
Abstract
Clonal dominance characterizes hematopoiesis during aging and increases susceptibility to blood cancers and common nonmalignant disorders. VEXAS syndrome is a recently discovered, adult-onset, autoinflammatory disease burdened by a high mortality rate and caused by dominant hematopoietic clones bearing somatic mutations in the UBA1 gene. However, pathogenic mechanisms driving clonal dominance are unknown. Moreover, the lack of disease models hampers the development of disease-modifying therapies. In the present study, we performed immunophenotype characterization of hematopoiesis and single-cell transcriptomics in a cohort of nine male patients with VEXAS syndrome, revealing pervasive inflammation across all lineages. Hematopoietic stem and progenitor cells (HSPCs) in patients are skewed toward myelopoiesis and acquire senescence-like programs. Humanized models of VEXAS syndrome, generated by inserting the causative mutation in healthy HSPCs through base editing, recapitulated proteostatic defects, cytological alterations and senescence signatures of patients’ cells, as well as hematological and inflammatory disease hallmarks. Competitive transplantations of human UBA1-mutant and wild-type HSPCs showed that, although mutant cells are more resilient to the inflammatory milieu, probably through the acquisition of the senescence-like state, wild-type ones are progressively exhausted and overwhelmed by VEXAS clones, overall impairing functional hematopoiesis and leading to bone marrow failure. Our study unveils the mechanism of clonal dominance and provides models for preclinical studies and preliminary insights that could inform therapeutic strategies.
Similar content being viewed by others


Main
DNA mutations are acquired by somatic cells during life1,2,3,4,5,6 and may increase cellular fitness, leading to clonal expansion and dominance7,8,9 in high-turnover tissues. Cell-extrinsic pressures, like inflammation, may further confer advantage to mutant clones10,11. Clonal dominance in hematopoiesis during aging is driven by positive selection of HSPCs and is associated with the risk of progression to hematological malignancies12,13,14,15,16.
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is a paradigm of an acquired, severe autoinflammatory and hematological disease of aging caused by clonal dominance17. VEXAS syndrome predominantly affects men aged >50 years (1 in 4,000)18 and is hallmarked by a prevalence of proinflammatory hematopoietic clones originating from HSPCs bearing peculiar mutations in the UBA1 (ubiquitin-like modifier activating enzyme 1) gene17,19. The main manifestations of VEXAS syndrome are cytopenia, macrocytic anemia, bone marrow (BM) failure, fever and multiorgan chronic inflammation20,21,22,23,24,25,26,27,28,29. About 50% of patients present with myelodysplastic syndrome (MDS) at diagnosis; clones bearing classic clonal hematopoiesis of indeterminate potential (CHIP) mutations are often detectable, albeit blood cancers in patients with VEXAS syndrome remain sporadic17,19,30,31. Allogeneic HSPC transplantation (allo-HSCT) could cure VEXAS syndrome, but its applicability is limited by toxicities in older patients32. Pharmacological treatments quenching inflammation and debulking mutant clones have poor efficacy and/or little evidence20,31,33,34,35,36,37. Consequently, patients’ quality of life and prognosis are dismal, with a 50% mortality rate in 5 years from diagnosis17,26,38.
UBA1 is the primary enzyme responsible for ubiquitin activation39,40,41,42. Two isoforms are known, UBA1b and UBA1a, which differ in their starting methionine (Met). The most prevalent VEXAS mutations affect Met41, the starting codon of UBA1b17,18, resulting in the expression of a dysfunctional isoform, UBA1c, loss of ubiquitylated proteins and activation of stress responses17,43. Patients with VEXAS syndrome show activation of inflammatory signatures, secretion of proinflammatory cytokines, high prevalence of UBA1-mutant myeloid cells and lack of UBA1-mutant lymphoid progeny17,44,45,46.
As a result of of its intriguing biology and remarkable clinical impact, VEXAS syndrome has sparked keen interest47,48,49. However, its recent discovery, the small and heterogeneous patient cohorts and the paucity of experimental models have precluded thorough investigations of the mechanisms underlying clonal dominance. It is unknown whether dominance of UBA1-mutant HSPCs over time ascribes to an enhanced expansion capacity and/or a paracrine detrimental effect on healthy hematopoiesis. Gaining insights into VEXAS syndrome pathology may instruct repurposing of approved treatments or designing therapeutic approaches.
Previous attempts to model VEXAS syndrome were based on pharmacological inhibition, RNA interference or CRISPR–Cas9 nuclease-mediated gene disruption17,45,50. Yet, these strategies lack specificity for the UBA1b isoform and fail to accurately recapitulate VEXAS pathophysiology because the specific pathogenic mutation is not introduced. Conversely, base and prime editors can insert a desired mutation into the genome with superior specificity and precision than CRISPR–Cas9 nuclease51,52,53 by converting C•G to T•A (cytosine base editor) or A•T to G•C (adenine base editor)54,55,56,57. As such, these editors are perfectly tailored to model acquisition of somatic mutations.
In the present study, we developed humanized models of VEXAS syndrome through base editing and capitalized on single-cell transcriptomic data from both the patient cohorts and the humanized model to unravel the mechanisms driving clonal dominance in VEXAS syndrome.
Results
Patients’ HSPCs are myeloid biased, less primitive and defective to engraft
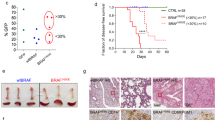
To address the heterogeneity of hematopoiesis in VEXAS syndrome, we performed multiparametric flow-cytometry analyses58 on fresh patients’ BM aspirates from an internal cohort of patients (Extended Data Table 1 and Supplementary Fig. 1) and compared data with those from BM samples of age-matched individuals not diagnosed with VEXAS syndrome and undergoing hip surgery59. Cellularity of patients’ BM was lower than controls, reaching significance in proerythroblasts (Fig. 1a and Extended Data Fig. 1a). Within the CD34− myeloid compartment, we found a decrease in polymorphonuclear neutrophils and an increase in immature myeloblasts in two patients (Fig. 1b and Extended Data Fig. 1b). Moreover, we observed a slightly lower abundance of natural killer (NK) cells and a dramatic reduction of all lymphoid committed progenitors and B cells (Fig. 1c and Extended Data Fig. 1c). Within CD34+ cells, we found a shrinkage of multi-lymphoid progenitors (MLPs), B and NK cell precursors (pre-BNKs) and megakaryocyte progenitors (MKPs) (Fig. 1d and Extended Data Fig. 1d). Conversely, common myeloid progenitors (CMPs), granulocyte–monocyte progenitors (GMPs) and early T cell progenitors (ETPs) were expanded. This unbalanced composition confirms a preferential myeloid differentiation of HSPCs in the BM of patients with VEXAS syndrome (Fig. 1e). Primitive hematopoietic stem cells (HSCs) and multipotent progenitors (MPPs) were reduced compared with controls (Fig. 1d and Extended Data Fig. 1d). As inflammation has been previously linked to enhanced egression of HSPCs into the bloodstream60,61,62,63, we analyzed peripheral blood samples with the same flow-cytometry panel. We observed a heightened proportion of circulating CD34+ HSPCs in patients with VEXAS syndrome compared with controls (Fig. 1f), with a composition reflecting the increase in myeloid progenitors and ETPs and the decrease of MLPs and pre-BNKs in the BM (Fig. 1g and Extended Data Fig. 1e–h). Genotyping analyses showed dominance of the UBA1-mutant (UBA1mut) cells within HSPCs and CD13+ myeloid cells, but not in CD19+ B cells and CD3+ T cells (Fig. 1h). CD56+ NK cells were heterogeneous among patients.

