
Abstract
CD8+ T cell exhaustion (Tex) limits immune control of cancer, but the underlying molecular drivers are unclear. In the present study, we identified the prostaglandin I2 (prostacyclin) receptor PTGIR as a cell-intrinsic regulator of T cell exhaustion. Transcriptomic profiling of terminally exhausted (Ttex) CD8+ T cells revealed increased activation of the nuclear factor erythroid 2-related factor 2 (NRF2) oxidative stress response pathway. Enhancing NRF2 activity (by conditional deletion of Kelch-like ECH-associated protein 1 (KEAP1)) boosts glutathione production in CD8+ T cells but accelerates terminal exhaustion. NRF2 upregulates PTGIR expression in CD8+ T cells. Silencing PTGIR expression enhances T cell effector function (that is, interferon-γ and granzyme production) and limits Ttex cell development in chronic infection and cancer models. Mechanistically, PTGIR signaling impairs T cell metabolism and cytokine production while inducing transcriptional features of Tex cells. These findings identify PTGIR as a NRF2-dependent immune checkpoint that regulates balance between effector and exhausted CD8+ T cell states.
Similar content being viewed by others
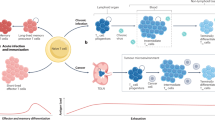


Main
Immune evasion is a hallmark of cancer. This is in part the result of progressive decline in CD8+ T cell function, referred to as T cell exhaustion, which arises due to sustained antigen exposure and immunosuppressive cues within the tumor microenvironment (TME)1. Exhausted CD8+ T (Tex) cells exhibit reduced proliferation, impaired production of effector molecules (for example, IFNγ, TNF and granzymes) and sustained expression of inhibitory receptors, including PD-1, CTLA-4 and TIM-3 (ref. 1). Immune checkpoint inhibitors (ICIs) target these inhibitory pathways to restore T cell effector activity2; however, clinical responses to ICIs are often limited3. To improve immunotherapy efficacy, a deeper understanding of the molecular programs driving CD8+ T cell exhaustion—beyond canonical checkpoint interactions such as PD-1:PD-L1—is critical.
Dysregulated metabolism is a hallmark of both cancer4 and exhausted T cells5,6. Although effector CD8+ T (Teff) cells rely on glycolysis and oxidative phosphorylation (OXPHOS)7 to fuel anabolic growth and function8,9,10, Tex cells exhibit defective mitochondrial activity and impaired OXPHOS11,12. Oxidative stress, arising from mitochondrial dysfunction or hypoxic conditions in the TME, contributes to terminal exhaustion of CD8+ T cells13,14. Reducing oxidative stress (by reducing reactive oxygen species (ROS)) can counteract chronic antigenic simulation and prevent this fate14. The antioxidant glutathione is critical for maintaining metabolic fitness and proliferation in activated CD8+ T cells15, highlighting the importance of minimizing oxidative stress to sustain effector responses.
The KEAP1–NRF2 signaling pathway regulates cellular adaptation to oxidative stress. Under homeostatic conditions, KEAP1 binds NRF2 in the cytosol, targeting NRF2 for ubiquitin-mediated proteasomal degradation16. In response to elevated ROS, KEAP1 releases NRF2 to translocate into the nucleus and activate antioxidant gene expression, such as promoting glutathione synthesis by inducing components of the glutamate cysteine ligase (GCL) complex (composed of catalytic (GCLC) and modifier (GCLM) subunits)17.
Leveraging a mouse model with conditional Keap1 deletion in T cells (Cd4-Cre; Keap1fl/fl, referred to as Keap1−/−), in the present study, we showed that NRF2 activation enhances glutathione production and reduces ROS in CD8+ T cells but, paradoxically, accelerates T cell exhaustion in chronic settings. Importantly, we identify the prostaglandin I2 receptor (PTGIR), a receptor for the eicosanoid prostacyclin, as a NRF2-dependent immune checkpoint that promotes terminal T cell exhaustion.
Results
NRF2 signaling promotes terminal exhaustion of CD8+ T cells
To define transcriptional programs associated with CD8+ T cell exhaustion, we performed a meta-analysis of published RNA sequencing (RNA-seq) data from murine CD8+ T cells isolated from Listeria monocytogenes-infected mice, lymphocytic choriomeningitis virus (LCMV)-infected mice or autochthonous liver tumors18,19,20. Based on expression of canonical markers of naive (Tn), effector (Teff) and exhausted (Tex) T cells (Fig. 1a), we conducted unsupervised gene set enrichment analysis (GSEA) to identify pathways enriched in Tex versus Teff cells (Fig. 1b and Supplementary Table 1). Among oncogenic gene sets in the MSigDB C6 collection, the NRF2 pathway emerged as the most enriched signature in Tex cells (Fig. 1b). Mechanistically, under homeostatic ROS levels, KEAP1 forms dimers that bind NRF2, targeting NRF2 for degradation (Fig. 1c). Under oxidative stress, KEAP1 thiols are covalently modified, releasing NRF2 to translocate to the nucleus and induce cytoprotective gene expression (Fig. 1c), such as genes involved in glutathione synthesis (for example, Gclc and Gclm)21.

a, Pearson’s correlation of RNA-seq data from Tn, Teff and Tex CD8+ T cells isolated from mice infected with LCMV or bearing liver tumors18,19,20. b, GSEA showing the top five oncogenic signatures (MSigDB C6) enriched in Tex versus Teff cell clusters from a. c, Schematic of KEAP1–NRF2 signaling. Oxidative stress disrupts KEAP1 stability, enabling NRF2 nuclear translocation. Keap1 deletion in T cells activates NRF2 independently of ROS. d, Immunoblots of KEAP1, NRF2 targets (HO-1, NQO1) and LAMIN B1 in CD8+ T cells and hepatocytes from WT and T cell Keap1−/− mice. e,f, Frequency of LCMV-specific (H-2Db-gp33+) CD8+ T cells in WT and Keap1−/− mice post-LCMV clone 13 (CL13) infection, as measured in blood over time (e; n = 6 mice per group), and in the spleen (f; n = 5–6 mice per group) 35 d.p.i. g,h, Expression of inhibitory receptors on gp33+ CD8+ T cells in WT and Keap1−/− mice during LCMV CL13 infection: PD-1hi cells in blood over time (g; n = 5–6 mice per group) and PD-1hiTIM-3hi cells in spleen at 35 d.p.i. (h; n = 5–6 mice per group). i, LCMV CL13 plaque assay from liver and kidney tissues at 35 d.p.i. (n = 5–6 mice per group). j, Competitive expansion of WT and Keap1−/− P14 CD8+ T cells in spleen after co-transfer into naive hosts followed by LCMV CL13 infection (n = 4 mice per group). k, Progenitor (Tpex cells; LY108+/TIM-3−) and terminally exhausted (Tex cells; LY108−/TIM-3+) Keap1−/− and WT splenic P14 cells after LCMV CL13 infection at 19 d.p.i. (n = 4–5 mice per group). l, Representative flow cytometry staining for IFNγ and TNF in PD-1+ splenic P14 cells at 19 d.p.i. with LCMV CL13 (n = 5–6 mice). Data are indicated as mean ± s.e.m. (e–i, k and l). Statistical significance was calculated via two-tailed, unpaired Student’s t-test (e–i, k and l) or two-tailed, paired Student’s t-test (j). *P = 0.05; **P = 0.01; ***P = 0.001; ****P = 0.0001.
To test the role of NRF2 in CD8+ T cell responses, we conditionally deleted Keap1 in T cells using Cd4-Cre transgenic mice (Fig. 1c). KEAP1 protein was absent in Keap1−/− CD8+ T cells, whereas the NRF2 targets Hmox1 (HO-1) and Nqo1 (NQO1) were increased compared with wild-type (WT) controls (Fig. 1d). Reverse transcription–PCR confirmed deletion of Keap1 exon 2 and increased expression of Nqo1, Hmox1 and Gclc messenger RNAs in Keap1−/− CD8+ T cells (Extended Data Fig. 1a–c and Supplementary Table 2). Gene expression of NRF2 targets was unaltered in hepatocytes from Keap1fl/fl and Cd4-Cre; Keap1fl/fl mice (Fig. 1d and Extended Data Fig. 1c), confirming T cell-specific deletion. Mice with T cell-specific Keap1 deletion showed only a modest reduction in peripheral CD3+ T cells (Extended Data Fig. 2a). The frequency of splenic CD8+ and CD4+ T cells was unchanged (Extended Data Fig. 2b,d), but we observed fewer effector T cells and also fewer CD8+ central memory T cells (Extended Data Fig. 2c–e). Frequencies of peripheral B and natural killer (NK) cells increased, and NK T cells, neutrophils and type 1 conventional dendritic cells (cDC1s) decreased with T cell-specific Keap1−/− (Extended Data Fig. 2f–h).
Our findings confirm that loss of KEAP1 leads to hyperactivation of NRF2 in CD8+ T cells (Fig. 1d), establishing a genetic model to study NRF2’s role in T cell function. We next challenged WT and Keap1−/− mice with LCMV clone 13 (CL13), a model of chronic infection that generates PD-1+ CD8+ Tex cells1. Circulating antigen-specific (H-2Db-gp33+) CD8+ T cells were significantly reduced approximately twofold in Keap1−/− mice as early as 7 d postinfection (d.p.i.), a defect that persisted through infection (Fig. 1e). Similarly, the frequency of antigen-specific CD8+ T cells in the spleen was approximately threefold lower in Keap1−/− versus WT mice at 35 d.p.i. (Fig. 1f). By 14 d.p.i., antigen-specific Keap1−/− T cells exhibited increased PD-1 expression, which persisted over time (Fig. 1g). By 35 d.p.i., ~65% of antigen-specific Keap1−/− CD8+ T cells co-expressed PD-1 and TIM-3, denoting terminal exhaustion (Fig. 1h). This correlated with poor viral control, with LCMV titers being elevated in both the kidney and the liver of T cell Keap1−/− animals compared with WT controls (Fig. 1i). These data indicate that KEAP1 loss and subsequent NRF2 activation impair antigen-specific CD8+ T cell expansion and promote a terminally exhausted phenotype.
To determine whether these effects were T cell intrinsic, we co-transferred WT and Keap1−/− P14 CD8+ T cells into congenic hosts, followed by CL13 infection. At 7 d.p.i., we observed fewer Keap1−/− P14 T cells compared with WT P14 T cells in the spleens of infected animals (Fig. 1j), confirming a cell-intrinsic defect in CD8+ T cell expansion caused by KEAP1 deficiency. Consistent with previous data, the number of Keap1−/− P14 cells were significantly reduced in the spleen at 19 d.p.i. (Fig. 1e,f,j and Extended Data Fig. 3a,b). Keap1−/− P14 cells also displayed altered Tex subset differentiation in response to LCMV CL13 infection. The proportion of progenitor exhausted cells (Tpex cells; LY108+/TIM-3−) of Keap1−/− P14 cells at 19 d.p.i. was markedly reduced, whereas the proportion of Ttex cells (LY108−/TIM-3+) increased ~20-fold compared with WT controls (Fig. 1k). Classification based on LY108 and CD69 expression22 showed a tenfold increase in LY108−/CD69+ Ttex cells in Keap1−/− mice (Extended Data Fig. 3c). Most Keap1−/− P14 T cells co-expressed PD-1 and TIM-3 (Extended Data Fig. 3d) and failed to produce IFNγ or TNF (Fig. 1l), consistent with a terminally exhausted phenotype.
Collectively, these results demonstrate that deletion of Keap1 (and subsequent NRF2 hyperactivation) drives CD8+ T cells toward dysfunction, marked by reduced expansion, diminished effector function and acquisition of a Ttex phenotype.
NRF2 activation promotes CD8+ T cell dysfunction in tumors
We next examined the impact of NRF2 hyperactivation on CD8+ tumor-infiltrating lymphocyte (TIL) function. We first examined NRF2 pathway activity in CD8+ TIL populations sorted from B16 melanoma tumors grown in C57BL/6 mice and analyzed by single-cell RNA-seq (scRNA-seq). A Uniform Manifold Approximation and Projection (UMAP) of cell-specific transcripts identified five major CD8+ T cell populations in B16 tumors based on previously reported markers23,24: proliferating (high expression of cyclin-dependent kinase 1 or Cdk1hi), progenitor exhausted (Tpex), exhausted (PD-1+ LAG3+) with expression of killer cell lectin-type receptors (TexKLR) or IFN-stimulated genes (TexISG) and PD-1+ TIM-3+ Ttex cells with elevated expression of thymocyte selection-associated high-mobility group box (Tox) (Fig. 2a and Extended Data Fig. 4a). GSEA analysis revealed that Toxhi Ttex TILs expressed the highest NRF2 signature and pathway activity score compared with all other T cell clusters (Fig. 2b,c and Extended Data Fig. 4b).

