
Abstract
Still, Zika virus (ZIKV) infection poses a substantial public health risk, especially for pregnant women and their fetuses, as it can result in congenital abnormalities and fetal mortality during pregnancy. Despite significant advances in understanding and combating ZIKV, considerable challenges remain in the fight against this flavivirus. A crucial component of this effort is the development of vaccines, none of which have yet been licensed for human use. Here, we present a comprehensive study of a novel ZIKV vaccine candidate based on virus-like particles (VLPs), designed to provide broad immunological protection against viral infection combined with safety, without the need for additional adjuvants. A self-adjuvanted VLPs-based vaccine displaying the envelope protein domain III (EDIII) of ZIKV was built. The EDIII protein was expressed in E. coli and chemically conjugated to QβVLPs. Immunization of C57BL/6 mice with two doses of the EDIII-QβVLPs vaccine elicited strong EDIII-specific Th1-based immune response. Notably, the vaccine induced neutralizing antibodies and conferred protection in type I IFN receptor-deficient (G129) mice against ZIKV challenge. Furthermore, vaccinated male mice were protected from ZIKV-induced cerebral and testicular damage, critical concerns for ZIKV pathogenesis. These findings suggest that the EDIII-QβVLP vaccine is a promising candidate for preventing ZIKV infection, with potential applications in combatting this and other emerging flaviviruses.
Similar content being viewed by others


Introduction
Zika virus (ZIKV), a member of the Flaviviridae family transmitted primarily by Aedes mosquitoes, emerged as a significant public health threat following its global spread from the Pacific Islands in 2007 to the Americas in 20151,2,3. Phylogenetic analyses revealed that strains from the Americas are closely related to Asian strains previously associated with causing neurological damage4. The epidemic highlighted the potential of ZIKV to cause severe outcomes, such as congenital Zika syndrome (CZS), spontaneous abortions, and Guillain–Barré syndrome5,6. Despite a decline in reported cases since 2016, sporadic outbreaks persist, presenting continual health risks, exemplified by recent resurgences in India and concerns regarding novel viral lineages in Brazil7,8,9,10,11.
The absence of approved vaccines or specific therapies for ZIKV complicates efforts to control the disease. This challenge is further exacerbated by difficulties in diagnosing mild cases and the risk of misdiagnosis, as these cases often resemble other co-circulating flavivirus infections in the same environment12,13. Importantly, ~2 billion individuals are currently at risk of ZIKV infection10, and this number is anticipated to increase based on projections indicating an expansion in the geographical distribution of mosquito vectors14. In addition to primary transmission of ZIKV to humans through mosquito bites, there are other non-vector-associated transmission routes. These include vertical transmission from mother to fetus, sexual contact, and blood transfusions. The diversity of these transmission pathways complicates control efforts, thereby increasing the risk of ZIKV outbreaks across various global regions4,15,16,17.
When ZIKV infects a pregnant woman, its strong affinity for neuronal progenitor cells and placental cells can result in CZS in the developing fetus15,18,19. This syndrome is characterized by microcephaly, structural brain abnormalities, and cognitive impairments19,20,21. Although progress has been made in developing vaccine candidates since the 2015 outbreak, no prophylactic vaccines have been licensed to date22. Pregnant women remain the most vulnerable group for ZIKV infection19,23, with persistent evidence of the virus circulating within the human population and remaining in semen for months24,25,26,27.
Structurally similar to other flaviviruses, such as dengue virus (DENV), West Nile virus (WNV), and yellow fever virus (YFV), ZIKV presents distinctive challenges for vaccine development28. While human antibodies against DENV have shown cross-protective responses to ZIKV, instances of antibody-dependent enhancement (ADE) were also described29,30,31,32. To address this issue, the envelope protein domain III (EDIII), critical for viral entry and immune response in hosts, emerges as a promising target for vaccine design due to its specificity across various flavivirus serotypes33,34,35,36. However, its inherently low immunogenicity necessitates strategies to enhance antigen presentation, such as optimized epitope display13,22,37,38.
In this study, we present a novel, and safe auto-adjuvanted vaccine candidate against ZIKV employing the virus-like particles (VLPs) technology, specifically utilizing QβVLPs39. The QβVLPs are nanoparticles characterized by an icosahedral capsid structure composed of 180 subunits from the Qβ bacteriophage coat protein40,41. This structure allows for repetitive, multivalent display of epitopes, enhancing immune system stimulation by directly activating B cells or being recognized by antigen-presenting cells (APCs). The optimal size of VLPs plays a crucial role in immune activation39,42, facilitating their passage into the lymphatic system through pores in lymphatic vessels, thereby ensuring efficient recognition by APCs and rapid presentation of antigens to follicular T cells within lymphoid organs43.
As a result, these characteristics of VLPs contribute to increased levels of IgG production and the development of more potent secondary plasma cells, which produce antibodies with increased neutralizing capacity. We established a favorable safety profile for a vaccine candidate using the type I IFNR mutant mouse strain, G129, against the Brazilian ZIKV strain (Brazil-ZKV2015). This approach demonstrates potential for combating ZIKV and advancing vaccine platforms aimed at addressing emerging infectious diseases.
Results
Production and characterization of a ZIKV vaccine candidate using the EDIII and VLPs
The vaccine antigen EDIII, incorporating a 6x His-tag and a small free cysteine linker at the C-terminus, was produced in the prokaryotic system E. coli BL21 (DE3), solubilized in urea, and purified using immobilized metal affinity chromatography (IMAC). The SDS–PAGE analysis revealed a 14-kDa band in the insoluble fraction corresponding to EDIII (Fig. 1a), and confirming the presence and purity of the protein post-purification (Fig. 1b). Recombinant protein expression was further validated by Western blot using antibodies anti-6xHis-tag (Fig. 1c) and anti- EDIII (Fig. 1d), which demonstrated both monomer and dimer forms of the recombinant protein. No detection was observed in control samples from non-transformed and non-induced bacteria. The average yield of the recombinant EDIII protein was 5 mg/L.

a The EDIII protein expression from E. coli BL21(DE3) cells and analysis on a Coomassie blue stained 15% SDS–PAGE under reducing conditions. T0 = not induced cellular extract, T3 = cellular extract after 3 h post-induction, S = soluble fraction of the bacterial lysate; I = insoluble fraction of the bacterial lysate. The calculated molecular weight of EDIII is 14 kDa. b Protein purification with a Ni-NTA column, EDIII was analyzed by 15% SDS–PAGE reducing conditions, eluted proteins were detected by Coomassie blue staining. c and d Western blot of the recombinant purified proteins using anti-His 6x and anti-EDIII antibodies. Recombinant proteins were assessed using SDS–PAGE and Western blot to determine the kinetics of expression. Culture samples were harvested at different times and the bacteria were lysed. e 3D structure of the EDIII protein (green) predicted by AlphaFold Software, the C-terminal His-6-tag is highlighted in red, the short C-terminal cysteine (GGC) is highlighted in blue. f Conjugation of the EDIII protein by modifying the QβVLPs with a chemical crosslinker (SMPH) and binding it to a modified protein (SATA) with sulfhydryl groups (–SH). g Transmission electron microscopy (TEM) image of the QβVLPs nanoparticle. h TEM image of the candidate vaccine. i SDS–PAGE stained with Coomassie blue was used for the characterization of the vaccine formulation, showing distinct bands corresponding to the protein and the VLPs. These bands confirm the presence and integrity of the key components in the vaccine formulation. j–l Western blot representing vaccine formulation j performed with anti-6x His antibody, k performed with anti-EDIII antibody, l performed with anti-QβVLPs antibody. The EDIII ZIKV (PDB 5VIG) (https://www.ebi.ac.uk/pdbe/entry/pdb/5vig/protein/3). The QβVLPs (PDB 1QBE) (https://doi.org/10.1016/s0969-2126(96)00060-3). Tridimensional structures were rendered on 3D protein imager (https://doi.org/10.1093/bioinformatics/btaa009). Complete gels and blots corresponding to this figure are provided in the Supplementary Material (Figs. 1–5).