a–d, Number of total hematopoietic cells (a), myeloid cells (b), lymphoid cells (c) and HSPCs (d) per microliter in BM aspirates of patients with VEXAS syndrome and age-matched controls assessed by multiparametric flow cytometry (n = 19 and 7). The median is shown with the interquartile range (IQR). Data on age-matched reference controls from ref. 59 are shown. e, Ratio between the number of myeloid and lymphoid committed progenitors from patients (n = 19 and 7). The median is shown with the IQR. Data on age-matched reference controls from ref. 59 are shown. f, Number of total hematopoietic circulating cells per microliter in the blood of patients with VEXAS syndrome and age-matched controls (n = 19 and 7). The median is shown with the IQR. Data on age-matched reference controls in e–g from ref. 59 are shown. g, Number of circulating HSPCs from different subpopulations per µl in the blood (n = 19 and 7). The median is shown with the IQR. h, Percentage of UBA1mut cells in the BM (left, n = 6, 5, 4 and 4) and peripheral blood (PB; right, n = 7, 6, 7 and 6) of different patients across hematopoietic subpopulations. Symbols and colors identify different patients. The median is shown with the IQR. i, Percentage of human cells in the BM of hematochimeric mice 12 weeks after transplantation of CD34+ HSPCs from healthy donors (n = 3) or patients with VEXAS syndrome (n = 3). Symbols and colors identify different donors of HSPCs. VEXAS HSPCs were transplanted without dilution with healthy donor HSPCs. j, Number of cells obtained on in vitro differentiation of HSPCs from patients with VEXAS syndrome (n = 2) and age-matched controls (n = 3). k, Number of erythroid and myeloid colonies obtained on seeding of 1,000 HSPCs from VEXAS PT1 (n = 1). The percentage represent the proportion of mutant colonies. For all panels, the Mann–Whitney U-test was used. *P < 0.5; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To investigate whether HSPCs from patients retain stem cell properties despite different immunophenotypes, CD34+ cells from patient 1 (PT1) and PT5 were transplanted into immunodeficient mice or seeded in culture for differentiation and clonogenic assays. Notably, patients’ HSPCs either failed to engraft (PT1) or showed a remarkably lower chimerism (PT5) and reduced in vitro differentiation potential compared with HSPCs from control individuals (Fig. 1i,j and Extended Data Fig. 1i). Moreover, HSPCs from PT1 generated mostly UBA1-wild-type (UBA1wt) myeloid colonies (Fig. 1k), which were much fewer than routinely obtained from older controls59.
Our data define myeloid bias, increased mobilization to the bloodstream and lower primitiveness of HSPCs as hallmarks of altered hematopoiesis in patients. However, the poor availability of HSPCs, their reduced engraftment capacity and the interpatient heterogeneity limit the use of patient-derived cells to build preclinical models and investigate the mechanisms of VEXAS syndrome.
Base editing efficiently installs VEXAS mutation in UBA1wt HSPCs
To overcome these constraints, we designed a base-editing strategy to insert the most common VEXAS mutation (p.Met41Thr) in male HSPCs from adult healthy donors by co-delivering ABE8.20-m56 and a guide RNA (gRNA) targeting Met41 (ref. 53) (Fig. 2a and Extended Data Fig. 2a). We obtained ∼90% UBA1mut HSPCs without differences in growth, cell cycle and viability compared with control edited (UBA1wt) HSPCs (Fig. 2b–e). UBA1mut HSPCs displayed expression of UBA1a, loss of UBA1b and appearance of UBA1c (Fig. 2f and Extended Data Fig. 2b), resulting in reduced ubiquitylated proteins (Fig. 2g and Extended Data Fig. 2c). UBA1mut HSPCs exhibited increased expression of BiP, a master regulator of the unfolded protein response64 (Fig. 2h and Extended Data Fig. 2c). Transmission electron microscopy (TEM) and optical microscopy showed altered organelle ultrastructure in UBA1mut HSPCs. Vacuoles were larger and less electrodense (Fig. 2i,j) and mitochondria showed aberrant morphology (that is, cristae disruption and higher occupied area) (Fig. 2k,l). This modeling strategy thus recapitulates the cellular hallmarks of VEXAS syndrome described in patients17,44,45.

a, Scheme of the base-editing strategy. b, Percentage of HSPCs carrying the intended Met41Thr VEXAS mutation 7 days after editing (n = 7 biological replicates). The median is shown with the IQR. c,d, Growth curve (c; n = 3 biological replicates; median with IQR) and cell-cycle phases (d; n = 2 biological replicates) of HSPCs after treatment. e, Percentage of live, early or late apoptotic and necrotic cells 24 h after treatment (n = 3 biological replicates). The values are the mean ± s.e.m. f–h, Western blot analysis of UBA1a, UBA1b and UBA1c protein expression (f), poly(ubiquitylated) proteins (poly(Ub)) (g) and BiP expression level (h) in UBA1wt and UBA1mut HSPCs. β-Actin or β-tubulin was used as a protein-loading control (n = 2 technical replicates). i, Representative TEM images showing vacuoles in cultured UBA1wt and UBA1mut HSPCs 7 days after editing. High (left) and low (right) magnification images are shown. j, Representative optical microscopy images (×100 magnification objective) of UBA1wt and UBA1mut HSPCs 7 days after editing. Cells were stained with modified Giemsa. Red arrows indicate macrovacuoles in UBA1mut HSPCs (n = 1). k, Representative TEM images showing mitochondria in UBA1wt and UBA1mut HSPCs cultured for 7 days after editing. High (left) and low (right) magnification images are shown (n = 1). l, Quantitative analysis of circularity and occupied mitochondrial area in UBA1wt and UBA1mut HSPCs (n = 20 cells per condition from one donor). m, Significantly positively (pos) (red) and top-ten negatively (neg) (blue) enriched gene ontology biological processes in the bulk transcriptomic analyses of UBA1mut versus UBA1wt HSPCs cultured for 7 days after editing (n = 3 technical replicates of a pool of independent donors). Wilcoxon’s rank-sum test with the Benjamini–Hochberg correction was used. n, Volcano plot showing upregulated (red) and downregulated (blue) metabolites 7 days after differentiation of UBA1mut or UBA1wt HSPCs in culture (n = 3 biological replicates). The median is shown with the IQR. Student’s t-test with the Benjamini–Hochberg correction. o,p, Number of cells (o) and number of myeloid, erythroid, megakaryocytic, NK and pre-T cells (p) obtained on differentiation of UBA1mut or UBA1wt HSPCs in culture (n = 3 technical replicates). The median is shown with the IQR. q, Number of erythroid and myeloid colonies obtained on seeding HSPCs edited as indicated (n = 4, 4, 3 and 4). The median is shown with the IQR. Act., activation; aggreg., aggregation; imm., immunity; med, mediated; MK, mekakaryocytes; resp., response; UT, untreated. Panel a was created with BioRender.com.
We performed bulk transcriptomic analysis of UBA1mut and UBA1wt HSPCs65. Although only one gene was differentially expressed 24 h after editing (Extended Data Fig. 2d, left), 264 genes were differentially expressed (differentially expressed genes (DEGs)) at 7 days (Extended Data Fig. 2d, right and Supplementary Table 1). Enrichment analysis showed upregulation of genes related to innate immunity and neutrophil function and downregulation of genes governing platelet activation (Fig. 2m). Metabolic analysis on the same samples showed decreased ATP and upregulation of AMP, and accumulation of the glycolytic intermediate d-glyceraldehyde 3-phosphate (Fig. 2n). We then compared the proficiency of UBA1mut and UBA1wt HSPCs to sustain multilineage differentiation in vitro66,67. The total cell output was lower for UBA1mut HSPCs (Fig. 2o), with impaired generation of megakaryocytes, erythroid cells and pre-T cells but preserved myeloid and NK cells (Fig. 2p). Concordantly, UBA1mut HSPCs showed an exclusive myeloid colony-forming potential (Fig. 2q). Conversely, UBA1-knockout (UBA1KO) HSPCs, generated by Cas9-mediated disruption, were unable to make colonies, strengthening the value of inserting the VEXAS mutation rather than disrupting the UBA1 gene. When inserting the VEXAS mutation in male T cells from healthy donors, we observed apoptosis-driven loss of viability and counterselection of UBA1mut cells (Extended Data Fig. 2e–g). These findings indicate that the VEXAS mutation p.Met41Thr constrains in vitro HSPC differentiation and hampers T cell survival, in agreement with anemia, thrombocytopenia and lack of UBA1mut lymphoid cells in patients17,29.
