
Abstract
The ability of influenza virus to undergo rapid antigenic shift to elude humoral immunity highlights the need for effective broad-spectrum influenza antivirals for treatment, prophylaxis and pandemic preparedness. Strategies providing durable, universal influenza protection in healthy and high-risk populations are urgently needed. Here we describe the design and preclinical characterization of CD388, a first-in-class antiviral drug–Fc conjugate (DFC), in mice and cynomolgus macaques. CD388 comprises a multivalent conjugate of the influenza virus neuraminidase inhibitor zanamivir, linked to a CH1–Fc hybrid domain of human IgG1 engineered for extended half-life. CD388 improves the antiviral activity of zanamivir, demonstrating potent, universal activity across influenza A and B viruses, including high pathogenicity and neuraminidase inhibitor resistant strains, a low potential for resistance development and potent efficacy in lethal mouse infection models. These results suggest that CD388 has the potential for universal prevention of influenza A and B in healthy and high-risk populations.
Similar content being viewed by others


Main
Two major surface glycoproteins, haemagglutinin (HA) and neuraminidase (NA), are targets for current influenza vaccines and antiviral therapies. HA binds sialic acids for viral entry and fusion, while NA cleaves sialic acids for viral egress1,2. Current vaccines primarily prevent infection by inducing antibodies against the immunodominant HA head domain3. However, pressure from the immune system, error-prone genome replication that results in antigenic drift of the virus4 and the use of strain-specific vaccines necessitate annual vaccination with multivalent vaccines.
Antibodies raised against the conserved but less immunogenic HA stalk domain provide coverage across influenza A virus that encompasses group 1 and, in some cases group 2 HAs. However, most HA antibodies lack influenza B coverage5,6. Only one HA stalk antibody has been reported that did not neutralize influenza B in vitro but demonstrated protective efficacy in mice7. To achieve broader coverage for prevention and treatment of influenza, NA has long been recognized as an attractive target8,9. NA plays two key roles in establishing influenza infection: releasing nascent virus particles from infected cells and clearing decoy sialic acids in mucin lining the respiratory tract8. NA active sites are highly conserved across all influenza A viruses and both influenza B lineages10. Genetic drift in NA is generally slower and discordant with that of HA11,12. These attributes have spurred efforts to discover NA-targeting vaccines and monoclonal antibodies (mAbs) with greater breadth of coverage than therapies targeting HA8,13. Indeed, the high degree of active site conservation and unique active site architecture of influenza NA compared with human sialidases14 have been successfully leveraged to develop selective small-molecule influenza neuraminidase inhibitors (NAIs) with broad-spectrum viral coverage that have become mainstays of influenza treatment for the past 20 years15.
Globally, three NAIs are licensed for influenza treatment: zanamivir (ZAN, Relenza)16, oseltamivir (OST, Tamiflu)17 and peramivir (Rapivab)18. ZAN, the first-in-class, was designed on the basis of the NA transition state analogue 2,3-dehydro-2-deoxy-N-acetylneuraminic acid decorated with a guanidine group at the C4 position to fill a negatively charged active site subpocket. ZAN demonstrated best-in-class potency against influenza A and B neuraminidases but poor oral absorption and rapid renal clearance following intravenous (i.v.) administration, necessitating dosing by buccal inhalation, increasing risk of bronchospasms in patients on ventilators or with asthma or chronic obstructive pulmonary disease19. Despite ZAN’s poor pharmacological properties, a high-dose twice-daily i.v. formulation of ZAN has been developed to treat patients with severe influenza on a compassionate-use basis20. OST was designed to improve oral absorption and drug-like properties17. As the only oral NAI, OST dominates uncomplicated influenza treatment. However, it is the least potent of the approved NAIs, with weaker influenza B activity and is most susceptible to escape mutations15. Unlike ZAN, OST induces a conformational change to accommodate a bulky lipophilic pentyl ether substituent21. Viral mutants that impede this conformational change confer resistance to OST and have emerged in the clinic with relatively high frequency15,22. As a transition state analogue, ZAN binds without inducing a conformational change in NA. Hence, most OST-resistant mutants have low cross-resistance to ZAN, and the incidence of ZAN resistance in the clinic is exceedingly rare15. Recently, baloxavir marboxil (BXM, Xofluza), a cap-dependent endonuclease inhibitor, was approved for influenza treatment. BXM exhibits modestly improved efficacy compared with OST, but treatment-emergent variants with reduced susceptibility to BXM were identified during clinical trials23.
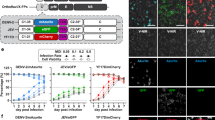
To enhance the best-in-class activity of ZAN and improve its pharmacological attributes, we developed a drug–Fc conjugate (DFC), represented by CD388, comprising a stable multivalent conjugate of a ZAN dimer with an N-terminal extended Fc domain of human IgG1 (Fig. 1a), engineered to prolong half-life. The homotetrameric structure of NA (Extended Data Fig. 1) and dense clustering of NA ‘islands’ on the influenza viral envelope24,25 make NA an ideal target for multivalent NAIs that crosslink NA active sites from different monomers.

a, Molecular surface representation of the full-length hIgG1 (PDB code 1HZH) structure redacted to show only the Fc construct boundaries used in CD388. Positions of lysine residues that were preferentially conjugated in CD388 are shown in green (based on proteolytic digestion and peptide mapping using a conjugate with wild-type hIgG1; Methods). CD388 is a multivalent conjugate of a ZAN dimer (shown in blowup) stably conjugated to lysines on the N-terminal extended hIgG1 Fc with an average drug:antibody ratio (DAR) of 4.5:1. The spatial disposition of conjugated lysine residues on the Fc domain surface and the length and flexibility of the PEG linkers result in ZAN dimer constellations on CD388 where sufficient separation (>90 Å) exists between ZAN dimers to allow simultaneous engagement of a single CD388 molecule with multiple active sites within an NA tetramer, between neighbouring NA tetramers on the same virus particle, or between NA tetramers on different virus particles. b–f, EC50 values [nM] of CD388, OST, ZAN or BXA against a panel of influenza strains A/H1N1 (b), A/H3N2 (c), B in a cell-based cytopathic effect assay (d), and A/H5N1 (e) and A/H7N9 (f) in a cell-based microneutralization assay. In b–f, data points in box and whiskers plots represent mean EC50 values for different influenza strains derived from dose-response curves measured in duplicate. The boxes bound the 25th and 75th percentile, with median values shown in the boxes. Whiskers highlight strains with the highest and lowest EC50s.
Previous research demonstrated that dimeric compositions of ZAN using linkers that optimally space the ZAN monomers by 18–22 Å exhibited enhanced antiviral activity compared with monomeric ZAN in cell-based assays and mouse infection models; antiviral activity is enhanced by promoting aggregation of virus via ‘head-to-head’ cross-linking of isolated NA tetramers on different virions26,27. Tetrameric ZAN compositions with larger spacing between the ZAN moieties, sufficient to bridge all four NA monomers within an NA tetramer, demonstrated substantial improvements over monomeric ZAN in binding affinity to NA and drove enhancements in antiviral activity28. However, in both instances mentioned above, the inhibitor candidates did not address the pharmacological limitations of ZAN to justify advancement to the clinic.
CD388 contains chemically and metabolically stable ZAN dimers conjugated to an N-terminally extended hIgG1 Fc domain. The Fc domain has two modifications: a C220S mutation near the N terminus to reduce the propensity for disulfide-bond-mediated aggregation and the M252Y/S254T/T256E (YTE) triple mutation to increase half-life29. The ZAN dimers are symmetrically fused through N-methyl carbamate moieties on the C7-hydroxyl of ZAN to a flexible 15-atom central-linker that separates the ZAN monomers by ~18 Å. The carbamate linkage to ZAN was N-methylated to prevent spontaneous rearrangement of the carbamate from the secondary hydroxyl on C7 to the primary hydroxyl at the C9 position. Multiple ZAN dimers are heterogeneously conjugated to surface-exposed lysine residues on the Fc domain through a 32-atom flexible polyethylene glycol (PEG)-based linker, which projects them out from the Fc surface by 35–40 Å when the linker is fully extended. Specific solvent-exposed lysine residues were preferred sites for conjugation (Fig. 1a), and the average ratio of ZAN dimers to Fc in CD388 of 4.5:1 provides potent antiviral activity and favourable solution properties. Syntheses of the ZAN dimer and final conjugate are described in Methods. The spatial distribution of preferred sites for conjugation coupled with the length and flexibility of the cross-linker allows CD388 to simultaneously interact with multiple NA active sites within a tetramer (~45 Å or 70 Å) (Extended Data Fig. 1), which has previously been shown to improve potency for tetrameric ZAN28. In addition, the spacing of ZAN dimers allows bridging of NA active sites across neighbouring NA tetramers on the same virion (25–30 Å) or across two virions (16 Å)26. Thus, CD388 has the potential for multivalent engagement of NA to sterically interfere with NA–host cell interactions and promote viral aggregation.
The unique attributes of CD388 conferred potent universal activity against a panel of over 50 influenza A and B viruses superior to OST or ZAN in cytopathic effect (CPE) assays, with retention of activity against OST- and ZAN-resistant influenza strains and a high barrier to resistance development in serial passage studies. The engineered Fc domain extended the circulating half-life in cynomolgus macaques and demonstrated robust distribution to the lungs and the epithelial lining fluid (ELF) in mice. These features delivered potent efficacy in lethal-challenge treatment and prophylaxis mouse models. CD388, currently in clinical development for seasonal prevention of influenza (NCT05285137, NCT05523089, NCT05619536 and NCT06609460), is a therapeutic modality with the potential to address the longstanding unmet need for durable universal treatment and long-term prevention of both seasonal and pandemic influenza.
Results
Universal in vitro activity of CD388 against influenza
We investigated CD388’s intrinsic target-based activity against viral NA in an NA inhibition assay. CD388 showed potent activity against all influenza A and B strains tested. It had uniform potent median half-maximal inhibitory concentrations (IC50s) of 1.29 nM against subtype A/H1N1, 2.24 nM against subtype A/H3N2 and 2.37 nM against influenza B (Supplementary Data Table 3). ZAN and OST were also highly potent against H1N1 and H3N2 strains, and in many cases demonstrated even higher sub-nM IC50s against H1N1 and H3N2 strains. Differences between CD388 and OST and ZAN were probably due to target binding interference between the linker at the C7 position of ZAN in CD388 (Fig. 1a) that was not fully compensated for by avidity in the multivalent conjugate. Overall, OST demonstrated weaker activity against influenza B strains than CD388 and ZAN (Supplementary Data Table 3). CD388 demonstrated potent activity against recombinant NA derived from highly pathogenic avian influenza (HPAI) viruses A/H5N1 and A/H7N9, with an IC50 of 3.72 nM and a median IC50 of 1.23 nM, respectively (Extended Data Table 1). OST and ZAN showed high potency against recombinant NA from A/H5N1 and A/Anhui/1/2013 (H7N9), but lost activity against NA from A/Shanghai/1/2013 (H7N9) with IC50 values of >1 µM and >100 nM, respectively (Extended Data Table 1).
CD388 showed potent activity against a wide range of influenza A and B viruses in cell-based CPE assays, with median half-maximal effective concentrations (EC50s) of 0.80 nM against subtype A/H1N1, 1.27 nM against subtype A/H3N2 and 1.72 nM against influenza B (Fig. 1b–d and Supplementary Data Table 4). Remarkably, the median EC50s for CD388 were over 2 logs lower than those for OST and ZAN against the same strains. Similar enhancements in antiviral activity with multimeric NAI compositions compared with monomers have been observed previously, attributed to inhibitor-mediated viral aggregation and increased viral immobilization on infected cells as confirmed by electron miscroscopy26,27. The increased activity observed with CD388 could also be attributed to the inhibition of NA activity via steric hindrance, akin to the effects noted with specific non-catalytic site antibodies30. CD388 showed improved activity against HPAI strains in cell-based microneutralization assays compared with OST and ZAN, with median EC50 values of 0.57 nM for H5N1 strains and 0.04 nM for an A/H7N9 strain (Fig. 1e,f and Extended Data Table 2). CD388 also demonstrated comparable activity to baloxavir acid (BXA, the active moiety of BXM) against most influenza A and B strains tested (Fig. 1b–d and Supplementary Data Table 4). BXA was exceptionally potent against HPAI strains, with EC50 values below 0.03 nM (Fig. 1e,f and Extended Data Table 2). In multiple human cell lines and Madin–Darby canine kidney cells transfected with human 2,6-sialtransferase (MDCK-SIAT1 cells), CD388 had a half-maximal cytotoxic concentration (CC50) of >10,000 nM (Supplementary Data Tables 5 and 6), resulting in a >1,000-fold selectivity index (SI) for CD388 (Supplementary Data Table 7). Collectively, these data demonstrate that CD388 is selective and has enhanced activity and spectrum compared with small-molecule NAIs in cell-based assays.
CD388 is efficacious in mouse treatment models
The efficacy of CD388 in lethal mouse infection models was evaluated. CD388 administered as a single intramuscular (i.m.) dose at 2 h post challenge was fully protective at 0.3 mg kg−1 against a lethal challenge with mouse-adapted influenza A/Puerto Rico/8/1934 (H1N1) (Fig. 2a). Unconjugated Fc or phosphate-buffered saline (PBS) vehicle controls succumbed to infection at similar times post challenge. Only limited transient body weight loss occurred after CD388 treatment at the minimal protective dose (0.3 mg kg−1) (Fig. 2b). CD388 reduced plaque-forming units (p.f.u.s) g−1 in lung tissue in a dose-dependent manner, from 0.83 logs at 0.03 mg kg−1 to below the limit of detection at 3 mg kg−1 (Fig. 2c). It also reduced lung pro-inflammatory cytokines IL-6 and MCP-1 dose-dependently, with levels declining to those in uninfected control mice at 3 mg kg−1 (Fig. 2d,e). Interestingly, OST administered twice a day (b.i.d.) for 5 days at the human equivalent dose (5 mg kg−1) was not protective, although it delayed time of death (Fig. 2f). OST at 10× the human equivalent dose (50 mg kg−1) resulted in 80% survival (Fig. 2f) but with substantial body weight loss (Fig. 2g). A dose response in viral burden reduction was not observed with OST at 5 mg kg−1 versus 50 mg kg−1 (b.i.d. for 4 days) and resulted in minimal, non-dose-dependent viral load reductions (Fig. 2h). However, OST at both doses significantly reduced lung IL-6 and MCP-1 cytokine levels (Fig. 2i,j). In conclusion, CD388 demonstrated significantly improved efficacy compared with OST in this model.

