
Abstract
Cellular plasticity is a principal feature of vertebrate adaptation, tissue repair and tumorigenesis1,2. However, the mechanisms that regulate the stability of somatic cell fates remain unclear. Here, we use the somatic plasticity of thymic epithelial cells, which facilitates the selection of a self-discriminating T cell repertoire3, as a physiological model system to show that fluctuations in background chromatin accessibility in nucleosome-dense regions are amplified during thymic epithelial maturation for the ectopic expression of genes restricted to other specialized cell types. This chromatin destabilization was not dependent on AIRE-induced transcription but was preceded by repression of the tumour suppressor p53. Augmenting p53 activity indirectly stabilized chromatin, inhibited ectopic transcription, limited cellular plasticity and caused multi-organ autoimmunity. Genomic regions with heightened chromatin accessibility noise were selectively enriched for nucleosome-destabilizing polymeric AT tracts and were associated with elevated baseline DNA damage and transcriptional initiation. Taken together, our findings define molecular levers that modulate cell fate integrity and are used by thymic epithelial cells for immunological tolerance.
Similar content being viewed by others

Main
The stability of somatic cell identities is essential for the coordination of specialized organ systems, and aberrant deviations from differentiated states can lead to disease4. However, alterations in somatic cell fates can promote tissue repair and enable adaptation to changing microenvironments1. How functional variation in a somatic lineage is constrained or promoted to regulate the balance between cellular stability and plasticity remains unclear. Here we investigate the underlying mechanisms in medullary thymic epithelial cells (mTECs), which express nearly the entire coding genome and adopt states that mirror specialized cells in disparate tissues for the selection of T cells that can protect the host but remain tolerant to self-constituents3,5,6. The breakdown of this selection in autoimmune polyglandular syndrome type 1 revealed the transcriptional activator AIRE to be an important determinant of thymic epithelial plasticity7. However, chromatin accessibility and transcriptional initiation at AIRE-regulated loci do not require AIRE8,9, indicating that there is an orthogonal mechanism that poises mTECs for cellular plasticity. We investigated the nature of this mechanism in individual mTECs by jointly profiling their transcriptome and chromatin accessibility landscapes along their developmental trajectory (Fig. 1a).

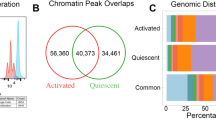
a,b, A scATAC-seq uniform manifold approximation and projection (UMAP) (b) visualization of 9,928 mTECs from Multiome profiling (a); the colours show the cluster annotation. c, Aire expression data overlaid on the UMAP. d, Number of expressed αTSGs overlaid on the UMAP. e, The indicated αTSG expressions overlaid on the UMAP. f, Sum expression of each αTSG (n = 3,184) in each UMAP cluster (colours as in a) as a fraction of the total. g, Histogram of minimum false discovery rate (FDR) values of differentially accessible scATAC-seq peaks (two-sided Mann–Whitney–Wilcoxon tests) for cells expressing a particular αTSG (αTSGpos) versus not expressing it (αTSGneg) (black, n = 3,184 αTSGs) or early AIRE+ versus immature mTECs (red arrow, Pmin = 8.04 × 10−17). h, Histogram of significant peaks detected (FDR ≤ 0.1) from αTSGpos versus αTSGneg (black) or early AIRE+ versus immature mTECs (red arrow, n = 35,204 peaks) comparisons. i, Schematic of differential chromatin accessibility analysis visualized as heatmaps at tissue-specific loci between αTSGpos versus αTSGneg mTECs. j, Heatmaps of differential OOP (left) or WIP (right) accessibility at indicated αTSGs between αTSGpos versus αTSGneg mTECs. k, Heatmaps of differential OOP (left) or WIP (right) accessibility at all 3,184 αTSGs detected between αTSGpos versus αTSGneg mTECs. l,m, Aggregate ratios of OOP or WIP scATAC-seq fragments from αTSGpos versus αTSGneg mTECs (l) or MIGpos versus MIGneg mTECs (m) (Extended Data Fig. 1u) at the indicated loci. n, scATAC-seq fragment size distributions from mature mTECs. o, Histogram of indicated P-values (two-sided likelihood ratio tests) for the probability of each αTSG being expressed as a function of local OOP or WIP fragments. p, Distribution of the logistic regression coefficient P-value ratios (OOP/WIP) from o. Ratio = 1 (black dotted line); geometric mean ratio = 0.007 (red dotted line, the indicated P-value from one-sided Mann–Whitney U-test). q, Volcano plot of regression coefficients β versus the P-values from o. Number of P-values ≤ 0.1 for non-zero coefficients β from out-of-peak (red) or within-peak (blue) analyses. r, Cumulative distribution of probabilities that within a αTSGpos versus αTSGneg mTEC, another αTSG within 50 kb (local) or a random distance is expressed (for 3,184 αTSGs). P-value (local αTSGpos versus αTSGneg) from one-sided Mann–Whitney–Wilcoxon test.
Epigenetic noise is linked to plasticity
To identify the molecular levers that give rise to mTEC plasticity, we used the 10X Genomics Chromium Multiome platform on mTECs from a four-week-old C57BL/6 mouse that were sorted by fluorescence-activated cell sorting (FACS). We obtained 9,928 high-quality cells after quality-control filtering and exclusion of further differentiated ‘mimetic’ mTECs that extinguished Aire expression5,6 to focus on AIRE-independent mechanisms that poise mTECs for somatic plasticity (Fig. 1a, Extended Data Fig. 1a–h and Supplementary Fig. 1). We delineated a developmental trajectory consisting of five clusters of cells that corresponded well with the known mTEC developmental program10 (Fig. 1b,c and Extended Data Fig. 1i–p). We next investigated the role of chromatin accessibility states in promoting mTEC plasticity and found that the expression of nearly all AIRE-dependent tissue-specific genes (αTSGs) was not confined to any particular chromatin state across the developmental axis (Fig. 1d–f and Extended Data Fig. 1q–t). Moreover, we rarely found differentially accessible peaks between mTECs expressing (αTSGpos) versus not expressing (αTSGneg) a particular αTSG (Fig. 1g,h), indicating that the repertoire of accessible elements does not confer the potential for ectopic transcription.
However, we often noticed differences in Tn5 inserts outside the single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) peaks at regions flanking αTSGs between αTSGpos and αTSGneg mTECs (Fig. 1i–k). The differential out-of-peak (OOP) signal (defined by the same union peak set applied to all cells) accumulated symmetrically about the transcriptional start site (TSS) across a characteristic length scale of around 100 kilobases (kb), eventually converging with the differential within-peak (WIP) signal to a common normalized baseline (Fig. 1l). The differential OOP signal was not observed at AIRE-independent loci induced during mTEC maturation, indicating that it is not a general feature of active chromatin (Fig. 1m and Extended Data Fig. 1u). Furthermore, we found that OOP scATAC-seq fragments consisted of longer nucleosomal lengths than WIP fragments (Fig. 1n), which is indicative of nucleosome-dense regions becoming more labile for Tn5 integration. The OOP ATAC-seq fragments are generally considered to be ‘noise’, and the fraction of reads within peaks is commonly used as a metric for ATAC-seq signal enrichment11,12. These results led us to hypothesize that the differential OOP signal is predictive of ectopic gene expression and indicates destabilization of chromatin barriers (Extended Data Fig. 1v).
To test this hypothesis, we performed a series of logistic regressions. We fit the probability (P) of expressing an αTSG to the normalized OOP fragments within the characteristic length scale of destabilized chromatin (L ≈ ±50 kb). We also included the number of scATAC-seq fragments (nFrags) in each cell as a regression covariate to control for the extent of sampling per cell:
As a comparative association, we fit the probability of expressing the αTSG to the normalized scATAC-seq WIP fragments:
We then profiled the magnitudes, directions and statistical significance of the fit regression coefficients for local OOP \(({\beta }_{{\rm{OOP}}}^{\pm L})\) and WIP \(({\beta }_{{\rm{WIP}}}^{\pm L})\) fragments. The regression coefficients \({\beta }_{{\rm{OOP}}}^{\pm L}\) for OOP fragments exhibited high levels of statistical significance compared with the coefficients \({\beta }_{{\rm{W}}{\rm{I}}{\rm{P}}}^{\pm L}\) for WIP fragments, with the regression P-values for \({\beta }_{{\rm{OOP}}}^{\pm L}\) being around 140-fold smaller than those for \({\beta }_{{\rm{WIP}}}^{\pm L}\) for the same αTSGs. These results indicate that variation in local OOP fragments was a much more reliable predictor of ectopic transcription than local WIP fragments (Fig. 1o,p). Furthermore, statistically significant regression coefficients \({\beta }_{{\rm{OOP}}}^{\pm L}\) for OOP fragments were almost always greater than zero, meaning that an increase in OOP fragments was associated with a higher probability of αTSG expression when controlling for the extent of sampling per cell (Fig. 1q). Moreover, the expression of a given αTSG by an mTEC (αTSGpos) substantially increased the likelihood of expressing a neighbouring αTSG within the same 100-kb region of destabilized chromatin compared with the likelihood in αTSGneg cells, at a significance level (P = 9.15 × 10−166) that was far greater than the difference in the likelihood of expressing a random subset of αTSGs (P = 4.34 × 10−4) (Fig. 1r). Together, these results indicate that enhanced fluctuations in background chromatin accessibility are strongly predictive of ectopic expression of the local tissue-specific genes.
Epigenetic noise is AIRE-independent
To identify when in mTEC development chromatin accessibility noise becomes amplified, we followed the proportion of scATAC-seq fragments within peaks genome-wide (the WIP fraction) and found a prominent decrease at the early mature stage that was maintained through the later stages (Fig. 2a,b and Extended Data Fig. 2a–c). This decrease was not dependent on the magnitude of the transcriptome detected, nor the number, size or significance of peaks called (Extended Data Fig. 2d–l). Furthermore, we did not observe similar decreases in WIP fraction in quiescent versus cycling cells from Multiome datasets of embryonic day 18 (E18) mouse brain, indicating that this decrease was not a general feature of postmitotic cells (Extended Data Fig. 2m–o).

a, Fraction of scATAC-seq fragments within scATAC-seq peaks (WIP) across the mTEC developmental axis defined in Fig. 1b. b, Violin and box (median, 25th and 75th percentile; whiskers show 1.5 × the interquartile range) plots depicting the distributions of the fraction of scATAC-seq WIP fragments across annotated clusters (n = 9,928: immature, 2,107; transitional, 1,790; early mature, 2,825; mid mature, 2,511; late mature, 695). P-values were calculated by one-sided Mann–Whitney U tests. c, Bulk ATAC-seq fragment size distributions from mature and immature mTECs from published datasets9. d, Mean number of expressed AIRE-dependent tissue-specific genes (αTSGs) (blue) and mean WIP fraction (purple) across mTEC development. e, Cluster annotations overlaid on a UMAP of merged scATAC-seq data5 from Aire+/+ and Aire−/− mTECs. f, WIP fraction overlaid on the scATAC-seq UMAP defined in e. g, Paired violin plots comparing the distributions of WIP fraction from the indicated genotypes across the annotated developmental clusters defined in e. P-values calculated by one-sided Mann–Whitney U tests. KO, knockout. h, Rank-sorted differences in motif prevalence within accessible genomes (chromVAR deviation scores) of mTECs between the indicated developmental stages for 884 known transcription factor (TF) motifs. i, Distributions of the prevalence of p53-target motifs in accessible genomes (chromVAR deviation scores) of mTECs within the indicated cluster. j, Transcription factor footprinting at p53-target motifs (highlighted region) within the indicated mTEC developmental cluster. k, Aggregate expression of p53-target genes across mTEC development (overlaid on the scATAC-seq UMAP from Fig. 1b). l, Scatter plot of differential expression (TPM, transcripts per million) of known p53 regulators for the indicated comparisons (n = 2 biological replicates). Highly significant differentially expressed genes (Benjamini–Hochberg FDR ≤ 1 × 10−9, fold-change ≥ 2 or ≤ 0.5) indicated as repressors (brown text) or promoters (green text) of p53 activity. m,n, Expression of the p53 regulators Cop1 (m) and Sirt1 (n) across mTEC development (scATAC-seq UMAP from Fig. 1b).
