
Abstract
H9N2 avian influenza virus (AIV) is a globally prevalent pathogen that causes economic losses in poultry and poses zoonotic threats. Due to antigenic drift and shift, traditional inactivated vaccines often show reduced efficacy. This study presents a novel subunit vaccine based on a conserved HA6 scaffold derived from the hemagglutinin stem domain and coupled with a fusion peptide epitope (fPE) via Snoopligase-mediated ligation. The HA6 protein was validated by its binding to the broad-spectrum antibody CR6261, and the fPE-HA6 fusion construct incorporated T- and B-cell epitopes. Immunization trials in a chicken demonstrated that fPE-HA6 induced stronger humoral and cellular immune responses than individual immunogens. Upon challenge with H9N2 strains YZ4 and SN, the fusion vaccine significantly reduced viral shedding, demonstrating broad-spectrum protection. These findings highlight the potential of HA6 as a modular scaffold for influenza vaccines and the utility of Snoopligase technology in developing broadly protective immunogens against antigenically variable viruses.

Similar content being viewed by others


Introduction
H9N2 avian influenza virus (AIV) is a significant pathogen in poultry, primarily causing respiratory distress and reduced egg production, with mortality rates escalating during co-infections1,2,3,4. Despite extensive vaccination campaigns in China, where over 20 commercial inactivated influenza vaccines are deployed, H9N2 remains endemic, causing significant economic losses5,6,7. The continued spread of the virus among vaccinated poultry highlights the limitations of current vaccines, particularly their vulnerability to antigenic drift and shift caused by the virus’s segmented genome and error-prone RNA polymerase8,9,10. These factors drive the rapid evolution of H9N2, complicating its control and underscoring the urgent need for innovative vaccine strategies capable of providing broad and durable protection.
The hemagglutinin (HA) protein, the primary antigen of AIV, consists of two distinct domains: the membrane-distal HA1 head and the membrane-proximal HA2 stem. While antibodies targeting the HA2 stem domain exhibit broad-spectrum neutralizing activity against diverse influenza subtypes11,12,13,14, the immunodominance of the HA1 head often limits the immune system’s ability to recognize and respond to conserved regions within the HA2 stem. Additionally, the structural shielding of the HA stem by the globular HA1 head further hampers immune accessibility10,15. Therefore, a critical focus of vaccine design is to develop immunogens that enhance immune responses against conserved HA stem epitopes, offering cross-protection against multiple AIV subtypes.
Subunit vaccines have gained recognition as a promising strategy for targeting conserved regions of viral antigens. By selectively expressing specific antigenic domains, these vaccines reduce off-target immune responses while maximizing immunological precision and resource utilization16,17,18,19,20. Bommakanti demonstrated that HA stem immunogens expressed in E. coli elicit broad protection against homologous influenza subtypes, supporting our design strategy16,17. Recent advancements in protein engineering have revolutionized antigen design and functionality. Among these, enzyme-mediated protein ligation systems like Snoopligase have emerged as powerful tools for site-specific antigen conjugation under mild reaction conditions, producing stable and homogenous products21,22. Buldun et al. demonstrated the efficacy of Snoopligase in vaccine preparation by enabling the efficient assembly of modular antigens, paving the way for customizable and adaptable vaccine platforms. By combining conserved immunogens with strain-specific epitopes, this approach offers a versatile solution to address the antigenic variability of rapidly evolving viruses23,24.
In our previous study, we identified several B-cell and T-cell epitopes of H9 subtype AIV HA protein25. In this study, we designed the conserved stem domain HA6 as an immunogen scaffold based on the HA2 region of H9 subtype AIV, and used the Snoopligase system for modular antigen assembly of B- and T-cell epitopes from H9 subtype AIV. Through comprehensive immunological evaluation, we demonstrate the efficacy and versatility of this combination of scaffold and epitope assembly, providing valuable insights for the design of next-generation influenza vaccines with enhanced cross-protective potential.
Results
The design of the immunogen HA6 protein targeting the stem region of H9 subtype AIV
The HA6 immunogen was designed to focus the immune response on the conserved stem region of the hemagglutinin (HA) protein. Based on the crystal structure of the H9 HA protein (PDB ID: 1JSD), the interaction interfaces between the HA1 and HA2 subunits were analyzed with PyMOL. A total of 13 interaction regions on HA1 and 25 on HA2 were identified (Fig. 1a and Supplementary Table 1).

a Protein interaction site selection, HA1 (red), HA2 (cyan), HA1-HA2 interaction sites (yellow). b Red represents the HA1 domain with non-interacting regions removed, exposing new amino acid sites. c After stability optimization, AlphaFold2 simulates the tertiary structure of HA6. Green represents the mutation sites optimized for stability using RosettaDesign, and yellow represents the F63D and L73D mutations. d Prokaryotic expression of HA2 protein from H9 subtype AIV: 1–3 supernatants after induction at 16, 25, and 37 °C, 4–6 pellets after induction at 16, 25, and 37 °C. e Purification of expressed HA2 protein: 1–2 supernatant and precipitate from bacterial lysis, 3–6 wash and dissolve the residue with 0, 2, 4, 8 M urea. f Prokaryotic expression of HA6 protein: 1–3 supernatants after induction at 16, 25, and 37 °C, 4–6 pellets after induction at 16, 25, and 37 °C. g. Purification of HA6 protein: 1 pellet lysate supernatant, 2 elution with 20 mM imidazole, 3–6 elution with 100 mM imidazole, 7 elution with 250 mM imidazole. h Indirect ELISA to determine the binding capacity of CR6261-SCFV to HA2 and HA6. i Indirect ELISA to determine the binding capacity of H9 subtype AIV immune serum to HA2 and HA6. j Competitive ELISA to measure the competitive ability of HA6 immune serum against CR6261-SCFV. k Competitive ELISA to evaluate the competitive ability of HA2 immune serum against CR6261-SCFV. Statistical analysis was performed using two-tailed t-tests. Error bar, mean ± SD.
Non-interacting regions of HA1 were excluded, retaining only residues 1–36 and 281–317. These selected segments were connected to HA2 via a flexible GSAGSA peptide linker. Additional HA1 regions (residues 99, 102–103, 255–258, and 260) were excluded from the HA6 design as they primarily comprise polar amino acids and are spatially distant from other HA1-HA2 interaction sites in the primary structure. The resulting HA6 construct focuses on critical HA stem epitopes while minimizing immunodominant regions unrelated to the HA stem.
To identify newly exposed sites, differences in ASA were calculated for individual amino acid positions in both the full-length HA and the truncated HA6 construct. The analysis identified 24 amino acid positions with significant changes in ASA (Fig. 1b and Supplementary Table 2). Increased ASA values at certain positions were predicted to potentially reduce protein stability. To address this, RosettaDesign was used to optimize amino acid residues with an ASA increase rate exceeding 100% and non-polar residues with a lower ASA increase rate. Targeted mutations were introduced at specific sites, including I385T, I386P, and F390I in HA2, as well as T281D, H286D, V288M, A292W, G294S, C296A, and P297A in HA1. These modifications aimed to enhance structural stability while preserving the functional conformation of HA6.
Building on the work of Bommakanti16,17, the amino acid mutations F63D and L73D were introduced into the loop region between α-helices 4 and 5 of HA2. These mutations were designed to stabilize the HA6 protein by weakening its low-pH structural transitions. Structural predictions generated using AlphaFold2 confirmed that the engineered HA6 construct closely resembled the pre-fusion conformation of HA2 (Fig. 1c). The complete amino acid sequences of HA6 and HA2, including the introduced mutations, are provided in the Supplementary Materials.
The expression and purification of HA6 and HA2 proteins
The HA2 and HA6 proteins were expressed optimally at 25 °C and were mostly found in the precipitate. To purify HA2, a gradient washing protocol was employed using an inclusion body washing buffer with increasing concentrations of urea, effectively removing contaminants and yielding purified HA2 protein. HA6 was purified using Ni-column chromatography. The HA2 and HA6 proteins displayed high purity, as confirmed by SDS-PAGE analysis (Fig. 1d–g).
Antigenicity and functional validation of CR6261-SCFV binding to HA6
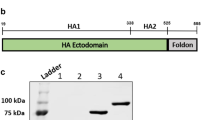
The pFUSE-mIgG2A-Fc2-CR6261 plasmid (Supplementary Fig. 1a) was transfected into 293T cells and cultured in selective medium containing zeocin. Western blot analysis confirmed the successful expression of CR6261-SCFV, yielding a target band of ~55 kDa in both supernatant and lysate (Supplementary Fig. 1b). Protein G resin purification resulted in a final yield of 0.8 mg/L of purified CR6261-SCFV.
