB RARPR-33 (Fig. 9B). This was not observed in the buffer control group. Very limited T cell proliferation was detected after SpA* stimulation (Fig. 9A). LukAB-specific T cell responses were detected in CD4+, CD8+, and CD4+/CD8+ T cell subsets (Fig. 9C), the latter representing an antigen-experienced T cell subset in minipigs33,34,35.

Minipig PBMCs were isolated pre- and post-immunization of animals immunized with 100 µg SpA* + 100 µg LukAB RARPR-33 adjuvanted with AS01B and frozen until use. To track vaccine-specific T cell responses, PBMCs were thawed, labelled with carboxifluorescein diacetate succinimidyl ester (CFSE) and ex vivo restimulated for 4 days in the presence of 8 µg/ml SpA* or LukAB RARPR-33. Subsequently, T-cell proliferation was tracked by CFSE dilution (A-C). The proliferation index represents the fold increase in CFSE dilution between stimulated and unstimulated cells of total T cells (CD3 + ) specific for A SpA*, B LukAB, C CD4 + T cells, CD8 + T cells, and CD4 + /CD8 + T cells specific for LukAB. D IFNγ and TNFα secreting cells from minipig PBMCs isolated after three immunizations with LukAB RARPR-33 + AS01B or AS01B alone was determined by ELISpot by direct ex vivo stimulation of the isolated fresh PBMCs with LukAB RARPR-33. Each point represents an individual animal and group mean ± SD is shown. AS01B Adjuvant System AS01B, IFN-γ interferon-gamma, LukAB leukocidin AB, SFU spot-forming units, SpA SpA Staphylococcal protein A, TNFα tumor necrosis factor alpha.
In an additional experiment, IFN‑γ and tumor necrosis factor alpha (TNFα) secreting cells were determined by ELISpot in animals immunized with LukAB RARPR‑33 + AS01B (Fig. 9D). Cells that secrete these cytokines were detected when PBMCs from minipigs were re-stimulated ex vivo with LukAB RARPR‑33. IFN‑γ and TNFα secreting cells were not observed in PBMCs isolated from the adjuvant-only group.
Together, these results show that upon immunization with LukAB RARPR‑33 + AS01B, specific T cell responses are induced that are able to produce effector cytokines, indicating that a functional cellular response is induced by the LukAB toxoid.
Added value of the TH1 adjuvant GLA-SE
Next, we addressed the added value of including a TH1 adjuvant in a S. aureus vaccine. For this experiment we used the TH1 adjuvant GLA-SE that showed comparable efficacy in the SSI compared to AS01B (Fig. 7A). Immunization with SpA* + LukAB RARPR-33 with TH1 adjuvant showed significantly greater reductions in CFUs in mid and deep muscle when TH1 adjuvant was present compared to SpA* + LukAB RARPR-33 without TH1 adjuvant (Fig. 10). Antibody responses to SpA* and LukAB were similar in the groups vaccinated with SpA* + LukAB RARPR-33 with or without GLA-SE adjuvant (Supplementary Fig 1).

Minipigs (3/group) were immunized with 100 µg SpA* and100 µg LukAB RARPR‑33 with or without Th1 adjuvant (GLA-SE, 10 µg GLA, 2% SE) and challenged with 106 CFU S. aureus CC398 in the minipig SSI model. Each point represents a single animal. Open symbols indicate censored values that are at the limit of detection of the plate reader. Statistical significance was determined using Tobit model with Bonferroni post hoc test to correct for multiple comparisons, *P < 0.05. GLA-SE synthetic toll-like receptor 4 agonist in squalene oil-in-water emulsion, CFU colony-forming units, LukAB leukocidin AB, SpA Staphylococcal protein A.
Efficacy of SpA* + LukAB RARPR-33 + GLA-SE in the minipig superficial wound infection (SWI) model
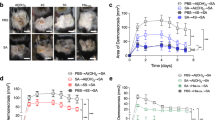
In the SSI model, no reduction in CFU was observed in the skin upon immunization with SpA*, LukAB RARPR-33 and a Th1 adjuvant. This could be due to the presence of sutures in the skin, where a biofilm could form. A S. aureus superficial wound infection model was developed in Göttingen minipigs to address efficacy of the S. aureus vaccine candidate in a different skin infection model. To generate skin wounds, a full thickness incision 2 cm long and positioned 2 cm from the spine was made in the skin of a minipig and S. aureus USA300 was pipetted into the wound (Fig. 2B).
