
Abstract
Lysosomes have crucial roles in regulating eukaryotic metabolism and cell growth by acting as signalling platforms to sense and respond to changes in nutrient and energy availability1. LYCHOS (GPR155) is a lysosomal transmembrane protein that functions as a cholesterol sensor, facilitating the cholesterol-dependent activation of the master protein kinase mechanistic target of rapamycin complex 1 (mTORC1)2. However, the structural basis of LYCHOS assembly and activity remains unclear. Here we determine several high-resolution cryo-electron microscopy structures of human LYCHOS, revealing a homodimeric transmembrane assembly of a transporter-like domain fused to a G-protein-coupled receptor (GPCR) domain. The class B2-like GPCR domain is captured in the apo state and packs against the surface of the transporter-like domain, providing an unusual example of a GPCR as a domain in a larger transmembrane assembly. Cholesterol sensing is mediated by a conserved cholesterol-binding motif, positioned between the GPCR and transporter domains. We reveal that the LYCHOS transporter-like domain is an orthologue of the plant PIN-FORMED (PIN) auxin transporter family, and has greater structural similarity to plant auxin transporters than to known human transporters. Activity assays support a model in which the LYCHOS transporter and GPCR domains coordinate to sense cholesterol and regulate mTORC1 activation.
Similar content being viewed by others


Main
The lysosomal membrane has emerged as a dynamic signalling platform that senses and responds to changes in the availability of nutrients, growth factors and other essential cellular components, including cholesterol3. A crucial element of cellular membranes, cholesterol defines membrane structure and fluidity and has a central role in many cellular processes, including signalling and membrane trafficking4. Cholesterol levels are regulated at lysosomes, which can degrade or recycle intracellular cholesterol as part of a broader homeostatic cycle that incorporates cholesterol synthesis, endocytosis and transport5. The lysosomal transmembrane protein LYCHOS is a key component of cholesterol signalling, functioning as a cholesterol sensor to enable the cholesterol-dependent activation of mTORC1-dependent cell growth pathways2.
The protein kinase mechanistic target of rapamycin (mTOR) orchestrates eukaryotic cell growth in response to nutrient availability and growth signals, including amino acids, insulin and glucose6. Lysosomes provide a platform for mTORC1 activity, with cell growth cues triggering the allosteric activation of mTORC1 after it binds to the small GTPase Rheb at the lysosomal membrane7. Activated mTORC1 phosphorylates downstream effectors, including ribosomal S6 kinase and eukaryotic initiation factor 4E binding protein 1 (4EBP1), which are crucial regulators of protein synthesis and cell growth8. Conversely, amino acid deficiency inhibits mTORC1-dependent cell growth through the activity of the GATOR1 complex9. GATOR1 is a GTPase-activating protein for the RagA GTPase, promoting conversion from the active GTP-bound state to the inactive GDP-bound state9,10. The RagAGTP–RagCGDP heterodimer recruits mTORC1 to the lysosomal surface, and the GATOR1-catalysed conversion of RagA to the GDP-bound state restricts mTORC1 lysosomal recruitment and cell growth signalling11,12. When cholesterol levels are sufficient to support cell growth, LYCHOS binds cholesterol at its N terminus and is thought to interact with GATOR1 by means of an elongated cytoplasmic-facing loop2. By sequestering GATOR1 (refs. 2,13), LYCHOS promotes the recruitment of mTORC1 to the lysosome to drive cell growth2. Therefore, LYCHOS enables lysosomes to integrate lysosomal cholesterol levels with mTORC1 signalling to coordinate cell metabolism and growth.
The cryo-EM structure of LYCHOS
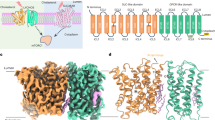
LYCHOS was at first classified as a GPCR (GPR155) but remains an orphan receptor with unknown GPCR-like activity14. To gain a deeper understanding of LYCHOS structure and function, we determined the 2.65 Å resolution cryo-electron microscopy (cryo-EM) structure of LYCHOS (Fig. 1a–c, Supplementary Video 1, Extended Data Figs. 1a–f, 2a,b and 3a–f and Extended Data Table 1). The structure revealed an N-terminal transporter-like domain fused to a central GPCR domain, with a Dishevelled, EGL-10 and Pleckstrin (DEP) domain located at the C terminus (Fig. 1a–d). The transporter-like domain forms a homodimeric assembly of ten transmembrane helices in a twofold repeating topology, with a further seven transmembrane helices contributed by the GPCR domain (Fig. 1b–d). The GPCR domain is homologous to the transmembrane region of the class B2 family of adhesion GPCRs, and is in the apo state, with no ligand or G protein bound (Extended Data Fig. 4a–g and Supplementary Discussion 1). The GPCR domain resides proximal to the dimer core with a relatively small interface (992 Å2 buried surface area) formed between the GPCR and the transporter-like domain (Fig. 1e,f). The dimerization interface is extensive (1,410 Å2 buried surface area) with conserved interactions mediated by the transporter-like domain (Fig. 1e,f and Extended Data Fig. 5a,b). In addition, the DEP domains dimerize on the cytosolic surface of the transporter-like domain, forming a second, less extensive dimer interface mediated by the symmetrical arrangement of DEP helices α18 and α19 (Extended Data Fig. 5c,d). Notably, both the scaffold and DEP interfaces are highly conserved, highlighting a strong selection pressure on the formation of dimeric LYCHOS (Extended Data Fig. 5a–d). These data suggest that the dimer has a crucial role in function. However, whether the DEP domains have a biological function beyond supporting dimer formation is at present unclear.

a, Schematic of LYCHOS domain layout. b, The 2.65 Å resolution cryo-EM density map of the LYCHOS homodimer in the membrane plane oriented with the cytoplasm at the top and the lysosomal lumen at the bottom. Orientation as previously determined2. A second LYCHOS cryo-EM reconstruction in thinner ice was determined to a more uniform 2.75 Å resolution across the transmembrane domains, but density for the DEP domains was missing from the map (Extended Data Figs. 2 and 3 and Extended Data Table 1). c, Model of the LYCHOS homodimer oriented as in b, with the transporter scaffold region in blue, the transporter region in purple, the GPCR domain in orange and the DEP domain in green. d, Topology diagram of LYCHOS oriented as in b, with key domains and secondary structural elements highlighted. e,f, LYCHOS model (e) and cryo-EM density map (f) viewed from the lysosomal lumen.
The GPCR third intracellular loop (ICL3) corresponds to a large intrinsically disordered loop (residues 551–641) that is not observed in the structure and extends from helices α15 and α16 (Fig. 1b,d and Extended Data Fig. 4c). The GPCR ICL3 was previously referred to as the LYCHOS effector domain (LED)2. The ICL3 (LED) is oriented into the cytoplasm and is thought to mediate interactions with the GATOR1 complex at the lysosomal membrane2. Consequently, this orientation of LYCHOS positions the GPCR orthosteric ligand-binding pocket into the lysosomal lumen with the G-protein-binding site facing the cytoplasm (Fig. 1b and Extended Data Fig. 4c). Comparison of the LYCHOS orthosteric binding pocket with the structures of active adhesion GPCRs reveals that LYCHOS α11 (GPCR domain transmembrane helix 1, TM1) and α16 (GPCR TM6) are positioned to occlude the pocket (Extended Data Fig. 4d and Supplementary Discussion 1). In addition, the LYCHOS lysosomal loop 2, corresponding to GPCR extracellular loop 2 (ECL2), buries itself within the orthosteric binding pocket, which is likely to further block ligand entry (Extended Data Fig. 4d). At the G-protein-binding site, alignments between LYCHOS and G-protein-coupled ADGR4 reveal that the LYCHOS α15 (GPCR TM5) and α16 (GPCR TM6) transmembrane helices, along with the DEP domains, would sterically preclude a canonical mode of G-protein coupling (Extended Data Fig. 4e). Furthermore, LYCHOS α16 (GPCR TM6) adopts an extended linear conformation, rather than the kinked conformation that opens the G-protein-binding pocket15 (Extended Data Fig. 4f and Supplementary Discussion 1). Together, these findings suggest that in the observed conformation, the LYCHOS GPCR domain does not engage with G proteins. However, considering the wide variation in the interaction angles between arrestins, G proteins and GPCRs, we cannot completely exclude the possibility of such couplings. Whether LYCHOS has the potential to signal through canonical GPCR pathways will require future verification through a comprehensive assessment of canonical GPCR effectors and signalling mechanisms. Overall, the LYCHOS domain arrangement provides a highly unusual structural example of a GPCR domain fused to additional domains within a larger transmembrane assembly.
The LYCHOS transporter-like domain
To further investigate the structure of the transporter-like domain, we undertook an extensive structural similarity search across the entire AlphaFold16 and Protein Data Bank (PDB)17 databases using FoldSeek18. Of note, the highest structural similarity for the LYCHOS transporter domain was to plant PIN auxin transporters (TM score ≈ 0.84; Extended Data Table 2). Sequence analysis further showed that the LYCHOS transporter-like domain was more closely related to the plant PIN transporters than to any human transporter family (Fig. 2a, Extended Data Fig. 6a–f and Extended Data Table 2).

a, Phylogenetic analysis of the LYCHOS transporter domain. Models of selected transporters (stars) are shown for structural comparison. For detailed analyses, see Extended Data Fig. 6 and Extended Data Table 2. b, Structural alignment of the LYCHOS transporter domain with plant PIN1 (root mean square deviation (RMSD) 2.86 Å), PIN3 (RMSD 3.35 Å) and PIN8 (RMSD 3.57 Å) structures19,20,21. c,d, The LYCHOS transporter domain crossover motif (c), aligned to the three plant PIN transporters (d). The conserved asparagine and proline residues are numbered as per LYCHOS. e, Surface plasmon resonance of IAA binding to LYCHOS wild type (WT) (n = 8) or after N145A (n = 5), A148W (n = 8) or L177W (n = 7) mutation (Extended Data Fig. 7a,h–j). Steady-state affinity fit assumes one-to-one binding. Symbols show n independent experimental replicates; bold line shows the mean. Kd, dissociation constant. f, The 2.40 Å resolution cryo-EM density map of the IAA-bound LYCHOS homodimer. IAA (cryo-EM density indicated in green) binds to LYCHOS in an inward-open state, reminiscent of the plant PIN efflux carriers bound to IAA or to NPA. g, Boxed region in f, highlighting conserved residues that coordinate IAA binding to the LYCHOS transporter channel. h, Time course of IAA efflux in HEK293 cells co-transfected with AuxSen, and empty vector, LYCHOS or PIN8. IAA efflux in PIN8-transfected cells was measured without or with 25 μM NPA. Mean ± s.e.m. of n = 3 independent biological replicates. i, Rate of efflux determined from h. Symbols show values from n = 3 independent biological replicates; the bold line shows the mean; error bars show s.e.m.; NS, not significant; P values determined using one-way ANOVA with Šidák’s multiple comparisons test (comparisons to empty vector) or unpaired two-sided t-test (comparison to +NPA).