Engrafted UBA1mut HSPCs lack multilineage potential and exclusively contribute to myelopoiesis
Next, we generated a humanized model of VEXAS syndrome by transplanting UBA1mut (>80%) or control base-edited HSPCs (UBA1wt) into immunodeficient mice (Fig. 3a). HSPCs were injected 1 day after editing, when the transcriptome was not perturbed and cells could home to the BM (Extended Data Fig. 3a). We observed a 10- to-50-fold reduction in human cell content in UBA1mut mice compared with controls (Fig. 3b). This was the result of a dramatic shrinkage of the B cell compartment (Fig. 3c), usually the most abundant in hematochimeric mice, whereas myeloid and NK cell outputs were more preserved (Fig. 3d,e). We also found a tendency toward lower relative abundance of human HSPCs in the BM of UBA1mut mice (Fig. 3f). More than 90% CD34+ HSPCs in the UBA1mut group co-expressed the myeloid marker CD13, supporting the acquisition of a myeloid-committed phenotype (Fig. 3g). Targeted sequencing showed >80% UBA1mut cells in HSPCs and the myeloid compartment, and predominance of UBA1wt cells in the lymphoid one (Fig. 3h, i). Recapitulation of a lineage repopulation pattern characteristic of patients with VEXAS syndrome was observed only when inserting the mutation by base editing. Indeed, transplantation of UBA1KO HSPCs resulted in the counterselection of UBA1KO cells and did not model the functional hematological consequences of VEXAS syndrome (Fig. 3j and Extended Data Fig. 3b).

a, Schematic representation of the in vivo transplantation experiment. b, Percentage of human cells in PB, spleen and BM of hematochimeric mice transplanted with UBA1mut or UBA1wt HSPCs (n = 12 and 11). The median is shown with the IQR. c–e, Percentage of human B cells (c), myeloid cells (d) and NK cells (e) within total cells in PB and hematopoietic organs of mice from b (n = 12 and 11). The median is shown with the IQR. f, Percentage of human HSPCs in the BM of mice from b (n = 12 and 11). The median is shown with the IQR. g, Percentage of cells within human CD34+ HSPCs expressing the CD19 or CD13 markers, or none of them, in mice from b (n = 12 and 11). Values given are mean ± s.e.m. h,i, Percentage of UBA1mut cells across hematopoietic populations in spleen (h) and BM (i) of mice from b (n = 12, 11). The median is shown with the IQR. j, Percentage of cells bearing insertions and deletions in the infused HSPC edited product (n = 1) and in the spleen and BM of hematochimeric mice 14 weeks after transplantation (n = 4 per group). The median is shown with the IQR. i, The Kruskal–Wallis test was used and, for all other panels, the Mann–Whitney U-test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Ctrl, control. Panel a created with BioRender.com.
UBA1mut triggers pervasive inflammatory signatures and cell-cycle slowdown in human hematopoiesis in mice
To characterize the VEXAS xenograft model, we performed single-cell RNA sequencing (scRNA-seq) on human BM cells from UBA1mut and UBA1wt mice. We annotated 18 clusters, including both progenitor and differentiated cells (Fig. 4a). Pseudotime analyses68 traced eight branches of differentiation. The lymphoid and erythroid trajectories were arrested prematurely in the UBA1mut xenograft compared with controls (Fig. 4b), suggesting impaired differentiation of lymphoid progenitors and ineffective erythropoiesis in the xenograft model of VEXAS syndrome.

a, Uniform manifold approximation and projection (UMAP) plot of human CD45+ cells, enriched in the CD34+ HSPC fraction, from the BM of mice transplanted with 100% UBA1wt or UBA1mut HSPCs. Clusters and associated cell types are indicated by name and color. b, B cell (top) and erythroid (bottom) pseudotime trajectories starting from the HSC or MPP cluster in the UMAP plot (left) and distribution of cells along the trajectory (right) from a. c, Heatmap showing normalized enrichment scores (NESs) for the GSEA performed within the differentiated clusters comparing 100% UBA1mut versus UBA1wt xenografts. Gray squares represent a mismatch between the DEGs of a selected cluster and the gene set as a result of the threshold for detecting the DEGs and the minimum gene set size for the calculation of the enrichment score. Wilcoxon’s rank-sum test with the Benjamini–Hochberg correction was used. d, Distribution of cells in G1, S or G2/M phases of the cell cycle within differentiated cell cluster and sample according to signatures from ref. 87. e, UMAP plot of human CD34+ HSPCs from the BM of mice transplanted with 100% UBA1wt or 40% UBA1mut HSPCs. f, Distribution of cells across HSPC clusters from scRNA-seq in e. g, Heatmap as in c showing NESs for the GSEA performed within HSPC clusters in e comparing 40% UBA1mut versus 100% UBA1wt xenografts. Wilcoxon’s rank-sum test with the Benjamini–Hochberg correction was used. h, Distribution of cells in G1, S or G2/M phases of the cell cycle within HSPC clusters in e. i, Density plots showing the distribution of cells from the 100% UBA1wt and 40% UBA1mut groups on the BM reference map from Extended Data Fig. 3e. j,k, AUCell scores based on a monocyte signature from ref. 69 (j) and aged HSC signatures from refs. 71,72 (k) within primitive HSCs or MPPs according to projection on the BM reference map (n = 1,728 and 532 cells). The whiskers are located at 1.5× the IQR and the dots represent outliers. Wilcoxon’s rank-sum test with the Benjamini–Hochberg correction was used. **P < 0.01; ****P < 0.0001. Baso, basophils; conv, conventional; EMT, epithelial-to-mesenchymal transition; imm, immature; junc, junction; lympho, lymphoid; mast, mast cells; met, metabolism; myelo, myeloid cells; prec, precursors; prog, progression; prolif, proliferative; rej, rejection; sign, signalling; UPR, unfolded protein response.
Profound alteration of gene expression was found between UBA1mut and UBA1wt mice within the differentiated clusters (Supplementary Table 2). Gene set enrichment analysis (GSEA) within myeloid cells in VEXAS mice showed robust upregulation of genes belonging to inflammatory categories (for example, interferon-α (IFNα)-, IFNγ- and tumor necrosis (TNF)-mediated nuclear factor-κB (NF-κB) signaling) (Fig. 4c), accompanied by upregulation of apoptotic genes and negative enrichment of cell-cycle-related signatures. Concordantly, UBA1mut cells were more frequently in the G1 phase than UBA1wt across all differentiated clusters (Fig. 4d). Despite UBA1mut cells being counterselected in B cell subpopulations, the differentiated lymphoid compartments showed transcriptomic changes comparable to myeloid cells, although to a milder extent (Fig. 4c).
Primitive UBA1mut HSCs are inflamed and myeloid biased and display signatures of senescence and accelerated aging
To deepen our analysis of HSPCs, we performed scRNA-seq on sorted human CD34+ HSPCs from the BM of UBA1mut and UBA1wt mice. We identified 14 HSPC clusters (Fig. 4e), including primitive (HSCs or MPPs) and committed progenitors (Supplementary Table 3). In agreement with patients’ data, we found a relative increase of myeloid progenitors, a loss of lymphoid and erythroid progenitors and a reduction of the HSC or MPP cluster by 1.6-fold in UBA1mut HSPCs (Fig. 4f). Pseudotime trajectories within HSPCs confirmed the premature block of lymphoid and erythroid differentiation and the enhancement of the myelo-GMP trajectory (Extended Data Fig. 3c).