The route of vaccine administration impacts the immune response36. Intradermal administration might be more beneficial to induce immune response in the skin. Minipigs received either three intramuscular or intradermal immunization with SpA* + LukAB RARPR-33 + TH1 adjuvant (GLA-SE) and were challenged with the S. aureus USA300 strain in the minipig SWI model (Fig. 2B). Eight days post-infection, the bacterial burden was determined in the superficial wound. A statistically significant reduction in CFU was found when the vaccine was administered intradermally, but not intramuscularly, compared with the adjuvant control (Fig. 11).

Minipigs (3/group) were immunized with SpA*, LukAB RARPR‑33, and GLA-SE either intramuscular (IM) or intradermal (ID) and challenged with 5 × 106 CFU S. aureus USA300 in the minipig superficial wound infection model. For IM vaccinations, 100 µg of each protein was used in combination with 10 µg GLA, 2% SE. For ID vaccinations 20 µg of each protein was used combined with 5 µg GLA, 2% SE. Eight days post challenge, the bacterial burden (log10 CFU per g of tissue) was determined at the skin. Each point represents a single animal. The fold difference between the control group and the ID immunized group is indicated. Statistical significance was determined using ANOVA with Dunnett post hoc test to correct for multiple comparisons, *P < 0.05. GLA-SE synthetic toll-like receptor 4 agonist in squalene oil-in-water emulsion, CFU colony-forming units, ID intra-dermal, IM intra-muscular, LukAB leukocidin AB, SpA Staphylococcal protein A.
Discussion
This study describes the development of a S. aureus vaccine candidate consisting of two genetically detoxified critical virulence factors, SpA and LukAB, targeting two major immune escape mechanisms, with a TH1 adjuvant included, that provided protection against deep tissue and skin infections. Leukocidin AB (LukAB) has been shown to be the primary toxin responsible for primary human polymorphonuclear leukocyte (PMN) cell death29 and we demonstrated that compared to the other S. aureus bicomponent leukocidins, LukAB displays the highest toxicity against primary human PMNs. LukAB is universally present in all S. aureus strains. The LukAB RARPR‑33 toxoid antigen was newly developed to provide cross-neutralizing activity against naturally occurring LukAB variants. Biological inactivation of LukAB RARPR-33 was confirmed by low PMN cytotoxicity compared with wild-type LukAB, and with the prototype LukAB CC8Δ10C toxoid previously assessed in the minipig SSI model8. Cross-neutralizing activity of LukAB RARPR‑33 was observed against LukAB toxins from numerous S. aureus clonal complexes CCs and sequence types. SpA* + LukAB RARPR-33 + commercially available AS01B induced antigen-specific antibodies and cellular responses with release of TH1 effector cytokines that were able to clear bacteria. Reductions in CFUs in mid- and deep muscle and distant sites (spleen) were observed in the minipig SSI model after vaccination with SpA* + LukAB RARPR-33 + AS01B, whereas no efficacy was demonstrated in animals who received a vaccine resembling SA4Ag, a vaccine that showed promise in rodent models but ultimately failed in the clinic despite robust immunogenicity32. This lack of protection is similar to the lack of protection observed in minipigs vaccinated with capsule antigens even in combination with an adjuvant, which also failed in clinical trials where they were tested without adjuvant8.