a–e, Efficacy of CD388 dose response on survival (a) and % body weight change (b), and the effect of CD388 on lung viral burden (c) (P values from left to right: >0.05, 0.0014, 0.0004, 0.0007, 0.0002, 0.0002) and pro-inflammatory lung cytokine levels of IL-6. bld, below levels that are detectable. (d) (P values from left to right: >0.05, <0.0001 for remaining columns) and MCP-1 (e) (P values from left to right: >0.05, <0.0001 for remaining columns) at 4 days post infection following lethal challenge with mouse-adapted influenza A/Puerto Rico/8/1934 (H1N1) in BALB/c mice. f–j, Efficacy of OST on survival (f) and % body weight change (g) in a lethal mouse model with mouse-adapted influenza A/Puerto Rico/8/1934 (H1N1) in BALB/c mice (n = 5 per group), and effect of OST on lung viral burden (h) (P values from left to right: 0.0041, >0.05) and pro-inflammatory lung cytokine levels of IL-6 (i) (P values from left to right: <0.0001 for all columns) and MCP-1 (j) (P values from left to right: <0.0001 for all columns) at 4 days post infection. For all viral burden and cytokine analyses, plots show mean ± s.d.; n = 5 per group. k–m, Efficacy of CD388 in lethal mouse models of A/Hong Kong/1/1968 (H3N2) (k), B/Florida/4/2006 (Yamagata) (l) and B/Malaysia/2506/2004 (Victoria) (m). n, Efficacy of CD388 against mouse-adapted A/Puerto Rico/8/1934 (H1N1) in immune-compromised BALB/c SCID mice (n = 5 per group). Statistical analyses were performed using one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test (**P < 0.01, ***P < 0.001; NS, not statistically significant) in GraphPad Prism v.9. Data distribution was assumed to be normal, but this was not formally tested.
The in vitro activity and spectrum of CD388 translated to efficacy against representative A/H1N1, A/H3N2, influenza B Victoria and Yamagata lineage viruses in lethal-challenge models. Full protection was achieved with single i.m. doses of 0.3 mg kg−1 against A/Hong Kong/1/1968 (H3N2), B/Florida/4/2006 (Yamagata) and B/Malaysia/2506/2004 (Victoria) (Fig. 2k–m and Extended Data Fig. 2a–c), while single i.m. doses of 1 mg kg−1 or lower were fully protective against A/WSN/1933 (H1N1), A/California/07/2009 (H1N1)pdm, A/California/12/2012 (H1N1)pdm09, A/North Carolina/04/2014 (H1N1)pdm09, A/Hawaii/70/2019 (H1N1)pdm09 and B/Colorado/6/2017 (Victoria) (Extended Data Fig. 2d–i and Table 3). Similar to the results observed with A/Puerto Rico/8/1934 (H1N1), CD388 treatment at the minimal protective dose corresponded with nominal transient body weight loss with the other strains tested (Extended Data Fig. 2). To assess the potential of CD388 to prevent infections in high-risk groups including the elderly and immune compromised, CD388 efficacy was evaluated in severe combined immune-deficient (SCID) mice, where CD388 provided full protection against A/Puerto Rico/8/1934 (H1N1) with a single 1 mg kg−1 dose, while OST and BXM were ineffective or only partially protective, respectively (Fig. 2n). Taken together, these data highlight the potential of CD388 to provide universal influenza protection in healthy and high-risk populations with similar doses.
CD388 provides long-acting protection in prophylaxis models
Pharmacokinetic (PK) profiling and prophylactic efficacy studies demonstrated CD388’s potential as a durable long-acting agent for universal influenza prevention. CD388’s half-life in mice and cynomolgus macaques was 106 h and 364 h, respectively, after a single i.v. dose (Supplementary Data Table 8). As expected, the engineered YTE-Fc used in CD388 demonstrated improved binding compared with unconjugated human wild-type (WT) Fc to human FcRn (in a pH-dependent manner) by biolayer interferometry (BLI) (Supplementary Data Table 9). CD388 exhibited dose-linear PK in mice across a wide (0.3–30 mg kg−1) i.m. dose range (Fig. 3a and Supplementary Data Table 10). Intact CD388 was stable in vivo, as measured by comparing exposures using two different ELISA capture methods: one that measured Fc levels with two distinct human Fc-capture antibodies, and another that measured intact CD388 using recombinant viral NA for CD388 capture and an anti-human Fc-capture antibody for detection. CD388 plasma levels were equivalent between methods, indicating that it remains intact in circulation. CD388 dosed i.m. at 10 mg kg−1 rapidly distributed to blood, extracellular body water and ELF, maintaining a consistent ratio to plasma (~34% of plasma levels on average) over time (Fig. 3b). To assess its efficacy in preventing influenza, mice were administered CD388 7 days before infection. Serum concentrations of ~1 µg ml−1 at time of infection, achieved from 1 mg kg−1 i.m. doses (Fig. 3a), provided complete protection against various mouse-virulent influenza strains, including influenza A/Puerto Rico/8/1934 (H1N1), A/Hong Kong/1/1968 (H3N2), B/Florida/4/2006 (Yamagata), B/Malaysia/2506/2004 (Victoria) (Fig. 3c–f, and Extended Data Fig. 3a–d and Table 3) and numerous other strains (Extended Data Fig. 4). The combination of low serum levels necessary for protection in preclinical lethal-challenge models, long half-life and the capacity to remain stable in high-concentration pharmaceutically acceptable formulations (≥150 mg ml−1) (Methods) underscores the potential of CD388 as a therapeutic option for the universal prevention of both seasonal and pandemic influenza.

a, Plasma levels of CD388 at doses of 0.3–30 mg kg−1 following i.m. administration in mice (mean; n = 2 per group) by NA capture. b, CD388 levels in plasma and lung epithelial lining fluid (ELF) of mice after i.m. dosing (mean ± s.d.; n = 6 per group) by NA capture. c–f, Protection provided by CD388 dosed as indicated i.m. 7 days before lethal challenge against influenza A/Puerto Rico/8/1934 (H1N1) (c), A/Hong Kong/1/1968 (H3N2) (d), B/Florida/4/2006 (Yamagata) (e) and B/Malaysia/2506/2004 (Victoria) (f) in BALB/c mice (n = 5 per group).
CD388 is active against NAI-resistant variants
The multivalent presentation of ZAN dimers in CD388 prevented loss of activity against viruses resistant to small-molecule NAIs. CD388 retained potent in vitro activity against the CDC NA inhibitor susceptibility reference virus panels (v.2.0 and 3.0), comprising clinically relevant variants with reduced or highly reduced activity to NAIs, including H275Y (A/H1N1), E119V (A/H3N2), D197E (B), D198N (B), R152K (B), H134N (B) and the clinical isolate R292K (A/H3N2). While OST and ZAN lost >100-fold potency against some highly resistant variants (Extended Data Table 4), CD388’s IC50 values were only marginally shifted against all tested resistant variants compared to parental strains. The efficacies of CD388 and OST were evaluated against the OST-resistant H275Y NA variant in lethal mouse infection models. CD388 retained a minimal protective dose of 0.3 mg kg−1, similar to protective dose levels against NA-sensitive viruses (Extended Data Table 3). OST dosed at the human equivalent dose of 5 mg kg−1 b.i.d. for 5 days did not confer survival benefit (Fig. 4a). CD388 and ZAN were compared in lethal-challenge models with paired ZAN-sensitive and ZAN-resistant NA H134N strains. CD388 demonstrated identical minimal protective doses of 0.3 mg kg−1 against both strains (Fig. 4b,c), while the minimal protective dose of ZAN shifted from 1 mg kg−1 against the ZAN-sensitive virus to 10 mg kg−1 against the ZAN-resistant variant (Fig. 4d,e). The retention of potency observed with CD388 against clinically observed NA mutants with reduced susceptibility to other approved small-molecule NAIs underscores CD388’s potential durability as a preventative agent against seasonal and pandemic influenza.