a, Weighted nearest neighbor UMAP (wnn_UMAP) of cell-specific transcripts for the five major activated (CD44+) CD8+ T cell clusters in B16 tumors (14 d.p.t.i.): Tpex, Ttex, TexKLR and TexISG cells and proliferating (prolif.) cells. Gene expression density plots for Cdk1, Pdcd1, Lag3, Tox and Havcr2 are shown. b, GSEA embedding of NRF2 transcriptional target signature (M2870) in the CD8+ TIL UMAP from a. c, Violin plot of NRF2 (M2870) pathway activity score for each CD8+ T cell cluster defined in a. d, B16-OVA melanoma tumor growth in mice after adoptive transfer (A.T.) of WT or Keap1−/− OT-I CD8+ T cells at 8 d.p.t.i. (n = 8–11 mice per group). e, PD-1 versus TIM-3 expression on WT and Keap1−/− OT-I CD8+ TILs isolated from B16 tumors at 15 d.p.t.i. (n = 5 mice per group). f,g, Histogram of PD-1 (f) and TIM-3 (g) expression on WT or Keap1−/− OT-I CD8 TILs isolated from B16 tumors as in e, with gMFI shown in the inset. the bar graphs quantify the gMFI of PD-1 and TIM-3 expression (n = 5 mice per group). h, Representative flow cytometry plot of IFNγ versus granzyme B (GZMB) expression for PD-1Hi WT and Keap1−/− TILs isolated as in e and re-stimulated with phorbol myristate acetate (PMA) plus ionomycin in vitro. i–k, Representative histograms and bar graph of gMFI (n = 5 mice per group) for IFNγ (i), GZMB (j) and TBET (k) expression by PD-1Hi WT or Keap1−/− OT-I CD8+ TILs isolated from B16 tumors as in e. l, B16-OVA melanoma tumor growth in mice after adoptive transfer of WT sgScr or WT sgNfe2l2 OT-I CD8+ T cells at 12 d.p.t.i. (n = 12–17 mice per group). m,n, Functional analysis of PD-1+ WT sgScr or WT sgNfe2l2 OT-I CD8+ TILs isolated from B16 tumors at 18 d.p.t.i. m, Representative flow cytometry plots of IFNγ versus GZMB expression for PD-1+ TILs. The bar graph quantifies the percentage of IFNγ+ GZMB+ donor TILs within tumors (n = 4 mice per group). n, Bar graphs quantifying gMFI of IFNγ, GZMB, TNF and TOX levels in PD-1+ WT sgScr versus WT sgNfe2l2 TILs (n = 4 mice per group). Data are indicated as mean ± s.e.m. (d–n). For tumor plots (d and l), statistical significance was calculated via two-way ANOVA. For all other bar plots (e–k, m and n) statistical significance was calculated via two-tailed, unpaired Student’s t-test. NS (nonsignificant), P ≥ 0.05; *P = 0.05; **P = 0.01; ***P = 0.001; ****P = 0.0001.
Given the pronounced NRF2 signature displayed by Ttex cells in tumors (Fig. 2b,c) and the increased features of exhaustion shown by Keap1−/− CD8+ T cells during chronic LCMV infection (Fig. 1), we hypothesized that NRF2 hyperactivation promotes CD8+ T cell dysfunction in tumors. To test this, we inoculated Cd4-Cre; Keap1fl/fl (Keap1−/−) and Keap1fl/fl control (WT) mice with MC38 colorectal cancer cells subcutaneously in their rear flank and tracked tumor growth. MC38 tumor growth was accelerated in T cell-specific, KEAP1-deficient mice compared with controls, as determined by accelerated tumor onset and experimental endpoint (tumor size >1,500 mm3) (Extended Data Fig. 4c). Notably, by day 40, all Keap1−/− animals had reached the experimental endpoint, whereas ~30% of control mice did not develop tumors (Extended Data Fig. 4c). Next, we assessed the impact of KEAP1 deficiency on T cell-intrinsic control of tumor growth via adoptive transfer of OT-I (CD45.2+) CD8+ T cells into mice bearing ovalbumin (OVA)-expressing B16 melanoma cells. CD45.1+ C57BL/6 mice inoculated with B16-OVA tumors received either WT or Keap1−/− OVA-specific OT-I CD8+ T cells (CD45.2+) 8 days post-tumor inoculation (d.p.t.i.). WT OT-I T cells significantly suppressed tumor growth, whereas Keap1−/− OT-I T cells failed to do so (Fig. 2d and Extended Data Fig. 4d).
We next assessed the impact of elevated NRF2 activity on the anti-tumor functions of CD8+ TILs. TILs were isolated from B16-OVA tumor-bearing mice that received either WT or Keap1−/− OT-I T cells (Fig. 2d). Mice that received Keap1−/− OT-I T cells displayed an approximately threefold increase in terminally exhausted (PD-1hi/TIM-3hi) TILs (Fig. 2e), with increased PD-1 and TIM-3 protein expression (Fig. 2f,g). Within the PD-1+ subset, Keap1−/− OT-I TILs showed reduced IFNγ, GZMB and TBET expression compared with WT controls (Fig. 2h–k), indicating impaired effector function.
To test whether reducing NRF2 expression in CD8+ T cells can improve anti-tumor T cell function, we deleted NRF2 in OT-I CD8+ T cells via CRISPR–Cas9 gene editing using single guide (sg)RNAs targeting the gene encoding NRF2 (Nfe2l2). NRF2 pathway activity, as measured by expression of the NRF2 target Hmox1, was reduced in OT-I T cells expressing Nfe2l2 sgRNAs compared with control cells expressing a scrambled sgRNA (sgScr) (Extended Data Fig. 4e and Supplementary Table 2). We next assessed the growth of B16-OVA tumors in mice that received no T cells (Hanks’ balanced salt solution (HBSS)) or adoptive transfer of control (sgScr) or NRF2-deficient (sgNfe2l2) OT-I T cells. Strikingly, tumor growth was significantly reduced in mice that received NRF2-deficient T cells compared with controls (Fig. 2l). Analysis of OT-I TILs isolated from these tumors (Fig. 2l) revealed a marked increase in effector function when NRF2 was deleted, with an ~20% increase in the number of tumor-infiltrating IFNγ+GZMB+ OT-I T cells in mice that received NRF2-deficient T cells compared with controls (Fig. 2m). Similarly, NRF2-deficient OT-I TIL from tumor-bearing mice displayed not only increased expression (that is, higher geometric mean fluorescence intensity (gMFI)) of IFNγ, GZMB and TNF, but also decreased expression of TOX—a central driver of exhaustion25 (Fig. 2n and Extended Data Fig. 4f). Collectively, these data indicate that reducing NRF2 levels improves CD8+ T cell effector function and anti-tumor control in vivo.
NRF2 drives antioxidant synthesis but promotes CD8+ T cell dysfunction
NRF2 binding is a hallmark interaction for KEAP1; however, KEAP1 also interacts with other regulators of cellular metabolism, including the autophagy receptor p62 and the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase26. To establish whether NRF2 is the mediator of exhaustion in the context of KEAP1 deficiency, we genetically ablated NRF2 in KEAP1-deficient CD8+ T cells using CRISPR–Cas9 gene editing (Supplementary Table 2)27. Proteomics analysis validated depletion of NRF2 in Keap1−/− CD8+ T cells that received sgNfe2l2 (NRF2-deficient) compared with sgScr sgRNAs (Fig. 3a and Extended Data Fig. 5a). NRF2 transcriptional activity was reduced in NRF2-deficient Keap1−/− CD8+ T cells, paralleling levels observed in control (sgScr WT) cells, as evidenced by reduced protein expression of GCLM—an enzyme that is transcriptionally regulated by NRF2 and is essential for glutathione synthesis28 (Fig. 3a and Extended Data Fig. 5a).

a, Relative abundance of NRF2 and GLCM protein levels in CRISPR–Cas9-edited Keap1−/− CD8+ T cells. WT or Keap1−/− P14 T cells were transduced with control (sgScr) or Nfe2l2-targeting (sgNfe2l2) sgRNAs and protein levels determined by proteomics (n = 3 mice per group). b, Schematic of [U-¹³C]glutamine incorporation into glutathione biosynthesis via glutaminase (GLS), GCL and glutathione synthase (GS); integration into the TCA cycle and transamination reactions are also shown. c, Isotopologue labeling (M + 5) and total pool size of glutathione, γ-glutamylcysteine and glutamine in edited CD8+ T cells. Activated CD8+ T cells of the indicated genotypes were cultured in VIM containing 0.5 mM [U-13C]glutamine for 4 h before isolation of intracellular metabolites (n = 4 replicates per group). d, DCF fluorescence in Keap1−/− (sgNfe2l2 or sgScr) and WT (sgScr) P14 CD8+ T cells (n = 4 replicates per group). e, Representative flow cytometry plots and quantification of CD45.2+ Keap1−/− (sgNfe2l2 or sgScr) and WT (sgScr) P14 T cells in the spleen of LCMV CL13-infected mice at 7 d.p.i. (n = 6–8 mice per group). f, PD-1 versus TIM-3 expression on donor CD45.2+ P14 T cells from e. The bar graph quantifies the percentage of PD-1+TIM-3+ P14 cells per genotype at 7 d.p.i. (n = 6–8 mice per group). g, TOX gMFI in PD-1+ P14 donor T cells (CD45.2+) at 7 d.p.i. from e (n = 3–4 mice per group). h, Frequency of IFNγHi cells among PD-1+ P14 donor T cells (CD45.2+) at 7 d.p.i. from e (n = 6–8 mice per group). i, IFNγ gMFI in PD-1+ P14 donor T cells from e (n = 3–4 mice per group). Data are indicated as mean ± s.e.m. in a and d–i and mean ± s.d. for technical replicates from a representative experiment (c). Statistical significance was calculated via one-way ANOVA with Tukey’s multiple comparison. NS, P ≥ 0.05; *P = 0.05; **P = 0.01; ***P = 0.001; ****P = 0.0001. Rel. pept. int., relative peptide intensity.
We next assessed the impact of silencing NRF2 on the metabolism of Keap1−/− CD8 T cells. NRF2 protects cells by regulating antioxidant defense systems, such as production of glutathione, which is synthesized de novo from glutamine, glycine and cysteine (Fig. 3b)29. We used metabolomics to track the production of glutathione from uniformly labeled [U-13C]glutamine in NRF2-deficient Keap1−/− T cells compared with control (sgScr) Keap1−/− and WT T cells. Silencing NRF2 increased intracellular glutamine in Keap1−/− T cells but had minimal impact on the contribution of [U-13C]glutamine to the tricarboxylic acid (TCA) cycle metabolites (that is, malate and citrate) (Extended Data Fig. 5b). The major metabolic impact of silencing NRF2 in Keap1−/− T cells was an approximately threefold reduction in glutathione abundance, as determined by both the percentage and the pool size of M + 5-labeled glutathione (Fig. 3c and Extended Data Fig. 5c). This reduction in glutathione production was linked to reduced GCL activity, because not only was GCLM protein expression decreased >90% (Fig. 3a), but M + 5 γ-glutamylcysteine production from [U-13C]glutamine was reduced >60% in NRF2-deficient Keap1−/− T cells (Fig. 3c and Extended Data Fig. 5c).
As glutathione serves as an antioxidant in its reduced form, we measured ROS levels in Keap1−/− and WT T cells using 2ʹ,7ʹ-dichlorodihydrofluorescein diacetate (DCFDA), a fluorogenic reporter for intracellular ROS when oxidized to 2ʹ,7ʹ-dichlorofluorescein (DCF)30. NRF2-deficient Keap1−/− CD8+ T cells showed a 30% increase in DCF fluorescence compared with controls, indicating higher ROS levels (Fig. 3d). Keap1−/− CD8+ T cells also displayed the lowest GSSG to GSH ratio (oxidized to reduced glutathione)—indicating reduced oxidative stress—compared with NRF2-deficient Keap1−/− and WT T cells (Extended Data Fig. 5d) Thus, NRF2 promotes glutathione synthesis, which restrains oxidative stress in CD8+ T cells.
We next assessed the impact of silencing NRF2 on the response of Keap1−/− T cells to chronic infection with LCMV CL13. KEAP1-deficient (Keap1−/− sgScr), NRF2-deficient Keap1−/− (sgNfe2l2) or control (WT sgScr) P14 T cells (CD45.2+ background) were transferred into congenic (CD45.1+) hosts, followed by LCMV CL13 infection 1 d later. Flow cytometry analysis of splenocytes at 7 d.p.i. revealed an approximately threefold increase in the percentage and number of P14 Keap1−/− T cells when NRF2 was deleted (Fig. 3e and Extended Data Fig. 6a). Silencing NRF2 led to both a reduction in inhibitory receptor expression and a restoration of CD8+ T cell effector function in Keap1−/− T cells. Deleting NRF2 reduced both the percentage of PD-1+ Keap1−/− P14 T cells (Extended Data Fig. 6b) and the overall PD-1 expression (Extended Data Fig. 6c) by Keap1−/− P14 T cells after LCMV CL13 infection. Moreover, there were fewer terminally exhausted (PD-1hi/TIM-3hi) Keap1−/− P14 T cells in the spleen of LCMV CL13-infected animals when NRF2 was deleted (sgNfe2l2 compared with sgScr) (Fig. 3f). Consistent with reduced features of terminal exhaustion, PD-1+ P14 Keap1−/− T cells displayed lower TOX expression and more closely resembled WT T cells on NRF2 deletion (Fig. 3g). Functionally, deleting NRF2 restored IFNγ production by PD-1+ Keap1−/− P14 T cells to levels displayed by WT P14 T cells, in terms of the percentage of IFNγ-producing cells (Fig. 3h and Extended Data Fig. 6d) and the amount of IFNγ produced per cell (Fig. 3i). The percentage of IFNγ+TNF+ PD-1+ Keap1−/− P14 T cells was similarly increased on NRF2 deletion (Extended Data Fig. 6e). However, no significant changes in expression of the TNF and transcription factors T cell factor 1 (TCF1) and T box transcription factor TBX21 (TBET) were observed across genotypes (Extended Data Fig. 6f–h). Together, these findings indicate that, although NRF2 enhances antioxidant capacity, its sustained activation skews CD8 T cells toward terminal exhaustion.