Our data show that the minipig SSI model has utility in preclinical S. aureus vaccine development by demonstrating efficacy in reduction of bacterial load at the mid-muscle and deep-seated surgical sites, and in reduction of bacterial dissemination8. Minipigs were selected based on strong similarities between humans and pigs in S. aureus infectious disease, colonization and transmission patterns, immune responses to infection, pre-existing antibodies to S. aureus, and structural and immunological similarity of the epidermis and dermis8. LukAB has been shown to bind pig CD11b to a similar level as human CD11b37. These similarities allow use of human S. aureus isolates in challenge experiments that produce a high bacterial burden in deep-seated sites with bacterial dissemination to other organs. The minipig SSI model showed differential efficacy in skin, muscle layers, and spleen, with the most consistent reductions in CFUs observed in mid and deep muscle, and spleen. The lack of efficacy induced by vaccination with a SA4Ag-like vaccine is consistent with clinical trial results. These data suggest that in the absence of a positive control that showed efficacy in humans the minipig SSI model mimics negative human responses to S. aureus infection more closely than rodents. Therefore, while efficacy in humans will have to be demonstrated, the preclinical assessment in the minipig surgical site infection model could support reducing the number of unsuccessful S. aureus vaccines moving past preclinical development into clinical trials.
The experiment conducted in the additional minipig SWI model suggests that intradermal immunization may be more effective in engaging the skin-based immune system and preventing S. aureus skin infections than intramuscular immunization. The dermis contains dendritic cells that can induce CD4+ TH1 T cells, and resident T cells that show a memory effector phenotype, being readily available to respond on antigen exposure38,39. The intradermal route is used for some authorized influenza and hepatitis B vaccines, and has been investigated for numerous candidate vaccines because of superior protective responses that can be achieved compared to intramuscular vaccination, as well as dose sparing, and ease of administration40,41. Pig skin is recognized as a relevant model for evaluation of intradermal vaccination of humans41.
There is an urgent clinical need for an effective S. aureus vaccine to prevent potentially lethal bloodstream infections and other community-acquired and nosocomial S. aureus infections. Our study provides compelling evidence that a multi-component vaccine containing detoxified proteins derived from the critical immune evasion factors SpA and LukAB, is a strong candidate for further development. Efficacy as demonstrated in the two minipig models may de-risk further development compared to testing in mouse models given the apparent applicability of the model to the human context. Further testing in humans of the safety and immunogenicity and ultimately the efficacy of the vaccine containing modified SpA* and LukAB RARPR-33 antigens would have to be conducted. We believe that the use of an appropriate adjuvant will be key to success. While we included a TH1 adjuvant in the prototype vaccine, addition of a TH17 adjuvant might also be necessary.
Methods
Antigen production
Protein vaccine antigens were produced recombinantly in E. coli at Genscript. The lukA and lukB genes for LukAB CC8Δ10C and LukAB RARPR-33 were cloned in a single expression vector behind a T7 promoter, and a non-cleavable N-terminal His-tag was added to LukA. The genes for the SpA* surface protein, MntC and ClfA were cloned in an expression vector behind a T7 promoter, and a cleavable N-terminal His-SUMO tag was added. All vaccine antigens were expressed in the E. coli cytosol and subsequently affinity purified from the supernatant of the whole cell lysate. After affinity purification of the LukAB RARPR-33 protein, the material was subjected to gel filtration to remove residual impurities. After affinity purification of the SpA* protein, MntC and ClFA, the cleavable His-SUMO tag was removed.
The wild-type LukAB toxins were produced in S. aureus. The lukA and lukB genes for LukAB toxins were cloned in a single expression vector behind a native lukAB promoter, and a non-cleavable N-terminal His-tag added to LukA. The toxins were purified from the S. aureus culture supernatant by affinity purification.
CP5 and CP8 were extracted from S. aureus biomass at Premas Biotech (Gurgaon, India) and conjugated to CRM197 (Reagent proteins, Pfenex) through thioether chemistry.
Adjuvants
Commercially available AS01B, which is provided as separate vial as part of the Shingrix vaccine (GSK) was used42. The liposome-based adjuvant contains 100 µg/ml 3-O-desacyl-4′-monophosphoryl lipid A (MPL), a TLR4 agonist, and 100 µg/ml QS-21, a natural saponin molecule. For animal studies, the adjuvant was mixed 1:1 (v/v) with antigens prior to vaccination, resulting in injection of a half human AS01B dose (25 µg MPL and 25 µg QS-21 in 500 µl). Liquid research grade GLA-SE, a synthetic TLR4 agonist in squalene oil-in-water emulsion, was manufactured by the Access to Advanced Health Institute (AAHI, Seattle, WA) formerly the Infectious Disease Research Institute (IDRI)43. The concentration of the GLA-SE stock was 250 µg/ml GLA, 10% SE. For preparation of the formulation with antigens, the GLA-SE stock was diluted with 10% SE and buffer (1.825% (v/v) glycerol and 25 mM ammonium phosphate pH 5.75) to 40 µg/ml GLA, 4% SE and mixed 1:1 (v/v) with the antigens to obtain a dose of 10 µg GLA, 2% SE for intramuscular vaccination. For intradermal vaccination, the GLA-SE stock was diluted with buffer to 100 µg/ml GLA, 4% SE and mixed 1:1 (v/v) with the antigens to obtain a 5 µg GLA dose with 2% SE.