The binding specificity of CR6261-SCFV was assessed using IFA. CR6261-SCFV exhibited strong fluorescence signals for H1, H5, and H9 subtype AIVs, with weaker signals for H7 subtype AIV (Supplementary Fig. 1c). Neutralizing assays further demonstrated CR6261-SCFV’s potent inhibitory activity, the results showed that CR6261-SCFV had the best inhibitory effect on the H5 subtype virus, with the lowest inhibitory concentration of 8.33 ± 2.87 μg/mL. The neutralization concentration on the H1 subtype virus was 12.5 ± 3.77 μg/mL. For H9 viruses, CR6261-SCFV exhibited strain-dependent neutralization, achieving greater efficacy against the F98 strain (16.67 ± 5.77 μg/mL) compared to the JY040218C strain (49.98 ± 4.38 μg/mL) (Supplementary Fig. 2).
To evaluate the antigenicity of the HA6 and HA2 proteins, indirect ELISA was employed. CR6261-SCFV and immune sera against various H9 AIVs effectively bound with both HA6 and HA2 proteins, with reactivity considerably higher than that of the negative control sera (Fig. 1h, i). Notably, CR6261-SCFV demonstrated significantly stronger binding affinity for HA6 compared to HA2 (p = 0.0099, Fig. 1h).
To further explore the relationship between HA6 and CR6261-SCFV, competitive ELISA assays were performed. CR6261-SCFV binding to HA6 was inhibited by 44.78 ± 2.47% when pre-incubated with HA6 immune sera, whereas the inhibition rate with HA2 immune sera was considerably lower at 23.63 ± 17.16% (Fig. 1j, k). These findings indicate that HA6 effectively induces antibodies that recognize epitopes similar to those targeted by CR6261.
Optimized Snoopligase ligation reaction
The optimal temperature for expressing of Snoopligase, SnoopTagjr-EGFP, and IMX-DogTag was determined to be 25 °C, with all proteins predominantly found in the supernatant (Supplementary Fig. 3a–c). Snoopligase and SnoopTagjr-EGFP were purified using Ni-NTA affinity chromatography, with proteins eluted through a gradient of imidazole (Supplementary Fig. 3d–f). The IMX-DogTag protein, owing to its high thermal stability, was subjected to incubation at 80 °C for 15 min, followed by a reduction step with β-mercaptoethanol at 25 °C for 5 min. Precipitation was performed using 500 mM glycine (pH 2.0), and the purified IMX-DogTag was re-suspended in Tris-HCl buffer (pH 8.0) (Supplementary Fig. 3e).
To evaluate the catalytic activity of Snoopligase in protein ligation, SnoopTagJr-EGFP and IMX-DogTag were used as substrates. The reaction mixture, containing both substrates and Snoopligase, was incubated at 4 °C for 24 h. SDS-PAGE analysis revealed distinct bands corresponding to dimeric products, confirming the successful catalysis and ligation of SnoopTagJr-EGFP and IMX-DogTag (Supplementary Fig. 4a).
Optimization of the ligation reaction identified pH 6.0–8.5 as the optimal range for efficient coupling, with maximum efficiency observed at pH 7 (37.35% ± 2.03%). The overall coupling efficiency across the range was 36.46% ± 1.22%. Reaction conditions were further optimized, demonstrating that the highest catalytic efficiency was achieved under the following conditions: 30–40% (v/v) glycerol concentration, 15 μM substrate concentration, and 30 μM Snoopligase concentration. These parameters yielded product efficiencies of 36.20% ± 1.88%, 37.31% ± 2.25%, and 48.36% ± 2.57%, respectively. Reaction time optimization indicated that peak efficiency was attained at 13 h (36.77% ± 0.41%), with no significant improvement upon further extension (Fig. 2a–g, Supplementary Fig. 4b–i).

a pH optimization. b Reaction time optimization. c Glycine concentration optimization. d SnoopTagjr substrate concentration optimization. e DogTag substrate concentration optimization. f SnoopTagjr and DogTag dual substrate concentration optimization. g Snoopligase concentration optimization. h Schematic representation of CL6B epoxidation and protein immobilization. i Schematic diagram of immobilized Snoopligase in the ligation reaction and purification of products. j Snoopligase immobilization: 1–2 before and after immobilization. k Purification of reaction products after ligation: 1 SnoopTagjr-EGFP, 2 IMX-DogTag, 3 Snoopligase, 4 unbound enzymes and substrates reaction, 5 immobilized enzymes and substrates reaction pH 2.0, elution 6–7 unreacted substrates. Error bar, mean ± SD.
Immobilization of SnoopLigase
Snoopligase was immobilized on cross-linked epoxy-activated Sepharose CL6B resin to facilitate the purification of ligation products (Fig. 2h, i). The concentration of Snoopligase in the supernatant was measured before and after immobilization, revealing a reduction from 5.15 to 3.09 mg/mL, corresponding to an immobilization efficiency of 40% (Fig. 2j). The concentration of Snoopligase on the CL6B resin post-immobilization was determined to be 6.18 mg/mL. Catalytic coupling reactions performed with the immobilized Snoopligase demonstrated that its enzymatic activity was retained following immobilization. SDS-PAGE analysis confirmed efficient coupling of substrates, with clear dimer bands observed (Fig. 2k). These results highlight the feasibility of Snoopligase immobilization for maintaining catalytic functionality while enabling practical applications in protein ligation.
The conjugation of DogTag-HA6 and fPE-SnoopTagjr
The expression results indicated that DogTag-HA6 and fPE-SnoopTagJr were primarily localized in the insoluble fraction. Expression levels of both proteins were enhanced when cultured at 25 °C in HB-PET auto-induction medium (Fig. 3a, b). Following the expression, the proteins were successfully purified using High-Affinity Ni-Charged Resin FF, as confirmed by SDS-PAGE analysis (Fig. 3c, d).

a DogTag-HA6 expression: 1–3 IPTG-induced lysate, 4–6 HB-PET auto-induction lysate, 7–9 IPTG-induced precipitate, 10–12 HB-PET auto-induction precipitate. b fPE-SnoopTagjr expression: 1–3 IPTG-induced lysate, 4–6 HB-PET auto-induction lysate, 7–9 IPTG-induced precipitate, 10–12 HB-PET auto-induction precipitate. c DogTag-HA6 purification: 1 elution peak, 2 20 mM imidazole elution, 3 100 mM imidazole elution, 4–5 250 mM imidazole elution. d fPE-SnoopTagjr purification: 1 elution peak, 2 20 mM imidazole elution, 3 100 mM imidazole elution, 4–5 250 mM imidazole elution. e Purification and conjugation of fPE-HA6: 1. Coupling in the free system 2. Purification of coupled enzymes 3. Washing of unreacted substrates in the coupled enzyme.
Ligation of DogTag-HA6 with fPE-SnoopTagJr was conducted under optimized conditions using immobilized Snoopligase. During the washing steps, incomplete reaction substrates were efficiently removed, yielding highly purified ligation products (Fig. 3e). These results demonstrate that the optimized ligation protocol, coupled with the immobilization strategy, effectively enhances product purity and consistency.
The determination of antibody titer
ELISA was used to determine the specific antibody titers against the SN and YZ4 viruses in sera collected from immunized chickens. For the SN virus, antibody titer analysis revealed no significant increase across all groups following the primary immunization. However, after the booster immunization, antibody titers generally increased, with the fPE-HA6 group showing higher titers compared to individual recombinant protein groups and significantly higher than the fPE group (p = 0.0115) on 42 days post-immunization (d.p.i.). Among all groups, the inactivated vaccine TX group exhibited the highest antibody titers on 42 d.p.i., considerably surpassing those of the protein-immunized groups (Fig. 4a, c).

a Specific antibody titer against the SN virus. b Specific antibody titer against the YZ4 virus. c Specific antibody titer against the SN virus at 42 d.p.i. d Specific antibody titer against the YZ4 virus at 42 d.p.i. e–h Cytokine level measurement after primary immunization. i–l Cytokine level measurement after booster immunization. m, n Detection of lymphocyte proliferation. o, p ELISpot detection after the YZ4 and SN viruses stimulation. Statistical analysis was performed using the one-way ANOVA. Error bar, mean ± SD.