Structural alignment of the LYCHOS transporter-like domain to the experimentally determined structures of plant PIN1 (ref. 19), PIN3 (ref. 20) and PIN8 (ref. 21) revealed a marked similarity between the tertiary folds (Fig. 2b and Extended Data Fig. 6a–f). Specifically, PIN transporters have a conserved crossover motif that enables a conformational change referred to as an elevator-type passive transport mechanism19,20,21 (Fig. 2c,d and Extended Data Fig. 6c,e,f). Alignment of the crossover motif between LYCHOS and the plant PIN transporters revealed highly similar structures (Fig. 2d and Extended Data Fig. 6e), sequences (Extended Data Fig. 6f) and transmembrane topology (Extended Data Fig. 6c). The conserved Asn145 and Pro324 residues are located at the centre of the motif (Fig. 2d and Extended Data Fig. 6f), with asparagine mediating contacts with auxin in the transport channel of plant PIN transporters19,20,21. Although human solute carrier transporters contain crossover motifs22, we observe notable distinctions in their primary sequence, which places the conserved proline and asparagine residues in a different order (Extended Data Fig. 6f). Compared with LYCHOS, human SLC transporters show greater sequence divergence (Extended Data Fig. 6f), exhibit clear differences in transmembrane topology and are poorly conserved relative to LYCHOS (Extended Data Fig. 6a–c,f). Conversely, there is clear sequence and topological conservation between the bacterial ASBT homologue (Neisseria meningitidis) and the SLC10 family (Homo sapiens), providing further evidence of the distinctiveness of LYCHOS from the SLC families (Extended Data Fig. 6a–c,f).
LYCHOS retains IAA binding
Auxins are a key class of plant hormones that regulate various aspects of plant growth, including stem elongation, root development and tropisms23. Indole-3-acetic acid (IAA), the main auxin produced by plants, is not classically known to have a central role in humans. However, studies have detected IAA in humans, produced by the microbiome24 and as a by-product of tryptophan metabolism25, and have suggested that IAA has a role in human physiology and disease, including altering the efficacy of chemotherapy26. To investigate whether LYCHOS retains the capacity to bind small-molecule indole derivatives, we used IAA as a surrogate molecule for further binding and structural analyses.
Surface plasmon resonance analysis determined that LYCHOS retained IAA binding, consistent with the similarity to plant PIN auxin transporters. However, the 1.6 mM binding affinity of IAA to LYCHOS was lower than the reported affinity of IAA for plant PIN auxin transporters, which ranges between 0.2 and 0.4 mM (refs. 19,20,21) (Fig. 2e and Extended Data Fig. 7a). Next, we conducted solid supported membrane (SSM)-based electrophysiology measurements of LYCHOS, which revealed that an order-of-magnitude-higher concentration of IAA was required to elicit electrogenic currents equivalent to those initiated by PIN8 (ref. 21) (Extended Data Fig. 7b,c). We suggest that this finding aligns with anion binding, but not transport. To elucidate the structural basis of IAA binding, we determined the 2.40 Å resolution cryo-EM structure of IAA-bound LYCHOS in an inward-open conformation (Fig. 2f,g, Extended Data Figs. 2a and 7d–g and Extended Data Table 1). Here, additional cryo-EM density consistent with IAA was observed within the recessed cavity defined by the transporter domain and the crossover motif (Fig. 2g and Extended Data Fig. 7d–g). The IAA binding pose is consistent with the plant PIN transporters19,20,21 and is mediated by the same conserved Asn145 and Pro324 residues at the centre of the motif (Extended Data Fig. 6f). The binding pocket is predominantly hydrophobic, mediating interactions with the indole group of IAA, with Pro327 directly contributing π–CH contacts. The carboxylate of IAA forms hydrogen bonds with Asn145, as well as amine groups of the crossover motif backbone (Fig. 2g and Extended Data Fig. 7f). Overall, the LYCHOS binding pocket shares several biochemical and structural features with the plant PINs. To validate the observed binding site, we produced three variants within the cavity of the LYCHOS transporter domain. Mutation of Asn145 to alanine or Ala148 or Leu177 to tryptophan significantly reduced IAA binding affinity, as measured by surface plasmon resonance (Fig. 2e and Extended Data Fig. 7a,h–j).
To confirm SSM electrophysiology measurements suggesting that LYCHOS does not readily transport IAA, we used a genetically encoded auxin biosensor (AuxSen27) to measure cellular IAA efflux. The AuxSen biosensor was transfected into HEK293 cells along with wild-type LYCHOS, or the plant IAA transporter PIN8, and the rate of IAA efflux was monitored (Fig. 2h,i and Extended Data Fig. 7k). Transfection with PIN8 resulted in a clear increase in the rate of IAA efflux, which could be reversed with the addition of the auxin PIN transporter inhibitor N-1-naphthylphthalamic acid (NPA) (Fig. 2h,i). By contrast, we observed no significant change in the rate of IAA efflux in cells transfected with LYCHOS compared with those transfected with empty vector (Fig. 2h,i). These data, combined with SSM measurements, indicate that under conditions equivalent to those used for the plant PIN8 transporter, LYCHOS does not exhibit the capacity to transport IAA.
Finally, the structural similarities between LYCHOS and plant PIN transporters suggest that LYCHOS senses or transports alternative auxin-like metabolites that, unlike IAA, are commonly produced in humans at high concentrations, such as tryptophan. Indeed, both LYCHOS structures were observed in an inward-open conformation, configured to bind a cytosolic ligand. However, surface plasmon resonance analysis of tryptophan binding revealed only linear association at high tryptophan concentrations typical of non-specific binding (Extended Data Fig. 7l). Furthermore, cryo-EM analysis of LYCHOS in the presence of excess tryptophan (10 mM) showed no evidence of the small molecule in the transport channel or other regions across LYCHOS (Extended Data Fig. 7d,e). Therefore, we suggest that tryptophan is likely to be too large, or has an incorrect charge distribution to enter the binding pocket efficiently.
In summary, phylogenetic and structural analyses reveal that the LYCHOS transporter domain is a previously unidentified human orthologue of the plant PIN transporter family. Although LYCHOS retains the ability to bind to IAA, its affinity is reduced, and no clear transport activity was observed in direct comparison with plant PIN transporters. Given the considerable diversity of indole-like and related small molecules in the human metabolome, extensive lysosomal metabolomic studies across a range of target cells and models, in combination with transport studies, are needed to identify endogenous substrates of LYCHOS and confirm its transport activity.
Structural basis of cholesterol binding
We next sought to gain insight into the structural basis of LYCHOS cholesterol sensing. Previous mass spectrometry studies identified that LYCHOS α1 residues Glu48 and Cys55 are photolabelled by cholesterol analogues2. Our structural analysis located these residues on LYCHOS α1, positioned at the interface of the transporter and GPCR domains (Extended Data Fig. 8a–f). In addition, we identified a cholesterol recognition amino acid consensus (CRAC28) motif on α1, which forms a highly conserved pocket on the LYCHOS surface (Extended Data Fig. 8d–f). The identified CRAC-motif pocket is consistent with previous studies that showed that mutations of Phe43 and Pro44 at the base of the pocket, or of the conserved Tyr57 residue at the top, resulted in a loss of LYCHOS cholesterol binding and activity2.
However, despite the addition of cholesterol hemisuccinate (CHS) during purification, we did not observe CHS molecules in the conserved CRAC-motif pocket in any of the wild-type LYCHOS reconstructions. Inspection of the CRAC-motif pocket in both wild-type structures revealed a closed conformation, in which the packing of helices α10 and α17 forms a tight junction (Fig. 3a). Here intimate π-stacking interactions between the conserved residues Phe352 (α10) and Phe705 (α17) form a conformational clamp that physically occludes access to the CRAC-motif and thereby blocks cholesterol entry (Fig. 3a). Comparison across three additional structures, each incubated with IAA for varying durations, revealed two distinct conformations of the CRAC-motif pocket (Fig. 3a,b, Extended Data Figs. 2a and 8g–l, Extended Data Table 1 and Supplementary Discussion 2). These included a closed conformation that matched the apo wild-type LYCHOS (Fig. 3a and Extended Data Fig. 8h), as well as a partially open conformation (Fig. 3b and Extended Data Fig. 8i). Partial opening of the CRAC-motif pocket is marked by the movement of helices α10 and α17, which is enabled by rotation and translation of the GPCR domain relative to the transporter domain (Fig. 3b and Extended Data Fig. 9a,b). This movement uncouples the Phe352–Phe705 clamp, promotes alternate rotamers and increases accessibility to the CRAC-motif pocket. Indeed, Phe352 adopts alternate rotamers across these structures without affecting GPCR movement. However, additional conformational relaxation of helices α10 and α17 was only observed in samples treated with IAA (Extended Data Figs. 8h,i and 9a,b). Despite these changes in conformation around the CRAC-motif pocket and CHS being in excess, we were still unable to observe any CHS molecules within the CRAC-motif pocket in the wild-type or in the IAA-bound LYCHOS reconstructions. We conclude, therefore, that CHS entry and binding to the CRAC-motif requires further opening of the GPCR domain relative to the transporter domain.

a, The cholesterol pocket is occluded owing to π-stacking interactions made between a triad of phenylalanine residues (F352, F705 and F708) at the interface of the GPCR and transporter domains. Both wild-type and IAA-treated (closed state) LYCHOS are observed in this conformation. b, Opening of the phenylalanine gate was observed in IAA-treated (partially open state) LYCHOS. c, Focused view of the two cholesterol molecules packed within the cholesterol pocket of LYCHOS(F352A/W678R). Cryo-EM density is shown as transparent overlay. The cholesterol-binding pocket is defined by the GPCR (orange) and transporter (magenta) domains. For detailed analyses, see Extended Data Fig. 8. d, Top-down (cytoplasmic side) and end-on (membrane plane) views of the cholesterol-induced conformational change of the LYCHOS GPCR domain. Lateral movement of the whole GPCR domain is associated with CHS binding. Helices α16 and α17 must move to accommodate CHS entry. Three conformational states outline a possible mechanism of cholesterol binding to LYCHOS. e, In the closed state, Phe352 and Phe705 form a closed gate that occludes cholesterol entry into the binding groove. A CHS molecule can be seen queuing at the gate, unable to enter. f, IAA binding was associated with the partial opening of α10 and α17, whereby the gating phenylalanine residues rearrange but do not fully open. Rigid docking of CHS indicates that the pocket remains sterically occluded (Extended Data Fig. 9c–f). g, Mutagenesis of Phe352 completely bypasses the phenylalanine gate, thereby allowing CHS entry. This is accompanied by an opening of the GPCR domain relative to the transporter domain. A high degree of CHS occupancy was observed in the conserved CRAC-motif pocket.
To promote cholesterol binding, we produced a LYCHOS double mutant (F352A/W678R) designed to induce the opening movement of the GPCR domain relative to the transporter domain. In addition to bypassing the F352 gate (F352A), W678R was introduced to form a salt bridge with Glu296 and promote an open state. Notably, the 2.68 Å resolution cryo-EM map of LYCHOS(F352A/W678R) revealed clear density for two CHS molecules (Fig. 3c, Extended Data Figs. 2a and 8a–f and Extended Data Table 1). The first CHS molecule is bound at the α1 CRAC-motif at the PIN and GPCR domain interface (Fig. 3c). Inspection of the CRAC-motif pocket revealed a considerable degree of conservation, with virtually every residue defining the groove being conserved (Extended Data Fig. 8d). The groove forms extensive contacts with the CRAC-motif CHS molecule (431 Å2 buried surface area), is defined by numerous hydrophobic residues of the CRAC-motif and has a location that is entirely consistent with previous photolabelling2, mutagenesis2, binding2,13 and activity assays2 (Extended Data Fig. 8d–f). The second CHS molecule packs against the CRAC-motif-bound CHS, residing within the open gate region and forming fewer protein contacts with LYCHOS overall (Fig. 3c). After CHS binding, we observed a more substantial translation and rotation of the GPCR domain, reminiscent of the switch from the closed to the partially open state observed for incubation with IAA (Fig. 3b–d and Extended Data Fig. 9a–f).