We also observed a reciprocal increase in the prevalence of nucleosomal fragments in mature mTECs compared with immature progenitors or peripheral T cells from previously published bulk ATAC-seq studies9,13 (Fig. 2c and Extended Data Fig. 2p–r). The progressive loss in WIP fraction and gain in OOP fragments mirrored the progressive increase in the number of αTSGs expressed per maturing mTEC (Fig. 2d). We also found these features to be conserved in human mTECs (Supplementary Fig. 2a–i and Supplementary Notes), further linking chromatin destabilization with ectopic transcription during mTEC maturation.
To determine whether this association is dependent on transcription, we analysed published scATAC-seq data5 from Aire−/− mTECs and found no substantial differences in WIP fraction between Aire+/+ and Aire−/− mTECs (Fig. 2e–g and Extended Data Fig. 2s–v). Rather, we observed a small increase in median nucleosomal fragments in the accessible genomes of mature Aire−/− versus Aire+/+ mTECs (Extended Data Fig. 2w), which may be associated with the previously reported repressive influence of AIRE on chromatin accessibility9,14. These data indicate that increased chromatin accessibility noise was not driven by AIRE-dependent expression of tissue-specific genes, nor by the AIRE-dependent facets of mTEC maturation.
mTECs repress p53 during maturation
To identify potential drivers of chromatin accessibility noise, we conducted transcription factor motif enrichment and found that the greatest differential feature was the depletion of p53-binding motifs in the accessible genome of mature versus immature mTECs (Fig. 2h,i and Extended Data Fig. 3a,b). We also observed losses in footprinting at sites containing p53-binding motifs and expression of validated p53 target genes15 in mature versus immature mTECs (Fig. 2j,k and Extended Data Fig. 3c). These data, in conjunction with the well-characterized roles of p53 in enforcing differentiation along committed lineage trajectories16 and inhibiting somatic reprogramming17, led us to hypothesize that mTECs repress p53 activity to amplify chromatin accessibility noise for cellular plasticity.
To investigate how p53 is repressed in mTECs, we assessed the differential expression of known p53 regulators and found highly significant induction of Mdm2 (the primary regulator of p53 that promotes its proteosomal degradation18) during mTEC maturation at both the transcript and protein levels (Fig. 2l and Extended Data Fig. 3d–g). Other negative regulators of p53 were also induced in mature versus immature mTECs, such as COP1, another E3 ubiquitin ligase of p53 (ref. 18), and SIRT1, which deacetylates p53 to inhibit its transactivation potential19 (Fig. 2l–n).
We also observed repression of p53 regulators that promote p53 activity, including genes that encode SET7/9 and PCAF (which respectively methylate20 and acetylate21 p53 to promote its transcriptional activity), c-ABL and CHK1 (which respectively phosphorylate p53 (ref. 22) and MDM2 (ref. 23) to inhibit p53 degradation), RASSF1A (which promotes MDM2 ubiquitination24) and ATF3 and p63 (which facilitate cooperative binding of p53 target genes25,26) in mature versus immature mTECs (Fig. 2l and Extended Data Fig. 3h–n). By contrast, we did not detect significant differences in the expression of Trp53 (which encodes p53), except for a brief induction in transit-amplifying mTECs that was extinguished before the AIRE+ state (Fig. 2l and Extended Data Fig. 3o). Importantly, we observed the systematic repression of p53 activity in human mTECs (Supplementary Fig. 2j–u and Supplementary Notes) and in mature AIRE-deficient mTECs (Extended Data Fig. 3p,q), which is consistent with the AIRE-independent nature of amplified chromatin accessibility noise.
Augmenting p53 activity stabilizes chromatin
To test the role of p53 in regulating chromatin accessibility noise, we generated Trp53LSL-QM/wt;Foxn1cre (p53-cHyper) mice that conditionally express in mTECs (from the endogenous Trp53 locus) a transactivation-dead p53 mutant that impairs MDM2 binding, allowing it to stabilize heteromeric wild type–mutant complexes to bring about modest increases in p53 activity27. Compared with sex-matched Trp53wt/wt;Foxn1cre (p53 wild-type) littermate controls, thymi from p53-cHyper mice had comparable total cellularity, frequency of AIRE+ mTECs, mean expression of AIRE, frequencies of FOXP3+ regulatory T cells and other thymocyte compartments, indicating that p53 hyperactivity did not disrupt mTEC or thymocyte differentiation in p53-cHyper mice (Extended Data Fig. 4a,b).
To investigate the effect of p53 hyperactivity, we FACS-sorted mTECs from sex-matched p53-cHyper and p53 wild-type littermates and jointly profiled the transcriptome and chromatin accessibility landscapes (Fig. 3a,b and Extended Data Fig. 4c). Differential enrichment of p53-binding motifs and enhanced footprinting at these sites in mature p53-cHyper versus p53 wild-type mTECs validated the perturbation (Extended Data Fig. 4d,e). We next quantified the proportion of scATAC-seq fragments within peaks (the WIP fraction) across the developmental trajectory and found highly significant increases in p53-cHyper compared with p53 wild-type mTECs (Fig. 3c and Extended Data Fig. 4f). Notably, the increase in WIP fraction became progressively larger across the developmental stages, such that the median WIP fraction of mature p53-cHyper mTECs was greater than that of immature p53 wild-type controls (Fig. 3c). Furthermore, we observed the reciprocal loss in prevalence of nucleosomal scATAC-seq fragments in mature p53-cHyper versus p53 wild-type mTECs, indicating greater stability of nucleosome-dense regions in p53-cHyper mTECs (Extended Data Fig. 4g).

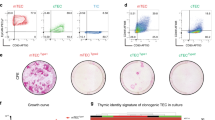
a, UMAP visualization of merged scATAC-seq profiles of mTECs from the indicated genotypes (colours) generated from Multiome datasets. b, Cluster annotations overlaid on the UMAP from a. c, Paired violin plots comparing distributions of the fraction of scATAC-seq fragments within scATAC-seq peaks (WIP) from the indicated genotypes across annotated clusters of mTEC maturation. P-values were calculated by one-sided Mann–Whitney U tests. d, Differences in the number (left) or sum of mRNA (right) of expressed αTSGs between neighbouring p53-cHyper and p53-WT mTECs overlaid on the UMAP from a. e, Scatter plot comparing the differential expression of genes by bulk RNA-seq resulting from mTEC maturation (horizontal axis) or p53 hyperactivity in mature mTECs (vertical axis) (n = 3 biological replicates). Fold-change densities are shown on the top and right margins. Statistically significant (Benjamini–Hochberg FDR ≤ 0.05) differential expression between p53-cHyper and p53-WT mTECs is highlighted in blue for all genes and red for αTSGs. f, UMAP of merged scRNA-seq profiles of p53-WT and p53-cHyper mTECs with cluster annotations overlaid. g, Differential density of genotypes (p53-cHyper versus p53-WT) across the mimetic mTEC clusters defined in f. h, Fraction of the indicated mimetic mTEC subtypes represented by each genotype. i, Comparison of the numbers of the indicated mimetic mTEC compartments between p53-WT and p53-cHyper sex-matched littermates (n = 6). P-values for two-sided paired ratio tests are shown.
To determine whether the suppressed chromatin accessibility noise in p53-cHyper mTECs affected their potential for ectopic transcription, we quantified the expression levels of αTSGs and found both the diversity and magnitude of ectopic gene expression to be compromised in mature p53-cHyper versus p53 wild-type mTECs (Fig. 3d). To assess the full scope of this effect, we conducted bulk RNA sequencing (RNA-seq) on FACS-sorted mature and immature mTECs from sex-matched p53-cHyper and p53 wild-type littermates. We found that p53 hyperactivity in mature p53-cHyper mTECs significantly affected the expression of 6,279 genes, 77% of which were repressed, which is consistent with the heightened stability of nucleosomal barriers (Fig. 3e). We also found that 1,653 tissue-specific genes, which were normally induced during mTEC maturation, were repressed in mature p53-cHyper versus p53 wild-type mTECs, with around 70% of these being AIRE-dependent, indicating a strong concordance (P < 2.3 × 10−308) between p53 repression and AIRE-mediated ectopic expression of tissue-specific genes (Fig. 3e). At the same statistical thresholds, this effect resulting from p53 hyperactivity was more than 3.4-fold more deleterious than the effect of dysregulated mTEC maturation resulting from p53 deficiency in Trp53fl/fl;Foxn1cre (p53-cKO) mice on AIRE-dependent ectopic gene expression (Supplementary Fig. 3 and Supplementary Notes).
Augmenting p53 activity limits plasticity
To determine whether p53 hyperactivity in mTECs affected the differentiation of downstream ‘mimetic’ phenotypes5,6, we re-integrated the mimetic populations into the p53-cHyper/wild-type Multiome analysis (Fig. 3f and Extended Data Fig. 4h–m). We detected a relative paucity of p53-cHyper mTECs in the microfold, enterocyte, tuft and secretory mimetic compartments (3.1-fold, 2.8-fold, 1.4-fold and 1.3-fold fewer than p53 wild-type mTECs, respectively), in contrast to the almost 1:1 ratios observed in the keratinocyte and ciliated compartments (Fig. 3g,h). To confirm and extend these findings to other mimetic compartments, we used previously established mimetic flow cytometry panels6 (Extended Data Fig. 4n–s) and found a significant decrease in numbers of keratinocyte (about 28% fewer), ciliated (about 44% fewer) and myoid (about 52% fewer) mimetic mTECs in p53-cHyper versus p53 wild-type thymi, along with confirmed decreases in tuft (about 29% fewer) and overall mimetic mTEC numbers (30% fewer) (Fig. 3i). These data indicate that suppression of chromatin accessibility noise by p53 hyperactivity constrained the potential of mTECs to deviate from the established state, preventing the activation of genes restricted to other tissues and compromising the differentiation of mimetic subtypes.
p53 stabilizes chromatin indirectly
To identify the mode by which p53 suppresses chromatin accessibility noise, we conducted p53-targeted cleavage under targets & release using nuclease28 (CUT&RUN) in sorted immature and mature mTECs. We detected little p53 occupancy near αTSGs with heightened chromatin accessibility noise compared with the focal signal within p53 CUT&RUN peaks and p53 target genes, consistent with the relative dearth of p53 target motifs within 50 kb of αTSGs (Fig. 4a,b and Extended Data Fig. 5a–c). Moreover, we observed a prominent loss of p53 occupancy at p53-binding sites (p53 CUT&RUN peaks) in mature versus immature mTECs (Fig. 4c,d), consistent with the depletion of p53-binding motifs within scATAC-seq peaks during mTEC maturation. These results indicate that p53 does not physically localize to genomic regions with elevated chromatin accessibility noise and instead imposes its suppressive influence indirectly.