For the YZ4 virus, the primary immunization similarly showed no significant increase in antibody titers across all protein groups. Following the booster immunization, antibody levels rose in all groups. On 42 d.p.i., the fPE-HA6 and HA6 groups displayed comparable titers, both significantly higher than those of the fPE group (p = 0.0242). Due to antigenic variation, the TX group did not achieve comparable titers against the YZ4 virus to SN (Fig. 4b, d).
Measurement of cytokine transcription levels
To comprehensively evaluate immune responses post-immunization, the transcription levels of key cytokines, including IL-2, IL-4, IL-5, and INF-γ, were assessed.
Following the primary immunization, IL-4 levels were significantly elevated in the HA6 and fPE-HA6 groups compared to the PBS group (2.d.p.i. HA6 p = 0.023 and fPE-HA6 p = 0.0464, 3.d.p.i. fPE-HA6 p = 0.001). INF-γ levels were markedly higher in the fPE (2.d.p.i. p = 0.0015, 3.d.p.i. p < 0.0001) group compared to the PBS group. Similarly, IL-2 levels significantly increased in the HA6 and fPE groups (2.d.p.i HA6 p = 0.0003, fPE p = 0.038, 3.d.p.i. p = 0.0008; Fig. 4e–h).
After the booster immunization, cytokine transcription levels further increased across multiple groups. IL-4 levels were significantly elevated in the HA6 and fPE groups (2.d.p.i. HA6 p = 0.0217, 3.d.p.i. HA6 p = 0.0072, fPE p = 0.0008). IL-5 levels showed significant increases in the HA6 and fPE-HA6 groups (1.d.p.i. HA6 p = 0.0054, fPE-HA6 p = 0.0105). INF-γ levels were markedly upregulated in the fPE, fPE-HA6, and HA6 groups (1.d.p.i. HA6 p = 0.0001, fPE-HA6 p = 0.001, 3.d.p.i. fPE p = 0.0001, fPE-HA6 p = 0.0299). Additionally, IL-2 levels significantly increased in the fPE-HA6, HA6 compared to the PBS group (1.d.p.i. HA6 p < 0.0001, 2.d.p.i. fPE-HA6 p = 0.0061; Fig. 4i–l). These results highlight the distinct cytokine profiles elicited by different immunization groups, with fPE-HA6 inducing robust Th1 and Th2 cytokine responses.
The results of lymphocyte proliferation
On day 14 following primary and booster immunizations, PBMCs were isolated and assessed for proliferation after stimulation with SN and YZ4 viruses. After the primary immunization, PBMCs from the fPE-HA6 group exhibited significantly enhanced proliferation in response to SN stimulation compared to the PBS group (p = 0.0057). However, YZ4 stimulation induced significant proliferation in PBMCs from the HA6, fPE, and fPE-HA6 groups compared to the PBS group (HA6 p = 0.0073, fPE p < 0.0001, fPE-HA6 p = 0.0305; Fig. 4m).
Following the booster immunization, the HA6, fPE-HA6, and inactivated TX groups showed a highly significant increase in PBMC proliferation under SN stimulation compared to the PBS group (HA6 p = 0.0025, fPE-HA6 p = 0.0002, TX p < 0.0001). Under YZ4 stimulation, all groups demonstrated a highly significant increase in proliferation (HA6 p = 0.0017, fPE p = 0.0005, fPE-HA6 p = 0.0005, TX p = 0.0116; Fig. 4n).
These results highlight the distinct proliferation responses elicited by the fPE-HA6 immunogen, demonstrating its ability to robustly activate PBMCs upon viral stimulation.
Measurement of INF-γ expression levels
Peripheral blood mononuclear cells (PBMCs) were collected on day 14 following primary and booster immunizations, and INF-γ expression levels were measured using ELISpot assays after stimulation with YZ4 and SN viruses.
Under YZ4 stimulation, both primary and booster immunizations with recombinant protein groups resulted in a significant increase in INF-γ expression compared to the PBS group (HA6 p = 0.0028 and 0.0349, fPE p < 0.0001, fPE-HA6 p < 0.0001). However, under SN stimulation, only the fPE and fPE-HA6 groups demonstrated significantly elevated INF-γ levels relative to the PBS group (fPE p < 0.0001 fPE-HA6 p < 0.0001 Fig. 4o, p and Supplementary Fig. 5).
These findings highlight the robust INF-γ responses elicited by recombinant proteins, especially those incorporating the fPE peptide, indicating their potential for inducing strong Th1-biased cellular immunity.
Immune protection
SPF chickens were challenged with SN and YZ4 viruses after booster immunization. Throat and cloacal swabs were collected on days 3, 5, and 7 post-challenge, and viral shedding was quantified using qRT-PCR. The critical thresholds for viral detection were established at 16.34 copies for SN and 24.76 copies for YZ4 at a dose of 10¹ EID50.
Following the YZ4 virus challenge, analysis of the throat virus shedding protection index revealed that the fPE-HA6 group demonstrated superior protective efficiency compared to the fPE group. Notably, there were no significant differences in protective efficiency between the HA6 and fPE-HA6 groups. In terms of cloacal virus shedding, the fPE group provided a comparable protection rate to other protein-immunized groups on day 3 post-challenge; however, it revealed a lower protection rate compared to the HA6 and fPE-HA6 groups on day 5 post-challenge (Table 1).
Following the SN virus challenge, the fPE-HA6 group exhibited superior throat virus shedding protection compared to the individually immunized fPE or HA6 groups. Analysis of cloacal virus shedding demonstrated a pattern consistent with the YZ4 challenge (Table 1). These data indicate that the fPE-HA6 group demonstrated broad-spectrum protection efficiency in substantial reducing both throat and cloacal virus shedding following challenge with either the SN or YZ4 strains.
Discussion
Inactivated influenza vaccines primarily induce antibodies targeting the immunodominant HA1 epitope, which limits their efficacy due to antigenic drift and shift. Ekiert et al. identified that the broadly neutralizing antibody CR6261 binds to the highly conserved α-helix region in the HA protein stem, suggesting that vaccines designed to target this epitope could elicit CR6261-like antibodies, providing broad-spectrum protection11. While direct expression of HA2 as a broad-spectrum immunogen is theoretically ideal, studies have shown that HA2 expressed in Escherichia coli without HA1 adopts a low-pH post-fusion conformation, which is unfavorable for inducing effective neutralizing antibodies26. Bommakanti et al. addressed this challenge by designing HA6 immunogens from H1 and H3 subtypes, preserving key HA1 residues interacting with HA2 under neutral pH conditions. This approach maintained the pre-fusion stem structure and demonstrated broad-spectrum protective effects16,17.
In this study, CR6261-SCFV was expressed to screen for conserved epitopes of the H9 subtype AIV. As expected, CR6261-SCFV demonstrated broad-spectrum binding activity against H5, H1, and H9 subtype influenza A viruses, consistent with epitope conservation among group 1 viruses27. Subsequently, an immunogen designated HA6 was designed based on the HA1 and HA2 regions of the H9 subtype AIV. ELISA results demonstrated significantly higher binding affinity between CR6261-SCFV and HA6 compared to HA2 alone. Competitive ELISA further revealed that CR6261-SCFV inhibited the binding of HA6 immune serum to the antigen by 44.78 ± 2.47%, which was greater than the inhibition observed for HA2 immune serum (23.63 ± 17.16%). These findings indicate that HA6 is more effective than HA2 at eliciting CR6261-like antibodies. Given that the HA2 stem region targeted by CR6261 is structurally conserved across group 1 influenza A viruses (including H1, H2, H5, and H9), these results suggest that the fPE-HA6 vaccine has the potential to offer cross-subtype protection within this group. Indeed, our IFA assays (Supplementary Fig. 1c) confirmed that CR6261-SCFV binds to both H1 and H5 subtypes, reinforcing the conservation of the targeted epitope. However, it is important to note that such cross-reactivity does not extend efficiently to group 2 viruses such as H3 and H7, due to significant structural divergence in the HA stem region28. Therefore, while fPE-HA6 may provide cross-protection against H5N1, its efficacy against H7N9 is likely limited. Further in vivo studies are ongoing to evaluate these possibilities in detail.
Although our primary focus was to evaluate the immunogenicity and protective efficacy of the fPE-HA6 construct in vivo, we acknowledge the value of detailed biophysical characterization. Techniques such as transmission electron microscopy (TEM) and dynamic light scattering (DLS) can provide useful insights into protein particle morphology, size distribution, and aggregation behavior. However, since the recombinant fPE-HA6 protein was expressed in E. coli and recovered through inclusion body solubilization and refolding, structural heterogeneity and transient aggregation are expected, which may limit the interpretability of TEM and DLS data29,30,31. Nevertheless, as we continue to optimize the refolding conditions for structural consistency, we plan to incorporate TEM and DLS in future studies to assess colloidal stability, formulation performance, and batch-to-batch reproducibility, particularly for downstream development and scale-up.