In summary, across the seven wild-type and IAA LYCHOS reconstructions, we observe both a closed and a partially open conformation of the CRAC-motif pocket (Fig. 3e,f and Extended Data Figs. 8h,i and 9a,b). In these reconstructions, no CHS was bound at the CRAC-motif pocket. Furthermore, rigid-body docking of the closed and partially open models to the LYCHOS(F352A/W678R) structure revealed steric clashes with CHS, which would preclude CHS binding to the CRAC-motif pocket (Extended Data Fig. 9c–f). Consistent with this, in two reconstructions (one wild-type, and the short IAA incubation) we observe partial and poorly coordinated density for a third CHS molecule positioned outside the conserved cholesterol CRAC-motif pocket (Fig. 3e and Extended Data Fig. 9g). The partial CHS density seemed to be in a queuing position restricted from entry by the gating Phe352 and Phe705 residues (Fig. 3e). Only after mutation of the gating Phe352 residue (F352A) do we observe the entry of CHS into the conserved CRAC-motif pocket (Fig. 3g). Together, these data suggest that entry of cholesterol into the conserved CRAC-motif pocket requires structural rearrangement of both the GPCR domain and the Phe352 gating residue of the transporter domain beyond the movements observed between the closed and partially open structures.
Coordination of mTORC1 regulation
To understand how LYCHOS regulates mTORC1 activity, we used a genetically encoded mTORC1 FRET biosensor targeted to the lysosomal membrane (LysoTORCAR29, a 4EBP1-based biosensor). Consistent with previous reports2, adding cholesterol caused a steady increase in the activity of mTORC1 in HEK293 cells transfected with LYCHOS, whereas depletion of cholesterol led to a decrease in mTORC1 activity (Fig. 4a,b, Extended Data Fig. 10a–c and Supplementary Discussion 3). This decrease in the activity of mTORC1 was reversed after cholesterol repletion (Fig. 4a,b, Extended Data Fig. 10a–c and Supplementary Discussion 3). Furthermore, mutation of the conserved Tyr57 (Y57A) of the CRAC-motif abolished mTORC1 activity in response to cholesterol2 (Extended Data Fig. 10a,d,e).

a,c,f,h, mTORC1 activity in response to cholesterol addition or depletion (dotted line) followed by repletion (arrow) in HEK293 cells co-transfected with LysoTORCAR and LYCHOS (a; n = 3), LYCHOS(F352A/W678R) (c; n = 3), f, LYCHOS(N145A) (f; n = 3) or LYCHOS(F352A/W678R/N145A) (h; n = 3). Mean ± s.e.m. of n independent biological replicates. b,d,g,i, Fold change in mTORC1 activity in response to cholesterol addition, depletion (average F/F0 at −5–0 min versus 35–40 min), or repletion (average F/F0 at 35–40 min versus 5 min average at peak after repletion) in HEK293 cells co-transfected with LYCHOS (b), LYCHOS(F352A/W678R) (d), LYCHOS(N145A) (g) or LYCHOS(F352A/W678R/N145A) (i). Symbols show n = 3 independent biological replicates; bold line shows the mean; error bars show s.e.m.; P values calculated by one-way ANOVA with Šidák’s multiple comparisons test (versus control) or two-sided paired t-test (depletion versus repletion). e,j, Control-subtracted area under the curve (AUC; 0–40 min). Symbols show n = 3 independent biological replicates; bold line shows the mean; error bars show s.e.m.; P values calculated by two-way ANOVA with Šidák’s multiple comparisons test. k, LYCHOS is oriented with the GPCR orthosteric binding pocket facing the lysosomal lumen, and DEP and ICL3 (LED) facing the cytoplasm. l, Silhouette of LYCHOS (side view) highlighting the two potential conformational positions of the PIN crossover motif during elevator transport. Inset, focused illustration of the conformational change associated with a transport cycle initiated by transport ligand interactions (PDB 7Y9V (ref. 19), 7XXB (ref. 20) and 7QPA (ref. 21)). Vertical elevator movement of the transporter domain displaces F352, uncoupling the conformational clamp and opening the Phe352 gate (Supplementary Discussion 4). m, Freely diffusing cholesterol in the lysosomal bilayer can enter the CRAC-motif pocket and bind to the open state of LYCHOS between the transporter and GPCR domains, signalling to mTORC1 through the GATOR1 complex. The highly dynamic conformational landscape of ICL3 might modulate this2. CHL, cholesterol.
We then examined the role of the Phe352 gate in the regulation of LYCHOS activity. Mutation of Phe352 to alanine (LYCHOS(F352A) or LYCHOS(F352A/W678R)) resulted in a rapid and sustained increase in mTORC1 activity in response to cholesterol addition that was significantly greater than that seen for wild-type LYCHOS (Fig. 4c–e and Extended Data Fig. 10a,f–h). In contrast to wild-type LYCHOS, in cells transfected with LYCHOS(F352A) or LYCHOS(F352A/W678R), there was no longer any change in mTORC1 activity in response to cholesterol depletion, although cholesterol repletion still rapidly increased mTORC1 activity (Fig. 4c–e and Extended Data Fig. 10f,g). These findings suggest that Phe352 functions as a cholesterol gate to regulate LYCHOS activity. Removing this gating mechanism allows cholesterol unrestricted access to the binding pocket, facilitating LYCHOS and mTORC1 activity. In the absence of cholesterol, LYCHOS(F352A) or LYCHOS(F352A/W678R) can no longer be locked in an inactive state, which explains the lack of effect of cholesterol depletion on mTORC1 activity.
The atypical combination of a transporter domain and a GPCR within a single transmembrane assembly, along with the positioning of the cholesterol-binding site wedged between these domains, suggests that transport and cholesterol sensing are functionally linked. To test this hypothesis, we measured the activity of mTORC1 in HEK293 cells transfected with LYCHOS in which the conserved asparagine of the transporter-domain crossover motif was mutated to alanine (N145A). We observed a complete loss of responsiveness of mTORC1 activity in response to either cholesterol addition or cholesterol depletion followed by repletion (Fig. 4f,g and Extended Data Fig. 10a,h). This suggests that transport is required to open the Phe352 gate to allow the entry of cholesterol and therefore the regulation of mTORC1 activity. If this is the case, removal of the Phe352 gate in the absence of transport should rescue mTORC1 activity in response to cholesterol addition. Indeed, in HEK293 cells transfected with LYCHOS(F352A/W678R/N145A), we observed a recovery of mTORC1 activity in response to cholesterol addition, to a level comparable with that seen for wild-type LYCHOS (Fig. 4h–j and Extended Data Fig. 10a,h). These data suggest that either the LYCHOS transport pocket, or transport itself, regulates the ability of LYCHOS to sense and respond to changes in cholesterol levels.
To further test this hypothesis, we used a quantitative bioluminescence resonance energy transfer (BRET) assay to measure LYCHOS and GATOR1 association. We transfected cells with a constant level of LYCHOS fused at the C terminus with nano luciferase (NLuc; donor) and increasing amounts of the GATOR1 component NPRL2 fused at the N terminus with cpmCitrine (acceptor) (Extended Data Fig. 10i). For wild-type LYCHOS, we observed a characteristic hyperbolic curve indicating that the two proteins specifically associate (BRET value at 50% of the maximal signal (BRET50) value of 0.04 ± 0.01) (Extended Data Fig. 10i–k). By contrast, LYCHOS(N145A) showed a right-shifted hyperbolic curve (BRET50 = 0.23 ± 0.06) (Extended Data Fig. 10j,k). The right-shifted acceptor/donor ratio indicates that there is a decrease in affinity between LYCHOS and NPRL2 after mutation of the LYCHOS transporter domain. To investigate further, we tested whether the right-shifted LYCHOS(N145A) mutant could be rescued by removing the Phe352 cholesterol-gating residue. Consistent with the mTORC1 activity assays, the LYCHOS(F352A/W678R/N145A) mutant exhibited a left-shifted hyperbolic curve (BRET50 = 0.03 ± 0.01), compared with LYCHOS(N145A), that was similar to wild-type LYCHOS (Extended Data Fig. 10j,k). Combined with mTORC1 activity assays, these data suggest that the conserved LYCHOS transporter domain works in concert with cholesterol binding to regulate mTORC1 activity.
Discussion
LYCHOS provides an unusual example of a GPCR functioning as a domain within a larger transmembrane assembly. This discovery broadens our understanding of the GPCR superfamily to include GPCR–transporter hybrids. Furthermore, LYCHOS extends examples of the PIN transporter family to humans, with its transporter domains showing greater similarity to plant PIN transporters than to human transporters.
The GPCR and transporter domains seem to jointly coordinate cholesterol sensing. The superposition of LYCHOS across the steps of the vertical elevator motion observed in plant PIN transporters19,20,21 reveals a possible model linking the transporter domain to cholesterol sensing (Fig. 4k–m). At rest, cholesterol is blocked from entering the cholesterol-binding site by the positioning of residues Phe352 and Phe705 (Fig. 4k). During the elevator motion of the PIN-like transporter domain, a translation of one complete helical turn would occur in the LYCHOS crossover motif (Fig. 4l). This translation would reposition the Phe352 gating residue by roughly 7 Å, allowing free cholesterol to enter the open cholesterol-binding site between the transporter and GPCR domains (Fig. 4m). As such, the elevator transport mechanism of the LYCHOS transporter domain, combined with the movement of the GPCR domain, opens the aromatic gate to fully accommodate the entry of cholesterol. Consequently, we hypothesize that metabolite and cholesterol binding combine to form a two-step relay mechanism (Supplementary Discussion 4).
Despite the high level of conservation in the LYCHOS transporter domain, it remains to be determined whether LYCHOS functions as an active transporter and whether the protein has endogenous substrates. Endogenous indole-like candidate substrates are common throughout the human metabolome, suggesting that detailed metabolomic studies across a range of cell lines will be required to characterize LYCHOS transport activity and confirm its links to cholesterol sensing. In addition, capturing structural snapshots of wild-type LYCHOS throughout the transport cycle will be important to characterize the structural basis of transport. These structural insights will also be crucial to delineate the precise molecular mechanisms that coordinate the opening of the Phe352 gate in wild-type LYCHOS to regulate cholesterol entry.
In addition, the mechanism by which cholesterol binding allows LYCHOS to sequester GATOR1 at the lysosomal membrane remains unclear. It is proposed that ICL3 (or LED) of the LYCHOS GPCR domain binds to and sequesters GATOR1 at the lysosomal membrane, enabling mTORC1 activation2. ICL3 is typically the largest intracellular loop in GPCRs, with a highly dynamic conformational landscape that modulates G-protein binding and selectivity30. We were unable to observe ICL3 in the open or closed LYCHOS conformations. Consequently, it is unclear whether parallels can be drawn between typical GPCR ICL3 activity and the proposed direct binding and sequestration of GATOR1 by the LYCHOS ICL3 (LED). Future structural and functional studies will be required to dissect the mechanism of LYCHOS–GATOR1 association and how it is regulated by cholesterol.