NRF2 regulates expression of the prostacyclin receptor PTGIR
To interrogate how NRF2 hyperactivation drives CD8+ T cell exhaustion, we analyzed changes in mRNA levels in Keap1−/− versus WT P14 cells after LCMV infection. WT or Keap1−/− P14 T cells (CD45.2+) were adoptively transferred into congenic CD45.1+ hosts, followed by infection with LCMV, and P14 T cells sorted via flow cytometry at 7 d.p.i. for RNA-seq analysis (Fig. 4a). Differential gene expression analysis verified that hallmark NRF2 targets including Nqo1 and Gclc were upregulated in Keap1−/− P14 T cells compared with controls, whereas WT cells displayed high expression levels of cell-division-associated genes and killer cell lectin-like receptor family genes (that is, Klra3, Klra4 and Klrk1; Fig. 4b and Supplementary Table 3). Pathway enrichment analysis identified NRF2 targets and oxidative stress response pathways as signatures of Keap1−/− P14 T cells, consistent with hyperactivation of NRF2 in these T cells (Extended Data Fig. 7a and Supplementary Table 4). Notably, the most upregulated transcript in Keap1−/− P14 cells was Ptgir (Fig. 4b), which encodes for PTGIR. PTGIR binds prostacyclin, a circulating eicosanoid family lipid derived from arachidonic acid. Using quantitative (q)PCR, we validated that Ptgir mRNA expression was increased >1,000-fold in Keap1−/− versus control P14 CD8+ T cells responding to LCMV infection in vivo (Fig. 4c). The relative induction of Ptgir mRNA in Keap1−/− P14 T cells in response to LCMV infection was greater than the conventional NRF2 target Nqo1, which was increased ~50-fold over control T cells (Fig. 4c).

a, Schematic of RNA-seq workflow for WT or Keap1−/− P14 T cells (CD45.2+) responding to LCMV Armstrong infection. P14 T cells were sorted via CD45.2+ selection at 7 d.p.i. (three biological replicates or genotypes). b, Volcano plot highlighting differentially expressed genes (DEGs) in WT and Keap1−/− P14 T cells at 7 d.p.i. with LCMV Armstrong. The x axis shows log2(fold-change) (log2(FC)) (Keap1−/− or WT) and the y axis −log10(FDR). Gray- and blue-shaded regions represent genes significantly enriched (−log10(FDR) > 1.30, corresponding to FDR < 0.05) in WT and Keap1−/− P14 T cells, respectively. Blue circles highlight conventional NRF2 targets from the NFE2L2.V2 gene set (M2870). Ptgir is shown in red. c, Relative expression levels of Nqo1 and Ptgir mRNA (normalized to Actb) in WT and Keap1−/− P14 T cells isolated from LCMV-infected mice (7 d.p.i.) as determined by qPCR (n = 3 mice per group). d,e, Relative expression of mRNA (d) and protein (e) levels for NAD(P)H quinone dehydrogenase 1 (Nqo1/NQO1) and Ptgir/PTGIR in activated WT and Keap1−/− P14 cells after CRISPR–Cas9 editing using control (sgScr) or Nfe2l2-targeting (sgNfe2l2) sgRNAs. RNA levels were normalized to Actnb. Protein levels were normalized relative to ACTN4 levels (n = 3 replicates per group). f, Data tracks for NRF2 peak enrichment at Ptgir (top) and Nqo1 (bottom) gene loci in mouse cells using NRF2 ChIP–seq. Left, NRF2 peak enrichment in Keap1−/− MEFs as determined by NRF2 ChIP–seq (accession no. GSE87357, ref. 31). Right, NRF2 peak enrichment in CD8+ T cells expressing a constitutively active form of NRF2 (CA-NRF2) (accession no. GSE224756, ref. 32). Data tracks for NRF2 ChIP–seq and input samples are shown. Data are indicated as mean ± s.e.m. (c) or mean ± s.d. (d and e) for technical replicates from a representative experiment. Statistical significance was calculated via Student’s t-test (c) or one-way ANOVA with Tukey’s test (d and e). NS, P ≥ 0.05; *P = 0.05; **P = 0.01; ***P = 0.001; ****P = 0.0001.
We next assessed whether the induction of Ptgir in Keap1−/− CD8+ T cells was dependent on NRF2. CRISPR–Cas9-mediated knockout of NRF2 in Keap1−/− T cells (sgNfe2l2) reduced expression of Ptgir mRNA (Fig. 4d) and PTGIR protein (as inferred by peptide intensity; Methods, Fig. 4e and Extended Data Fig. 7b–d) to levels observed in control (WT sgScr) T cells, indicating that PTGIR expression in Keap1−/− CD8+ T cells was dependent on NRF2 expression. To determine whether NRF2 can localize to the Ptgir gene, we first analyzed NRF2 chromatin immunoprecipitation sequencing (ChIP–seq) data from Keap1−/− mouse embryonic fibroblasts (MEFs) that display constitutive NRF2 activity31. Based on optimized predictions of NRF2-binding motifs derived from Keap1−/− and Nfe2l2−/− MEFs31, we identified a putative NRF2 consensus binding motif (5ʹ-TGACNNNGC-3ʹ) upstream of the promoter region of the Ptgir gene. NRF2 ChIP–seq of Keap1−/− MEFs revealed enrichment of NRF2 binding at this putative NRF2 consensus sequence in the Ptgir locus (Fig. 4f). NRF2 binding was also observed upstream of the promoter for Nqo1, a bona fide NRF2 target (Fig. 4f). Finally, we analyzed NRF2 ChIP–seq data from CD8+ T cells expressing a constitutively active form of NRF2 (ref. 32). Similar to Keap1−/− MEFs, NRF2 ChIP–seq revealed enrichment of NRF2 binding upstream of the promoter regions of the Ptgir and Nqo1 genes in CD8+ T cells (Fig. 4f). Collectively, these data suggest that the prostacyclin receptor PTGIR is transcriptionally regulated by NRF2 in CD8+ T cells.
NRF2-mediated PTGIR expression drives CD8+ T cell exhaustion
PTGIR is normally expressed on the surface of vascular smooth muscle cells, where it engages with prostacyclin to promote vasodilatation and platelet aggregation33. Although our scRNA-seq data indicate that NRF2 activity is enriched in CD8+ Ttex cells (Fig. 2b,c), Ptgir expression in CD8+ T cell subsets is not known. Comparing Ptgir expression versus NRF2 pathway enrichment scores for the datasets from Fig. 1a, we found a strong positive correlation (R2 = 0.92) between NRF2 pathway activity and Ptgir mRNA expression across CD8+ T cell subsets (Fig. 5a). The highest Ptgir expression was found in CD8+ TILs isolated from established tumors (Fig. 5a).

a, Correlation between NRF2 activity and Ptgir expression in CD8+ T cell subsets. Relative Ptgir mRNA expression (log2(fold-change)) versus NRF2 pathway enrichment scores (NFE2L2.V2) in CD8 T cells from L. monocytogenes infection (Tn, Teff and Tmem cells) or CD8+ TILs isolated from early (<21 d) versus late (>21 d) stage liver tumors (data from Philip et al.19). Expression values were normalized to Teff cells. Linear regression indicates goodness of fit (R2 = 0.92). b, Relative PTGIR expression in WT CD8+ T cells transduced with either an EV control or PTGIR-expressing plasmid (+PTGIR) and Keap1−/− CD8+ T cells (transduced with EV). PTGIR peptide (GFTQAIAPDSR) levels were determined by mass spectrometry and normalized to ACTN4 levels (n = 3 technical replicates). c, B16-OVA melanoma tumor growth in mice that received either no T cells (HBSS) or WT control (WT + EV), PTGIR-expressing (WT + PTGIR) or Keap1−/− (Keap1−/− + EV) OT-I T cells at 7 d.p.t.i. (n = 12 mice per group). d, Kaplan–Meier (KM) survival curves for time to tumor endpoint (>1,500 mm3) for B16-OVA-bearing mice from c. e, Relative expression of Hmox1 and Ptgir mRNA in CRISPR–Cas9-edited WT or Keap1−/− OT-I T cells. Control WT OT-I cells were edited with an sgScr and transduced with an EV construct (WT sgScr + EV). Keap1−/− OT-I cells were engineered to express control constructs (Keap1−/− sgScr + EV) or to delete NRF2 without (Keap1−/− sgNfe2l2 + EV) or with ectopic PTGIR expression (Keap1−/− sgNfe2l2 + PTGIR). The mRNA levels were determined by qPCR and normalized to Actnb levels (n = 3 technical replicates). f, B16-OVA melanoma tumor growth in mice that received (on 6 d.p.t.i.) no T cells (HBSS) or adoptive transfer of OT-I cells of the following genotypes: WT control (sgScr + EV), Keap1−/− (sgScr + EV), NRF2-deficient Keap1−/− (sgNfe2l2 + EV) or PTGIR-expressing and NRF2-deficient Keap1−/− (sgNfe2l2 + PTGIR) (n = 12–20 mice per group). g, KM survival curves for time to tumor endpoint (>1,500 mm3) for B16-OVA-bearing mice from f. The data are indicated as mean ± s.d. in b and e for technical replicates from a representative experiment. The data in c and f are indicated as mean ± s.e.m. Statistical significance was calculated via one-way ANOVA with Tukey’s test for b and e, two-way ANOVA for c and f and log(rank test) for d and g. NS, P ≥ 0.05; *P = 0.05; **P = 0.01; ***P = 0.001; ****P = 0.0001.
To examine the function of PTGIR in CD8+ T cell anti-tumor responses, we assessed the impact of ectopic PTGIR expression on the ability of OT-I CD8+ T cells to control growth of B16-OVA tumors in vivo. We expressed PTGIR in WT OT-I CD8+ T cells via retroviral transduction and confirmed PTGIR expression via proteomics (Fig. 5b, Extended Data Fig. 8a–d and Supplementary Table 2). B16-OVA tumor-bearing mice that received PTGIR-expressing OT-I T cells (WT + PTGIR) displayed increased tumor growth compared with animals that received empty vector (EV)-expressing OT-I cells (WT + EV) (Fig. 5c and Extended Data Fig. 8e). Tumor growth and overall survival for animals that received PTGIR-expressing OT-I T cells were comparable to mice that received no T cells (HBSS) or KEAP1-deficient (Keap1−/− + EV) OT-I T cells (Fig. 5c,d).
Next, we engineered Keap1−/− OT-I T cells to ablate NRF2 expression and simultaneously express PTGIR (Keap1−/− + sgNfe2l2 + PTGIR). This approach allowed us to assess the impact of PTGIR expression in Keap1−/− T cells independent of NRF2 activity. We validated this model by qPCR, establishing that NRF2 activity (via Hmox1 mRNA levels) was highest in Keap1−/− OT-I cells and reduced in Keap1−/− OT-I cells with NRF2 knockout (Fig. 5e). Knocking out NRF2 (sgNfe2l2 + EV) ablated PTGIR expression in Keap1−/− OT-I cells (Fig. 5e) and restored their ability to control B16-OVA tumor growth (Fig. 5f and Extended Data Fig. 8f). Finally, we showed that PTGIR expression limited T cell-mediated tumor controlling the absence of NRF2 signaling. Ectopic PTGIR expression (sgNfe2l2 + PTGIR) was sufficient to ablate the anti-tumor activity of NRF2-deficient Keap1−/− (sgNfe2l2 + EV) OT-I cells, with tumors growing at similar rates as in mice that received no (HBSS) or Keap1−/− (sgScr + EV) OT-I cells (Fig. 5f and Extended Data Fig. 8f). Survival of tumor-bearing mice was similar between mice that received Keap1−/− OT-I T cells and NRF2-deficient Keap1−/− cells expressing PTGIR (Fig. 5g), pointing to PTGIR as the critical determinant of NRF2 signaling in T cells that regulate tumor growth. These data establish KEAP1–NRF2 signaling as a negative regulator of anti-tumor CD8 T cell function and identify PTGIR as a critical factor downstream of NRF2 that mediates this process.
PTGIR signaling promotes metabolic and transcriptional features of Tex cells
Given the inhibitory effect of PTGIR on CD8+ T cell anti-tumor function (Fig. 5), we investigated the mechanisms by which prostacyclin signaling drives T cell dysfunction. Using RNA-seq, we investigated two complementary models of active PTGIR: WT CD8+ T cells with ectopic PTGIR expression (Fig. 6a) and Keap1−/− CD8+ T cells, which exhibit naturally elevated PTGIR expression (Fig. 6b). In vitro activated CD8+ T cells from these two models were treated with the prostacyclin mimetic iloprost (5 μM) or vehicle (dimethyl sulfoxide (DMSO)) for 24 h.