The Snoopligase system has proven highly versatile in various biological applications23,24,32,33. For instance, Andersson successfully utilized it to produce a stability-enhanced recombinant vaccine by combining the thermostable protein IMX with a malaria antigen24. Hills employed the SpyTag/SpyCatcher system to assemble quartets of receptor-binding domains (RBDs) from SARS-like beta coronaviruses, inducing robust neutralizing antibodies against multiple coronavirus strains34. Similarly, Yang used self-assembling ferritin nanocages as scaffolds to display combinations of PEDV S protein antigens, demonstrating the potential of modular vaccine platforms for interchangeable, efficient, and safe vaccine development against PEDV variant strains35.In this study, we leveraged the Snoopligase system to develop an “insert-display” assembly method for an H9 subtype avian influenza vaccine, using H9 HA6 as a scaffold. During development, we observed that adding SnoopTagJr to either the N- or C-terminus of HA6, even with extended flexible linker sequences, failed to facilitate coupling with substrate proteins carrying DogTag. However, when DogTag was fused to HA6, the coupling reaction proceeded efficiently, enabling HA6 to participate in ligation. These findings highlight the need to consider the potential masking effects of substrate proteins on the tag during the practical application of the Snoopligase system. While the modular Snoopligase-mediated conjugation enhances the vaccine’s flexibility and immune presentation, the current purification process involves an additional step using agarose-immobilized Snoopligase to remove unbound components. This adds some complexity and potential cost at scale. To address this, ongoing work focuses on process simplification, such as exploring single-step or ligand-free purification alternatives. Importantly, Andersson et al. demonstrated that low levels of residual Snoopligase did not impair vaccine efficacy in a similar subunit vaccine context24, suggesting that in field applications, complete removal may not be necessary, potentially further reducing costs. Compared to traditional inactivated vaccines that rely on high-biosafety virus culture, our recombinant system provides a safer, scalable, and economically viable alternative for mass production in the poultry industry.
Phylogenetic analysis of HA genes revealed that the inactivated vaccine strain TX was more closely related to the challenge strain SN, but exhibited significant divergence from YZ4. In contrast, the origin strain (HK98) of HA6 demonstrated greater homology with YZ4 (Supplementary Fig. 6, Supplementary Table 3). Notably, HA6 shares over 94% amino acid sequence identity across multiple H9N2 lineages, including F98-like, Y280-like, and the G1-like strain A/quail/Hong Kong/G1/97, suggesting potential for broad cross-lineage protection (Supplementary Fig. 7). The fPE component was designed based on highly conserved B-cell and T-cell epitopes from diverse H9 subtype AIVs25, further enhancing the spectrum of immune coverage. Immunogenicity analysis showed that the TX vaccine group induced higher antibody titers against the SN strain than the YZ4 strain, consistent with phylogenetic proximity. By contrast, both HA6 and fPE-HA6 groups elicited robust and comparable antibody responses against SN and YZ4 viruses, significantly outperforming the fPE group. These findings demonstrate that HA6 and fPE-HA6 elicit broad-spectrum humoral immunity, with fPE-HA6 providing synergistic enhancement due to multivalent epitope inclusion. Although G1-like strains were not included in the present challenge experiments due to their limited circulation in China36,37, their high sequence identity with HA6 indicates potential cross-reactivity. This supports the modular design of the fPE-HA6 platform as a promising strategy for broader protection against diverse H9N2 sublineages, including those not directly tested in this study. However, the duration of the immune response induced by the fPE-HA6 vaccine remains to be determined. In the present study, antibody titers were monitored only within a defined time window post-vaccination, and no long-term serological follow-up was conducted. Future studies will aim to evaluate the longevity of protective immunity and determine whether booster immunizations are necessary to maintain effective protection in poultry populations.
The HA6 domain was derived from the HA2 stem region of the HK98 strain, which remains the only H9N2 virus with a high-resolution HA crystal structure available in the Protein Data Bank (PDB ID: 1JSD). This provided a structurally reliable and widely accepted template for vaccine design. To enhance stability and immunogenicity, rational modifications were introduced, including truncation of HA1 to expose the stem, redesign of hydrophobic patches, and introduction of two mutations (F63D, L73D) to prevent the low-pH fusion conformation16. Structural stability of the optimized construct was evaluated through in silico Gibbs free energy calculations (ΔG ≈ 1.25) and 3D structural alignment with the native HA (RMSD ≈ 4.74 Å), indicating preservation of the HA2 stem conformation (Supplementary Fig. 8). These strategies collectively ensured that the HA6 design was not only structurally stable but also broadly representative.
Cellular immunity plays a vital role in combating viral infections, with lymphocyte responses varying significantly based on the immunogen and viral strain38. In this study, cytokine transcription analysis revealed that IL-4 levels were significantly elevated in the HA6 and fPE-HA6 groups, underscoring their ability to stimulate strong Th2-type immune responses. Similarly, IL-5, which is essential for early B cell activation, was also markedly increased in the HA6 and fPE-HA6 groups following booster vaccination, further supporting their capacity to enhance humoral immunity. Th1-type immune responses were robustly induced as well, with notable increases in INF-γ or IL-2 transcription levels observed in the HA6, fPE, and fPE-HA6 groups following primary or booster vaccination. Cell proliferation assays reinforced these findings, showing that HA6 and fPE-HA6 effectively induced lymphocyte proliferation upon stimulation with both the SN and YZ4 viruses after booster vaccination. ELISpot analysis further demonstrated that all recombinant protein groups exhibited significant increases in INF-γ levels under YZ4 stimulation, while only the fPE and fPE-HA6 groups showed significant elevation under SN stimulation. These results highlight the superior performance of the recombinant protein vaccines, particularly fPE-HA6, in inducing robust cellular immune responses. In contrast, the TX inactivated vaccine group displayed limited cytokine transcription and INF-γ expression, reflecting its weaker cellular immunity induction.
In terms of humoral immunity, fPE-HA6 elicited significantly higher IgG titers against both challenge strains (YZ4 and SN) than the TX group and exhibited greater cross-lineage reactivity, likely due to its design targeting conserved epitopes within the HA2 stem region. Unlike the TX vaccine, which primarily induces antibodies against the variable HA1 head domain and therefore provides strain-specific protection, the fPE-HA6 vaccine directs immune responses toward conserved, subdominant epitopes. Although these HA2-targeted antibodies do not exhibit hemagglutination inhibition (HI) or in vitro neutralization titers, they can interfere with membrane fusion and viral entry, offering broader protection in vivo through Fc-mediated mechanisms such as antibody-dependent cellular cytotoxicity. This is consistent with previous findings showing that non-neutralizing stem-specific antibodies can confer full protection in animal models despite lacking in vitro neutralizing activity39.
The inclusion of fPE, incorporating conserved T- and B-cell epitopes from multiple H9N2 lineages, further enhances both breadth and immunological recall. This is reflected by stronger IFN-γ responses and elevated lymphocyte proliferation in fPE-HA6 immunized birds. The immune protection assay results demonstrated that the fPE group only modestly reduced viral shedding in cloacal samples at 3 and 5 days post-challenge, with no significant effect in throat samples. In contrast, the fPE-HA6 group conferred superior protection against viral shedding from both sites, comparable to the TX group. These results suggest that, despite the lack of traditional neutralizing antibodies, the protective efficacy of fPE-HA6 is mediated through a synergistic mechanism involving HA2-targeted humoral immunity and robust cellular responses. Collectively, these findings support the development of fPE-HA6 as a next-generation H9N2 poultry vaccine with broader cross-lineage efficacy and improved cellular immune induction compared to current inactivated formulations16,39.