Minipig immunization
Five to eight-month-old male Göttingen minipigs (Marshall Biosciences, North Rose, NY) were group-housed and maintained on a 12-hour light/dark cycle with access to water ad libitum. Animals were vaccinated by 3 injections separated by 3 weeks with antigen(s) combined with the adjuvant 30 min prior to each vaccination. On the morning of vaccination, fasted minipigs were sedated with Telazol (Zoetis, Parsippany-Troy Hills, NJ; 3.5–4 mg/kg) given intramuscularly and bled before each vaccination. For intramuscular injections, the vaccines were administered in a 500 µl volume in the left hind leg using a 23 G ¾” needle. For intradermal injections, the vaccines were administered in a 100 µl volume behind the ear using the Tropis® needle-free injection system (PharmaJet, Golden, CO). Surgery and infection occurred 3 weeks after the last immunization.
Deep seated SSI model
The deep-seated SSI model was described earlier8. On the morning of surgery, fasted minipigs were sedated with a mixture of ketamine (8–10 mg/kg) and dexmedetomidine (Dexdomitor; Zoetis, Parsippany-Troy Hills, NJ; 0.08 to 0.1 mg/kg) given intramuscularly, away from the surgical site. Once intubated, the animals were placed on isoflurane inhalant anesthesia and maintained for the duration of the surgery. Before surgery, animals received buprenorphine (0.02 to 0.05 mg/kg) intramuscularly away from the surgical site.
Surgery was performed on the left thigh whereby the muscle layer was exposed and a 5-mm bladeless trocar (ENDOPATH® Xcel, Ethicon Endo-Surgery; Guaynabo, Puerto Rico) was advanced to the depth of the femur. A bacterial challenge consisting of 20 µL inoculum (approx. 106 CFU S. aureus) was injected into the wound (top of femur) via a 6-inch MILA spinal needle (Mila International, Inc.; Florence, KY) through the trocar, which was then removed. After administration of the bacterial challenge, the muscle was closed with a silk suture, and the skin was closed with an absorbable polydioxanone suture. Eight days later while under sedation, minipigs were euthanized with a barbiturate. Samples were homogenized in saline using a Bead Ruptor Elite (Omni International; Kennesaw, GA, USA), then diluted and plated on tryptic soy agar plates using an Autoplate 5000 Spiral Plater (Spiral Biotech; Norwood, MA, USA). Plates were incubated 18–24 h at 37 °C, then read on a QCount colony counter (Spiral Biotech). Clearance of bacteria from the surgical site was our primary endpoint. We also examined body weight and body temperature of the animals. The animals appeared bright and alert following surgery/infection and there was no effect on body weight as all animals continued to gain weight after bacterial challenge. Body temperature was assessed, and all animals experienced an increase in body temperature following infection but generally this was resolved within 24 h, regardless of the treatment. All animal studies were reviewed and approved by the Janssen Spring House Institutional Animal Care and Use Committee and housed in an AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care International)-accredited facility.
Minipig SWI model
Fasted minipigs were sedated, intubated, and placed under anesthesia for the duration of the surgery. A full thickness incision 2 cm long and positioned 2 cm from the spine was made in the skin and 50 µl of the inoculum (5 × 106 CFU of S. aureus USA300) was pipetted into the incision. The incision remained open for the 8-days infection period. Skin was collected and homogenized. Tissue was collected from both edges and weighed and normalized to tissue weight. Samples were processed as described above for the SSI model.