a,b, Schematic and volcano plot of WT CD8+ T cells transduced with PTGIR (WT + PTGIR) (a) or Keap1−/− CD8+ T cells treated with vehicle (veh.; DMSO) or iloprost (ilo.) 5 µM for 24 h (b). DEGs are shown as log2(fold-change) (x axis) versus −log₁₀(FDR) (y axis). Shaded regions denote significant enrichment (FDR < 0.05) in iloprost- or vehicle-treated cells. c,d, Normalized enrichment scores (NESs; q < 0.05) of the top ten Hallmark pathways downregulated by iloprost treatment in WT + PTGIR (c) and Keap1−/− T cells (d). e,f, NESs of the top ten Hallmark pathways upregulated by iloprost in WT + PTGIR (e) and Keap1−/− CD8+ T cells (f). g, Relative growth and viability of activated WT and Keap1−/− CD8+ T cells cultured with vehicle or iloprost (5 µM) for 24 h. Cell counts for growth assays were normalized to initial seeding density (n = 3–4 mice per group). h, Bioenergetic profiling of WT and Keap1−/− CD8+ T cells after 24 h of vehicle (+veh) or iloprost (+ilo) treatment. OCR (left) and ECAR (right) for T cells over time are shown (n = 8–12 technical replicates per group). Time of addition of oligomycin (O), fluorocarbonyl cyanide phenylhydrazone (FCCP) (F), rotenone and antimycin A (R/AA) and monensin (M) are indicated. i, NESs of Tex cell transcriptional signatures (accession no. GSE188670, ref. 23) in iloprost-treated Keap1−/− T cells using DEG values from b. Heatmap shows expression of TexISG genes. Inset highlights top contributing genes to the TexISG signature in iloprost-treated Keap1−/− T cells. j,k, Cytokine production by WT and Keap1−/− CD8+ T cells after 24 h of treatment with vehicle control or iloprost (5 μM). j, Representative flow cytometry plots for IFNγ versus TNF expression for CD8+ T cells. k, Quantification of IFNγ+/TNF+ T cells after treatment (n = 4 mice per group). The data in h are indicated as mean ± s.d. and display technical replicates from a single representative experiment. The data in g, j and k are indicated as mean ± s.e.m. Statistical significance was calculated via one-way ANOVA with Tukey’s test. NS, P ≥ 0.05; *P = 0.05; **P = 0.01; ***P = 0.001; ****P = 0.0001. Cholest. homeost.; cholesterol homeostasis; FA, fatty acid; Inflamm., inflammatory; rej., rejection; resp., response; sig., signaling.
Ectopic PTGIR expression in WT T cells (WT + PTGIR) had minimal impact on gene expression (Extended Data Fig. 9a and Supplementary Tables 5 and 6). Although iloprost treatment caused few transcriptional changes in WT and vector-only (WT + EV) T cells (Extended Data Fig. 9b,c and Supplementary Tables 5 and 6), iloprost triggered broad transcriptional remodeling in PTGIR-expressing WT T cells (2,414 upregulated and 1,849 downregulated genes) and Keap1−/− T cells (434 up, 213 down; Fig. 6a,b, Extended Data Fig. 9d and Supplementary Tables 5 and 6). Shared downregulated pathways included cholesterol, sterol and secondary alcohol biosynthesis, whereas shared upregulated pathways included cyclic 3′,5′-AMP (cAMP) and nucleotide phosphodiesterase activity (Extended Data Fig. 9d and Supplementary Table 6). This is consistent with PTGIR’s known function as a suppressor of intracellular cAMP signaling in platelets and smooth muscle cells33, which impairs DNA binding of SREBP2, a key transcription factor controlling cholesterol homeostasis34.
GSEA of Hallmark pathways revealed that iloprost suppressed programs linked to CD8+ T cell proliferation (E2F targets, G2M checkpoint, mitotic spindle), anabolic growth (MYC, MTORC1) and energy metabolism (OXPHOS, glycolysis) in both models (Fig. 6c,d and Supplementary Table 6). In contrast, inflammatory response pathways such as TNF signaling via NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) and interleukin 6 (IL-6)/JAK/STAT3 signaling were upregulated (Fig. 6e,f and Supplementary Table 6). As iloprost dampened transcription of genes involved in DNA replication and cell division, we tested its impact on proliferation. Iloprost selectively impaired expansion of PTGIR-expressing Keap1−/− T cells in vitro, probably as a result of effects on cell viability (Fig. 6g). This could be explained by disrupted cellular metabolism: iloprost reduced both oxygen consumption rate (OCR; OXPHOS) and extracellular acidification rate (ECAR; glycolysis) of PTGIR-expressing Keap1−/− T cells, leading to an ~50% decrease in total ATP output (Fig. 6h and Extended Data Fig. 9e).
To explore the effect of PTGIR signaling on the T cell state, we compared transcriptomes of iloprost-treated cells with gene signatures derived from antigen-specific CD8+ T cells during LCMV CL13 infection23. Proliferative signatures (Prolif. I and II) were diminished in Keap1−/− T cells after iloprost exposure (Extended Data Fig. 9f), consistent with the observed reduction in T cell proliferation (Fig. 6g). Strikingly, iloprost induced expression profiles characteristic of terminally exhausted T cells, including TexISG and Ttex cells (Fig. 6i and Extended Data Fig. 9g). GSEA of additional CD8+ T cell datasets9,24 further confirmed enrichment of IFN-stimulated gene (ISG) programs in iloprost-treated Keap1−/− T cells (Extended Data Fig. 9f). Iloprost elevated canonical ISGs, such as Isg15, Isg20 and Ifit1 and increased expression of IFN-I signaling mediators, including Ifi204, Ifi209, Stat1, Stat2, Socs1 and Irf9 (Fig. 6i)35,36. Notably, Cd274, encoding PD-L1, was also robustly upregulated after PTGIR stimulation (Fig. 6i).
We next assessed how PTGIR signaling affected cytokine production. Consistent with previous findings showing that NRF2 can boost cytokine production37, Keap1−/− CD8+ T cells produced more IFNγ and TNF than WT controls under in vitro activating conditions (Fig. 6j). However, activation of PTGIR signaling with iloprost significantly reduced cytokine production by Keap1−/− CD8+ T cells in vitro (Fig. 6j,k). CD8+ T cells with low PTGIR levels were refractory to iloprost treatment (Fig. 6j,k). Together, these findings demonstrate that PTGIR signaling suppresses CD8+ T cell function by reprogramming transcriptional and metabolic states, promoting features of terminal exhaustion and reducing effector cytokine production.
PTGIR is a checkpoint for CD8+ T cell function
Given that increasing PTGIR expression is sufficient to blunt CD8 T cell anti-tumor responses (Fig. 5), we assessed whether PTGIR functions as an immune checkpoint. We silenced Ptgir expression in P14 T cells (CD45.2+) using short hairpin RNAs (shRNAs) targeting Ptgir (Supplementary Table 2), adoptively transferred these cells into congenic (CD45.1+) hosts, then challenged mice with LCMV CL13 infection. At 7 d.p.i., flow cytometry of P14 cells revealed greater expansion of Ptgir-silenced cells compared with controls (Fig. 7a and Extended Data Fig. 10a). Silencing Ptgir also reversed several features of T cell dysfunction in vivo, including a reduced frequency of PD-1+ or TIM-3+ cells (Fig. 7b and Extended Data Fig. 10b) and increased production of IFNγ by PD-1+ T cells (Fig. 7c). Moreover, PD-1+ Ptgir-silenced cells were more polyfunctional, displaying higher co-expression of IFNγ with GZMB (Fig. 7d) and TNF (Extended Data Fig. 10c) than shCtrl-expressing counterparts.

a, Representative flow cytometry plots of CD8+ versus CD45.2 expression on splenocytes harvested from LCMV CL13-infected mice (7 d.p.i.) that received P14 CD8+ T cells expressing control (shCtrl) or Ptgir-targeting (shPtgir) shRNAs. The percentage of donor CD45.2+ P14 T cells of total CD8+ splenocytes at 7 d.p.i. is quantified (n = 4–5 mice per group). b, PD-1 versus TIM-3 expression by donor CD45.2+ P14 T cells (shCtrl versus shPtgir) after LCMV CL13 infection from a. The percentage of PD-1hiTIM-3hi CD45.2+ P14 T cells in the spleen of infected mice at 7 d.p.i. is shown (n = 4–5 mice per group). c, IFNγ expression (gMFI) for PD-1hi shCtrl- or shPtgir-expressing CD45.2+ P14 T cells at 7 d.p.i. from a. d, IFNγ versus GZMB levels in shCtrl- or shPtgir-expressing CD45.2+ P14 T cells at 7 d.p.i. from a. Left, representative flow cytometry plots for IFNγ and GZMB expression by PD-1hi P14 CD8+ T cells. Right, bar graph quantifying the percentage and number of polyfunctional (IFNγ+GZMB+) PD-1hi P14 T cells in the spleen at 7 d.p.i. (n = 5 mice per group). e,f, Growth of MC38-OVA (e) and B16-OVA (f) tumors in mice with no T cell transfer (HBSS) or adoptive transfer of shCtrl- or shPtgir-expressing OT-I T cells at 7 d.p.t.i. (n = 8–12 mice per group). g–i, Functional analysis of shCtrl- or shPtgir-expressing OT-I TILs from B16-OVA tumors in f: representative histograms and bar graphs of PD-1 (g), TCF1 (h) and TOX (i) expression levels (gMFI) in shCtrl- or shPtgir-expressing OT-I TILs from B16-OVA tumors (n = 4–5 mice per group). gMFI levels are shown inset. j, IFNγ versus TNF expression in shCtrl- or shPtgir-expressing OT-I TILs from B16-OVA tumors in f. The bar graph quantifies the percentage of IFNγ+TNF+ OT-I TILs from B16-OVA tumors (n = 4–5 mice per group). The data are indicated as mean ± s.e.m. The statistical significance was calculated via two-tailed, unpaired Student’s t-test (a–d and g–j) and two-way ANOVA (e and f). NS, P ≥ 0.05; *P = 0.05; **P = 0.01; ***P = 0.001; ****P = 0.0001.
Finally, we assessed the impact of Ptgir knockdown on CD8+ T cell-mediated, anti-tumor immunity in the context of adoptive cell therapy. WT OT-I CD8+ T cells were transduced with control (shCtrl) or Ptgir-targeting (shPtgir) shRNAs, followed by adoptive transfer into mice bearing MC38-OVA or B16-OVA tumors. Silencing PTGIR in OT-I T cells resulted in a robust attenuation of tumor growth and increased overall survival compared with tumor-bearing mice that received control OT-I T cells for both MC38 (Fig. 7e and Extended Data Fig. 10d,e) and B16 (Fig. 7f and Extended Data Fig. 10f,g) tumor models.
Analysis of TILs from B16-OVA tumors revealed reduced PD-1 and TOX expression and increased TCF1 expression in PTGIR-silenced OT-I TIL compared with control cells (Fig. 7g,i), suggesting a shift in CD8+ TILs toward a more functional ‘progenitor exhausted’ state when PTGIR expression was reduced. Consistent with this, silencing PTGIR in OT-I T cells boosted the percentage of polyfunctional T cells in B16 tumors, marked by a twofold increase in the percentage of IFNγ+ or TNF+ CD8+ TILs compared with controls (Fig. 7j). Collectively, our data demonstrate that reducing PTGIR expression in CD8 T cells promotes increased effector function and reduced exhaustion, leading to more robust tumor control.
Discussion
Altered cellular metabolism is a hallmark of exhausted T cells in cancer5,6. Yet, how metabolism influences T cell fate between functional and dysfunctional states is not fully understood. In the present study, we identified the prostacyclin receptor PTGIR as a downstream mediator of KEAP1–NRF2 signaling that drives terminal exhaustion in CD8+ T cells. Transcriptomic analyses of CD8+ T cells from chronic infection and tumors revealed elevated NRF2 activity in terminally exhausted (PD-1⁺ or TIM-3⁺) cells. Genetic ablation of NRF2 restored anti-tumor T cell responses, whereas enforced expression of PTGIR—even in NRF2-deficient cells—promoted T cell dysfunction and poor tumor control. Mechanistically, PTGIR limited T cell proliferation, cytokine production and mitochondrial metabolism, while inducing transcriptional programs of terminal exhaustion. Conversely, silencing PTGIR enhanced effector responses and reduced tumor growth in adoptive cell therapy models, establishing PTGIR as a NRF2-regulated immune checkpoint.
Although NRF2 activation via metformin38, auranofin39 or enforced expression of constitutively active NRF2 (ref. 37) enhances CD8+ T cell function in vitro, we found that chronic NRF2 activation via KEAP1 loss leads to T cell dysfunction in vivo. Unlike drug-induced ROS responses, KEAP1 loss causes persistent NRF2 signaling and increased glutathione synthesis via GCLC, GCLM and GSS17. Despite mitigating oxidative stress, persistent NRF2 signaling paradoxically promotes terminal exhaustion—suggesting that NRF2-mediated metabolic adaptations to stress cannot alone preserve T cell effector function.
Among NRF2 targets, Ptgir was the most upregulated gene in Keap1−/− CD8+ T cells and its expression was NRF2 dependent both in vitro and in vivo. Activating PTGIR signaling in CD8+ T cells with the prostacyclin mimetic iloprost reduced ATP production from both OXPHOS and glycolysis, which may contribute to both secondary effects on cholesterol biosynthesis—also downregulated by iloprost—and T cell dysfunction40. A key finding from our work is that PTGIR signaling is sufficient to promote gene signatures of terminal exhaustion in the absence of chronic stimulation. Keap1−/− T cells exposed to iloprost in vitro displayed prominent features of TexISG cells, marked by genes involved in IFN-I production (that is, Ifi204 and Ifi209) and signaling (Stat1, Stat2 and Irf9). Type I IFNs are essential for antiviral immunity but also contribute to immune evasion in cancer by promoting T cell exhaustion41. Enrichment of ISGs within CD8+ Tex cells correlates with reduced effector function and poor responses to ICI therapy42,43. Thus, PTGIR may promote T cell dysfunction in part through changes in IFN signaling.