The immune protection assay results demonstrated that the fPE group only induced partially reduced virus shedding in cloacal samples on days 3 and 5 post-challenge, with no significant effect observed in throat samples. In contrast, the fPE-HA6 group demonstrated superior protection against virus shedding in throat and cloacal samples compared to the HA6 group, achieving a level of efficacy comparable to that of the TX group. These findings suggest that, despite the fPE-HA6 group’s inability to induce hemagglutination inhibition (HI) and microneutralization antibodies, its effective inhibition of viral shedding is likely due to the combined action of stalk-specific antibodies and cellular immunity. The superior immune protection offered by fPE-HA6 highlights its ability to elicit balanced humoral and cellular responses. These results underscore the potential of fPE-HA6 as a next-generation vaccine candidate capable of addressing antigenic variability in H9N2 avian influenza viruses16,40. Throughout the immunization phases, no adverse clinical signs, behavioral abnormalities, or mortality were observed in vaccinated birds, indicating a favorable safety profile. Although no toxic effects were observed in this study, we acknowledge the importance of comprehensive safety evaluation and plan to conduct detailed toxicity analyses in future preclinical studies to support large-scale application.
In summary, the designed HA6 demonstrated significantly enhanced binding activity to CR6261-SCFV compared to the HA2 protein of H9 subtype AIV and exhibited broad-spectrum immunogenic properties. The Snoopligase-constructed fPE-HA6 fusion protein elicited balanced Th1/Th2 responses and enhanced protection against viral shedding post-challenge. These findings offer valuable insights into designing vaccines that integrate broad-spectrum HA6 as a scaffold in conjunction with other T- and B-cell epitopes, thereby providing comprehensive protection against antigenically variable H9N2 viruses, with both immunogenic efficacy and biosafety in mind.
Methods
Ethics and animal welfare
All animal experiments were conducted in strict accordance with the ARRIVE Essential 10 guidelines and institutional animal welfare protocols. Specific-pathogen-free (SPF) White Leghorn chickens, aged 3 weeks and with a mean body weight of 150–200 g, were obtained from Lihua Agriculture (Zhejiang, China). A total of n = 8 chickens per group were used, with mixed-sex groups randomly allocated unless otherwise specified. Chickens were housed in individually ventilated negative-pressure isolators under controlled environmental conditions (22–24 °C, 45–55% humidity, 12-h light/dark cycle), with ad libitum access to commercial feed and water. The experimental protocol was reviewed and approved by the Jiangsu Administrative Committee for Laboratory Animals (Permit No.: SYXKSU-2016-0020). All personnel involved in animal handling were certified in laboratory animal science and welfare. To reduce bias, animals were randomly assigned to experimental groups using a computer-generated randomization list. Outcome assessors were blinded to group allocation during sample collection and immunological assays.
No unexpected adverse events or mortality were observed throughout the study. At the end of the experimental period, animals were humanely euthanized by manual cervical dislocation (CD) performed by trained personnel. The method followed the AVMA Guidelines for the Euthanasia of Animals (2020) and was verified by the absence of corneal reflex, heartbeat, and spontaneous respiration. This method is widely accepted for poultry research and ensures immediate and irreversible loss of brain function41,42,43. No animals or data points were excluded from the study.
Viruses, sera, and cells
Two H9N2 AIV strains, A/chicken/Fujian/Shengnong/2014/H9N2 (SN, Y280-like) and A/chicken/Jiangsu/YZ4/2012/H9N2 (YZ4, F98-like), The selected challenge strains exhibiting significant antigenic divergence and poor cross-protection, thus providing a stringent model for evaluating broad-spectrum efficacy44. Additional AIV strains, including A/chicken/Shanghai/F/98/H9N2 (F98), A/chicken/Taixing/10/2010/H9N2 (TX), A/PR/8/34/H1N1 (PR8), A/mallard/Huadong/S/2005/H5N1 (SY), and A/Chicken/Guangdong/GD4/2017/H7N9 (GD4), were maintained in our laboratory for comparative studies. All viruses were propagated in 9-11-day-old embryonated SPF chicken eggs (Lihua Agriculture, Zhejiang, China) at 37 °C, and allantoic fluid was harvested 72 h post-inoculation and stored at −70 °C.
Madin-Darby canine kidney (MDCK) cells were cultured in Minimum Essential Medium (MEM, HyClone, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and incubated at 37 °C with 5% CO2. Human embryonic kidney (293T) cells were maintained in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM, HyClone, USA) with 10% FBS under identical conditions.
Virus concentration and purification
The viruses YZ4 and SN were inoculated into chicken embryos for amplification. Allantoic fluid was collected, which was then centrifuged at 4 °C and 12,000 rpm for 10 min. The resulting supernatant was collected and filtered through a 0.22 μm membrane. Next, centrifugation was carried out using a 32 Ti centrifuge tube at 4 °C and 30,000 rpm for 90 min. Using a syringe, various sucrose concentrations (20%, 40%, 60%) were layered into a 41 Ti centrifuge tube. The concentrated virus solution was slowly added to the top layer, followed by centrifugation at 4 °C and 30,000 rpm for 90 min. The virus bands between 40 and 60% sucrose concentrations were carefully extracted, and the separated virus was resuspended in a 10 times volume 0.1 M PBS. The suspension was then subjected to centrifugation in a 32 Ti centrifuge tube at 4 °C and 30,000 rpm for 90 min. The supernatant was discarded, and the precipitate was resuspended in 0.1 M PBS, followed by overnight dissolution at 4 °C. Finally, the virus was stored at −70 °C.
Phylogenetic and amino acid conservation analysis
Genetic evolution analysis of H9N2 AIVs was performed using IQ-TREE (v.1.6.8)45. Visualization was performed using ITOL (v.6.5.2) (https://itol.embl.de/), wherein the key antigenic sites of the HA protein underwent multiple sequence alignments and were correlated with the evolutionary tree46. Inter-branch genetic distances were calculated using MEGA5.0.
Expression of CR6261-SCFV
The variable regions of the heavy and light chains of CR6261-SCFV (GenBank ID: HI919029.1, HI919031.1) were synthesized (GenScript in Nanjing, China). After the synthesis, these sequences were attached to the Plasmid pFUSE-mIgG2a-FC2 (Supplementary Fig. 1a) using the corresponding primers in Supplementary Table 4.
The plasmid pFUSE-mIgG2a-FC2-CR6261-SCFV was transformed into 293T cells for antibody expression. The 293T cells were seeded in a 6-well plate and cultured to 80% confluence. After a gentle wash with PBS, 1 mL of serum-free DMEM medium was added and incubated at 37 °C in a 5% CO2 incubator. Next, 4 μg of pFUSE-mIgG2a-FC2-CR6261-SCFV was mixed with 100 μL of serum-free DMEM, and then 3 μL of PolyJet (SignaGen, Frederick, USA) was added and mixed. The mixture was incubated at room temperature for 15 min and then dropwise added to the 293T cells, followed by a 12-h incubation at 37 °C in a 5% CO2. The medium was replaced with DMEM containing 200 μg/mL of zeocin and 4% fetal bovine serum, and the cells were further cultured for 60 h. The cell culture supernatant was collected, and cells were lysed for western blot analysis to assess antibody expression. The 293T cells expressing CR6261-SCFV were expanded under zeocin-containing media, and the cell culture supernatant was collected to purify the CR6261-SCFV antibody using Protein G Resin (GenScript, Nanjing, China).
Characteristic evaluation of CR6261-SCFV
To evaluate the characteristics of the CR6261-SCFV, MDCK cells were seeded into 96-well plates at ~80% confluence and washed three times with phosphate-buffered saline (PBS). The cells were then inoculated with different AIV subtypes, including F98, TX, SY, PR8, and GD4, at a multiplicity of infection (MOI) of 0.01. After 1 h of virus adsorption, the cells were washed and cultured in MEM supplemented with 2 μg/mL TPCK trypsin (Sigma-Aldrich, USA) without antibiotics or serum for 12 h at 37 °C with 5% CO2. The cells were subsequently fixed with 4% paraformaldehyde and blocked with 3% bovine serum albumin (BSA) in PBS containing 0.05% Tween-20 (PBST). CR6261-SCFV (10 ng/mL) was added to each well, followed by incubation with Alexa Fluor™ 488-conjugated goat anti-mouse IgG (Thermo Fisher Scientific, USA) for fluorescence detection using a microscope.
The neutralization activity of CR6261-SCFV was further assessed using a microneutralization (MN) assay47. Serial 10-fold dilutions of CR6261-SCFV were mixed with virus solutions containing 2000 TCID50/mL and incubated at 37 °C for 1 h. The mixture was then added to 96-well plates seeded with MDCK cells and cultured in MEM containing 2 μg/mL TPCK trypsin for 72 h. Hemagglutination assays were performed to quantify positive infections and determine the neutralization capacity of CR6261-SCFV.