To assess the conformational structure of the protein, the EDIII sequence was analyzed using AlphaFold (Fig. 1e). Additionally, we evaluated the endotoxin levels in the EDIII protein, yielding an endotoxin activity of 0.9 EU/mL, well below the acceptable threshold of 20 EU/mL for preclinical evaluations44. Next, the protein was analyzed to determine its recognition by hyperimmune serum containing a high concentration of specific antibodies, using samples from mice previously vaccinated with a commercial E DIII protein (ProSpecBio, USA) formulation. An ELISA was performed, revealing that the purified protein was strongly recognized by the antibodies present in the serum samples. The EDIII protein is crucial for effective immunization against ZIKV due to its potential for enhancing neutralization and minimizing cross-reactivity with other flaviviruses38.
QβVLPs were produced in a prokaryotic system, purified, and refolded according to standardized protocols39,45,46. Transmission electron microscopy (TEM) images (Fig. 1g) and SDS–PAGE (Fig. 1i) confirmed the successful production of particles, with Western blot analysis using an anti-Qβ antibody (Fig. 1l) verifying the presence of the 14 kDa monomer band. The particles measured approximately 27 nm in diameter, with a yield of soluble particles around 50 mg/L. Endotoxin levels were quantified at 1.4 EU/mL, which is within acceptable limits for preclinical assays44.
Following the production of the protein and VLPs, EDIII was covalently conjugated to the surface of QβVLPs using the chemical crosslinker SMPH (Fig. 1f). To optimize the protein conjugation on the VLPs, we employed a molar ratio of 1:2. Various experiments conducted by our group indicate that increased exposure of VLPs molecules facilitates protein conjugation through natural conjugation sites, such as the amino groups on exposed lysines on the VLPs surface, which are suitable for conjugation to cysteine-containing protein antigens using bifunctional crosslinkers.46,47,48,49. TEM analysis confirmed the correct spatial configuration of the EDIII-QβVLPs vaccine, which exhibited an increased average diameter of 47 nm (Fig. 1g) compared to the 27 nm diameter of the unmodified QβVLPs (Fig. 1h). SDS–PAGE under denaturating and reducing conditions were used to confirm the presence of both VLPs and EDIII components in the vaccine candidates (Fig. 1i), followed by Western blotting analysis using specific antibodies. The first analysis utilized an anti-6xHis-tag antibody, confirming the presence of the recombinant EDIII protein (Fig. 1j). The second assessment used antibodies against EDIII (Fig. 1k) derived from the serum of mice immunized with the commercial EDIII protein. The final analysis employed anti-QβVLPs antibodies (Fig. 1l), confirming the presence of both the EDIII protein and VLPs in the EDIII-QβVLPs formulations, as expected.
The vaccine candidate induces strong humoral immune responses with neutralizing antibodies
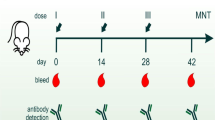
To assess the immunogenicity of the vaccine, female C57BL/6 mice, approximately six weeks of age, received two doses of the formulated vaccine EDIII-QβVLPs (Fig. 3a). The mice were stratified into five distinct groups: one group receiving only EDIII-20 µg, a second group administered Phosphate Buffer Saline (PBS), a third group receiving only QβVLPs, and two groups corresponding to the vaccine formulations (Fig. 2). Based on initial studies aimed at identifying the optimal vaccine dosing38, we maintained the 20 µg group and introduced an additional group at 50 µg, which provided enhanced protection when applied in accordance with strategies similar to those outlined in this study39. A booster dose was administered to the animals 21 days after the initial vaccination. Blood samples were collected 14, 21, 28, 35, and 42 post prime immunization (animals received the boost after the day 21 sample collection) (Fig. 3a).

The animal experiments were conducted in six independent studies: three to evaluate the humoral and cellular immune responses and neutralizing antibodies in C57BL/6 mice, and three to perform viral challenge experiments in male and female G129 mice. Samples were analyzed using ELISA, flow cytometry, CPE-VNT, qPCR, and histological studies to assess immune response, viral neutralization, and tissue integrity. Created with BioRender.

a Schematic representation of the prime-boost immunization protocol in female C57BL/6 WT mice, with weekly blood collection and euthanasia on day 42. Binding IgG and neutralizing antibody titers were measured at selected time points. b Detection of anti-EDIII IgG antibodies in sera by ELISA. Sera were collected before (days 14 and 21) and after (days 35 and 42) the boost immunization, and diluted 1:200 for analysis. The number of animals per group was: PBS (n = 9), only QβVLP (n = 3), only EDIII-20 µg (n = 8), EDIII-QβVLP-20 µg (n = 20), and EDIII-QβVLP-50 µg (n = 10). c Ratio of IgG2b/IgG1 antibody titers measured in serum collected 3 weeks after the second immunization. Mean values are indicated above each group. d Anti-ZIKV neutralizing antibody endpoint titers at day 42, measured by cytopathic effect-based virus neutralization test (CPE-VNT), and calculated as the dilution at which the percentage of ZIKV-positive cells was reduced to 100% of negative controls. LOD = 20. The numbers after the up arrow indicate the median fold increase in antibody endpoint titer induced by the prime-boost immunization regimen compared with only EDIII- 20 µg immunization. The number of animals per group was: only EDIII-20 µg (n = 3), EDIII-QβVLP-20 µg (n = 5), and EDIII-QβVLP-50 µg (n = 5). e Representative dot plot and frequency of plasma cells (CD138+). f Representative dot plot and frequency of CD45R/B220+ (activation B cells) among CD19+. g Frequency of IL-10 among CD19+ B cells. All graphs show the distribution of values using boxplots, where the central line represents the median, the boxes represent the interquartile range (25th–75th percentiles), and the whiskers indicate the range of the data. Individual data points are also shown. The number of animals per group was: only EDIII-20 µg (n = 8), EDIII-QβVLP-20 µg (n = 10), and EDIII-QβVLP-50 µg (n = 10). Data were analyzed by Kruskal–Wallis, and post-hoc by Dunn *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
We conducted an ELISA with the EDIII protein to quantify the specific antibody response. Sera from the groups of mice were analyzed for the reactivity of total IgG and its subclasses, IgG1 and IgG2b. We observed an increase in total IgG production following the booster immunization (Fig. 3b). Both vaccinated groups exhibited a similar profile, they induced strong IgG response after boost immunization (days 35 and 42) with a statistically significant difference compared to the group of EDIII alone.
Analysis of IgG1 and IgG2b revealed that both vaccinated groups (20 and 50 µg) suppressed IgG1 and enhanced IgG2b responses (Fig. 3c). In contrast, the group vaccinated with only EDIII induced IgG1 but failed to generate IgG2b response (Supplementary Fig. 6).
Next, we investigated whether the immune response in vaccinated mice could generate protective neutralizing antibodies capable of preventing viral infection. Virus neutralization titers were determined as the highest dilution of serum that effectively neutralized viral growth, as indicated by the absence of cytopathic effects (dark blue; Supplementary Fig. 7). Mice vaccinated with EDIII-QβVLPs at doses of 20 or 50 µg developed antibody responses that effectively neutralized ZIKV after two vaccinations (Day 42), exhibiting inhibition of cytopathic effects at a dilution of 1:80 in most groups (Supplementary Fig. 7). In contrast, mice vaccinated with EDIII alone did not generate humoral immunity with virus neutralization capability (Fig. 3d). These results indicate that the vaccine candidate, at both 20 and 50 µg doses, successfully elicited antibodies that effectively neutralized ZIKV.