a–c, Efficacy of CD388 administered i.m. at the indicated doses at 2 h post infection against influenza A/Texas/23/2012 (H1N1)pdm09 NA H275Y variant (a), ZAN-susceptible influenza B/Laos/0080/2016 (Victoria) (b) and ZAN-resistant B/Laos/0654/2016 NA H134N variant (Victoria) (c) in BALB/c mice (n = 5 per group). d,e, Comparisons of dose responses of ZAN administered intranasally starting at 2 h post infection for 5 days and CD388 administered i.m. once at 2 h post infection at 0.3 mg kg−1 to ZAN-susceptible influenza B/Laos/0080/2016 (d) and ZAN-resistant B/Laos/0654/2016 NA H134N (e) variants in BALB/c mice (n = 5 per group).
CD388 shows low potential for development of resistant strains
To assess the potential of influenza resistance development against CD388, in vitro serial passage experiments in MDCK cells infected with influenza A/H1N1 (n = 2), A/H3N2 (n = 2) and B (n = 2) viruses were conducted using static sub-inhibitory and incremental dose-escalation methodologies. Using both methodologies, minimal susceptibility changes were observed in all strains after 10 rounds of passaging compared to their isogenic parental strains. Genotypic analyses identified a single NA residue (S247 or A246 using N1 or N2 numbering, respectively) substituted to arginine or valine following prolonged exposure to CD388. This residue is near the CD388 binding site, and mutations were found in representative strains of A/H1N1 (S247R; S231R based on A/WSN/1933 strain numbering, derived from static methods) and A/H3N2 (A246V, A/Victoria/3/75, derived from escalating-dose methods). These amino acid substitutions have been documented previously31,32 and occur at very low frequencies in clinical isolates.
We assessed the cross-resistance of the identified S247R and A246V NA variants in a cell-based plaque reduction assay (PRA). CD388 lost less than 3-fold activity in PRA, demonstrating equivalent or higher potency compared with other NAIs (Fig. 5a,b and Supplementary Data Table 11). Importantly, the EC50 values for CD388 against these variants were within the median EC50 ranges against susceptible subtypes.

