
Abstract
Chimeric antigen receptor (CAR) natural killer (NK) cell immunotherapy offers a promising approach against cancer1,2,3. However, the molecular mechanisms that regulate CAR-NK cell activity remain unclear. Here we identify the transcription factor cyclic AMP response element modulator (CREM) as a crucial regulator of NK cell function. Transcriptomic analysis revealed a significant induction of CREM in CAR-NK cells during the peak of effector function after adoptive transfer in a tumour mouse model, and this peak coincided with signatures of both activation and dysfunction. We demonstrate that both CAR activation and interleukin-15 signalling rapidly induce CREM upregulation in NK cells. Functionally, CREM deletion enhances CAR-NK cell effector function both in vitro and in vivo and increases resistance to tumour-induced immunosuppression after rechallenge. Mechanistically, we establish that induction of CREM is mediated by the PKA–CREB signalling pathway, which can be activated by immunoreceptor tyrosine-based activation motif signalling downstream of CAR activation or by interleukin-15. Finally, our findings reveal that CREM exerts its regulatory functions through epigenetic reprogramming of CAR-NK cells. Our results provide support for CREM as a therapeutic target to enhance the antitumour efficacy of CAR-NK cells.
Similar content being viewed by others


Main
NK cells have a crucial role in immune surveillance and are able to eliminate abnormal cells independently of major histocompatibility complex class I restriction or antigen priming4. Despite their inherent antitumour activity, NK cells often exhibit functional impairment in the tumour microenvironment secondary to immunosuppressive factors and other tumour escape mechanisms5. The targeting of checkpoints predominantly linked to T cell dysfunction has resulted in substantial advances in immuno-oncology6. However, detailed investigations into the mechanisms that govern NK cell dysfunction are limited. Although the suppressive roles of checkpoints such as TIGIT, TIM3, NKG2A and LAG3 in NK cells are well established7, the effects of others such as PD-1 and CTLA4 on NK cells remain controversial8. Moreover, research has predominantly focused on the targeting of surface receptors, with limited studies of the role of transcription factors that negatively regulate immune responses.
CAR-NK cells have emerged as a promising immunotherapy for both haematological and solid tumours1. Most clinically advanced CAR-NK cell constructs incorporate interleukin-15 (IL-15) to extend the short lifespan of NK cells3. This approach facilitates strong NK cell activation through CAR-associated immunoreceptor tyrosine-based activation motif (ITAM) signalling after antigen binding9, whereas IL-15 promotes proliferation and persistence10. Encouraging clinical outcomes have been reported with NK cells that express both a CD19-directed CAR and IL-15 (CAR19–IL-15)2,3, and CAR-NK cells that target other antigens are in development1. Although some checkpoints of IL-15-mediated NK cell activation have been identified11,12,13, the mechanisms that regulate NK cell function downstream of combined CAR-associated ITAM and cytokine signalling remain largely unknown.
Here we identify the PKA–CREB–CREM axis as a key regulatory pathway in CAR-NK cells that are activated by both IL-15 and CAR signalling. Targeting CREM significantly enhances the antitumour efficacy of CAR-NK cells, thereby offering a new strategy to improve cell therapy.
CREM is induced in CAR-NK cells in vivo
We have previously shown that CAR19–IL-15 NK cells derived from cord blood exert potent activity in vivo14. We also analysed their single-cell transcriptional signatures after adoptive transfer in a non-curative mouse model of lymphoma developed using Raji cells14 (Fig. 1a). To further investigate the regulatory processes that govern the function of CAR19–IL-15 NK cells in vivo, we delved deeper into these single-cell RNA sequencing (scRNA-seq) data. Uniform manifold approximation and projection (UMAP) clearly distinguished the transcriptional profiles of CAR19–IL-15 NK cells before and after infusion (Fig. 1b). Analyses of differentially expressed genes in CAR19–IL-15 NK cells after infusion revealed notable differences (Extended Data Fig. 1a), with the top upregulated genes including those that encode transcription factors such as CREM, FOS and ASCL2, among others. We focused our attention on CREM given the well-established immunomodulatory role of the cAMP pathway and recent growing interest in targeting it to augment immunotherapy15,16,17,18. Although previous research has highlighted the inhibitory effects of CREM in T cells and macrophages15,19,20, its specific role in NK cells, especially in the context of CAR-NK cell antitumour activity, remains underexplored.

a, Schematic of the in vivo experiment for the scRNA-seq data14 from the Raji mouse model treated with CAR19–IL-15 NK cells (n = 2 mice each, euthanized on day 7 and day 14). i.v., intravenous. Created in BioRender. Rafei, H. (2025) https://BioRender.com/9xq1zl4. b, UMAP depicting CAR19–IL-15 NK cells before (green) and after (red) infusion (data pooled from day 7 and day 14 after infusion). c, Quantification of CREM expression before and after infusion. d,e, Expression of select genes encoding NK cell activation markers (d) or inhibitory markers (e) before (green) and after (red) infusion. f, Schematic of the constructs encoding CAR70, CAR70.3ζ.Y6F and CD27(ECD). ICD, intracellular domain; TMD, transmembrane domain. g, CREM expression in NT and CAR-transduced NK cells with or without CD70 antigen stimulation as assessed by qPCR (n = 4 donors). h, CREM expression in NT NK cells stimulated with IL-15 as assessed by qPCR (n = 7 donors). i, Schematic of the constructs encoding IL-15, CAR70–IL-15 and CAR70. j, t-SNE CUDA analysis of the phenotypic signature of NK cells from NT, IL-15, CAR70–IL-15 and CAR70 NK cells after co-culture with UMRC3 cells at an effector-to-target (E/T) ratio of 1:1 as assessed by mass cytometry (n = 3 donors). k, Contour plots showing t-SNE CUDA cluster prevalence in the indicated NK cell conditions. l, Heatmap of expression levels of NK cell markers across NK cell clusters. The expression of each marker is represented by the robust z score for expression normalized across the clusters, with a colour scale ranging from blue (z = −1) to orange (z = +1) and the size of the circle for percentage of expression. Statistical comparisons were performed using two-sided Wilcox rank-sum tests (c–e), two-way analysis of variance (ANOVA) with Tukey’s correction (g), one-way ANOVA with Tukey’s correction (h) or one minus Pearson’s hierarchical clustering (l). Data are presented as the mean ± s.e.m. NS, not significant.
Elevated CREM expression in post-infusion samples (Fig. 1c and Extended Data Fig. 1b,c) correlated with optimal tumour control in vivo14. This response was characterized by an increased expression of effector genes such as GZMB (which encodes granzyme B), GZMH and GNLY, and genes related to calcium signalling, including S100A10, S100A6 and S100A4 (Fig. 1d and Extended Data Fig. 1d). Of note, these cells showed upregulation of genes associated with NK cell inhibition, including ADGRG1, KIR2DL3 and KLRG1 (Fig. 1e). CREM expression in CAR19–IL-15 NK cells remained consistent across all cell cycle stages (Extended Data Fig. 1e,f). By contrast, analyses of IL-15 activity using CytoSig showed that it was significantly elevated after infusion (Extended Data Fig. 1g,h) and varied by cell cycle, with the highest levels observed in S phase (Extended Data Fig. 1i). We also observed a significant positive correlation between IL-15 activity and CREM expression after infusion, whereby both were increased (Extended Data Fig. 1j). Collectively, these data suggest that CREM is induced in CAR-NK cells during peak antitumour activity and is associated with a transcriptional profile that balances both activation and regulatory functions.
CAR and IL-15 stimulation induce CREM
We next asked whether CREM is also induced in CAR-NK cells that target other antigens. To test this possibility, we engineered NK cells to express a CD70-targeting CAR using the CD27 receptor extracellular domain (ECD) (the natural receptor for CD70) fused to CD28 and CD3ζ signalling endodomains without an IL15 transgene (CAR70; Fig. 1f). We also designed and tested CAR variants with altered signalling: one with mutated CD3ζ ITAM phosphorylation sites (CAR70.3ζ.Y6F) and another expressing only the CD27 ECD and therefore without intracellular signalling (CD27(ECD)); Fig. 1f and Extended Data Fig. 2a–c). Stimulation with CD70 antigen increased CREM expression only in CAR70 NK cells, but not in non-transduced (NT) NK cells or those expressing CARs that lack intracellular signalling (Fig. 1g). This result confirmed the important role of ITAM signalling in CREM induction.
We also investigated the effect of IL-15 on CREM induction in NK cells after cytokine starvation. IL-15 treatment resulted in a dose-dependent (50–5,000 pg ml–1) increase in CREM expression (Fig. 1h) that was more substantial than with equivalent doses of IL-2 (Extended Data Fig. 2d) and was abrogated by IL-15 blockade (Extended Data Fig. 2e). Moreover, stimulation of CAR70 NK cells with both CD70 antigen and exogenous IL-15 resulted in an additive increase in CREM expression (Extended Data Fig. 2f). Flow cytometry analyses revealed that both IL-15 receptor (IL-15R) expression and CREM levels increased with elevated IL-15 concentrations (Extended Data Fig. 2g), with significantly greater CREM expression in IL-15R+ than in IL-15R− NK cells (Extended Data Fig. 2h).
We next studied the kinetics of CREM induction after IL-15 stimulation and its relationship with Ki67. Although Ki67 increased at 48 h, CREM expression rose earlier and increased by 24 h (Extended Data Fig. 2i,j). CREM mRNA was upregulated as early as 6 h, peaked at 24–48 h and returned to baseline by 96 h (Extended Data Fig. 2k). This finding indicated the occurrence of transient IL-15-induced expression. However, in our CAR–IL-15 system, in which IL-15 is constitutively secreted, CREM remained consistently high after transduction and after expansion with feeder cells and IL-2 treatment, which indicated that it was stably expressed (Extended Data Fig. 2l).
CREM exists in more than 40 isoforms in humans, generated through distinct promoters, transcription factor activity and alternative splicing21. Some isoforms act as activators (for example, CREMτ, CREMτ1, CREMτ2 and CREMτα), whereas others function as repressors (for example, CREMα, CREMβ and CREMγ) depending on the presence or absence of transactivation domains22. By contrast, the inducible cAMP early repressor (ICER) isoforms arise from an alternative promoter, lack transactivation domains and function exclusively as repressors23 (Extended Data Fig. 3a).
We quantified CREM-specific and ICER-specific transcripts in CAR-NK cells and after IL-15 stimulation (Methods). At baseline, NK cells expressed higher levels of CREM-specific transcripts than ICER-specific transcripts (Extended Data Fig. 3b). Compared with NT NK cells, CAR70–IL-15 NK cells showed upregulation of CREM-specific transcripts and, to a lesser extent, ICER-specific exon 2 transcripts (Extended Data Fig. 3b). Similarly, IL-15 induced CREM-specific transcripts in a dose-dependent manner (Extended Data Fig. 3c). Using an antibody that recognizes all CREM gene products24,25, we confirmed expression of both CREM and ICER isoforms in NK cells, with CAR–IL-15 NK cells showing increased expression of multiple CREM isoforms above 20 kDa, including CREMα, CREMβ, CREMγ and CREMτ isoforms (Extended Data Fig. 3d,e).
Collectively, these findings indicate that both CAR ITAM signalling and IL-15 are potent inducers of CREM in NK cells, thereby highlighting their complex interplay in regulating NK cell activity.
Cytokines and receptor signalling induce CREM
We examined the effects of various cytokines on CREM expression. IL-2, IL-12, IL-18 and IL-15 induced a dose-dependent increase in CREM, with IL-15 producing the strongest response. IL-10 and IL-21 had minimal effects on CREM levels (Extended Data Fig. 4a). To assess the role of ITAM signalling through endogenous NK cell receptors, we stimulated NK cells with anti-CD16, anti-NKp30 and anti-NKp46 antibodies (Extended Data Fig. 4b), which similarly upregulated CREM expression (Extended Data Fig. 4c). These results suggest that CREM upregulation is a general feature of NK cell activation that is driven by both cytokine and ITAM-based signalling and with stimulus-specific differences in strengths and kinetics.
CREM is upregulated in tumour-infiltrating NK cells
We next explored CREM expression in the context of NK cell exhaustion in the tumour microenvironment. Analyses of scRNA-seq datasets from the TISCH2 database revealed broad CREM expression in tumour-infiltrating immune cells, especially NK cells, T cells and monocytes and macrophages, but minimal expression in malignant or stromal cells. Tumour-infiltrating NK (TI-NK) cells showed higher CREM expression than NK cells in peripheral blood mononuclear cells (PBMCs), especially those from bladder cancer, cholangiocarcinoma and lung cancer (Extended Data Fig. 5a). This pattern held across multiple cancer types in the TI-NK cell atlas, in which CREM was consistently increased in TI-NK cells compared with PBMC NK cells26 (Extended Data Fig. 5b).
To further validate these findings, we analysed TI-NK cells from nine patients with pancreatic cancer treated at the MD Anderson Cancer Center, including five primary tumours and four metastatic lesions27. CREM expression was highest in metastatic TI-NK cells compared with PBMCs or primary tumours (Extended Data Fig. 5c), and ranked among the top 50 differentially active regulons in the transcriptome of metastatic TI-NK cells (Extended Data Fig. 5d,e).
Analyses of data from The Cancer Genome Atlas (TCGA) showed that high CREM expression generally correlated with poor survival in several cancers, including low-grade glioma, lung adenocarcinoma, stomach adenocarcinoma and breast invasive carcinoma (Extended Data Fig. 5f).
Finally, CREM was enriched in mature NK1 clusters from an atlas of NK cell heterogeneity28 (Extended Data Fig. 5g). Collectively, our findings indicate that CREM is prominently expressed in TI-NK cells across various cancers and that, in a healthy context, its expression is associated with a more mature NK cell phenotype.
CREM shapes the NK cell phenotype
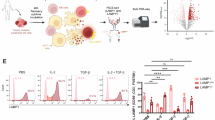
Next, we performed mass cytometry to determine the phenotype of CREM-expressing NK cells. NK cells expressing IL-15 without a CAR (IL-15 NK cells), CAR70 (no IL-15) or IL-15-secreting CAR70 (CAR70–IL-15; Fig. 1i) were cultured together with CD70+ UMRC3 cells. CAR70–IL-15 NK cells consistently expressed the highest levels of CREM, followed by CAR70 and IL-15 NK cells (Extended Data Fig. 6a–c). Six distinct clusters emerged (clusters 1–6), with clusters 1 (highest CREM expression), 2 and 4 predominantly found in CAR70–IL-15, CAR70 and IL-15 NK cells, respectively (Fig. 1j,k and Extended Data Fig. 6d–g).
Cluster 1 was characterized by higher levels of activation markers (NKG2D, CD25, CD2, GZMB and perforin) and inhibitory markers (NKG2A, LAG3, TIGIT and TIM3; Fig. 1l). Similar but more moderate patterns were observed in cluster 2 (most abundant in CAR70 NK cells) and cluster 4 (most abundant in IL-15 NK cells). By contrast, cluster 5 (most abundant in NT cells) had a signature of suppression of both activation and inhibitory markers (Fig. 1l and Extended Data Fig. 6h). CREM was associated with higher expression of both activation and inhibitory markers across all conditions (Extended Data Fig. 6i), a result that supports the link between CREM induction and a phenotype of activation-induced exhaustion.
Collectively, our data support the notion that CREM is preferentially induced by strong NK cell activation, such as through CAR signalling and IL-15 stimulation, and its expression is associated with a complex immunophenotype characterized by both activation and exhaustion.
CREM is induced through the PKA–CREB axis
CREM is regulated through the cAMP signalling pathway29, in which increases in intracellular cAMP levels activate protein kinase A (PKA), which in turn phosphorylates transcription factors such as CREB and CREM. These factors then bind cAMP-response elements to regulate the transcription of target genes30. Thus, to determine whether the canonical PKA–CREB axis is involved in CREM induction after NK cell activation, we measured phosphorylated CREB (pCREB) levels in NK cells after CAR or IL-15 stimulation. CAR stimulation resulted in marked CREB phosphorylation in CAR70 NK cells but not in NT or CAR-NK cells with mutated or absent CD3ζ signalling (CAR70.3ζ.Y6F and CD27(ECD), respectively; Fig. 2a,b). These data paralleled previous observations of CREB phosphorylation by PKA downstream of T cell receptor (TCR) signalling31.