The design of immunogenic HA6 targeting the stalk domain of the H9 HA
The trimeric structure of the hemagglutinin (HA) protein from the H9N2 AIV strain A/swine/Hong Kong/9/98 (HK98) (PDB ID: 1JSD) was analyzed using PyMOL to identify interaction domains between the HA1 and HA2 subunits. Regions of HA1 not involved in HA2 interactions were removed, resulting in a truncated structure termed HA6. The final construct retained HA1 residues 1–36 and 281–317, linked to HA2 through a flexible GSAGSA linker. To assess the structural exposure of amino acid residues, the accessible surface area (ASA) was calculated using PyMOL before and after truncation.
Newly exposed residues with significant ASA changes were further analyzed for potential destabilizing effects. RosettaDesign was employed to introduce stabilizing mutations, targeting residues with hydrophobic or polar character changes, including I385T, I386P, F390I in HA2, and T281D, H286D, V288M, A292W, G294S, C296A, and P297A in HA1. Additionally, mutations F63D and L73D were introduced into the HA2 loop region to weaken low-pH structural transitions and preserve the pre-fusion conformation48.
AlphaFold2 modeled the designed HA6 protein structure49, confirming its alignment with the pre-fusion HA stem conformation. The optimized sequence of HA6 was further validated for stability and functionality, with the corresponding amino acid sequence provided in the Supplementary Materials.
Expression of immunogenic HA6 targeting the stalk domain of the H9 HA
The nucleotide sequence of HA6 was optimized for Escherichia coli and synthesized (GenScript in Nanjing, China). The HA2 and HA6 genes were then added to the pET-28a using the primers in Supplementary Table 5 and transformed into Escherichia coli BL21 (DE3) competent cells for expression. The BL21-pET28a-HA6 and BL21-pET28a-HA2 cultures were grown in LB medium at 37 °C and 220 rpm. When the OD600 reached ~0.6, IPTG was added to induce protein expression, after which the bacterial cells were harvested. The collected cells were washed twice with PBS and then suspended in LE buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0). After being subjected to ultrasound treatment, they were centrifuged at 12,000 rpm for 10 min. The resulting pellet was washed with Inclusion Body Wash Buffer I (50 mM Tris HCl, 100 mM NaCl, 10 mM EDTA, and 0.5% (v/v) TritonX-100, pH 8.0). After incubating the pellet at room temperature for 10 min, it was centrifuged again at 12,000 rpm to separate the supernatant from the precipitate. This washing process was repeated using Inclusion Body Wash Buffer II (2 M urea, prepared in inclusion body wash buffer I) and inclusion body wash buffer III (4 M urea, prepared in inclusion body wash buffer I). Finally, the cells were resuspended and precipitated using LE buffer containing 8 M urea (100 mM NaH2PO4, 10 mM Tris HCl, 8 M urea, pH 8.0).
To purify the protein, use the High-Affinity Ni-Charged Resin FF Prepacked Column (GenScript, Nanjing, China) for affinity chromatography. Samples after expression and purification were validated through SDS-PAGE analysis.
Validation of the binding affinity between CR6261-SCFV and HA6
The binding affinity between HA6 and CR6261-SCFV was assessed using an indirect enzyme-linked immunosorbent assay (ELISA). HA6 and HA2 proteins were diluted to 1 μg/mL in carbonate buffer (pH 9.6) and coated onto 96-well ELISA plates overnight at 4 °C. After washing three times with PBST and blocking with 5% skimmed milk, CR6261-SCFV, as well as chicken sera against H9N2 AIVs, and control groups, were diluted and added to the wells. After 1 h of incubation at 37 °C, HRP-conjugated goat anti-mouse or anti-chicken antibodies (Abcam, Cambridge, UK) were used as secondary antibodies. Detection was performed using TMB substrate, and absorbance was measured at OD450.
A competitive ELISA was conducted to evaluate whether HA6 induces antibodies that mimic CR6261 binding. HA6 and HA2 proteins were coated on ELISA plates as described above. CR6261-SCFV (100 ng/mL) was pre-incubated with equal volumes of chicken immune sera from HA6- or HA2-immunized groups. The resulting mixture was added to the plates, and bound CR6261-SCFV was detected using HRP-conjugated secondary antibodies. The inhibition rate was calculated to assess competitive binding. Finally, the inhibitory rate was calculated using a predetermined formula.
Expression and purification of SnoopLigase and building blocks
The nucleotide sequences of Snoopligase (GenBank: MG867372.1) and IMX-DogTag (GenBank: MH798875.1) were synthesized (GenScript, Nanjing, China). To amplify the EGPF sequence, pEGFP-C1 was used as a template and SnoopTagjr was added through overlap extension PCR. The amplified gene fragments were then inserted into the pET-28a plasmid and transformed into Escherichia coli BL21 (DE3) for expression. The primer sequences can be found in Supplementary Table 6. Finally, a High-Affinity Ni-Charged Resin FF Prepacked Column was used for the purification of the samples.
Evaluation and optimization of Snoopligase reaction conditions
To assess the catalytic capability of Snoopligase, Snoopligase and IMX-DogTag were synthesized, while SnoopTagJr-EGFP was amplified from plasmid pEGFP-1 using overlap extension PCR in conjunction with and SnoopTagir linker. 20 μM of three proteins were individually introduced into a 50 mM Tris-borate buffer (pH 8.0). The reaction mixtures were incubated with rotation at 4 °C for 24 h. After confirming Snoopligase catalytic activity, the catalytic conditions were further optimized by adjusting various factors in the reaction system. The pH values (ranging from 6 to 8.5), glycerol proportions (0–60% v/v), reaction time (1–24 h), and substrate and Snoopligase concentration (5–60 μM) were systematically varied to determine the optimal conditions for the reaction. The results were analyzed through SDS-PAGE and quantified using grayscale quantitative analysis performed with Image J and the formula.
SnoopLigase immobilization
Protein immobilization is used to purify the products following the ligation reaction33,50. For immobilize Snoopligase, epoxy-activated Sepharose CL6B gel was employed. Briefly, 10 mL of Sepharose CL6B gel was washed thoroughly with distilled water (ddH2O). The CL6B gel was then resuspended in a 50 mL conical flask with 12 mL of 0.35 M NaOH and stirred. While stirring, 8 mL of 1, 4-butanediol diglycidyl ether was slowly added dropwise, and the mixture was incubated with agitation at 170 rpm at 37 °C for 3 h. The activated gel was then subjected to a series of acetone washes in the following sequence: 25%-50%-75%-100%-75%-50%-25%, followed by rinsing with ddH2O to remove residual acetone. The gel was stored in a 1:1 ratio with ddH2O at 4 °C.
To immobilize Snoopligase, prepare a mixture of 3 mL of Snoopligase (10 mg/mL) and 2 mL of immobilization buffer (1 M Na2SO4, 200 mM NaHCO3, pH 10.0). Wash 2 mL of CL6B epoxy-activated resin with immobilization buffer, then add the resin to the Snoopligase solution. Incubate the mixture with agitation at 170 rpm at 37 °C for 12 h. After incubation, assess the protein concentration in the supernatant to determine the immobilization efficiency, thoroughly wash the gel with ddH2O. Next, treat the gel with an equal volume of 4% ethanolamine (pH 8.5) with agitation at 170 rpm at room temperature for 4 h. Finally, wash the gel again with ddH2O, resuspend it with PBS, stored at 4 °C.
The product was purified using a column immobilized Snoopligase. First, the CL6B gel matrix containing immobilized Snoopligase was placed in a chromatography column and washed with 50 mM Tris-boric acid. To reduce nonspecific adsorption, the optimal substrate system was prepared with 0.02% Tween-20. The 50 mM Tris-boric acid was then mixed with the Snoopligase gel in the column and incubated at 4 °C for 24 h. The Snoopligase gel was thoroughly washed with 50 mM Tris-boric acid to remove any unreacted substrates. This was followed by washed with a wash 50 mM glycine (pH 3.5) for 5 min using an oscillator. The column was then allowed to stand, and the washed products were collected by gravity sedimentation. The matrix was further washed with a product elution buffer (50 mM glycine, pH 1.5) for 5 min using an oscillator, followed by a static collection of eluted products by gravity sedimentation. Finally, the product was neutralized with 500 mM glycine (pH 9.0) to an appropriate pH and identified using SDS-PAGE.