a,b, Aggregate histograms of p53 occupancy at αTSGs (blue), p53 CUT&RUN (C&R) peaks (green) (a) or p53-induced target genes (red) (b) in total mTECs from wild-type mice. c, Heatmaps of p53 occupancy in immature (left) or mature (right) mTECs at p53-binding sites (CUT&RUN peaks). d, Aggregate histograms of p53 occupancy at p53 CUT&RUN peaks in immature (purple) or mature (blue) mTECs. e, MA plot of the differential expression of known p53 target genes between mature p53-cHyper and p53-WT mTECs by bulk RNA-seq (n = 3 biological replicates). Point colours show Benjamini–Hochberg FDR levels. Red text is used for genes known to promote cell death. Inset bar plots (right) indicate the most enriched Gene Ontology (GO) terms for p53-induced (top) or p53-repressed (bottom) genes in p53-cHyper mTECs. Neg reg DDR, negative regulators of DNA damage response. f,g, Differences in the expression of the genes Bax (f) and Perp (g) for neighbouring p53-cHyper and p53-WT mTECs overlaid on the UMAP from Fig. 3b. h, MA plot of the differential expression of genes encoding proteins of the intrinsic apoptosis pathway between mature and immature p53-cHyper mTECs by bulk RNA-seq (n = 3 biological replicates). Point colours show the Benjamini–Hochberg FDR levels; red text indicates genes that encode pro-apoptotic proteins and green text indicates pro-survival proteins. i,j, Differences in the expression of the genes Bcl2l11 (i) and Mcl1 (j) between neighbouring p53-cHyper and p53-WT mTECs overlaid on the UMAP from Fig. 3b. k–n, Aggregate histograms of OOP ChIP–seq dyads over the indicated loci in mature mTECs from the indicated genotypes; γH2AX (k), TOP2α (l), TOP1 (m) and POL2 (n). o,p, Comparison of transcription factor motif enrichment within scATAC-seq fragments from mature mTECs mapping to OOP regions within 100 kb of αTSGs (o) or silent genes (p) versus the adenine/thymine content of each motif. The trend line (red) with two-sided 95% confidence intervals (t-distribution) for the linear regression fit (grey) is indicated.
To understand how p53 stabilizes chromatin indirectly, we conducted differential expression analysis of validated p53 target genes15 between p53-cHyper and p53 wild-type mature mTECs and found p53 hyperactivity-induced genes encoding effectors of cell death (Fig. 4e–g and Extended Data Fig. 5d,e). The connection between p53 activity and BAX (an effector of the intrinsic apoptosis pathway29) was particularly notable because the conditional deletion of Bax in mTECs (on a Bak−/− background) caused a selective increase in the number of immature mTECs30, the only developmental subset with substantial p53 activity in wild-type mice (Fig. 2h–k and Extended Data Fig. 3a–c). We also found significant upregulation of other genes encoding pro-apoptotic BH3-only proteins31 (such as NOXA, BIM, BIK and BLK) and Hrk, which blocks the pro-survival function of Bcl2l1 (encoding BCL-XL)32, as well as downregulation of pro-survival Bcl2 in mature versus immature p53-cHyper mTECs (Fig. 4h).
Skewed levels of pro-apoptotic versus pro-survival genes was also observed in p53 wild-type mature versus immature mTECs (Extended Data Fig. 5f), indicating that mature mTECs are inherently predisposed to BAX/BAK-driven apoptosis31. Thus, the differential upregulation of Bax and other genes encoding the pro-apoptotic proteins BIM, BAD and BID by p53 hyperactivity, and the downregulation of the essential pro-survival protein MCL-1 (ref. 33), differentially sensitized p53-cHyper versus p53 wild-type mTECs to apoptosis (Fig. 4e,f,h–j and Extended Data Fig. 5g,h). Together with the data indicating normal thymic cellularity and frequencies of mTEC and thymocyte compartments in p53-cHyper mTECs, these results indicate that p53 hyperactivity imposes a selective advantage for more-stable p53-cHyper mTECs with low chromatin accessibility noise by triggering apoptosis of those exhibiting cellular plasticity, which is potentially similar to the modes by which p53 eliminates premalignant cancers34.
Epigenetic noise is linked to DNA damage
Because DNA damage is the primary trigger for p53 activation in many contexts34, we reasoned that it may serve as a connection between chromatin accessibility noise and p53-mediated apoptosis in p53-cHyper mTECs. To investigate this possibility, we profiled the deposition of γH2AX, which marks sites of DNA double-strand breaks35, is the substrate for ataxia telangiectasia mutated (ATM) kinase (which also phosphorylates p53 for activation35), and is upregulated in mature versus immature p53-cHyper mTECs, along with a cohort of other genes encoding DNA damage-response proteins (Extended Data Fig. 5i). Using published chromatin immunoprecipitation followed by sequencing (ChIP–seq) datasets from mTECs36, we found elevated levels of γH2AX at OOP regions flanking αTSGs compared with those at unexpressed silent loci with low levels of chromatin accessibility noise (Fig. 4k and Extended Data Fig. 5j). The elevated deposition of γH2AX was largely maintained in Aire−/− mTECs, indicating that AIRE-induced transcription and AIRE-mediated recruitment of topoisomerases36,37 did not contribute significantly to the DNA damage at destabilized regions near αTSGs (Fig. 4k). However, we did observe AIRE-independent differential localization of both TOP2α and TOP1 near αTSGs compared with silent loci, indicating the recruitment of enzymatic activity that causes DNA breaks (Fig. 4l,m).
To understand how topoisomerases are recruited to genomic regions with increased chromatin accessibility noise, we focused on their role in generating long-lived DNA breaks at transcription units to resolve the torsional stress caused by RNA polymerase II (Pol II)38. We explored the possibility that stochastic loading of Pol II may be enhanced at destabilized chromatin flanking αTSGs, because nucleosome density has a role in preventing spurious transcriptional initiation39. Towards this goal, we profiled global Pol II occupancy in mTECs using published ChIP–seq datasets36 and found elevated levels of OOP Pol II recruitment at destabilized regions near αTSGs compared with those near silent loci (Fig. 4n). The elevated Pol II occupancy was not a consequence of AIRE-induced transcription because the levels were largely maintained in Aire−/− mTECs (Fig. 4n). Furthermore, annotated distal enhancers40 in destabilized regions near αTSGs also exhibited elevated loading of Pol II, γH2AX deposition and topoisomerase recruitment compared with enhancers near silent loci (Supplementary Fig. 4a–p and Supplementary Notes). Together, these results indicate a concordance between amplified chromatin accessibility noise, spurious transcriptional initiation, recruitment of topoisomerases, increased DNA damage and p53-mediated triggering of apoptosis in p53-cHyper mTECs versus cellular plasticity in wild-type mTECs.
Epigenetic noise is genome-encoded
To identify the determinant that initially causes chromatin accessibility noise in mTECs, we conducted de novo motif enrichment on the destabilized chromatin fragments and found the enrichment of a near-homopolymeric 10-mer adenine tract to be different from the other motifs (Fig. 4o). This was notable because poly(dA:dT) tracts are known to destabilize nucleosomes at magnitudes proportional to the length and perfection of the tracts41; and an 18-mer poly-A tract was the most-enriched motif in genomic regions associated with allelic imbalances in chromatin accessibility and gene expression of nearby αTSGs in mTECs from NOD × B6 F1 hybrid mice37. We observed a highly significant positive correlation between motif enrichment in OOP fragments flanking αTSGs and the motif adenine/thymine (AT) content (R = 0.42, P = 4.9 × 10−24), with the top three enriched motifs consisting of an average of 93.3% AT (Fig. 4o). By contrast, the motif enrichment in fragments within peaks overlapping the same genomic regions was negatively correlated with motif AT content (R = −0.14, P = 1.9 × 10−3) with the top three motifs containing an average of 16.1% AT (Extended Data Fig. 5k). Similarly, neither OOP fragments from silent loci (R = –0.14, P = 1.8 × 10−3; top three enriched motifs, around 16.6% AT) nor highly expressed housekeeping loci (R = 0.05, P = 0.10; top three enriched motifs, around 25.0% AT) showed a substantial correlation between motif enrichment and motif AT content (Fig. 4p and Extended Data Fig. 5l). This association between chromatin accessibility noise and enrichment of low-complexity AT-rich motifs suggests that the intrinsic resistance of AT-rich sequences to adopt nucleosome-favourable conformations41,42 may be a determinant of chromatin accessibility noise in vivo.
The selective enrichment of low-complexity AT-rich motifs at OOP fragments near αTSGs could be a product of transcription factor activity and/or an inherent prevalence of these motifs in the genomic sequence. To investigate the first possibility, we assessed the expression levels of the transcription factors that target these motifs in maturing versus immature mTECs. We found their expression to be around 1–3 orders of magnitude lower than those encoding known regulators of mTEC differentiation (such as Foxn1 or Relb) and even lower than that for insulin (Ins2) transcripts in mature AIRE+ mTECs (Extended Data Fig. 5m), indicating that they were unlikely to contribute substantially to chromatin accessibility noise.
To investigate the inherent prevalence of poly-AT tracts in the genomic sequence near αTSGs, we generated a set of 500-base pair contiguous tiles spanning 100-kb regions flanking αTSGs minus any tile that overlapped with scATAC-seq peaks. We observed no positive correlation between motif enrichment and motif AT content; however, of the top 5% of the enriched motifs (n = 26), 4 had more than 75% AT content, 3 of which consisted of imperfect 10–12-mer AT tracts (Extended Data Fig. 5n). Another motif that met the top 10% threshold was 91.6% AT with an imperfect 10-mer AT tract (Extended Data Fig. 5n). By contrast, none of the significantly enriched motifs at the silent loci had an AT content of 75% or more, or long imperfect AT tracts (Extended Data Fig. 5o). These results reinforce a concordance between the genomic prevalence of nucleosome-disfavourable poly-AT tracts and amplified chromatin accessibility noise at regions flanking αTSGs, including at nearby tissue-specific enhancers (Supplementary Fig. 4q,r and Supplementary Notes).
Augmenting p53 activity in mTECs causes autoimmunity
To determine whether the suppression of chromatin accessibility noise and mTEC plasticity by p53 hyperactivity affected T cell repertoire selection, we assayed for evidence of the escape of self-reactive T cells in the organs of p53-cHyper mice. We found significant increases in activated T cells that became progressively larger as a function of age, as well as reciprocal decreases in naive T cells compared with their p53 wild-type sex-matched littermates (Fig. 5a,b and Extended Data Fig. 6a–c). We also found substantial lymphocytic infiltration in multiple organs of 6–12-month-old p53-cHyper mice, most frequently in the salivary gland, lung, kidney, lacrimal gland, pancreas, liver and prostate/ovaries, that was not present or less severe in p53 wild-type mice (Fig. 5c–e and Extended Data Fig. 6d), indicating autoreactive tissue damage by the activated effector T cells. Together, these results indicate that enhanced p53 activity in mTECs causes a failure in central tolerance induction, leading to systemic hyperactivity of peripheral T cells and autoimmune manifestations in multiple organs.