a, Schematic of experiments for b–d. Created in BioRender. Rafei, H. (2025) https://BioRender.com/cczoc8f. b, pCREB expression measured by phospho-flow in NT, CAR70, CAR70.3ζ.Y6F and CD27(ECD) NK cells that were treated with (+) or without (–) CD70 antigen for stimulation, H89 (PKA inhibitor) or EGTA (calcium chelator) (n = 5 donors). c, FACS plots of pCREB in CAR70 NK cells under the indicated conditions. Forskolin (FSK) was used as the positive control. d, CREM expression by qPCR in the indicated NK cell conditions treated with or without CD70 antigen, H89 or EGTA for 24 h (n = 3 donors). e, Schematic of the experiments in f–j. Created in BioRender. Rafei, H. (2025) https://BioRender.com/j6djzbh. f,g, FACS plots (f) and quantification (g) of pCREB in NT NK cells that were unstimulated or stimulated for 30 min with increasing concentrations of IL-15 (n = 14 donors). h, Whole-cell lysates from NT NK cells that were treated as in f and g were analysed by western blotting for pCREB and total CREB. β-actin served as the loading control. A representative blot is shown. i, Densitometry analysis quantifying the relative band intensity of pCREB normalized to total CREB (n = 5 donors). j, CREM expression by qPCR in NT NK cells that were stimulated or not for 24 h with increasing concentrations of IL-15 in the presence or absence of H89 (n = 3 donors). k, ChIP–qPCR for the enrichment of CREB in the promoter region of CREM and a region of no enrichment (negative control) in the indicated conditions (n = 3 donors). l, ChIP–qPCR for the enrichment of pCREB in the promoter region of CREM in NT NK cells incubated for 6 h in the absence or presence of increasing concentrations of IL-15 (n = 3 donors). Statistical comparisons were performed using two-way ANOVA with Bonferroni correction (b,d), one-way ANOVA with Tukey’s correction (g,i) or two-sided multiple t-tests with Holm–Šídák correction (j–l). Data are presented as the mean ± s.e.m.
Pretreatment of NK cells with H89, a PKA inhibitor, strongly inhibited CAR-induced CREB phosphorylation and CREM induction after CAR ligation (Fig. 2b–d). Given the established function of calcium as an activator of PKA32,33 and its pivotal role in the immune cell activation cascade34, we also chelated calcium with EGTA. Treatment with EGTA led to a consistent suppression of CAR-induced CREB phosphorylation and CREM induction (Fig. 2b–d). Similarly, we observed activation of this pathway in NK cells stimulated through ITAM-associated CD3ζ downstream of CD16, NKp30 and NKp46, which was effectively inhibited by PKA or calcium blockade (Extended Data Fig. 7a,b). Moreover, IL-15 resulted in a dose-dependent increase in PKA activity (Extended Data Fig. 7c) and pCREB levels in NK cells (Fig. 2e–i), whereas CREB phosphorylation and the subsequent increase in CREM expression were effectively blocked by H89 (Fig. 2j and Extended Data Fig. 7d,e) and EGTA (Extended Data Fig. 7f,g). Together, these data provide support for a pivotal role of the PKA–CREB axis in CREM upregulation in response to CD3ζ or IL-15 stimulation.
The parallel patterns of CREB phosphorylation and CREM induction after CAR ligation or IL-15 stimulation suggested that CREM might be a direct target of CREB. Chromatin immunoprecipitation and quantitative PCR (ChIP–qPCR) assays confirmed strong enrichment of CREB on the CREM promoter (promoter 1; Extended Data Fig. 3a) in CAR70–IL-15 NK cells (Fig. 2k). Similarly, treatment of NK cells with increasing doses of IL-15 led to a dose-dependent increase in binding of pCREB to the CREM promoter (Fig. 2l). These patterns of CREB and pCREB enrichment correlated with CREM transcription levels in these cells under the same conditions (Fig. 1h and Extended Data Fig. 6a), which strongly indicated that CREM is directly regulated by CREB.
IL-15 activates NK cells through the JAK–STAT pathway, specifically STAT3 and STAT5. IL-15 induced a dose-dependent increase in pSTAT3 and pSTAT5 in NT NK cells as assessed by western blot analysis, with pSTAT5 showing greater sensitivity (Extended Data Fig. 7h,i). By contrast, CAR activation alone did not induce pSTAT3 or pSTAT5. At baseline, only CAR70–IL-15 NK cells showed pSTAT3 and pSTAT5 bands, which was probably driven by autocrine IL-15 signalling (Extended Data Fig. 7j,k). To determine the role of STAT3 and STAT5 in IL-15-mediated CREM induction, we assessed CREM expression after IL-15 stimulation in wild-type (WT), STAT3 knockout (KO; Extended Data Fig. 7l), STAT5A and STAT5B (STAT5A/B) KO (Extended Data Fig. 7m) and CREM KO NK cells. CREM levels were partially reduced in both STAT3 KO and STAT5A/B KO NK cells (Extended Data Fig. 7n), which indicated that both these factors are involved in CREM induction. ChIP analysis did not reveal significant pSTAT3 binding at the CREM promoter (promoter 1; Extended Data Fig. 3a), whereas STAT5B binding was significantly enriched after IL-15 treatment (Extended Data Fig. 7o,p).
Our findings indicate that both STAT3 and STAT5 regulate CREM expression, either indirectly (STAT3) or directly through promoter binding (STAT5). Notably, CREM induction also occurred independently of STAT signalling, specifically after CAR stimulation in the absence of pSTAT3 and pSTAT5, and instead depended on the PKA–pCREB axis (Fig. 2b–d and Extended Data Fig. 7j,k). These findings collectively highlight a dual mechanism of CREM regulation in CAR-NK cells through the PKA–CREB signalling axis and through STAT3–STAT5 downstream of IL-15.
CREM KO enhances CAR-NK cell potency
Our analyses indicated a complex interplay among CAR activation, cytokine signalling and CREM induction. To determine whether CREM functions as a negative regulator in CAR-NK cells, we used CRISPR–Cas9 to KO CREM (an exon shared by most of the non-ICER isoforms) in two CAR-NK cell models: (1) CAR70–IL-15 NK cells (KO efficiency is shown in Extended Data Fig. 3d) against CD70+ tumour models (the renal cell carcinoma cell line UMRC3, the patient-derived xenograft (PDX) cell line BCX.010 of breast cancer and the Raji cell line of Burkitt lymphoma) and (2) TROP2-targeting CAR-NK cells (CAR.TROP2–IL-15; KO efficiency is shown in Extended Data Fig. 3e) against TROP2+ cancer lines (the pancreatic cancer cell line PATC148 and the ovarian cancer cell line SKOV3). CREM KO significantly enhanced the cytotoxicity of CAR–IL-15 NK cells in long-term two-dimensional and three-dimensional tumour spheroid cultures and in tumour rechallenge assays, particularly at low effector-to-target ratios (Fig. 3a–d, Extended Data Fig. 8a–d and Supplementary Videos 1–4).

a, Spheroid killing assay of GFP+ UMRC3 cells by CREM WT NT and CREM WT or KO CAR70–IL-15 NK cells (n = 3 donors). The spheroid green image mean determines spheroid growth over time. The black arrow indicates the time of NK cell addition. The bar graph shows the area under the curve (AUC) for the normalized spheroid green image mean. b, Representative images over time of the spheroid assay in a. c, Tumour rechallenge assay with CREM WT NT and CREM WT or KO CAR70–IL-15 NK cells against Raji–mCherry. Tumour cells were added every 2–3 days (black arrows; n = 3 donors). d, Impedance killing assay of PATC148 cells by CREM WT NT and CREM WT or KO CAR.TROP2–IL-15 NK cells over time (n = 3 donors). The cell index (tumour growth) was normalized to the NK cell addition time (black arrow). e, Tumour rechallenge assay with CREM WT or KO NT, IL-15, CAR70–IL-15 and CAR70 NK cells against UMRC3 cells using xCELLigence (n = 3 donors). The black arrow indicates the time of NK cell addition. RC, tumour rechallenge, indicated by vertical black lines. The cell index was normalized to the time of NK cell addition, and negative values were transformed to 0. f, Percentage of tumour necrosis factor (TNF) and interferon γ (IFNγ) (intracellular staining) response of the indicated CREM WT or KO NK cells after 6 h of incubation with UMRC3 cells (1:1 E/T ratio; n = 3 donors). g,h, Spheroid killing assay of GFP+ UMRC3 cells by CREM WT or KO NT, IL-15, CAR70–IL-15, CAR70, CAR70.3ζ.Y6F and CD27(ECD) NK cells (2:1 E/T ratio; n = 3 donors). Data were quantified and normalized as in a. Images in h were taken on day 3 of the assay. Statistical comparisons were performed using two-way ANOVA with Tukey’s correction (a (left),c,d,e,g), one-way ANOVA with Tukey correction (a, right) or one-way ANOVA (Fisher’s least significance difference test; f). Data are presented as the mean ± s.e.m. Scale bars, 400 µm (b,h).
Furthermore, CREM KO enhanced the cytotoxicity of IL-15, CAR70 and CAR70–IL-15 NK cells in tumour rechallenge assays (Fig. 3e and Extended Data Fig. 8e) and their ability to produce cytokines (Fig. 3f), with no effect on NT NK cells. Together, these data suggest that NK cell activation through CAR and/or IL-15 stimulation drives CREM expression to act as an intracellular checkpoint.
To further elucidate the contributions of CAR signalling and IL-15 on CREM induction, we tested the activity of NK cells expressing different constructs, including IL-15, CAR70 and CAR70–IL-15, as well as those with impaired (CAR70.3ζ.Y6F) or absent (CD27(ECD)) CD3ζ endodomain signalling, on UMRC3 spheroids. CREM KO enhanced CAR-NK cell cytotoxicity in conditions in which CAR activation or IL-15 signalling was present, with the greatest effect observed when both pathways were active (CAR70–IL-15; Fig. 3g,h).
We tested CAR-NK cell cytotoxicity against CD70 KO UMRC3 cells (Extended Data Fig. 8f). CREM KO enhanced the effector function of IL-15-secreting NK cells, regardless of tumour expression of the cognate antigen, but had no effect on CAR70 NK cells without IL-15 (Extended Data Fig. 8g). This result suggests that CREM acts as an inhibitory checkpoint downstream of IL-15 stimulation or CAR signalling after antigen engagement. CREM KO did not have a significant effect on the function of NT NK cells; however, after exposure to increasing concentrations of exogenous IL-15, CREM KO led to enhanced cytotoxicity against tumour targets (Extended Data Fig. 8h).
Together, these data point to an important role for CREM as a potent intracellular immune checkpoint that restrains NK cell antitumour activity in response to either CAR or IL-15 stimulation.
CREM KO improves CAR-NK cell in vivo activity
We evaluated the impact of CREM KO on CAR-NK cell function in vivo using three different NOD/SCID IL-2Rγnull (NSG) mouse models of haematological and solid tumours. To model NK cell exhaustion, we used a mouse model of aggressive CD70+ Raji Burkitt lymphoma and treated these mice with long-term cultured (23-day-old) NT, CREM WT or KO CAR70–IL-15 NK cells at a subtherapeutic dose (Fig. 4a). Treatment with CREM KO CAR70–IL-15 NK cells led to significantly enhanced tumour control compared with CREM WT CAR70–IL-15 NK cells, NT NK cells or untreated controls (Fig. 4b,c), and resulted in improved survival (Fig. 4d). Moreover, CREM KO led to increased proliferation of CAR70–IL-15 NK cells in peripheral blood (Fig. 4e) and greater infiltration into the lungs, liver, bone marrow and spleen (Fig. 4f). Notably, the administration of CREM KO CAR70–IL-15 NK cells did not cause toxicity, as evidenced by stable body weights in treated mice (Extended Data Fig. 9a). We also studied the dynamics of ex vivo function and cytokine production of CREM WT and KO CAR70–IL-15 NK cells. By day 10 after infusion, CREM WT CAR70–IL-15 NK cells acquired a dysfunctional phenotype, with reduced cytotoxicity against targets in Raji and K562 cells compared with the original infusion product (Extended Data Fig. 9b–e). By contrast, CREM KO CAR70–IL-15 NK cells maintained their cytotoxic function at this early time point (Extended Data Fig. 9b–e). However, by day 20, CREM KO CAR70–IL-15 NK cells also showed signs of dysfunction (Extended Data Fig. 9b–e). Serum analyses for human cytokines revealed increased levels of TNF, IFNγ, granzyme family members, perforin and other effector molecules in mice treated with CREM KO CAR70–IL-15 NK cells, particularly at later time points, which indicated different dynamics of cytotoxicity and cytokine production (Extended Data Fig. 9f).

a, Schematic of the experimental plan for the Raji mouse model. Created in BioRender. Rafei, H. (2025) https://BioRender.com/83ud7ql. b,c, Bioluminescence imaging (b) and quantification (c) of tumour burden over time (n = 5 mice per group). d, Kaplan–Meier survival curves. e,f, Quantification of CAR+ NK cells (human CD45+ (hCD45+)CD56+CD16+CD27+) in the blood (e) and the indicated organs (f) of mice 10 or 20 days after NK cell infusion by flow cytometry (n = 5 mice per group). g, Schematic of the experiment for the BCX.010 metastatic PDX model (the timed euthanasia and survival experiments were performed independently using three donors, one for the timed euthanasia and two for survival). Created in BioRender. Rafei, H. (2025) https://BioRender.com/8dgkmca. h, Number of metastatic nodules in mice in the indicated groups at day 35 (n = 5 mice per group). i, Representative FACS plots of human NK cells (hCD45+) at day 35 after NK cell treatment in the blood of mice treated with CREM WT or KO CAR70–IL-15 NK cells. j, Flow cytometry analysis of hCD45+CD56+CD16+CD27+ cells in the blood of mice 10, 20 or 35 days after NK cell infusion (n = 5 mice per group). k,l, Representative fused and deconvoluted images (k) and quantification (l) of immunohistochemistry (IHC) staining of hCD45 (green) and GZMB (red) in adjacent serial lung sections (k) or sections from metastatic sites (lung and liver) (l) of mice at day 35 after NK cell infusion (n = 5 mice per group). Scale bars, 100 µm. m, Kaplan–Meier survival curves. Data were pooled from two donors (n = 4 mice in BCX.010 alone group and n = 10 mice in each of the NT, CREM WT CAR70–IL-15 and CREM KO CAR70–IL-15 groups with 5 mice per donor). Statistical comparisons were performed using two-way ANOVA with Tukey’s correction (c), log-rank test (Mantel–Cox; d,m), two-way ANOVA with Šídák’s correction (e,f,j), one-way ANOVA (uncorrected Dunn’s test; h) or two-sided Mann–Whitney test (l). Red P values indicate CREM WT versus KO CAR70–IL-15 NK cell group comparisons. Data are presented as the mean ± s.e.m.
In a metastatic CD70+ breast cancer PDX model, NSG mice were engrafted with BCX.010 cells and received intravenous treatment of NT, CREM WT CAR70–IL-15 or CREM KO CAR70–IL-15 NK cells 7 days later or were left untreated (Fig. 4g). All mice were euthanized 35 days after NK cell infusion, and tumour burden in the lungs and liver was assessed by histopathology. Mice treated with CREM KO CAR70–IL-15 NK cells showed significantly lower tumour burden than all control groups (Fig. 4h and Extended Data Fig. 9g). CREM KO also led to increased proliferation of CAR-NK cells in peripheral blood and greater infiltration into the lungs and liver (Fig. 4i–l), accompanied by elevated GZMB expression, a result consistent with an activated NK cell phenotype (Fig. 4k,l). In an independent experiment using the same model, CREM KO CAR70–IL-15 NK cells led to increased survival of mice compared with controls (Fig. 4m). Notably, treatment with CREM KO cells was well tolerated, with no weight loss observed (Extended Data Fig. 9h), and again led to increased CAR-NK cell proliferation in peripheral blood (Extended Data Fig. 9i).
These results were further validated in an orthotopic TROP2+ PATC148 mouse model of pancreatic cancer (Extended Data Fig. 9j). Mice were euthanized 36 days after NK cell infusion for analyses. Intraperitoneal injection of CREM KO CAR.TROP2–IL-15 NK cells led to improved tumour control (Extended Data Fig. 9k) and in vivo systemic persistence (Extended Data Fig. 9l).
Targeting inhibitory checkpoints in NK cell activation raises the possibility of unwanted toxicity. We further investigated the safety of CREM KO CAR-NK cells using our BCX.010 mouse model. In brief, mice with BCX.010 tumours were treated intravenously with either CREM WT or KO CAR70–IL-15 NK cells. As a control, we infused CREM KO CAR70–IL-15 NK cells into mice without tumours. Two weeks later, during active NK cell proliferation, mice were euthanized and histological analyses of major organs (liver, lungs and kidneys) did not show notable abnormalities attributable to NK cell therapy in mice treated with either CREM WT or KO CAR70–IL-15 NK cells (Extended Data Fig. 10a). In a separate cohort, blood collected from mice with BCX.010 tumours 30 days after infusion of NK cells did not show significant differences in haematological parameters or serum chemistry between mice treated with CREM WT or KO CAR70–IL-15 NK cells (Extended Data Fig. 10b–d).