Bacterial strains for in vivo challenge and growth conditions
Two clinical blood isolates strains of S. aureus were used and are described in Table 1: ST398 (OC 26263, MSSA) was used for the minipig challenge studies (ST398 strains typically colonize pigs but can cause disease in humans) and ST8/USA300 (OC26260, MRSA) were included because of their high global prevalence among healthcare-associated and community-acquired MRSA infections. Strains were grown in tryptic soy broth overnight prior to use.
Enzyme-linked immunosorbent assay
To determine total IgG in minipig serum, antigens were coated onto Nunc 384 -well Maxisorp plates (VWR, Amsterdam, Netherlands) for 1 h (LukAB, 1 µg/ml), or overnight (SpA*, 0.25 µg/ml), at 2 °C–8 °C. Plates were blocked for 1 h at room temperature (RT) with 2.5% (w/v) skimmed milk in PBS prior to washing and subsequent addition of serial dilution of serum. The sera were incubated for 1 h at RT. After washing, secondary antibody (rabbit antipig IgG horseradish peroxidase [HRP], Sigma Aldrich, St. Louis, MO, USA) was added at 1:10,000 dilution and incubated at RT for 1 h. After further washing, 3,3′,5,5′-tetramethylbenzidine was added to detect the HRP. The reaction was stopped after 30 min with 1 M sulfuric acid, and absorbance was read at 450 nm.
Cytotoxicity assays
To evaluate the cytotoxicity of LukAB protein complexes, freshly isolated primary human PMNs were intoxicated with S. aureus toxins. Prior to intoxication, all toxins were normalized to 100 µg/ml (per subunit) and then serially diluted 2-fold in 10 µl of 1× PBS. PMNs were isolated and normalized to 200,000 cells per 90 µl RPMI (10 mM HEPES + 0.1% HSA). 90 µl of PMNs were added to each well and incubated at 37 °C + 5% CO2 for 1 hr. 10 µl of CellTiter 96 Aqueous One Solution (CellTiter; Promega, Madison, WI, USA) was added, and the mixture was incubated at 37 °C in 5% CO2 for 1.5 h. PMN viability was assessed with a PerkinElmer EnVision 2103 Multilabel Reader at an absorbance of 492 nm. % Dead cells were calculated by subtracting out background (healthy cells + PBS) and normalizing to Triton X-100-treated cells which were set at 100% dead.
Toxin neutralization assays
Neutralization of LukAB toxin with sera from minipigs was described earlier8. In brief, THP1 cells were incubated for 2 h with serially diluted serum samples and a fixed concentration of each LukAB toxin. The supernatant was harvested, and the release of lactate dehydrogenase (LDH) was measured via a fluorometric reaction. The amount of LDH released in the supernatant is directly proportional to the damage inflicted by the toxin. A percentage cytotoxicity was calculated for each serum dilution. Toxin neutralization results were reported as IC50 values calculated from 4-PL curves of the cytotoxicity values, which indicate the half maximal inhibitory concentration effective in inhibiting the LukAB toxin function.
PBMC assays
Minipig blood was collected in cell preparation tubes (CPT) with sodium heparin. Within two hours after collection, tubes were centrifuged for 20 min at 1700 g at RT. The mononuclear cell layer was collected and washed twice with PBS (10 min at 300 g). Cells were frozen at concentration of 5 × 106 cells/ml in 10% DMSO, 25% FBS and 65% RPMI. After overnight storage at −80 °C, the cells were moved to liquid nitrogen until further use. For the T cell proliferation assay, cells were thawed at 37 °C and washed with IMDM with Glutamax + 10% FBS and penicillin-streptomycin. Cells were rested for 2 h at 37 °C, after which they were labelled with CellTrace CFSE (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s protocol. 2 × 105 PBMCs were stimulated per well, and depending on cell availability multiple wells were used per condition. PBMCs were stimulated for 4 days with 8 µg/ml LukAB RARPR-33 or SpA*. Medium only was used as a negative control. After 4 days, PBMCs were stained with conventional T cell markers and analyzed using a LSR Fortessa flow cytometer. For ELISpot assay, fresh PBMCs were isolated from the blood three weeks post the third immunization. 500,000 cells were stimulated for 18–20 h with 8 µg/ml LukAB RARPR-33 or medium in ELISpot plates (Mabtech, Nacka Strand, Sweden). ELISpot plates were revealed for IFN-γ and/or TNF-α secreting cells according to manufacturer’s protocol (Mabtech).