a, Representative flow-cytometry plots of the effector memory T (TEM) cell and naive T (TN) cell compartments of splenic CD4+ T cells from 9-month-old p53-WT (left) and p53-cHyper (right) sex-matched littermates. b, Frequency of CD4+ TEM cells in the spleen from p53-WT and p53-cHyper mice as a function of age and lines of best fit for each genotype. c, Histological analysis of the indicated tissues for infiltrating lymphocytes through haematoxylin and eosin (H&E) staining (filled triangles indicate a histopathology score ≥ 2) from 7–12-month-old p53-WT and p53-cHyper mice. Each heptagon represents an individual mouse. d, Comparison of the mean histopathology scores (based on the number and size of lymphocytic infiltrates) from the organs assessed in c and in Extended Data Fig. 6d for sex-matched littermates of the indicated genotypes. The P-value was calculated by a one-sided t-test. e, Representative H&E staining of the indicated organs from p53-WT and p53-cHyper mice (n = 8). The arrows indicate lymphocytic infiltration. Scale bars, 100 μm.
Epigenetic noise is regulated by p53 in cancer
The importance of p53 in tumour suppression34 and the role of somatic plasticity in tumour progression2 prompted us to explore whether the link between chromatin accessibility noise, p53 repression and cellular plasticity in mTECs can be extended to tumour contexts. Towards this goal, we investigated the well-characterized lung adenocarcinoma (LUAD) model43 by using published Multiome datasets of lung epithelial cells from Trp53wt/wt (p53 wild-type), Trp53−/− (p53-knockout) and Trp53LSL-F53Q,F54S (p53-Hyper) backgrounds44 10 weeks after oncogenic KRAS-G12D activation. We based our analysis on the established stages of LUAD development in p53 wild-type and knockout backgrounds43, and identified four main transcriptional states of LUAD progression43: the native alveolar type-2 (AT2) state; a mixed AT1/AT2 state mimicking alveolar progenitors; an embryonic liver-like state representing the loss of alveolar identity; and an epithelial-to-mesenchymal transition (EMT) state representing the loss of epithelial identity (Extended Data Fig. 7a–f).
We identified cells in four clusters (5, 8, 6 and 7) with both a low-WIP fraction and a high prevalence of nucleosomal scATAC-seq fragments indicating destabilized chromatin (Extended Data Fig. 7b,e–j). A common feature across all these cells was the loss of AT2-specific gene expression, including the loss in expression and activity of the lung lineage-defining transcription factor NKX2.1 (refs. 43,45) (Extended Data Fig. 7c,k,l). Furthermore, cells from cluster 6 exhibited high expression and activity levels of RUNX2, a driver of LUAD metastatic transition46 (Extended Data Fig. 7m,n). Moreover, ‘transitional’ cells coexpressing Nkx2-1 and Runx2 (cluster 3), or exhibiting transcription factor activity for both NKX2.1 and RUNX2 (cluster 4), had intermediate WIP fractions with a low prevalence of nucleosomal fragments (Extended Data Fig. 7g–n). Together, these results indicate a concordance between the magnitude of chromatin destabilization and the degree to which cells deviated from the native lung epithelial lineage.
We next re-integrated the p53-Hyper cells on the developmental axis we established with p53 wild-type and p53-knockout cells (Extended Data Fig. 8a–j) and quantified the differential WIP fraction and prevalence of nucleosomal scATAC-seq fragments as a function of the Trp53 genotype for each cluster (Extended Data Fig. 8l–o). For all clusters except the embryonic liver-like cluster 5 (perhaps owing to the paucity of p53-Hyper cells in this cluster), we observed significant increases in WIP fraction in p53-Hyper versus p53 wild-type cells, indicating that p53 hyperactivity suppressed chromatin accessibility noise (Extended Data Fig. 8n). Reciprocally, we observed significant decreases in the WIP fraction in p53-knockout cells compared with p53 wild-type cells in all clusters except the knockout-enriched Runx2posNkx2-1pos cluster 3, indicating less stable nucleosomal barriers in p53-deficient cells (Extended Data Fig. 8n). By and large, we also observed consistent associations between the Trp53 genotype and the prevalence of nucleosomal scATAC-seq fragments (Extended Data Fig. 8o). Together, these results indicate that the distinct roles of p53 in suppressing chromatin accessibility noise and promoting lineage fidelity work in concert to inhibit phenotypic plasticity during LUAD tumorigenesis.
Discussion
The mechanistic basis of somatic plasticity has remained enigmatic despite its broad relevance to organismal adaptation, tissue repair and tumorigenesis1,2. Our findings indicate a central role for amplified fluctuations in background chromatin accessibility that increase the entropy of the epigenetic landscape, which is similar to raising a ‘statistical temperature’ to facilitate a more permissive state. In the developmental framework of cellular differentiation, these results indicate that somatic plasticity is not initially driven by the induction of alternative cell fates, but rather by the stochastic erosion of chromatin barriers.
Our findings show that amplified accessibility noise at chromatin barriers contributes to a highly mixed transcriptional state that lacks clear regulatory logic, as observed in the coexpressed genes in individual AIRE+ mTECs47,48,49 or high-plasticity states of LUAD progression43 that are not related by tissue-specific function, lineage-specific regulation or developmental origin. This high-plasticity state then promotes heterogeneity in downstream phenotypes, as observed in the differentiation of mimetic mTECs3,5,6 or the emergence of primordial gut programs and the EMT state in advanced LUAD tumours43,44 (Supplementary Discussion).
Notably, the destabilization of chromatin barriers and the repression of p53 in mTECs did not require AIRE, in concordance with previously published studies demonstrating that AIRE acts late in the transcription cycle8,9,14. Considering the affinity of AIRE to positive elongation factors of transcription50,51,52, it may function to facilitate Pol II pause release at sites of destabilized chromatin, increasing the probability and frequency of transcriptional bursts. AIRE could target these sites through multiple modes, including: interactions with topoisomerases and DNA repair machinery36,37,53,54; the formation of Z-DNA, resulting from the negative supercoils generated by nascent transcription37,38,55; AIRE’s histone-binding module specific for unmodified amino-terminal histone H356,57,58,59; and the interaction of AIRE with the ATF7ip–MBD1 complex, which targets repressive chromatin enriched in H3K9me3 and DNA methylation60.
Our findings identify molecular levers that regulate fluctuations in nucleosome dynamics at chromatin barriers that influence a cell’s potential for alternative fates. Understanding the molecular and biophysical mechanisms that govern noise in the chromatin landscape may illuminate principles of gene regulation and cellular differentiation, and enable therapeutic innovation for diseases linked to chromatin instability.
Methods
Mice
The mice used in this study were housed in pathogen-free facilities at the University of Chicago and Stanford University. All mice were housed in positively pressurized, individually ventilated cage racks and changed in biological safety cabinets. Cage supplies were sanitized using hot water (82 °C). Bedding and shredded-paper enrichment were autoclaved and cages were provided with irradiated food. Reverse Osmosis water was provided by an automated watering system directly to each cage. Rodent housing rooms were maintained at a 12 h:12 h light:dark cycle. Temperature and humidity were within the Guide for the Care and Use of Laboratory Animals recommended ranges: 20–26 °C and 30–70% humidity. All experiments and animal-use procedures were conducted in compliance with the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Chicago. B6.129-Trp53LSL-L25Q,W26S,F53Q,F54S heterozygous mice27,61 were provided by Laura Attardi (Stanford University) and were bred with B6-Foxn1cre homozygous mice62 purchased from Jackson Laboratories to generate Trp53LSL-L25Q,W26S,F53Q,F54S/wt;Foxn1cre/wt and Trp53wt/wt;Foxn1cre/wt littermates. Trp53fl/fl mice were purchased from Jackson Laboratories and bred with B6-Foxn1cre mice to generate Trp53fl/fl;Foxn1cre/wt mice. C57BL/6J mice were purchased from Jackson Laboratories. mTECs and thymocytes were collected from mice 4–5 weeks old. Sex-matched littermates were used for all comparisons of genetic perturbations.
Isolation, sorting and analysis of mouse mTECs
Thymic epithelial cells were isolated as previously described63 with minor modifications. In brief, thymi from 4–6-week-old mice were removed and connective tissue was removed. Stromal tissue was perforated using scissors and incubated with rotation in DMEM-F12 (Gibco) at room temperature for 10 min to liberate the thymocytes. The remaining stromal tissue was enzymatically digested (0.5 mg ml−1 Collagenase D (MilliporeSigma), 0.2 mg ml−1 DNaseI (MilliporeSigma), 0.5 mg ml−1 Papain (Worthington Biochemical)). Cells were stained with anti-EpCAM antibodies conjugated to APC-Cy7 (clone G8.8, BioLegend, 3 µl per 100 million cells) and EpCAM+ cells were enriched by positive selection using magnetic anti-Cy7 beads (Miltenyi, 10 µl per 100 million cells). The enriched fraction was stained with the appropriate panel of fluorochrome-conjugated antibodies to CD45 (clone 30-F11, Invitrogen, 1:100), Ly-51 (clone 6C3, BioLegend, 1:100), MHC-II I-A/I-E (clone M5/114.15.2, Invitrogen, 1:100), CD104 (clone 346-11A, BD Biosciences, 1:200), GP2 (clone 2F11-C3, MBL, 1:10), CD177 (clone 1171 A, R&D, 1:25), Ly-6D (clone 49-H4, Invitrogen, 1:200), Sca-1 (clone D7, BioLegend, 1:200), AIRE (clone 5H12, Invitrogen, 1:500), Ki-67 (clone SolA15, Invitrogen, 1:100), SynCAM (clone 3E1, MBL, 1:100), CD171/L1CAM (clone 555, Miltenyi, 1:25) along with fluorescein-labelled UEA-I (Vector Labs, 1:100), Zombie Aqua (BioLegend, 1:500) and DAPI (Invitrogen, 1:20). Intracellular staining for AIRE and Ki-67 was subsequently done using the eBioscience FoxP3 transcription factor staining kit (Invitrogen) according to the manufacturer’s instructions. Intracellular staining for MDM2 (clone EPR22256-98, Abcam, 1:25) was also done using the eBioscience FoxP3 transcription factor staining kit (Invitrogen) according to the manufacturer’s instructions with the addition of a 1-h incubation in blocking buffer (eBioscience permeabilization buffer with 5% normal donkey serum) before a secondary stain (BV412 donkey anti-rabbit, Jackson Immuno, 1:50). Cells were sorted using FACS Symphony S6, FACSAria Fusion or FACSAria II equipped with a 100-μm nozzle (BD Biosciences). Flow-cytometry data for thymic mimetic cells were acquired using a Cytek Aurora. All other flow-cytometry data were acquired using a BD LSRII or Fortessa. All flow-cytometry data were analysed using FlowJo (v.10).