In summary, our findings support the therapeutic potential of CREM KO to enhance the efficacy of CAR-NK cells in vivo while maintaining a favourable safety profile, thereby highlighting the translational relevance of this approach.
CREM alters the CAR-NK cell genetic landscape
To understand how CREM modulates CAR-NK cell function, we performed ChIP–seq using an antibody that recognizes all CREM isoforms to identify genome-wide CREM-binding sites in NK cells. CREM binding was detected across numerous genomic sites, and antibody specificity was confirmed by the enrichment of CREM motifs under ChIP–seq peaks (Fig. 5a). CREM occupancy was higher in IL-15, CAR70–IL-15 and CAR70 NK cells than in NT NK cells (Fig. 5b and Extended Data Fig. 11a), a pattern that was also observed for core CREM-binding target genes (Fig. 5c). CREM-bound genes included those relevant to NK cell activity (chemokines and cytokines), cytotoxicity molecules (IFNG, LAMP1 and GZMB), calcium signalling (CAMK1D, CAMK2D and CAMLG) and genes with regulatory function (FOS, FOSB, NFKB2, NFKBIA, STAT3 and EGR1). Some binding targets were unique to specific conditions or shared between similar conditions. For example, DUSP23, DUSP28, EGR4, GZMK, KLRG1, SOCS2 and JUND were common to CAR70 and CAR70–IL-15, whereas CCR1, TMEM family members, EOMES and IL13 were shared between IL-15 and CAR70–IL-15. CISH, SOCS7, NCR1, NCR3, EGR3 and TMEM members were found in all three activated conditions (IL-15, CAR70 and CAR70–IL-15). Hallmark pathway enrichment analysis of CREM target genes identified the MYC targets V1 as the top pathway enriched among all conditions (Fig. 5d and Extended Data Fig. 11b). Other pathways, such as TNF signalling through NF-κB, MTORC1 signalling and G2M checkpoint pathways, were also enriched among the targets in CAR70–IL-15 NK cells (Fig. 5d).

a, CREM-binding motif enrichment in CREM ChIP–seq in NT, IL-15, CAR70–IL-15 and CAR70 NK cells (n = 3 donors for NT, IL-15 and CAR70 and one donor for CAR70–IL-15 NK cells; Methods). b, UpSet plot of CREM-binding sites in CAR70–IL-15 NK cells from one representative donor. c, Venn diagram of CREM target genes (within 1 kb vicinity of the transcription start site (TSS) of a gene) in NT, IL-15, CAR70–IL-15 and CAR70 NK cells, with colours referring to the number of targets (blue, lowest to yellow, highest count). d, Hallmark gene sets enriched among the CREM targets in CAR70–IL-15 NK cells and proportion of genes in each set that were recovered among the targets. e, GSEA enrichment plots of upregulated and downregulated Hallmark pathways (only direct targets of CREM from ChIP–seq were considered in each Hallmark pathway) in CREM KO versus WT CAR70–IL-15 NK cells by RNA-seq (n = 2 donors); the red dashed lines indicate the top and bottom of the enrichment score. f, Averaged line graphs and heatmaps showing the ATAC–seq signal intensities surrounding the TSS of all genes in CREM KO and WT CAR70–IL-15 NK cells cultured with or without UMRC3 cells for 24 h (1:1 E/T ratio; n = 2 donors). g, Chromatin accessibility tracks for select genes in CREM WT and KO CAR70–IL-15 NK cells in culture with UMRC3 cells from one representative donor. h, GSEA of transcription factor motif families differentially accessible in CREM KO versus WT CAR70–IL-15 NK cells cultured with UMRC3 cells. i, Motif analyses of top open and closed peaks in CREM KO CAR70–IL-15 NK cells cultured with UMRC3 cells. Motif activities were quantified using ChromVar. j, Schematic of CREM-induction pathways in CAR–IL-15 NK cells, and its mechanism as an activation checkpoint. Created in BioRender. Rafei, H. (2025) https://BioRender.com/9ifmaz6. Statistical comparisons were performed using one-sided Fisher’s method with false-discovery rate (FDR) correction (a), one-sided hypergeometric test with FDR correction (d) or GSEA modelling one-sided Kolmogorov–Smirnov test with FDR correction (e,h).
Given the dual function of CREM as both a transcriptional repressor and activator with tissue-specific and cell-specific expression35, we performed integrative analysis of our ChIP–seq and RNA-seq data from CREM WT and KO CAR70–IL-15 NK cells. CREM KO led to both the upregulation and downregulation of direct CREM target genes, a result that underscores its context-specific regulatory functions (Extended Data Fig. 11c). Although the specific changes in the direct target genes varied among donors (Extended Data Fig. 11d), consistently downregulated target genes in CREM KO CAR70–IL-15 NK cells included those related to exhaustion and stress responses, including BTG1 (ref. 36), DUSP2 (ref. 37), SMAD3, NFKB2 (ref. 3) and RGS1 (ref. 8). Conversely, CREM KO upregulated genes involved in NK cell cytotoxicity such as GZMB and IFNG, and transcription factors implicated in T cell and NK cell development (KLF7 and RUNX2)38,39. Gene set enrichment analysis (GSEA) of direct CREM targets revealed that CREM KO upregulated pathways crucial for NK cell effector function and metabolism, including MYC and E2F targets, MTORC1 signalling and oxidative phosphorylation. In parallel, it downregulated immunosuppressive pathways such as hypoxia and TGFβ signalling (Fig. 5e and Extended Data Fig. 11e,f). Together, these data suggest that CREM contributes to NK cell dysfunction by directly binding and upregulating exhaustion and stress-related genes while suppressing pathways essential for NK cell function and proliferation.
To assess the overall impact of CREM on the epigenome, we analysed differences in chromatin accessibility between CREM WT and CREM KO CAR70–IL-15 NK cells cultured alone or with UMRC3 cells using assay for transposase-accessible chromatin with sequencing (ATAC–seq). Overall, CREM WT CAR70–IL-15 NK cells displayed reduced chromatin accessibility when cultured together with tumour cells, whereas CREM KO CAR70–IL-15 NK cells maintained a more open chromatin landscape under the same conditions (Fig. 5f). Notably, CREM KO CAR70–IL-15 NK cells cultured together with tumour cells retained accessibility in most cellular metabolic and effector pathways, including MTORC1, PI3K–AKT–MTOR signalling and the mitotic spindle (Extended Data Fig. 11g). Compared with CREM WT, CREM KO CAR70–IL-15 NK cells showed enrichment of chromatin accessibility at loci associated with activation of E2F targets, oxidative phosphorylation, MYC targets, G2M targets, MTORC1 signalling and the mitotic spindle, a result consistent with an effector, activated and proliferating NK cell phenotype (Extended Data Fig. 11h). Indeed, genes associated with effector function (for example, GZMA and PRF1) and transcription factors essential for NK cell maturation and function (for example, EOMES and TBX21) showed increased accessibility in CREM KO CAR70–IL-15 NK cells (Fig. 5g and Extended Data Fig. 11i).
Motif enrichment analysis revealed that binding motifs for JUN-related factors (JUND, JUNB, FOSL1–JUND and FOSL1), STAT factors and CEBP factors were highly enriched in CREM KO compared with WT CAR70–IL-15 NK cells, whereas ETS-related factors were among the most differentially enriched in CREM WT compared with KO cells (Fig. 5h,i and Extended Data Fig. 11j). These patterns of enriched AP-1 and closed ETS motifs mirror epigenetic signatures associated with long-lived memory T cells40 and may underlie the enhanced persistence and function of CREM KO CAR-NK cells.
To further dissect the mechanisms by which CREM KO improves NK cell function, we first assessed IL-15R proximal signalling. Expression of IL-15R signalling components was comparable between CREM WT and CREM KO NK cells, with some donor-to-donor variability (Extended Data Fig. 12a,b). Similarly, levels of negative regulators of IL-15R signalling, including SOCS1 and CISH, were not significantly different (Extended Data Fig. 12a,c). We next investigated whether CREM KO mirrors the biological effects of IL-15R signalling in NK cells, including enhanced proliferation, extended lifespan and improved metabolic fitness. In keeping with our transcriptomic data of upregulated cell cycle pathways, CREM KO CAR-NK cells showed increased proliferation and extended lifespan, particularly in vivo (Fig. 4e,f,i,j and Extended Data Fig. 9i,l). Moreover, metabolic functional assays revealed enhanced glycolytic capacity and a trend towards increased mitochondrial respiration in CREM KO CAR70–IL-15 NK cells (Extended Data Fig. 12d–i).
Taken together, these findings indicate that CREM regulates NK cell function by modulating gene expression and chromatin states and by promoting a dysfunctional state (Fig. 5j). This state may be reversed by CREM KO, which leads to increased chromatin accessibility for key transcription factors involved in NK cell activation, especially members of the AP-1 complex, STAT family and CEBP family41,42,43.
Discussion
The discovery of T cell immune checkpoints has substantially advanced cancer treatment. However, there remains a need to identify alternative checkpoints that can be exploited across diverse immune cell subsets. Although most immunomodulatory approaches have focused on augmenting T cell responses, recent attention has expanded to harnessing the therapeutic potential of NK cells1. CAR-NK cell therapies, in particular, have demonstrated promising clinical results in B lymphoid malignancies and show promise in preclinical models of solid tumours1,3,43,44,45.
In this study, we identified the role of the PKA–CREB–CREM axis as a key regulator of CAR-NK cell activation. Notably, we showed that CREM can be upregulated by CAR activation and serves as an important checkpoint in CAR-NK cells. These results are in keeping with previous findings that TCR stimulation can induce CREM and ICER46, and that PKA activation and CREB phosphorylation can attenuate TCR signalling31.
Consistent with findings in other cell types, our results demonstrated that NK cells express various CREM and ICER isoforms at baseline. After CAR activation or IL-15 stimulation, several of these isoforms are strongly upregulated, most prominently CREMα, CREMβ, CREMγ and CREMτ. Although our study did not aim to dissect the individual contributions of each isoform, we focused on establishing the overall function of CREM in CAR-NK cells and evaluating its potential as a therapeutic target to enhance CAR-NK cell efficacy.
Mechanistically, we showed that CAR ligation and downstream CD3ζ signalling activate the PKA pathway, which leads to CREB phosphorylation in NK cells. pCREB then binds to the CREM promoter to initiate its transcription. This sequence of events occurs rapidly and can be disrupted through both PKA inhibition and calcium chelation. IL-15 induces CREM expression in NK cells through a similar mechanism, which is further regulated by STAT3 and STAT5. These findings reveal a complex regulatory axis activated by IL-15 that integrates the canonical JAK–STAT signalling pathway with the cAMP–PKA signalling.
Although CREM has been described as a suppressor of T cell and macrophage function20,47, its role in NK cell biology, especially in the context of CAR therapy, is unclear. Recent studies have reported distinct subsets of NK cells with high CREM expression8,48, although the functional implications were not explored. Here we identified CREM as an immunoregulatory checkpoint in NK cells. CRISPR–Cas9-mediated CREM KO significantly enhanced the antitumour efficacy of CAR-NK cells, with the greatest effects observed under dual activation by CAR and IL-15. Notably, CREM KO CAR70–IL-15 NK cells exhibited a favourable safety profile in vivo. Nonetheless, further studies will be needed to fully assess long-term safety, particularly in light of potential risks associated with removing intrinsic inhibitory mechanisms.
Our research extends the understanding of NK cell biology by uncovering the role of the CREM transcription factor in epigenetic and transcriptional regulation. In T cells, CREM can bind the IL2 promoter and regulate its expression46 as well as other target genes, such as those encoding IL-17A49, IL-17F50, SYK51, the CD3ζ chain52 and MIP-1β53. In CAR–IL-15 NK cells, our integrated ChIP–seq and RNA-seq analyses revealed that CREM directly binds and represses a broad set of crucial NK cell effector genes. Notably, CREM KO preserved global chromatin accessibility after tumour co-culture, with AP-1 motifs remaining highly accessible, a finding consistent with the previously reported inhibition of JUN–AP-1 activity by CREM54,55. Furthermore, CREM KO increased accessibility at loci encoding key effector genes, including PRF1, GZMA and GZMB.
In summary, our study established CREM as a key transcriptional checkpoint in NK cell activation. Targeting CREM may offer a promising strategy to enhance CAR-NK cell activity and persistence. Our findings pave the way for future research into the complex pathways that regulate NK cell function, with potential implications for next-generation immunotherapeutic strategies.
Methods
Cell lines and culture conditions
Human cancer cell lines of Raji (CCL-86), K562 (CCL-243), SKOV3 (HTB-77) and 293T (CRL-3216) were obtained from the American Type Culture Collection (ATCC). Raji cells were cultured in RPMI-1640 medium with 10% FBS, 2 mM l-glutamine (Invitrogen) and 1% penicillin–streptomycin (Invitrogen). SKOV3 and 293T cells were cultured in DMEM medium (Invitrogen) supplemented with 10% FBS, 1% penicillin–streptomycin and 2 mM l-glutamine (Invitrogen). The UMRC3 cell line was obtained from Sigma-Aldrich (08090512) and cultured in DMEM medium with 10% FBS, 2 mM l-glutamine (Invitrogen) and 1% penicillin–streptomycin (Invitrogen). The PDX cell line BCX.010 of breast cancer was provided by F. Meric-Bernstam at MD Anderson Cancer Center (MDACC), and PATC148 cells were provided by A. Maitra (MDACC). Both BCX.010 and PATC148 cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS, 1% penicillin–streptomycin and 2 mM l-glutamine (Invitrogen). Cells were passaged every 3–4 days. The CD70 gene in UMRC3 cells was deleted using CRISPR–Cas9 methods (detailed below) to generate CD70 KO UMRC3 cells. UMRC3 and BCX.010 cells were transduced to express GFP for fluorescence monitoring. Raji cells were transduced to express mCherry or GFP, and K562 cells were transduced to express mCherry for fluorescence monitoring by microscopy. Raji cells were modified to express firefly luciferase to enable bioluminescence in vivo imaging. All cells were maintained in 5% CO2 at 37 °C and were authenticated by STR profiling at the MDACC Cell Line Characterization Core Facility. All cell lines were tested regularly for mycoplasma contamination using a MycoAlert Mycoplasma Detection kit (Lonza) and were only used when tested negative for contamination.
Cord blood NK cell isolation and expansion
Cord blood (CB) units were provided by the MDACC CB Bank under a protocol approved by the Institutional Review Board (Lab04-0249). Lymphocytes from CB were isolated through density gradient (Ficoll-Histopaque, Sigma). NK cells (CD56+CD3–) were purified from lymphocytes by negative selection using a NK cell isolation kit (Miltenyi Biotec) following the manufacturer’s instructions. Isolated NK cells were preactivated for 16 ± 2 h using recombinant human (rh) IL-12 (BioLegend; 10 ng ml–1), rhIL-18 (MBL International; 50 ng ml–1) and rhIL-15 (BioLegend; 50 ng ml–1) as previously described56, washed 2 times with PBS to remove cytokines and expanded with irradiated (100 Gy) universal antigen presenting cells (uAPCs) at a feeder cell-to-NK cell ratio of 2:1 and cultured in 50% Click’s medium (EHAA, Fujifilm) and 50% RPMI-1640 (referred to hereafter as NK cell medium) supplemented with rhIL-2 (Proleukin, 200 U ml–1, Chiron). Medium was changed every 2–3 days, and irradiated uAPCs were added every week to the NK cell culture to support expansion. Transduction of NK cells with retroviral constructs (detailed below) was performed on days 5–6 after expansion with uAPCs. An equal number of NK cells from each condition was electroporated for CRISPR–Cas9 KO and expanded on day 2 after transduction. Functional experiments were performed on days 14–15 (7–8 days after second expansion), unless mentioned otherwise. Cell counts were recorded using ViaStain AOPI staining solution (Nexcelom) and a cellometer Auto 2000 (Nexcelom) instrument on a regular basis to monitor NK cell proliferation or at the time of functional assays to ensure equal cell numbers between conditions.
Generation of retroviral constructs, virus production and CAR-NK cells
The construct for the CD70-targeting CAR (CD27(ECD).CD28.ζ.2 A.IL-15), referred to as CAR70–IL-15, incorporates the ECD of CD27 (the natural receptor for CD70 ligand) linked to the CD28 co-stimulatory domain and the CD3ζ signalling domain. It also includes the IL15 transgene. The CAR70 construct was generated through deletion of IL-15 by restriction digestion. The six tyrosine (Y) residues of the ITAMs in the CD3ζ region of CAR70 construct were changed to phenylalanine (F) and termed CAR70.3ζ.Y6F44. The entire CD3ζ region of the CAR70 construct was deleted and termed CD27(ECD).