a,b, Fold change of a plaque-purified clone isolated at passage 10 versus parental strains by EC50 [nM] to CD388-selected NA variants S231R (S247R) in A/WSN/1933 (H1N1) (a) or A246V in combination with HA N113H/T453I in A/Victoria/3/75 (H3N2) (b) for CD388, OST and ZAN in cell-based PRA. EC50 values were derived from 3 independent PRA replicates.
Discussion
Seasonal influenza persists, causing severe illness and mortality. The emergence of novel, highly pathogenic, zoonotic strains, particularly H5N1, pose significant concerns. After two seasons of being reduced by SARS-CoV-2 pandemic precautions, influenza infections rebounded and now continue at pre-pandemic levels. From 1 October 2023 to 18 May 2024, the CDC estimates 35–64 million influenza infections in the United States, leading to 390,000–820,000 hospitalizations and 25,000–71,000 deaths33. Limited vaccination coverage in the United States (~50%), low vaccine efficacy (10–60% depending on season) and poor immune responses in immune-senescent populations leave many vulnerable34. In addition, waning immunity reduces overall vaccine efficacy within a season35,36. Next-generation vaccines or novel therapeutic agents with broad coverage are urgently needed to reduce severe cases, hospitalizations, deaths and economic burden37.
In recent years, mAbs have emerged as promising strategies for treating and preventing viral diseases, including influenza38. Antibodies raised against HA and NA have been shown to correlate with protection39,40. However, most efforts to develop mAbs against influenza have focused on the conserved stalk domain of HA. No HA antibodies have advanced clinically, as highlighted by disappointing results in a recent clinical trial41, and none provide coverage against influenza B. Influenza B causes, on average, 25% of influenza infections42 and spiked to 60% in the early 2019–2020 season in the United States43, underscoring the need for universal protection against influenza A and B.
NA has long been recognized as an attractive alternative target for anti-influenza therapies with broader coverage2,9. A recent human challenge study demonstrated a superior correlation between reduced disease severity and high anti-NA titres compared with high anti-HA titres44. Anti-NA mAbs isolated from patients infected with influenza have broader coverage than anti-HA mAbs with fewer spectrum gaps13,45,46,47. However, targeting the strictly conserved NA active site with small molecules has achieved the best success in expanding coverage. CD388 enhances the exceptional potency and broad-spectrum activity of ZAN, combining it with the duration of action of a long-acting mAb therapeutic. Furthermore, CD388 breadth of coverage encompasses clinically observed NAI-resistant influenza variants and demonstrated low potential for resistance in serial passage studies. The low predisposition of influenza A and B to develop reduced susceptibility to CD388, the lack of novel mutations recovered and equivalent or higher levels of reduced susceptibility of approved NAIs to the selected resistance substitutions at NA residue A246/S247, suggest a low potential for clinical resistance, potentially outperforming that of ZAN. Of note, the current serial passage studies were not specifically designed to identify non-plaque-forming strains that may be associated with reduced susceptibility. Genotypic screening will be conducted in ongoing clinical trials to detect any strains that may emerge with reduced susceptibility to CD388.
A preclinical evaluation of the potential immunogenicity of CD388 in human participants was not conducted because human proteins are well known to generate anti-drug antibodies (ADAs) in preclinical species48. However, the immunogenicity potential for CD388 has been evaluated in the clinic and will be reported in due course.
The combined attributes of CD388, including its small size (~65 kDa) and multivalent ZAN presentation, resulted in superior distribution to the lung compared with mAbs49, dose-dependent reductions in lung viral burden and robust activity in treatment and prophylactic efficacy models. Importantly, CD388 efficacy was retained in an immune-compromised mouse model, suggesting that efficacy is driven by the intrinsic antiviral activity of CD388, with minimal reliance on Fc-mediated immune effector function. Taken together, the preclinical data reported here highlight the potential for the use of CD388 as a universal long-acting therapeutic for the treatment and prevention of seasonal and pandemic influenza in both healthy and high-risk individuals. CD388 has advanced to clinical development, where it is being evaluated in a Phase 2b study for universal prevention of influenza during the 2024–2025 flu season in the Northern Hemisphere (NCT06609460).
Methods
Mice and macaques
In all studies, animals were housed and cared for in accordance with local, state, federal and institute policies in facilities accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International, under standards established in the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals. Mouse studies were performed at Explora BioLabs and approved by the Institutional Animal Care and Use Committee (IACUC) of Explora BioLabs (Protocol EB17-033-00). Mice were monitored for physical health, food consumption and body weight. All mice were euthanized at the end of efficacy experiments or after PK study bleeds in compliance with the American Veterinary Medical Association Guidelines for the Euthanasia of Animals 2020 edition, using CO2 exposure and cervical dislocation. The cynomolgus macaque study was performed by BTS Research and approved by the IACUC of BTS Research (Protocol 20P-CDR-001). Healthy female Chinese-origin macaques (Macaca fascicularis) were individually housed and monitored for physical health, food consumption and body weight.
Mouse and primate pharmacokinetic assays
CD388 PK experiments were conducted using i.v. bolus administration in BALB/c mice (N = 3 female mice, 6–8 weeks old, ~0.02 kg each; 3 mg kg−1 of 0.6 mg ml−1 dosing solution in PBS; PK sampling from 0.25 to 168 h post dose) or macaques (N = 3 females, 7–12 years old, ~4 kg each; 10 mg kg−1 of 20 mg ml−1 dosing solution in PBS; PK sampling from 0.25 to 1,334 h post dose).
Levels of CD388 in serum and in vivo stability of the intact conjugate were evaluated by comparing concentrations at matched timepoints measured using two sandwich ELISA assays. Nunc MaxiSorp 96-well plates were coated with 0.1 U per well NA from A/California/04/2009 (H1N1) (Sino Biological) for NA capture in 1× KPL coating buffer, or with 0.1 µg per well of mouse anti-human IgG (CH2 domain) clone R10Z8E9 (BioRad) for Fc capture in carbonate buffer. Plates were incubated at room temperature (r.t.) for 1 h on an orbital plate shaker at 500 r.p.m. for NA capture, or stationary at 4 °C overnight for Fc capture. Plates were washed 5× with PBST and blocked with 1% bovine serum albumin (BSA, MilliporeSigma) for NA capture or 5% non-fat dry milk in PBST (Cell Signalling) for Fc capture for 1 h at r.t. with shaking. NA sample diluent was PBS with 0.5% BSA and 0.025% Tween 20. Fc diluent was PBS with 2.5% non-fat dry milk and 0.025% Tween 20. Duplicate serial dilutions of the plasma samples were plated at 100 µl per well and incubated at r.t. for 2 h. Sample diluent was prepared with species-matched plasma (cynomolgus macaque at final dilution of 1:2,500; mouse at final dilution of 1:100). CD388 standard curves ranged from 0.230 to 500 ng ml−1 for NA capture and 0.03–55 ng ml−1 for Fc capture. Curves were run on each plate in duplicate. Following the 2 h incubation, plates were washed 5× with 300 µl PBST per well. Conjugate bound to NA or anti-Fc antibody on the plates was then probed with an HRP-conjugated anti-human IgG Fc F(ab’)2 (Jackson ImmunoResearch) diluted 1:2,000 in sample diluent for 1 h at r.t. with shaking. Plates were then washed 8× with 300 µl PBST per well and developed with 100 µl TMB substrate per well for 7–8 min. The reaction was stopped with 100 µl 1 N H2SO4 per well. Absorbance was read at 450 nm with an EnSpire multimode plate reader. Plasma samples were interpolated following nonlinear regression analysis (Sigmoidal, 4PL analysis) of the standard curves in GraphPad Prism v.8. Plasma concentrations measured by ELISA were used to calculate pharmacokinetic parameters by non-compartmental analysis using Phoenix WinNonlin (v.6.4, Pharsight).
In vivo efficacy
Mouse efficacy studies utilized female BALB/c or BALB/c SCID mice (Jackson Laboratories, strain 000651 or 001803; 6–8 weeks old; n = 5 per group for all studies). No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications50. Animals were anaesthetized with ketamine/xylazine (100/10 mg kg−1, intraperitoneal) and challenged intranasally with 3× an inoculum that is lethal to 95% of the animals (LD95) of influenza A or B suspended in 30 µl PBS. After infection, mice were randomized into dosing groups. A single i.m. dose of CD388 was administered via the flank at a dose volume of 10 ml kg−1 either 2 h after or 7 days before viral challenge. The general health of animals was monitored and body weight recorded daily for up to 21 days after viral challenge. Moribund animals, or animals exhibiting 20% or more of body weight loss, were recorded as a mortality. Statistical significance was determined from survival graphs relative to vehicle using the log-rank (Mantel–Cox) test in GraphPad Prism v.6.0.
Lung burden and cytokine analyses
Female BALB/c mice (6–8 weeks old) were challenged intranasally with 3 × 102 p.f.u.s of mouse-adapted influenza A/Puerto Rico/8/1934 (H1N1). At 2 h post challenge, mice were administered either CD388 as a single i.m. dose (0.01–3 mg kg−1) or oral OST at the human equivalent dose of 5 mg kg−1 or 10× human equivalent dose (50 mg kg−1) b.i.d. for 4 days. At 4 days post infection, mice were killed and both lung lobes were collected. Lungs were homogenized with 1-mm silica beads in 1 ml PBS using a MagNA Lyser (Roche). Homogenization was carried out at 6,000 r.p.m. for 60 s, and homogenized lungs were chilled on ice for 5 min between runs. After lung homogenization, tubes were centrifuged for 10 min at 600 × g and supernatant was transferred into a new tube. For p.f.u. determination, supernatants of lung homogenate were diluted in infection buffer ranging from 10−1 to 10−6. P.f.u.s were calculated relative to the weight of the collected lung (p.f.u.s g−1 lung). For cytokine analyses, supernatants of lung homogenate were serially diluted 2-fold in 96-well plates. Cytokine levels for IL-6 and MCP-1 were determined by ELISA according to manufacturer instructions (R&D Systems).
Viral strains
All virus experiments were performed in approved biosafety level 2 (BSL 2) (Cidara, Explora) or biosafety level 3 (BSL 3) (Viroclinics) facilities. Influenza viruses were obtained through BEI Resources, NIAID, NIH, the International Reagent Resource, CDC’s WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza; and Microbiologics. Influenza strains A/WSN/1933 (H1N1) and B/Malaysia/2506/2004 (Victoria) were kindly provided by Dr Sumit Chanda (Scripps Research, CA). A complete list of viruses used herein is shown in Supplementary Data Table 1. Influenza strains were propagated in Madin–Darby Canine Kidney (MDCK) or MDCK-SIAT1 (Sigma, 05071502) at 37 °C and 5% CO2 until 80% of CPE. Cell-free supernatant was obtained by centrifugation at 500 × g for 5 min and virus was stored at −80 °C. P.f.u.s were determined by plaque assay. Briefly, 100 µl of virus dilutions were added to confluent monolayer of MDCK cells in 24-well plates and incubated for 1 h at room temperature with rocking every 15 min. After removing the virus, liquid overlay media containing Avicel were added to MDCK cells. Cells were incubated at 37 °C and 5% CO2 for 40–72 h. After incubation and media removal, cells were stained with crystal violet to enumerate plaques.
Recombinant neuraminidase
Recombinant NAs from cell lysates of A/H5N1 and AH7N9 viruses (Sino Biological) are summarized in Supplementary Data Table 2.
Neuraminidase inhibition
CD388, OST or ZAN were tested at 0.001–1,000 nM (25 µl per well) in 96-well plates (Corning). Influenza virus or NA from cell lysate (Sino Biological) was diluted to be within the linear range of detection. Virus and test article were incubated for 20 min before the addition of NA substrate at 37 °C and 5% CO2. NA inhibition activity was determined using the commercial NA-Fluor kit (Applied Biosystems) according to manufacturer instructions. Fluorescence was measured with the EnSpire multimode plate reader (PerkinElmer). IC50 values were calculated with GraphPad Prism v.8 using nonlinear regression analysis (dose-response inhibition).
Cytopathic effect assay
MDCK or MDCK-SIAT1 cells were seeded in 96-well plates at 1–2 × 104 cells per well in DMEM medium (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco). Cells were incubated for 18–24 h at 37 °C and 5% CO2. The next day, the medium of a confluent cell monolayer (>90%) was aspirated and washed with PBS. CD388, OST, ZAN or BXA were tested at concentrations ranging from 0.001 pM to 10,000 nM. Subsequently, cells were infected with influenza A or B virus in DMEM in the presence of 1 µg ml−1 TPCK-trypsin to yield >70% CPE in the untreated control. CPE was determined after fixation and staining with 0.1% crystal violet after 3 days for influenza A and after 5 days for influenza B. The absorbance was read at 595 nm on an EnSpire multimode plate reader. EC50 values were calculated with GraphPad Prism v.8 using nonlinear regression analysis (dose-response inhibition).
Plaque reduction assays
PRAs were performed on starting and final-passage viruses in MDCK or MDCK-SIAT1 cells for sub-inhibitory H3N2 passaged viruses. Cells were seeded in 24-well plates at 5 × 105 cells per well in 0.5 ml of DMEM containing 10% FBS and incubated for ~24 h at 37 °C and 5% CO2. CD388, OST and ZAN were diluted in infection buffer containing PBS, 0.35% BSA, 1.105 mM CaCl2 and 2.24 mM MgCl2. Test articles were pre-incubated with virus for 30 min at room temperature before addition to cell monolayers. The multiplicity of infection (MOI) for each drug–virus combination was selected to produce 30 plaques in the PBS control well. Adsorption was carried out for 1 h, the virus–test article mix was removed, and the infected cells were incubated for 48 h in the presence of test article diluted in a mixture of 1.25% Avicel, DMEM, 0.01% DEAE-dextran and 2 μg ml−1 TPCK-trypsin at 37 °C and 5% CO2. After 48 h, the Avicel mixture was then removed, and cells were fixed with paraformaldehyde and stained with 1% crystal violet to count the plaques. EC50 values were calculated using GraphPad Prism v.8.
Microneutralization
Microneutralization experiments were performed at Viroclinics at a BSL 3 facility, as previously described51. Briefly, a 2-fold dilution series of CD388, OST, ZAN or BXA was performed and mixed with virus, aiming for 50–100 p.f.u.s per well. The virus–test article mixture was immediately added to MDCK cells and incubated for 90 min at 37 °C and 5% CO2. After incubation, the inoculum was removed, cells were washed and replaced with medium containing the same dilutions of test articles. The plates were incubated for ~24 h at 37 °C and 5% CO2. After completion of the incubation period, the cells were formalin fixed and permeabilized with 70% ethanol. They were then incubated with a primary mouse antibody directed against the viral influenza A nucleoprotein (clone Hb65, EVL), followed by incubation with a secondary goat anti-mouse IgG HRP peroxidase conjugate. TrueBlue substrate was added, resulting in the development of a blue precipitate on virus-positive cells. Images of all wells were acquired by a CTL Immunospot analyser equipped with software to quantitate the virus-positive cells (=virus signal). Quantification was done using ‘well area covered’ (WAC) counts. The EC50 was calculated according to the method previously described52.
Sub-inhibitory static serial passage
Sub-inhibitory static serial passage studies were conducted in MDCK cells infected with influenza A/WSN/1933 (H1N1), A/Washington/01/2007 (H3N2) or B/Malaysia/2506/2004 (Victoria) at an MOI of 0.05 at 37 °C and 5% CO2 for 24 h. The concentrations for each test article were optimized to result in a ~2-log reduction in titre as compared with the vehicle control while maintaining sufficient virus for subsequent passages. CD388 was tested at 0.003125–0.25 nM, BXA at 4–20 nM and OST at 4–200 nM, or with PBS as a control. Following the addition of test articles, cells were incubated for 24 h. Next, viral supernatants were collected and the viral titre was determined by plaque assay. Freshly seeded cells were infected with viral supernatant adjusted to MOI 0.05. This process was repeated for 10 passages.
Dose-escalation serial passage
Dose-escalation serial passage studies were conducted in MDCK cells infected with influenza A/Hawaii/70/2019 (H1N1)pdm09, A/Victoria/3/75 (H3N2) or B/Phuket/3073/2013 (Victoria) at an MOI of 0.01–0.05 and incubated at 37 °C and 5% CO2 for 24 h (influenza A) or 48 h (influenza B). The concentrations for each test article were optimized to result in a ~2-log reduction in titre as compared with the vehicle control while maintaining sufficient virus for subsequent passages. CD388 was tested at 2–8 nM, BXA at 4–20 nM and OST at 4–200 nM. A PBS control was also included. Following the addition of test articles, cells were incubated for 24 h. After 24 h, viral supernatants were collected and the viral titres were determined by plaque assay. Freshly seeded cells were infected with viral supernatant adjusted to MOI 0.05. Each virus was serially passaged in the presence of 2-fold increasing concentrations of the test article until viruses could not be propagated further. The susceptibility of individual clones isolated from final passage was determined against test articles using a PRA.
Sequencing of viral RNA
Viral RNA was extracted from influenza strains using a QIAamp Viral RNA mini kit (QIAGEN). To prepare samples for whole genome sequencing (WGS), RNA was reverse transcribed and the entire genome of influenza was amplified in a single RT–PCR reaction using the Uni/Inf primer set53. PCR products were cleaned and concentrated using a DNA Clean and Concentrator-25 kit (Zymo Research). DNA concentrations were determined using a NanoDrop spectrophotometer (ThermoFisher) before WGS analysis. WGS was performed on individual starting and final-passage plaque-purified strains at BATJ. Library construction and quantification for each sample started with ~500 ng viral complementary (c)DNAs that were then enzymatically fragmented to ~200–500-bp pieces. NGS libraries were then constructed using the TruSeq Nano DNA library prep kit (Illumina), which included steps of end repair, library size selection, 3′-end adenylation, ligation of indexed adaptors and PCR enrichment. The constructed libraries were then quantified with a Qubit fluorometer (ThermoFisher). Sequencing was performed via MiSeq (Illumina). Libraries were diluted to 2 nM individually, then pooled for denaturation and further dilution to a final concentration of 10 pM. A MiSeq Reagent Nano Kit v2 (500 cycles) was then used for setting up the sequencing run (2 × 250 bp, paired-end; up to 2 × 1 million reads). A variant of the SPAdes genome assembly pipeline (St Petersburg State University, Center for Algorithmic Biotechnology) was used for assembling the fastQ raw sequencing data to contigs with optimized settings. WGS mutation analyses were performed at Cidara using Sequencher 5.4.6 (Gene Codes).
For NGS deep sequencing of passage 10 total population (TP) samples, samples were prepared in the same manner as those used in WGS (DNA samples were generated at Cidara and provided to BATJ for sequencing). Library construction and quantification were also similar to the WGS protocol except that ~1,000 ng viral cDNAs per sample were used in fragmentation. The MiSeq sequencing run followed the WGS protocol except that a MiSeq Reagent Kit v2 (500 cycles) was used for the sequencing run (2 × 250 bp, paired-end, generating up to 2 × 10 million reads). Alignment analysis utilized a variant of the breseq sequence alignment pipeline (Barrick Lab) for identification of mutations relative to the reference sequences. For metagenomic samples that contained a mixed population, the polymorphism mode (≥1% frequency threshold) was used to predict polymorphic (mixed) mutations. Final analyses (performed by Cidara) utilized GenBank sequences for alignment that extended beyond the reading frame in both directions; thus, clipping extraneous sequences and renumbering mutation positions were required.
Synthesis of ZAN dimer
The route for synthesis of the ZAN dimer is described in detail below.
Synthesis of intermediate 6