GSEA revealed pervasive upregulation of genes belonging to inflammation-related categories and downregulation of cell-cycle-related ones in UBA1mut versus UBA1wt HSPCs, with the more committed subsets showing significant enrichment for proapoptotic pathways (Fig. 4g and Supplementary Table 4). Concordantly, UBA1mut clusters were mostly in the G1 phase (Fig. 4h). Among GMP clusters, GMP-2 was enriched in UBA1mut cells and showed an inflammatory profile and lower expression of cell-cycle genes (Extended Data Fig. 3d). Conversely, GMP-1 was enriched in UBA1wt cells and exhibited a higher abundance of proliferating cells (Fig. 4h). Finally, the transcriptional perturbations in UBA1mut erythroid progenitors mainly involved upregulation of genes related to ineffective erythropoiesis (‘heme metabolism’, Fig. 4g).
We projected the scRNA-seq HSPC dataset on to a reference map of human BM69 (Extended Data Fig. 3e), confirming the differences in cell distribution between UBA1mut and UBA1wt xenografts (Fig. 4i and Extended Data Fig. 3f). When probing HSC or MPP cells with a monocyte-derived signature69, we observed positive enrichment in UBA1mut (Fig. 4j), suggesting early priming of VEXAS HSCs or MPPs toward myeloid differentiation. Again, genes belonging to inflammatory and apoptotic pathways were more expressed in primitive cells in UBA1mut xenografts, whereas cell-cycle-related genes were downregulated (Extended Data Fig. 3g,h). As inflammation has been associated with accelerated HSC aging70, we probed HSCs or MPPs and bulk HSPCs with two signatures from aged human HSCs71,72 and found significant positive enrichment in UBA1mut compared with the control (Fig. 4k and Extended Data Fig. 3i). Senescence-associated signatures73,74,75 were also enriched in VEXAS xenografts (Extended Data Fig. 3j).
These data show that the VEXAS mutation rewires the transcriptional programs of hematopoiesis, activating pervasive inflammatory signatures. Primitive UBA1mut HSCs are less prone to engage cell-cycle progression and display signatures of senescence and are primed toward myelopoiesis.
Proinflammatory signatures in patients are phenocopied by the xenograft model
To dissect to what extent the VEXAS xenograft recapitulates the perturbations of hematopoiesis observed in patients, we performed scRNA-seq on BM mononuclear cells (BMMCs) from six patients, pre-enriched in CD34+ HSPCs, and integrated and harmonized data with published datasets from patients (n = 9) and age-matched controls (n = 4)44. We identified 23 subsets corresponding to various stages of hematopoietic differentiation, including one GMP and one NK cell patient-specific cluster (Fig. 5a).

a, UMAP plot of hematopoietic cells from the BM of patients with VEXAS syndrome from the San Raffaele (PT1, PT2, PT3, PT5, PT8 and PT9) and Wu et al.44 cohorts and age-matched controls. Clusters and associated cell types are indicated by name and color. b, Heatmap as in Fig. 4c showing the NESs for the GSEA performed within the CD34− clusters from a. Wilcoxon’s rank-sum test with the Benjamini–Hochberg correction was used. c, Volcano plots showing fold-changes of upregulated (red) and downregulated (green) genes comparing patients with VEXAS syndrome and controls within the ‘late erythroid cell’ cluster. Nonsignificant genes are shown in gray. Wilcoxon’s rank-sum test with the Benjamini–Hochberg correction was used. d, UMAP plot of human CD34+ cells from the BM of patients with VEXAS syndrome (San Raffaele and Wu et al.44 cohorts) and age-matched controls44,72. Clusters and associated cell types are indicated by name and color. ‘VEXAS-enr’ labels cluster with predominance of cells from patients with VEXAS syndrome. e,f, Heatmap as in Fig. 4c showing NESs for the GSEA performed within the CD34+ clusters from d in all patients (e) or in patients with the threonine mutation (f). Wilcoxon’s rank-sum test with the Benjamini–Hochberg correction was used. g,h, UCell scores based on the ‘VEXAS xenograft signature’ within CD34+ (g) and CD34− (h) clusters from patients with VEXAS syndrome and controls. The signature was built considering the top-50 upregulated genes in UBA1mut monocytes compared with the UBA1wt counterpart. Wilcoxon’s rank-sum test with the Holm–Bonferroni correction was used. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. NS, nonsignificant; ery, erythroid; hemato, hematopoietic; hom, homeostasis.
GSEA on patients’ CD34− clusters (Extended Data Fig. 4a) highlighted striking similarities to the xenograft model. Inflammatory categories were significantly upregulated across all myeloid subpopulations (Fig. 5b and Supplementary Table 5). Bulk transcriptomic and metabolomic analyses on circulating monocytes confirmed activation of inflammation-related gene signatures and immunometabolic pathways (Extended Data Fig. 4b,c). Apoptotic transcriptional signatures were also enriched in the myeloid clusters, paralleled by downregulation of cell proliferation categories (Fig. 5b) and cell-cycle slowdown (Extended Data Fig. 4d). A concordant, albeit milder, pattern was observed even in T cells and B cells despite being predominantly UBA1wt (Fig. 1h), hinting at a paracrine effect of proinflammatory mutant cells on the wild-type counterpart in patients (Fig. 5b). Similarly, endothelial cells, assumed to be UBA1wt and carried on as contaminants during BMMC processing, showed upregulation of genes related to inflammatory categories, suggesting that nonhematopoietic cells might also be affected by a proinflammatory milieu (Fig. 5b). Moreover, we found robust enrichment of the ‘heme metabolism’ category in the late stages of erythroid differentiation in patients driven by elevated expression of genes involved in erythroid differentiation and hemoglobin synthesis (for example, HBG1/2, HBB and HBD), iron homeostasis (NCOA4 and SLC25A37) and regulation of stress responses (GLRX5 and EIF2AK1) (Fig. 5b,c). These alterations, reproduced in the xenograft model, further support a putative attempt to compensate ineffective erythropoiesis and anemia caused by the impaired ubiquitin–proteasome system in patients. Transcriptomic changes were consistent between the San Raffaele and Wu et al.44 cohorts, albeit more exacerbated in the latter (Extended Data Fig. 4e), possibly as a result of different treatments and/or disease stage.
To gain insights into genotype–phenotype correlation possibly explaining heterogeneous clinical manifestations, we segregated patient data according to the UBA1-mutation and compared them with controls. The p.Met41Thr and p.Met41Leu mutations contributed the most to the inflammatory signatures, whereas p.Met41Thr and p.Met41Val manifested more robust activation of responses related to altered erythropoiesis (Extended Data Fig. 4f). This evidence suggests that distinct UBA1-mutations may selectively affect the amplitude and the nature of pathogenic manifestations across lineages.
To comprehensively analyze HSPCs in patients, we selected cells from the clusters associated with HSPC signatures from the two VEXAS datasets above, and we included three additional age-matched controls from another dataset72. We annotated 19 populations, 5 of which predominantly, but not exclusively, comprised VEXAS cells (Fig. 5d) and belonged to different stages of erythroid differentiation. GSEA revealed pervasive upregulation of inflammatory responses, negative enrichment of cell-cycle categories in some clusters and upregulation of apoptosis in the more committed subsets (Fig. 5e). It is interesting that NR4A1 and SOCS3, previously described as promoting resilience and driving dominance of asxl1-mutant HSPCs in a zebrafish model of CHIP76, were among the significantly upregulated genes in patients compared with controls (Extended Data Fig. 4g). All transcriptomic changes were exacerbated in patients with p.Met41Thr (Fig. 5f). Intriguingly, senescence gene profiles73,74,75, as well as a VEXAS xenograft signature, were enriched in the patients across most clusters (Fig. 5g,h and Extended Data Fig. 4h).
Overall, these results demonstrate that the VEXAS xenograft model phenocopies most transcriptomic perturbations of hematopoiesis found in patients.