The construct for the TROP2-targeting CAR (TROP2scFv (clone hRS7).CD28.ζ.2 A.IL-15), referred to as CAR.TROP2–IL-15, consists of a scFv targeting TROP2 (derived from the sequence of the TROP2-targeting antibody–drug conjugate sacituzumab govitecan, human RS7 (hRS7)) linked to the CD28 co-stimulatory domain and the CD3ζ signalling domain. Similarly, the construct includes the IL15 transgene.
All CAR constructs and the retroviral construct encoding IL-15 were cloned into a SFG retroviral backbone by GeneArt Gene Synthesis (Thermo Fisher Scientific) to generate viral vectors. Transient retroviral supernatants were produced from transfected 293T cells with CAR plasmids along with packaging and envelope plasmids. CAR transduction efficiency was measured 48–72 h after transduction by flow cytometry.
CAR, CD16, NKp30, NKp46 and cytokine-stimulation assays
For CAR-stimulation assays, human CD27 ligand–CD70 protein (ACROBiosystems, CDL-H82D7) was plated in PBS in appropriate plates (96-well ELISA plates or 6-well plates) at a concentration of 1.2 μg ml–1. Plates were kept at 4 °C on a plate shaker overnight. NK cells from the indicated conditions were placed in NK cell medium without any cytokine support for 24–72 h before stimulation. The next day, PBS was aspirated, and an equal number of NK cells from the indicated conditions was stimulated in the plates at 37 °C after a brief centrifugation step to promote uniform interaction of NK cells from the indicated conditions with the surface-coated CD70. NK cells were then collected at appropriate intervals according to the downstream assays (30 min for pCREB detection and 24 h for qPCR). For stimulation assays with CD16, NKp30 and NKp46, the following antibodies were used: anti-human CD16 (BD Biosciences, 3G8, 556617), anti-human NKp30 (R&D systems, 210845, MAB1849) and anti-human NKp46 (eBioscience, 9E2, 16-3359-85). Similar to the CAR-stimulation assays, antibodies were plated in PBS in appropriate plates at a concentration of 1 μg ml–1. The assays were then performed as described for CAR stimulation.
For cytokine stimulation, NK cells were placed in NK cell medium without any cytokine support for 24–72 h before stimulation. The next day, NK cell medium containing various cytokine concentrations (50 pg ml–1, 500 pg ml–1 and 5,000 pg ml–1) were prepared by serial dilution. NK cells were counted, pelleted and then resuspended in equal numbers in medium with the corresponding cytokine concentrations and incubated for an appropriate interval (30 min for pCREB detection and PKA calorimetric assay, and 24 h for qPCR). The following cytokines were used: rhIL-2 (Proleukin), rhIL-10 (StemCell Technologies, 78024.1), rhIL-12 (P70, BioLegend, 573004), rhIL-15 (BioLegend, 570304), rhIL-18 (MBL International, B003-5) and hIL-21 (Miltenyi Biotec, 130-095-767). For IL-15 antagonism experiments, a functional-grade IL-15 monoclonal antibody (eBioscience, 16-0157-82, clone ct2nu) was used at a concentration of 100 ng ml–1 where indicated to neutralize the bioactivity of human IL-15.
Flow cytometry
NK cells or tumour cells were collected for flow cytometry, washed with PBS, pelleted and stained with Live/Dead Fixable Aqua Dead cell stain (ThermoFisher, 1:200) to determine their viability. After washing with FACS buffer (PBS with 1–2% FBS), cells were centrifuged and pellets were resuspended in antibody cocktail, mixed and incubated for 20 min at room temperature protected from light for surface staining. The transduction efficiency of IL-15 NK cells was measured using a conjugated goat F(ab′)2 anti-human lgG (H+L; Jackson ImmunoResearch) that recognizes the IgG hinge portion of the construct (as previously described)3. CAR expression on the CAR-NK cells transduced with the various CD70-targeting constructs was measured using anti-CD27 antibody. CAR expression on the TROP2-targeting CAR-NK cells was measured by incubating the cells in human TROP2–TACSTD2 protein, His tag (ACROBiosystems, TR2-H5223) at a concentration of 1.2 μg ml–1 for 30 min at room temperature, after which they were washed with PBS and stained with anti-His antibody and the rest of the surface antibodies for 20 min at room temperature. The following antibodies were used for flow cytometry experiments: APC-Cy7 anti-human CD3 (BioLegend, HIT3a, 300318, 1:100); BV605 anti-human CD56 (BioLegend, HCD56, 318334, 1:50); BV650 anti-human CD16 (BD Biosciences, 3G8, 563173, 1:50); PerCP anti-human CD45 (BioLegend, HI30, 304026, 1:50); APC-Cy7 anti-human CD45 (BioLegend, HI30, 304014, 1:50); PE-CF594 anti-human CD27 (BD Biosciences, M-T271, 562297, 1:50); PE-Cy7 anti-human CD70 (BioLegend, 113-16, 355112, 1:50); AF-700 anti-mouse CD45 (BioLegend, QA17A26, 157616, 1:50); PE anti-human TROP2 (BioLegend, NY18, 363804, 1:50); anti-His-APC (BioLegend, J095G46, 362605, 1:50); PE anti-human CD326 (EpCAM; BioLegend, 9C4, 324206, 1:50); anti-human CD215 (IL-15Rα; BD Biosciences, JM7A4, 566589, 1:40); and anti-Ki67 (BD Biosciences, B56, 563462, 1:33). Staining for Ki67 required additional fixation and permeabilization steps as detailed below. Data were acquired using LSRFortessa (BD Biosciences), and analysis was performed using FlowJo software (v.10.8.2). All sorting was performed on a BD Biosciences Aria II Cell Sorter or a Beckman Coulter CytoFLEX SRT Cell Sorter at the MDACC North Campus Flow Cytometry and Cellular Imaging Core Facility. Representative gating strategies of NK cells are shown in Supplementary Fig. 1.
Intracellular staining
NK cells from the indicated conditions were stimulated by culturing them with cancer cells at an E/T ratio of 1:1 for 5–6 h in the presence of brefeldin A (10 μg ml–1, BD Biosciences GolgiPlug, 555029) and monensin (1×, BioLegend, 420701) to inhibit protein transport. Unstimulated NK cells were used as negative controls, and NK cells stimulated with phorbol 12-myristate 13-acetate (Sigma-Aldrich, P8139) and inomycin (Sigma-Aldrich, I0634) were used as positive controls. Cells were collected after incubation, washed with PBS and stained using a Live/Dead Fixable Aqua Dead cell stain kit (ThermoFisher, 1:200) to identify viable cells. Cells were then stained with surface antibodies for 20 min at room temperature in the dark. Cells were then fixed and permeabilized using a BD Cytofix/Cytoperm kit (BD Biosciences, 554714) following the manufacturer’s instructions. Intracellular staining was performed with PE anti-human IFNγ (BioLegend, 506507, 1:40) and PE-Cy7 anti-human TNF (BioLegend, 502930, 1:40) antibodies for 30 min at 4 °C. Expression of IFNγ and TNF was assessed by flow cytometry and expressed as a percentage of CD56+CD3– NK cells.
pCREB and CREM detection by flow cytometry
A FOXP3 transcription factor staining kit (eBioscience) was used for pCREB and CREM staining. In brief, cells (deprived of cytokine support for 24 h) were washed with PBS and stained with Live/Dead Fixable Aqua Dead cell stain (ThermoFisher, 1:200) as described above. After washing, cells were resuspended in NK cell medium and stimulated in 96-well plates with IL-15 or on antigen-coated plates as described above at 37 °C for 30 min. Following the stimulation step, cells were pelleted and permeabilized with FOXP3 Fix/Perm buffer at 4 °C for 45 min on a plate shaker. Cells were then washed twice with 1× Perm Wash buffer, after which they were stained with PE anti-human pCREB (pS133)/pATF-1 (pS63) antibody (BD Phosflow, J151-21, 558436, 1:20) or anti-human CREM (Santa Cruz Biotechnology, 22, sc-101530, 1:20) and appropriate surface antibodies, depending on the experiment, at room temperature for 30 min. Cells were then washed twice and data were acquired on a flow cytometer. Cells treated with FSK (Sigma-Aldrich, F6886) at a concentration of 20 μM were used as positive controls. In some experiments, cells were treated with the PKA inhibitor H89, dihydrochloride (Cell Signaling, 9844) at a concentration of 30 μM for 1–2 h before stimulation. For calcium chelation, a 500 mM stock solution of EGTA was prepared as follows: 6.645 g of 10 N NaOH with 19.0175 g of EGTA (Millipore, 324626) and MilliQ dH2O to 100 ml. Cells in the corresponding conditions were treated with 10 mM EGTA for 1–2 h before stimulation.
Western blotting
NK cells as indicated were lysed using IP lysis buffer (Pierce IP Lysis Buffer, Thermo Scientific, 87788) supplemented with protease and phosphatase inhibitors (Halt protease and phosphatase inhibitor single-use cocktail, EDTA-free, Thermo Scientific, 78443) and incubated for 30 min on ice. Whole-cell lysates were obtained after centrifugation. A BCA Protein Assay kit (Pierce) was used to measure protein concentrations. Cell lysates from the indicated conditions were subjected to electrophoresis in equal amounts using SDS–PAGE and transferred to polyvinylidene difluoride membranes (Bio-Rad). Membranes were blocked with 5% milk (in PBST) or 5% BSA (in PBST) for 30 min, followed by primary antibody incubation overnight at 4 °C. The following primary antibodies were used: pCD3ζ Tyr142 (Abcam, EP265(2)Y, ab68235, 1:1,000); CD247/CD3ζ (Bethyl Laboratories, A305-212A, 1:2,000); CREM (Creative Diagnostics, CABT-B10032, 1:2,000); pCREB (Ser133; Cell Signaling, 87G3, 9198, 1:1,000); total CREB (Cell Signaling, 48H2, 9197, 1:1,000); STAT3 (Cell Signaling, 9139, 1:1,000); pSTAT3 (Cell Signaling, 9145, 1:1,000); STAT5A (Cell Signaling, 4807, 1:1,000); STAT5B (Cell Signaling, 3466, 1:1,000); pSTAT5A/B (EMD Millipore, 04-886, 1:1,000); STAT5-PY694 (Cell Signaling, 4322S, 1:1,000); STAT5 (Cell Signaling Technology, 94205S, 1:1,000); JAK1-PY1034/1035 (Cell Signaling, 74129S, 1:1,000); JAK1 (Cell Signaling, 3344S, 1:1,000); JAK2-PY1008 (Cell Signaling, 8082S, 1:500); JAK2 (Cell Signaling, 3230S, 1:1,000); S6-PS240/244 (Cell Signaling, 5364S, 1:1,000); S6 (Cell Signaling, 2217S, 1:1,000); SOCS1 (Cell Signaling, 55313S, 1:1,000); and β-actin (Sigma-Aldrich, AC-15, A5441, 1:5,000). The membranes were washed 3 times with PBST (5 min each) and then incubated with secondary antibody (donkey anti-rabbit IgG, HRP-linked whole Ab, Genesee Scientific, 84-854, and sheep anti-mouse IgG, HRP-linked whole antibody, Genesee Scientific, 84-848; both 1:3,000, 5% milk in PBST) for 60 min. Where applicable, the membranes were stripped to remove bound antibodies using Restore Western Blot Stripping buffer (Thermo Scientific) for further protein analyses. Protein signals were detected using ECL (Amersham) following the manufacturer’s instructions. Densitometry analyses were performed by evaluating the band intensity mean grey value of the indicated protein and normalizing it with the mean grey value of the total protein of the corresponding lane or the loading control (β-actin) using ImageJ 1.53t software.
PKA enzymatic assay
NK cells deprived of cytokines for 24 h were stimulated with the corresponding IL-15 concentrations as described above at 37 °C for 30 min. Cell lysates were then prepared as above. PKA activity was measured using a PKA Colorimetric Activity kit (Invitrogen, EIAPKA) following the manufacturer’s instructions.
CRISPR gene editing
For CRISPR–Cas9-mediated KO, pre-designed sgRNAs targeting CREM, ICER, CD70, STAT3, STAT5A and STAT5B were obtained from Integrated DNA Technologies (IDT). The gRNA sequences used are presented below.
For CREM-specific exon KO (Extended Data Fig. 3a,d,e): sgRNA-1: ACCACCTAGTATTGCTACCA; sgRNA-2: TCTTCAATCTTGGGAACACC.
For ICER-specific exon KO (encompassing both ICER-specific exon 1 and exon 2 (Extended Data Fig. 3a,d,e)): sgRNA-1: GCTGTAACTGGAGATGACAC; sgRNA-2: GCTCGATCTTACCACTAAGC.
For CD70: sgRNA-1: TCACCAAGCCCGCGACCAAT; sgRNA-2: GGCCATCTGCTCCTCCACGA.
For STAT3: sgRNA-1: GCAGAAAACTCTCACGGACG; sgRNA-2: TCTTCTGCCTGGTCACTGAC.
For STAT5A: sgRNA-1: CAAGTAGTGCCGGACCTCGA; sgRNA-2: CATTGACTTGGACAATCCCC.
For STAT5B: sgRNA-1: CATCAGATGCAAGCGTTATA; sgRNA-2: AAATAATGCCGCACCTCAAT.
To form the ribonucleoprotein (RNP) complex, sgRNAs (100 µM) were combined with Cas9 (Alt-R S.p. Cas9 Nuclease V3, 1081059) in the presence of P3 primary solution (P3 Primary Cell 4D-Nucleofector X Kit S, Lonza) at a sgRNA-to-P3-to-Cas9 ratio of 0.22:0.48:0.3 µl. The RNP complex was incubated for 20 min at room temperature. One million NK cells were collected, washed twice with PBS, pelleted and resuspended in 20 µl of the RNP complex in the presence of an electroporation enhancer (IDT, Alt-R Cas9 Electroporation Enhancer, 1075916) and P3 primary solution for electroporation. The final concentration for each electroporation was 2.2 µM sgRNA, 1.9 μM Cas9 nuclease and 5 µM Cas9 electroporation enhancer. The cells were electroporated in Nucleocuvette strips using the X unit of a 4D-Nucleofector device (Lonza) using the CM-137 electroporation program. After electroporation, the cells were left to rest for 10–15 min in a 37 °C incubator, after which they were transferred into prepared flasks with NK cell medium and feeder cells and IL-2, and cultured in a 37 °C incubator. The KO efficiency of CREM was evaluated by western blotting or by qPCR or PCR followed by gel electrophoresis using the following primers: forward: 5′-TGAATGAACTGTCCTCTGATGTG-3′; reverse: 5′-CCTGAGTTGCTTCAATATAACTAGAGA-3′. The KO efficiency for STAT3 and STAT5A/B was tested by western blotting.
CD70 KO in UMRC3 cells was performed using a Neon transfection system (Invitrogen). The sgRNAs were first diluted with nuclease-free water to a concentration of 44 µM. Cas9 was diluted with R buffer (Neon Electroporation kit, Invitrogen) at a Cas9 to R buffer ratio of 3:2. Diluted sgRNA and Cas9 were mixed to form the RNP complex at a 1:1 ratio and incubated for 20 min at room temperature. UMRC3 cells were collected and washed twice with PBS in aliquots of 100,000 cells each. The supernatant was removed, and the cells were resuspended in 12 µl of the RNP complexes along with an electroporation enhancer in R buffer. The final concentration for each electroporation was 1.8 µM sgRNAs, 1.6 µM Cas9 nuclease and 2.25 µM Cas9 electroporation enhancer. The cells were electroporated using a Neon Transfection system at 1,700 V, 20-ms pulse width and 1 pulse with 10 µl electroporation tips (Thermo Fisher Scientific, MPK5000). After electroporation, the cells were transferred into pre-warmed DMEM medium and cultured in a 37 °C incubator. The KO efficiency of CD70 was evaluated by flow cytometry.
Incucyte live cell spheroid and tumour rechallenge assays
GFP+ UMRC3 or BCX.010 spheroids were formed by plating 20,000 single cells in 100 μl medium in a 96-well clear round-bottom ultralow attachment spheroid microplate (Corning, 4520) followed by a 10-min centrifugation step at 120g. The plates were then placed in a 37 °C incubator for 24–48 h to allow for the spheroids to form. Once formed, the spheroids were treated with various NK cell conditions at the specified E/T ratios in technical duplicate or triplicate wells or left untreated as controls. Frames were captured with a ×10 objective at 4-h intervals over a number of days (as specified in each experiment). The green signal was quantified using an Incucyte S3 live-cell analysis system (Sartorius) in real-time and reported as the spheroid green image mean normalized to the green image mean immediately before the time of NK cell addition when the spheroid had already formed. Videos or images were exported using the same analysis system.