The expression and purification of DogTag-HA6, and fPE-SnoopTagjr
A fusion peptide epitope (fPE, Supplementary Materials) was synthesized by combining molecular adjuvants, DC-induced peptide, and cIL-2 with B-cell epitopes (39ENNVPVTHAKELLHTEHNGM59, 219RIFKPLIGPRPLVNGLMGRI239, 269SGESHGRILKTDLKMGSCTV289, 309YAFGNCPKYIGVKSLKLAVG329) and T-cell epitopes (159MRWLTRKDGNYPTQDAQYTN179, 229PLVNGLMGRIDYYWSVLKPGQTLRIKSDGN259) from the H9 HA1 influenza protein (GenScript, Nanjing, China)25. The DogTag sequence was fused to the N-terminus of HA6, while SnoopTagJr was appended to the C-terminus of fPE using the overlap extension PCR method. The DogTag-HA6 construct was ligated into the pET-28a plasmid, while fPE-SnoopTagJr was cloned into the pET-32a plasmid. Protein expression was induced in Escherichia coli BL21 (DE3), and the expressed proteins were purified using high-affinity Ni-charged resin FF prepacked columns (GenScript, Nanjing, China). The primer sequences are provided in Supplementary Table 7.
The ligation of DogTag-HA6, and fPE-SnoopTagjr
Catalytic ligation reactions involving DogTag-HA6 and fPE-SnoopTagJr were carried out under optimized SnoopLigase reaction conditions. The optimized parameters included a reaction pH of 8.0, 30% glycerol (v/v), a substrate concentration of 15 μM, and a SnoopLigase concentration of 30 μM. Reactions were incubated for 13 h using SnoopLigase immobilized on CL6B Sepharose resin. The resulting ligation products were purified using a wash/elution buffer and identified by SDS-PAGE.
Immuno-protective efficacy of fPE-HA6
A total of 80 3-week-old SPF chickens were randomly assigned to 10 groups as described in Supplementary Table 8. Vaccines were prepared by emulsifying fPE-HA6 with the adjuvant MONTANIDE ISA 71 VG (SEPPIC, France) in a 3:7 ratio. Control groups, including individual proteins, inactivated vaccine TX, and PBS, were similarly formulated. The vaccines were administered subcutaneously, followed by a booster immunization 3 weeks later. Notably, the TX group received a single immunization when the booster immunization of the protein group.
Peripheral blood mononuclear cells (PBMCs) were collected (n = 3) on days 1, 2, and 3 post-primary and booster immunizations to evaluate cytokine transcription levels. On day 14, both immunizations, PBMCs were isolated (n = 3) for lymphocyte proliferation assays and interferon-gamma (IFN-γ) expression analysis. Serum samples were collected weekly to measure humoral immune responses.
Three weeks after the booster immunization, chickens were challenged intranasally with YZ4 and SN at a dose of 106 EID50 per chicken. Viral shedding was monitored via throat and cloacal swabs collected on days 3, 5, and 7 post-challenge. Viral load in swabs was quantified using real-time quantitative PCR (qPCR). Primer and probe sequences used for qPCR are detailed in Supplementary Table 9. An overview of the immunization and challenge protocol is provided in Supplementary Fig. 9.
Antibody level determination
To evaluate the antibody levels post-immunization, purified YZ4 and SN viruses were diluted to 6 μg/mL and coated onto 96-well ELISA plates (100 μL/well). The plates were then blocked with PBST containing 3% BSA. Immune sera were initially diluted 100-fold, followed by two-fold serial dilutions, and 100 μL of each dilution was added to the wells, and incubation at 37 °C for 1 h. After three washes, 100 μL of appropriately diluted HRP-conjugated anti-chicken IgY was added to each well, followed by a 30-min incubation at 37 °C. The plates were washed five times with PBST, 50 μL TMB substrate was added to develop the color reaction for 10 min at room temperature. The reaction was stopped by adding 50 μL of 2 M H2SO4, and the absorbance at OD450 was measured using a microplate reader.
The antibody titer was defined as the highest serum dilution at which the absorbance of the test well exceeded 2.1 times the absorbance of the blank control well (P/N ratio). These titer values were used to assess the antibody potency elicited by the immunization protocols.
Lymphocyte separation
Chicken PBMCs were isolated using a chicken PBMC separation reagent kit (Solarbio, Beijing, China). To start, the anticoagulant whole blood was collected and then diluted with pre-warmed PBS in equal amounts. Next, 10 mL of lymphocyte separation reagent was added to the bottom of a centrifuge tube, and the diluted whole blood was gently layered on top of the separation reagent. The tube was centrifuged at 700 g for 30 min in a horizontal rotor at room temperature. After centrifugation, the lymphocyte layer was aspirated, and the cells were washed with 10 mL of PBS and frozen.
lymphocyte proliferation and ELISpot
PBMCs were collected from immunized chickens at 2 and 5 weeks post-immunization. The cells were seeded into a 96-well plate at a density of 2 × 106 cells per well and incubated overnight at 37 °C with 5% CO2. Following incubation, cells were stimulated with YZ4 or SN viruses at a multiplicity of infection (MOI) of 0.01 for 72 h. Subsequently, 10 μL of CCK-8 solution was added to each well, and the plates were incubated for 2 additional hours at 37 °C. The OD450 was measured using a microplate reader. The proliferation rate was calculated using the formula.
To evaluate IFN-γ expression levels, ELISpot assays were conducted using the ELISpot Flex: Chicken IFN-γ (ALP) assay kit (Mabtech, Sweden). MSIP plates (Merck, Germany) were activated with 15 μL of 35% ethanol, washed five times with ddH2O, and coated with 100 μL of the capture antibody (MT6C2, 15 μg/mL). Plates were incubated overnight at 4 °C. Following PBS washes, the wells were blocked with RPMI 1640 medium supplemented with 10% FBS for 1 h at room temperature. PBMCs (1 × 106 cells per well) were stimulated with YZ4 or SN viruses at an MOI of 0.01. Positive controls (20 ng/mL Phorbol 12-myristate 13-acetate (PMA) and 1 μM ionomycin), negative controls (unstimulated cells), and blank controls (medium only) were included. Plates were incubated at 37 °C with 5% CO2 for 48 h. After incubation, wells were washed five times with PBS, followed by the addition of 100 μL biotinylated detection antibody (MT7C10-biotin, 1 μg/mL). After a 2-h incubation at room temperature, plates were washed again and incubated with 100 μL of streptavidin-alkaline phosphatase (diluted 1:1000) for 1 h. Following another washes, 100 μL BCIP/NBT-plus substrate was added to develop the color reaction, which was terminated by rinsing the plates with ddH2O (5–30 min). Plates were air-dried in a dark environment, and spots were analyzed using the S6 Entry system (CTL, USA).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (version 9.0; GraphPad Software, USA). The mean and standard deviation (SD) were calculated for each dataset. Use a two-tailed Student’s t test for comparisons between two groups and one-way ANOVA for comparisons involving three or more groups. Differences were considered statistically significant at p ≤ 0.05 and highly significant at p ≤ 0.01. All experiments were conducted in triplicate, and representative data were presented.
Data availability
All the data generated or analyzed during the study are included in this published article. The datasets used and/or analyzed during the present research project are available from the corresponding authors upon reasonable request. The HA6 and fPE sequences used in this study have been submitted to GenBank with the IDs: PV679987 and PV679988. The sequence is publicly accessible via the following URL: https://www.ncbi.nlm.nih.gov/nuccore/PV679987 and https://www.ncbi.nlm.nih.gov/nuccore/PV679988.
Code availability
The underlying code for this study is not publicly available but may be made available on reasonable request from the corresponding author.
References
Lai, V. D. et al. First report of field cases of Y280-like LPAI H9N2 strains in South Korean poultry farms: pathological findings and genetic characterization. Avian Pathol.50, 327–338 (2021).
Kishida, N., Sakoda, Y., Eto, M., Sunaga, Y. & Kida, H. Co-infection of Staphylococcus aureus or Haemophilus paragallinarum exacerbates H9N2 influenza A virus infection in chickens. Arch. Virol. 149, 2095–2104 (2004).
Kye, S. J. et al. Pathogenicity of H9N2 low pathogenic avian influenza viruses of different lineages isolated from live bird markets tested in three animal models: SPF chickens, Korean native chickens, and ducks. Poult. Sci. 100, 101318 (2021).
Bano, S., Naeem, K. & Malik, S. A. Evaluation of pathogenic potential of avian influenza virus serotype H9N2 in chickens. Avian Dis. 47, 817–822 (2003).
Sun, Y. & Liu, J. H9N2 influenza virus in China: a cause of concern. Protein Cell 6, 18–25 (2015).
Bi, Y. et al. Dominant subtype switch in avian influenza viruses during 2016-2019 in China. Nat. Commun. 11, 5909–5909 (2020).