Synthesis of 9

Methyl 5-acetamido-7,8,9-tri-O-acetyl-2,6-anhydro-4-azido-3,4,5-trideoxy-d-glycero-d-galacto-non-2-enonate (10.0 g, 22 mmol) was dissolved in 60 ml dry methanol, then treated with sodium methoxide in methanol (8.8 ml of 0.5 M in methanol, 4.4 mmol) while cooling in an ice-water bath. Progress of the reaction was monitored by LC–MS, which was complete after 2 h. The pH of the reaction solution was then adjusted to a value of 5–6 by using Amberlite IRN-77 ion exchange resin. The mixture was filtered to remove the resin and evaporated to dryness under vacuum. The resulting oil was used in the next step without further purification. 1H-NMR (300 MHz, MeOD): δ 5.93 (d, J = 2.6 Hz, 1H), 4.39-4.09 (m, 3H), 3.92-3.82 (m, 2H), 3.80 (s, 3H), 3.71-3.58 (m, 2H), 2.04 (s, 3H). 13C-NMR (75 MHz, MeOD): δ 174.4, 163.7, 146.8, 108.4, 78.1, 71.1, 69.6, 64.8, 59.8, 53.0. 49.5, 22.7. HRMS (m/z): [M + H]+ calculated for C12H19N4O7, 331.1248; found, 331.1251.
Synthesis of 10

To a solution of crude 9 (0.22 mmol) in acetone (70 ml), we added 2,2-dimethoxypropane (30 ml) and p-toluenesulfonic acid monohydrate (400 mg, 2.0 mmol). The resulting solution was stirred at room temperature overnight, at which time, sodium bicarbonate (170 mg, 2.0 mmol) was added and the mixture was concentrated to dryness. The resulting material was used in the next step without further purification. 1H-NMR (300 MHz, MeOD): δ 5.93 (d, J = 2.5 Hz, 1H), 4.39-4.26 (m, 2H), 4.22-3.99 (m, 4H), 3.80 (s, 1H), 3.62 (dd, J = 1.0 Hz, 7.8 Hz, 1H), 3.35 (s, 3H), 2.03 (s, 3H), 1.37 (s, 3H), 1.33 (s, 3H). 13C-NMR (75 MHz, MeOD): δ 174.2, 163.5, 146.7, 110.3, 108.5, 78.4, 76.1, 70.5, 67.9, 59.6, 53.0, 49.5, 27.2, 25.6, 22.7. HRMS (m/z): [M + H]+ calculated for C15H23N4O7, 371.1561; found, 371.1557.
Synthesis of 11 and 12

To a solution of crude 10 (0.22 mmol) in methanol (60 ml), we added Lindlar catalyst (5.0 g). The resulting mixture was vacuum flushed with hydrogen every 30 min to remove nitrogen gas and stirred for a total 5 h. After complete reaction as determined by LC–MS, the catalyst was removed by filtration through celite. The filtrate was concentrated and used in the next step without further purification. 1H-NMR (300 MHz, MeOD): δ 5.95 (d, J = 2.5 Hz, 1H), 4.36-4.28 (m, 1H), 4.18-4.11 (m, 1H), 4.04-3.97 (m, 2H), 3.88 (t, J = 9.0 Hz, 1H), 3.78 (s, 3H), 3.62-3.56 (m, 2H), 2.06 (s, 3H), 1.37 (s, 3H), 1.33 (s, 3H). 13C-NMR (75 MHz, MeOD): δ 175.0, 164.2, 145.0, 113.9, 110.3, 78.6, 76.1, 71.2, 68.0, 52.7, 52.1, 50.4, 27.2, 25.6, 22.7. HRMS (m/z): [M + H]+ calculated for C15H25N2O7, 345.1656; found, 345.1653.
Crude 11 (0.22 mmol) in tetrahydrofuran (60 ml) was treated with N,N’-Di-Boc-1H-pyrazole-1-carboxamidine (9.30 g, 30.0 mmol) and N,N-diisopropylethylamine (DIEA) (9.9 ml, 57.0 mmol). The resulting solution was stirred at room temperature until guanylation was complete as determined by LC–MS (4 h). The solution was concentrated and purified by flash chromatography, eluting with 20% to 80% ethyl acetate/dichloromethane. Yield 8.8 g, 59% for four steps. 1H-NMR (300 MHz, CDCl3): δ 11.32 (s, 1H) 8.63 (d, J = 7.7 Hz, 1H), 8.04 (s, 1H), 5.74 (d, J = 1.9 Hz, 1H), 5.23-5.09 (m, 2H), 4.37-4.28 (m, 1H), 4.14-3.85 (m, 4H), 3.72 (s, 3H), 3.47-3.41 (m, 1H), 1.95 (s, 3H), 1.45 (s, 9H), 1.43 (s, 9H), 1.36 (s, 3H), 1.30 (s, 3H). 13C-NMR (75 MHz, CDCl3): δ 174.0, 161.9, 161.8, 157.4, 152.6, 146.8, 109.1, 106.7, 84.5, 80.4, 78.4, 74.0, 69.7, 67.4, 53.4, 51.8, 48.5, 28.2, 28.0, 27.1, 25.2, 22.9. HRMS (m/z): [M + H]+ calculated for C26H43N4O11, 587.2923; found, 587.2917.
Synthesis of 6

A reaction flask containing 12 (24.7 g, 42.14 mmol) dissolved in anhydrous dichloromethane (500 ml) was vacuum flushed with nitrogen. The solution was cooled in an ice-water bath, then treated with DIEA (22.1 ml, 126.42 mmol), 4-dimethylaminopyridine (5.15 g, 42.14 mmol), followed by 4-nitrophenylchloroformate (17.0 g, 84.27 mmol) in portions. The solution was stirred at 0 °C for 1 h, then allowed to warm to room temperature for 12 h. The reaction was concentrated and purified by flash chromatography, eluting with 5% to 50% ethyl acetate/dichloromethane. Yield 19.0 g, 60%. 1H-NMR (300 MHz, CDCl3): δ 11.32 (s, 1H), 8.50 (d, J = 6.8 Hz, 1H), 8.21 (d, J = 9.4 Hz, 2H), 7.49 (d, J = 9.4 Hz, 2H), 6.48 (d, J = 8.9 Hz, 1H), 5.86 (d, J = 2.1 Hz, 1H), 5.25-5.20 (m, 1H), 5.18-5.09 (m, 1H), 4.44-4.31 (m, 2H), 4.24-4.06 (m, 3H), 3.76 (s, 3H), 1.87 (s, 3H), 1.45-1.41 (m, 18H), 1.36 (s, 3H), 1.32 (s, 3H). 13C-NMR (75 MHz, CDCl3): δ 171.5, 162.7, 161.6, 157.2, 155.8, 152.7, 152.5, 145.4, 145.3, 125.2, 122.4, 109.2, 108.9, 84.0, 79.8, 77.5, 75.1, 74.2, 65.6, 52.5, 48.6, 48.5, 28.2, 28.0, 26.4, 25.5, 23.1. HRMS (m/z): [M + H]+ calculated for C33H46N5O15, 752.2985; found, 752.2983.
Synthesis of 8

Synthesis of 1

To a well-stirred solution of N-Boc-N-Me-glycinol (3.5 g, 20 mmol) in dimethylsulfoxide (20 ml) cooled with an ice-water bath, we added allyl bromide (3.6 g, 30.0 mmol), followed by finely ground KOH powder (3.5 g, 30.0 mmol) over 15 min. The resulting solution was stirred overnight at room temperature. The resulting mixture was partitioned between 5% aq. HOAc (50 ml) and ethyl acetate (200 ml). The organic layer was separated, washed with brine, dried with sodium sulfate, filtered and concentrated, then purified by flash chromatography, eluting with 10% to 80% ethyl acetate/hexane. Yield of product 4.1 g, 95%. 1H-NMR (300 MHz, MeOD): δ 5.96-5.83 (m, 1H), 5.29 (dq, J = 17.2, 1.8 Hz, 1H), 5.17 (d, J = 10.4 Hz, 1H), 3.98 (d, J = 5.9 Hz, 2H), 3.56 (t, J = 5.6 Hz, 2H), 3.40 (t, J = 5.5 Hz, 2H), 2.90 (s, 3H), 1.47 (s, 9H). 13C-NMR (75 MHz, MeOD): δ 157.7, 136.2, 117.0, 81.1, 72.9, 69.4, 36.1, 35.7, 28.9. HRMS (m/z): [M+Na]+ calculated for C11H21NO3, 238.1419; found, 238.1415.
Synthesis of 2

Ozone was bubbled through a solution of 1 (8.0 g, 37 mmol) in MeOH (50 ml) and dichloromethane (50 ml) at −78 °C until the appearance of a light blue colour. Unreacted ozone was removed by bubbling with oxygen for 10 min before the addition of NaBH4 (1.6 g, 40 mmol) in small portions over 10 min. After all NaBH4 was added, the mixture was gradually warmed to room temperature. The resulting solution was partitioned between ethyl acetate (100 ml) and brine (50 ml). The organic layer was separated, washed with brine, dried with sodium sulfate, filtered, concentrated to an oil and then purified by flash chromatography, eluting with 10% to 80% ethyl acetate/dichloromethane. Yield of product 5.0 g, 62%. 1H-NMR (300 MHz, MeOD): δ 3.70 (t, J = 4.7 Hz, 2H), 3.60-3.53 (m, 4H), 3.39 (t, J = 5.6 Hz, 2H), 2.87 (s, 3H), 2.47 (bs, 1H), 1.44 (s, 9H). 13C-NMR (75 MHz, MeOD): δ 156.2, 155.9, 79.7, 72.4, 69.5, 69.1, 61.8, 48.7, 48.2, 35.3, 28.5. HRMS (m/z): [M + H]+ calculated for C10H21NO4, 220.1549; found, 220.1543.
Synthesis of 3