Additionally, the vaccine formulations significantly enhanced B cell activation (CD19+/CD45R/B220+) and plasma cell proliferation (CD138+), compared to the group that received only EDIII protein (Fig. 3e and f). We also observed moderate production of IL-10 in CD19+ cells (Fig. 3g), highlighting its critical role in immune regulatory response.
The vaccine candidate elicits antigen-specific T-cell immune response
To evaluate the capacity of the vaccine formulation to induce a specific T cell immune response, we established immune profiles in C57BL/6 mice and in mouse strains deficient in IFN signaling50,51 (Fig. 2). In vaccinated mice deficient in IFN signaling, serum samples were collected at 2, 5, and 8 days post-infection (dpi). Additionally, brains, livers, kidneys, ovaries, and spleens were obtained after euthanasia at 28 dpi to analyze the T cell immune response in both sera and organs. This analysis was conducted concurrently with the challenge experiment, as detailed in the following section.
The results from the experiment involving immunized C57BL/6 mice, whose splenocytes were stimulated with the EDIII antigen, demonstrated that vaccination with the EDIII-QβVLPs formulation significantly enhanced the secretion of INF-γ and TNF-α. These secretion levels were notably higher compared to those observed in the EDIII-only and VLPs-only groups.
Additionally, we analyzed the activation profile of lymphocyte subpopulations in the spleens of interferon-gamma knockout (G129) mice 28 days post-infection52,53. The proportion of effector T cells was markedly higher in the vaccinated animals (20 and 50 µg), as indicated by the CD38+/high, CD62L/low and CD3+ markers (Fig. 4a, b) when compared to the only-EDIII protein group. Moreover, vaccinated animals exhibited an expansion of CD4+ T cells expressing TNF-α and IL-2, but not IL-4 (Fig. 4c). Furthermore, a greater proportion of CD8+ T cells from vaccinated mice produced IL-2 and TNF-α (Fig. 4d). Although we did not assess IFN-γ production in mice lacking IFN receptors, these results indicate an early T cell response following vaccination in G129 mice. Moreover, in vitro stimulation of splenocytes obtained from vaccinated mice 28 days after ZIKV infection suggests that the vaccination induced a robust immune response mediated by CD8+ and CD4+ T cells, favoring a pro-inflammatory profile with the potential to confer protection against ZIKV infection. The absence of increased IL-4 expression may suggest a polarization of the immune response toward a Th1 profile, characterized by the production of inflammatory cytokines. This pattern was also observed in previous data, where we noted suppressed IgG1 responses and enhanced IgG2b responses induced by the candidate vaccine (Fig. 3c).

Flow cytometry was performed on splenocytes from EDIII-QβVLPs (20 and 50 µg) or EDIII-only at 28 dpi. For cytokines detection, splenocytes were restimulated in vitro with EDIII protein in the presence of brefeldin A for 4 h. a Representative dot plot and frequency of CD38+ (activation T cells) among CD3+. b Representative dot plot and frequency of CD62L- (effector) among CD3+. c Representative counter plot and frequency of IL-2, IL-4, and TNF-∝ among CD4+ T cells. d Representative counter plot and frequency of IL-2, and TNF-∝ among CD8+ T cells. All graphs show the distribution of values using boxplots, where the central line represents the median, the boxes represent the interquartile range (25th–75th percentiles), and the whiskers indicate the range of the data. The number of animals per group was: only EDIII-20 µg (n = 8), EDIII-QβVLP-20 µg (n = 10), and EDIII-QβVLP-50 µg (n = 10). Data are representative of three independent experiments. Data were analyzed by Kruskal–Wallis, and post-hoc by Dunn. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001.
The vaccine candidate provides protection against ZIKV infection and prevents cerebral and testicular damage
To investigate the impact of ZIKV infection and replication on the central nervous system as well as other organs such as the kidneys, liver, ovaries, and testis, we performed histopathological analyses and assessed viral replication in various mouse tissues using quantitative PCR (qPCR). Additionally, we examined the effects of viral infection on the development of clinical signs of disease. It is important to note that a non-lethal dose of the virus was employed for the infection challenge experiments, allowing us to study the consequences of ZIKV infection without causing fatality.
Throughout the experiment, we observed a significant weight loss in the sham group for several days post-challenge (Fig. 5a), in contrast to the animals immunized with EDIII-QβVLPs, which maintained their weight. Notably, despite these differences in weight, no severe clinical signs of disease were evident in any of the groups.

a Body weight measured up to 28 dpi, expressed as the mean percentage of initial weight ± standard deviation; N = 5 mice/group. b Testis weight at day 28 post-infection. c Testis image. d Viral titer in various organs of mice was measured by qRT-PCR (relative expression). e Viremia after challenge measurements by qRT-PCR (relative expression) were performed on days 2, 5, and 8 post-infection. f–i Brains in distinct regions (midbrain and cerebral cortex) from ZIKV-infected was processed for histological staining with hematoxylin and eosin (HE). f Representative uninfected animals with no abnormalities observed. g White arrows indicate necrotic areas in sham-immunized animals. h and i Vaccinated groups show no abnormalities. j–m Histological analysis of the testis from ZIKV-infected G129 mice. j Abnormalities were not observed from uninfected G129 mice. k Disrupted seminiferous tubules with disorganized cells (large black arrows) and various degenerating germinal epithelial cells (black arrows) were observed inside the seminiferous tubules of ZIKV-infected testes. Additionally, intratesticular varicoceles/congestion (white arrow) and lymphocyte infiltration (small black arrows) were present in the interstitial areas of sham ZIKV-infected G129 testes. l and m Normal seminiferous tubules with different steps of germinal epithelial cells (arrow) in order and normal intratesticular capillary (arrow) in immunization G129 mice testis. The 4 μm-thick sections were stained with HE and images were captured with Slide Digitizer-3D Histech scanner. The number of animals per group was: only EDIII-20 µg, n = 8 (females), Sham (PBS) n = 4 (2 females, 2 males), EDIII-QβVLP-20 µg, n = 15 (11 females, 4 males), and EDIII-QβVLP-50 µg, n = 15 (11 females, 4 males). Data were analyzed by Kruskal–Wallis, and post-hoc by Dunn in b, d, e. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
We evaluated the capacity of the vaccine candidate EDIII–QβVLPs to prevent testis infection and associated injury in G129 mice. Given that male mice reach sexual maturity at 8 weeks of age54, we assessed vaccine efficacy in three groups: a 20 µg formulation (n = 5), a 50 µg formulation (n = 5), and a PBS-sham group (n = 3) (Fig. 2). Male mice were vaccinated with two doses of the vaccine, as previously described, and were subsequently challenged via intraperitoneal injection with 105 PFU/mL of the ZIKV strain (100 µL) 2 weeks after the booster vaccination. Following the strategy outlined in previous studies52,53, we assessed the viral levels on day 28 post-challenge to investigate potential protection in the male reproductive organs. Immunized mice with EDIII–QβVLPs showed little ZIKV levels (relative expression) in the testes compared to the infected control group (sham-immunized mice), indicating effective protection conferred by the vaccine. (Fig. 5d).
In line with these findings, ZIKV-challenged sham mice displayed notable reductions in testicular weight (Fig. 5b) and size (Fig. 5c), as well as significant histopathological abnormalities, including disrupted seminiferous tubules, lymphocyte infiltration in interstitial areas, and varicoceles (Fig. 5k).