Resilient, proinflammatory UBA1mut cells poison hematopoiesis and drive clonal dominance
We leveraged our model to investigate whether UBA1mut HSPCs expand over time owing to a cell-intrinsic proliferative surplus and/or whether bystander effects triggered by the proinflammatory progeny of UBA1mut HSPCs promote clonal dominance. We performed competitive transplantations of UBA1mut and UBA1wt HSPCs at different ratios, mimicking the stages of disease progression (Fig. 6a). We found that the absolute number of human cells was reduced when transplanting ≥40% UBA1mut HSPCs (Fig. 6b,c). Conversely, the frequencies of human on live (murine and human) cells were reduced only in the 100% UBA1mut group (Fig. 6d,e). These data suggest a detrimental bystander effect of human UBA1mut cells on mouse hematopoiesis (Fig. 6f). The myeloid:lymphoid ratio within human cells progressively increased with the rising percentage of infused UBA1mut HSPCs (Fig. 6g). Although lymphoid subpopulations were reduced in UBA1mut groups, the myeloid subpopulations and CD34+ HSPCs shrank only and progressively in the 40% and 100% UBA1mut groups (Fig. 6h–j). The analysis of the HSPC compartment showed a progressive skewing of its composition toward CMPs, GMPs and ETPs at an increasing input of UBA1mut HSPCs (Fig. 6k). These data suggest a nonlinear, nearly exponential dose–response with mutant clones destabilizing human hematopoiesis when accounting for >25% UBA1mut HSPCs.

a, Schematics of the in vivo competitive transplantation experiment mixing UBA1mut and UBA1wt HSPCs at different ratios. b,c, Number of human cells from the different hematopoietic compartments in the BM (b) and PB (c) of mice transplanted as shown in a (n = 5 per group). The median is shown with the IQR. d,e, Percentage of human hematopoietic cells within total live cells in the BM (d) and PB (e) of mice from b (n = 5 mice per group). The values are the mean ± s.e.m. f, Number of human and mouse cells in the BM of mice from b (n = 5 mice per group). The median is shown with the IQR. g, Ratio between the number of myeloid and lymphoid cells (n = 5 mice per group). The median is shown with the IQR. h,i, Number of lymphoid (h) and myeloid (i) cell subpopulations in mice from b (n = 5 mice per group). The median is shown with the IQR. j, Number of CD34+ HSPCs in mice from b (n = 5 mice per group). The median is shown with the IQR. k, Percentage of HSPC subpopulations in the BM of mice from b (n = 5 mice per group). The right panel zooms in the most primitive HSPC compartment. The values are the mean ± s.e.m. l–n, Number of UBA1mut and UBA1wt HSPCs (l), myeloid cells (m) and B cells (n) (n = 5 mice per group). The median is shown with the IQR. o, Percentage of UBA1mut cells within each hematopoietic subpopulation in the mixed groups (n = 5 mice per group). Dotted squares indicate the percentage of infused UBA1mut HSPCs. The median is shown with the IQR. p, Concentrations of human cytokines in the BM of UBA1mut and UBA1wt mice 4 weeks after transplantation (n = 3, 4). q, UMAP plot of human CD45+ cells, enriched in the CD34+ HSPC fraction, from the BM of mice transplanted with 50% UBA1wt or UBA1mut HSPCs. Clusters and associated cell types are indicated by name and color. r, Heatmap as in Fig. 4c showing NESs for the GSEA performed within clusters in q comparing UBA1wt female cells from VEXAS mice versus UBA1wt female cells from wild-type mice. Wilcoxon’s rank-sum test with the Holm–Bonferroni correction was used and, for all other panels, the Kruskal–Wallis test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Panel a created with BioRender.com.
In the 25% UBA1mut group, UBA1 genotyping of sorted subpopulations showed a stable proportion of UBA1mut HSPCs, a tendency toward enrichment of UBA1mut myeloid cells and a decrease in lymphoid cells consistent with the loss of lymphoid differentiation potential by the transplanted 25% UBA1mut HSPCs (Fig. 6l–o). Intriguingly, both HSPC and myeloid compartments of the 40% UBA1mut group showed dominance, through remarkable enrichment of UBA1mut cells owing to the loss of the UBA1wt counterpart (Fig. 6l,m,o). The B cell compartment, comprising only UBA1wt cells, was shrunk despite the initial proportion of transplanted UBA1wt HSPCs (Fig. 6n,o). These data suggest that (1) the loss of UBA1wt contributes to graft shrinkage at an increasing proportion of UBA1mut cells and (2) the higher resilience of UBA1mut cells to a ‘poisonous’ cell-extrinsic effect, rather than a cell-autonomous proliferative surplus of UBA1mut progenitors, leads to clonal dominance.
To uncover the ‘poisoning’ factors, we compared the abundance of proinflammatory cytokines in the BM of 40% UBA1mut and UBA1wt mice 4 weeks after transplantation, when the BM niche is being repopulated (Extended Data Fig. 5a). We found increased levels of human interleukin-1β (IL-1β), IL-18, CXCL10 and IL-1 receptor antagonist (IL-1RA) in UBA1mut mice (Fig. 6p), which is suggestive of inflammasome activation, as also reported in patients46. Despite the evidence of inflammatory hematopoiesis, histopathological analyses did not show signs of organ inflammation (Supplementary Fig. 2).
To confirm that this milieu can affect human bystander cells in the BM, we profiled the transcriptome of human hematopoiesis in xenotransplanted mice 6 weeks after competitive transplantation (Fig. 6q). Mice were transplanted with female UBA1wt HSPCs in competition with either male UBA1mut or male UBA1wt HSPCs to distinguish mutant and wild-type cells a posteriori by XIST expression. In agreement with the results above, we found robust activation of inflammatory programs and downregulation of cell-cycle-related genes in VEXAS versus wild-type mice (Extended Data Fig. 5b,c). It is interesting that female UBA1wt cells engrafted with male UBA1mut cells showed robust activation of inflammatory pathways and downregulation of cell-cycle-related genes (Fig. 6r), proving a paracrine effect of mutant hematopoiesis. The IFN signature was lower in UBA1mut than UBA1wt HSCs, MPPs or GMPs (Extended Data Fig. 5d,e), suggesting that UBA1mut progenitors might be less responsive to inflammatory stimuli. Of note, both UBA1mut and UBA1wt HSPCs harvested from the BM of VEXAS mice 7 weeks after transplantation retained the capacity to engraft into secondary recipients (Extended Data Fig. 5f–h), suggesting that HSPC function was not fully compromised by the inflammatory milieu.
We then assessed the clonogenic capacity of UBA1wt HSPCs on prolonged exposure to the inflammatory milieu. UBA1wt HSPCs labeled with a green fluorescent protein (GFP)-expressing lentiviral vector were co-transplanted with either UBA1mut or UBA1wt HSPCs labeled with a blue fluorescent protein (BFP)-expressing lentiviral vector (Extended Data Fig. 5i). After 14 weeks, BM CD34+ GFP+ cells were sorted and assayed for the ability to form colonies in vitro. The clonogenic yield was ∼50% lower for UBA1wt HSPCs from VEXAS versus wild-type mice (Extended Data Fig. 5j), pointing to partially impaired clonogenicity of UBA1wt HSPCs on exposure to the VEXAS milieu. Notably, the detrimental microenvironment also affected bystander murine HSPCs in VEXAS mice (Extended Data Fig. 5k,l and Supplementary Table 6), leading to upregulation of the inflammatory response and activation of proapoptotic and p53 target genes (Extended Data Fig. 5l). Cell proliferation-related signatures were upregulated in bystander, wild-type murine HSCs or MPPs exposed to human UBA1mut cells.
Finally, transplantation of allogenic UBA1wt GFP+ HSPCs in conditioned VEXAS mice to model HSCT showed efficient engraftment of donor-derived cells and eradication of UBA1mut cells (Extended Data Fig. 6a–d), proving that (1) allo-HSCT can eradicate VEXAS clones and (2) our xenograft model can be leveraged to investigate therapeutic approaches for VEXAS syndrome.
Overall, we uncovered the poisonous paracrine activity of UBA1mut cells against wild-type HSPCs as a major driver of clonal dominance in VEXAS pathogenesis (Extended Data Fig. 7 and Extended Data Table 2).