For the Raji tumour rechallenge assay, NK cells were cultured with Raji tumour cells labelled with mCherry and fresh tumour cells were added to this culture every 2–3 days in 96-well flat clear-bottom black microplates (Corning, 3904). Each well received 25,000 single Raji cells at each rechallenge, and duplicate or triplicate wells were used for each different condition. Images of each well were captured in real-time in five distinct regions per well. The counts of tumour cells for which the mCherry fluorochrome was detected were analysed using an Incucyte S3 live-cell analysis system that measures the number of target cells (fluorochrome labelled) in real-time.
xCELLigence killing and tumour rechallenge assays
Tumour cells were plated in 96-well RTCA E-plates (Agilent) for 24 h (to allow tumour cell adherence to the plate and growth) before NK cells were added at the designated E/T ratios. Only medium was added for tumour alone controls. Impedance was monitored over a number of days (specified individually for each experiment) at intervals of 15 min in a xCELLigence machine (Agilent) and reported as the normalized cell index, which was normalized to the cell index at the time NK cells were added using RTCA immunotherapy module software (Agilent).
For rechallenge assays using xCELLigence, the first killing assay was set up as described above. After that, new tumour cells were plated in new 96-well RTCA E-plates every 2–4 days and a new killing assay was started in the xCELLigence machine using RTCA immunotherapy module software. NK cells of the indicated conditions were transferred from the previous killing assay plate onto the newly plated tumours (after 24 h), and impedance was monitored over the next 2–4 days before the next rechallenge. Tumour growth is reported as the normalized cell index.
Mass cytometry and data analysis
Mass cytometry experiments and primary antibody conjugation were performed as previously described57. In brief, cells were collected, washed with cell staining buffer (0.5% BSA in PBS) and incubated with 2.5 µM cisplatin (Sigma Aldrich) for viability assessment. Cells were washed, incubated with 5 µl human Fc receptor blocking solution (Trustain FcX, BioLegend) for 10 min at room temperature, and then stained for cell surface markers with a freshly prepared antibody mix for 30 min at room temperature on a shaker. After washing with cell staining buffer, samples were fixed and permeabilized using BD Cytofix/Cytoperm solution for 30 min in the dark at 4 °C, washed twice with Perm/Wash buffer and stained with antibodies directed against intracellular markers. Samples were then washed and stored overnight in 500 µl of 1.6% paraformaldehyde (EMD Biosciences) and PBS with 125 nM iridium nucleic acid intercalator (Fluidigm). The next day, samples were washed, filtered, counted and resuspended in MilliQ dH2O supplemented with EQTM four element calibration beads at a concentration of 0.5 × 105 per ml. Samples were acquired on a Helios instrument (Fluidigm) using Helios (v.6.5.358) acquisition software (Fluidigm). The following antibodies and corresponding metal tag isotopes were used: CD45 (Standard Biotools, 3089003B, HI30, 89Y, 1:167); CCR6 (Miltenyi Biotec, 130-108-023, REA190, 141Pr, 1:250); Eomes (Invitrogen, 14-4877-82, WD1928, 142Nd, 1:167); CD127 (Standard Biotools, 3143012B, A019D5, 143Nd, 1:200); GFP (BioLegend, 338002, FM264G, 144Nd, 1:250); CD70 (BioLegend, 355102, 113-16, 145Nd, 1:500); CD8a (Miltenyi Biotec, 130-122-281, REA734, 146Nd, 1:167); NKG2C (Miltenyi Biotec, 130-122-278, REA205, 147Sm, 1:125); TRAIL (Miltenyi Biotec, 130-126-490, REA1113, 148Nd, 1:125); CD25 (Standard Biotools, 3149010B, 2A3, 149Sm, 1:337); CD69 (Miltenyi Biotec, 130-124-326, REA824, 150Nd, 1:5,000); 2B4 (Miltenyi Biotec, 130-124-523, REA122, 151Eu, 1:5,000); CD95 (Miltenyi Biotec, 130-124-328, REA738, 152Sm, 1:500); panKIR (R&D systems, MAB1848, 180704, 153Eu, 1:337); CX3CR1 (Miltenyi Biotec, 130-122-286, REA385, 154Sm, 1:125); CD27 (Standard Biotools, 3155001B, L128, 155Gd, 1:250); CXCR3 (Standard Biotools, 3156004B, G025H7, 156Gd, 1:167); OX40 (Miltenyi Biotec, 130-095-212, REA621, 158Gd, 1:125); CD11c (Standard Biotools, 3159001B, Bu15, 159Tb, 1:250); T-bet (Standard Biotools, 3160010B, 4B10, 160Gd, 1:250); TIGIT (Miltenyi Biotec, 130-122-310, REA1004, 161Dy, 1:125); Ki67 (Standard Biotools, 3162012B, B56, 162Dy, 1:250); BTLA (Standard Biotools, 3163009B, MIH26, 163Dy, 1:67); CD73 (Miltenyi Biotec, 130-120-066, AD2, 164Dy, 1:125); TIM3 (Miltenyi Biotec, 130-122-333, REA635, 165Ho, 1:10); NKG2D (Standard Biotools, 3166016B, ON72, 166Er, 1:334); CREM (Creative Diagnostics, CABT-B10032, 4C6, 167Er, 1:500); KLRG1 (Miltenyi Biotec, 130-126-458, REA261, 168Er, 1:125); NKG2A (Standard Biotools, 3169013B, Z199, 169Tm, 1:500); CD161 (Miltenyi Biotec, 130-122-347, REA631, 170Er, 1:500); DNAM (Standard Biotools, 3171013B, DX11, 171Yb, 1:250); CD38 (Miltenyi Biotec, 130-122-307, REA572, 172Yb, 1:500); CXCR4 (Standard Biotools, 3173001B, 12G5, 173Yb, 1:167); PD1 (Miltenyi Biotec, 130-096-168, PD1.3.1.3, 174Yb, 1:125); LAG3 (Miltenyi Biotec, 130-124-529, REA351, 175Lu, 1:125); ICOS (Miltenyi Biotec, 130-122-304, REA192, 176Yb, 1:167); CD16 (Standard Biotools, 3209002B, 3G8, 209Bi, 1:167); CD57 (Miltenyi Biotec, 130-124-525, REA769, 115In, 1:500); CD39 (Miltenyi Biotec, 130-093-506, MZ18-23C8, Pt195, 1:100); perforin (Standard Biotools, 3196002B, B-D48, Pt196, 1:167); GZMB (Standard Biotools, 3198002B, GB11, Pt198, 1:167); CD56 (Miltenyi Biotec, 130-108-016, REA196, 106Cd, 1:167); CD2 (Miltenyi Biotec, 130-122-348, REA972, 111Cd, 1:334); HLA-DR (Miltenyi Biotec, 130-122-299, REA805, 112Cd, 1:250); NKp30 (Miltenyi Biotec, 130-092-554, AF29-4D12, 113Cd, 1:167); NKp46 (Miltenyi Biotec, 130-124-522, REA808, 114Cd, 1:125); and NKp44 (Miltenyi Biotec, 130-126-465, REA1163, 116Cd, 1:125).
Mass cytometry data were analysed using Cytobank. FCS files were first processed using FlowJo by removing beads and then gating singlets in Ir-191 and Ir-193 double-positive cells. NK cell populations were identified by Pt-195 (cisplatin)low GFP–CD45+CD56+. The gating strategy was applied to all files. Representative gating strategies of NK cells are shown in Supplementary Fig. 1. Data from 20,000 NK cell events for each condition were randomly subsampled in FlowJo using the DownSample plugin. Normalized data from donors representing the same condition were concatenated. Downstream analysis was performed on randomly sampled 10,000 events from each condition. NK cells from the various conditions were merged to create a single t-SNE CUDA map. The positive population for each marker was gated in Cytobank. The mean expression of each marker was normalized using the z score, then hierarchically clustered, and plotted as a heatmap, with the percentage positivity overlayed for each marker using Morpheus matrix visualization and analysis software (Broad Institute).
qPCR assay
NK cells were stimulated as described above. RNA was extracted using a RNeasy Plus Mini kit (Qiagen, 74134) according to the manufacturer’s protocol. cDNA was synthesized using an Iscript DNA Synthesis kit (Bio-Rad, 1708891). A qPCR reaction mix of 20 μl included 10 μl TaqMan Advanced Fast PCR master mix (Applied Biosystems, 4444557), 2 μl CREM or ICER PrimeTime Std qPCR assay (IDT), 2 μl cDNA and 6 μl nuclease-free water. The following primers were used.
CREM-specific exon (ENSE00003481920; Ensembl GRCh38.p14; Extended Data Fig. 3a) shared by the majority of the CREM non-ICER isoforms: primer 1 (FWD): 5′-ACTGAATGAACTGTCCTCTGATG-3′; primer 2 (REV): 5′-GTACTGCCATGGTAGCAATACT-3′.
ICER-specific exon 1 (ENSE00001890657, ENSE00001427126 or ENSE00001925783; Ensembl GRCh38.p14; Extended Data Fig. 3a): primer 1 (FWD): 5′-GATGTCAGTCCTCCTGCTTATC-3′; primer 2 (REV): 5′-CCTGTGTCATCTCCAGTTACAG-3′.
ICER-specific exon 2 (ENSE00001923116, ENSE00001835159 or ENSE00001384382; Ensembl GRCh38.p14; Extended Data Fig. 3a): primer 1 (FWD): 5′-TCAGTTCCTTTCCGCTTTGTA-3′; primer 2 (REV): 5′-TCAAGCAGACAACCACTTCA-3′.
qPCR was performed on an ABI 7500 Fast Real Time PCR system (Applied Biosystems). ΔCT was calculated as the CT of the target gene – the CT of the corresponding internal control 18S. Relative expression was determined by normalizing the amount of each gene of interest to the experimental control using the ΔΔCT method.
scRNA-seq
scRNA-seq data for CAR19–IL-15 NK cells from a non-curative in vivo model of lymphoma was obtained from a previous study14.
In data from the in vivo model, CAR19–IL-15 NK cells from time points with greater than 100 NK cells were retained (before and after infusion (day 7 and day 14)) and were processed using a standard Seurat workflow58. In brief, data were normalized and scaled using the functions NormalizeData() with scale.factor = 10,000 and ScaleData(), respectively. A total of 2,000 variable genes were identified using FindVariableFeatures(). RunPCA() and RunUMAP() were run to generate a UMAP embedding using 20 principal components selected by inspecting the elbow plots. FindAllMarkers() was used to identify differentially expressed genes between cells before and after infusion, with log fold change threshold = 0 using Wilcox tests. Differentially expressed genes were identified at adjusted P values < 0.01 and absolute average log2 fold change > 0.5. All analyses were performed in R (v.4.0.1) with Seurat (v.4.1.1). IL-15 activity was inferred from scRNA-seq using CytoSig59.
Analysis of publicly available scRNA-seq datasets
scRNA-seq data were obtained from publicly available datasets and analysed for CREM expression. First, normalized CREM expression levels in various cell types from scRNA-seq cancer datasets were downloaded from the TISCH2 database (available at http://tisch.comp-genomics.org)60,61. Datasets with available data for NK cell expression were included for this analysis.
To study CREM gene expression in TI-NK cells in the microenvironment of different types of cancer, we used the preprocessed scRNA-seq data and models of TI-NK cells prepared as previously described26, which have been made available on Zenodo (https://doi.org/10.5281/zenodo.10139343) and as an online resource (http://nk-scrna.malmberglab.com/).
For the analysis of CREM gene expression in different types of NK cells, we studied the scRNA-seq datasets provided in previous study28 that can be downloaded from the associated website (https://collections.cellatlas.io/meta-nk). The analysis of the above data and corresponding plotting were conducted using the Python based software scanpy. We followed the analytical steps detailed in the official website for scanpy software (https://scanpy.readthedocs.io/en/stable/tutorials/index.html).
Analysis of TCGA data
Gene expression and clinical data were obtained from TCGA (https://gdc.cancer.gov/about-data/publications/pancanatlas). Association of CREM with survival was performed using Cox regression implemented in the R package survival. Survival analysis was performed at the level of the entire cohort and individual cancers (with >400 samples) while controlling for tumour stage and age at diagnosis. For the survival analysis with the full cohort, we also controlled for the tumour type.
Regulon analysis
To identify key transcription factors and quantify the biological activity of their corresponding regulons in the pancreatic ductal adenocarcinoma scRNA‐seq dataset27, we applied the single‐cell regulatory network inference and clustering (SCENIC) workflow, as previously described62. In brief, we used the Python implementation pySCENIC (with default parameters) on a high‐performance computing cluster. Regulatory interactions between a curated list of transcription factors and candidate target genes were inferred from scRNA‐seq co‐expression patterns using the GRNBoost2 algorithm implemented in Arboreto63. This procedure generated an adjacency matrix linking each transcription factor to putative target genes along with an importance score. Next, we assembled candidate modules consisting of each transcription factor and its associated target genes. To distinguish direct from indirect regulatory targets, these modules were refined by selecting only those genes for which promoters contained the relevant transcription factor‐specific DNA motifs, as determined by RcisTarget motif enrichment analysis. We then quantified the relative biological activity of each refined module (regulon) at single‐cell resolution by calculating the AUC for every cell. Differential regulon activity across experimental groups was identified using the FindMarkers function in Seurat, and we visualized the top 50 differentially active regulons (adjusted P < 0.01) by generating a heatmap of the scaled AUC matrix with the DoHeatmap function.
ChIP assay
ChIP assays were performed at the MDACC Epigenomics Profiling Core as previously described64,65 with some modifications. In brief, 20–30 million NK cells of the indicated conditions (NT, IL-15, CAR70–IL-15 and CAR70) were crosslinked with 1% formaldehyde for 10 min, quenched with 125 mM glycine for 5 min followed by chromatin preparation and sonication to obtain fragment sizes of 200–600 bp. In other experiments, NT NK cells were treated for 1 or 6 h with increasing concentrations of IL-15 (0, 500 and 5,000 pg ml–1). Similarly, NK cells were then crosslinked with 1% formaldehyde for 10 min, quenched with 125 mM glycine for 5 min followed by chromatin preparation and sonication to obtain fragment sizes of 200–600 bp. ChIP was performed overnight with antibodies specific to pCREB (9198, Cell Signaling Technology, 1:50) and pSTAT3-Y705 (9145, Cell Signaling Technology, 1:100), STAT5B (13-5300, Invitrogen, 1:50), CREB (sc-240, 1:175), CREM (CABT-B10032, Creative Diagnostics, 1:100) and IgG (2729, Cell Signaling Technology). The immunocomplexes were collected the following day using DiaMag Protein A-coated magnetic beads (Diagenode, C03010020), washed and reverse-crosslinked overnight followed by DNA extraction. The DNA regions of interest were detected by SYBR real-time qPCR using oligonucleotides in putative promoter regions of CREM and STAT3 using primer pairs listed in Supplementary Table 1. For CREM ChIP–seq, input and CREM ChIP DNA libraries were prepared using a NEB Ultra II DNA library prep kit (New England Biolabs, E7645) following manufacturer’s instructions and subjected to next-generation sequencing to obtain about 30 million 50 bp paired-end reads per sample. Sequencing was performed at the MDACC Advanced Technology Genomics Core (ATGC) facility.
Bulk RNA-seq and analysis
NT NK cells, CREM WT CAR70–IL-15 NK cells and CREM KO CAR70–IL-15 NK cells were either cultured alone or incubated for 24 h with UMRC3 cells at an E/T ratio of 1:1. After that, NK cells were collected and sorted on a BD Biosciences Aria II cell sorter or a Beckman Coulter CytoFLEX SRT cell sorter at the MDACC North Campus Flow Cytometry and Cellular Imaging Core Facility. NK cells were sorted on single cells+live+GFP–CD45+CD56+ cells. RNA was isolated from NK cells using a RNeasy Plus Mini kit (Qiagen, 74134) according to the manufacturer’s protocol. Barcoded, Illumina compatible, stranded mRNA libraries were prepared using a KAPA Stranded mRNA-Seq kit (Roche). In brief, 250 ng total RNA from each NK cell condition was captured using magnetic Oligo-dT beads. After bead elution and clean-up, the resultant PolyA RNA was fragmented using heat and magnesium. First-strand synthesis was performed using random priming followed by second-strand synthesis with the incorporation of dUTP into the second strand. The ends of the resulting double-stranded cDNA fragments were repaired, 5′-phosphorylated and 3′-A tailed, and Illumina-specific indexed adapters were ligated. The products were purified and enriched to generate the full-length library with nine cycles of PCR. The strand marked with dUTP was not amplified, which resulted in a strand-specific library. The libraries were quantified using a Qubit dsDNA HS Assay kit and assessed for size distribution using 4200 TapeStation High Sensitivity D1000 ScreenTape (Agilent Technologies) and then multiplexed, with 24 libraries per pool. The library pool was quantified by qPCR using a KAPA Library Quantification kit (Roche) and sequenced on a NovaSeq6000 SP flow cell (Illumina) using the 100 nucleotide paired-end format.