Conversely, vaccinated mice exhibited no reductions in testicular size or weight and maintained normal histological structure (Fig. 5l and m). Collectively, these results indicate that immunization with EDIII–QβVLPs effectively protects the testes from infection and injury in male mice.
A progressive increase in viral titer levels was observed in the brains of the sham group four weeks post-infection (Fig. 5d). Furthermore, histopathological examination of brain tissue revealed distinct areas of necrosis and micro-hemorrhagic lesions within the midbrain and cerebral cortex in sham-immunized mice (Fig. 5g), indicating that the virus preferential viral tropism for these brain regions. These observations are supported by previous studies that have shown neural progenitor cells as primary ZIKV targets, particularly in interferon-deficient mice55,56, emphasizing the vulnerability of neural tissue to ZIKV infection.
In contrast, vaccinated mice displayed no signs of necrosis or histological abnormalities in the brain, similar to uninfected animals (Fig. 5f), highlighting the protective efficacy of the candidate vaccine in safeguarding cerebral structures from ZIKV-induced damage (Fig. 5h, i). The absence of abnormalities in the brain tissue of vaccinated mice highlights the vaccine’s favorable safety profile and its potential to prevent neurological complications associated with ZIKV infection.
Further assessment of viral titer levels in several other tissues was conducted 12 days post-challenge34,50,56,57 in G129 female mice and revealed high viral titers in the liver, kidneys, and ovaries of ZIKV-infected, sham-immunized mice, indicating widespread viral dissemination in these organs (Fig. 5d). Notably, in the ovaries, sham-infected mice retained detectable viral titers, whereas EDIII–QβVLPs-vaccinated mice had little ZIKV titer levels detected. These findings highlight the protective efficacy of the vaccine and its ability to reduce viral titers in critical tissues. Additionally, viremia levels (Fig. 5e) were markedly reduced in vaccinated mice, indicating effective systemic control of ZIKV replication following immunization.
Discussion
The advancement of vaccine technology is critical for addressing emerging and re-emerging diseases, enabling broader distribution and application across diverse populations, including children, the elderly, pregnant and breastfeeding women, and immunocompromised individuals58. Traditional vaccines, such as attenuated or inactivated formulations, balance efficacy and safety but face challenges; attenuated vaccines generate robust immunity but are unsuitable for vulnerable groups, while inactivated vaccines are safer but less immunogenic59. Adjuvants, like aluminum-based compounds, enhance vaccine efficacy60 but may not meet all immunological needs, particularly in inducing Th1-type responses61, and their high costs limit accessibility for neglected diseases like Zika. Recent innovations, including mRNA and viral vector-based vaccines, offer transformative potential with rapid development and scalability. However, mRNA vaccines face logistical challenges, such as stringent storage requirements62, while viral vector-based vaccines must overcome the risk of immune responses against the vector itself63. Addressing these obstacles is essential for improving universal vaccine access and achieving effective, equitable immunization strategies.
Our study introduces a vaccine formulation based on QβVLPs, we harnessed the antigen delivery and robust immunogenic capabilities of QβVLPs to formulate a vaccine utilizing EDIII as the antigen. A significant concern in this context is the risk of ADE, particularly in regions where ZIKV co-circulates with other flaviviruses, such as DENV31,32. In these environments, the presence of cross-reactive antibodies may facilitate enhanced viral entry into host cells, potentially exacerbating disease severity30,64. Therefore, selecting an appropriate antigen—one that is specific to ZIKV and minimizes cross-reactivity with DENV—is crucial for the development of a safe and effective vaccine65. Recent studies conducted by our group have demonstrated that EDIII derived from ZIKV can effectively induce neutralizing antibodies while preventing the enhancement of DENV infection38,45. This specific immune response is vital, as it mitigates the risks associated with ADE, which has been a significant challenge in the design of vaccines for flavivirus infections66,67.
The EDIII protein was successfully expressed in the prokaryotic system E. coli BL21 (DE3). Employing E. coli as a production system offers several key advantages that enhance its suitability for large-scale protein production. Firstly, this system allows for rapid scalability through the use of bioreactors, enabling efficient production in response to varying demands. Additionally, the cost-effectiveness of E. coli as a host organism significantly reduces the overall expenses associated with protein synthesis, making it an attractive option, particularly in resource-limited environments. Furthermore, the well-established genetic and biochemical tools available for E. coli facilitate the optimization of expression conditions and the purification process, ultimately leading to increased production yields and the generation of high-purity antigens. However, E. coli recombinant expression also has clear disadvantages that must be considered. One major limitation is its inability to perform post-translational modifications, such as glycosylation, which are essential for the proper folding and functionality of many eukaryotic proteins. Additionally, high-level protein leads to the formation of insoluble inclusion bodies, requiring additional steps for protein refolding and putification34,68,69,70. These factors highlight the strategic advantages of using E. coli for protein production, particularly for applications in vaccine development and other biotechnological endeavors where rapid scalability and cost-effectiveness are critical.
Building on the known immunogenic properties of QβVLPs and EDIII proteins, our study evaluated the potential of an EDIII–QβVLP-based vaccine candidate. In a mouse model, immunization with this formulation elicited a strong humoral immune response and offered protection against ZIKV-induced tissue damage. These results suggest the induction of an immune response capable of limiting viral replication and reducing pathology, particularly in organs relevant to ZIVK pathogenesis.
Further investigations into the neutralizing capacity of antibodies generated by the EDIII-QβVLPs vaccine formulation demonstrated its effectiveness in inhibiting ZIKV infection. This finding was corroborated by challenge experiments conducted in type I interferon receptor-deficient G129 mice, where the vaccine candidate provided robust protection against ZIKV. The vaccine demonstrated a capacity to protect male mice from testicular damage. This is particularly relevant in light of the known risks associated with the sexual transmission of ZIKV and its potential to cause testicular injury, which can negatively impact spermatogenesis and overall reproductive health52,53,54. These findings are consistent with previous studies linking ZIKV infection to inflammation, disrupted spermatogenesis, and potential impacts on fertility53,71. These emphasizes the need for effective vaccination strategies that inhibit viral spread and preserve male reproductive integrity. Recent studies have indicated that ZIKV can lead to inflammation and apoptosis in testicular tissue, potentially resulting in long-term fertility issues53,71. Furthermore, as the epidemiology of ZIKV continues to evolve, particularly in regions where the virus co-circulates with other flaviviruses, the implementation of comprehensive vaccination programs becomes increasingly critical.
Another important result demonstrated in this study was the histopathological tests, which revealed areas of necrosis and micro-hemorrhagic lesions in the midbrain and cerebral cortex of unvaccinated animals. However, the vaccine conferred immunity following a booster immunization, exhibiting a favorable in vivo safety profile, as no abnormalities were observed in the brain cells of vaccinated mice. These findings underscore the potential application of such vaccines in vulnerable populations, particularly pregnant women, to protect infants from the neurological damage associated with ZIKV infection. Given that congenital Zika syndrome has been linked to severe brain defects, the administration of a safe and effective vaccine could significantly mitigate the risk of adverse developmental outcomes in neonates, aligning with current recommendations for maternal immunization strategies to enhance both maternal and infant health72,73. These results are encouraging, our model did not include pregnant animals or vertical transmission studies. Further research is required to assess the efficacy and safety of this vaccine in maternal–fetal contexts.
In conclusion, our findings support the potential of the EDIII-QβVLP vaccine formulation as a promising candidate for ZIKV immunization. The induction of neutralizing antibodies, protection against tissue pathology, and favorable in vivo safety profile provide a strong rationale for continued preclinical development. Nevertheless, further studies are necessary to validate these findings in larger cohorts and more translational models, including non-human primates, and eventually in human clinical trials, which will be crucial to confirm its safety and efficacy. Additionally, the methodologies and insights gained from this research offer a valuable foundation for the development of vaccines against other emerging infectious diseases, thereby broadening the impact of this work in the field of vaccinology.