Human thymic tissue acquisition and processing
Thymus fragments were obtained from a 12-week-old human patient with no known genetic abnormalities undergoing standard-of-care cardiac surgery. The patient was de-identified on receipt with written informed consent for the release of genomic sequence data in accordance with IRB protocol 20–1392 approved by the Biological Sciences Division and University of Chicago Medical Center Institutional Review Boards at the University of Chicago and protocol 2020-203 approved by the Advocate Aurora Health Research Subject Protection Program and Advocate Aurora Health Care Institutional Review Board. Connective tissue was removed and the remaining tissue was minced, then incubated with rotation in DMEM-F12 (Gibco) at 4 °C for 20 min to liberate the thymocytes. Stromal tissue was enzymatically digested using 0.5 mg ml−1 Collagenase D (MilliporeSigma) and 0.2 mg ml−1 DNase I (MilliporeSigma) at 37 °C for 20 min. The remaining fragments were incubated with rotation in 0.5 mg ml−1 Papain (Worthington), 0.25 mg ml−1 Collagenase D and 0.1 mg ml−1 DNase I at 37 °C for 20 min. Cells were stained with anti-EpCAM antibodies conjugated to APC-Cy7 (clone 9C4, BioLegend, 1:100) and EpCAM+ cells were enriched by positive selection with magnetic anti-Cy7 beads (Miltenyi). The enriched fraction was stained with DAPI (Invitrogen, 1:20), CD45 (clone 2D1, BioLegend, 1:100), LY51/CD249 (clone 2D3/APA, BD Biosciences, 1:00) and HLA-DRA (clone L243, BioLegend, 1:100) and sorted on a Symphony S6 (BD Biosciences).
Flow cytometry of thymocytes and splenocytes
Thymi from 4–6-week-old mice were removed and small cortical incisions were made before mechanical agitation with wide-bore glass pipettes in DMEM/F-12 (Gibco) to liberate the thymocytes. Spleens from mice aged 4 weeks to 12 months old were isolated in RPMI (Gibco) supplemented with 10% FCS. Cells were liberated by mincing with a syringe plunger and filtered through a 40-μm strainer. Following red blood cell lysis (BD PharmLyse), cells were stained with fluorochrome-conjugated antibodies specific for mouse CD4 (GK1.5, 1:100), CD8α (53-6.7, 1:100), CD25 (PC61, 1:100), CD44 (IM7, 1:100), CD69 (H1.2F3, 1:100), CD62L (MEL-14, 1:100), TCRβ (H57-597, 1:100) and DAPI (Invitrogen, 1:20). Intracellular staining for FoxP3 (clone FJK-16s, eBioscience, 1:100) was done using an eBioscience FoxP3 transcription factor staining kit (Invitrogen) according to the manufacturer’s instructions. Flow-cytometry data were acquired using a BD LSRII or Fortessa and analysed using FlowJo (v.10).
Bulk RNA-seq sample preparation
We FACS-sorted 75,000 primary mTECs directly into RULT lysis buffer (Qiagen RNEasy UCP Micro Kit) and total RNA was extracted following the manufacturer’s instructions. The mRNA was enriched and RNA-seq libraries were constructed using an Illumina TruSeq Stranded mRNA kit. Paired-end, dual-index sequencing was performed on an Illumina NovaSeq 6000 platform.
Bulk RNA-seq data processing
RNA-seq reads were mapped to the mm10 mouse genome assembly using TopHat (v.2.1.1) with the setting –microexon-search. Unmapped, unpaired and low-quality reads (MAPQ ≤ 5) were removed using samtools (v.1.9) view with settings -q 5 -f 2. Paired reads were counted for each gene using featureCounts from Subread (v.2.0.1). TPM values were calculated for each gene to quantify the relative abundance of transcripts for clustering analysis. The trimmed mean of M values was calculated for each gene for differential comparisons across samples using edgeR (v.4.0.2) (calcNormFactors()). Common dispersions were estimated using estimateCommonDisp() and Benjamini–Hochberg FDRs were calculated for pairwise comparisons using the exactTest(). Genes with FDR ≤ 0.05 were regarded as significant.
Definition of tissue-specific and AIRE-dependent genes
Previously published transcriptional data64 from Aire wild-type and Aire-knockout mTEChi were analysed according to the bulk RNA-seq pipeline outlined above. Genes that exhibited at least 1.5-fold induction in Aire wild type relative to Aire knockout and had Benjamini–Hochberg FDR ≤ 0.05 were regarded as Aire-induced. TSGs were classified as previously64, and αTSGs were taken to be the intersection of these two gene sets. For human TSGs, GTEx65 expression counts (median TPM), Shannon entropy \(\left(S=-\sum p{\log }_{2}p\right)\) across tissues was calculated for each gene. Genes with an entropy S ≤ 3 were included for downstream analyses.
Multiome sample preparation and sequencing
For all Multiome experiments, we used an ATAC + GEX single-cell kit and protocol (10X Genomics 1000236 with protocol CG000338 RevE) with minor modifications to sample preparation. In brief, 40,000 mTECs were FACS-sorted into 1× PBS supplemented with 2% BSA and centrifuged at 300g for 5 min. Cells were gently washed in 50 μl lysis buffer (10 mM Tris, 10 mM NaCl, 3 mM MgCl2 in nuclease-free water) and centrifuged at 300g for 5 min. Cells were resuspended in 50 μl permeabilization buffer (10 mM Tris, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween20, 0.01% digitonin and RNase inhibitor (Invitrogen) in nuclease-free water) and incubated for 5 min on ice. Nuclei were gently washed with wash buffer (10 mM Tris, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween20 and RNase inhibitor in nuclease-free water) and centrifuged at 500g for 5 min. Finally, nuclei were resuspended in 5 μl chilled diluted nuclei buffer (10X Genomics) and added to the transposition mix. Paired-end, dual-index sequencing was performed on an Illumina NovaSeq 6000 platform.
Multiome data quality control
After sequencing, bcl files were converted to fastq using cellranger-arc (v.2.0.2) mkfastq. FASTQ files were aligned to the mm10 or hg38 genome assembly using cellranger-arc count. ATAC-seq fragment files were used as inputs to the ArchR66 (v.1.0.2) analysis pipeline in R (v.4.3.2). Transcript count matrices were used as inputs to the Seurat (v.5.1.0) gene expression analysis pipeline. For gene expression quality control, cells with nFeature_RNA ≥ 250 and ≤ 6,000, nCount_RNA ≤ 25,000 and percent_mitochondrial ≤ 25 were included for downstream analyses. Transcript counts were log-normalized. For scATAC-seq quality control, cells with n_ATAC_Frags ≥ 3,000 and TSS_Score ≥ 10 were included for downstream analyses. Doublet inference was conducted using ArchR addDoubletScores(), and presumed doublets were excluded. Cells that passed each filter were admitted for downstream analyses. Finally, based on gene expression markers, contaminating cells (thymocytes) and putative mTEC mimetic cells were excluded from analysis (except for targeted analyses of mimetic compartments). In the wild-type multiome (Fig. 1), a further cluster of cells that exhibited uncharacteristically low TSS enrichment scores was excluded.
Multiome data processing
Dimensionality reduction, scATAC-seq clustering, projections, pseudotime, transcription factor motif enrichment (except for scATAC-seq fragments or genomic tiles, which was computed using HOMER2 (v.5.1) findMotifsGenome.pl with settings -size given), and transcription factor footprinting were performed using the ArchR pipeline with default parameters. For UMAP plots overlaid with continuous colour scales, MAGIC67 (v.2.0.3) imputation was used for data smoothing to facilitate better visualization. MAGIC-imputed values were used for UMAP display purposes only; imputed values were not used anywhere else in the analysis of scATAC-seq or scRNA-seq datasets (such as violin plots or heatmaps). For scATAC-seq peak calling, the standard ArchR workflow was used using MACS2 (v.2.2.9.1). To maximize the detection of open chromatin regions specific to each sample and stage in the mTEC developmental trajectory, fixed-width 501-bp scATAC-seq peaks were called (extendSummits = 250) on the Tn5-corrected single base insertions (shift = −75, extsize = 150, –nomodel) for each scATAC-seq cluster identified per sample (groupBy = Clusters, reproducibility = 1) using the ArchR wrapper function addReproduciblePeakSet(). The significance of each called peak was calculated as a false discovery rate (q-value) comparing the observed number of Tn5 insertions in the sliding window (300 bp) and the expected number of insertions (total number of insertions/genome size (–nolambda)). A q-value cutoff (cutOff = 0.1) and an upper limit for the number of peaks called per cell (peaksPerCell = 1,000, minCells = 100) were applied to prevent consideration of low-quality peaks. We also excluded peaks that mapped to the mitochondrial or Y chromosomes (excludeChr = c(chrM, chrY)). Peak sets called from each scATAC-seq cluster from respective samples were combined and trimmed for overlap using an iterative procedure that discarded any peak that directly overlapped with the most significant peak66. The resultant ‘union peak set’ was applied to all cells for WIP and OOP count-based and motif-based analyses. The fraction of fragments within peaks was computed automatically as a product of the addReproduciblePeakSet() function. Subnucleosomal and mononucleosomal fractions for each cell or sample were computed as the fraction of the cell’s scATAC-seq fragments whose length L ≤ 100 bp (subnucleosomal) or 100 < L ≤ 200 bp (mononucleosomal). To ensure reproducibility of bioinformatic analysis results, for each dataset, a single script was used for all the quality control and pre-processing, including purging of low-quality cells, doublet removal, peak calling, motif enrichment, dimensionality reduction and clustering. A file representing the full processed data was saved using saveArchRProject() and loaded for all subsequent analyses (this file was not edited after pre-processing). More individual scripts were used to load processed data and perform specific analyses or generate specific figures.
Peak-centric differential accessibility analysis
Differential chromatin accessibility analysis across peaks was done using ArchR getMarkerFeatures() with the following arguments: useMatrix = PeakMatrix, bias = c(TSSEnrichment, log10(number of scATAC-seq fragments)), testMethod = wilcoxon.
Processing of OOP scATAC-seq fragments
For each Multiome dataset, WIP and OOP fragments near genes of interest (such as αTSGs, housekeeping genes and maturation-induced genes) were retrieved using the ArchR and GenomicRanges R packages. For each gene: first, a search window, search_window, was established around the \({\rm{TSS}}({\rm{search}}\_{\rm{window}}={\rm{TSS}}\pm {\ell })\); and second, scATAC-seq fragments intersecting the search_window were retrieved from cells of interest, cell_subset, using the ArchR getFragmentsFromProject() function with arguments subsetBy = search_window and cellNames = cell_subset. Fragments were then partitioned based on whether they overlapped the data’s union peak set using subsetByOverlaps() with arguments invert = FALSE to retrieve WIP fragments, or invert = TRUE to retrieve OOP fragments. Finally, fragments were binned and/or tallied for the specific application (see below).
Analyses comparing αTSGpos and αTSGneg mTECs
Cells from early mature, mid mature and late mature clusters expressing any αTSGi > 0 were selected as the αTSGpos cohort and a size-matched cohort of αTSGneg cells was sampled randomly from the remaining cells from the same three clusters. These cohorts were then used as inputs to getMarkerFeatures()in ArchR for differential accessibility of peaks between αTSGpos and αTSGneg mTECs. For local OOP and WIP analysis, ATAC-seq fragments within peaks and outside of peaks from αTSGpos and αTSGneg cohorts were intersected with a ±5 kb sliding window with 1 kb increments, normalized to the total number of ATAC-seq fragments per cell, and tallied in each window within a region flanking αTSGi . For αTSG coexpression analysis, the probability of detecting each αTSGi neighbouring αTSG0 within the specified length scale (or a randomly selected alternative αTSG as a control) was computed for each of the αTSGpos and αTSGneg cohorts.