In summary, these molecular findings provide a structural framework for delineating the crucial role of LYCHOS as a mTORC1 cholesterol sensor. More broadly, GPCRs and transporters are among the most therapeutically targeted protein families. These discoveries offer new avenues to modulate mTORC1-dependent signalling for disease treatment.
Methods
Protein expression and purification
The gene encoding LYCHOS (GPR155) (Uniprot Q7Z3F1), was synthesized and subcloned into pTwist CMV BG WPRE Neo (Twist Bioscience) with a C-terminal Flag tag for mammalian expression. For insect cell expression, LYCHOS (Uniprot Q7Z3F1) was synthesized and cloned (Genscript) into pFastBac1 (Thermo Fisher Scientific) with a C-terminal Flag tag. All mutants were generated by site-directed mutagenesis (GenScript or in-house) and sequences verified by whole-plasmid sequencing. Throughout our investigation, we routinely compared mammalian-derived and insect-derived recombinant LYCHOS in an attempt to capture different protein conformations using cryo-EM. However, our assessments with cryo-EM, surface plasmon resonance, mass photometry and size-exclusion chromatography revealed no significant differences in LYCHOS behaviour between the two expression systems. SLC10A1 was synthesized and cloned (GenScript) into pFASTBac1 (Thermo Fisher Scientific) with a C-terminal Twin-Strep-tag for insect cell expression.
For mammalian expression, LYCHOS was expressed in Expi293F cells (Thermo Fisher Scientific) and passaged in Expi293 Expression Medium (Thermo Fisher Scientific). Cells were transfected with polyethylenimine (PEI) Max (Polysciences) at a PEI:DNA ratio of 5:1, and a final DNA amount of 1.25 μg ml−1. Recombinant proteins were expressed for 72 h at 37 °C, and cells were supplemented with 5% CO2. Cells were collected by centrifugation and frozen at –80 °C before purification. For insect cell expression, LYCHOS or indicated mutants were expressed in Sf9 cells for three days after infection of each 1-l culture with 4 ml of baculovirus, produced by following the manufacturer’s protocol (Bac-to-Bac, Invitrogen). Cells were collected by centrifugation, washed in phosphate-buffered saline (PBS) and stored at −80 °C until use.
Cells were resuspended in buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 1% (w/v) lauryl maltose neopentyl glycol (LMNG), 0.1% (w/v) CHS and one EDTA-free protease inhibitor cocktail tablet (Roche) per litre of cell culture. The cells were incubated at 4 °C for 3 h with stirring to solubilize the membranes. Subsequently, the solubilized material was centrifuged at 20,000 rpm for 1 h, followed by filtration of the supernatant through a 0.8 µm filter. Anti-Flag G1 affinity resin (GenScript) was added and incubated for 30 min at 4 °C on a horizontal roller. Flag resin was collected by centrifugation, washed and transferred to a gravity flow column (BioRad). The resin was washed with 20 column volumes (CVs) of 0.1% (w/v) LMNG and 0.01% (w/v) CHS buffer, followed by 20 CVs of 0.05% (w/v) LMNG and 0.005% (w/v) CHS buffer. Protein was eluted with 0.1 mg ml−1 Flag peptide (Genscript) in 0.002% (w/v) LMNG and 0.0002% (w/v) CHS buffer. Fractions containing protein were concentrated and further purified by size-exclusion chromatography using a Superose 6 10/300 column (Cytiva) equilibrated with 20 mM HEPES (pH 7.4), 150 mM NaCl, 2 mM DTT, 0.002% (w/v) LMNG and 0.0002% (w/v) CHS. Peak fractions were concentrated by a factor of 40−50 to approximately 20 µM using a centrifugal concentrator with a 100-kDa molecular weight cut-off and snap-frozen before storage at −80 °C. SLC10A1–STREP was expressed in Sf9 cells and purified as described for LYCHOS except that the Flag-affinity step was substituted for Strep-tag purification using a 1-ml StrepTrap XT prepacked column (Cytiva) and the protein was eluted with 50 mM biotin.
Surface plasmon resonance
Surface plasmon resonance experiments were performed using Sf9 cell expressed recombinant LYCHOS on a Biacore T200 (GE Healthcare) in running buffer containing 10 mM HEPES (pH 7.4), 300 mM NaCl, 0.005% (w/v) LMNG, 0.0005% CHS (w/v), 0.005% NP-40 (v/v) and 2% (v/v) dimethyl sulfoxide (DMSO) at 25 °C with a flow rate of 30 µl min−1. To measure the binding of IAA (175.18 g mol−1) and tryptophan (204.23 g mol−1) to LYCHOS, we generated high-density LYCHOS immobilized on a CM7 sensor chip (GE Healthcare). In brief, 1-ethyl-3-(-3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) standard amine coupling was performed by passing LYCHOS at 200 µg ml−1 (1 µM, 193.8 kDa) in sodium acetate buffer (100 mM, pH 4.5) at 10 µl min−1. Immobilization levels of 25,000–30,000 response units (RU) were reached to measure the small-molecule ligands (theoretical RMax ≈ 36–54, respectively, assuming two binding sites). Here, SLC10A1, a known sodium/taurocholate co-transporter with a similar protein fold was chosen as the reference to improve subtraction of baseline and non-specific effects. Immobilized LYCHOS was allowed to stabilize for two to four hours in running buffer at 30 µl min−1 before measurements to ensure a stable baseline. To minimize buffer mismatch effects, stock solutions of tryptophan and IAA were made to 500 mM. The pH was adjusted to 7.4 and twofold serial dilutions of each ligand were made into running buffer. Serial dilutions of IAA and tryptophan (20 mM to 156.3 µM) were injected in ascending order for 60 s and dissociation was monitored for 180 s. Carry-over control injections and needle pre-dipping were performed to prevent sample cross-contamination. Each injection was measured at 10 kHz with a 50% (v/v) DMSO wash after each cycle. Reference and blank subtractions were performed to account for drift, bulk and solvent effects. Affinity, measured from steady-state curves, is expressed as the mean of four to seven experimental replicates. RMax values of 19.8 ± 1.3, 52.4 ± 3.0, 42.8 ± 2.5 and 36.4 ± 2.0 were obtained for immobilized WT (19,544 RU), L177W (30,981 RU), N145A (26,191 RU) and A148W (28,808 RU) variants of LYCHOS, respectively. These values are in line with the expected theoretical RMax, suggesting that the vast majority of immobilized LYCHOS remains in an active state. At least two separate protein preparations were used to conduct affinity measurements, and between five and eight experimental replicates were conducted for each LYCHOS variant. Each experimental replicate consisted of an independent concentration series, which was modelled with a one-to-one binding curve (as defined previously31). Affinity measurements were in excellent agreement between independent protein preparations, sensor chips and IAA solutions.
Cryo-EM sample preparation and data collection
Quantifoil R1.2/1.3 200 mesh copper grids were glow discharged at 30 mA for 30 s using a Pelco easiGlow instrument. Freshly purified LYCHOS (3.5 μl at 10 µM) was immediately applied to grids and rapidly vitrified in liquid ethane. To analyse IAA and tryptophan interactions, solutions of each ligand were made at 100 mM and diluted into the sample to a final concentration of 10 mM. In comparison, an IAA concentration of 15 mM was used to determine the IAA-bound PIN8 structure21. IAA and tryptophan were left to incubate with LYCHOS for 60 s before freezing. In one instance, LYCHOS–IAA was left to incubate for 2 h on ice. The vitrification process was performed with a Vitrobot Mark IV (Thermo Fisher Scientific) after hand blotting with Fisherman Grade 1 filter paper. The temperature was maintained at 4 °C with 100% relative humidity. Data were collected on a Titan Krios G1 (Thermo Fisher Scientific) electron microscope operating at 300 kV with a 50 μm C2 aperture. Micrographs were obtained using a Gatan K3 direct electron detector in counting mode at a nominal EF-TEM magnification of 105,000×, corresponding to a calibrated physical pixel size of 0.8234 Å. A Gatan GIF Quantum energy filter was used with a slit width of 10 eV. The electron dose rate was set to 10.7 electrons pixel−1 s−1, with a total exposure time of 3.77 s and a cumulative dose of 60 electrons Å−2 distributed across 60 frames. Automated collection was performed using EPU (v.2.12.1.2782) with beam shift, capturing 21 images per stage movement. The nominal defocus range was set between −0.5 and −2.0 µm.
Multiple wild-type LYCHOS samples were prepared from either Expi293F or Sf9 tissue cultures for electron microscopy. Overall, these samples were indiscernible. We note that during these comparisons, grid variation and ice thickness (for example, when looking at the same preparation of LYCHOS) affected the stability of the DEP domains, which were prone to denaturing. We performed direct side-by-side comparisons of Sf9 wild-type LYCHOS with (PDB 8U5N) and without (PDB 8U54) IAA, both originating from the same protein preparation. Likewise, direct comparisons were performed between Expi293F-produced wild-type LYCHOS (same batch) with tryptophan (PDB 8U58), with IAA (PDB 8U5Q, 8U5V and 8U5X) or without ligand (PDB 8U56). Here, we report two reconstructions of wild-type LYCHOS, which yielded the highest resolution (PDB 8U56; Expi293F) and the most complete sequence coverage (PDB 8U54; Sf9). We collected an additional two datasets after brief incubations with IAA (PDB 8U5Q, 8U5V and 8U5X; Expi293F) and tryptophan (PDB 8U58; Expi293F). Finally, we investigated the W678R/F352A LYCHOS mutation (PDB 8U5C; Sf9) and LYCHOS–IAA after prolonged incubation (PDB 8U5N; Sf9).
Cryo-EM data processing
A total of six datasets were collected with movies ranging from 4,000 to 16,000. Dose-fractionated movies were corrected for beam-induced motion and compensated for radiation damage within MotionCor2 (v.1.1.0)32. Aligned, dose-weighted averages were subsequently imported into cryoSPARC33 for all further processing. The contrast transfer function (CTF) parameters were estimated with CTFFIND (v.4.1.8)34 or by patch-based CTF estimation. Micrographs lacking Thon rings at 5 Å or better were discarded. In the first instance, multiple rounds of autopicking and blob picking in cryoSPARC33 were performed, followed by particle duplicate removal and two-dimensional (2D) classification. Clean classes were used to train a Topaz35 model. This model was used subsequently for all datasets. In the case of monomeric LYCHOS (long incubation with IAA), an extra round of blob picking was performed to ensure no particles were omitted.
All particles were at first extracted in a 400 × 400-pixel box and downsampled by Fourier cropping to 64 corresponding to a pixel size of around 5 Å pixel−1. These were subjected to multiple rounds of 2D classification in both RELION (v.3.1, 4.0b)36 and cryoSPARC33, yielding particles of sufficient quality and homogeneity for three-dimensional (3D) classification. Typically, this was performed once only to remove clear false positives and contamination. These were re-extracted, centred and downsampled to 128 × 128, for a pixel size of around 2.5 Å pixel−1. This set was subjected to two rounds of ab initio classification, using four classes (maximum and minimum resolution set to 5 Å and 12 Å, respectively; Fourier radius step, 0.08; initial and final minibatch, 1,500) to filter particles. Typically, a single volume was selected for further refinement. Particles were re-extracted at around 1.1 Å pixel−1 and subjected to non-uniform refinement with C2 symmetry, yielding 3.3 Å maps on average.