Methods
Expression and purification of the recombinant EDIII protein
The customized pET-21a(+) plasmid, encoding the ZKV2018 strain EDIII sequence was selected from GenBank, reference number AZS35340.1, and was purchased from GenScript (USA). This plasmid was used to transform the chemically competent E. coli BL21(DE3) cells. A pre-inoculum was performed with the colonies and subsequently added to the Terrific Broth medium containing ampicillin (100 μg/mL). The recombinant expression of the protein was conducted at 37 °C with shaking at 200 rpm until the optical density at 600 nm (OD) reached 0.6–0.8. Subsequently, 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG, Sigma-Aldrich) was added to the culture. The incubation was then continued at 18 °C with shaking at 200 rpm for 18 h. Culture samples were collected for analysis before and after IPTG induction. The cells were centrifuged at 4032×g for 10 min and transferred to a buffer containing Tris–HCl 100 mM, NaCl 200 mM, SigmaFast Protease Inhibitor (Sigma-Aldrich), 10 μg/mL RNaseI (Thermo Scientific). Cell lysis was carried out by sonication for a total of 10 min, with 20 cycles of 15 s sonication and 30 s pause. The cell lysate was centrifuged at 8000×g for 20 min, and the soluble and insoluble fractions were recovered. The same amount of noninduced and induced culture whole extracts, as well as the soluble and insoluble fractions, were dissolved in a loading buffer and subjected to SDS–PAGE and Western blot analysis. The insoluble fraction was solubilized in a sodium Tris–HCl buffer with 8 M urea, pH 8, and subjected to nickel affinity chromatography on a 500 μL Ni Sepharose 6 Fast Flow resin (Cytiva Life Sciences). The resin was pre-equilibrated with Tris–HCl 100 mM, NaCl 200 mM, urea 8 M, and pH 8 buffer in Poly-Prep chromatography columns (BioRad). After equilibration, the extract was added. Two washes were performed using the initial buffer supplemented with Imidazole 30 mM. Recombinant proteins were eluted using a gradient concentration of imidazole (100-, 200-, 300-, 400 and 500 mM) in Tris–HCl 100 mM, NaCl 200 mM, urea 8 M (pH 8). The eluted fractions containing protein were then pooled and subjected to a refolding process through successive dialyses, gradually reducing the urea concentration (6-, 4- and 2-M) in Tris–HCl 100 mM, NaCl 200 mM, and glycerol 10% (pH 8), with buffer exchanges at each step. Finally, the samples were dialyzed against a urea-free buffer [Tris buffer 100 mM (pH 8.0) with NaCl 200 mM and glycerol 10%]. Purified protein samples were quantified using the Bradford assay (Thermo Scientific, USA). Endotoxin levels in recombinant protein were determined by the Limulus amebocyte lysis method, using the Thermo ScientificTM PierceTM Chromogenic Endotoxin Quant Kit, according to the manufacturer’s instructions.
Western blot analysis
The SDS–PAGE was performed using 20 μL of the protein sample, which was subsequently transferred via blotting. A duplicate gel was stained with Coomassie Brilliant Blue R-250 for visualization. After the transfer, nitrocellulose membranes were blocked in a blocking buffer consisting of 5% skimmed milk in tris-buffered saline (TBS) with 0.05% Tween 20 overnight at 4 °C. Western blot analysis was conducted by incubating the membranes for 2 h with a specific anti-6x His tag monoclonal antibody (Invitrogen, USA) diluted 1:3000 in blocking buffer, or with serum samples from hyperimmune mice containing high levels of anti-EDIII antibodies, at room temperature. Subsequently, the membranes were washed and incubated with a goat anti-mouse horseradish peroxidase–IgG conjugate (1:10,000; Southern Biotechnology) for 1 h at room temperature. The membranes were then developed using a chemiluminescence detection kit (ECL kit; Thermo Fisher, USA).
Production of virus-like particle
The QβVLPs construction was made by Creative Biostructure (USA). The coat protein (CP) gene from Qβ bacteriophage was cloned into pET28a(+) without a 6x-His tag. The constructed plasmid was then used to transform BL21 (DE3) E. coli competent cells (Thermo Fisher Scientific, USA). Recombinant expression of the QβVLPs was carried out in Hannahan’s Broth (SOB medium) for 16 h at 30 °C and 200 rpm. When the optical density at 600 nm (OD600 nm) reached 0.6, 1 mM IPTG was added to induce protein expression. After induction, the bacterial cell suspension was centrifuged at 5000 rpm for 15 min, and the resulting cellular sediment was resuspended in PBS. Subsequently, the cells were lysed by sonication for 5 min using 15 s on and 30 s off. The lysate was then centrifuged, and both the supernatant and pellet fractions were analyzed using SDS–PAGE to assess protein expression. For purification of the QβVLPs, 40% (w/v) PEG 8000 was added to the supernatant fraction, and the mixture was incubated overnight on a nutating mixer at 4 °C to precipitate total proteins. The precipitate was collected by centrifugation at 9000 rpm for 30 min. Afterward, 70% ammonium sulfate was added to the fraction and incubated for two hours. The pellet was then resuspended in PBS and filtered through a 0.22 µm membrane. Pure protein was obtained by removing residual contamination with size-exclusion chromatography (SEC) using a Superdex 200 Increase 10/300 column (GE Healthcare, Glattbrugg Switzerland).
Vaccine formulation: coupling QβVLPs with EDIII
Following protocols previously standardized by our group for conjugating vaccine antigens to VLPs38,39,45, the protein was incubated with a 7.5-fold molar excess of N-succinimidyl-S-acetylthioacetate (SATA) (Thermo Fisher Scientific, USA). Excess reagent was removed by diafiltration using 3 kDa Amicon Ultra centrifugal filters (Millipore, USA). Subsequently, the derivatized protein was deacetylated with hydroxylamine–HCl, resulting in the addition of reactive thiol residues to the protein. Concurrently, QβVLPs were incubated with a 10-fold molar excess of the heterobifunctional chemical cross-linker, succinimidyl 6-(b-maleimidopropionamido) hexanoate (SMPH) (Thermo Fisher Scientific, USA), and excess reagent was also removed by diafiltration. Following the chemical modifications, the EDIII was covalently conjugated to the QβVLPs at a 1:2 molar ratio.