Regression analysis
For each αTSGi, the total number of OOP and WIP scATAC-seq fragments within the characteristic window of instability \(({\ell }=\pm 50\,{\rm{kb}})\) was computed for each mTEC in the early mature, mid mature and late mature clusters. A logistic regression framework was used (glm() with family = binomial) to estimate the probability of expressing a given αTSG based on the number of log10(OOP + 1) or log10(WIP + 1) fragments using log10(n_ATAC_Frags) per cell as a covariate. P-values for regression coefficients were generated using the Wald-χ2 test (anova(test = ‘LR’)).
CUT&RUN sample preparation
CUT&RUN was performed as previously described28 with minor modifications. In brief, 350,000–500,000 cells were washed 3 times in wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine, 1× EDTA-free protease inhibitor cocktail (Roche)) then bound to Concanavalin-A beads (Bangs Laboratories) according to the manufacturer’s instructions. Cells were incubated with 1:100 dilution of anti-p53 antibody (Leica NCL-L-p53-CM5p) for 2 h or overnight at 4 °C in permeabilization buffer (1× permeabilization buffer (eBioscience), 0.5 mM spermidine, 1× EDTA-free protease inhibitor cocktail, 2 mM EDTA). The sample was then incubated with 700 ng ml−1 pA-MNase (S. Henikoff) in permeabilization buffer at 4 °C for 1 h. Digestion was done in 0.5× permeabilization buffer supplemented with 2 mM CaCl2 at 4 °C for 1 h. The reaction was stopped by the addition of 2× stop buffer (final concentration 100 mM NaCl, 10 mM EDTA, 2 mM EGTA, 20 μg ml−1 glycogen, 25 μg ml−1 RNase A (Thermo Fisher)) and the sample was incubated at 37 °C for 20 min. Protein in the sample was then digested in 0.1% SDS and 250 μg ml−1 Proteinase K (New England Biolabs) for 2 h at 56 °C, shaking gently. CUT&RUN fragments were purified by phenol chloroform extraction. CUT&RUN libraries were generated using NEBNext UltraII DNA Library Prep Kit for Illumina coupled with NEBNext Multiplex Oligos for Illumina (New England Biolabs) with modifications optimized for small fragments, as detailed in https://doi.org/10.17504/protocols.io.wvgfe3w. Paired-end, dual-index sequencing was performed on the Illumina NextSeq500 platform.
CUT&RUN data processing
CUT&RUN reads were mapped to mm10 mouse genome assembly using Bowtie2 (v.2.2.9) with settings --local --very-sensitive-local –no-unal –no-mixed –no-discordant –phred33 -I 10 -X 700. PCR duplicates were removed using Picard (v.2.21.8) MarkDuplicates REMOVE_DUPLICATES=true VALIDATION_STRINGENCY = LENIENT. Reads with MAPQ scores below 30 were purged and excluded from downstream analysis using samtools (v.1.9) view -b -q 30 -f 2 -F 1804. Peaks were called for each sample using MACS2 (v.2.2.7.1) with settings --shift 0 --extsize 200 --nomodel --call-summits --keep-dup all -p 0.01. For each sample, a 301-bp fixed-width peak set was generated by extending the MACS2 summits by 150 bp in both directions. Peaks were ranked by significance (MACS2 peak score) and overlapping peaks with lower peak scores were removed iteratively to create non-overlapping sample peak sets. Peaks mapping to chrY, as well as any that spanned genomic regions containing “N” nucleotides, were removed. Robust peaks were defined by a score per million (SPM) (each peak score divided by the sum of all peak scores in the sample, divided by 1 million), and we retained only those peaks with SPM ≥ 5. We defined p53 CUT&RUN peaks by further filtering for peaks that overlapped with known p53-binding motifs (HOMER2, v5.1) from samples with characterized p53 activity (mTEClo samples). CUT&RUN fragment counts across regions of interest were normalized by the number of unique fragments in the sample library.
ChIP–seq data processing
ChIP–seq reads were mapped to mm10 mouse genome assembly using Bowtie2 (v.2.2.9) with settings --very-sensitive -X 2000. PCR duplicates were removed using Picard (v.2.21.8) MarkDuplicates REMOVE_DUPLICATES=true VALIDATION_STRINGENCY = LENIENT. Reads with MAPQ scores below 30 were purged and excluded from downstream analysis using samtools (v.1.9) view -b -q 30 -F 1796. ChIP–seq read counts were normalized by the number of unique reads in the sample library.
Histopathology
Histopathology experiments were carried out as previously described9. In brief, tissues were fixed in buffered 10% formalin and paraffin-embedded. H&E staining was done by the standard methods. Histopathology scores were assigned using a four-tier system based on the degree and distribution of lymphocytic infiltration observed in the tissue sections. A score of 0 was assigned when no lymphocyte infiltration was detected; a score of 1 corresponded to minimal infiltration, characterized by very few small, isolated clusters; a score of 2 corresponded to moderate infiltration, in which several small to moderately sized clusters of lymphocytes were observed; a score of 3 corresponded to severe, diffuse infiltration, indicated by the presence of numerous large clusters distributed throughout the tissue.
Statistical analysis
De novo and known transcription factor motif P-values were determined using HOMER2 (v.5.1). For bulk RNA-seq, P-values for differentially expressed genes were computed using edgeR (v.4.0.2) (estimateCommonDisp()) and corrected for multiple testing using the Benjamini–Hochberg FDR method. For scATAC-seq and scRNA-seq, FDR-corrected Wilcoxon test P-values for differentially accessible ATAC peaks and differentially expressed genes were computed using ArchR (v.1.0.2) (getMarkerFeatures(testMethod = “wilcoxon”)). Logistic regression coefficient estimate P-values were computed using analysis of variance (ANOVA; anova(test = “Chisq”)) to compare the regression results from glm(). Box plots show the median (centre line), 25th and 75th percentiles (edges), and whiskers show ±1.5 times the interquartile range. Outliers beyond the interquartile range are represented as individual dots. All other P-values and statistical tests were computed in R or Prism and are specified in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Original raw scATAC-seq, scRNA-seq and bulk RNA-seq data have been deposited at the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus: accession numbers GSE274320, GSE274324, GSE290716 and GSE301724. Further Gene Expression Omnibus accession numbers for published datasets used in this study include GSE53111, GSE102526, GSE234331, GSE194253, GSE231681 and GSE92597. All other data are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
This study did not generate any new code. Analysis scripts are available from the corresponding author upon reasonable request.
References
Merrell, A. J. & Stanger, B. Z. Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 17, 413–425 (2016).
Gupta, P. B., Pastushenko, I., Skibinski, A., Blanpain, C. & Kuperwasser, C. Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 24, 65–78 (2019).
Kyewski, B. & Klein, L. A central role for central tolerance. Annu. Rev. Immunol. 24, 571–606 (2006).
Flavahan, W. A., Gaskell, E. & Bernstein, B. E. Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380 (2017).
Michelson, D. A., Hase, K., Kaisho, T., Benoist, C. & Mathis, D. Thymic epithelial cells co-opt lineage-defining transcription factors to eliminate autoreactive T cells. Cell 185, 2542–2558 (2022).
Givony, T. et al. Thymic mimetic cells function beyond self-tolerance. Nature 622, 164–172 (2023).
Anderson, M. S. et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 (2002).
Giraud, M. et al. Aire unleashes stalled RNA polymerase to induce ectopic gene expression in thymic epithelial cells. Proc. Natl Acad. Sci. USA 109, 535–540 (2012).
Koh, A. S. et al. Rapid chromatin repression by Aire provides precise control of immune tolerance. Nat. Immunol. 19, 162–172 (2018).
Abramson, J. & Anderson, G. Thymic epithelial cells. Annu. Rev. Immunol. 35, 85–118 (2017).
Hitz, B. C. et al. The ENCODE uniform analysis pipelines. Preprint at bioRxiv https://doi.org/10.1101/2023.04.04.535623 (2023).
Landt, S. G. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012).
Gamble, N. et al. PU.1 and BCL11B sequentially cooperate with RUNX1 to anchor mSWI/SNF to poise the T cell effector landscape. Nat. Immunol. 25, 860–872 (2024).
Goldfarb, Y. et al. Mechanistic dissection of dominant AIRE mutations in mouse models reveals AIRE autoregulation. J. Exp. Med. 218, e20201076 (2021).
Fischer, M. Census and evaluation of p53 target genes. Oncogene 36, 3943–3956 (2017).
Puisieux, A., Pommier, R. M., Morel, A.-P. & Lavial, F. Cellular pliancy and the multistep process of tumorigenesis. Cancer Cell 33, 164–172 (2018).
Kawamura, T. et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460, 1140–1144 (2009).
Kruse, J.-P. & Gu, W. Modes of p53 regulation. Cell 137, 609–622 (2009).
Vaziri, H. et al. hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 (2001).
Chuikov, S. et al. Regulation of p53 activity through lysine methylation. Nature 432, 353–360 (2004).
Liu, L. et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 19, 1202–1209 (1999).
Shieh, S.-Y., Ahn, J., Tamai, K., Taya, Y. & Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 14, 289–300 (2000).
Goldberg, Z. et al. Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. EMBO J. 21, 3715–3727 (2002).
Song, M. S., Song, S. J., Kim, S. Y., Oh, H. J. & Lim, D.-S. The tumour suppressor RASSF1A promotes MDM2 self‐ubiquitination by disrupting the MDM2–DAXX–HAUSP complex. EMBO J. 27, 1863–1874 (2008).
Catizone, A. N. et al. Locally acting transcription factors regulate p53-dependent cis-regulatory element activity. Nucleic Acids Res. 48, 4195–4213 (2020).
Karsli Uzunbas, G., Ahmed, F. & Sammons, M. A. Control of p53-dependent transcription and enhancer activity by the p53 family member p63. J. Biol. Chem. 294, 10720–10736 (2019).
Bowen, M. E. et al. The spatiotemporal pattern and intensity of p53 activation dictates phenotypic diversity in p53-driven developmental syndromes. Dev. Cell 50, 212–228 (2019).
Skene, P. J. & Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856 (2017).
Miyashita, T. & Reed, J. C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80, 293–299 (1995).
Jain, R. et al. How do thymic epithelial cells die? Cell Death Differ. 25, 1002–1004 (2018).
Adams, J. M. & Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337 (2007).
Inohara, N., Ding, L., Chen, S. & Núñez, G. harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival‐promoting proteins Bcl‐2 and Bcl‐XL. EMBO J. 16, 1686–1694 (1997).
Jain, R. et al. A critical epithelial survival axis regulated by MCL-1 maintains thymic function in mice. Blood 130, 2504–2515 (2017).
Kastenhuber, E. R. & Lowe, S. W. Putting p53 in context. Cell 170, 1062–1078 (2017).
Fernandez-Capetillo, O., Lee, A., Nussenzweig, M. & Nussenzweig, A. H2AX: the histone guardian of the genome. DNA Repair 3, 959–967 (2004).
Bansal, K., Yoshida, H., Benoist, C. & Mathis, D. The transcriptional regulator Aire binds to and activates super-enhancers. Nat. Immunol. 18, 263–273 (2017).
Fang, Y., Bansal, K., Mostafavi, S., Benoist, C. & Mathis, D. AIRE relies on Z-DNA to flag gene targets for thymic T cell tolerization. Nature 628, 400–407 (2024).
Puc, J., Aggarwal, A. K. & Rosenfeld, M. G. Physiological functions of programmed DNA breaks in signal-induced transcription. Nat. Rev. Mol. Cell Biol. 18, 471–476 (2017).
Core, L. & Adelman, K. Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes Dev. 33, 960–982 (2019).