Particle polishing was performed in RELION36, re-extracting particles for a final pixel size of 1.108 Å pixel−1, and the particles were re-refined. Here, an additional round of ab initio particle classification was performed. In the case of LYCHOS–IAA (short incubation), two populations were evident in which the GPCR domain seemed to sample from two distinct conformations. Symmetry expansion and focused classification were performed, followed by reconstruction without an additional angular search. Conversions between software were performed with EMAN (v.2.2)37, with code written in-house, or by pyem. The Fourier shell correlation (FSC) was used to estimate resolution at the 0.143 threshold. Local resolution was estimated by the windowed blocres FSC method (0.5 threshold) as implemented in cryoSPARC33. Map sharpening in cryoSPARC33 and EMReady38 was performed to assist in residue assignment and model building.
Model building
In the first instance, a computed model of LYCHOS with C2 symmetry was generated by AlphaFold16 using an A6000 GPU with 48 Gb of VRAM. Domains and appropriate interfaces from this model were split and rigid-body fitted into the EMReady38 sharpened map. Several regions were manually rebuilt and adjusted using a combination of Coot39, ISOLDE40 and ChimeraX41. Finally, real space refinement was conducted in PHENIX (v.1.20.1)42 using harmonic potential restraints. All figures, analysis, video renders and visualizations were produced in ChimeraX41 or with Python and Matplotlib.
SSM-based electrophysiology
SSM electrophysiology was performed on a SURFE2R N1 (Nanion Technologies). Electrode sensors (3 mm) were purchased (Nanion Technologies) and prepared according to established protocols21,43. First, to confirm sensor fidelity, capacitance and conductance measurements were performed in non-activating buffer (20 mM HEPES, pH 8.5, 150 mM NaCl and 1 mM MgCl2). Ideal sensors have capacitance between 60 nF and 80 nF and conductivity between 10 nS and 50 nS. Next, sensors were rinsed with ultrapure water, dried under a gentle N2 (g) stream and treated with 50 µl of 0.5 mM 1-octadecanethiol (Sigma) for 30 min at 25 °C or overnight at 4 °C. To prevent evaporation, an inverted Petri dish with a small volume of distilled water was used to form an airtight seal. Thiol solution was removed by inverting the sensor and gently tapping the sensor to filter paper, followed by generous rinsing with 100% (v/v) isopropanol and ultrapure water. The sensor was again dried under a gentle N2 (g) stream. To prepare the SSM, a 2.0 µl droplet of a 7.5 mg ml−1 solution of 1,2-diphytanoylphosphatidylcholine (Avanti) dissolved in n-decane was applied directly to the clean and dry gold surface using a Hamilton pipette, ensuring a uniform coverage of the 3 mm sensor surface. Immediately, 50 µl of non-activating buffer was applied and the SSM was left to assemble at 25 °C for 1 h in an airtight chamber. Sensors were again validated for capacitance and conductance.
To prepare LYCHOS embedded vesicles, 10 mg of soy phospholipid mixture (38% w/v phosphatidylcholine, 30% w/v phosphatidyl ethanolamine, 18% w/v phosphatidyl inositol, 7% w/v phosphatidic acid and 7% w/v other soy lipids; Avanti) was dispended into glass vials and dried under argon (g). Lipid films were then resuspended in activating buffer to 3 mg ml−1 and extruded through a polycarbonate filter (ø 200 nm) to form unilamellar vesicles. Proteoliposomes containing LYCHOS were assembled by the LAiR method44. Here, 5 µg wild-type LYCHOS (Sf9; 3.6 mg ml−1, 17 µM) was diluted into 10 µl liposomes and allowed to incubate for 15 min, before further dilution with 40 µl non-activating buffer. Next, 50 µl proteoliposomes was applied to prepared SSMs and sensors were centrifuged at 4,000g for 30 min at 25 °C in a custom 3D-printed apparatus. Sensors were subsequently washed with non-activating buffer.
Typical single solution exchange experiments were conducted. In brief, a single measurement at a given concentration consisted of a 1-s pulse of non-activating buffer at 200 µl s−1 to establish a baseline, immediate (<30 ms) injection of a 1-s pulse of activating buffer to induce transient currents, and a 1-s pulse of non-activating buffer to exchange the system back to a resting state (total of 3 s). After each measurement, a 2-s pulse with non-activating buffer was performed to equilibrate the sensor. Current transients were recorded for a full concentration series of IAA (0–30 mM). For each concentration series, a corresponding blank sensor measurement series was performed. Measurements were performed on at least two individual sensors. Each concentration series was conducted independently for a total of four experimental replicates (as described previously21). Finally, to describe the peak currents in response to IAA, we fitted a Michaelis–Menten curve to the peak currents, after blank subtraction and normalization (I/IMax, to account for different quantities of LYCHOS across sensors).
Mass photometry
To assess the stoichiometry and mass distribution of LYCHOS samples, standard mass photometry landing assays were conducted at 20 °C using the TwoMP instrument (Refeyn) on an active anti-vibration platform. Silicone gaskets and glass coverslips were purchased from Refeyn. Glass coverslips were cleaned by washing in sonication baths of 100% (v/v) isopropanol, ultrapure water and, finally, by plasma glow discharge. To avoid noise originating from LMNG micelles, each sample was maintained in 20 mM HEPES, 150 mM NaCl, 0.002% (w/v) LMNG and 0.0002% (w/v) CHS until measurement. Samples were then diluted 20-fold into detergent-free buffer before immediately measuring a 60–120 s image series. The final protein concentration at the time of measurement was 20 nM. Standards of bovine serum albumin (Merck) and apoferritin (Merck) were measured on the same day to calibrate extracted particle contrast to mass. Image series were acquired with an 8 ms exposure (128 Hz) at 488 nm, with a set field of view of 12 × 17 µm. Frame and pixel binning were applied, with a factor of 3 and 6, respectively, for an effective pixel size of 72 nm. Analysis and acquisition were performed using the Refeyn AquireMP and DiscoverMP packages (v.2.5). To investigate the effect of prolonged incubation of LYCHOS with IAA, samples (Sf9 or Expi293F-derived) were incubated with or without IAA for 2 h at room temperature. Here, we observed no difference between Sf9 or Expi293F-derived protein after incubation with IAA. Time-zero samples were obtained by measuring each reaction immediately before the addition of 10 mM IAA.
Cell culture and cDNAs for transfection
HEK293 cells (ATCC; negative for mycoplasma contamination, not authenticated) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% (v/v) fetal bovine serum (FBS). The sequence for the IAA FRET biosensor, AuxSen, was a gift from M. Kolb27 and the biosensor was commercially synthesized (Genscript) into pcDNA3.1 (Thermo Fisher Scientific). The pcDNA3-LysoTORCAR construct was a gift from J. Zhang (Addgene plasmid 64929; http://n2t.net/addgene:64929; RRID: Addgene_64929). For LysoTORCAR assays, LYCHOS or LYCHOS mutants were cloned into pcDNA 3.1 (Thermo Fisher Scientific) with a C-terminal Flag tag. For AuxSen efflux assays, LYCHOS was synthesized and cloned into pTwist CMV BG WPRE Neo (Twist Bioscience) with a C-terminal Flag tag. As a positive control for AuxSen assays, the gene encoding PIN8 (Uniprot Q9LFP6), was synthesized and subcloned into pTwist CMV BG WPRE Neo (Twist Bioscience) with an N-terminal Flag tag. LYCHOS-Flag-NLuc and HA-cpmCitrine-NPRL2 constructs were generated in-house and cloned into pcDNA 3.1 (Thermo Fisher Scientific) by Gibson assembly. The genes for cpmCitrine and NLuc were subcloned from the cAMP BRET biosensor, CAMYEN45, a gift from B. Hoare. LYCHOS genes were obtained from pFASTBac1 expression vectors (see ‘Protein expression and purification’) and NPRL2 was subcloned from pRK5 HA-NPRL2, a gift from D. Sabatini and K. Shen (Addgene plasmid 99709; http://n2t.net/addgene:99709; RRID :Addgene_99709). All LYCHOS-NLuc variants were generated in-house using overlap extension PCR, Gibson assembly or QuickChange mutagenesis. The construct for β2-adrenoceptor-NLuc was a gift from C. W. White and S. J. Hill.
AuxSen efflux assay
HEK293 cells were transfected and seeded in suspension in six-well plates using 25 kDa linear PEI at a ratio of 1:6 DNA:PEI. The cells were co-transfected with a FRET biosensor for IAA, AuxSen27 (200 ng per well) and empty vector control, LYCHOS–Flag (WT) or Flag–PIN8 at 1 μg per well, to mislocalize LYCHOS and PIN8 to the plasma membrane (Extended Data Fig. 7k; as described previously19,21). After 24 h, cells were reseeded into a poly-d-lysine-coated black, optically clear 96-well plate (Perkin Elmer ViewPlate), and left to adhere for 24 h. On the day of the experiment, medium was removed and cells were equilibrated in Hank’s balanced salt solution (HBSS; Invitrogen) at room temperature. Fluorescence imaging was performed using a high-content Perkin Elmer Operetta with an Olympus LUCPlanFLN 20× NA 0.45 objective and Harmony software (v.4.8) as previously described46, with some modifications. For emission ratio analysis, cells were excited sequentially (410–430 nm excitation filter) with emission measured using 520–560 nm and 460–500 nm emission filters. Cells were imaged every 1 min. Before imaging, 25 μM NPA was added to PIN8-transfected cells, as stated. Baseline emission ratio images were captured for 5 min before the addition of 10 μM IAA, and influx emission ratio images were captured for 15 min (to allow IAA influx to reach a stable plateau). IAA was then removed, and cells were placed into HBSS without or with 25 μM NPA (as stated), before efflux ratio images were captured for 25 min. Data were analysed as described previously46 using in-house automated macros within the Fiji distribution of ImageJ (v.2.14.0/1.54f)47. Cells with a greater than 10% change in F/F0 (FRET ratio relative to baseline for each cell) after IAA influx were selected for analysis using Microsoft Excel (v.16.45). Data are expressed as the average emission ratio relative to the total IAA influx (15 min after IAA addition) and efflux (25 min after buffer exchange) for each cell (F/FInflux), and averaged over each biological replicate. To determine the rate of efflux (K), data were fitted using a plateau followed by one-phase decay non-linear regression model in GraphPad Prism (v.10.0.3), with X0 constrained to 0, Y0 constrained to 1 and plateau constrained to 0. Data shown were obtained from three independent biological replicates.