Procedures involving mice
Female wild-type (WT) C57BL/6 mice, aged 6–8 weeks, were obtained from the Central Animal House of the Faculty of Medicine of the University of São Paulo (FMUSP, Brazil) and maintained under specific pathogen-free conditions according to animal care guidelines. Additionally, adult (female and male) C57BL/6 mice with interferon α and β receptor depletion (IFNabR−/−) (B6.129S7-Ifngr1tm1Agt/J, https://www.jax.org/strain/003288#), a known susceptible model for ZIKV infection (Brazil-ZKV2015), were purchased from the Institute of Biomedical Sciences (ICB-USP, Brazil) for infection assays. All reported experiments and protocols on live vertebrates were approved by the Research and Ethics Committee of the University of São Paulo, Institute of Biomedical Sciences (CEUA, https://ww3.icb.usp.br/ceua/) according to protocol numbers: 1332900720 and 284310822. All experiments were performed in accordance with the Brazilian Federal laws no. 11.794 which establishes procedures for the scientific use of animals and human studies and in State law no. 11.977 which establishes the Code of Protection to Animals of the State of São Paulo. Mice were immunized with two doses administered via intramuscular injection, and ZIKV infection was animal-challenged two weeks after the booster vaccination via intraperitoneal injection with 105 PFU/mL of the Brazilian ZIKV strain (Brazil-ZKV2015). In the scientific studies, a combination of ketamine (100 mg/kg) and xylazine (10 mg/kg) was employed for anesthesia, providing effective sedation and analgesia. This approach facilitates safe handling and minimizes stress during experimental procedures. Additionally, isoflurane was used as an inhalational anesthetic for both short and extended procedures, ensuring consistent anesthesia maintenance and stable physiological conditions throughout the experiments. At the end of each study, the animals were humanely euthanized using an approved Schedule 1 method (cervical dislocation). These procedures adhere to ethical standards and ensure compliance with animal welfare regulations throughout the research. All methods were reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Assessment of humoral immunity
EDIII-specific antibodies (IgG) in serum samples from immunized mice were titrated using enzyme-linked immunosorbent assay (ELISA), following well-established protocols with minor modifications39,45. The recombinant EDIII protein was diluted in 50 mM carbonate buffer (pH 9.6) and used to coat microplates (KASVI) at a concentration of 200 ng per well. The plates were then incubated overnight at 4 °C. The following day, the plates were washed three times with PBS containing 0.05% Tween 20 (PBST) and then blocked with 1×PBS Tween containing 0.5% BSA (bovine serum albumin) and incubated for 2 h at room temperature. After a new wash cycle, serum samples were serially diluted (log2) starting at 1:200 and incubated at room temperature for 2 h. After a new wash cycle, the anti-mouse IgG antibody, conjugated to peroxidase (Sigma-Aldrich, USA), was added to wells and incubated again for 1 h. After final washing, plates were developed with Tetramethylbenzidine (TMB) (Sigma-Aldrich), and the reaction was stopped after 10 min with the addition of 50 μL of H2SO4 at 2 N. The optical density reading was performed at 450 nm with a plate reader (Synergy/HTX–Microplate Reader-Biotek). The antibody titers are shown as dilutions leading to half-maximal OD (OD50) and the values observed in the negative control group were subtracted from the titres of the vaccinated groups.
Spleen cell isolation and flow cytometry
The spleen was macerated using a Potter homogenizer. The splenocytes were suspended and washed once with RPMI 1640 medium (Sigma-Aldrich, MO) with 2% fetal bovine serum (FBS; Gibco). Red blood cells were lysed with 3 mL of ACK solution (NH3Cl 150 mM, KHCO3 10 mM, and EDTA 0.1 mM) per spleen for 3 min at room temperature. After two additional washes with RPMI and 2% FBS, the spleen cells were resuspended in R10 medium (RPMI supplemented with 10% fetal bovine serum). Cell viability was assessed using 0.1% trypan blue dye exclusion, and cells were counted using a Neubauer chamber. And then, plated in 96-well microplate at a concentration of 3 × 106 cells/mL in RPMI-1640 medium supplemented with 10% FBS. To detect cytokines, the cells were incubated with 2 µg of EDIII + brefeldin A Stop Golgi. All samples were then incubated for 4 h at 37 °C in a 5% CO2 environment. After incubation, the cells were then stained with Live/Dead (1:500) for 20 min at 4 °C, followed by washing with FACS buffer. Next, the cells were divided into separate plates for staining with B cell and T cell panels. T cell staining: The cells were stained with the following anti-mouse antibodies: CD3-APC (BD Biosciences, USA), CD4-AF700 (BD Biosciences, USA), CD8-PE, CD38-PE/Cy7, TNF-PerCP-Cy5.5, IL-2-FITC, IL-4-BV605, and IFNγ-BV421 (BD Biosciences, USA). B cell staining: The cells were stained with anti-mouse CD138-BV421, CD19-PerCP, CD45R-APC/Cy7, CD62L-PE, and IL-10-FITC antibodies. Following staining, the cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences, USA). Flow cytometry was performed on a BD LSRFortessa™ X-20 (BD Biosciences, USA), and data analysis was conducted using FlowJo™ v.10.8 software (FlowJo, USA, https://www.flowjo.com/solutions/flowjo).
Tissue collection and preparation
Mice were anesthetized (100 mg/kg ketamine and 10 mg/kg xylazine, i.p.) before euthanasia. Brains and testes from each animal were collected and preserved in 10% phosphate-buffered formalin for 72 h. A portion of the tissues was then extracted for RNA isolation and qPCR analysis. The remaining brain and testis tissues were embedded in paraffin and processed using a tissue processor (PT05 TS, LUPETEC, UK). Histological paraffin (Histosec, Sigma-Aldrich) was used to embed the samples. Thin sections of 4 μm thickness were prepared and stained with hematoxylin and eosin. The sections were digitized using a 3D Histech Slide Digitizer scanner, and images were analyzed with CaseViewer 2.4, 64-bit version (3D Histech, Hungary, https://www.3dhistech.com/solutions/caseviewer/). Additionally, the liver, kidneys, and ovaries were collected and stored in RNAlater™ Stabilization Solution (Invitrogen, USA) for subsequent RNA extraction and qPCR analysis.
Propagation of ZIKV
The virus stock was prepared by infecting a C6/36 cell monolayer in 75 cm2 tissue culture flasks at 75–85% confluence. When the infected monolayer showed cytopathic effects, the cells and supernatant were homogenized and diluted in a 40% polyethylene glycol solution in 2 M NaCl (Sigma-Aldrich, MO) and incubated at 4 °C overnight. The suspension was centrifuged at 6000 rpm for 1 h. The virus was suspended in 1/15 of the total volume with a glycine buffer (Tris 50 mM, Glycine 200 mM, NaCl 100 mM, EDTA 1 mM) and 1/30 of the total volume of FBS. The virus was homogenized, aliquoted, and frozen at −80 °C until use.
Virus neutralization test
The cytopathic effect (CPE)-based virus neutralization test (VNT) was performed in 96-well plates using 1 × 104 VERO cells per well, seeded 24 h before the experiment. Previously heat-inactivated serum samples were serially diluted by a factor of two (1:20–1:2560). Subsequently, 102 PFU of ZIKV were added to the diluted serum samples and the mixture was incubated for 1 h at 37 °C/5% CO2. The serum and virus mixtures were transferred to the cell monolayer and incubated for 72 h at 37 °C/5% CO2. After this period, each well was analyzed by microscopy to evaluate the presence of cytopathic effects. Virus containing medium was removed from the plates and the cells were fixed/stained with naphthol blue-black dye (0.1% amido black solution [w/w] with 5.4% acetic acid, 0.7% sodium acetate) for 30 min at RT. The neutralizing antibody titers correspond to the highest serum dilution capable of neutralizing virus particles (absence of cytopathic effects). Uninfected and ZIKV-infected cells (no serum) were included as controls. A heatmap was generated based on the measurement of image absorbance on cell culture plates. Absorbance levels were higher in areas where the cells remained intact (i.e., without cytopathic effects) and low absorbance (indicating cytopathic effects). In parallel, anti-ZIKV neutralizing antibody endpoint titers were determined. Titers were calculated as the highest serum dilution at which the percentage of ZIKV-positive cells was reduced to the level of negative controls (100%) and are presented as log10-transformed values. LOD = 20.
Viral load quantification by qRT-PCR
Viral load quantification was performed in sera and other tissues of ZIKV-infected mice through qRT-PCR/Taqman. Briefly, viral RNA was extracted from serum and tissues using the Trizol reagent, according to the manufacturer’s instructions. ZIKV RNA was amplified using the TaqMan Fast Virus 1–8Step Master Mix reagent (Applied Biosystems, USA) in a 7500 Fast Dx Real-Time PCR Instrument (Applied Biosystems, USA). The following primers and probes were used to quantify viral RNA: 5′- CCGCTGCCCACACAAG-3′ (forward primer); 5′-CCACTAACGTTCTTTTGCAGACAT-3′ (reverse primer), and FAM-AGCCTACCTTGACAAGCAGTCAGACACTCAA-TAMRA (probe). The qRT-PCR data were presented as 1/\(\triangle\)Ct and were performed in triplicate. Positive and negative controls corresponded to purified viruses (from cell culture) and water, respectively.