Importantly, our data suggest that exposure to prostacyclin in the TME may—via PTGIR—drive transcriptional reprogramming of CD8+ T cells toward a dysfunctional or exhausted phenotype, independent of tumor antigens. This aligns with reports that elevated levels of 6-keto-prostaglandin F1α—a stable breakdown product of prostacyclin—have been measured in advanced-stage tumors, including pancreatic ductal adenocarcinoma, which are commonly resistant to immunotherapy44. In addition, scRNA-seq analyses show that cancer-associated fibroblasts express prostacyclin synthase (PTGIS) in both murine and human pancreatic ductal adenocarcinoma44, suggesting that prostacyclin production by stromal cells may suppress local immunity in the TME. Last, PTGIR may also regulate CD4⁺ Treg cell function by enhancing FOXP3 and IL-10 expression45, further enforcing its immunosuppressive potential. Other eicosanoids beyond prostacyclin—such as prostaglandin E2—similarly suppress CD8+ T cells through EP2 or EP4 signaling46,47,48. In chronic infection and tumor models, genetic deletion or pharmacological blockade of the prostaglandin signaling axis restored CD8+ T cell function and reduced tumor burden. These results, combined with our data linking prostacyclin signaling to suppression of anti-tumor T cell responses, support a broader role for lipid-derived signaling molecules in tuning T cell fate in the TME.
Together, our findings establish PTGIR as a lipid-sensing immune checkpoint and position KEAP1–NRF2 signaling as a key axis in the regulation of CD8+ T cell dysfunction in cancer. Given that up to 70% of patients do not respond to current ICIs3, targeting the KEAP1–NRF2–PTGIR axis may offer new therapeutic options for immunotherapy. Potential strategies to reduce the suppressive effects of prostacyclin signaling on T cell function include PTGIS inhibition, PTGIR-blocking antibodies or silencing PTGIR expression in CAR-T cell products. Identifying patterns of NRF2 and PTGIR expression in human TILs may help stratify patients likely to benefit from such interventions.
Methods
RNA-seq meta-analysis
Raw sequences from RNA-seq of CD8+ T cells from three previously published studies (Gene Expression Omnibus (GEO); accession nos. GSE89307, GSE84820 and GSE86881) were downloaded and analysis was conducted as described in the methods section by Luda et al.9, with the following exceptions: differential gene expression analyses of all Tex (early, intermediate, late) cells versus Teff cells were conducted on raw counts using DESeq2, with a covariate to adjust for batch, and Benjamini–Hochberg adjusted P (Padj) values to maintain a 5% false discovery rate (FDR). Analysis was conducted by the Van Andel Institute (VAI) Bioinformatics and Biostatistics Core (RRID:SCR_024762).
Mouse models
All experiments were performed using either male or female C57BL/6J background mice aged 8–16 weeks, weighing between 18 g and 25 g. Mice were maintained under specific pathogen-free conditions at the VAI under approved protocols. Mice were housed under a 12-h light:dark cycle with continuous access to water and standard 5010 chow provided freely (LabDiet). Genotyping was performed using DNA extracted from tail or ear biopsies. C57BL/6J, CD90.1 (Thy1.1+), B6.SJL-Ptprca Pepcb/BoyJ (CD45.1+), Tg(TcraTcrb)1100Mjb (OT-I) and B6.Cg-Tg(Cd4-Cre)1Cwi/BfluJ mice (Cd4-Cre) were purchased from the Jackson Laboratory (JAX) and B6(Cg)-Keap1tm1.1Sbis/J (Keap1flox/flox or Keap1fl/fl) mice were purchased from Taconic Biosciences. Under the VAI Institutional Animal Care and Use Committee (IACUC)-approved protocols, Keap1fl/fl mice were crossed to Cd4Cre mice, followed by subsequent breeding of Keap1fl/+ heterozygotes to generate a stable Cd4-Cre; 4Keap1fl/fl mouse colony. Cd4-Cre; Keap1fl/fl mice were crossed to P14 or OT-I mice followed by subsequent breeding of heterozygotes to generate stable Cd4-Cre; Keap1fl/fl P14 or Cd4-Cre; Keap1fl/fl OT-I colonies. No statistical methods were used to predetermine sample sizes for experiments with these models, but our sample sizes are similar to those reported in previous publications8,10. For mouse experiments, data distribution was assumed to be normal but this was not formally tested. Mouse husbandry was facilitated by the VAI Vivarium (RRID:SCR_023211).
RNA isolation, sequencing and qPCR analysis
Total RNA was isolated from murine T cells via RNeasy Kit (QIAGEN, cat. no. 74104) with DNase digestion (QIAGEN, cat. no. 79254) following the manufacturers’ instructions. For qPCR analysis, total RNA was reverse transcribed using a High-Capacity cDNA Reverse Transcriptase kit (Life Technologies, cat. no. 4368814) and qPCR performed using universal SYBR green supermix (BioRad, cat. no. 1725150). Results were normalized to Actb mRNA levels and control conditions using standard ∆∆Ct methods. A list of qPCR primers used in the present study can be found in Supplementary Table 2. RNA preparation and library construction for RNA-seq was conducted by the VAI Pathology and Biorepository Core (RRID:SCR_022912) and VAI Genomics Core (RRID:SCR_022913) as previously described49. Libraries were sequenced using a NovaSeq 6000 (Illumina) with 50 bp paired-end sequencing (5 × 107 reads per sample).
Immunoblotting
Protein samples were prepared according to the methods section in ref. 9. Membranes were blocked for 1 h in 5% nonfat milk in 1× Tris-buffered saline with Tween 20 (TBST) at room temperature (RT) and incubated with primary antibodies against KEAP1 (Cell Signaling, cat. no. 8047, 1:500 v:v), NQO1 (Proteintech, cat. no. 11451-1-AP), HMOX1 (Proteintech, cat. no. 66743-1-ig) and LAMIN B1 (Cell Signaling, cat. no. 13435) overnight at 4 °C. Membranes were washed 3× for 5 min with 1× TBST and then incubated for 1 h at RT with the corresponding anti-rabbit immunoglobulin G (IgG; Cell Signaling, cat. no. 7074) or anti-mouse IgG (Cell Signaling, cat. no. 7076), horseradish peroxidase (HRP)-conjugated secondary antibody diluted in 5% nonfat milk. Membranes were washed 3× with 1× TBST and then developed using ECL solution (Thermo Fisher Scientific, cat. no. P132106). All primary and secondary antibodies were used at 1:1,000 (v:v) dilution unless otherwise stated.
Flow cytometry
Mice were sacrificed and spleens were obtained—or blood was obtained via submental bleed of live mice—and lymphocytes were isolated using red blood cell (RBC) lysis buffer containing 0.15 M NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA, followed by neutralization with 3 volumes of TCM. Lymphocyte suspensions were surface stained with a cocktail of fluorescently labeled antibodies: GP33 allophycocyanin (APC) tetramer (customized order, Baylor College of Medicine), KLRG1 AF532 (eBioscience, cat. no. 58-5893-80, clone 2F1), CX3CR1 BB700 (BD Biosciences, cat. no. 149036, clone SA011F11), CD8A BUV395 (BD Biosciences, cat. no. 563786, clone 53-6.7), CD44 BV650 (BioLegend, cat. no. 103049, clone 1M7), CD127 BV785 (BioLegend, cat. no. 135037, clone A7R34), TIM-3 BV711 (BioLegend, cat. no. 119727, clone RMT3-23), PD-1 BUV737 (BD Biosciences, cat. no. 749306, clone RMPI-30), IFNγ FITC (eBioscience, cat. no. 17-7311-82, clone XMG1.2), TNF phycoerythrin (PE)-Cy7 (eBioscience, cat. no. 35-7321-83, clone MP6-X1222), TOX PE (Miltenyi, cat. no. 130-120-785, clone REA473), TCF1 BV421 (BD Biosciences, cat. no. 566692, clone S33-966 RUO), Granzyme B PE/Dazzle594 (BioLegend, cat. no. 372216, clone QA16A02), TBET BV605 (BioLegend, cat. no. 644817, clone 4B10), CD45.2 BUV805 (BD Biosciences, cat. no. 741957, clone 104) and Thy1.1 APC (eBioscience, cat. no. 17-0900-82, clone HIS51). Cell viability was assessed using Fixable Viability or FVD eFluor 780 (Invitrogen, cat. no. 65-0865-18) according to the manufacturer’s protocols. To assess cytokine production, splenic preps or tumor preps were incubated (37 °C, 5% CO2 incubator) in TCM in the presence of 1 μg ml−1 of GP3333–41 (KAVYNFATM) peptide (Anaspec, cat. no. AS-61669), phorbol myristate acetate (PMA; 50 ng ml−1) or ionomycin (500 ng ml−1), respectively, for 4 h. GolgiStop protein transport inhibitor (1:1,500 dilution; BD Biosciences, cat. no. 554724) was added for the last 2 h of stimulation before surface staining. After re-stimulation, cells were surface stained, fixed and permeabilized using Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, cat. no. 00-5523-00), followed by intracellular staining using fluorescently labeled antibodies. Flow cytometry was performed on Cytoflex (Beckman Coulter) or Aurora Cytek cytometers and cell sorting on Astrios (Beckman Coulter) or BD FACSAria Fusion cell sorters. Flow cytometry analysis was performed using FlowJo software (Tree Star). VAI Flow Cytometry Core (RRID:SCR_022685).
LCMV preparation and infection
For preparing viral particles, 10 × 106 BHK21 cells (American Type Culture Collection (ATCC), cat. no. CCL-10) in a T-175-cm2 flask were infected with either LCMV CL13 or Armstrong virus stocks at a multiplicity of infection of 0.1 in 5 ml BHK21 medium (see formulation below) and incubated at 37 °C, 5% CO2 for 1 h, rocking every 15 min. The supernatant was then removed and replaced with 15 ml of fresh medium. Cells were next incubated for 48 h at 37 °C, 5% CO2 and medium, collected and centrifuged at 500g for 10 min and 4 °C. The supernatant was then aliquoted into fresh tubes and frozen at −80 °C until further use after quantification of plaque. BHK21 medium formulation: 1% (v:v) of 200 mM l-glutamine (Gibco, cat. no. 25030164), 2.8% (v:v) 20% glucose (Thermo Fisher Scientific, cat. no. A2494001), 7% tryptose phosphate broth (Thermo Fisher Scientific, cat. no. 18050039), 1% (v:v) sodium pyruvate (Thermo Fisher Scientific, cat. no. 11360070) and 1% (v:v) penicillin–streptomycin (pen/strep; Gibco, cat. no. 15070063). LCMV Armstrong and LCMV CL13 were diluted to 2 × 105 and 2 × 106 plaque-forming units, respectively, in 1× d-PBS (Wisent, cat. no. 311-425) and administered intraperitoneally (Armstrong) or via the tail vein (CL13) into host mice.
LCMV plaque assays
Kidneys or liver lobes were weighed and homogenized in 1 ml of extraction buffer (HBSS + 2% FBS + 1% pen/strep) with a stainless-steel bead using a QIAGEN TissueLyser (30 Hz, 3 min × 2 rounds). Homogenates were centrifuged at 17,000g for 10 min and supernatants were transferred to fresh tubes on ice (1–2 h). Extracts were serially diluted (1:10 to 1:333) in Dulbecco’s modified Eagle’s medium (DMEM) + 2% FBS to 300 µl total. For infection, 200 µl of diluted extract was added to 24-well plates, followed by 200 µl of MC57G cells (1.4 × 10⁵ cells per well) in DMEM with 10% FBS, 1% pen/strep, 1% l-glutamine and 0.1% 2-mercaptoethanol. Plates were tapped gently and incubated at 37 °C, 5% CO₂ for 3–6 h to allow monolayer formation. Then, 200 µl of pre-warmed 2× DMEM with 2% methylcellulose was added per well and incubated for 48 h. After incubation, wells were washed with HBSS, fixed in 4% formalin (in PBS) for 20 min, then permeabilized with 0.5% Triton X-100 in HBSS for 20 min. For staining, cells were blocked with 10% FBS in PBS (60–90 min at RT), followed by incubation with VL-4 anti-LCMV antibody (1:10 in PBS + 1% FBS, 30–60 min, RT) and three PBS washes. Secondary staining was done using HRP-conjugated goat anti-rat antibody (1:200 in PBS + 1% FBS, 30–60 min, RT), followed by three PBS washes. Plaques were developed using 200 µl of o-phenylenediamine dichloride (OPD)-based color reaction buffer (0.1 M citric acid, 0.2 M Na2HPO4·2H2O, OPD and 100 µl of 30% H2O2), incubated for 10–30 min. Visible plaques were counted and normalized to tissue weight and dilution to calculate the plaque-forming units.
CD8+ T cell purification and culture
P14, OT-I or total CD8+ T cells were purified from spleen and peripheral lymph nodes by negative selection using the EasySep Mouse CD8+ T cell isolation kit (STEMCELL Technologies, cat. no. 19753), as previously described.
Cell lines
Cells, 293T (ATCC, cat. no. CRL-3216), B16-OVA (Sigma-Aldrich, cat. no. SCC420) and MC38-OVA (provided by J. Stagg), were cultured in DMEM supplemented with 10% v:v FBS, 1% v:v pen/strep and l-glutamine at a final concentration of 6 mM. All cells were cultured at 37 °C in a humidified 5% CO2 incubator.