LysoTORCAR mTORC1 activity assay
HEK293 cells were co-transfected in six-well plates with a lysosomally localized FRET biosensor for mTORC1, LysoTORCAR29 (500 ng per well), and empty vector control, LYCHOS–Flag (WT), LYCHOS–Flag (Y57A), LYCHOS–Flag (F352A), LYCHOS–Flag (F352A/W678R), LYCHOS–Flag (N145A) or LYCHOS–Flag (F362A/W678R/N145A) (500 ng per well), using X-tremeGENE at a 1:3 DNA:reagent ratio. After 24 h, cells were replated into a black, optically clear 96-well plate (Perkin Elmer ViewPlate), pre-coated with poly-d-lysine. After six hours, cells were serum starved overnight in DMEM (no FBS). On the day of the experiment, cells were amino acid starved for 1 h in HBSS at 37 °C before fluorescence imaging using a high-content Perkin Elmer Operetta with an Olympus LUCPlanFLN 20× NA 0.45 objective and Harmony software (v.4.8). Cells were excited sequentially (410–430 nm excitation filter) with emission measured using 520–560 nm and 460–500 nm emission filters. Cells were imaged every one minute. Baseline emission ratio images were captured for 5 min before complete buffer exchange to phenol red-free DMEM alone (control), or containing 1.3% (w/v) methyl-β-cyclodextrin (MβCD) (cholesterol depletion) or 0.1% (w/v) MβCD/50 μM cholesterol complexes (cholesterol addition)2,48. Emission ratio images were captured for 40 min, before complete buffer exchange for MβCD-treated cells only, to 0.1% (w/v) MβCD/50 μM cholesterol complexes (cholesterol repletion). Emission ratio images were captured for an additional 30 min. Data were analysed as for the AuxSen FRET biosensor using Fiji (v.2.14.0/1.54f) and in-house automated macros. Addition of phenol red-free DMEM caused a change in auto-fluorescence which was corrected using Microsoft Excel (v.16.45). Data are expressed as the average change in FRET ratio relative to baseline for each cell (F/F0), and averaged over each biological replicate. The AUC and the fold change induced by a stimulus (average of five time points at peak response relative to baseline, or relative to the last five time points of MβCD treatment for repletion) were calculated using GraphPad Prism (v.10.0.3). Data shown were obtained from three or four independent biological replicates.
LYCHOS–GATOR1 saturation BRET assay
HEK293 cells were transfected in six-well plates using 25-kDa linear PEI at a ratio of 1:6 DNA:PEI. Cells were co-transfected with 25 ng per well LYCHOS-NLuc WT, N145A, F352A/W678R or F352A/W678R/N145A, or 25 ng per well β2-adrenoceptor-NLuc (negative control), and increasing amounts of cpmCitrine-NPRL2 (0 ng, 135 ng, 270 ng, 405 ng, 810 ng or 1,200 ng). The total amount of DNA was made up to 1,225 ng per well using empty vector. After 24 h, cells were reseeded in quadruplicate into a poly-d-lysine-coated white opaque 96-well plate (Perkin Elmer CulturePlate) at 5 × 104 cells per well. After another 24 h, cells were washed and equilibrated with assay buffer (10 mM HEPES in HBSS, pH 7.4 at 37 °C) for 25 min. Cells were then incubated for 5 min at 37 °C with the NanoGlo substrate (1:1,000; Promega N1120). BRET measurements were obtained with the LUMIstar microplate reader (BMG Labtech; Omega control software v.6.20), which enabled simultaneous measurement of luminescence (NLuc; 445–505 nm) and cpmCitrine emission (505–565 nm) in individual wells. At the end of the experiment, total cpmCitrine fluorescence was measured with an excitation of 482–512 nm, dichroic filter set at 517.2 nm and emission at 520–560 nm using the CLARIOstar microplate reader (BMG Labtech, SMART Control software v.4.20) to determine the relative amount of cpmCitrine-NPRL2 expressed in cells. BRET ratios were calculated as follows: cpmCitrine emission (505–565 nm)/NLuc emission (445–505 nm). Data are expressed as the net BRET ratio relative to the ratio of cpmCitrine/NLuc emission for each transfection condition (relative expression of each protein in the cells), and were fitted using a non-linear regression one-site-specific binding model in GraphPad Prism (v.10.0.3) to obtain BRET50 (Kd) and BRETMax (Bmax) values. For comparisons between LYCHOS mutants, data are expressed as the net BRET relative to the BRETMax for each condition, determined from the non-linear regression one-site-specific binding model. Data are from three to five independent biological replicates.
Western blot expression analysis
To confirm equivalent expression of LYCHOS–Flag WT or mutants for the LysoTORCAR experiments, HEK293 cells were transfected with the same amount of DNA in six-well plates (500 ng per well LYCHOS–Flag WT, Y57A, F352A or N145A) using X-tremeGENE (as described above). After 48 h, cells were collected in ice-cold PBS, and pellets were resuspended in ice-cold RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% v/v Triton X-100, 0.5% w/v sodium deoxycholate, 0.1% w/v SDS, protease inhibitor cocktail, phosphatase inhibitor cocktail, 1 mM benzamidine and 1 mM PMSF). Cells were incubated on ice with agitation for 20 min, then lysates were centrifuged at 14,000g for 10 min at 4 °C, and supernatants were recovered. Total protein concentration was quantified using a Pierce BCA Protein Assay Kit, and 35 μg was run on 4–15% Mini-PROTEAN pre-cast gels (BioRad). Proteins were transferred to nitrocellulose membranes by electroblotting using the Trans-Blot Turbo Transfer System (BioRad) and the STANDARD SD transfer protocol. The membranes were blocked with 5% (w/v) BSA, 0.02% (w/v) sodium azide in Tris-HCl-buffered saline (TBS) with 0.1% (v/v) Tween-20 (TBS-T) for 1 h. The blots were incubated overnight at 4 °C with mouse anti-Flag (Merck, F3165; 1:5,000), or rabbit anti-β-tubulin (Cell Signaling Technology 2146; 1:1,000) in 5% (w/v) BSA, 0.02% (w/v) sodium azide in TBS-T. Blots were washed three times with TBS-T before incubation with IRDye 680RD goat anti-mouse and IRDye 800CW goat anti-rabbit fluorescent secondary antibodies (LICOR 926-68070 and 926-32211) at a 1:15,000 dilution in 5% (w/v) BSA, 0.02% (w/v) sodium azide in TBS-T for 1 h. Blots were then washed three times in TBS-T and once in TBS, and bands were visualized with an Amersham Typhoon 5 (control software v.3.0.0.2). Densitometry of bands was determined with Fiji (v.2.14.0/1.54f). Data are expressed relative to the loading control (β-tubulin) and are from three independent biological replicates.
Confocal imaging for plasma-membrane localization of Flag-tagged LYCHOS and PIN8
To confirm that some LYCHOS–Flag and Flag–PIN8 were mislocalized to the plasma membrane for the AuxSen experiments (and not under normal transfection conditions), HEK293 cells were transfected and seeded in six-well plates with LYCHOS–Flag (WT) or Flag–PIN8 (both 500 ng per well or 1 μg per well) in suspension using 25-kDa linear PEI at a ratio of 1:6 DNA:PEI. After 24 h, cells were reseeded into a black optically clear 96-well plate (Perkin Elmer PhenoPlate). After 24 h, cells were washed three times in ice-cold PBS, before fixation in 4% (v/v) paraformaldehyde for 15 min at room temperature. After three washes with PBS (5 min each), cells were blocked using blocking buffer (5% v/v normal goat serum in PBS with 0.3% v/v Triton X-100) for one hour at room temperature, before incubation at room temperature for one hour with mouse anti-Flag antibody (Merck, F3165; 1:1,000) in antibody dilution buffer (1% w/v BSA, 0.3% v/v Triton X-100 in PBS). Cells were washed three times with PBS (5 min each), then incubated for one hour at room temperature with goat anti-mouse Alexa488 secondary antibody (Abcam, ab150113; 1:1,000) in antibody dilution buffer. After three final washes in PBS, cells remained in PBS until imaging. Imaging was performed using a Leica TCS SP8 confocal microscope equipped with a water immersion 40× HC PL APO CS2 1.10 NA objective. An OPSL 488 laser (498–622 nm emission) was used to image the anti-mouse AlexaFluor 488 secondary antibody bound to the anti-Flag M2 primary antibody. All transfection conditions were performed in duplicate and three fields of view were captured per well. Representative single-cell images are shown.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The three-dimensional cryo-EM density maps have been deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb/) under accession numbers EMD-41913, EMD-41912, EMD-41916, EMD-41929, EMD-41930, EMD-41934, EMD-41935 and EMD-41914, The coordinates are deposited in the PDB (https://www.rcsb.org) with accession numbers 8U56, 8U54, 8U5C, 8U5N, 8U5Q, 8U5V, 8U5X and 8U58. All other data supporting the findings of this study are available within the paper and its Extended Data. Source data are provided with this paper.
References
Settembre, C. & Perera, R. M. Lysosomes as coordinators of cellular catabolism, metabolic signalling and organ physiology. Nat. Rev. Mol. Cell Biol. 25, 223–245 (2024).
Shin, H. R. et al. Lysosomal GPCR-like protein LYCHOS signals cholesterol sufficiency to mTORC1. Science 377, 1290–1298 (2022).
Liu, G. Y. & Sabatini, D. M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203 (2020).
Luo, J., Yang, H. & Song, B. L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 21, 225–245 (2020).
Meng, Y., Heybrock, S., Neculai, D. & Saftig, P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 30, 452–466 (2020).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Duran, R. V. & Hall, M. N. Regulation of TOR by small GTPases. EMBO Rep. 13, 121–128 (2012).
Huang, J. & Manning, B. D. The TSC1–TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 412, 179–190 (2008).
Bar-Peled, L. et al. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106 (2013).
Shen, K. et al. Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature 556, 64–69 (2018).
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P. & Guan, K. L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10, 935–945 (2008).
Sancak, Y. et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008).
Xiao, J. et al. 25-Hydroxycholesterol regulates lysosome AMP kinase activation and metabolic reprogramming to educate immunosuppressive macrophages. Immunity 57, 1087–1104 (2024).
Alexander, S. P., Mathie, A. & Peters, J. A. Guide to Receptors and Channels (GRAC), 3rd edition. Br. J. Pharmacol. 153, S1–S209 (2008).
Wootten, D. & Miller, L. J. Structural basis for allosteric modulation of class B G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 60, 89–107 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Berman, H., Henrick, K. & Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 10, 980 (2003).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2024).
Yang, Z. et al. Structural insights into auxin recognition and efflux by Arabidopsis PIN1. Nature 609, 611–615 (2022).
Su, N. et al. Structures and mechanisms of the Arabidopsis auxin transporter PIN3. Nature 609, 616–621 (2022).
Ung, K. L. et al. Structures and mechanism of the plant PIN-FORMED auxin transporter. Nature 609, 605–610 (2022).
Goutam, K., Ielasi, F. S., Pardon, E., Steyaert, J. & Reyes, N. Structural basis of sodium-dependent bile salt uptake into the liver. Nature 606, 1015–1020 (2022).
Teale, W. D., Paponov, I. A. & Palme, K. Auxin in action: signalling, transport and the control of plant growth and development. Nat. Rev. Mol. Cell Biol. 7, 847–859 (2006).
Roager, H. M. & Licht, T. R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 9, 3294 (2018).
Sadik, A. et al. IL4I1 Is a metabolic immune checkpoint that activates the AHR and promotes tumor progression. Cell 182, 1252–1270 (2020).
Tintelnot, J. et al. Microbiota-derived 3-IAA influences chemotherapy efficacy in pancreatic cancer. Nature 615, 168–174 (2023).