Statistical analysis
ANOVA with Dunnett post hoc test, or in case of censored values, a Tobit Model with a Bonferroni correction, was used to test statistical significance between multiple groups within one study. The model contained group and surgery date as explanatory factors. P values < 0.05 were considered significant.
Data availability
The data that support the findings of this study are available from Janssen Vaccines and Prevention B.V but restrictions apply to the availability of sequence data for intellectual property reasons and so are not publicly available. Data are however available from the corresponding author upon reasonable request.
References
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327 (2018).
Tong, S. Y., Davis, J. S., Eichenberger, E., Holland, T. L. & Fowler, V. G. Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 28, 603–661 (2015).
GBD 2019 Antimicrobial Resistance Collaborators Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 400, 2221–2248 (2022).
World Health Organization. Global tuberculosis report 2023. https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023 (accessed 15 May 2024).
Kern, W. V. & Rieg, S. Burden of bacterial bloodstream infection-a brief update on epidemiology and significance of multidrug-resistant pathogens. Clin. Microbiol. Infect. 26, 151–157 (2020).
Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Clegg, J. et al. Staphylococcus aureus vaccine research and development: The past, present and future, including novel therapeutic strategies. Front. Immunol. 12, 705360 (2021).
Fernandez, J. et al. Vaccination with detoxified leukocidin AB reduces bacterial load in a Staphylococcus aureus minipig deep surgical wound infection model. J. Infect. Dis. 225, 1460–1470 (2022).
Poolman, J. T. Expanding the role of bacterial vaccines into life-course vaccination strategies and prevention of antimicrobial-resistant infections. NPJ Vaccines 5, 84 (2020).
Bear, A. et al. The immune evasion roles of Staphylococcus aureus protein A and impact on vaccine development. Front. Cell Infect. Microbiol.13, 1242702 (2023).
Howden, B. P. et al. Staphylococcus aureus host interactions and adaptation. Nat. Rev. Microbiol. 21, 380–395 (2023).
Wong Fok Lung, T. et al. Staphylococcus aureus adaptive evolution: recent insights on how immune evasion, immunometabolic subversion and host genetics impact vaccine development. Front. Cell Infect. Microbiol. 12, 1060810 (2022).
Pauli, N. T. et al. Staphylococcus aureus infection induces protein A-mediated immune evasion in humans. J. Exp. Med. 211, 2331–2339 (2014).
Sasso, E. H., Silverman, G. J. & Mannik, M. Human IgM molecules that bind staphylococcal protein A contain VHIII H chains. J. Immunol. 142, 2778–2783 (1989).
Graille, M. et al. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. USA 97, 5399–5404 (2000).
Falugi, F., Kim, H. K., Missiakas, D. M. & Schneewind, O. Role of protein A in the evasion of host adaptive immune responses by Staphylococcus aureus. mBio 4, e00575–00513 (2013).
Spaan, A. N., van Strijp, J. A. G. & Torres, V. J. Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 15, 435–447 (2017).
DuMont, A. L. et al. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc. Natl. Acad. Sci. USA 110, 10794–10799 (2013).
DuMont, A. L. et al. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect. Immun. 81, 1830–1841 (2013).
Stulik, L. et al. Preventing lung pathology and mortality in rabbit Staphylococcus aureus pneumonia models with cytotoxin-neutralizing monoclonal IgGs penetrating the epithelial lining fluid. Sci. Rep. 9, 5339 (2019).
Berends, E. T. M. et al. Staphylococcus aureus impairs the function of and kills human dendritic cells via the LukAB toxin. mBio 10, e01918 (2019).
Messina, J. A., Thaden, J. T., Sharma-Kuinkel, B. K. & Fowler, V. G. Jr. Impact of bacterial and human genetic variation on Staphylococcus aureus infections. PLoS Pathog. 12, e1005330 (2016).
Picard, C. et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine 89, 403–425 (2010).