The raw FASTQ files were processed using the nf-core/rnaseq pipeline (v.3.14.0). All samples passed the built-in quality control. The resulting read mapping data in bigwig format and the expression quantification matrix in transcripts per million (TPM) were used for downstream analyses and visualization plots.
Bulk ATAC–seq and analysis
NK cells were prepared and sorted as for bulk RNA-seq preparation. ATAC–seq library preparation was performed at the MDACC Epigenomics Profiling Core following a previously described protocol3,66 with minor modifications. Nuclei isolated from NK cells were tagmented using Tagment DNA TDE1 enzyme (Illumina) and the resulting libraries were purified using SPRISelect beads (Beckman Coulter). Libraries were sequenced on a NovaSeq6000 SP flow cell using the 100 nucleotide PE format with an 8 nucleotide single index.
For each bulk ATAC–seq sample, the pair-end reads from raw FASTQ files were aligned to the human genome (GRCh38) using bwa67 mem mode with duplicated reads removed. The 5′ ends of ATAC–seq reads were shifted to the actual cut site of the transposase using the alignmentSieve module implemented in DeepTools. Peaks were called using MACS2 (ref. 68) using the pair-end read information. The MACS2 outputs from multiple samples were loaded using DiffBind (v.3.8.4)69. The peak sets from multiple samples were identified as the overlapping ones among samples using the UseSummarizeOverlaps function in DiffBind. Overall chromatin openness was visualized using Deeptools (v.3.5.2)70. Motif activities for each sample were calculated using the function RunChromVAR in Signac (v.1.12.0)71 with the JASPAR motif database (2020 version)72. GeneActivities of each sample were calculated using the aggregation method implemented in Signac73.
ChIP–seq analysis
The read processing of ChIP–seq data was the same as for ATAC–seq. We performed quality control through visualization of coverage enrichment along a gene body and filtered two samples (from two donors in the CAR70–IL-15 NK cell condition) that showed no enrichment of TSS regions. Thus, these samples were excluded from the analysis. Target genes were called for each sample when peaks were detected within 1 kb of the gene TSS using ChIPseeker (v.1.38.0)74 and were removed if overlapping with the paired input (control) sample. Top targets (when aligned with RNA-seq) were called when the fold change was greater than 0.3 in log[TPM] between conditions. Functional enrichment of gene sets was performed using a hypergeometric test (HypeR package (v.2.0.0)75). Differential enrichment of gene sets between conditions was performed using gsea (fgsea package (v.1.28.0)76). Genome track visualization was performed using epiwraps (https://github.com/ETHZ-INS/epiwraps) using bigWig files as the original input.
Figure 5e was prepared using GSEA of upregulated and downregulated pathways in CREM KO versus WT CAR70–IL-15 NK cells as assessed by bulk RNA-seq. For this GSEA, only direct targets of CREM from CREM ChIP–seq were considered in each Hallmark pathway. Fig. 5h also represents GSEA. GSEA was applied as previously described77 to test the enrichment of a given gene set by assessing whether the gene set members commonly rank at the top or bottom of a ranked gene list, with a model related to one-sided Kolmogorov–Smirnov tests.
Animal experiments
All procedures and experimental protocols involving mice were performed in accordance with the American Veterinary Medical Association and National Institutes of Health recommendations under protocols approved by the MD Anderson Cancer Center Institutional Animal Care and Use Committee (protocol number 00001263-RN01). We used 8–10-week-old NSG mice for the xenograft model for the mouse experiments to assess the antitumour activity of CREM KO CAR-NK cells in vivo. Where applicable, mice were randomized prior to therapy according to tumour size to ensure an equal tumour burden among all groups. Mice were injected and treated by an operator who was blinded to the treatment groups. Pathology analyses were performed by a pathologist who was blinded to the differences between the treatment groups and expected outcomes.
For the Burkitt lymphoma Raji model, NSG mice were irradiated with 300 cGy. The following day, mice received 20,000 firefly luciferase-labelled Raji cells intravenously. Mice were then injected the same day with NK cells of the indicated conditions through the tail vein. To model exhaustion, we used older CAR70–IL-15 NK cells (23 days old) at a subtherapeutic dose (4 × 106 CAR+ cells). Tumour growth was monitored using weekly bioluminescence imaging (Spectral Instrument Imaging (SII) system). Signal quantitation as the average radiance (p s–1 cm–2 sr–1) was determined using Aura (v.4.0.7). Mice were either followed for survival or underwent timed euthanasia at days 10 and 20 after treatment. To study NK cell engraftment, blood was collected on days 10 and 20 after NK cell injection and analysed by flow cytometry. For the mice that underwent timed euthanasia, liver, lung, spleen and bone marrow were collected. The organs were processed and either stained to assess NK cell engraftment by flow cytometry or pooled for ex vivo functional assays. The following kits were used for tissue processing according to the manufacturer’s protocols: Lung Dissociation kit, mouse (Miltenyi Biotec, 130-095-927), Liver Dissociation kit, mouse (Miltenyi Biotec, 130-105-807) and Spleen Dissociation Kit, mouse (Miltenyi Biotec, 130-095-926).
In the metastatic PDX mouse model of breast cancer, female NSG mice received an injection of 300,000 BCX.010 cells through the tail vein on day −7, followed by irradiation on day −1. NK cells were injected on day 0 through the tail vein. We performed two independent experiments: one with timed euthanasia of the mice and one for survival using three CB donors infused at subcurative doses. One donor was used for the timed euthanasia experiment and two donors were used for the survival experiment. Flow cytometry analysis of NK cell kinetics was conducted by drawing blood on days 10 and 20 after NK cell injection. To investigate the effectiveness and migration of NK cells to tumour sites in the BCX.010 model, mice were euthanized on day 35 after NK cell injection in the timed euthanasia experiment. Lungs and livers were collected, fixed and paraffin-embedded. Tumour nodules were quantified by a pathologist through haematoxylin and eosin (H&E) staining of lung and liver sections using bright-field microscopy. Immunohistochemistry staining for hCD45 and GZMB was performed on lung and liver sections to identify NK cells and cytotoxic NK cells, respectively. Whole slide digital imaging was performed using an Aperio AT2 after immunohistochemical staining on a Leica Bond RX autostainer for hCD45 and GZMB on adjacent serial sections. Images were deconvoluted in HALO (v.3.6) Deconvolution module (v.1.1.8). Deconvoluted images were subsequently registered and fused using the same HALO software.
To investigate safety concerns with CREM KO CAR-NK cells, we performed an in vivo experiment based on the BCX.010 infusion model. As described above, mice engrafted with the BCX.010 cells were treated with CREM WT or CREM KO CAR70–IL-15 NK cells 1 week later. As a control, CREM KO CAR70–IL-15 NK cells were infused in mice without tumours. Two weeks later, at the peak of NK cell engraftment, mice were euthanized and the histology of sections of vital organs (liver, lungs and kidneys) was evaluated by a veterinary pathologist who is board certified by the American College of Veterinary Pathologists and the American Board of Toxicology. Blood was also collected from another group of mice 30 days after treatment with either CREM WT or CREM KO CAR70–IL-15 NK cells and analysed for haematological parameters and chemistry to evaluate for acute and subacute toxicity. For the BCX.010 model in vivo experiments, NK cells were infused at a dose of 1 or 3 × 106 cells based on the pre-infusion in vitro activity for each donor.
For the orthotopic mouse model of human pancreatic cancer, NSG mice underwent surgical orthotopic implantation of 300,000 PATC148 cells directly into the pancreas. One week later, mice were intraperitoneally treated with the indicated NK cell products (5 × 106 cells) after irradiation one day before. Blood was similarly drawn on days 10 and 20 after NK cell treatment. To assess tumour burden by histology, mice were euthanized on day 36 after NK cell treatment. Pancreases were collected, fixed and paraffin embedded. Tumour nodules were quantified after H&E staining by a pathologist.
Seahorse assays
Glycolysis measured on the basis of the extracellular acidification rate (ECAR) and mitochondrial respiration measured on the basis of the oxygen consumption rate (OCR) were assayed using an Agilent Seahorse XF Analyzer (Agilent) following the manufacturer’s protocol. Seahorse glycolysis stress tests were performed using 2 g l−1 d-glucose, 2.5 μM oligomycin and 50 mM 2-deoxyglucose mixed with Hoechst 33342 (Invitrogen) dye. Seahorse Mito stress tests were performed using 2.5 μM oligomycin, 0.5 μM FCCP and 0.5 μM rotenone–antimycin A mixed with Hoechst 33342 (Invitrogen) dye. Technical triplicates for each biological donor replicate were used. Following the assays, viable cells were counted using live-cell imaging and counting in a Cytation 1 machine. Normalized OCR or ECAR data per 1,000,000 live NK cells are shown. The basal respiration was calculated using the following formula: last rate measurement before first injection − non-mitochondrial respiration rate. This value represents the minimum rate measurement after rotenone–antimycin A treatment. The maximal respiration was calculated using the following formula: maximum rate measurement after FCCP injection − non-mitochondrial respiration. The baseline glycolysis represents the non-glycolytic acidification (the last rate measurement before glucose injection). The glycolytic capacity was calculated using the following formula: maximum rate measurement after oligomycin injection − last rate measurement before glucose injection.
Statistics
Data were collected and organized using Microsoft Excel for Mac 2023. Statistical analyses were conducted with GraphPad Prism (Prism 9 and 10, GraphPad Software). Quantitative differences were assessed using either ANOVA for multiple groups (with multiple comparisons when applicable) or a t-test for two groups. Statistical significance was defined as P < 0.05. Mean values ± s.e.m. were used to represent the data, unless otherwise specified. The specific statistical tests used and corresponding sample sizes (n) are detailed in each figure legend. No statistical methods were used to predetermine sample sizes.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq data are available through the Gene Expression Omnibus (GEO) under accession number GSE190976. The bulk ATAC–seq, RNA-seq and ChIP–seq data are available through the GEO under accession number GSE267523. The following publicly available datasets from the GEO were reanalysed: GSE245690 and GSE156405. The TISCH2 database (available at http://tisch.comp-genomics.org) was used to analyse available datasets (indicated in the corresponding heatmap). The datasets in ref. 28 were downloaded from the associated website (https://collections.cellatlas.io/meta-nk). The human gene database GeneCard was used to design the CAR constructs. Source data are provided with this paper.
Code availability
No custom code was used.
References
Li, Y., Rezvani, K. & Rafei, H. Next-generation chimeric antigen receptors for T- and natural killer-cell therapies against cancer. Immunol. Rev. 320, 217–235 (2023).
Liu, E. et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 382, 545–553 (2020).
Marin, D. et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19+ B cell tumors: a phase 1/2 trial. Nat. Med. 30, 772–784 (2024).
Lanier, L. L. Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 9, 495–502 (2008).
Cózar, B. et al. Tumor-infiltrating natural killer cells. Cancer Discov. 11, 34–44 (2021).
Sharma, P. & Allison, J. P. The future of immune checkpoint therapy. Science 348, 56–61 (2015).
Cao, Y. et al. Immune checkpoint molecules in natural killer cells as potential targets for cancer immunotherapy. Signal Transduct. Target. Ther. 5, 250 (2020).
Tang, F. et al. A pan-cancer single-cell panorama of human natural killer cells. Cell 186, 4235–4251 (2023).
Lindner, S. E., Johnson, S. M., Brown, C. E. & Wang, L. D. Chimeric antigen receptor signaling: functional consequences and design implications. Sci. Adv. 6, eaaz3223 (2020).
Rautela, J. & Huntington, N. D. IL-15 signaling in NK cell cancer immunotherapy. Curr. Opin. Immunol. 44, 1–6 (2017).
Delconte, R. B. et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 17, 816–824 (2016).
Zhu, H. et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell 27, 224–237 (2020).
Daher, M. et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 137, 624–636 (2021).
Li, L. et al. Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Sci. Adv. 9, eadd6997 (2023).
Globig, A.-M. et al. The β1-adrenergic receptor links sympathetic nerves to T cell exhaustion. Nature 622, 383–392 (2023).
Morotti, M. et al. PGE2 inhibits TIL expansion by disrupting IL-2 signalling and mitochondrial function. Nature 629, 426–434 (2024).
Wu, V. H. et al. The GPCR–Gαs–PKA signaling axis promotes T cell dysfunction and cancer immunotherapy failure. Nat. Immunol. 24, 1318–1330 (2023).
Zhu, J. et al. Tumour immune rejection triggered by activation of α2-adrenergic receptors. Nature 618, 607–615 (2023).
Bodor, J. & Habener, J. F. Role of transcriptional repressor ICER in cyclic AMP-mediated attenuation of cytokine gene expression in human thymocytes. J. Biol. Chem. 273, 9544–9551 (1998).
Bohn, T. et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 19, 1319–1329 (2018).
Foulkes, N. S., Borrelli, E. & Sassone-Corsi, P. CREM gene: use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell 64, 739–749 (1991).
Subramanyam, S. H. & Tenbrock, K. The cAMP responsive element modulator (CREM) is a regulator of CD4+ T cell function. Biol. Chem. 402, 1591–1596 (2021).
Molina, C. A., Foulkes, N. S., Lalli, E. & Sassone-Corsi, P. Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell 75, 875–886 (1993).
Luo, Z. et al. Differential expression of CREM/ICER isoforms is associated with the spontaneous control of HIV infection. mBio 13, e0197921 (2022).
Yoshida, N. et al. ICER is requisite for Th17 differentiation. Nat. Commun. 7, 12993 (2016).
Netskar, H. et al. Pan-cancer profiling of tumor-infiltrating natural killer cells through transcriptional reference mapping. Nat. Immunol. 25, 1445–1459 (2024).
Lee, J. J. et al. Elucidation of tumor–stromal heterogeneity and the ligand–receptor interactome by single-cell transcriptomics in real-world pancreatic cancer biopsies. Clin. Cancer Res. 27, 5912–5921 (2021).
Rebuffet, L. et al. High-dimensional single-cell analysis of human natural killer cell heterogeneity. Nat. Immunol. 25, 1474–1488 (2024).
Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 4, a011148 (2012).
Delmas, V. & Sassone-Corsi, P. The key role of CREM in the cAMP signaling pathway in the testis. Mol. Cell. Endocrinol. 100, 121–124 (1994).
Abrahamsen, H. et al. TCR- and CD28-mediated recruitment of phosphodiesterase 4 to lipid rafts potentiates TCR signaling. J. Immunol. 173, 4847–4858 (2004).
Melville, Z. et al. The activation of protein kinase A by the calcium-binding protein S100A1 is independent of cyclic AMP. Biochemistry 56, 2328–2337 (2017).
Dunn, T. A., Storm, D. R. & Feller, M. B. Calcium-dependent increases in protein kinase-A activity in mouse retinal ganglion cells are mediated by multiple adenylate cyclases. PLoS ONE 4, e7877 (2009).
Trebak, M. & Kinet, J. P. Calcium signalling in T cells. Nat. Rev. Immunol. 19, 154–169 (2019).
Foulkes, N. S., Borjigin, J., Snyder, S. H. & Sassone-Corsi, P. Transcriptional control of circadian hormone synthesis via the CREM feedback loop. Proc. Natl Acad. Sci. USA 93, 14140–14145 (1996).
Bulliard, Y., Andersson, B. S., Baysal, M. A., Damiano, J. & Tsimberidou, A. M. Reprogramming T cell differentiation and exhaustion in CAR-T cell therapy. J. Hematol. Oncol. 16, 108 (2023).
Sun, F. et al. The MAPK dual specific phosphatase (DUSP) proteins: a versatile wrestler in T cell functionality. Int. Immunopharmacol. 98, 107906 (2021).
Wahlen, S. et al. The transcription factor RUNX2 drives the generation of human NK cells and promotes tissue residency. eLife 11, e80320 (2022).
Burns, A. et al. The role of KLF7 in hematopoiesis and lymphopoiesis (160.2). J. Immunol. 186, 160.2 (2011).
McCutcheon, S. R. et al. Transcriptional and epigenetic regulators of human CD8+ T cell function identified through orthogonal CRISPR screens. Nat. Genet. 55, 2211–2223 (2023).
Dean, I. et al. Rapid functional impairment of natural killer cells following tumor entry limits anti-tumor immunity. Nat. Commun. 15, 683 (2024).
Bernard, K. et al. Engagement of natural cytotoxicity programs regulates AP-1 expression in the NKL human NK cell line. J. Immunol. 162, 4062–4068 (1999).
Shanley, M. et al. Interleukin-21 engineering enhances NK cell activity against glioblastoma via CEBPD. Cancer Cell 42, 1450–1466 (2024).
Acharya, S. et al. CD28 costimulation augments CAR signaling in NK cells via the LCK/CD3ζ/ZAP70 signaling axis. Cancer Discov. 14, 1879–1900 (2024).