Herud-Sikimic, O. et al. A biosensor for the direct visualization of auxin. Nature 592, 768–772 (2021).
Fantini, J., Epand, R. M. & Barrantes, F. J. Cholesterol-recognition motifs in membrane proteins. Adv. Exp. Med. Biol. 1135, 3–25 (2019).
Zhou, X. et al. Dynamic visualization of mTORC1 activity in living cells. Cell Rep. 10, 1767–1777 (2015).
Sadler, F. et al. Autoregulation of GPCR signalling through the third intracellular loop. Nature 615, 734–741 (2023).
Rich, R. L. & Myszka, D. G. in Label‐Free Technologies for Drug Discovery (eds Cooper, M. & Mayr, L. M.) 1–25 (2011).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
He, J., Li, T. & Huang, S. Y. Improvement of cryo-EM maps by simultaneous local and non-local deep learning. Nat. Commun. 14, 3217 (2023).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D 74, 519–530 (2018).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Bazzone, A. & Barthmes, M. Functional characterization of SLC transporters using solid supported membranes. Methods Mol. Biol. 2168, 73–103 (2020).
Godoy-Hernandez, A. et al. Rapid and highly stable membrane reconstitution by LAiR enables the study of physiological integral membrane protein functions. ACS Cent. Sci. 9, 494–507 (2023).
Monnich, D. et al. Activation of multiple G protein pathways to characterize the five dopamine receptor subtypes using bioluminescence technology. ACS Pharmacol. Transl. Sci. 7, 834–854 (2024).
Halls, M. L., Poole, D. P., Ellisdon, A. M., Nowell, C. J. & Canals, M. Detection and quantification of intracellular signaling using FRET-based biosensors and high content imaging. Methods Mol. Biol. 1335, 131–161 (2015).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Klein, U., Gimpl, G. & Fahrenholz, F. Alteration of the myometrial plasma membrane cholesterol content with β-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 34, 13784–13793 (1995).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Pandy-Szekeres, G. et al. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 46, D440–D446 (2018).
Xiao, P. et al. Tethered peptide activation mechanism of the adhesion GPCRs ADGRG2 and ADGRG4. Nature 604, 771–778 (2022).
Dates, A. N. et al. Heterogeneity of tethered agonist signaling in adhesion G protein-coupled receptors. Cell Chem. Biol. 31, 1542–1553 (2024).
Ashkenazy, H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016).
Acknowledgements
C.B.-J. is an Australian Research Council (ARC) DECRA Fellow (DE240100992). M.L.H. is a Viertel Senior Medical Research Fellow supported by The Cross Family and The Frank Alexander Charitable Trusts. A.M.E. is a Victorian Department of Health and Human Services Victorian Cancer Agency Mid-Career Fellow (MCRF21036). We thank M. Papaioannou and B. Liu for assistance with tissue culture; X. Xu for assistance with cholesterol addition and depletion experiments; M. Belousoff for GPCR structural advice; and A. Bazzone and F. Tadini-Buoninsegni for discussion and advice on SSM electrophysiology measurements. This research was supported by equipment funded by an Australian Research Council Grant (LE170100016). The authors acknowledge the use of instruments and assistance at the Monash Ramaciotti Centre for Cryo-Electron Microscopy, a Node of Microscopy Australia; the use of the services and facilities of Micromon Genomics; and the use of instruments at the Monash Institute of Pharmaceutical Sciences Imaging and FACS Facility at Monash University. This work was supported by the MASSIVE HPC facility. We also acknowledge the office of the Vice-Provost for Research and Research Infrastructure (VPRRI) at Monash University and of Bioplatforms Australia (BPA) as part of the National Collaborative Research Infrastructure Strategy (NCRIS).
Author information
Authors and Affiliations
Contributions
C.B.-J., C.J.L., M.L.H. and A.M.E. conceived the study. C.B.-J., C.J.L., Y.-G.C., A.C.K., G.D.J., W.L., C.Q. and A.M.E. performed cloning, protein expression, purification and mass photometry. C.J.L. prepared cryo-EM grids. C.J.L. and H.V. collected cryo-EM data. C.B.-J. processed cryo-EM data. C.B.-J., C.J.L. and A.M.E. built, refined and analysed cryo-EM models. C.B.-J. performed and analysed surface plasmon resonance experiments. C.B.-J. and R.J.C. designed, performed and analysed SSM-based electrophysiology experiments. C.M. and M.L.H. performed and analysed AuxSen biosensor and confocal imaging experiments. S.D. and M.L.H. performed and analysed LysoTORCAR biosensor and western blotting experiments. A.C.K. performed and analysed BRET assays. All authors wrote and drafted the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 LYCHOS purification and characterization.
a, Size-exclusion chromatography (Superose 6 10/300; Cytiva) of recombinant LYCHOS. Elution volume is approximately 14.3 ml. The peak fraction is indicated with grey shading. b, Gradient SDS-polyacrylamide gel electrophoresis of recombinant purified LYCHOS. Representative of >10 independent purifications. c, Trypsin digest and MS/MS of purified recombinant LYCHOS. Sequence coverage (~75%) of experimentally detected peptides is highlighted (coloured according to Fig. 1b) with some larger peptides being undetected. d, Mass photometry frequency distribution of wild-type LYCHOS. e, The 2.75 Å resolution reconstruction of wild-type LYCHOS. A single subunit is coloured from N- to C-terminus (yellow to purple), and the symmetrically related subunit is shown in white. f, Pipes and planks representation of e with numbered transmembrane helices.
Extended Data Fig. 2 Cryo-EM analysis.
a, High-level overview of the image analysis pipeline including key parameters and analysis variables. b, Overview of representative 2D class averages, ab initio classification volumes, refinement mask and final reconstructed cryo-EM volumes.
Extended Data Fig. 3 Cryo-EM volumes and validation.
a, Raw and denoised cryo-EM micrograph of vitrified LYCHOS. Scale bar 100 nm. Representative of >50,000 micrographs. b, Two-dimensional class averages calculated using prior assigned Euler angles without additional alignments. Scale bar 100 Å. c, Selected regions of cryo-EM map and model agreement showing side-chain density (grey) and corresponding model (blue: scaffold, magenta: transporter, orange: GPCR). d, Per-voxel local resolution variation. All volumes are coloured by the same uniformly sampled (0.5 Å increments) resolution range (2.5 to 4.5 Å). e, Map-to-model (solid green), unmasked (grey dashed line), and noise-substituted corrected (solid purple) gold-standard half-map Fourier shell correlation plots. f, Polar plots of normalized angular distribution frequency for the final refined particle sets.
Extended Data Fig. 4 The LYCHOS GPCR domain is most closely related to class B2 adhesion GPCRs.
a, Multiple sequence alignment of the LYCHOS GPCR domain and its related homologues. Residues with high identity are shaded magenta. Here, for clarity, the loop sequences are omitted. b, Structural comparison of class B2 adhesion GPCRs and the LYCHOS GPCR domain (oriented by normal GPCR convention). Three related GPCR paralogues were identified by performing pBLAST49 against human GPCR sequences (GPCRdb50), namely ADGRG4 (E-value 3.1 × 10−7), ADGRG6 (E-value 4.3 × 10−6), and ADGRC1 (E-value 9.1 × 10−5). No other significant matches were identified. c, Pipes and planks representation of the LYCHOS GPCR domain labelled with the overall structural helical numbering. The standard GPCR transmembrane numbering convention is included in parentheses. The LYCHOS GPCR domain has been resolved in the apo state in an apparently inactive conformation. d, View from the lysosomal lumen perpendicular to the membrane plane of the LYCHOS GPCR orthosteric ligand-binding site showing a superimposed cartoon of LYCHOS (orange) and ADGRG4 (white) (PDB: 7WUJ51; RMSD 4.6 Å). ADGRG4 is in the active state, the tethered agonist binding footprint is shown in dark shading. See also Supplementary Discussion 1. e, Alignment of the cytoplasmic-facing LYCHOS G protein-binding pocket with the ADGRG1/2/4 active states reveals likely steric clashes between Gβ and the DEP domains and between Gα and LYCHOS α15/α16 (GPCR TM5/6). LYCHOS α15 (GPCR TM5) and α16 (GPCR TM6) are comparatively larger than TM5/6 of the adhesion GPCRs and extend beyond the membrane boundary to define the ICL3 (LED) region. f, LYCHOS α16 (GPCR TM6) and α17 (GPCR TM7) do not adopt the kinked conformation necessary to accommodate G protein binding. g, The glycine of the PXXG motif required to support the kinked TM6 conformation to enable G protein binding15,52 is conserved in LYCHOS (LYCHOS α16, GPCR TM6). See also Supplementary Discussion 1.
Extended Data Fig. 5 LYCHOS scaffold and DEP domain interfaces.
a, Surface representation of the interface between LYCHOS scaffold domains coloured by per-residue conservation (ConSurf53). Buried surface area (BSA) of 1410 Å2 indicated by the dotted line. b, Cartoon representation of a, key interface residues are labelled. The interface is predominantly hydrophobic with key conserved glycine residues that symmetrically arrange to form tight contact interfaces (Gly72, Gly247, Gly254). c,d, Surface representation (c) of the DEP domain interface, and cartoon representation (d). BSA of 469 Å2. Key residues are labelled that define a conserved hydrophobic core. Surface residues Tyr805, His758, Leu761, Trp789, Gly794 are conserved and define the DEP interface.
Extended Data Fig. 6 The LYCHOS scaffold and transporter domains are PIN orthologues.
a, Comparison of LYCHOS and superpositions of the Arabidopsis thaliana PIN transporters (PINs), H. sapiens SLC9 and SLC10 homologues and N. meningitdis ASBT homologue. The scaffold domain is divergent between LYCHOS and the SLC9 and SLC10 families with poor overall structural superposition. In contrast, the scaffold domain of PINs and LYCHOS is conserved. b, The transporter unit is structurally conserved between all families, each with a canonical crossover motif. Greatest similarity of LYCHOS is to the PIN transporter family. c, Simplified helical topology schematic for LYCHOS compared to the PIN, SLC9 and SLC10 families, and N. meningitidis ASBT. The overall topological arrangement and sequence of helices is conserved between LYCHOS and the PINs, while the SLC families achieve the same overall fold with a different arrangement. d, The overall Coulomb surface of LYCHOS and PIN8 is conserved. Surface representation coloured by electrostatic potential charge ( − 10 to 10 kT/e). e, Structural superposition of the conserved crossover motif between LYCHOS and PIN1, PIN3, and PIN8 (PDB 7Y9V19, 7XXB20, and 7QPA21). The experimentally determined binding pose of IAA in the four structures is shown. The overall position of IAA is consistent and mediated by a key conserved asparagine (LYCHOS Asn145). f, Multiple sequence alignment of the crossover motif between LYCHOS, PINs, SLC9, and SLC10 families, and N. meningitidis ASBT. The conserved motif in LYCHOS is most closely related to the PINs. While the conserved motif is also present in the SLC families (including N. meningitidis ASBT), the conserved asparagine and proline are contributed by different helices owing to distinct topological arrangements (as shown in c). In LYCHOS and the PIN family, the conserved asparagine and proline are contributed by the first and second helices of the crossover motif (asparagine first, then proline). While the SLC9 and 10 families possesses the same conserved residues, the sequence order is inverted.