Park, B. & Liu, G. Y. Staphylococcus aureus and Hyper-IgE Syndrome. Int. J. Mol. Sci. 21, 9152 (2020).
Levy, R. et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc. Natl Acad. Sci. USA113, E8277–E8285 (2016).
Broker, B. M., Mrochen, D. & Peton, V. The T cell response to Staphylococcus aureus. Pathogens 5, 31 (2016).
Kim, H. K., Cheng, A. G., Kim, H. Y., Missiakas, D. M. & Schneewind, O. Nontoxigenic protein A vaccine for methicillin-resistant Staphylococcus aureus infections in mice. J. Exp. Med. 207, 1863–1870 (2010).
Shi, M. et al. A protein A based Staphylococcus aureus vaccine with improved safety. Vaccine 39, 3907–3915 (2021).
Dumont, A. L. et al. Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol. Microbiol. 79, 814–825 (2011).
DuMont, A. L. et al. Identification of a crucial residue required for Staphylococcus aureus LukAB cytotoxicity and receptor recognition. Infect. Immun. 82, 1268–1276 (2014).
Perelman, S. S. et al. Genetic variation of staphylococcal LukAB toxin determines receptor tropism. Nat. Microbiol. 6, 731–745 (2021).
Hassanzadeh, H. et al. Efficacy of a 4-antigen Staphylococcus aureus vaccine in spinal surgery: the STaphylococcus aureus suRgical Inpatient Vaccine Efficacy (STRIVE) randomized clinical trial. Clin. Infect. Dis. 77, 312–320 (2023).
Gerner, W., Käser, T. & Saalmüller, A. Porcine T lymphocytes and NK cells-an update. Dev. Comp. Immunol. 33, 310–320 (2009).
Pescovitz, M. D., Sakopoulos, A. G., Gaddy, J. A., Husmann, R. J. & Zuckermann, F. A. Porcine peripheral blood CD4+/CD8+ dual expressing T-cells. Vet. Immunol. Immunopathol. 43, 53–62 (1994).
Saalmüller, A., Hirt, W. & Reddehase, M. J. Phenotypic discrimination between thymic and extrathymic CD4-CD8- and CD4+CD8+ porcine T lymphocytes. Eur. J. Immunol. 19, 2011–2016 (1989).
Rotrosen, E. & Kupper, T. S. Assessing the generation of tissue resident memory T cells by vaccines. Nat. Rev. Immunol. 23, 655–665 (2023).
Boguslawski, K. M. et al. Exploiting species specificity to understand the tropism of a human-specific toxin. Sci. Adv. 6, eaax7515 (2020).
Clark, R. A. et al. The vast majority of CLA+ T cells are resident in normal skin. J. Immunol. 176, 4431–4439 (2006).
Klechevsky, E. et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity 29, 497–510 (2008).
Kis, E. E., Winter, G. & Myschik, J. Devices for intradermal vaccination. Vaccine 30, 523–538 (2012).
Co-Rives, I., Chen, A. Y. & Moore, A. C. Skin-based vaccination: a systematic mapping review of the types of vaccines and methods used and immunity and protection elicited in pigs. Vaccines 11, 450 (2023).
Lecrenier, N. et al. Development of adjuvanted recombinant zoster vaccine and its implications for shingles prevention. Expert Rev. Vaccines 17, 619–634 (2018).
Dubois Cauwelaert, N. et al. The TLR4 agonist vaccine adjuvant, GLA-SE, requires canonical and atypical mechanisms of action for TH1 induction. PloS One 11, e0146372 (2016).
Acknowledgements
This study was funded by Janssen Vaccines & Prevention B.V. The funder was involved in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.The authors thank Craig McLahan, Veterinary Medical Doctor Jolaine Wilson, Doctor of Veterinary Medicine for surgical assistance and clinical care; Jessica Henn, Leslie Buehler, Matthew Willms, Danielle Malone, and Kaitlyn Grubb for performing in vivo models; Jeroen Zeijpveld, Joan van Kregten, Timothy van Eijl, Martijn Kremer, Pepijn Wijnands, Kirstin Jansen and Maria Rodriguez-Gomez for performing in vitro assays; Roxanne Douwes, Gerrard Perdok and Marco Glandrup for LukAB toxin production; Hanna Hanzha for preparing and analyzing the antigen and adjuvant formulations and Jan Serroyen for the statistical analysis. Writing and production assistance was provided by Joanne Wolter (independent on behalf of Janssen Vaccines & Prevention B.V.). The authors thank Christopher Fox and Anne Pierson from Access to Advanced Health Institute (AAHI) for providing the GLA-SE adjuvant and their expert scientific support. The authors thank Carmen Ledesma-Feliciano and Kira Elma from PharmaJet for providing training for the Tropis needle-free injection system for intradermal injections.