Discussion
Our study deciphers the hallmarks and mechanisms of clonal dominance in VEXAS syndrome, showing that the homeostasis of VEXAS hematopoiesis is subverted by inflammation-resilient, UBA1-mutant HSPCs, which bear dysfunctional differentiation properties and, concurrently, undermine contribution and long-term maintenance of wild-type HSPCs through their proinflammatory progeny.
Clonal dominance has often been ascribed to cell-intrinsic expansion capacity caused by proliferative advantage and enhanced self-renewal. Our data challenge this hypothesis by supporting the establishment of a cell-extrinsic inflammatory circuit more harmful to wild-type than to mutant cells in VEXAS syndrome. VEXAS syndrome may thus represent a paradigm of acquired disorders driven by mutant proinflammatory clones attaining the ability to partially protect themselves from the poisoning microenvironment that they have shaped. The mechanism of clonal dominance described for VEXAS syndrome may underly evolution and/or resistance in acquired BM failure syndromes77, CHIP78, MDS79, inflammatory myeloid neoplasms80,81 and other hematological malignancies, and be portable to physiological aging of other tissues. The VEXAS paradigm of clonal dominance shares similarities with a zebrafish model of ASXL1-driven CHIP76, wherein mutant clones displayed more enhanced fitness than wild-type cells and malignant histiocytosis, wherein senescence establishment and non-cell autonomous inflammation poison hematopoiesis81. UBA1-mutant clones may acquire a resilient state by entering dormancy70 or undergoing senescence, which may shield mutant cells from inflammation-induced apoptosis and immune clearance82,83. The reduced propensity of UBA1-mutant progenitors to proliferate may explain the relatively low incidence of hematological cancers in patients with VEXAS syndrome, despite the high prevalence of MDS and co-occurrence of CHIP mutations19,30,31.
Besides driving clonal dominance, the VEXAS inflammatory milieu probably orchestrates early acquisition of the myeloid bias and drives premature aging of hematopoiesis. Exposure to IL-1β or chronic inflammatory stimuli is known to prime primitive HSCs toward myelopoiesis, erode their self-renewing capacity and accelerate aging70,84,85. IL-1β is also a prominent component of the senescence-associated secretory phenotype, the factors of which can affect bystander cells82. Notably, our findings are consistent with the accelerated pace of aging found by DNA methylation clock analyses in patients19.
Single-cell transcriptomic and immunophenotypic analyses described progenitor- and lineage-specific perturbations and adaptive responses to the pleiotropic effect of the UBA1-mutation within the VEXAS hematopoietic landscape. Being pathognomonic of ineffective hematopoiesis, investigation in larger patient cohorts of parameters, such as the myeloid:lymphoid ratio, the abundance of primitive cells in the BM and the increased circulating HSPCs, may identify prognostic factors of disease severity and progression. Enhanced egression of HSPCs from the BM may contribute to the progressive BM failure in VEXAS syndrome by shrinking the reservoir of long-term HSCs. Future studies may further investigate the underlying mechanisms and their role in disease progression.
Our study holds translational implications for the treatment of VEXAS syndrome and, possibly, of other conditions associated with acquired BM failure and clonal hematopoiesis. Combined targeted therapies blunting systemic inflammation and erasing the resilient mutant clones might be more effective than single treatments because they would address both pathogenic components of the disease. Coupling early molecular diagnosis and effective therapies before the mutant clones subvert hematopoiesis may slow down disease progression and reduce its clinical burden. Early combined anti-inflammatory and clone-eradicating treatment strategies might reset the inflammatory milieu, deplete the mutant clones and prevent secondary mutations19 and progression to MDS. Future studies elucidating when UBA1-mutations are acquired during life and the pace of hematopoietic erosion will help to tailor the type, timing and strength of therapeutic interventions.
Despite the humanized model of VEXAS syndrome faithfully phenocopying molecular, cellular and hematological hallmarks of the disease, it lacks overt inflammation and tissue damage in nonhematopoietic murine organs. This limitation might be ascribed to poor infiltration of murine organs by human cells, incomplete maturation of human myeloid cells, partial cytokine-receptor crosstalk between human hematopoiesis and murine parenchymal cells and/or limited experiment duration. Prospectively, the use of other mouse strains or HSPC sources, as well as more prolonged exposure to UBA1mut cells, might overcome this limitation. Despite engraftment of HSPCs from patients with MDS having been recently reported86, our strategy overcomes paucity of patient samples and interpatient variability. Moreover, it allows the establishment of diseased hematopoiesis by transplanting HSPCs early after acquisition of the somatic mutation, allowing full deployment of its pathogenic consequences after the cells have homed to the BM. The latter aspect, together with the partial exhaustion or differentiation of residual UBA1wt HSPCs, probably explains why CD34+ HSPCs from patients showed poorer engraftment capacity in mice than VEXAS-like HSPCs generated by editing. At variance with other in vitro modeling strategies45, the development of humanized models offers the opportunity to expand the breadth of studies for VEXAS syndrome and beyond, with the goal of identifying disease-modifying therapies through preclinical testing of small molecules, biologic agents and gene therapies.
Methods
Study participants and ethics oversight
Collection of clinical data from patients was approved by the San Raffaele Ethical Committee (PanImmuno protocol). Biological samples from male patients with VEXAS syndrome and male age-matched controls were collected on written informed consent, in compliance with the TIGET09 ethical protocol, approved by San Raffaele Ethical Committee. Sex was determined by physical examination. Gender was not considered because it was not relevant for the purpose of the study. As VEXAS syndrome is a somatic X-linked disease that predominantly, but not exclusively, affects men, male cells were used throughout the study, unless otherwise specified in the text and needed for experimental purposes. Buffy coats were obtained in accordance with the Declaration of Helsinki, as anonymized residues of blood donations, used on signature of specific institutionally informed consent for blood product donation by healthy blood donors. Purchase and isolation of human HSPCs from healthy donors were done according to the TIGET-HPCT protocol, approved by OSR Ethical Committee. All experiments and procedures involving animals were performed with the approval of the Animal Care and Use Committee of the San Raffaele Hospital (IACUC nos. 1206, 1358, and 1535) and authorized by the Italian Ministry of Health.
Plasmids and gRNAs
The ABE8.20-m plasmid was a gift from N. Gaudelli (Addgene plasmid, cat. no. 136300; http://n2t.net/addgene:136300). The plasmid for mRNA in vitro transcription (IVT) was generated in ref. 53. The AAVS1 and B2M gRNAs used as mock-edited controls were previously reported53. The UBA1 gRNA used to insert the p.Met41Thr mutation and the UBA1 gRNA used for gene knockout are listed in Supplementary Table 7.
IVT of mRNA
The ABE8.20-m plasmid was linearized with SpeI and purified by phenol–chloroform extraction. Different preps of mRNAs were in vitro transcribed using the commercial 5X MEGAscript T7 kit (Thermo Fisher Scientific). The ABE8.20-m mRNA was capped with 8 mM CleanCapAG (Trilink). The mRNAs were purified using RNeasy Plus Mini Kit (QIAGEN), followed by high-performance liquid chromatography (HPLC; ADS BIOTEC WAVE System) and concentration with Amicon Ultra-15 (30-kDa) tubes (Millipore). To assess quality and integrity, we denatured and resolved the mRNAs by capillary electrophoresis on 4200 TapeStation System (Agilent) according to the manufacturer’s instructions. The mRNA was then aliquoted and stored at −80 °C.
Isolation of patients’ cells
BMMCs were purified using SepMate PBMC Isolation Tubes (STEMCELL Technologies), according to the manufacturer’s instructions. In brief, density gradient medium (Lymphoprep) was added through the SepMate insert and whole BM blood, diluted at a 1:1 v:v ratio with sterile PBS 1× containing 2% fetal bovine serum (FBS), was laid on it. On centrifugation for 10 min at 2,500 rpm (brake on), BMMCs were transferred into a new tube and washed with PBS 1×. The red blood cells were lysed using ammonium–chloride–potassium lysing buffer and the BMMCs resuspended in specific buffer according to the subsequent experimental procedure.