Extended Data Fig. 7 LYCHOS binds IAA but not tryptophan.
a, Surface plasmon resonance of IAA binding to wild-type LYCHOS (apparent Kd 1.62 mM, pKd 0.19 ± 0.04, n = 8), showing raw sensorgrams and concentration-response curves. Steady-state affinity fit assumes one-to-one binding. b, Superimposed sequence of electrogenic transient peak currents induced by wild-type LYCHOS embedded proteoliposomes upon injection of increasing concentrations of IAA (0 to 30 mM). c, Normalized peak current versus concentration of IAA, corresponding to (b). LYCHOS has a Michaelis–Menten constant of 13.7 ± 3.4 mM (95% CI; n = 4 independent experimental replicates), consistent with anion binding but likely not transport across the bilayer. d, Sliced top-down (cytoplasmic side) view of two reconstructions of LYCHOS that were incubated with 10 mM IAA or tryptophan, respectively. e, Focused view of the recessed cavity in the LYCHOS transporter domain showing additional density corresponding to IAA. No density was observed for tryptophan. f, Side-chain level contacts between IAA and LYCHOS are predominantly hydrophobic van der Waals interactions. The key asparagine forms hydrogen bonds with the carboxylate of IAA. Additional hydrogen bonds are observed between IAA and the main-chain amine groups. g, Cartoon representation of the LYCHOS transporter domain coloured by per-residue conservation (ConSurf53) highlighting the IAA binding pocket. Inset: Focused surface representation view of (f), showing that the IAA binding pocket is highly conserved. h–j,Site-directed mutagenesis of the conserved IAA binding pocket reduces the affinity of IAA for LYCHOS, with an increase in the apparent Kd for N145A (h; apparent Kd 5.49 mM, pKd 0.74±0.02, n = 5, p = 0.0003 vs wild-type), L177W (i; apparent Kd 3.93 mM, pKd 0.59±0.02, n = 8, p = 0.000001 versus wild-type) and A148W (j; apparent Kd 3.38 mM, pKd 0.52±0.03, n = 8, p = 0.0023 vs wild-type). RU, response units (n = 5–8 independent experimental replicates). One-way ANOVA with Tukey’s multiple comparisons test; pKd reported as mean±SEM. All RMax values are within the expected theoretical limit assuming two binding sites per LYCHOS molecule. k, Representative images of HEK293 cells transfected with LYCHOS–Flag or Flag–PIN8 at either 500 ng/well or 1 μg/well. Scale bar shows 10 μm. Transfection with 500 ng/well (as for LysoTORCAR assays, Fig. 4a–j and Extended Data Fig. 10b–g) leads to patterning consistent with a limited localization to intracellular membranes, whereas over-transfection with 1 μg/well (as for AuxSen efflux assays, Fig. 2h,i) caused a mislocalization of LYCHOS and PIN8 to additional cellular membranes, allowing us to measure transport of IAA by PIN8 across the plasma membrane. l, Tryptophan was unable to specifically bind LYCHOS wild-type or mutant variants as determined using surface plasmon resonance (n = 4 independent experimental replicates).
Extended Data Fig. 8 Cholesterol recognition is mediated by a CRAC-motif in a highly conserved pocket.
a, Sliced top-down (cytosolic face) view of two LYCHOS reconstructions with CHS (red) bound (F352A/W678R) and without CHS (wild-type). Two CHS molecules populate the interface between the GPCR and transporter domains. b, Focused view of the cryo-EM density of the cholesterol-binding region of LYCHOS (F352A/W678R) positioned at the interface of the GPCR and transporter domains. c, Wild-type LYCHOS is in a closed conformation with the GPCR domain clamped shut, occluding the LYCHOS cholesterol sensing pocket. Relaxation of the GPCR domain from the closed state to the open state is necessary to accommodate cholesterol entry. d, Surface representation of the LYCHOS cholesterol sensing pocket coloured by per-residue conservation (ConSurf53). A region of high surface conservation is apparent, with virtually all conserved residues defining a deep groove that accommodates cholesterol (innermost CHS molecule; white) e, Cartoon representation of d with the single innermost CHS moiety shown. Neighbouring key residues are labelled with colours according to conservation (as in d). The CRAC-motif (L/V-X1-5-Y-X1-5-K/R28) is defined by three residues (Val53, Tyr57, Arg61) on LYCHOS α1. Key residues Cys55 and Glu48 identified by photolabelling2 are indicated (yellow stars). Residues Phe43, Tyr57 and Pro44, previously identified through mutagenesis studies2 are indicated. f, A two-dimensional ligand interaction diagram showing a projected view of the key residues that define interactions in the cholesterol-binding pocket in e. Here a cholesterol molecule is modelled. g, After long incubation with IAA (2 h) a strictly monomeric LYCHOS population was observed by cryo-EM (see Supplementary Discussion 2). The 3.0 Å resolution cryo-EM reconstruction of the LYCHOS monomer in complex with IAA is shown. LYCHOS is coloured according to Fig. 1a and the micelle is shown as a white overlay. h,i, Short incubation (1 min) with IAA resulted in a mixed distribution of a closed (h) and partially open (i) conformational states by cryo-EM. In the closed state, a single CHS molecule with partial denisty is observed in a queued position, where due to Phe352, it is blocked from entering the LYCHOS cholesterol sensing pocket. j, In the monomeric state, α2 of the scaffold domain undergoes a subtle conformational rearrangement resulting in steric clashes at the dimer interface incompatible with dimer formation. k, Focused cartoon of α2 of the scaffold domain (monomer is coloured) superimposed with the same region of dimeric wild-type LYCHOS (white). l, No change was observed in the mass photometry histogram, indicating that IAA did not clearly promote the monomeric conformation of LYCHOS seen under cryo-EM. See Supplementary Discussion 2.
Extended Data Fig. 9 Conformational changes of the GPCR–transporter interface and Phe352 gate.
a, IAA-bound LYCHOS undergoes a conformational relaxation of the GPCR domain, characterized by rotation and partial opening. The transition between the closed and partially open state is only observed for wild-type LYCHOS upon incubation of IAA. b, Lateral movement of the whole GPCR domain (rigid-body transformation) is associated with the relaxation transition. c, Superposition of the middle and CRAC-motif CHS molecules from the LYCHOS (F352A/W678R) structure into the closed conformation (wild-type LYCHOS). Clear steric clashes (pink) are present due to the closed Phe352 gate. A single queuing CHS molecule is blocked from entry. d, As in c, showing the middle and CRAC-motif CHS molecules in the partially open conformation. Visible clashes suggest that alternate rotamer positions alone are not sufficient to allow cholesterol entry. e, The LYCHOS (F352A/W678R) structure showing that both CHS molecules (middle and CRAC-motif) are accommodated by bypassing the Phe352 gate and a lateral movement of α17. f, Focused illustration of F705 and F352 residues that contribute the most significant steric clashes in the closed (c) and partially open (d) conformations. These clashes are resolved in the fully open and Phe352 bypassed (F352A/W678R) structure (e). g, All three observed CHS molecules are shown with the wild-type and F352A/W678R LYCHOS structures superimposed. Cryo-EM density is shown for each CHS molecule. More complete density, potentially due to more extensive contacts, is visible for the CRAC-motif CHS molecule. By contrast, the queuing CHS is only partially resolved.
Extended Data Fig. 10 The LYCHOS transporter domain coordinates a potential gating mechanism for cholesterol.
a, Relative expression of LYCHOS–Flag WT, Y57A, F352A and N145A compared to empty vector control, and of LYCHOS–Flag WT compared to F352A/W678R or F352A/W678R/N145A in HEK293 cells after transfection with 500 ng/well as for the LysoTORCAR assay (this figure and Fig. 4a–j). Upper panel shows quantified densitometry of the LYCHOS–Flag band relative to β-tubulin loading control. Symbols show values from independent biological replicates (n = 3), the bold line shows the mean and error bars show SEM. Lower panel shows a representative western blot. For western blot source data, see Supplementary Figs. 1 and 2. b,d,f, mTORC1 activity in response to cholesterol addition or depletion (at 0 min, dotted line) followed by repletion (at 40 min, arrow) in HEK293 cells co-transfected with LysoTORCAR and empty vector control (b; n = 4), LYCHOS(Y57A) (d; n = 3), or LYCHOS(F352A) (f; n = 4). Symbols show the mean and error bars show SEM from independent biological replicates. c,e,g, Fold change in mTORC1 activity in response to cholesterol addition, depletion (average F/F0 at -5-0 min vs 35–40 min for red and blue curves, respectively), or repletion (average F/F0 at 35–40 min vs 5 min average at the peak after repletion) calculated from b,d,f. h, Control-subtracted area under the curve (AUC) in response to cholesterol addition, calculated from 0–40 min from f and Fig. 4a,c,f,h. For bar graphs, symbols show values from independent biological replicates, the bold line shows the mean and error bars show SEM; p-values were calculated by one-way ANOVA with Šidák’s multiple comparisons test (for h, and comparisons to control in c,e,f) or two-sided paired t-test (for comparisons between depletion and repletion in c,e,f). See also Supplementary Discussion 3. Saturation BRET in HEK293 cells transfected with increasing amounts of cpmCitrine-NPRL2 (BRET acceptor) and a low, constant amount of the BRET donors i, LYCHOS-NLuc (n = 3) or β2-adrenoceptor-NLuc (β2AR-NLuc; a G protein-coupled receptor that we used as a negative control; n = 3), or j, LYCHOS-NLuc WT (n = 5), N145A (n = 3), F352A/W678R (n = 3), or F352A/W678R/N145A (n = 3). Symbols show values from n independent biological replicates. k, BRET50 values calculated from the curves shown in (j). Symbols show values from independent experiments, the bold line shows the mean and error bars show SEM; p-value was calculated by one-way ANOVA with Šidák’s multiple comparisons test. The large and saturating increase in BRET between LYCHOS and NPRL2 (i) is consistent with specific interactions between LYCHOS and GATOR1 under growth conditions (normal cell culture medium). In contrast, there is minimal BRET observed between NPRL2 and the β2-adrenoceptor which, when not activated by ligand, predominantly resides at the plasma membrane.
Supplementary information
Supplementary Information
This file contains Supplementary Discussions 1–4 and Supplementary Figures 1&2.
Supplemantary Video 1
Summary video of LYCHOS structure and function.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bayly-Jones, C., Lupton, C.J., Keen, A.C. et al. LYCHOS is a human hybrid of a plant-like PIN transporter and a GPCR. Nature 634, 1238–1244 (2024). https://doi.org/10.1038/s41586-024-08012-9
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41586-024-08012-9
This article is cited by
-
Molecular architecture of human LYCHOS involved in lysosomal cholesterol signaling
Nature Structural & Molecular Biology (2025)
-
Cholesterol finds its pocket in LYCHOS
Nature Structural & Molecular Biology (2025)
-
Structural insights into cholesterol sensing by the LYCHOS-mTORC1 pathway
Nature Communications (2025)
-
Mechanisms of auxin action in plant growth and development
Nature Reviews Molecular Cell Biology (2025)
-
Structural basis for cholesterol sensing of LYCHOS and its interaction with indoxyl sulfate
Nature Communications (2025)