Retrovirus and lentivirus production and T cell transduction
For retrovirus production corresponding to PTGIR knockdown in CD8 T cells, 293T cells were first co-transfected with pCL-Eco and either pLMPd-Ametrine shFF (control vector, shCtrl) or pLMPd-Ametrine shPtgir (Supplementary Table 2)—developed according to the cloning pipeline by Dow et al.50—using Lipofectamine 2000 transfection reagent (Invitrogen, cat. no. 11668019). Viral supernatants were harvested 48 and 72 h after transfection, pooled and concentrated using Lenti-X Concentrator (Takara, cat. no. 631232), according to the manufacturer’s protocol. The concentrated retrovirus was added to 24-h-activated T cells with 8 ng ml−1 of polybrene (Sigma-Aldrich. cat. no. TR-1003), 200 U ml−1 of IL-2 (Thermo Fisher Scientific, cat. no. 212-12) and 20 mM Hepes, and the cells were centrifuged at 1,180g, 30 °C for 90 min. Ametrine-positive cells were sorted by FACS 72 h after transduction, cultured for 48 h in T cell medium, phenotypically validated for PTGIR knockdown and adoptively transferred into LCMV CL13-infected or tumor-bearing hosts. For retrovirus production corresponding to PTGIR overexpression in CD8+ T cells, 293T cells were first co-transfected with pCL-Eco and either MIGR1 (green fluorescent protein (GFP) control vector, kindly provided by W. Pear; Addgene, cat. no. 27490) or MIGR1-PTGIR (customized PTGIR-overexpressing vector, VectorBuilder), using Lipofectamine 2000 transfection reagent and subsequent procedures as mentioned above, with the following exception: 72 h after transduction GFP+ cells were flow sorted, cultured for 48 h in T cell medium, phenotypically validated for PTGIR overexpression and adoptively transferred into tumor-bearing hosts.
B16-OVA and MC38-OVA tumor models
Male or female 8- to 12-week-old Cd4-Cre; Keap1fl/fl (Keap1−/−) mice or Keap1fl/fl (WT) control littermates were injected with 5 × 105 MC38-OVA cells or 5 × 105 B16-OVA cells subcutaneously in the right abdominal flank. Once palpable tumors were present, tumor measurements were obtained every 2–3 d using calipers. Mice were euthanized as they reached humane endpoints, which included a maximum tumor volume ≥1,500 mm3 or when tumors became necrotic. For adoptive transfer experiments, mice were inoculated with 5 × 105 MC38-OVA or B16-OVA cancer cells and, on day 7 postinoculation, when palpable tumors were present, 1 × 106 OT-I T cells were adoptively transferred into tumor-bearing hosts. All tumor experiments were conducted according to the VAI IACUC-approved protocols in conjunction with the VAI Vivarium (RRID:SCR_023211).
TIL isolation
On day 7 after adoptive transfer (days 14–15 after cancer inoculation), mice were sacrificed and respective B16-OVA tumors were measured and excised using a scalpel. In a 6-well plate, tumors were homogenized and passaged 3× through a 100-μm filter, followed by a 40-μm filter. Tumor extracts were incubated with RBC lysis buffer for 1 min at RT and three volumes of TCM were added on top to neutralize the RBC lysis reaction. TILs were then centrifuged at 500g for 5 min, medium was aspirated and the TILs resuspended in 0.5–1 ml of TCM and used for downstream flow cytometry experiments.
CRISPR–Cas9-mediated gene editing
Electroporation of WT and Keap1−/− P14 and OT-I CD8+ T cells was adapted from the method developed by Nussing et al.27. First, antigen-specific (P14 or OT-I) CD8+ T cells were isolated from respective mice, assessed for purity (>95%) and activated for 48 h with plate-bound antibodies in the same fashion as in the above CD8+ T cell purification and culture section. After 48 h of culture, cells were counted and 2.5 × 106 cells were centrifuged at 500g for 5 min and washed twice with PBS. During the washes, 40 pmol of SpCas9 2NLS (Synthego) was complexed with 300 pmol of sgRNA (Synthego; Supplementary Table 2) in a total volume of 5 μl for 10 min. After the final PBS wash, cells were resuspended in 20 μl of P3 buffer mix (Lonza, cat. no. V4XP-3032) according to the manufacturer’s protocol and mixed with the 5 μl of pre-incubated Cas9–sgRNA ribonucleoprotein (RNP) complex. The cell–RNP mixture was then immediately added to a 16-well nucleofector strip and electroporated using program DN-100 of 4D-nucleofector X system (Lonza, cat. no. AAF-1003X). After electroporation, the cell–RNP mixture was topped with 130 μl of pen/strep-free TCM, then placed in a final volume of 2.5 ml of (pen/strep free) TCM containing 50 U ml−1 of IL-2 in a 12-well plate, and incubated at 37 °C (5% CO2) overnight. The following day, medium was changed to fresh TCM containing pen/strep and 50 U ml−1 of IL-2. Cells were moved to a 6-well plate and diluted to 0.4 × 106 cells ml−1 and cultured for 48 h. Knockout efficiency was assessed via qPCR for NRF2 target genes or via proteomics for PTGIR.
[13C]Glutamine stable isotope labeling
After initial T cell isolation, activation for 2 d and expansion in TCM for 4 d, WT or Keap1−/− CD8+ T cells were traced according to the method developed by Kaymak et al.8 and Luda et al.9. For 13C tracing, cells were resuspended in 0.8 ml of Van Andel Institute-modified Iscove’s Medium (VIM), (Wisent, cat. no. 319-266-CL) as a base solution, supplemented with 10% (v:v) dialyzed FBS, 1% (v:v) pen/strep and 0.1% (v:v) 2-mercaptoethanol and 12C-labeled isotopologues of 5 mM glucose, 0.1 mM glycine, 0.2 mM alanine, 0.01 mM aspartate, 0.02 mM glutamate, 0.04 mM methionine, 0.1 mM serine and 0.1 mM threonine. For [U-13C]glutamine (Cambridge Isotope Laboratories, cat. no. CLM-1822) tracing, 0.2 ml of 5× base solution containing 5 mM [U-13C]glutamine (0.5 mM final concentration)—containing all 12C-labeled metabolites at aforementioned concentrations with 250 U ml−1 of IL-2—was added to top the 0.8-ml cell suspension and incubated for 4 h under standard culture conditions.
13C-labeled metabolomics analysis
Metabolites were extracted in ice-cold acetonitrile:methanol:water (40%:40%:20%, v:v:v) at a concentration of 2 × 106 cells ml−1. Crude extracts were vortexed for 30 s, sonicated in a water bath sonicator for 5 min and incubated on wet ice for 1 h. Extracts were precipitated by centrifugation at 14,000g for 10 min at 4 °C and 760 µl of the soluble supernatant (1.52 × 106 cell equivalents) was dried in a vacuum evaporator. Dried extracts were resuspended in 50 µl of LCMS-grade water (water for chromatography), resulting in a final concentration of 3.0 × 107 cell equivalents ml−1. Stable isotope tracing metabolomics data were collected on an Orbitrap Exploris 240 mass spectrometer (Thermo Fisher Scientific) using a tributylamine ion-paired reversed-phase chromatograph. The column was a ZORBAX Rapid Resolution HD (2.1 × 150 mm2, 1.8 µm; Agilent, cat. no. 759700–902). Mobile phase A was 3% methanol (A) and mobile phase B was 100% methanol (B). Both mobile phases contained 10 mM tributylamine (Sigma-Aldrich, cat. no. 90780), 15 mM acetic acid and 2.5 µM medronic acid (Agilent Technologies, cat. no. 5191–4506). The LC (liquid chromatography) gradient was: 0–2.5 min 0% B, 2.5–7.5 min ramp to 20% B, 7.5–13 min ramp to 45% B, 13–20 min ramp to 99% B and 20–24 min hold at 99% B. The flow rate was 0.25 ml min−1 and the column compartment was heated to 35 °C. The column was then backflushed with 100% acetonitrile for 4 min (ramp from 0.25 to 0.8 ml min−1 in 1.5 min) and re-equilibrated with A for 5 min at 0.4 ml min−1. The H-ESI source was operated at spray voltage of −2,500 V, sheath gas: 60 a.u.; aux gas: 19 a.u.; sweep gas: 1 a.u.; ion transfer tube: 300 °C; vaporizer: 250 °C. Full-scan MS1 data were collected from 70 m/z to 800 m/z at a mass resolution of 240,000 FWHM (full width at half-maximum) with RF lens at 35% and standard automatic gain control (AGC). The data-dependent tandem mass spectrometry (ddMS2) data were collected on unlabeled control samples using HCD (higher energy collisional dissociation) fragmentation at stepped collision energies of 20%, 35% and 50%, with Orbitrap detection at 15,000 FWHM. Peak picking and integration were completed in Skyline (v.23.3) using an in-house curated compound database from analytical standards. Natural abundance correction was performed using IsoCorrectoR51. Metabolomics analysis was conducted by the VAI Mass Spectrometry Core (RRID:SCR_024903).
RNA-seq analysis
WT and Keap1−/−CD45.2+/+;P14+ cells were either activated for 2 d in vitro and treated with DMSO or iloprost (5 µM) for 1 d then harvested for RNA-seq, or naïve cells were adoptively transferred into B6.SJL-PtprcaPepcb/BoyJ (CD45.1+/+) hosts 1 d before acute LCMV Armstrong infection. The Armstrong strain was utilized to obtain sufficient cell numbers for downstream RNA-seq, because Keap1−/− P14 cells minimally expand on chronic (CL13) infection. P14 cells were isolated on 7 d.p.i. using the EasySep Mouse APC Positive Selection Kit II (STEMCELL Technologies, cat. no. 17667) with a CD45.2 APC antibody (eBioscience, cat. no. 17-0454-82, clone 104), followed by FACS for additional purification (~100% purity). Sorted cells were then processed for RNA-seq by the VAI Pathology and Biorepository Core and libraries for RNA-seq were prepared by the VAI genomics core. The VAI Bioinformatics and Biostatistics Core (RRID:SCR_024762) conducted differential expression analysis from Keap1 knockout and WT P14 cells (Fig. 4b, Extended Data Fig. 7a and Supplementary Tables 3 and 4). Specifically, adapter sequences and low-quality reads of RNA-seq reads were trimmed using Trim Galore52. Trimmed reads were aligned to the mm10 reference genome using STAR53, with parameter ‘--quantMode GeneCounts’. Differential gene expression (DGE) analyses were conducted using edgeR54,55,56, based on gene counts generated from STAR. Batch information was included in the edgeR model as a covariate. For DGE, Benjamini–Hochberg Padj values were set to 0.05. ClusterProfiler (v.4.0.5)57 was used to perform GSEA on gene collections C2, C5, C6, C7, NFE2L2.v2 and H. Data are available on the National Center for Biotechnology Information’s (NCBI’s) GEO at accession nos. GSE244465 or GSE290976.
Proteomics sample preparation, LC and tandem MS analysis
Cell lysates were processed using EasyPrep MS Sample Prep Kits (Thermo Fisher Scientific) and dried peptides were resuspended in 0.1% trifluoroacetic acid. Synthetic peptides (GFTQAIAPDS, EMGDLLAFR, DDPVTNLNNAFEVAEK and LVSIGAEEIVDGNAK; Biosynth) were used to confirm endogenous peptide identity and for absolute quantification. Global proteome quantitation was performed via data-independent acquisition (DIA) on an Orbitrap Eclipse with a Vanquish Neo LC and FAIMS Pro interface. Peptide, 2 μm, was separated on a 20 cm × 75 μm, 1.7-μm C18 column (CoAnn) at 300 nl min−1 with a 120-min gradient. Mobile phases were 0.1% formic acid in water (phase A) and 80% acetonitrile with 0.1% formic acid (phase B). FAIMS voltages of −40 V, −55 V and −70 V were used. MS1 and MS2 were collected at 120,000 and 30,000 resolution (FWHM), respectively, using standard AGC settings, a 380–980 m/z range, 50-m/z isolation windows (1 m/z overlap) and 32% HCD. A hybrid spectral library for directDIA (Spectronaut v.18) was built from pooled samples acquired in DDA (data-dependent acquisition) mode with 11 FAIMS compensation voltage (CV) settings (−30 to 80 V). DDA scans used 1.6 m/z isolation windows, 2–5+ ions and 32% HCD. Targeted quantification via parallel reaction monitoring (PRM) was conducted on an Exploris 480 with Vanquish Neo LC. Peptide, 2 μg, was separated using the same column and flow rate as above, with a 60-min gradient. MS1 (375–1,200 m/z) and MS2 scans were at 120,000 and 30,000 resolution, respectively. PRM used a scheduled inclusion list with a 32% HCD energy. PTGIR was quantified using two unique peptides, with α-actinin-4 peptides as internal standards.
For data analysis, DIA data were processed in Spectronaut using the Mus musculus UniProt database (Taxon ID: 10090). Trypsin/P was the enzyme (two missed cleavages allowed), with fixed carbamidomethylation and variable methionine oxidation or amino-terminal acetylation. Peptides and proteins were filtered at 1% FDR. Fragment ion XIC areas were used for quantification. Library Extension Runs incorporated DDA data to enhance coverage. PRM data were analyzed in Skyline, requiring co-elution and intensity rank match with synthetic peptides. All MS was conducted by the VAI MS core (RRDD:SCR_024903).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data mined for the meta-analysis in Fig. 1 are available at NCBI’s GEO, accession nos. GSE86881 (ref. 18), GSE89307 (ref. 19) and GSE84820 (ref. 20). Bulk ex vivo RNA-seq data from WT and Keap1−/− CD8+ P14+ corresponding to Fig. 4 and Extended Data Fig. 7 have been deposited in the NCBI’s GEO under accession no. GSE244465. Bulk RNA-seq data shown in Fig. 6 have been deposited in the NCBI’s GEO under accession no. GSE290976. Proteomic data corresponding to Figs. 3–5 and Extended Data Fig. 7 are uploaded on to the PRIDE database via login with accession no. PXD052688. All other data are provided in the paper and Supplementary Information. Source data are provided with this paper.