Primary cell culture
Human primary T cells were isolated from male healthy donors’ peripheral blood mononuclear cells (PBMCs) freshly purified from buffy coats with SepMate PBMC Isolation Tubes, according to the manufacturer’s instructions. CD3+ T cells were cultured in X-Vivo 15 (Lonza) supplemented with 0.5% human serum albumin (HSA, Baxalta), 100 IU ml−1 of penicillin, 100 µg ml−1 of streptomycin, 100 IU ml−1 of IL-7 and 200 IU ml−1 of IL-15 (both from Miltenyi Biotec) and stimulated with T Cell TransAct (Miltenyi Biotec) at 2 × 106 cells ml−1.
Granulocyte–colony-stimulating factor (G-CSF) + plerixafor mobilized (m)PB CD34+ HSPCs from male, unless otherwise specified, healthy donors were purified in house with the CliniMACS CD34 Reagent System (Miltenyi Biotec) from Mobilized Leukopak (AllCells), following the manufacturer’s instructions. HSPCs were seeded at the concentration of 5 × 105 cells ml−1 in serum-free StemSpan SFEM supplemented with 100 IU ml−1 of penicillin, 100 μg ml−1 of streptomycin, 2% glutamine, 300 ng ml−1 of human stem cell factor (hSCF), 300 ng ml−1 of human Fms-related tyrosine kinase 3 ligand (hFLT3L), 100 ng ml−1 of human thyroid peroxidase (hTPO) and 10 μM 16,16-dimethylprostaglandin E2 (added at the beginning of the culture), 1 μM SR1 and 35 nM UM171.
BM HSPCs from male healthy donors (Lonza) and patients with VEXAS syndrome were not cultured before infusion in immunodeficient mice.
All cells were cultured in a 5% CO2 humidified atmosphere at 37 °C.
Clonogenic assay
A colony-forming cell assay was performed 24 h after the editing procedure by plating 400–800 cells in methylcellulose-based medium (MethoCult H4434, STEMCELL Technologies) supplemented with 100 IU ml−1 of penicillin and 100 µg ml−1 of streptomycin. Three technical replicates were performed for each condition. Two weeks after plating, colonies were counted.
Mice
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) and NOD.Cg-KitW-41J Tyr+ Prkdcscid Il2rgtm1Wjl/ThomJ (NBSGW) 7- to 10-week-old female mice (Jackson Laboratory) were held in specific pathogen-free conditions with a 12 h:12 h dark:light cycle and standardized temperature (22 ± 2 °C) and humidity (55 ± 5%). Only female mice were used because they allow better human cell engraftment.
Gene editing of human T cells and analyses
Approximately 1.0 × 106 human T cells, were washed with 10 volumes of Dulbecco’s PBS (DPBS) without Ca2+ and Mg2+ and electroporated using the P3 Primary Cell 4D-Nucleofector X Kit (Lonza) and program DS-130. Cells were electroporated with 75 pM UBA1 or B2M gRNA (Synthego) and 1.5 μg of mRNAs unless otherwise specified. Cells were cultured for approximately 2 weeks. Cell pellets were harvested and the gDNA extracted for molecular analysis.
Gene editing of human HSPCs
From 2.0 × 105 to 7.5 × 105 mPB HSPCs were washed with 10 volumes of DPBS without Ca2+ and Mg2+ and electroporated using the P3 Primary Cell 4D-Nucleofector X Kit and program EO-100 after 3 days of culture. HSPCs were electroporated with 75 pmol of gRNA and 3.5 μg of ABE8.20-m or Cas9 mRNAs. Then, 7 days after electroporation, HSPCs were collected for flow-cytometry analyses and to extract genomic gDNA for molecular analysis.
Western blot analysis
HSPCs and T cells were cultured as described above for 7 days and 4 days, respectively before being lysed in lysis buffer (10 mM Tris-HCl, 150 mM NaCl and 1% sodium dodecylsulfate). Proteins (25 μg) were separated on poly(acrylamide) gels and transferred to nitrocellulose (iblotTM Transfer Stack, Thermo Fisher Scientific) for western blotting. UBA1a, UBA1b, UBA1c, ubiquitin, BiP and cleaved caspase-3 were detected by western blotting with the following antibodies: rabbit polyclonal anti-UBE1a/b (Cell Signaling Technology, cat. no. 4891), mouse monoclonal anti-ubiquitin (Cell Signaling Technology, cat. no. 14049 (P4D1)), rabbit monoclonal anti-Bip/GRP78 (Cell Signaling Technology, cat. no. 3177 (C50B12)) and rabbit monoclonal anti-cleaved caspase-3 (Cell Signaling Technology, cat. no. 9664 (5A1E)). Western blotting with β-tubulin (Sigma-Aldrich, cat. no. T4026) or β-actin (Sigma-Aldrich, cat. no. A2228 (AC-74)) antibodies was used to verify equal protein loading. Chemiluminescence detection was performed using the SuperSignal West Pico PLUS substrate (Thermo Fisher Scientific).
Modified Giemsa staining and optical microscopy
UBA1wt and UBA1mut HSPCs were centrifuged on glass slides and stained with the differential quick stain kit (modified Giemsa) following the manufacturer’s instructions (‘Diff-Quick’ kit; Polysciences). Stained UBA1wt and UBA1mut HSPCs were examined by optical microscopy. Images were taken with a ×100 objective on a Zeiss AxioImagerM2m (AxioCam MRc5).
Transmission electron microscopy
Cells for TEM analysis were fixed using a solution containing 2.5% glutaraldehyde in 0.1 M cacodylate buffer at pH 7.3 for 1 h, washed, post-fixed with 1% buffered osmium tetroxide for 1 h and stained overnight with 0.5% Millipore-filtered uranyl acetate. Dehydration was then performed using increasing concentrations of ethanol, followed by infiltration and embedding in Epon. Polymerization was carried out at 60 °C for 2 days. Ultrathin sections were obtained using a Leica EM FC7 ultramicrotome. After staining with uranyl acetate and lead citrate, the sections were analyzed using a TALOS L120C transmission electron microscope (Thermo Fisher Scientific) and images were acquired with a CETA 4 × 4k CMOS camera (Thermo Fisher Scientific). The Microscopy Image Browser (MIB) was used for analyzing mitochondria.
Ultra-HPLC–MS metabolomics and tracing experiments
Approximately 1 × 106 cells were pelleted and extracted in 1,000 µl of ice-cold extraction solution (methanol:acetonitrile:water, 5:3:2 v:v:v)88. Suspensions were vortexed continuously for 30:min at 4 °C. Insoluble material was removed by centrifugation at 18,000g for 10 min at 4 °C and supernatants were isolated for metabolomics analysis by UHPLC–mass spectrometry (MS)
Analyses were performed using a Vanquish UHPLC coupled online to a Q Exactive mass spectrometer (Thermo Fisher Scientific). Sample extracts, 10 μl, were loaded on to a Kinetex XB-C18 column (150 × 2.1 mm2, inner diameter 1.7 µm; Phenomenex). Samples were analyzed using a 5-min method as described88,89. Metabolite assignments were performed using MAVEN90. Graphs and statistical analyses (principal component analysis (PCA), hierarchical cluster analysis (HCA) or metabolomics pathway analysis (MetPA)) were prepared with GraphPad Prism 10.0 (GraphPad Software), GENE-E (Broad Institute) and MetaboAnalyst 6.0 (ref. 91).