Data availability
The EDIII ZIKV sequence and structure (PDB 5VIG) are available at the Protein Data Bank (https://www.ebi.ac.uk/pdbe/entry/pdb/5vig/protein/3). The QβVLPs sequence and structure (PDB 1QBE) can be accessed at (https://doi.org/10.1016/s0969-2126(96)00060-3).
References
Faria, N. R., et al. Zika virus in the Americas: early epidemiological and genetic findings. Science (1979). 352, 345–349 (2016).
Song, B. H., Yun, S. I., Woolley, M. & Lee, Y. M. Zika virus: history, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 308, 50–64 (2017).
Pielnaa, P. et al. Zika virus-spread, epidemiology, genome, transmission cycle, clinical manifestation, associated challenges, vaccine and antiviral drug development. Virology 543, 34–42 (2020).
Fajardo, Á, Cristina, J. & Moreno, P. Emergence and spreading potential of Zika virus. Front. Microbiol.7, 1667 (2016).
Weaver, S. C., et al. Zika virus: history, emergence, biology, and prospects for control. Antivir. Res. 130, 69–80 (2016).
de Oliveira, W. K., et al. Infection-related microcephaly after the 2015 and 2016 Zika virus outbreaks in Brazil: a surveillance-based analysis. Lancet 390, 861–870 (2017).
Akhtar, N., Gupta, S. K. & Singh, H. Surveillance of Zika and Dengue viruses in field-collected Aedes aegypti mosquitoes from different states of India. Virology 574, 96–101 (2022).
Cardona-Ospina, J. A., et al. Susceptibility to endemic Aedes-borne viruses among pregnant women in Risaralda, Colombia. Int. J. Infect. Dis. 122, 832–840 (2022).
PAHO/WHO. Actualización Epidemiológica: Dengue y otras Arbovirosis 1–15 (Pan American Health Organization, Washington, DC, USA, 2020).
Saxena, S. K. et al. Zika virus disease in India - Update October 2018. Travel Med. Infect. Dis. 27, 121–122 (2019).
Yadav, P. D., et al. Detection of Zika virus disease in Thiruvananthapuram, Kerala, India 2021 during the second wave of COVID-19 pandemic. J. Med. Virol. 94, 2346–2349 (2022).
Jorge, F. A. et al. Evolutions and upcoming on Zika virus diagnosis through an outbreak: a systematic review. Rev. Med. Virol. 30, e2150 (2020).
Lee, S. & Nguyen, M. T. Recent advances of vaccine adjuvants for infectious diseases. Immune Netw. 15, 51 (2015).
Albuquerque, D. C., et al. Combined detection of molecular and serological signatures of viral infections: the dual assay concept. Biosens. Bioelectron. 210, 114302 (2022).
Pomar, L., et al. Zika virus during pregnancy: from maternal exposure to congenital Zika virus syndrome. Prenat. Diagn. 39, 420–430 (2019).
Ferraris, P., Yssel, H. & Missé, D. Zika virus infection: an update. Microbes Infect. 21, 353–360 (2019).
Baud, D., Gubler, D. J., Schaub, B., Lanteri, M. C. & Musso, D. An update on Zika virus infection. Lancet 390, 2099–2109 (2017).
Martines, R. B., et al. Pathology of congenital Zika syndrome in Brazil: a case series. Lancet 388, 898–904 (2016).
Braga, C., et al. Seroprevalence of Dengue, Chikungunya and Zika at the epicenter of the congenital microcephaly epidemic in Northeast Brazil: a population-based survey. PLoS Negl. Trop. Dis. 17, e0011270 (2023).
Lopes Moreira, M. E., et al. Neurodevelopment in infants exposed to Zika virus In utero. N. Engl. J. Med. 379, 2377–2379 (2018).
Johansson, M. A., Mier-y-Teran-Romero, L., Reefhuis, J., Gilboa, S. M. & Hills, S. L. Zika and the risk of microcephaly. N. Engl. J. Med. 375, 1–4 (2016).
Côrtes, N. et al. Integrated control strategies for dengue, Zika, and Chikungunya virus infections. Front. Immunol. 14, 1281667 (2023).
de Araújo, T. V. B., et al. Association between microcephaly, Zika virus infection, and other risk factors in Brazil: final report of a case-control study. Lancet Infect. Dis. 18, 328–336 (2018).
Atkinson, B., et al. Detection of Zika virus in semen. Emerg. Infect. Dis. 22, 940–940 (2016).
Yang, W., et al. Single-cell RNA sequencing reveals the fragility of male spermatogenic cells to Zika virus-induced complement activation. Nat. Commun. 14, 2476 (2023).
Tang, W. W., et al. A mouse model of Zika virus sexual transmission and vaginal viral replication. Cell Rep. 17, 3091–3098 (2016).
Mansuy, J. M., et al. Zika virus: high infectious viral load in semen, a new sexually transmitted pathogen? Lancet Infect. Dis. 16, 405 (2016).
Gaunt, M. W. et al. Phylogenetic relationships of flaviviruses correlate with their epidemiology, disease association and biogeography. J. Gen. Virol. 82, 1867–1876 (2001).
Swanstrom, J. A. et al. Dengue virus envelope dimer epitope monoclonal antibodies isolated from dengue patients are protective against Zika virus. mBio 7, e01123–16 (2016).
Barba-Spaeth, G., et al. Structural basis of potent Zika–dengue virus antibody cross-neutralization. Nature 536, 48–53 (2016).
Bardina, S. V., et al. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 356, 175–180 (2017).
Priyamvada, L., et al. Human antibody responses after dengue virus infection are highly cross-reactive to Zika virus. Proc. Natl Acad. Sci. USA 113, 7852–7857 (2016).
Yang, C., Gong, R. & de Val, N. Development of neutralizing antibodies against zika virus based on its envelope protein structure. Virol. Sin. 34, 168–174 (2019).
Liang, H. et al. Recombinant Zika virus envelope protein elicited protective immunity against Zika virus in immunocompetent mice. PLoS ONE 13, https://pubmed.ncbi.nlm.nih.gov/29590178/ (2018)
Conlan, S. et al. Role of Zika virus envelope protein domain iii as a target of human neutralizing antibodies. mBio 10, https://pubmed.ncbi.nlm.nih.gov/31530669/ (2019)
Yang, M., Dent, M., Lai, H., Sun, H. & Chen, Q. Immunization of Zika virus envelope protein domain III induces specific and neutralizing immune responses against Zika virus. Vaccine 35, 4287–4294 (2017).
Pérez, O. et al. Adjuvants are key factors for the development of future vaccines: lessons from the Finlay adjuvant platform. Front. Immunol. 4, 407 (2013).
Cabral-Miranda, G. et al. Zika virus-derived E-DIII protein displayed on immunologically optimized VLPs induces neutralizing antibodies without causing enhancement of Dengue virus infection. Vaccines (Basel) 7, 72 (2019).
Vuitika, L., et al. A self-adjuvanted VLPs-based Covid-19 vaccine proven versatile, safe, and highly protective. Sci. Rep. 14, 24228 (2024).
Kozlovska, T. M., et al. Recombinant RNA phage qβ capsid particles synthesized and self-assembled in Escherichia coli. Gene 137, 133–137 (1993).
Brown, S. D., Fiedler, J. D. & Finn, M. G. Assembly of hybrid bacteriophage Qβ virus-like particles NIH Public Access. Biochemistry 48, 11155–11157 (2009).