The ENCODE Project Consortium et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Segal, E. & Widom, J. Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr. Opin. Struct. Biol. 19, 65–71 (2009).
Ioshikhes, I. P., Albert, I., Zanton, S. J. & Pugh, B. F. Nucleosome positions predicted through comparative genomics. Nat. Genet. 38, 1210–1215 (2006).
Marjanovic, N. D. et al. Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 38, 229–246 (2020).
Kaiser, A. M. et al. p53 governs an AT1 differentiation programme in lung cancer suppression. Nature 619, 851–859 (2023).
Winslow, M. M. et al. Suppression of lung adenocarcinoma progression by Nkx2-1. Nature 473, 101–104 (2011).
LaFave, L. M. et al. Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 38, 212–228 (2020).
Meredith, M., Zemmour, D., Mathis, D. & Benoist, C. Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat. Immunol. 16, 942–949 (2015).
Brennecke, P. et al. Single-cell transcriptome analysis reveals coordinated ectopic gene-expression patterns in medullary thymic epithelial cells. Nat. Immunol. 16, 933–941 (2015).
Dhalla, F. et al. Biologically indeterminate yet ordered promiscuous gene expression in single medullary thymic epithelial cells. EMBO J. 39, e101828 (2020).
Saltis, M. et al. Evolutionarily conserved and divergent regions of the autoimmune regulator (Aire) gene: a comparative analysis. Immunogenetics 60, 105–114 (2008).
Oven, I. et al. AIRE recruits P-TEFb for transcriptional elongation of target genes in medullary thymic epithelial cells. Mol. Cell. Biol. 27, 8815–8823 (2007).
Yoshida, H. et al. Brd4 bridges the transcriptional regulators, Aire and P-TEFb, to promote elongation of peripheral-tissue antigen transcripts in thymic stromal cells. Proc. Natl Acad. Sci. USA 112, E4448–E4457 (2015).
Abramson, J., Giraud, M., Benoist, C. & Mathis, D. Aire’s partners in the molecular control of immunological tolerance. Cell 140, 123–135 (2010).
Guha, M. et al. DNA breaks and chromatin structural changes enhance the transcription of autoimmune regulator target genes. J. Biol. Chem. 292, 6542–6554 (2017).
Ravichandran, S., Subramani, V. K. & Kim, K. K. Z-DNA in the genome: from structure to disease. Biophys. Rev. 11, 383–387 (2019).
Chakravarty, S., Zeng, L. & Zhou, M.-M. Structure and site-specific recognition of histone H3 by the PHD finger of human autoimmune regulator. Structure 17, 670–679 (2009).
Koh, A. S. et al. Aire employs a histone-binding module to mediate immunological tolerance, linking chromatin regulation with organ-specific autoimmunity. Proc. Natl Acad. Sci. USA 105, 15878–15883 (2008).
Org, T. et al. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep. 9, 370–376 (2008).
Koh, A. S., Kingston, R. E., Benoist, C. & Mathis, D. Global relevance of Aire binding to hypomethylated lysine-4 of histone-3. Proc. Natl Acad. Sci. USA 107, 13016–13021 (2010).
Waterfield, M. et al. The transcriptional regulator Aire coopts the repressive ATF7ip-MBD1 complex for the induction of immunotolerance. Nat. Immunol. 15, 258–265 (2014).
Van Nostrand, J. L. et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 514, 228–232 (2014).
Gordon, J. et al. Specific expression of lacZ and cre recombinase in fetal thymic epithelial cells by multiplex gene targeting at the Foxn1 locus. BMC Dev. Biol. 7, 69 (2007).
Kim, M.-J. & Serwold, T. Isolation of highly viable thymic epithelial cells for use in in vitro and in vivo experiments. Methods Mol. Biol. 1899, 143–156 (2019).
Sansom, S. N. et al. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 24, 1918–1931 (2014).
Lonsdale, J. et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
van Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729 (2018).
Acknowledgements
We are grateful to L. Attardi for the Trp53LSL-L25Q,W26S,F53Q,F54S mice; E. Pena for supporting IRB applications for the acquisition of human thymic tissue; C. Ciszewski for FACS support; and T. Kreslavskiy, L. Klein, D. Schatz and members of the Koh Lab for critical reading of the manuscript. This work was supported by grants from the National Institute of Health (R35-GM138150, 2UL1TR002389-06 and 5UL1TR002389-04 to A.S.K.; T32-AI07090 to N.G.; and T32-CA009594 and R35-GM138150-S1 to J.M.), the Chan Zuckerberg Biohub (to W.J.G.) and the National Science Foundation (PHY-2317138 to A.R.D.). Fellowship support was provided by the University of Chicago Women’s Board (to C.K.), the Stamps Scholarship (to A.B.) and the Stanford Genome Training Program (NIH/NHGRI) (to S.K.). Flow cytometry was supported by the University of Chicago Human Disease and Immune Discovery Facility (RRID:SCR_022936) and the Cytometry and Antibody Technology Facility (RRID:SCR_017760). Genomics sequencing was supported by the University of Chicago Genomics Facility (RRID:SCR_019196), which receives support from a Cancer Center Support Grant (P30-CA014599).
Author information
Authors and Affiliations
Contributions
A.S.K. and N.G. conceived the study. N.G. did the murine Multiome experiments, mimetic profiling, T cell and mTEC compartment profiling and histopathology experiments, and assisted with the human Multiome experiment, bulk RNA-seq studies and CUT&RUN experiments. J.A.C. did bulk RNA-seq experiments and assisted on the T cell and mTEC compartment profiling and histopathology experiments. C.K. did the human Multiome experiment, established the CUT&RUN protocol and assisted on the murine Multiome and histopathology experiments. J.M. did CUT&RUN experiments and assisted on the histopathology experiments. A.B. assisted on the histopathology experiments. P.J.D. established the mimetic profiling protocol. A.S.K. assisted on the bulk RNA-seq studies and histopathology experiments. N.G. and A.S.K. did bioinformatic analyses. A.S.K. supervised all the experiments and analyses. S.K. and W.J.G. provided resources. N.H. facilitated the acquisition of human thymic tissue. A.R.D. provided conceptual support. A.S.K. and N.G. wrote the manuscript. All authors reviewed and provided comments on the manuscript.
Corresponding author
Ethics declarations
Competing interests
W.J.G. is a consultant and equity holder for 10X Genomics, Guardant Health, Quantapore, Erudio Bio and Lamar Health, is a co-founder of Protillion Biosciences and is named on patents describing ATAC-seq. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Golnaz Vahedi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Metrics of single-mTEC Multiome profiling and working hypothesis.
a, Scatter plot depicting scATAC-seq library complexity versus quality of Tn5 transposition for 9,928 filtered mTECs. b, scATAC-seq fragment size distribution from 9,928 filtered mTECs. c, Aggregate histogram of Tn5 insertions across transcription start sites. d, scRNA-seq UMAP of mTECs colored by cluster annotation defined in Fig. 1b. Yellow=mimetic mTECs or cells that did not pass quality-control filters. Numbers=mimetic subtypes in (f–h). e, scATAC-seq UMAP mTECs colored by cluster annotation defined in Fig. 1b. Yellow= mimetic mTECs from (d,f–h) or cells that did not pass quality-control filters. f, Expression levels of genes encoding mimetic-defining transcription factors for indicated subtypes overlayed on scRNA-seq UMAP from (d). g,h, Heatmap of Z-scores of differentially expressed genes (g) or differentially enriched transcription factor motifs (h) across mimetic mTEC clusters defined in (d,f). i, Bar plot depicting number of cells per annotated scATAC-seq cluster from Fig. 1b. j, Bar plot depicting number of pseudo-bulk ATAC-seq peaks and associations to indicated genomic regions (colors) per annotated scATAC-seq cluster from Fig. 1b. k, Pseudotime trajectory analysis overlayed on scATAC-seq UMAP from Fig. 1b. l, Violin plots depicting the distributions of the number of unique scRNA-seq reads (UMIs) per cell within each annotated cluster (n = 9,928: Immature=2,107, Transitional=1,790, Early Mature=2,825, Mid Mature=2,511, Late Mature=695) defined in Fig. 1b. Box plots depict median, 25th and 75th percentile, whiskers=1.5 times interquartile range. m,n, Expression levels of indicated genes overlayed on scATAC-seq UMAP from Fig. 1b. o,p, Mean aggregate expression of genes associated with S- (o) or G2/M (p) cell cycle stages overlayed on scATAC-seq UMAP from Fig. 1b. q, Sum of mRNA from all AIRE-dependent tissue-specific genes (αTSGs) overlayed on scATAC-seq UMAP from Fig. 1b. r,s, Indicated αTSG expression overlayed on scATAC-seq UMAP from Fig. 1b. t, Sum expression of αTSGs within indicated clusters as a cumulative fraction of the total sum across all clusters. u, MA plot comparing differential expression of genes by bulk RNA-seq64 (n = 2 biological replicates) upon mTEC maturation and the mean gene expression. Fold-change density on right margin. Statistically significant (Benjamini-Hochberg FDR ≤ 0.05) differential gene expression between mature and immature mTECs is highlighted for indicated genes. v, Schematic depiction of hypothesis that out-of-peak scATAC-seq fragments represent fluctuations in nucleosome dynamics within inaccessible chromatin, compromising chromatin barriers flanking αTSGs.
Extended Data Fig. 2 Chromatin noise is independent of transcriptome size, peak definition, postmitotic state, and AIRE.
a–c, Mean aggregate expression of genes associated with the S- (a) or G2/M (b) phase of cell cycle (aqua) or Aire expression (c) and fraction of scATAC-seq fragments within scATAC-seq peaks (WIP) (purple) across mTEC developmental axis. d, Transcriptome size versus WIP fraction detected per mTEC. Trendlines with two-sided 95% confidence intervals (t-distribution) for the linear regression fit (gray) for each annotated cluster defined in Fig. 1b. e,f, Comparison of the minimum FDR (q-value) cutoff for scATAC-seq peak-calling versus total number of peaks called (e) or median WIP fraction (f). g,h, WIP fractions calculated with indicated q-value cutoff for peak-calling, overlayed on scATAC-seq UMAP defined in Fig. 1b. i,j, Comparison of the width of scATAC-seq peaks versus total number of peaks called (i) or the median WIP fraction (j). k,l, WIP fractions calculated with indicated peak width, overlayed on scATAC-seq UMAP defined in Fig. 1b. m–o, WIP fraction (m) or mean aggregate expression of genes associated with S- (n) or G2/M (o) phase of the cell cycle, overlayed on scATAC-seq UMAP of cells from embryonic E18 mouse brain 10X Multiome dataset. p, Comparisons of the nucleosome ratio (∑mononucleosomal versus ∑subnucleosomal bulk ATAC-seq fragments) as a function of size of the most prevalent nucleosomal ATAC-seq fragment across ATAC-seq libraries from mature, immature mTECs and splenic T cells from published datasets9,13. q, Fraction of mononucleosomal scATAC-seq fragments overlayed on scATAC-seq UMAP defined in Fig. 1b. r, Violin plots depicting the distributions of the fraction of mononucleosomal scATAC-seq fragments across annotated clusters of cells defined in Fig. 1b (n = 9,928: Immature=2,107, Transitional=1,790, Early Mature=2,825, Mid Mature=2,511, Late Mature=695). Box plots depict median, 25th and 75th percentile, whiskers represent 1.5 times interquartile range. P-values from one-sided Mann-Whitney U-tests. s, scATAC-seq UMAP of merged Aire+/+ and Aire-/- mTEC data5 colored by indicated genotype. t,u, Imputed ‘gene score’ (ArchR chromatin accessibility proxy of gene expression) for Aire (t) or H2-Ab1 (u) overlayed on scATAC-seq UMAP from Fig. 2e. v, Fraction of mononucleosomal scATAC-seq fragments overlayed on scATAC-seq UMAP defined in Fig. 2e. w, Paired violin plots comparing distributions of the fraction of mononucleosomal scATAC-seq fragments from indicated genotypes (left = Aire+/+, right = Aire-/-) across annotated clusters defined in Fig. 3e. P-values from one-sided Mann-Whitney U-tests.