Author information
Authors and Affiliations
Contributions
J.T.P.: Writing, review and editing, supervision, conceptualization, resources; V.J.T.: involved in the design of LukAB CC8Δ10C and LukAB RARPR-33 antigens used in proof-of-concept studies, advice for the design of the study, edited the manuscript; D.M.: Conceived and provided the SpA* antigen used in proof-of-concept studies, advice for the design of the study and edited the manuscript; M.S.: Conceived and provided the SpA* antigen used in proof-of-concept studies, advice for the design of the study, edited the manuscript; S.W.: Was involved in study design, coordinating in vivo activities and immunogenicity readouts, data collection, review and editing of the manuscript. J.F.: input into the study design, developed the infection models, supervised the study and participated in the in vivo studies; S.K.: advice for the design of the study and was involved in the study coordination, revised and edited the manuscript; A.L.D.: Was involved in study design and LukAB RARPR-33 antigen design and in vitro characterization, immunogenicity readouts, data collection, review and editing of the manuscript; A.O.K.: Purified LukAB toxin variants for in vitro studies, involved in LukAB RARPR-33 in vitro characterization, immunogenicity readouts, and data collection; B.M.: Contributed to the design, characterization, and production of the RARPR-33 antigen and reviewed the manuscript; P.B.: Was involved in antigen production, study design, and reviewed and edited the manuscript; J.Gr: Was involved in antigen production, the study design, and reviewed and edited the manuscript; MvB: Was involved in preparing the antigen and adjuvant formulations for in vivo studies and study design and reviewed and edited the manuscript. A.C.: Was involved in preparing the antigen and adjuvant formulations for in vivo studies, study design, reviewed and edited the manuscript; M.B.: Reviewed and edited the manuscript; J.Ge.: Was involved in RARPR-33 design/characterization, reviewed the manuscript; O.K.: Coordinated study, involved in writing, reviewed and edited the manuscript; P.R.: Provided the figures, coordinated, reviewed and edited the manuscript; G.vdD.: Involved in design of the experiments, supervised the study, reviewed, revised and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
J.T.P., S.W., J.F., S.K., B.M., P.B., J.Gr., M.vB., A.C., M.B., J.Ge., O.K., P.R. and G.vdD. are employees of Janssen Vaccines & Prevention B.V., J.T.P., P.B., J.Gr., M.vB., A.C., M.B., J.Ge., P.R., G.vdD., S.W. and J.F. hold stock/shares in Johnson & Johnson Pte Ltd. J.T.P. and J.F. are inventors on patents describing LukAB variants as vaccine against S. aureus. V.J.T. and A.L.D. are inventors on patents and patent applications filed by New York University, which are currently under the commercial license to Janssen Biotech Inc. (JBIO). JBIO provides research funding and other payments associated with exclusive licensing agreements. A.O.K. no conflict. Dominique Missiakas and Miaomiao Shi are the inventors of patents describing SpA variants as vaccines against S. aureus. D.M. is the founder of ImmunArtes LLC, a University of Chicago startup company that aims to develop vaccines and therapies against S. aureus infections.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Poolman, J.T., Torres, V.J., Missiakas, D. et al. A SpA+LukAB vaccine targeting Staphylococcus aureus evasion factors restricts infection in two minipig infection models. npj Vaccines 10, 78 (2025). https://doi.org/10.1038/s41541-025-01119-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-025-01119-8
This article is cited by
-
Methicillin-resistant and susceptible Staphylococcus aureus: tolerance, immune evasion and treatment
Nature Reviews Microbiology (2025)