Li, Y. et al. KIR-based inhibitory CARs overcome CAR–NK cell trogocytosis-mediated fratricide and tumor escape. Nat. Med. 28, 2133–2144 (2022).
Bodor, J., Fehervari, Z., Diamond, B. & Sakaguchi, S. ICER/CREM-mediated transcriptional attenuation of IL-2 and its role in suppression by regulatory T cells. Eur. J. Immunol. 37, 884–895 (2007).
Yu, K. et al. CREM is correlated with immune-suppressive microenvironment and predicts poor prognosis in gastric adenocarcinoma. Front. Cell Dev. Biol. 9, 697748 (2021).
Smith, S. L. et al. Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv. 4, 1388–1406 (2020).
Rauen, T., Hedrich, C. M., Juang, Y.-T., Tenbrock, K. & Tsokos, G. C. cAMP-responsive element modulator (CREM) α protein induces interleukin 17 A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J. Biol. Chem. 286, 43437–43446 (2011).
Hedrich, C. M., Rauen, T., Kis-Toth, K., Kyttaris, V. C. & Tsokos, G. C. cAMP-responsive element modulator α (CREMα) suppresses IL-17F protein expression in T lymphocytes from patients with systemic lupus erythematosus (SLE). J. Biol. Chem. 287, 4715–4725 (2012).
Ghosh, D., Kis‐Toth, K., Juang, Y. T. & Tsokos, G. C. CREMα suppresses spleen tyrosine kinase expression in normal but not systemic lupus erythematosus T cells. Arthritis Rheum. 64, 799–807 (2012).
Tenbrock, K. et al. The cyclic AMP response element modulator regulates transcription of the TCR ζ-chain. J. Immunol. 175, 5975–5980 (2005).
Barabitskaja, O., Foulke, J. S., Pati, S., Bodor, J. & Reitz, M. S. Suppression of MIP-1β transcription in human T cells is regulated by inducible cAMP early repressor (ICER). J. Leukoc. Biol. 79, 378–387 (2006).
Masquilier, D. & Sassone-Corsi, P. Transcriptional cross-talk: nuclear factors CREM and CREB bind to AP-1 sites and inhibit activation by Jun. J. Biol. Chem. 267, 22460–22466 (1992).
Schnoegl, D., Hiesinger, A., Huntington, N. D. & Gotthardt, D. AP-1 transcription factors in cytotoxic lymphocyte development and antitumor immunity. Curr. Opin. Immunol. 85, 102397 (2023).
Kerbauy, L. N. et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30+ malignancies. Clin. Cancer Res. 27, 3744–3756 (2021).
Li, L. et al. A novel immature natural killer cell subpopulation predicts relapse after cord blood transplantation. Blood Adv. 3, 4117–4130 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Jiang, P. et al. Systematic investigation of cytokine signaling activity at the tissue and single-cell levels. Nat. Methods 18, 1181–1191 (2021).
Han, Y. et al. TISCH2: expanded datasets and new tools for single-cell transcriptome analyses of the tumor microenvironment. Nucleic Acids Res. 51, D1425–D1431 (2022).
Sun, D. et al. TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 49, D1420–D1430 (2020).
Van de Sande, B. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247–2276 (2020).
Moerman, T. et al. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 35, 2159–2161 (2018).
Jain, A. K. et al. LncPRESS1 is a p53-regulated LncRNA that safeguards pluripotency by disrupting SIRT6-mediated de-acetylation of histone H3K56. Mol. Cell 64, 967–981 (2016).
Mahadevan, K. K. et al. Elimination of oncogenic KRAS in genetic mouse models eradicates pancreatic cancer by inducing FAS-dependent apoptosis by CD8+ T cells. Dev. Cell 58, 1562–1577 (2023).
Corces, M. R. et al. An improved ATAC–seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Ross-Innes, C. S. et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Fornes, O. et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92 (2019).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Yu, G., Wang, L.-G. & He, Q.-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Federico, A. & Monti, S. hypeR: an R package for geneset enrichment workflows. Bioinformatics 36, 1307–1308 (2019).
Korotkevich, G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv https://doi.org/10.1101/060012 (2021).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Acknowledgements
We thank staff at the MD Anderson Cancer Center Epigenomics Profiling Core for assistance with the ChIP and ATAC–seq assays. This work was supported in part by philanthropic contributions to the University of Texas MD Anderson Cancer Center Institute for Cell Therapy Discovery and Innovation; The Sally Cooper Murray endowment; The Marcus Foundation; Vijay and Marie Goradia; by grants (1 R01 CA211044-01, 5 P01CA148600-03 and 1 R01 CA288617-01) from the National Institutes of Health (NIH); by an ASCO Young Investigator Award from the American Society of Clinical Oncology Conquer Cancer Foundation; by a grant from the University Cancer Foundation through the Institutional Research Grant program at the University of Texas MD Anderson Cancer Center; by a Specialized Program of Research Excellence (SPORE) grant in Ovarian Cancer (5P50CA281701-02); by a grant from the NIH to the MD Anderson Cancer Center Advanced Technology Genomics Core (ATGC) Facility (CA016672 and 1S10OD024977-01); and by a grant (P30CA16672) from the NIH to the MD Anderson Flow Cytometry and Cellular Imaging Core Facility, which assisted with the CyTOF studies in this project. X.R.J. and Y.L. are supported by CPRIT Research Training Award RP210028. The Baylor College of Medicine Mass Spectrometry Proteomics Core, which provided critical confirmatory data, is supported by a Dan L. Duncan Comprehensive Cancer Center NIH award (P30 CA125123), a CPRIT Core Facility Award (RP210227) and NIH High-End Instrumentation Award (S10 OD026804). This work was partly supported by the Deutsche Forschungsgemeinschaft (DFG), SFB1292 TP01 (to T. Bohn and T. Bopp).
Author information
Authors and Affiliations
Contributions
H.R. and K.R. conceptualized the study. H.R., R.B., S.A., Y.-S.H., P. Liu, D.Z., T. Bohn, R.U., P.P., P. Li, C.M.J., S.K., M.S., B.L., S.M.M., D.F., Y.L., P.B., N.U., R.S., X.W., H.S., V.W., A.L.G., S. Rawal, M.K., X.R.J., A.K.N.C., I.B., B.K., S.T., M.M., J.J., R.Z.Y., S. Rosemore, J.-M.P., F.R.S. and A.K.J. performed experiments and interpreted and analysed data. Q.L., V.M., M. Dede, D.X., H.F., Y.T., C.Z. and J.D. performed bioinformatic analyses. N.W.F. performed the pathology analyses on mouse samples. K.R., R.B., S.A., P. Liu, P. Li, M.S., M. Daher, B.L., Y.L., A.B., P. Lin, G.M.D., L.M.-F., D.M., A.K.J., A.M., K.C., T. Bopp and E.J.S. provided advice on experiments. H.R. and K.R. wrote the manuscript. S.A., M.S., G.M.D., L.M.-F., A.K.J. and E.J.S. provided comments on the manuscript. K.R. supervised the study.
Corresponding author
Ethics declarations
Competing interests
H.R., R.B., S.A., M.S., M. Daher, P.B., N.U., Y.L., P. Lin, D.M., E.J.S. and K.R. and The University of Texas MD Anderson Cancer Center have an institutional financial conflict of interest with Takeda Pharmaceutical. S.A., R.B., D.M., E.J.S. and K.R. have an institutional financial conflict of interest with Affimed. A.M. is listed as an inventor on a patent that has been licensed by Johns Hopkins University to ThriveEarlier Detection. A.M. serves as a consultant for Tezcat Biotechnology. A.M. receives royalties from a patent that is licensed to Exact Sciences. K.R. participates on the Scientific Advisory Board for Avenge Bio, Virogin Biotech, Navan Technologies, Caribou Biosciences, Bit Bio, Replay, oNKo Innate, The Alliance for Cancer Gene Therapy ACGT, Innate Pharma and Shinobi Therapeutics. K.R. is the scientific founder of Syena. E.J.S. participates on the Scientific Advisory Board for Adaptimmune Limited, Axio Research, Celaid Therapeutics, FibroBiologics, Navan Technologies, New York Blood Center and Zelluna Immunotherapy. M. Daher participates on the Scientific Advisory Board of Cellsbin. The remaining authors declare that they have no competing interests.
Peer review
Peer review information
Nature thanks Adelheid Cerwenka, Klaus Tenbrock, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 CREM is upregulated in CAR-NK cells following adoptive transfer.
(a) Heatmap of differentially expressed genes (DEGs; adjusted P value < 0.01 and absolute log2 fold change (FC) > 0.5) in CAR19/IL-15 NK cells post-infusion (day 7 and day 14) vs. pre-infusion from scRNA-seq data (Li et al. dataset14) based on Raji mouse model presented in Fig. 1; (b) UMAP expression plot of CREM; (c) CREM expression in the pre-infusion CAR-NK cell product and in CAR-NK cells isolated from mice over time; (d) Expression of select genes encoding calcium binding proteins that were identified to be overexpressed in post-infusion NK cells relative to pre-infusion NK cells; (e) UMAP distribution of cell cycle stages in CAR19/IL-15 NK cells; (f) Quantification of CREM expression pre- and post-infusion by stages of the cell cycle; (g) UMAP expression plot of IL-15 activity inferred by CytoSig (refer to Methods); (h) Quantification of IL-15 activity pre- and post-infusion; (i) Quantification of IL-15 activity pre- and post-infusion by stages of the cell cycle; (j) Scatter plot depicting the correlation between CREM and IL-15 activity pre- and post-infusion. Statistical comparisons were performed using one-way ANOVA followed by Tukey’s test for pairwise comparisons (c), one-way ANOVA (f,i), Wilcox Rank Sum test (d,h), and Spearman correlation (j).
Extended Data Fig. 2 CREM is induced by CAR-CD3ζ and interleukin 15 (IL-15) stimulation.
(a) Transduction efficiency of CAR70, CAR70.3ζ.Y6F and CD27 ECD NK cells; ECD: extracellular domain; (b) Whole cell lysates from NT, CAR70, CAR70.3ζ.Y6F and CD27 ECD NK cell groups were analyzed by western blot for phospho-CD3ζ (Y142; pCD3ζ). NK cells were unstimulated (−) or stimulated (+) with CD70 antigen (Ag) for 30 min. β-actin was used as loading control. Representative blot is shown; (c) Densitometry analysis quantifying the relative band intensity of CAR-specific pCD3ζ normalized to loading control (n = 3 donors); (d) CREM expression in NK cells that were either unstimulated or stimulated with IL-2 or IL-15 at increasing concentrations (50, 500, and 5000 pg/ml) for 24 h as assessed by qPCR (n = 3 donors); (e) CREM expression in NK cells stimulated with IL-15 (500 pg/ml) for 24 h in the presence or absence of an IL-15 antagonist (IL-15 Ab) as assessed by qPCR (n = 4 donors); (f) CREM expression in NT and CAR70 NK cells that were either unstimulated or stimulated with either IL-15 (500 pg/ml) or CD70 antigen (Ag) or both for 24 h as assessed by qPCR (n = 3 donors); (g) Representative FACS plot of CREM and IL-15R expression in NK cells stimulated with IL-15 (5000 pg/ml) for 24 h compared to unstimulated cells; (h) Percentage CREM expression in IL-15R+ vs. IL-15R- NK cells stimulated with increasing concentrations of IL-15 (n = 3 donors); (i,j) Longitudinal analysis of CREM (i) and Ki67 (j) expression in NK cells following stimulation with IL-15 (5000 pg/ml), assessed by flow cytometry (n = 3 donors); gMFI FC: geometric mean fluorescence intensity fold change; (k) Longitudinal analysis of CREM expression in NK cells following stimulation with IL-15 (5000 pg/ml), as assessed by qPCR (n = 6 donors); (l) CREM expression in NT and CAR70/IL-15 NK cells both 48 h post-transduction (Before) as well as one week later following expansion with universal antigen presenting cells (uAPCs) and IL-2 (After) as assessed by qPCR (n = 2 donors). ns: non-significant. Statistical comparisons were performed using one-way ANOVA with Tukey correction (a,c,e), two-way ANOVA with Tukey correction (d), and two-way ANOVA (Fisher’s LSD test, f,h,i,j,k,l). Data are represented as mean ± SEM.
Extended Data Fig. 3 Differential upregulation of the various splicing isoforms of CREM in NK cells.
(a) Overview of the CREM gene. Created in BioRender. Rafei, H. (2025) https://BioRender.com/q33z837; (b) CREM expression in NT and CAR70/IL-15 NK cells using primers designed against CREM-specific exon, ICER-specific exon #1 or ICER-specific exon #2 as assessed by qPCR (n = 11 donors); (c) CREM expression in NK cells stimulated with increasing concentrations of IL-15 using primers designed against CREM-specific exon, ICER-specific exon #1 or ICER-specific exon #2 as assessed by qPCR (n = 10 donors); (d,e) whole cell lysates from CREM wild-type (WT) NT, CREM WT CAR70/IL-15 (d) or CAR.TROP2/IL-15 (e), and CAR-NK cells following CRISPR/Cas9 knockout (KO) using guides targeting the CREM-exon or ICER-exon or both as analyzed by western blot for CREM with β-actin serving as a loading control. ns: non-significant. Statistical comparisons were performed using t-tests (b) and two-way ANOVA with Tukey correction (c).
Extended Data Fig. 4 CREM is induced in NK cells stimulated by certain cytokines and through CD16, NKp30, and NKp46.
(a) CREM expression in NK cells stimulated with increasing concentrations of indicated cytokines as assessed by qPCR (n = 3 donors); (b) Whole cell lysates from NK cells that were either unstimulated or stimulated with plate-bound anti-CD16, anti-NKp30, or anti-NKp46 antibodies for 30 min were analyzed by western blot for pCD3ζ as well as total CD3ζ. β-actin was used as loading control for the pCD3ζ gel (n = 2 donors); (c) CREM expression in NK cells that were either unstimulated or stimulated with plate-bound anti-CD16 (αCD16), anti-NKp30 (αNKp30), or anti-NKp46 (αNKp46) antibodies for 24 h, assessed by qPCR (n = 9 donors). unstim: unstimulated; ns: non-significant. Statistical comparisons were performed using one-way ANOVA individually for each cytokine (a) and one-way ANOVA (Fisher’s LSD test, c). Data are represented as mean ± SEM.
Extended Data Fig. 5 CREM expression in NK cells from patient samples and the TCGA.
(a) Normalized CREM expression in various cell types from scRNA-seq cancer datasets available from the TISCH2 database (available at http://tisch.comp-genomics.org)60,61. Datasets with available data for NK cell expression were included; (b) CREM expression in tumor-infiltrating NK cells in patients with various cancer types compared to PBMC NK cells from the publicly available dataset GSE245690 (ref. 26); (c) CREM expression in tumor-infiltrating NK cells in patients with pancreatic adenocarcinoma in PBMC, primary or metastatic samples from the publicly available dataset GSE156405 (ref. 27); (d) Heatmap for the top 50 differentially active regulons (adjusted P < 0.01) for tumor-infiltrating NK cells in metastases samples compared to those in primary samples or PBMC NK cells; (e) CREM regulon activity in the various NK cells; (f) Forest plot depicting CREM overall survival hazard ratios (HRs) across TCGA cancers; (g) Heatmap showing CREM expression across various NK cell clusters from the publicly available dataset (Rebuffet et al.)28 Statistical comparisons were performed using pairwise Wilcoxon rank‐sum tests followed by Bonferroni correction for multiple comparisons (c,e).
Extended Data Fig. 6 CREM is expressed in CAR70/IL-15 NK cells upon encounter with CD70+ tumor cells and is associated with an activated phenotype.
(a) CREM expression in non-transduced (NT), IL-15, CAR70/IL-15, and CAR70 NK cells as assessed by qPCR (n = 8 donors); (b) Percentage of CREM positive cells in live CD45+ GFP- CD56+ NK cells from NT, IL-15, CAR70/IL-15, and CAR70 NK cells following 24 h coculture with UMRC3 tumor cells at an effector-to-target (E:T) ratio of 1:1 as assessed by mass cytometry (n = 3 donors); (c) Violin plots of CREM expression in the various NK cell conditions at the single cell level as assessed by mass cytometry (n = 3 donors); the gray dashed line represents the median expression in NT NK cells; (d) Violin plots of CREM expression at the single cell level compared between the various clusters of the merged tSNE_CUDA analysis of the various conditions (n = 3 donors); (e) Percentage of CREM positive NK cells in the tSNE_CUDA clusters (n = 3 donors); (f) Frequencies of NK cells expressing CREM in the tSNE_CUDA clusters (colored portion); the degree of the color reflects the robust z-score of CREM expression in the corresponding cluster on a scale ranging from dark orange (z = +1) to dark blue (z = −1); (g) Total cells (blue), cells expressing the CAR (based on CD27 staining; red) and CREM (green) are shown on the tSNE_CUDA plots. Inset numbers indicate the percentages (%) of CAR expression on and CREM expression in the corresponding NK cell conditions; (h) Violin plots of expression of select markers of activation, inhibition as well as checkpoints in NK cells from clusters 1 (most abundant in CAR70/IL-15 NK cells) and 5 (most abundant in NT NK cells) at the single cell level as assessed by mass cytometry (n = 3 donors); (i) Heatmap showing expression of key NK cell phenotypic markers on fractions of positive CREM expression (CREM+) and negative CREM expression (CREM-) in the various NK cell conditions. Expression of each marker is represented by the robust z-score for the expression normalized across the conditions with a color scale ranging from blue (z = −1) to orange (z = +1) and the size of the circle for percentage expression. GZMB: granzyme B. ns: non-significant. Statistical comparisons were performed using one-way ANOVA (Fisher’s LSD test, a-e), t-tests (h), and one minus pearson hierarchical clustering (i). Data are represented as mean ± SEM.