References
Wherry, E. J. & Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Roy, D. G., Kaymak, I., Williams, K. S., Ma, E. H. & Jones, R. G. Immunometabolism in the tumor microenvironment. Annu. Rev. Cancer Biol. 5, 137–159 (2021).
Franco, F., Jaccard, A., Romero, P., Yu, Y.-R. & Ho, P.-C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2, 1001–1012 (2020).
Bacigalupa, Z. A., Landis, M. D. & Rathmell, J. C. Nutrient inputs and social metabolic control of T cell fate. Cell Metab. 36, 10–20 (2024).
Kaymak, I. et al. Carbon source availability drives nutrient utilization in CD8+ T cells. Cell Metab. 34, 1298–1311.e6 (2022).
Luda, K. M. et al. Ketolysis drives CD8+ T cell effector function through effects on histone acetylation. Immunity 56, 2021–2035.e8 (2023).
Ma, E. H. et al. 13C metabolite tracing reveals glutamine and acetate as critical in vivo fuels for CD8 T cells. Sci. Adv. 10, eadj1431 (2024).
Bengsch, B. et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 45, 358–373 (2016).
Yu, Y.-R. et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol. 21, 1540–1551 (2020).
Scharping, N. E. et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021).
Vardhana, S. A. et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 21, 1022–1033 (2020).
Mak, T. W. et al. Glutathione primes T cell metabolism for inflammation. Immunity 46, 1089–1090 (2017).
McMahon, M., Itoh, K., Yamamoto, M. & Hayes, J. D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 278, 21592–21600 (2003).
Asantewaa, G. & Harris, I. S. Glutathione and its precursors in cancer. Curr. Opin. Biotechnol. 68, 292–299 (2021).
Pauken, K. E. et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165 (2016).
Philip, M. et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017).
Man, K. et al. Transcription factor IRF4 promotes CD8+ T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity 47, 1129–1141.e5 (2017).
Yamamoto, M., Kensler, T. W. & Motohashi, H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 98, 1169–1203 (2018).
Beltra, J.-C. et al. Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 52, 825–841.e8 (2020).
Daniel, B. et al. Divergent clonal differentiation trajectories of T cell exhaustion. Nat. Immunol. 23, 1614–1627 (2022).
Giles, J. R. et al. Shared and distinct biological circuits in effector, memory and exhausted CD8+ T cells revealed by temporal single-cell transcriptomics and epigenetics. Nat. Immunol. 23, 1600–1613 (2022).
Khan, O. et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019).
Kopacz, A., Kloska, D., Forman, H. J., Jozkowicz, A. & Grochot-Przeczek, A. Beyond repression of Nrf2: an update on Keap1. Free Radic. Biol. Med. 157, 63–74 (2020).
Nüssing, S. et al. Efficient CRISPR/Cas9 gene editing in uncultured naive mouse T cells for in vivo studies. J. Immunol. 204, 2308–2315 (2020).
Yang, H. et al. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappaB and AP-1. Mol. Cell. Biol. 25, 5933–5946 (2005).
Telorack, M. et al. A glutathione-Nrf2-thioredoxin cross-talk ensures keratinocyte survival and efficient wound repair. PLoS Genet. 12, e1005800 (2016).
Eruslanov, E. & Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 594, 57–72 (2010).
Malhotra, D. et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734 (2010).
Zhu, Z. et al. The redox sensor KEAP1 facilitates adaptation of T cells to chronic antigen stimulation by preventing hyperactivation. Sci. Immunol. 9, eadk2954 (2024).
Moncada, S. & Vane, J. R. Prostacyclin: its biosynthesis, actions and clinical potential. Philos. Trans. R. Soc. Lond. B Biol. Sci. 294, 305–329 (1981).
Shimizu-Albergine, M. et al. SCAP/SREBP pathway is required for the full steroidogenic response to cyclic AMP. Proc. Natl Acad. Sci. USA 113, E5685–E5693 (2016).
Storek, K. M., Gertsvolf, N. A., Ohlson, M. B. & Monack, D. M. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J. Immunol. 194, 3236–3245 (2015).
Snell, L. M., McGaha, T. L. & Brooks, D. G. Type I interferon in chronic virus infection and cancer. Trends Immunol. 38, 542–557 (2017).
Gnanaprakasam, J. N. R. et al. Asparagine restriction enhances CD8+ T cell metabolic fitness and antitumoral functionality through an NRF2-dependent stress response. Nat. Metab. 5, 1423–1439 (2023).
Nishida, M. et al. Mitochondrial reactive oxygen species trigger metformin-dependent antitumor immunity via activation of Nrf2/mTORC1/p62 axis in tumor-infiltrating CD8T lymphocytes. J. Immunother. Cancer 9, e002954 (2021).
Renken, S. et al. Targeting of Nrf2 improves antitumoral responses by human NK cells, TIL and CAR T cells during oxidative stress. J. Immunother. Cancer 10, e004458 (2022).
Yan, C. et al. Exhaustion-associated cholesterol deficiency dampens the cytotoxic arm of antitumor immunity. Cancer Cell 41, 1276–1293.e11 (2023).
Boukhaled, G. M., Harding, S. & Brooks, D. G. Opposing roles of type I interferons in cancer immunity. Annu. Rev. Pathol. 16, 167–198 (2021).
Chen, W. et al. Chronic type I interferon signaling promotes lipid-peroxidation-driven terminal CD8+ T cell exhaustion and curtails anti-PD-1 efficacy. Cell Rep. 41, 111647 (2022).
Jacquelot, N. et al. Sustained type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 29, 846–861 (2019).
Gubbala, V. B. et al. Eicosanoids in the pancreatic tumor microenvironment—a multicellular, multifaceted progression. Gastro. Hep. Adv. 1, 682–697 (2022).
Norlander, A. E. et al. Prostaglandin I2 signaling licenses Treg suppressive function and prevents pathogenic reprogramming. J. Clin. Invest. 131, e140690 (2021).
Chen, J. H. et al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat. Med. 21, 327–334 (2015).
Morotti, M. et al. PGE2 inhibits TIL expansion by disrupting IL-2 signalling and mitochondrial function. Nature 629, 426–434 (2024).
Lacher, S. B. et al. PGE2 limits effector expansion of tumour-infiltrating stem-like CD8+ T cells. Nature 629, 417–425 (2024).
Roy, D. G. et al. Methionine metabolism shapes T helper cell responses through regulation of epigenetic reprogramming. Cell Metab. 31, 250–266.e9 (2020).
Dow, L. E. et al. A pipeline for the generation of shRNA transgenic mice. Nat. Protoc. 7, 374–393 (2012).
Heinrich, P. et al. Correcting for natural isotope abundance and tracer impurity in MS-, MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with IsoCorrectoR. Sci. Rep. 8, 17910 (2018).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Chen, Y., Lun, A. T. L. & Smyth, G. K. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 5, 1438 (2016).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Camb.) 2, 100141 (2021).
Acknowledgements
We thank S. E. LeBoeuf and T. Papagiannakopolous for providing sgRNA sequences to target NRF2 and R. DeBerardinis for scientific discussions contributing to this paper. We thank J. Wedberg, M. Brewer, M. Minard and C. Kraus for administrative assistance and support. We thank members of the VAI Core Facilities (Metabolomics and Bioenergetics, Genomics, Bioinformatics and Biostatistics, Flow Cytometry, Vivarium) for technical assistance. C.M.K. is supported by the National Institute of Allergy and Infectious Diseases (NIAID, grant no. R21AI153997) and VAI. R.G.J. is supported by the Paul G. Allen Frontiers Group Distinguished Investigator Program, Chan Zuckerberg Initiative, NIAID (grant no. R01AI165722) and VAI. J.L. is supported by the VAI Metabolism & Nutrition (MeNu) Program Pathway-to-Independence (P2i) Award and Canadian Institutes of Health Research (CHIR) Fellowship (grant no. MFE-181903). M.S.D. is supported by the VAI MeNu Program P2i Award, Fonds de recherche du Québec-Santé Postdoctoral Fellowship (no. 0000289124) and CIHR Fellowship (no. MFE-403514). This research was supported in part by a grant from the National Institutes of Health (NIH; grant no. AI154450), Cancer Prevention and Research Institute Texas (grant no. RR210035) and startup funds from University of Texas Southwestern (UTSW) to C. Yao, grants from the NIH (grant nos. AG056524 and AI158294), a V Scholar Award, an AFAR Grant for Junior Faculty, a Clinic & Laboratory Integration Program Grant (Cancer Research Institute) and startup funds from UTSW and CU to T. Wu and funding from VAI to R.G.J.
Author information
Authors and Affiliations
Contributions
M.S.D. and R.G.J. conceived the study, designed the methodology and wrote the original draft with S.M.K.-G., R.D.S. and H.L. M.S.D., L.M.D., B.M.O., S.E.C., J.L. and N.F. performed the experiments. R.G.J., R.D.S., H.L., A.J., A.E.E., M.V., C.M.K. and I.S. contributed to experimental design and methodology. M.V. and Z.F. performed data visualization. K.S.W. managed the project. R.G.J., C.Y. and T.W. supervised the study. M.S.D. and R.G.J. acquired the funding. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
R.G.J. is a scientific advisor for Servier Pharmaceuticals and a member of the Scientific Advisory Board of Immunomet Therapeutics. R.G.J. and M.S.D. are inventors on a provisional patent application titled ‘Compositions of leukocytes with resistance to prostanoids and related methods of use’ (no. IP-00237). The other authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks Annette Oxenius, Ruoning Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Nick Bernard, in collaboration with the rest of the Nature Immunology team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Examination of the transcriptional effects of Keap1 deletion in CD8+ T cells.
(a) qPCR of naïve versus in vitro activated CD8+ T cells from WT (Keap1fl/fl, Cd4 Cre-) and Keap1-/- (Keap1fl/fl, Cd4 Cre + ) mice. Shown are relative mRNA expression levels for NRF2 transcriptional targets Nqo1, Gclc, and Slc7a11 (b) Relative mRNA expression for Keap1, and NRF2 transcriptional targets Nqo1, Hmox1, and Gclc in CD8+ T cells and (c) hepatocytes from WT (Keap1fl/fl, Cd4 Cre-) and Keap1-/- (Keap1fl/fl, Cd4 Cre + ) mice (mean±SEM, n = 3). *P < 0.05, **P < 0.01, ****P < 0.0001.
Extended Data Fig. 2 Baseline immunophenotyping of splenocyte populations from Keap1fl/fl (WT) and Cd4Cre+ Keap1fl/fl (Keap1-/-) mice.
(a-b) Percent (top) and number (bottom) of splenic (A) CD3+ T cells and (b) CD8+ (gated on CD3+) T cells from WT and Keap1-/- mice aged 8-12 weeks (mean±SEM, n = 6). (c) Percent (top) and number (bottom) of naïve, effector, central memory, and CD44-/CD62L- (double negative) subsets of CD8+ T cells in the spleens of mice as in (a). (d-e) CD4+ T cells populations in WT and Keap1-/- mice. (d) Percent (top) and number (bottom) of CD4+ (gated on CD3+) T cells from WT and Keap1-/- mice aged 8-12 weeks (mean±SEM, n = 6). (e) Percent (top) and number (bottom) of naïve, effector, central memory, and CD44-/CD62L- (double negative) subsets of CD4+ T cells in the spleens of mice as in (d). (f-h) Percent (top) and number (bottom) of (f) B cells, Natural Killer (NK), and Natural Killer (NKT) T cells, (g) neutrophils, and (h) total CD11c+ dendritic cells (DCs) and conventional DC1 (cDC1) and DC2 (cDC2) subsets from WT and Keap1-/- mice aged 8-12 weeks (mean±SEM, n = 6). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Extended Data Fig. 3 Flow cytometry analysis of exhausted CD8+ T cell subsets.
(a) Schematic of P14 cell adoptive transfer with LCMV infection. (b) Representative flow cytometry plots of donor CD8+/CD45.2+ WT and Keap1-/- P14 cells harvested from spleen of LCMV CL13-infected mice at 19 dpi. Bar graph quantifies the percent of CD8+/CD45.2+ WT and Keap1-/- P14 T cells in the spleen (mean±SEM, n = 4-5/sample). (c) Representative flow cytometry plots of progenitor exhausted 1 (Texprog1; LY108+/CD69+), progenitor exhausted 2 (Texprog2; LY108+/CD69-), intermediate (Texint; LY108-/CD69-) and terminally exhausted (Texterm; LY108-/CDD69+) donor CD8+/CD45.2+ WT and Keap1-/- P14 cells harvested from spleen of LCMV CL13-infected mice at 19 dpi. Bar graphs quantify the percent of respective subsets (mean±SEM, n = 4-5/sample). (d) Representative flow cytometry plots of PD-1+ versus TIM-3+ donor CD8+/CD45.2+ WT and Keap1-/- P14 cells harvested as in (a-c). Bar graphs quantify the percent of PD-1+/ TIM-3+ CD8+/CD45.2+ WT and Keap1-/- P14 T cells in the spleen (mean±SEM, n = 4-5/sample). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Extended Data Fig. 4 scRNA-seq cluster analysis and impact of Keap1 deletion in T cells on tumor growth and survival.