To a solution of 2 (4.4 g, 20 mmol) and CBr4 (10.0 g, 30.0 mmol) in dichloromethane (50 ml) cooled in an ice bath, we added PPh3 (8.0 g, 30 mmol) slowly over 15 min (exothermic). During the course of the addition, the internal temperature was kept below 30 °C. After addition of PPh3, the reaction was stirred overnight at room temperature. The crude reaction was concentrated to an oil, then purified by normal phase chromatography, eluting with 10% ethyl acetate/hexanes to 80% ethyl acetate/hexanes. Fractions containing oil droplets on the inside of the collection tubes were checked by LC–MS, then combined and concentrated to a colourless oil. Yield 4.0 g, 70.5%. 1H-NMR (300 MHz, MeOD): δ 3.76 (t, J = 6.1 Hz, 2H), 3.60 (bs, 2H), 3.44 (t, J = 6.1 Hz, 2H), 3.90 (bs, 2H), 2.92 (s, 3H), 1.45 (s, 9H). 13C-NMR (75 MHz, MeOD): δ 155.9, 79.5, 70.9, 69.7, 48.8, 48.4, 35.9, 35.5, 30.6, 28.6. HRMS (m/z): [M + H]+ calculated for C10H20BrNO3, 282.0750; found, 282.0699.
Synthesis of 4

A solution of 3 (4.4 g, 15.5 mmol), propargyl-PEG4-amine (1.5 g, 6.4 mmol) and DIPEA (3.3 g, 25.8 mmol) in dimethylformamide (DMF) (20 ml) was heated in an oil bath at 75 °C for 18 h. The mixture was filtered, concentrated and purified by reverse phase liquid chromatography (RPLC) (5% acetonitrile/water to 100% acetonitrile). Yield 3.85 g, 92%. 1H-NMR (300 MHz, CDCl3): δ 4.17 (d, J = 2.5 Hz, 2H), 3.87-3.79 (m, 6H), 3.67-3.61 (m, 10H), 4.59 (d, J = 3.7 Hz, 2H), 3.55-3.43 (m, 10H), 3.35 (bs, 4H), 2.84 (s, 6H), 2.44 (t, J = 2.4 Hz, 1H), 1.42 (s, 18H). 13C-NMR (75 MHz, CDCl3): δ 156.0, 79.8, 79.7, 74.9, 70.7, 70.6, 70.5, 70.4, 70.3, 69.2, 69.1, 66.7, 66.5, 65.9, 65.7, 65.5, 58.5, 54.5, 54.1, 48.6, 48.0, 35.3, 28.6. HRMS (m/z): [M + H]+ calculated for C31H59N3O10, 634.4279; found, 634.4269.
Synthesis of 5

Intermediate 4 (3.85, 6.1 mmol) was treated with HCl (4 N in dioxane, 15 ml) for 2 h at room temperature. The solvent was removed by rotary evaporation, and the remaining oil was dissolved in deionized water (20 ml), frozen and lyophilised to afford the product as a light yellow oil. Yield was quantitative. 1H-NMR (300 MHz, MeOD): δ 4.21 (d, J = 2.4 Hz, 2H), 3.97-3.94 (m, 6H), 3.83 (t, J = 4.7 Hz, 4H), 3.69-3.56 (m, 18H), 3.29-3.25 (m, 4H), 2.92 (t, J = 2.4 Hz, 1H), 2.76 (s, 6H). 13C-NMR (75 MHz, MeOD): δ 80.8, 76.4, 71.7, 71.6, 71.5, 71.4, 70.3, 67.0, 65.9, 59.2, 55.1, 54.3, 49.8, 33.8. HRMS (m/z): [M + H]+ calculated for C21H43N3O6, 434.3230; found, 434.3218.
Synthesis of 7

To a solution of 5 (0.68 g, 1.34 mmol) and DIPEA (0.87 g, 6.7 mmol) dissolved in anhydrous DMF (5 ml), we added 6 (2.1 g, 2.8 mmol) in portions over 1 h. The reaction was stirred at room temperature overnight, then concentrated and purified by flash chromatography, eluting with 0% to 10% methanol/dichloromethane. Yield 1.45 g, 67%. 1H-NMR (300 MHz, MeOD): δ 5.98-595 (m, 2H), 5.34 (m, 2H), 4.98-4.95 (m, 2H), 4.44 (q, J = 5.1 Hz, 2H), 4.37-4.32 (m, 2H), 4.24-4.15 (m, 4H), 4.19 (d, J = 2.5 Hz, 2H), 4.06-4.03 (m, 2H), 3.92-3.84 (m, 6H), 3.81 (s, 6H), 3.72-3.63 (m, 18H), 3.62-3.57 (m, 8H), 3.03-2.92 (m, 6H), 2.89 (t, J = 2.4 Hz, 1H), 1.91 (s, 6H), 1.52 (s, 18H), 1.47 (s, 18H), 1.36-1.31 (m, 12H). 13C-NMR (75 MHz, MeOD): δ 173.6, 173.4, 165.0, 164.2, 163.5, 158.0, 157.3, 156.7, 154.0, 146.1, 145.9, 111.9, 111.6, 110.1, 110.0, 85.0, 81.0, 79.0, 76.7, 76.6, 76.4, 72.2, 72.0, 71.7, 71.5, 70.9, 70.3, 66.9, 65.8, 59.2, 55.1, 53.2, 53.1, 51.6, 50.3, 48.1, 47.1, 37.1, 36.9, 36.1, 31.8, 28.6, 28.4, 28.3, 27.0, 25.7, 25.4, 23.2, 23.0. HRMS (m/z): [M + H]+ calculated for C75H123N11O30, 1658.8515; found, 1658.8505.
Synthesis of 8