Tegerstedt, K., et al. Murine pneumotropic virus VP1 virus-like particles (VLPs) bind to several cell types independent of sialic acid residues and do not serologically cross react with murine polyomavirus VP1 VLPs. J. Gen. Virol. 84, 3443–3452 (2003).
Gomes, A. C. et al. Early transcriptional signature in dendritic cells and the induction of protective t cell responses upon immunization with VLPs containing TLR ligands—a role for CCL2. Front. Immunol. 10, 1679 (2019).
Brito, L. A. & Singh, M. Acceptable levels of endotoxin in vaccine formulations during preclinical research. J. Pharm. Sci. 100, 34–37 (2011).
Cabral-Miranda, G., Cardoso, A. R., Ferreira, L. C. S., Sales, M. G. F. & Bachmann, M. F. Biosensor-based selective detection of Zika virus specific antibodies in infected individuals. Biosens. Bioelectron. 113, 101–107 (2018).
Cabral-Miranda, G., et al. Virus-like particle (VLP) plus microcrystalline tyrosine (MCT) adjuvants enhance vaccine efficacy improving t and b cell immunogenicity and protection against Plasmodium berghei/vivax. Vaccines (Basel) 5, 10 (2017).
Cabral-Miranda, G., et al. DOPS adjuvant confers enhanced protection against malaria for VLP-TRAP based vaccines. Diseases 6, 107 (2018).
Cabral-Miranda, G., et al. Microcrystalline tyrosine (MCT®): a depot adjuvant in licensed allergy immunotherapy offers new opportunities in malaria. Vaccines (Basel) 5, 32 (2017).
Roesti, E. S., et al. Vaccination against amyloidogenic aggregates in pancreatic islets prevents development of type 2 diabetes mellitus. Vaccines (Basel) 8, 116 (2020).
Dowall, S. D., et al. Lineage-dependent differences in the disease progression of Zika virus infection in type-I interferon receptor knockout (A129) mice. PLoS Negl. Trop. Dis. 11, e0005704 (2017).
Lazear, H. M., et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730 (2016).
Clancy, C. S., Van Wettere, A. J., Siddharthan, V., Morrey, J. D. & Julander, J. G. Comparative histopathologic lesions of the male reproductive tract during acute infection of Zika virus in AG129 and Ifnar À/À mice. Am. J. Pathol. 188, 904–915 (2018).
Shan, C. et al. A single-dose live-attenuated vaccine prevents Zika virus pregnancy transmission and testis damage. Nat. Commun. 8, 676 (2017).
Letícia, M. et al. Early infection of Zika virus in the male reproductive system of AG129 mice: molecular and immunohistochemical evaluation. Clin. Microbiol. 53, https://doi.org/10.1007/s42770-022-00761-x (2022).
Li, H., et al. Zika Virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell 19, 593–598 (2016).
Figueiredo, C. P., et al. Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun. 10, 3890 (2019).
Aliota, M. T. et al. Characterization of Lethal Zika Virus Infection in AG129 Mice. PLoS Negl. Trop. Dis. 10, e0004682 (2016).
Shan, C., et al. A live-attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat. Med. 23, 763–767 (2017).
Stephenson, K. E., et al. Safety and immunogenicity of a Zika purified inactivated virus vaccine given via standard, accelerated, or shortened schedules: a single-centre, double-blind, sequential-group, randomised, placebo-controlled, phase 1 trial. Lancet Infect. Dis. 20, 1061–1070 (2020).
He, P., Zou, Y. & Hu, Z. Advances in aluminum hydroxide-based adjuvant research and its mechanism. Hum. Vaccin Immunother. 11, 477–488 (2015).
Oleszycka, E., et al. The vaccine adjuvant alum promotes IL-10 production that suppresses Th1 responses. Eur. J. Immunol. 48, 705–715 (2018).
Uddin, M. N. & Roni, M. A. Challenges of storage and stability of mRNA-based COVID-19 vaccines. Vaccines (Basel). 9, 1033 (2021).
D’Alise, A. M., et al. Phase I trial of viral vector-based personalized vaccination elicits robust neoantigen-specific antitumor T-cell responses. Clin. Cancer Res. 30, 2412–2423 (2024).
Stettler, K., et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353, 823–826 (2016).
Premkumar, L. et al. Development of envelope protein antigens to serologically differentiate Zika virus infection from dengue virus infection. J. Clin. Microbiol. 56, e01504 (2018).
Slon Campos, J. L., Mongkolsapaya, J. & Screaton, G. R. The immune response against flaviviruses. Nat. Immunol. 19, 1189–1198 (2018).
Balmaseda, A., et al. Antibody-based assay discriminates Zika virus infection from other flaviviruses. Proc. Natl Acad. Sci. USA 114, 8384–8389 (2017).
Baneyx, F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 10, 411–421 (1999).
Baneyx, F. & Mujacic, M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 22, 1399–1408 (2004).
Kanno, A. I., et al. Optimization and scale-up production of Zika virus ΔNS1 in Escherichia coli: application of response surface methodology. AMB Express 10, 1–13 (2020).
Schiller, J. T. & Lowy, D. R. Immunogenicity testing in human papillomavirus virus-like-particle vaccine trials. J. Infect. Dis. 200, 166–171 (2009).
Rasmussen, S. A., Jamieson, D. J., Honein, M. A. & Petersen, M. A. Zika virus and birth defects—reviewing the evidence for causality. N. Engl. J. Med. 374, 1981–1987 (2016).
Shan, C., et al. Maternal vaccination and protective immunity against Zika virus vertical transmission. Nat. Commun. 10, 5677 (2019).
Acknowledgements
We would like to express our gratitude to the Laboratory of Cellular Biology at the Butantan Institute (São Paulo, Brazil) for its assistance with negative-stain electron microscopy. We would also like to express our sincere gratitude to Dr. Lucio Holanda Freitas Junior for generously providing laboratory space for Zika virus cultivation and to Dr. Luis Carlos de Souza Ferreira for kindly supplying aliquots of the virus. The authors acknowledge the support provided by The São Paulo Research Foundation–FAPESP: G.C.-M.: 2019/14526-0, 2020/04667-3; NOSC 2023/04762-8; R.D-C 2019/01255-9, 2021/03684-4; N.C. and G.C.-M.: 2021/03508-1 (Ph.D. Scholarship); A.L. and G.C.-M.: 2021/03102-5 (Ph.D. Scholarship); W.A.P.S. and G.C.-M. 2021/08468-8 (Ph.D. Scholarship); E.C. and G.C.-M.: 2023/13991-7 (Scholarships—Master); B.H. and G.C.-M.: 2024/13269-2 (Scholarships—Technical Training Program). This work has also been supported by CAPES, financial code (001) and CNPq.
Author information
Authors and Affiliations
Contributions
N.C. has implemented all experiments, data analysis, and wrote the paper draft. A.L., J.D.Q.S., E.C., W.A.P.-S., and B.H. have contributed to the experiments and revising of the paper. R.D.-C., A.B., N.O.S.C., O.C.-M., N.P., E.C.S., and J.E.K. have contributed to the writing and revising of the paper. G.C.-M. has supervised all experiments and contributed to the writing and revising of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Côrtes, N., Lira, A., Silva, J.D.Q. et al. A VLPs based vaccine protects against Zika virus infection and prevents cerebral and testicular damage. npj Vaccines 10, 107 (2025). https://doi.org/10.1038/s41541-025-01163-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-025-01163-4
This article is cited by
-
Zika virus-like particle production in a stirred tank bioreactor using a baculovirus/insect cell system
Bioprocess and Biosystems Engineering (2025)