Extended Data Fig. 7 IL-15 and CD16, NKp30, and NKp46 stimulations of NK cells lead to CREB phosphorylation; and CREM regulation by STAT3 and STAT5.
(a) Representative FACS plots of NK cells showing pCREB levels under the denoted conditions; (b) pCREB expression by phospho-flow cytometry in NK cells that were either untreated (−) or treated (+) with plate-bound anti-CD16, anti-NKp30, or anti-NKp46 for stimulation for 30 min, the PKA inhibitor H89, or the calcium chelator EGTA as indicated prior to profiling (n = 3 donors); (c) PKA activity assessed in whole cell lysates from NT NK cells that were either unstimulated or stimulated for 30 min with increasing concentrations of IL-15 (n = 3 donors); (d,e) pCREB expression by phospho-flow cytometry in NT NK cells that were either unstimulated or stimulated for 30 min with increasing concentrations of IL-15 (50, 500, and 5000 pg/ml) in the presence or absence of the PKA inhibitor H89 for up to 2 h prior to stimulation (n = 5 donors); FSK: forskolin; gMFI FC: geometric mean fluorescence intensity fold change; control: unstained; (f) pCREB expression by phospho-flow cytometry in NT NK cells that were either unstimulated or stimulated for 30 min with increasing concentrations of IL-15 in the presence or absence of the calcium chelator EGTA for up to 2 h prior to stimulation (n = 3 donors); (g) CREM expression as assessed by qPCR in NT NK cells that were either unstimulated or stimulated for 24 h with IL-15 (500 pg/ml) in the presence or absence of EGTA during the stimulation time (n = 3 donors); (h,i) Whole cell lysates from NT NK cells following 30 min stimulation with increasing doses of IL-15 as analyzed by western blot for phospho- and total STAT3 (h) and STAT5 (i) with β-actin serving as a loading control (n = 3 donors); (j,k) Whole cell lysates from NT, CAR70/IL-15, and CAR70 NK cells following stimulation of the CAR for 30 min as analyzed by western blot for phospho- and total STAT3 (j) and STAT5 (k) with β-actin serving as a loading control (n = 2 donors); (l,m) Whole cell lysates from NK cells that were either wild-type (WT) or knockout (KO) for STAT3 (l) or STAT5a/STAT5b (m) as analyzed by western blot for total STAT3 (l) and STAT5 (m) with β-actin serving as a loading control for STAT3 (l) and STAT5a (m) gels (n = 3 donors); (n) CREM expression in NK cells that were either WT, STAT3 KO, STAT5a/b KO, or CREM KO that were either unstimulated or stimulated with IL-15 (5000 pg/ml), assessed by qPCR (n = 5 donors); (o,p) ChIP-qPCR for the enrichment for pSTAT3 (o) and STAT5b (p) in the promoter region of CREM as well as a positive control promoter (STAT3 for both) in NT NK cells incubated for 1 h in the absence or presence of IL-15 (n = 4 donors). ns: non-significant; unstim: unstimulated. Statistical comparisons were performed using two-way ANOVA with Tukey correction (b,n), one-way ANOVA (Fisher’s LSD test, c), multiple t-tests with Holm-Šídák correction (e,f), one-way ANOVA with Tukey correction (g), and t-tests (o,p). Data are represented as mean ± SEM.
Extended Data Fig. 8 CREM KO improves CAR-NK cell function in multiple tumor models.
(a,b) Spheroid killing assay of GFP+ UMRC3 cells by CREM WT NT, CREM WT CAR70/IL-15, and CREM KO CAR70/IL-15 NK cells at an E:T ratio of (a) 1:1 and (b) 2:1 (n = 3 donors per group; the assays at the different E:T ratios were performed as part of the same experiment using the same donors as the ones used in Fig. 3a). The spheroid green image mean determines the spheroid growth over time. Data were normalized to the spheroid green image mean at T = 0 when the spheroid was formed and before adding the NK cells (black arrow); the bar graphs show the area under the curve (AUC) for the normalized spheroid green image mean (n = 3 donors); (c) Spheroid killing assay of GFP+ BCX.010 cells by CREM WT NT, CREM WT CAR70/IL-15, and CREM KO CAR70/IL-15 NK cells (n = 2 donors). Data were quantified and normalized as in (a,b); (d) Impedance killing assay of SKOV3 cells by CREM WT NT, CREM WT CAR.TROP2/IL-15, and CREM KO CAR.TROP2/IL-15 NK cells over time by xCELLigence device (n = 2 donors). The cell index which reflects tumor growth was normalized to the time of NK cell addition (black arrow); (e) KO efficiency of CREM in NT, IL-15, CAR70/IL-15 and CAR70 NK cells as assessed by PCR followed by gel electrophoresis; (f) Histograms of CD70 expression by flow cytometry in CD70 WT vs. CD70 KO UMRC3 cells; FMO: fluorescence minus one (negative control); (g) Impedance killing assay of CD70 WT or CD70 KO UMRC3 cells by NT, IL-15, CAR70/IL-15, or CAR70 NK cells that are either CREM WT or CREM KO (n = 2 donors). The cell index which reflects tumor growth was normalized to the time of NK cell addition (black arrow); (h) Impedance killing assay of UMRC3 cells by CREM WT or CREM KO NT NK cells that were either unstimulated or stimulated by incubation with increasing concentrations of IL-15 (50, 500, or 5000 pg/ml) for 24 h prior to adding them to UMRC3 cells plated the day before in xCELLigence device (n = 3 donors). The cell index which reflects tumor growth was normalized to the time of NK cell addition (black arrow). ns: non-significant. Statistical comparisons were performed using one-way (a-right,b-right) or two-way ANOVA with Tukey correction (a-left,b-left,c,d,g,h), Data are represented as mean ± SEM.
Extended Data Fig. 9 CREM KO improves CAR-NK cell function in multiple in vivo models.
(a) Body weight of the mice over time in the various groups in experiment depicted in Fig. 4a (n = 5 mice/group); (b) Ex vivo cytotoxicity of CREM WT and CREM KO CAR70/IL-15 NK cells harvested from mice sacrificed at day 10 and day 20 following NK cell infusion compared to the infusion products against mCherry+ K562 (n = 5 mice in each group); (c) Representative images from the assay in (b) at day 10; (d) Ex vivo cytotoxicity of CREM WT and CREM KO CAR70/IL-15 NK cells harvested from mice sacrificed at day 10 and day 20 following infusion compared to the infusion products against GFP+ Raji (n = 5 mice in each group); (e) Representative images from the assay in (d) at day 10; (f) Heatmap for the concentrations of the various cytokines in the serum of mice from day 10 and day 20 post-infusion in pg/ml; (g) Representative H&E staining of full lung sections from mice injected intravenously with BCX.010 PDX cell line and treated with CREM WT NT, CREM WT CAR70/IL-15, or CREM KO CAR70/IL-15 NK cells from the timed sacrifice experiment depicted in Fig. 4g; (h) Body weight of the mice over time in the various groups of the survival experiment depicted in Fig. 4g; (i) Quantification of flow cytometry analysis of the number of hCD45 + CD56 + CD16 + CAR+ (CD27 + ) cells (a marker of CAR+ NK cell engraftment) in the blood of mice 10 or 20 days after NK cell infusion in the various groups; data in (h) and (i) were pooled from two CB donors (n = 4 mice in BCX.010 alone group and n = 10 mice in each of the NT, CREM WT CAR70/IL-15 and CREM KO CAR70/IL-15 groups with 5 mice/CB donor); (j) Schematic illustration of the in vivo experimental plan for the PATC148 orthotopic mouse model. Created in BioRender. Rafei, H. (2025) https://BioRender.com/ryvr1mz; (k) Number of pancreatic tumor nodules in mice that were treated with CREM WT or KO CAR.TROP2/IL-15 NK cells (n = 4-5 mice per group); (l) Graph depicting flow cytometry analysis of the number of hCD45+ CD56+ CD16+ cells (a marker of NK cell engraftment) in the blood of mice 10 or 20 days after NK cell infusion in the CREM WT or KO CAR.TROP2/IL-15 NK cell groups (n = 4-5 mice per group). ns: non-significant. Statistical comparisons were performed using two-way ANOVA with Tukey correction (a,b-left,d-left,h,i), two-way ANOVA (b-right,d-right); two-tailed t-test (k), and two-way ANOVA (Fisher’s LSD test, l). Data are represented as mean ± SEM.
Extended Data Fig. 10 CREM KO CAR70/IL-15 NK cells do not cause toxicity in mice in a patient-derived xenograft (PDX) mouse model of metastatic breast cancer.
(a) Hematoxylin and eosin [H&E] stained tissues from mice treated with either CREM KO CAR70/IL-15 NK cells (in BCX.010 tumor-bearing (n = 5 mice) or non-tumor bearing (n = 3 mice) mice) or CREM WT CAR70/IL-15 NK cells in BCX.010 bearing mice (n = 3 mice). Mice were euthanized on day 14 post-treatment. Magnification: 10x. Scale bar: 100 μm; (b-d) Assessment of blood cell populations (b), kidney (c) and liver (d) function parameters in the blood of mice engrafted with BCX.010 tumors, at day 30 post-treatment with CREM WT CAR70/IL-15 NK cells (n = 4 mice) or CREM KO CAR70/IL-15 NK cells (n = 5 mice). WBC, white blood cells; HGB, hemoglobin; BUN, blood urea nitrogen; ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate aminotransferase; LDH, lactate dehydrogenase; ns, non-significant. Statistical comparisons were performed using Student’s t-test (b-d). Data are represented as mean ± SEM.
Extended Data Fig. 11 CREM modulates NK cell function through its transcriptional and epigenetic activities.
(a) UpSet plots of CREM binding sites in NT, IL-15, and CAR70 NK cells from one representative donor (same donor used to generate CAR70/IL-15 NK cells in Fig. 5b) as analyzed by CREM ChIP-seq; (b) Hallmark gene sets enriched in the CREM targets in NT, IL-15, and CAR70 NK cells and the proportion of genes in each set that was recovered among the targets (n = 3 donors per group); (c) Gene set enrichment analysis (GSEA) enrichment plot of upregulated and downregulated direct targets of CREM (based on CREM ChIP-seq data from CAR70/IL-15 NK cells) in CREM KO vs. CREM WT CAR70/IL-15 NK cells as assessed by bulk RNA-seq (n = 2 donors); (d) Venn diagram of the overlap between 2 donors in CREM ChIP-seq targets in CAR70/IL-15 NK cells that are downregulated (top) or upregulated (bottom) in CREM KO CAR70/IL-15 NK cells; (e) GSEA bar plots of downregulated (top) and upregulated (bottom) pathways (only direct targets of CREM from CREM ChIP-seq in CAR70/IL-15 NK cells were considered in each pathway) in CREM KO CAR70/IL-15 NK cells as assessed by bulk RNA-seq (n = 2 donors); (f) GSEA enrichment plots of upregulated and downregulated pathways (only direct targets of CREM from CREM ChIP-seq in CAR70/IL-15 NK cells were considered in each pathway) in CREM KO vs. CREM WT CAR70/IL-15 NK cells as assessed by bulk RNA-seq (n = 2 donors); (g) Hallmark gene sets reflecting enriched pathways in gained and lost accessible chromatin peaks in CREM KO CAR70/IL-15 NK cells in coculture with UMRC3 tumor cells as analyzed by ATAC-seq (n = 2 donors); GSEA was used for pathway enrichment between gained and lost accessible chromatin peaks; (h) GSEA analyses reflecting enriched Hallmark gene sets in accessible peaks in CREM KO vs. WT CAR70/IL-15 NK cells in coculture with tumor cells; (i) Chromatin accessibility tracks for select genes in CREM WT (top) vs. CREM KO (bottom) CAR70/IL-15 NK cells in coculture with UMRC3 cells from one representative donor; (j) Heatmap and hierarchical clustering of differentially accessible transcription factor motifs in CREM WT and CREM KO CAR70/IL-15 NK cells with UMRC3 tumor cell coculture. Heatmap was generated by merging data from two donors; NES: normalized enrichment score. Statistical comparisons were performed using one-sided hypergeometric test with FDR correction (b), and GSEA modeling one-sided Kolmogorov-Smirnov test with FDR correction (c,e-h).
Extended Data Fig. 12 CREM KO does not impact IL-15 receptor (IL-15R) proximal signaling but enhances the metabolic fitness of CAR70/IL-15 NK cells.
(a) Whole cell lysates from NK cells that were either wild-type (WT) or knockout (KO) for CREM 0, 30 min and 2 h following stimulation with IL-15 (5000 pg/ml) as analyzed by western blot for IL-15R proximal signaling components. β-actin serves as a loading control (n = 5 donors); (b) Densitometry analysis quantifying the relative band intensity of pSTAT3 and pSTAT5 normalized to total STAT3 and STAT5, respectively (n = 5 donors); (c) CISH expression in NK cells that were either WT or CREM KO that were either unstimulated or stimulated with IL-15, assessed by qPCR (n = 3 donors); (d-f) Measures of extracellular acidification rate (ECAR) upon addition of glucose, oligomycin (oligo), and 2-deoxy-D-glucose (2-DG) (d) and quantified basal glycolysis (e) and glycolytic capacity (f) of NK cells in the indicated groups (n = 3 donors); (g-i) Measures of oxygen consumption rate (OCR) upon addition of oligo, FCCP, and rotenone and antimycin A (R/A) (g) and quantified basal respiration (h) and maximal respiration (i) of NK cells in the various conditions (n = 3 donors). ns: non-significant. Statistical comparisons were performed using two-way ANOVA with Šídák correction (b,c), and one-way ANOVA with Tukey correction (e,f,h,i). Data are represented as mean ± SEM.
Supplementary information
Supplementary Information
This file contains Supplementary Fig. 1, Supplementary Table 1 and legends for Supplementary Videos.
Supplementary Video 1
CREM WT NT NK cell function against UMRC3 cells. Video depicting the cytotoxicity in real-time of CREM WT NT NK cells against GFP+ UMRC3 cells in a spheroid-killing assay. Time-lapse imaging was performed using the Incucyte system.
Supplementary Video 2
CREM WT CAR70–IL-15 NK cell function against UMRC3 cells. Video depicting the cytotoxicity in real-time of CREM WT CAR70–IL-15 NK cells against GFP+ UMRC3 cells in a spheroid-killing assay. Time-lapse imaging was performed using the Incucyte system.
Supplementary Video 3
CREM KO CAR70–IL-15 NK cell function against UMRC3 cells. Video depicting the cytotoxicity in real-time of CREM KO CAR70–IL-15 NK cells against GFP+ UMRC3 cells in a spheroid-killing assay. Time-lapse imaging was performed using the Incucyte system.
Supplementary Video 4
Untreated UMRC3 spheroid growth over time. Video depicting the growth over time of an untreated spheroid of UMRC3 cells as a control. Time-lapse imaging was performed using the Incucyte system.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rafei, H., Basar, R., Acharya, S. et al. CREM is a regulatory checkpoint of CAR and IL-15 signalling in NK cells. Nature 643, 1076–1086 (2025). https://doi.org/10.1038/s41586-025-09087-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09087-8
This article is cited by
-
Beyond CRISPR: next-gen precision engineering of CAR-NK cells for enhanced persistence, trafficking, and tumor eradication
Cancer Cell International (2026)
-
Overcoming barriers in CAR-NK immunotherapy: CRISPR-Driven advances in checkpoint editing and allogeneic design
Functional & Integrative Genomics (2026)
-
A new era in CAR-NK cell therapy: from technological innovations to clinical applications
World Journal of Pediatrics (2026)
-
Boosting immune cells to combat cancer using CRISPR engineering and large-scale in vivo testing
Nature (2025)
-
Disarming a molecular brake: cAMP-responsive element modulator deletion supercharges CAR-NK cells
Signal Transduction and Targeted Therapy (2025)