Extended Data Fig. 3 mTECs repress p53 upon maturation independently of AIRE.
a, Aggregate prevalence of p53-target motifs across accessible mTEC genomes (chromVAR deviation scores) overlayed on scATAC-seq UMAP from Fig. 1b. b,c, Mean chromVAR deviation scores of p53-target motifs (red) and mean fraction of scATAC-seq fragments within scATAC-seq peaks (WIP) (purple) (b) or mean aggregate expression of p53-target genes (green) (c) in mTECs across developmental axis. d, Representative flow cytometry plots of the frequencies of MDM2hi cells in singlets from thymic digest and indicated mature and immature mTEC compartments. e,f, Comparison of frequencies of MDM2lo and MDM2hi cells (e) or ratio of MDM2 mean fluorescence intensity (MFI) (f) between immature and mature mTECs. P-values for two-sided paired t-tests displayed. g–o, Expression levels of indicated genes overlayed on scATAC-seq UMAP from Fig. 1b. p, Rank-sorted differences in motif prevalence within accessible genomes (chromVAR deviation scores) of AIRE-deficient mTECs between indicated mTECs defined in Fig. 2e (and UMAP inset) for 884 known transcription factor motifs. q, Scatter plot comparing differential gene expression (transcripts per million=TPM) of 51 known regulators of p53 activity between AIRE-deficient mature and immature mTECs versus AIRE-sufficient mature and immature mTECs. Benjamini-Hochberg FDR for AIRE-deficient mature versus immature comparison indicated as point colors with highly significant differentially expressed genes (FDR ≤ 1e-9, fold-change ≥ 2 or ≤ 0.5) indicated as repressors (brown text) or promoters (green text) of p53 activity.
Extended Data Fig. 4 Effects of p53 hyperactivity on thymic compartments.
a, Representative flow cytometry plots of the frequencies of AIRE+ mTECs from 3-4 week-old p53-WT or p53-cHyper sex-matched littermates. b, Comparison of frequencies or mean fluorescence intensity (MFI) of indicated thymus compartments between p53-WT and p53-cHyper sex-matched littermates (n = 6). P-values from two-sided paired t-tests. c, Aire expression overlayed on scATAC-seq UMAP from Fig. 3b. d, Rank-sorted differences in transcription factor motif prevalence within accessible genomes (chromVAR deviation scores) of p53-cHyper versus p53-WT Early Mature mTECs as defined in Fig. 3b. e, Transcription factor footprinting at p53-target motifs within p53-cHyper and p53-WT Early Mature mTECs. f, Differences in fraction of scATAC-seq fragments within scATAC-seq peaks (WIP) between neighboring p53-cHyper and p53-WT mTECs overlayed on scATAC-seq UMAP from Fig. 3b. g, Paired violin plots comparing distributions of the ratio of the normalized sum of mononucleosomal vs. subnucleosomal scATAC-seq fragments from indicated genotypes across annotated clusters defined in Fig. 3b. P-values from one-sided Mann-Whitney U-tests. h,i, Heatmap of Z-scores of differentially expressed genes (h) or differentially enriched transcription factor motifs (i) across mimetic mTEC clusters defined in Fig. 3f. j–l, Gene expression levels of Aire (j), H2-Ab1 (k) or module of S-phase genes (l) overlayed on scRNA-seq UMAP from Fig. 3f. m, Expression levels of genes encoding mimetic-defining transcription factors for indicated subtypes overlayed on scRNA-seq UMAP from Fig. 3f. n–s, Representative flow cytometry plots for the definition of mimetic mTEC compartments.
Extended Data Fig. 5 Nucleosome-destabilizing motifs underlie chromatin accessibility noise.
a,b, Aggregate histograms of p53 occupancy at AIRE-dependent tissue-specific genes (αTSGs) (blue), p53 CUT&RUN peaks (green) or p53-induced target genes (red) in total mTECs from WT mice. c, Motif enrichment of indicated motifs within 500 bp genomic windows (“tiles”) spanning +/−50 kb of αTSGs (blue) or sites of p53 occupancy (CUT&RUN peaks). d,e, Differences in gene expression of indicated genes between neighboring p53-cHyper and p53-WT mTECs overlayed on scATAC-seq UMAP from Fig. 3b. f, MA plot of the differential expression of genes encoding proteins of the intrinsic apoptosis pathway between mature and immature p53-WT mTECs by bulk RNA-seq (n = 3). Red text=pro-apoptotic, green text=pro-survival proteins. g,h, Differences in gene expression of indicated genes between neighboring p53-cHyper and p53-WT mTECs overlayed on scATAC-seq UMAP from Fig. 3b. i, MA plot of the differential expression of genes encoding DNA damage response proteins between mature and immature p53-cHyper mTECs by bulk RNA-seq. Point colors=Benjamini-Hochberg FDR levels. j, Aggregate histogram of out-of-peak (OOP) Tn5 inserts over αTSGs (blue) or Silent genes (orange) in mature mTECs. k,l, Comparison of transcription factor motif enrichment within scATAC-seq fragments from mature mTECs mapping to within-peak (WIP) (k) or out-of-peak (OOP) (l) at indicated regions versus the adenine/thymine content of each motif. Trendlines (red) with two-sided 95% confidence intervals (t-distribution) for the linear regression fit (gray) indicated. m, MA plot of the expression levels of genes encoding known transcription factors (transcripts per million=TPM) versus the fold-change in expression between transitional and immature mTECs. Motif enrichments (-log10 P value) within scATAC-seq fragments from mature mTECs mapping to out-of-peak regions (OOP frags) +/−100 kb of αTSGs are indicated as point colors. Red dotted line=Ins2 expression in mature mTECs (AIREpos mTEChi). n,o, Comparison of transcription factor motif enrichment within scATAC-seq fragments from mature mTECs mapping to out-of-peak (OOP) regions at indicated loci versus the adenine/thymine content of each motif. Trendlines (red) with two-sided 95% confidence intervals (t-distribution) for the linear regression fit (gray) indicated.
Extended Data Fig. 6 p53 hyperactivity in mTECs causes systemic defects in immune tolerance.
a, Representative flow cytometry plots of T effector memory (TEM) and naïve (TN) compartments of splenic CD8+ T cells from 10 month-old p53-WT and p53-cHyper sex-matched littermates. b,c, Comparison of frequencies of indicated splenic T cell compartments between p53-WT and p53-cHyper sex-matched littermates (n = 12). P-values from two-sided paired t-tests. d, Comparison of histopathology scores (based on number and size of lymphocytic infiltrates) from the indicated organs between sex-matched littermates (n = 8) of indicated genotypes.
Extended Data Fig. 7 Chromatin accessibility noise is elevated in high plasticity states of lung adenocarcinoma.
a,b, UMAP of merged scATAC-seq data from lung adenocarcinomas44 (LUAD) in p53-KO and p53-WT mice colored by genotype (a) and cluster annotation (b). c–f, LUAD progression depicted by aggregate expression of genes defining alveolar type-2 (AT2) lung epithelial state43 (c), mixed alveolar type-1 and type-2 lung epithelial state43 (d), embryonic liver state43 (e) or epithelial-to-mesenchymal transition (EMT) state43 (f) overlayed on scATAC-seq UMAP from (b). g, Fraction of scATAC-seq fragments within scATAC-seq peaks (WIP) overlayed on UMAP from (b). h, Fraction of mononucleosomal scATAC-seq fragments overlayed on UMAP from (b). i,j, Violin plots depicting WIP fraction (i) or the fraction of mononucleosomal scATAC-seq fragments (j) within cells (n = 3,172: C1 = 612, C2 = 511, C3 = 112, C4 = 581, C5 = 425, C6 = 188, C7 = 190, C8 = 553) across annotated LUAD clusters defined in (b–f). Box plots depict median, 25th and 75th percentile, whiskers represent 1.5 times interquartile range. P-values from one-sided Mann-Whitney U-tests. k, Nkx2-1 gene expression levels overlayed on scATAC-seq UMAP from (b). l, Prevalence of NKX2.1-binding motifs across accessible LUAD genomes (chromVAR deviation scores) overlayed on scATAC-seq UMAP from (b). m, Runx2 expression overlayed on scATAC-seq UMAP from (b). n, Prevalence of RUNX2-binding motifs across accessible LUAD genomes (chromVAR deviation scores) overlayed on scATAC-seq UMAP from (b).
Extended Data Fig. 8 p53 regulates chromatin accessibility noise in lung adenocarcinoma.
a, UMAP of merged scATAC-seq data from lung adenocarcinomas44 (LUAD) in p53-KO, p53-WT and p53-Hyper mice colored by genotype. b, Cluster annotations overlayed on UMAP of merged scATAC-seq data from (a). c–f, LUAD progression depicted by aggregate expression of genes defining alveolar type-2 (AT2) lung epithelial state43 (c), mixed alveolar type-1 and type-2 lung epithelial state43 (d), embryonic liver state43 (e) or epithelial-to-mesenchymal transition (EMT) state43 (f) overlayed on scATAC-seq UMAP from (b). g, Nkx2-1 gene expression levels overlayed on scATAC-seq UMAP from (a). h, Prevalence of NKX2.1-binding motifs across accessible LUAD genomes (chromVAR deviation scores) overlayed on scATAC-seq UMAP from (a). i, Runx2 expression overlayed on scATAC-seq UMAP from (b). j, Prevalence of RUNX2-binding motifs across accessible LUAD genomes (chromVAR deviation scores) overlayed on scATAC-seq UMAP from (a). k, Fraction of indicated LUAD clusters represented by each genotype. l, Fraction of scATAC-seq fragments within scATAC-seq peaks (WIP) overlayed on UMAP from (b). m, Fraction of mononucleosomal scATAC-seq fragments overlayed on UMAP from (b). n,o, Bar plots depicting WIP fraction (n) or the fraction of mononucleosomal scATAC-seq fragments (o) within cells (n = 5,556: C1 = 813, C2 = 745, C3 = 153, C4 = 1091, C5 = 495, C6 = 434, C7 = 369, C8 = 1456) of indicated genotypes across annotated LUAD clusters defined in (a–j). Error bars represent mean ± SEM. P-values from one-sided Mann-Whitney U-tests comparing p53-cHyper or p53-cKO cells to WT cells within each cluster.
Supplementary information
Supplementary Information
This file contains Supplementary Notes, Supplementary Figures, Supplementary Discussion and Supplementary References.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gamble, N., Caldwell, J.A., McKeever, J. et al. Thymic epithelial cells amplify epigenetic noise to promote immune tolerance. Nature (2025). https://doi.org/10.1038/s41586-025-09424-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41586-025-09424-x