Intermediate 7 (1.45 g, 0.87 mmol) was dissolved into 3 ml MeOH, then treated with a solution of lithium hydroxide (90 mg, 3.8 mmol) dissolved in deionized water (6 ml). The reaction was stirred for 15 min at room temperature at which time LC–MS showed the reaction was complete. The pH of the reaction solution was adjusted to a value of 5–6 by using Amberlite IRN-77 ion exchange resin, then filtered to remove the resin. The filtrate was concentrated to dryness by rotary evaporation and used in the next step without further purification. Ion found by LC-MS: [(M + 2H)/2]+ = 815.9.
The hydrolysis product was dissolved in dichloromethane (5 ml) and trifluoroacetic acid (TFA) (10 ml), and stirred at room temperature. The progress of the reaction was monitored by LC–MS. After complete Boc-removal (~4 h), the solution was concentrated to dryness with a rotary evaporator and then dissolved in 8 ml water. The resulting solution was stirred for another 2 h at room temperature at which time LC–MS showed complete removal of the acetonide protecting groups. This mixture was concentrated and purified by RPLC using an Isco CombiFlash liquid chromatograph eluted with 5–40% acetonitrile/water with 0.1% TFA as the modifier. Yield for three steps 780 mg, 65.0%. 1H-NMR (300 MHz, MeOD): δ 5.94 (d, J = 2.5 Hz, 2H), 5.05 (dt, J = 8.9, 3 Hz, 2H), 4.59 (d, J = 9.9 Hz, 2H), 4.46 (dt, J = 8.9, 2.4 Hz, 2H), 4.29-4.19 (m, 2H), 4.22 (d, J = 2.4 Hz, 2H), 4.08-3.98 (m, 2H), 3.93-3.84 (m, 6H), 3.79-3.47 (m, 28H), 3.30-3.21 (m, 2H), 3.02-2.93 (m, 6H), 2.92 (t, J = 2.4 Hz, 1H), 1.97 (s, 6H). 13C-NMR (75 MHz, MeOD): δ 173.7, 173.6, 165.1, 157.7, 157.3, 147.3, 109.3, 80.8, 77.8, 76.3, 72.3, 71.6, 71.5, 71.4, 70.7, 70.5, 70.2, 70.0, 66.7, 65.9, 65.7, 64.6, 59.2, 59.1, 52.7, 52.6, 36.4, 35.6, 23.1, 23.0. HRMS (m/z): [M + H]+ calculated for C47H179N11O22, 1150.5479; found, 1150.5497.
Cloning, expression and purification of the N-terminal extended Fc construct
On the basis of European Union (EU) numbering, a DNA sequence encoding residues 201–447 of the human IgG1 (with N-terminal extension) was used to generate the data described herein (see peptide sequence below). The unpaired cysteine at position 220 in human IgG1 was mutated to serine and an M252Y/S254T/T256E (YTE) triple mutation (both are highlighted in bold in the sequence) to alleviate issues with aggregation of the final product and extend the circulating half-life in humans, respectively. The CH1 domain residues included in the construct are underlined. The expression vector, which included an N-terminal mouse IgG VH region secretion signal sequence for expression in Chinese hamster ovary (CHO, Gibco) cells, was cloned into the pcDNA3.1(+) vector (GenScript). The constructed plasmid was transfected into suspension ExpiCHO cells (Gibco) per manufacturer protocol and collected after 13 days. The Fc protein was purified using protein A affinity chromatography (MabSelect Prism A, Cytiva) and dialysed into PBS (pH 7.4).
Sequence
NVNHKPSNTKVDKKVEPKSSDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLYITREPEVCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Purification of CD388
CD388 was purified via protein A affinity chromatography, dialysed into 100 mM EDTA in PBS (pH 7.4) to remove copper and then dialysed into PBS (pH 7.4) to remove EDTA. The conjugate was purified using size exclusion chromatography (SEC) (HiLoad 26/600 Superdex, Cytiva) in PBS (pH 7.4).
Synthesis of azido-Peg Fc
Azido-PEG4-NHS ester (0.361 ml 0.050 M solution in DMF, 0.0181 mmol, 16 equivalents) was added to a solution of the Fc domain described above (5.3 ml, 0.0657 g, 0.00113 mmol) in PBS adjusted to pH 8.5 with 0.10 M sodium bicarbonate. LC–MS (ESI) analysis was performed to monitor azide incorporation (~2 h) to achieve drug:antibody ratio (DAR) values typically between 4.5 and 5.0. Acetic acid (2.0 M) was added to lower the pH to 5.5. The resulting solution was dialysed into 20 mM MES buffer with 100 mM NaCl at pH 6.1 (3 times, total 8-million-fold dilution). The yield of azido functionalized Fc was typically >90% as determined by UV–Vis spectrophotometry.
Synthesis of CD388
In a separate vial, an aqueous solution of 2-(4-((bis((1-((text-butyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-1H-1,2,3-triazol-1-yl)acetic acid (BTTAA) (6.91 ml, 1.727 mmol, 125 equivalents, 0.25 M) was mixed with aqueous solutions of CuSO4 (3.45 ml, 0.345 mmol, 25 equivalents, 0.100 M), ZnCl2 (1.04 ml, 1.036 mmol, 75 equivalents, 1.00 M) and 2-aminoguanidine hydrochloride (3.45 ml, 3.45 mmol, 250 equivalents, 1.00 M). In a 50-ml polypropylene conical centrifuge tube charged with a MES buffer (pH 6.1) solution of azido Fc at DAR between 4.5 and 5.0 (63.0 ml, 0.800 g, 0.0138 mmol, 1.0 equivalent), we added 8 as a solid (237 mg of triple TFA salt, 0.159 mmol, 11.5 equivalents), which dissolved readily. The copper/ligand solution was added to freshly prepared sodium ascorbate solution (3.45 ml, 3.45 mmol, 1.00 M). The pH of the homogeneous reaction mixture after combining all the reagents was ~4.0. The reaction was gently rotated for 3 h and then quenched by adding an aqueous solution of EDTA (14 ml, 7.0 mmol, 0.5 M, pH 8.0). The reaction was dialysed into 100 mM EDTA in PBS (pH 7.4) to remove copper, and then into PBS (pH 7.4) to remove EDTA. The crude conjugate was purified by affinity chromatography over a protein A column. The desired fractions were combined, sterile filtered and then further purified by SEC (HiLoad 26/600 Superdex, Cytiva), eluting with PBS buffer (pH 7.4). Matrix-assisted laser desorption ionization-time of flight mass spectrometry analysis of the purified final conjugate showed an average mass of 64,520 Da (DAR ~ 4.5; typical DAR ranges of 4.2–4.7 are reproducibly achieved using this method), as determined by the equation DAR = (mass of conjugate − mass of Fc)/(mass of TM and azido linker) (Supplementary Fig. 1).
The final product was aggregate free (99.6% monodisperse) as determined by analytical SEC. The final solution was concentrated to 24 mg ml−1 in PBS (pH 7.4) in a total volume of 16.7 ml. The yield was 50%. Endotoxin levels were below detectable limits (<0.5 EU ml−1) and a qualitative streaking on a culture dish showed no bacterial contamination. CD388 was determined to possess the required physical and chemical attributes necessary to support its proposed use as a protective prophylaxis treatment for influenza. CD388 was formulated in aqueous buffer to 150 mg ml−1 without issue, and the characteristics of formulated CD388 support its suitability for its intended therapeutic use (data summarized in tabular format in Supplementary Data Table 12).
The stability of formulated CD388 product was studied extensively. Differential scanning calorimetry (DSC) was utilized to assess thermal stability and the points of protein folding/unfolding. As shown below, the exothermic onset temperature (TOnset) was determined to be favourable at 50.5 °C and the two transition mid-point temperatures (Tm1 and Tm2) were determined as 61.2 °C and 83.4 °C, respectively (Supplementary Data Fig. 2).
As part of ongoing clinical development activity, formulated CD388 product stability at practical storage conditions was studied and data indicate that the drug product is stable up to 24 months under storage conditions at −20 °C and 5 °C. There were no significant changes in colour, clarity, visible particles, pH, protein concentration, CE–SDS reduced, CE–SDS non-reduced, SEC–HPLC, DAR–MALDI, subvisible particles and Fc binding assay (ELISA) at −20 °C and 5 °C storage conditions for up to 24 months.
Peptide mapping
For enzymatic digestion, utilizing both unconjugated Fc or conjugated Fc using WT hIgG1, 200 µg protein conjugate was diluted to a final volume of 15 µl with deionized water, and 45 µl 10 mM ammonium bicarbonate (pH 8) was added. After mixing, samples were incubated at 80 °C for 1 min. Samples were allowed to cool down before addition of 5 µl of a 1% solution of Promega ProteaseMAX (Promega) along with 6 µl of water. Trypsin/lys-c enzyme (Promega) was reconstituted to 2 µg µl−1 and 4 µl was added to samples at an enzyme:substrate ratio of 1:25. Samples were mixed and incubated at 50 °C for 3 h, then quenched with 10 µl of formic acid. After sample cleanup using a Waters Oasis MCX μElution SPE plate (Waters), the sorbent was conditioned with 200 µl methanol and then equilibrated with 200 µl water. The acidified sample was loaded and washed, first with 200 µl 2% formic acid in water, followed by 5% methanol in water. Samples were eluted from the sorbent with 2% ammonium hydroxide in 60:40 acetonitrile:water. Samples were analysed by HRES–LC–MS using a Waters Acquity H-Class UPLC coupled to a Waters Q-TOF Premier mass spectrometer. The UPLC conditions included Waters Acquity CSH C18 column, 150 × 2.1 mm, 1.7 u at 40 °C with a flow rate of 0.5 ml min−1 using a gradient of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. The following gradient profile was used: 0–1 min 5% B, 1–50 min 5–35% B, 50–50.5 min 35–95% B, 50.5–55 min 95% B. MSe high and low energy mass spectral data were acquired over the mass range of 90–2,400 Da. Data analysis was performed using Waters MassLynx and BiopharmaLynx software.
FcRn binding
Human FcRn (Precision Antibody) at 10 µg ml−1 was captured on preconditioned biosensors. Sensors were exposed to test article at pH 5.8 or pH 7.4. The association rate constant (Kon) and dissociation rate constant (Koff) were measured on an Octet RED96 system (Octet) and the dissociation constant (KD) was determined by global fitting analysis based on the Kon and Koff.
Cytotoxicity of CD388 in human cells
The cytotoxicity of CD388 was determined in primary human cells (stationary cells) after 24 h and in dividing cell lines after 5 days. We chose 5 days to allow for multiple rounds of cell division and also because it is the longest incubation time used in the cell-based CPE assay. To determine the CC50 of CD388 in primary human cells, human lung fibroblast cells (HLFs, ATCC, PCS-201-013) were seeded at 1 × 104 cells per well and incubated at 37 °C and 5% CO2 for 18–24 h. Freshly isolated peripheral blood mononuclear cells (PBMCs, purchased from BioIVT) were added at 6 × 105 cells per well. The next day, test articles (TAs) were added at concentrations ranging from 0.0001 nM (HLFs) or 1 nM (PBMCs) to 10,000 nM to a confluent monolayer (>90%). Cytotoxicity was determined after 24 h using the CellTiter-Glo cell viability kit according to manufacturer instructions (Promega). Luminescence was read on an EnSpire (PerkinElmer) plate reader.
To determine the CC50 of CD388 in human cell lines, HEp-2 cells (ATCC, CCL-23) were seeded at 5 × 103 cells per well and A549 cells (ATCC, CRM-CCL-185) at 1 × 104 cells per well and incubated at 37 °C and 5% CO2 for 18–24 h. The next day, TAs were added at concentrations ranging from 0.00001 nM to 10,000 nM to a confluent monolayer (<65%). Cytotoxicity was determined after 5 days using the CellTiter-Glo kit according to manufacturer instructions (Promega). Luminescence was read on the EnSpire (PerkinElmer) plate reader. The % cytotoxicity was calculated using the following equation: % Cytotoxicity = (1−(RLU with TA)/(RLU cells only)) × 100% (RLU, relative light units). CC50 [nM] was determined on the basis of % cytotoxicity with nonlinear regression analysis ([inhibitor] vs normalized response with variable slope) using GraphPad Prism software v.8.
Statistics and reproducibility
No statistical method was used to predetermine sample size. No animals or data were excluded from the analyses. Except for the mouse efficacy studies, where mice were randomized into dosing groups after infection, experiments were not randomized. Data collection and analysis in pharmacokinetic studies were not performed blind to the conditions of the experiments. Quantitative analysis of viral burden and lung pro-inflammatory cytokines in mouse studies were conducted by scientists blinded to the treatment group of analysed samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All relevant data supporting the key findings of this study are available within the paper and its Supplementary Information. Structures used to generate Fig. 1a in the main text and Extended Data Fig. 1 were published elsewhere54,55 with PDB accession numbers 1HZH and 3TI5. Sequencing data from the CD388-selected serial passage mutants described herein have been deposited in GenBank (accession numbers PV081919, PV081921, PV081923, PV081924, PV081925 and PV081926) Source data are provided with this paper.