Ma, M. J. et al. Avian influenza A virus infection among workers at live poultry markets, China, 2013-2016. Emerg. Infect. Dis. 24, 1246–1256 (2018).
Xia, J. et al. Evolution of prevalent H9N2 subtype of avian influenza virus during 2019 to 2022 for the development of a control strategy in China. Poult. Sci. 102, 102957 (2023).
Zhang, N. et al. The emergence of new antigen branches of H9N2 avian influenza virus in China due to antigenic drift on hemagglutinin through antibody escape at immunodominant sites. Emerg. Microbes Infect. 12, 2246582 (2023).
Khanna, M., Sharma, S., Kumar, B. & Rajput, R. Protective immunity based on the conserved hemagglutinin stalk domain and its prospects for universal influenza vaccine development. BioMed. Res. Int. 2014, 546274 (2014).
Ekiert, D. C. et al. Antibody recognition of a highly conserved influenza virus epitope. Science 324, 246–251 (2009).
Sui, J. et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16, 265–273 (2009).
Fu, Y. et al. A broadly neutralizing anti-influenza antibody reveals ongoing capacity of haemagglutinin-specific memory B cells to evolve. Nat. Commun. 7, 12780 (2016).
Chai, N. et al. Two escape mechanisms of influenza A virus to a broadly neutralizing stalk-binding antibody. Plos Pathog. 12, e1005702 (2016).
Laursen, N. S. & Wilson, I. A. Broadly neutralizing antibodies against influenza viruses. Antivir. Res. 98, 476–483 (2013).
Bommakanti, G. et al. Design of an HA2-based Escherichia coli expressed influenza immunogen that protects mice from pathogenic challenge. Proc. Natl. Acad. Sci. USA 107, 13701–13706 (2010).
Bommakanti, G. et al. Design of Escherichia coli-expressed stalk domain immunogens of H1N1 hemagglutinin that protect mice from lethal challenge. J. Virol. 86, 13434–13444 (2012).
Lee, J.-S. et al. The highly conserved HA2 protein of the influenza a virus induces a cross protective immune response. J. Virol. Methods 194, 280–288 (2013).
To, K. K. W. et al. Recombinant influenza A virus hemagglutinin HA2 subunit protects mice against influenza A(H7N9) virus infection. Arch. Virol. 160, 777–786 (2015).
Zeng, D. et al. A hemagglutinin stem vaccine designed rationally by AlphaFold2 confers broad protection against influenza B infection. Viruses 14, https://doi.org/10.3390/v14061305 (2022).
Banerjee, A. & Howarth, M. Nanoteamwork: covalent protein assembly beyond duets towards protein ensembles and orchestras. Curr. Opin. Biotech. 51, 16–23 (2018).
Permana, D., Putra, H. E. & Djaenudin, D. Designed protein multimerization and polymerization for functionalization of proteins. Biotechnol. Lett. 44, 341–365 (2022).
Buldun, C. M., Jean, J. X., Bedford, M. R. & Howarth, M. SnoopLigase catalyzes peptide–peptide locking and enables solid-phase conjugate isolation. J. Am. Chem. Soc. 140, 3008–3018 (2018).
Andersson, A. C., Buldun, C. M., Pattinson, D. J., Draper, S. J. & Howarth, M. SnoopLigase peptide-peptide conjugation enables modular vaccine assembly. Sci. Rep.9, 4625 (2019).
Quan, K. et al. Identification of broad-spectrum B-cell and T-cell epitopes of H9 subtype avian influenza virus HA protein using polypeptide scanning1. J. Integr. Agr. https://doi.org/10.1016/j.jia.2024.07.005 (2024).
Chen, J. et al. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. USA 92, 12205–12209 (1995).
Lingwood, D. et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature 489, 566–570 (2012).
Corti, D. et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856 (2011).
Burgess, R. R. Refolding solubilized inclusion body proteins. Methods Enzymol. 463, 259–282 (2009).
Lebendiker, M. & Danieli, T. Production of prone-to-aggregate proteins. FEBS Lett. 588, 236–246 (2014).
Jaenicke, R. Protein stability and molecular adaptation to extreme conditions. Eur. J. Biochem 202, 715–728 (1991).
M, D. & S, B. Adenoviral vectors and vaccines thereof. WO 2022234276 A1 (2022).
Chen, J. et al. SnoopLigase enables highly efficient generation of C–C-linked bispecific nanobodies targeting TNF-α and IL-17A. Bioconjugate Chem. 33, 1446–1455 (2022).
Hills, R. A. et al. Proactive vaccination using multiviral Quartet Nanocages to elicit broad anti-coronavirus responses. Nat. Nanotechnol. https://doi.org/10.1038/s41565-024-01655-9 (2024).
Yang, D. et al. Combination of S1-N-terminal and S1-C-terminal domain antigens targeting double receptor-binding domains bolsters protective immunity of a nanoparticle vaccine against porcine epidemic diarrhea virus. Acs Nano 18, 12235–12260 (2024).
Sun, H. et al. Genetic, molecular, and pathogenic characterization of the H9N2 avian influenza viruses currently circulating in South China. Viruses 11, https://doi.org/10.3390/v11111040 (2019).
Gao, X. et al. Sequence characteristics and phylogenetic analysis of H9N2 subtype avian influenza A viruses detected from poultry and the environment in China, 2018. PeerJ 9, e12512 (2021).
Chen, X. et al. Host immune response to influenza A virus infection. Front. Immunol. 9, 320 (2018).
Sutton, T. C. et al. In vitro neutralization is not predictive of prophylactic efficacy of broadly neutralizing monoclonal antibodies CR6261 and CR9114 against lethal H2 influenza virus challenge in mice. J. Virol. 91, https://doi.org/10.1128/jvi.01603-17 (2017).
de Vries, R. D., Hoschler, K. & Rimmelzwaan, G. F. ADCC: an underappreciated correlate of cross-protection against influenza?. Front. Immunol. 14, 1130725 (2023).
Underwood, W. & Anthony, R. J. R. O. M. AVMA guidelines for the euthanasia of animals: 2020 edition. (2020).
Jacobs, L., Bourassa, D. V., Harris, C. E. & Buhr, R. J. Euthanasia: manual versus mechanical cervical dislocation for broilers. Animals 9, https://doi.org/10.3390/ani9020047 (2019).
Ripplinger, E. N. et al. Efficacy of a novel cervical dislocation tool for humane euthanasia of broilers and broiler breeders. Poult. Sci. 103, 103449 (2024).
Wang, Z. et al. Enhanced cross-lineage protection induced by recombinant H9N2 avian influenza virus inactivated vaccine. Vaccine 37, 1736–1742 (2019).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–w296 (2021).
Steel, J. et al. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. mBio 1, https://doi.org/10.1128/mBio.00018-10 (2010).
Miklos, A. E. et al. Structure-based design of supercharged, highly thermoresistant antibodies. Chem. Biol. 19, 449–455 (2012).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Buldun, C. M., Khairil Anuar, I. N. A. & Howarth, M. SnoopLigase-mediated peptide-peptide conjugation and purification. Methods Mol. Biol. 2208, 13–31 (2021).
Acknowledgements
This research was funded by the National Key R&D Project (2022YFD1801001-4), the Jiangsu Provincial Key R&D Project (BE2022329), the “Jie Bang Gua Shuai” Project at Yangzhou University (YZUXK202316), the Agricultural Science and Technology Independent Innovation Fund of Jiangsu Province (CX(23)3071), the Outstanding Technological Innovation Team of College and University in Jiangsu Province ([2021] No.1), 111 Project (D18007), and the Priority Academic Program Development of Jiangsu Higher Education (PAPD).
Author information
Authors and Affiliations
Contributions
S.C., D.P., and K.Q. conceived and designed the experiments. K.Q., N.Z., M.L., and Y.L. performed the experiments. K.Q., N.Z., Q.H., and T.Q. analyzed the data. N.Z., Y.L., and M.S. contributed reagents/materials/analysis tools. S.C., K.Q., and D.P. wrote the paper. X.L., T.Q., and D.P. offered suggestions and performed some of the experiments. All authors reviewed the manuscript. No potential conflict of interest was reported by the author(s).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Quan, K., Zhang, N., Lin, M. et al. Development of a broad-spectrum subunit vaccine against H9N2 avian influenza using HA stem domain scaffold and snoopligase system. npj Vaccines 10, 136 (2025). https://doi.org/10.1038/s41541-025-01191-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41541-025-01191-0