(a) Heatmap of gene expression markers for progenitor exhausted (Texprog), terminally exhausted (Texterm), exhausted killer cell lectin-like receptor-expressing (TexKLR), exhausted IFN-I stimulated gene expressing (TexISG), and proliferating (prolif) CD8+ TIL isolated from B16-OVA tumors (related to Fig. 2a). (b) Weighted nearest neighbor UMAP for CD8+ TIL clusters described in (a) and embedding of GSEA for NRF2 transcriptional target signature (M2662) across clusters. (c) Individual growth curves for MC38-OVA tumors in WT and Keap1-/- mice. Right, Kaplan-Meier (KM) survival curves for time to tumor endpoint ( > 1500 mm3) for MC38-OVA tumors grown in WT and Keap1-/- mice. (d) Individual growth curves for B16-OVA tumors following adoptive transfer of WT or Keap1-/- OT-I CD8+ T cells at 8 dpti. Right, Kaplan-Meier (KM) survival curves for time to tumor endpoint ( > 1500 mm3) for B16-OVA tumors. (e) Relative mRNA expression (versus Actb housekeeping gene) of NRF2 target Hmox1 in WT sgScr and sgNfe2l2 OT-I CD8+ T cells. (f) Representative flow cytometry histograms of GZMB, IFN-γ, TNF-α and TOX, gated on Thy1.1+/PD-1+ OT-I donor TILs, re-stimulated with PMA/ionomycin in vitro. Geometric mean fluorescence intensity (gMFI) values are displayed next to sample names.
Extended Data Fig. 5 Proteomic and metabolic profiling of Keap1-/- and WT CD8+ T cells.
(a) Raw peptide intensities for NRF2, GCLM, and ACTN4 in Keap1-/- CD8+ P14 T cells that received Nfe2l2-targeting (sgNfe2l2) or scrambled control (sgScr) sgRNAs, and WT CD8+ P14 T cells that received sgScr sgRNA. (mean±SEM, n = 3). (b-c) Mass isotopologue distribution (MID) for intracellular metabolites from sgNfe2l2 and sgScr Keap1-/-, and sgScr WT P14 T cells following culture with [U-13C]-Glutamine (mean±SEM, n = 4). Shown are the percent labelling (top) and intensities (bottom) for indicated isotopologues for (b) Glutamine, Malate and Citrate, and (c) Glutathione (reduced), γ-glutamyl-cysteine, and Glutamate. (d) Ratio of GGSG:GSH across conditions, taken from total pool values. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Extended Data Fig. 6 Immunophenotyping of Keap1-/- sgScr and sgNfe2l2, and WT sgScr P14 cells following LCMV CL13 infection.
(a-e) Bar graphs quantifying (a) the number of P14 (CD45.2+) donor cells and (b) percentage of PD-1hi donor P14 cells. (c) Representative histograms and bar graph of gMFI for PD-1 in CD45.2+ donor cells from (a) at 7 dpi (mean±SEM, n = 3-4/sample). (d) Bar graph quantifying the number of IFN-γhi (CD45.2+/PD-1+) donor cells, corresponding to Fig. 3h. (e) Representative flow cytometry plot of IFN-γ+/TNF-α+ sgNfe2l2 and sgScr Keap1-/-, and sgScr WT CD45.2+/PD-1+ P14 T cells at 7 dpi with quantification of the percentage and number of polyfunctional (IFN-γ+/TNF-α+) PD-1hi sgNfe2l2 and sgScr Keap1-/-, and WT sgScr PD-1+ P14 donor CD8 T cells from the spleen of LCMV CL13-infected mice (7 dpi). (f) Representative flow cytometry plots and representative histograms and bar graph of gMFI for TNF-α, (g) TCF1, and (h) TBET expression by PD-1+ donor cells from (b) at 7 dpi (mean±SEM, n = 3-4/sample). Plots d-f correspond to cells re-stimulated with gp33 (1 μg/mL) peptide in vitro *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.001.
Extended Data Fig. 7 Enriched C6 gene sets in Keap1-/- versus WT P14 cells and mass spectrometry validation of PTGIR peptides.
(a) Top 10 enriched pathways for MSigDB C6 (oncogenic signature) gene sets in Keap1-/- versus WT P14 T cells following LCMV infection (7 dpi). Data are plotted as Normalized Enrichment Score (NES) for each pairwise comparison. (b) Representative PRM transition traces for the synthetic (top) and endogenous (bottom) PTGIR peptides GFTQAIAPDSR and (c) EMGDLLAFR. (d) Representative MS2 spectra of the synthetic (top) and endogenous (bottom) peptides (from b) GFTQAIAPDSR and (from c) EMGDLLAFR of PTGIR.
Extended Data Fig. 8 PTGIR expressing T cells poorly control tumor growth.
(a) Relative PTGIR peptide (EMGDLLAFR) levels (normalized to ACTN4) from WT CD8+ T cells transduced with either an empty vector control (EV), or PTGIR expressing plasmid ( + PTGIR), and Keap1-/- CD8+ T cells transduced with EV control (mean±SEM, n = 3). (b) Raw intensity levels of PTGIR peptides GFTQAIAPDSR and (c) EMGDLLAFR, and (d) α-actinin-4—or ACTN4—peptide LVSIGAEEIVDGNAK from engineered cells described in (a) (mean±SEM, n = 3). (e) Individual growth curves, displayed vertically, for B16-OVA melanoma tumor growth in mice that received adoptive transfer of either WT empty vector control (WT + EV), PTGIR-expressing (WT + PTGIR), Keap1-/- empty vector (Keap1-/- + EV) OT-I CD8+ T cells, or HBSS at 7 dpti (mean±SEM, n = 12/group). (f) Individual growth curves, displayed vertically, for B16-OVA melanoma tumor growth in mice that received adoptive transfer of WT sgScr + EV, Keap1-/- sgScr + EV, Keap1-/- sgNfe2l2 + EV, Keap1-/- sgNfe2l2 + PTGIR OT-I cells, or HBSS at 6 dpti (mean±SEM, n = 12-20/group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Extended Data Fig. 9 Fig. S9. Transcriptional and bioenergetic profiling genetically engineered WT and Keap1-/- cells upon iloprost treatment.
(a) Schematic for WT empty vector (EV) and WT CD8 T cells with enforced PTGIR expression (WT + PTGIR) treated with vehicle (DMSO) for 24 h, and corresponding volcano plot (horizontal axis: Log2 fold change (WT + EV/WT + PTGIR), vertical axis: -Log10 False Discovery Rate (FDR)) highlighting differentially expressed genes (DEGs) in WT + EV versus WT + PTGIR cells. Light green and light red shaded regions represent genes significantly enriched (-Log10 FDR > 1.30, corresponding to FDR < 0.05) in vehicle-treated WT + EV and WT + PTGIR CD8 T cells, respectively. Normalized enrichment scores (NES) of one hallmark (curated) pathway upregulated in WT + EV cells. Only one NES score had a q-value < 0.05, and was displayed. (b) Schematic for WT (Keap1fl/fl Cd4 Cre-) CD8 T cells treated with vehicle (DMSO) or iloprost (5 μM, 24 h), and corresponding volcano plot (horizontal axis: Log2 fold change (iloprost/vehicle), vertical axis: -Log10 False Discovery Rate (FDR)) highlighting differentially expressed genes (DEGs) in iloprost versus vehicle treated WT (Keap1fl/fl Cd4 Cre-). Darker and lighter gray shaded regions represent genes significantly enriched (-Log10 FDR > 1.30, corresponding to FDR < 0.05) in iloprost and vehicle-treated WT (Keap1fl/fl Cd4 Cre-) CD8+ T cells, respectively. Normalized enrichment scores (NES) of four hallmark (curated) pathways downregulated upon iloprost treatment of WT (Keap1fl/fl Cd4 Cre-) cells. Only NES scores with q-value < 0.05 are displayed. (c) Schematic for WT empty vector (EV) CD8+ T cells treated with vehicle (DMSO) or iloprost (5 μM, 24 h), and corresponding volcano plot (horizontal axis: Log2 fold change (iloprost/vehicle), vertical axis: -Log10 False Discovery Rate (FDR)) highlighting differentially expressed genes (DEGs) in iloprost versus vehicle treated WT + EV cells. Darker and lighter green shaded regions represent genes significantly enriched (-Log10 FDR > 1.30, corresponding to FDR < 0.05) in iloprost and vehicle-treated WT + EV CD8 T cells, respectively. Normalized enrichment scores (NES) of two hallmark (curated) pathways downregulated and five upregulated upon iloprost vs. vehicle treatment of WT + EV cells. Only NES scores with q-value < 0.05 are displayed. (d) Venn diagram demonstrating number of significantly (FDR < 0.05) downregulated and upregulated mRNAs upon iloprost treatment of WT + PTGIR (vs veh) and Keap1-/- (vs veh) CD8+ T cells. Common (curated) biological process and molecular function pathways downregulated and upregulated upon iloprost treatment are displayed. (e) Total ATP from OXPHOS and glycolysis from seahorse bioenergetic measurements corresponding to Fig. 6h. (f) Normalized enrichment scores (NESs) of transcriptional signatures associated with CD8+ T cell subsets corresponding to GSE199565, and GSE212048, compared with the differentially expressed genes in iloprost-treated versus vehicle-treated Keap1-/- CD8+ T cells. (g) Heatmap of differentially expressed genes in TexTerm signature (GSE188670) in iloprost versus vehicle treated Keap1-/- CD8+ T cells. Inset displays top 5 genes driving the Texterm enrichment in Keap1-/- CD8+ T cells: Vps37b, Prrt1, Abcb9, Frmd4b, Abcb1a.
Extended Data Fig. 10 Ptgir knockdown improves anti-viral CD8+ T cell responses and attenuates tumor growth.
(a) Bar graph quantifying the number of control (shCtrl) and Ptgir knockdown (shPtgir) P14 donor cells in the spleen of LCMV CL13-infected mice at 8 dpi (mean±SEM, n = 4-5/sample). CD45.2 is a donor cell marker expressed by P14 T cells. (b) Bar graph of PD-1hi/TIM-3hi shCtrl- versus shPtgir-expressing P14 T cells from cells in (a). (c) Quantification of the percentage and number of polyfunctional (IFN-γ+/TNF-α+) PD-1hi shCtrl- versus shPtgir-expressing P14 T donor cells from LCMV CL13-infected mice at 8 dpi, and re-stimulated with gp33 peptide (1 μg/mL) in vitro (mean±SEM, n = 4-5/sample). (d) Individual growth curves, displayed vertically, for MC38-OVA tumors following adoptive transfer of OT-I CD8+ T cells expressing control (shCtrl) and Ptgir (shPtgir) shRNAs at 7 dpti. (e) Kaplan-Meier (KM) survival curves for time to tumor endpoint ( > 1500 mm3) for MC38-OVA tumors. (f) Individual growth curves, displayed vertically, for B16-OVA tumors following adoptive transfer of OT-I CD8+ T cells expressing control (shCtrl) and Ptgir (shPtgir) shRNAs at 7 dpti. (g) Kaplan-Meier (KM) survival curves for time to tumor endpoint ( > 1500 mm3) for B16-OVA tumors. **P < 0.01.
Supplementary information
Supplementary Table 2
The qPCR, shRNA and sgRNA oligonucleotide sequences. The 5ʹ- to 3ʹ-oligonucleotide sequences used for qPCR, CRISPR–Cas9-mediated knockout and shRNA-mediated knockdown. Q denotes primers used for qPCR, and F and R denote forward and reverse primers, respectively.
Supplementary Table 3
DEG analysis from RNA-seq of adoptively transferred Keap1−/− versus WT P14 cells, isolated 7 d post-LCMV Armstrong infection. Refer to the first tab of the Excel sheet for detailed description of column headings in subsequent data tab.
Supplementary Table 4
C2, C5, C6, C7, NFE2L2.v2 and H GSEA from RNA-seq of Keap1−/− versus WT P14+ CD8+ T cells on LCMV Armstrong infection. A positive or negative NES indicates a gene set enrichment in Keap1−/− cells or WT cells, respectively. Refer to the first tab of the Excel sheet for detailed description of column headings in subsequent data tabs.
Supplementary Table 5
DEG analysis from RNA-seq of in vitro cultured CD8+ T cells with variable expression of PTGIR, treated with vehicle or iloprost. Refer to the first tab of the Excel sheet for detailed description of column headings in subsequent data tabs.
Supplementary Table 6
Pathway analysis from RNA-seq of in vitro cultured CD8+ T cells with variable expression of PTGIR, treated with vehicle or iloprost. Refer to the first tab of the Excel sheet for detailed description of column headings in subsequent data tabs.
Source data
Source Data Fig. 1
Unprocessed immunoblots (annotated) for Lamin B1, NQO1, HO-1 and KEAP1 from liver extracts.
Source Data Fig. 2
Unprocessed immunoblots (annotated) for KEAP1 from T cells.
Source Data Fig. 3
Unprocessed immunoblots (annotated) for NQO1 from T cells.
Source Data Fig. 4
Unprocessed immunoblots (annotated) for HO-1 and Lamin B1 from T cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dahabieh, M.S., DeCamp, L.M., Oswald, B.M. et al. The prostacyclin receptor PTGIR is a NRF2-dependent regulator of CD8+ T cell exhaustion. Nat Immunol 26, 1139–1151 (2025). https://doi.org/10.1038/s41590-025-02185-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41590-025-02185-9
This article is cited by
-
Lipids rewire T cell exhaustion
Nature Immunology (2025)
-
Regulation of T cell exhaustion and stemness: molecular mechanisms and implications for cancer immunotherapy
Cellular & Molecular Immunology (2025)