References
Krammer, F. et al. Influenza. Nat. Rev. Dis. Primers 4, 3 (2018).
Krammer, F. et al. NAction! How can neuraminidase-based immunity contribute to better influenza virus vaccines? mBio https://doi.org/10.1128/mBio.02332-17 (2018).
Krammer, F. The human antibody response to influenza A virus infection and vaccination. Nat. Rev. Immunol. 19, 383–397 (2019).
Heaton, N. S., Sachs, D., Chen, C. J., Hai, R. & Palese, P. Genome-wide mutagenesis of influenza virus reveals unique plasticity of the hemagglutinin and NS1 proteins. Proc. Natl Acad. Sci. USA 110, 20248–20253 (2013).
Kallewaard, N. L. et al. Structure and function analysis of an antibody recognizing all influenza A subtypes. Cell 166, 596–608 (2016).
Ekiert, D. C. et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333, 843–850 (2011).
Dreyfus, C. et al. Highly conserved protective epitopes on influenza B viruses. Science 337, 1343–1348 (2012).
Wohlbold, T. J. & Krammer, F. In the shadow of hemagglutinin: a growing interest in influenza viral neuraminidase and its role as a vaccine antigen. Viruses 6, 2465–2494 (2014).
Eichelberger, M. C. & Monto, A. S. Neuraminidase, the forgotten surface antigen, emerges as an influenza vaccine target for broadened protection. J. Infect. Dis. 219, S75–S80 (2019).
Burmeister, W. P., Ruigrok, R. W. & Cusack, S. The 2.2 A resolution crystal structure of influenza B neuraminidase and its complex with sialic acid. EMBO J. 11, 49–56 (1992).
Sandbulte, M. R. et al. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc. Natl Acad. Sci. USA 108, 20748–20753 (2011).
Kilbourne, E. D., Johansson, B. E. & Grajower, B. Independent and disparate evolution in nature of influenza A virus hemagglutinin and neuraminidase glycoproteins. Proc. Natl Acad. Sci. USA 87, 786–790 (1990).
Stadlbauer, D. et al. Broadly protective human antibodies that target the active site of influenza virus neuraminidase. Science 366, 499–504 (2019).
Chavas, L. M. et al. Crystal structure of the human cytosolic sialidase Neu2. Evidence for the dynamic nature of substrate recognition. J. Biol. Chem. 280, 469–475 (2005).
Yates, P. J. et al. Lessons from resistance analysis in clinical trials of IV zanamivir. Virus Res. 325, 199039 (2023).
von Itzstein, M. et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 363, 418–423 (1993).
Kim, C. U. et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 119, 681–690 (1997).
Babu, Y. S. et al. BCX-1812 (RWJ-270201): discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J. Med. Chem. 43, 3482–3486 (2000).
Burch, J. et al. Prescription of anti-influenza drugs for healthy adults: a systematic review and meta-analysis. Lancet Infect. Dis. 9, 537–545 (2009).
Marty, F. M. et al. Intravenous zanamivir or oral oseltamivir for hospitalised patients with influenza: an international, randomised, double-blind, double-dummy, phase 3 trial. Lancet Respir. Med. 5, 135–146 (2017).
Collins, P. J. et al. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature 453, 1258–1261 (2008).
Nguyen, H. T., Fry, A. M. & Gubareva, L. V. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir. Ther. 17, 159–173 (2012).
Hayden, F. G. et al. Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N. Engl. J. Med. 379, 913–923 (2018).
Calder, L. J., Wasilewski, S., Berriman, J. A. & Rosenthal, P. B. Structural organization of a filamentous influenza A virus. Proc. Natl Acad. Sci. USA 107, 10685–10690 (2010).
Harris, A. et al. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl Acad. Sci. USA 103, 19123–19127 (2006).
Macdonald, S. J. et al. Potent and long-acting dimeric inhibitors of influenza virus neuraminidase are effective at a once-weekly dosing regimen. Antimicrob. Agents Chemother. 48, 4542–4549 (2004).
Tarbet, E. B. et al. A zanamivir dimer with prophylactic and enhanced therapeutic activity against influenza viruses. J. Antimicrob. Chemother. 69, 2164–2174 (2014).
Fu, L. et al. Structure-based tetravalent zanamivir with potent inhibitory activity against drug-resistant influenza viruses. J. Med. Chem. 59, 6303–6312 (2016).
Dall’Acqua, W. F., Kiener, P. A. & Wu, H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 281, 23514–23524 (2006).
Wan, H. et al. Structural characterization of a protective epitope spanning A(H1N1)pdm09 influenza virus neuraminidase monomers. Nat. Commun. 6, 6114 (2015).
Gubareva, L. V. et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2015–2016. Antiviral Res. 146, 12–20 (2017).
Mandal, R. S., Panda, S. & Das, S. In silico prediction of drug resistance due to S247R mutation of influenza H1N1 neuraminidase protein. J. Biomol. Struct. Dyn. 36, 966–980 (2018).
Preliminary Estimated Flu Disease Burden 2023–2024 Flu Season (CDC, 2024).
Chow, E. J. et al. Respiratory and nonrespiratory diagnoses associated with influenza in hospitalized adults. JAMA Netw. Open 3, e201323 (2020).
Ray, G. T. et al. Intraseason waning of influenza vaccine effectiveness. Clin. Infect. Dis. 68, 1623–1630 (2019).
Tokars, J. I. et al. Waning of measured influenza vaccine effectiveness over time: the potential contribution of leaky vaccine effect. Clin. Infect. Dis. 71, e633–e641 (2020).
Sah, P. et al. Future epidemiological and economic impacts of universal influenza vaccines. Proc. Natl Acad. Sci. USA 116, 20786–20792 (2019).
Salazar, G., Zhang, N., Fu, T. M. & An, Z. Antibody therapies for the prevention and treatment of viral infections. npj Vaccines 2, 19 (2017).
Couch, R. B. et al. Antibody correlates and predictors of immunity to naturally occurring influenza in humans and the importance of antibody to the neuraminidase. J. Infect. Dis. 207, 974–981 (2013).
Monto, A. S. et al. Antibody to influenza virus neuraminidase: an independent correlate of protection. J. Infect. Dis. 212, 1191–1199 (2015).
Tan, S. K. et al. A randomized, placebo-controlled trial to evaluate the safety and efficacy of VIR-2482 in healthy adults for prevention of influenza A illness (PENINSULA). Clin. Infect. Dis. 79, 1054–1061 (2024).
Tan, J., Asthagiri Arunkumar, G. & Krammer, F. Universal influenza virus vaccines and therapeutics: where do we stand with influenza B virus? Curr. Opin. Immunol. 53, 45–50 (2018).
Borchering, R. K. et al. Anomalous influenza seasonality in the United States and the emergence of novel influenza B viruses. Proc. Natl Acad. Sci. USA 118, e2012327118 (2021).
Memoli, M. J. et al. Evaluation of antihemagglutinin and antineuraminidase antibodies as correlates of protection in an influenza A/H1N1 virus healthy human challenge model. mBio 7, e00417-16 (2016).
Chen, Y. Q. et al. Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell 173, 417–429.e10 (2018).
Momont, C. et al. A pan-influenza antibody inhibiting neuraminidase via receptor mimicry. Nature 618, 590–597 (2023).
Yasuhara, A. et al. A broadly protective human monoclonal antibody targeting the sialidase activity of influenza A and B virus neuraminidases. Nat. Commun. 13, 6602 (2022).
van Meer, P. J. K. et al. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. mAbs 5, 810–816 (2013).
Magyarics, Z. et al. Randomized, double-blind, placebo-controlled, single-ascending-dose study of the penetration of a monoclonal antibody combination (ASN100) targeting Staphylococcus aureus cytotoxins in the lung epithelial lining fluid of healthy volunteers. Antimicrob. Agents Chemother. 63, e00350-19 (2019).
Honce, R. et al. Efficacy of oseltamivir treatment in influenza virus-infected obese mice. mBio 14, e0088723 (2023).
van Baalen, C. A. et al. ViroSpot microneutralization assay for antigenic characterization of human influenza viruses. Vaccine 35, 46–52 (2017).
Zielinska, E. et al. Development of an improved microneutralization assay for respiratory syncytial virus by automated plaque counting using imaging analysis. Virol J. 2, 84 (2005).
Zhou, B. et al. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J. Virol. 83, 10309–10313 (2009).
Saphire, E. O. et al. Crystal structure of a neutralizing human IgG against HIV-1: a template for vaccine design. Science 293, 1155–1159 (2001).
Vavricka, C. J. et al. Structural and functional analysis of laninamivir and its octanoate prodrug reveals group-specific mechanisms for influenza NA inhibition. PLoS Pathog. 7, e1002249 (2011).
Acknowledgements
We thank K. Bartizal (Cidara Therapeutics), P. De Jesus, L. Martin-Sancho, S. Chanda (Scripps Research, CA) and S. Schultz-Cherry (St Jude Children’s Research Hospital, TN) for their assistance, expert insight and for kindly providing reagents. We also thank B. Bassler (Princeton, NJ) for critical review of this manuscript and editorial suggestions. The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
Experiments were designed and conceived by S.D., J.L., J.N.C, J.B.L., V.O., G.H., S.F., R.M.H., A.N., T.H., W.J., J.L.S. and L.W.T. CD388 was invented and synthesized by A.B., D.C.B., Z.-Y.C., T.L., T.P.B., J.M.B. and L.W.T. Protein production was done by J.F. In vitro experiments were performed and analysed by S.D., J.N.C., J.D., A.A., E.A., R.G., D.Z. and N.D. Peptide mapping experiments and analysis were conducted and analysed by G.H. The in vivo studies were designed by J.L. and carried out and analysed by K.A., S.D., A.A. and E.A. Figures were prepared by S.D., J.L. and L.W.T. The manuscript was written by S.D., J.L., J.N.C., J.B.L, R.M.H. and L.W.T. All authors contributed to editing and reviewing the manuscript, and support the conclusions.
Corresponding author
Ethics declarations
Competing interests
All authors listed are current or past shareholders and/or employees of Cidara Therapeutics, Inc.
Peer review
Peer review information
Nature Microbiology thanks Daniel Perez, Kris White and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 The NA tetramer with tetrameric ZAN.
Crystal structure of tetrameric ZAN (green) complexed to an NA tetramer (PDB code 3TI5) from A/California/04/2009 H1N1. The C7 position on ZAN that was chosen for attachment to the Fc carrier (via an NHS ester) in the work presented here is highlighted. C7 on ZAN is solvent exposed and unencumbered sterically, allowing conjugation to the C7 position with minimal influence on binding affinity to NA.
Extended Data Fig. 2 Universal activity of CD388 against influenza A and B in lethal mouse models.
CD388 efficacy against lethal challenge with influenza in BALB/c mice. Shown is dose-response % body weight change in response to CD388 ranging from 0.03 – 1 mg/kg administered IM at 2 h post-infection against lethal challenge with influenza (a) A/Hong Kong/1/1968 (H3N2), (b) B/Florida/4/2006 (Yamagata), (c) B/Malaysia/2506/2004 (Victoria), and % body weight change and survival against (d) A/WSN/1933 (H1N1), (e) A/California/07/2009 (H1N1)pdm, (f) A/California/12/2012 (H1N1)pdm09, (g) A/North Carolina/04/2014 (H1N1)pdm09, (h) A/Hawaii/70/2019 (H1N1)pdm09, and (i) B/Colorado/6/2017 (Victoria) in BALB/c mice.
Extended Data Fig. 3 CD388 prophylactic activity – body weight change.
% body weight change in response to CD388 administered IM at 7 days prior to lethal challenge against influenza (a) A/Puerto Rico/8/1934 (H1N1), (b) A/Hong Kong/1/1968 (H3N2), (c) B/Florida/4/2006 (Yamagata) and (d) B/Malaysia/2506/2004 (Victoria).
Extended Data Fig. 4 CD388 prophylactic activity – Survival and body weight change.
Survival and % body weight change in response to CD388 administered IM at 7 days prior to lethal challenge against influenza (a) A/WSN/1933 (H1N1), (b) A/California/07/2009 (H1N1)pdm, (c) A/California/12/2012 (H1N1)pdm09, (d) A/North Carolina/04/2014 (H1N1)pdm09, (e) A/Hawaii/70/2019 (H1N1)pdm09, and (f) B/Colorado/6/2017 (Victoria) in BALB/c mice.
Supplementary information
Supplementary Information
Supplementary Data Tables 1–12, and Figs. 1 and 2.
Source data
Source Data Fig. 1–5
Source data for Fig. 1b–f. Source data for Fig. 2a–n. Source data for Fig. 3a–f. Source data for Fig. 4a–e. Source data for Fig. 5a,b.
Source Data Extended Data Fig. 2–4
Source data for Extended Data Fig. 2a–i. Source data for Extended Data Fig. 3a–d. Source data for Extended Data Fig. 4a–f.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Döhrmann, S., Levin, J., Cole, J.N. et al. Drug–Fc conjugate CD388 targets influenza virus neuraminidase and is broadly protective in mice. Nat Microbiol 10, 912–926 (2025). https://doi.org/10.1038/s41564-025-01955-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41564-025-01955-3