In vitro multilineage differentiation assay
In vitro differentiation assay was performed in a nontissue culture-treated, 96-well, flat-bottomed plate (Falcon). Then, 2 h before cell seeding, plates were coated with StemSpan Differentiation Coating Material (STEMCELL Technologies) according to the manufacturer’s specifications. CD34+ cells, 500, were seeded in SFEM II medium (STEMCELL Technologies) supplemented with hSCF (100 ng ml−1), hFLT3 (10 ng ml−1), human (h)IL-7 (100 ng ml−1), hTPO (75 ng ml−1), hIL-6 (40 ng ml−1), hIL-3 (10 ng ml−1), hIL-11 (50 ng ml−1), human erythropoietin (hEPO); 0.1 U ml−1 (Peprotech), hIL-2 (10 ng ml−1; Novartis), hIL-4 (10 ng ml−1; Miltenyi Biotec) and human low-density lipoprotein (4 μg ml−1; STEMCELL Technologies). Three technical replicates were performed for each condition and medium change was performed every 3–4 days. After 1, 2 and 3 weeks of culture, cells were collected and labeled with anti-CD235a, anti-CD1a, anti-CD5, anti-CD19, anti-CD42b, CD33, anti-CD7, anti-CD71 and anti-CD11c (BD Bioscience) and anti-CD41, anti-CD10, anti-CD15, anti-CD3, anti-CD56 and anti-CD34 (BioLegend) anti-human antibodies. The stained samples were acquired through a BD FACS Symphony A5 (BD Biosciences) cytofluorimeter after Rainbow bead (Spherotech) calibration. Raw FACS data were collected through DIVA software (BD Biosciences) and subsequently analyzed with FlowJo software v.10.5.3. Technically validated results were always included in the analyses and we did not apply any exclusion criteria for outliers.
CD34+ HSPC xenotransplantation experiments
For gene-editing experiments, the outgrowths of 1.0 × 106 mPB HSPCs at the start of the culture (t0 equivalent) were injected intravenously 24 h after editing into sublethally irradiated NSG mice (180–200 cGy) or nonconditioned NBSGW mice. The number of culture-initiating cells were matched among groups for each experiment. Mice were randomly distributed to each experimental group. Human CD45+ cell engraftment was monitored by serial collection of blood (approximately every 2–3 weeks) from the retro-orbital plexus and at the end of the experiment (14 weeks after transplantation, unless otherwise specified). BM and spleen were collected for endpoint analyses, including FACS of hematopoietic lineages and cell types.
For untreated BM HSPCs from healthy donors or patients with VEXAS syndrome, 1.0 × 105 CD34+ HSPCs were injected intravenously immediately after isolation or cell thawing into nonconditioned NBSGW mice. Mice were monitored as per above.
WBD, immunophenotyping, cell cycle and apoptosis analysis by flow cytometry
BM and peripheral blood samples and xenotransplanted mice from the competitive experiment were analyzed using the whole blood dissection (WBD) protocol58, labeling samples with fluorescent antibodies against CD3, CD56, CD14, CD41 or -61, CXCR4, CD34 and CD45RA (BioLegend) and CD33, CD66b, CD38, CD45, CD90, CD10, CD11c, CD19, CD7 and CD71 (BD Biosciences). Samples from xenotransplants were preincubated with a mouse FcR-blocking reagent (BD, dilution 1:100). Absolute cell quantification was performed by adding precision count beads (BioLegend) to samples before the WBD procedure. All stained samples were acquired through BD Symphony A5 cytofluorimeter after Rainbow bead calibration. Raw data were collected through DIVA software.
The cell-cycle analysis was performed using a Click-iT EdU Alexa Fluor Pacific Blue Imaging Kit (Thermo Fisher Scientific) and Hoechst (Thermo Fisher Scientific) staining. Then, 3–5 × 104 cells were treated with 2 µM 5-ethynyl-2′-deoxyuridine (EdU) for 4 h in culture. Cells were washed with 2% FBS in DPBS, stained for surface markers and fixed with 100 µl of Click-iT fixative for 15 min at room temperature. Cells were washed again and permeabilized with 100 µl of 1× Click-iT saponin-based permeabilization for 15 min at room temperature. EdU was marked by incubating cells with 500 μl of Click-iT Plus reaction cocktail for 30 min at room temperature. Finally, cells were washed and stained with DNA with 2 µM Hoechst overnight at 4 °C. After the incubation, sample acquisition was performed with a FAC Symphony A5 cytofluorimeter.
Other flow-cytometry analyses were performed using Canto II (BD Pharmingen). Cells were stained with antibodies listed in Supplementary Table 8 in a final volume of 100–200 μl and then washed with DPBS + 2% heat-inactivated FBS. The Live/Dead Fixable Dead Cell Stain Kit (Thermo Fisher Scientific) or 7-aminoactinomycin D (7AAD; Sigma-Aldrich) were included during sample preparation according to the manufacturer’s instructions to identify dead cells. Analysis of apoptosis was performed using Pacific Blue-conjugated Annexin V (BioLegend) and an apoptosis detection kit with 7AAD (BD Pharmingen) according to the manufacturers’ instructions. Percentages of live (7AAD−Annexin V−), early apoptotic (7AAD−Annexin V+), late apoptotic (7AAD+Annexin V+) and necrotic (7AAD+Annexin V−) cells were reported. Compensation beads (BD Biosciences) were used as single-color controls.
Gating strategies are provided in Supplementary Fig. 3. Technically validated results were always included in the analyses; we did not apply any exclusion criteria for outliers. Data were analyzed with FlowJo software v.10.5.3 (BD Biosciences) or FCS Express 7 Flow.
Cell sorting
Cell sorting was performed on a BD FACS Aria Fusion (BD Biosciences) instrument using BD FACS Diva software and equipped with four lasers: blue (488 nm), yellow/green (561 nm), red (640 nm) and violet (405 nm). Cells were sorted with an 85-µm nozzle. Sheath fluid pressure was set at 310.3 kPa. A highly pure sorting modality (four-way purity sorting) was chosen. Cell sorting was performed on a MoFlo Astrios EQ (Beckman Coulter) using Summit software and equipped with four lasers: blue (488 nm), yellow/green (561 nm), red (640 nm) and violet (405 nm). Cells were sorted with a 100-µm nozzle. Sheath fluid pressure was set at 172.4 kPa. A highly pure sorting modality (purify-1 sorting) was chosen. Sorted cells were collected in 1.5-ml Eppendorf tubes containing 500 μl of DPBS.
Cell sorting before scRNA-seq in Fig. 4 was performed on a MACSQuant Tyto Cell Sorter (Miltenyi Biotec) using the MACSQuantify Tyto Software and equipped with blue (488 nm), red (640 nm) and violet (405 nm) lasers. Sheath fluid pressure was set at 117–124 kPa.
Molecular analyses
For molecular analyses, gDNA was isolated with QIAamp DNA Micro Kit (QIAGEN) according to the manufacturer’s instructions. Base editing and Cas9 efficiencies, as well as the abundance of UBA1mut and UBAwt cells in edited samples and in patients, were measured by PCR amplification at the target locus, followed by amplicon Sanger sequencing (Eurofins Scientific), the results of which were then analyzed using EditR software (http://baseeditr.com)92 with default parameters or TIDE software (https://tide.nki.nl/)93.
Histopathological analyses
Mice were terminated by cervical dislocation and organs were collected and fixed in 10% buffered formalin. After trimming, samples were embedded in paraffin wax, sectioned and stained with hematoxylin and eosin following OECD Good Laboratory Practices principles, principles of data integrity and applicable GLP SR-TIGET standard operating procedures. Histopathological changes were evaluated by an experienced pathologist and graded on a scale of 1–5 as minimal (1), mild (2), moderate (3), marked (4) or severe (5), minimal referring to the least extent discernible and severe to the greatest extent possible. The slides were independently reviewed by another experienced pathologist and a consensus reached on the findings and scores. Slides were digitalized using the scanner Pannoramic MIDI (3DHISTECH) at ×20 magnification.
Total RNA library preparation and sequencing
Whole transcriptomic analyses were performed on edited HSPCs from a pool of male healthy donors (24 h or 7 days after editing) and on monocytes from patients with VEXAS syndrome. All conditions were performed in technical triplicate. Total RNA was purified using an RNeasy Micro Kit (QIAGEN). DNase treatment was performed using RNase-free DNAse Set (QIAGEN), according to the manufacturer’s instructions. RNA was quantified with a Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and its quality assessed by a 2100 Agilent Bioanalyser (Agilent Technologies). Minimum quality was defined as the RNA integrity number >8. Total RNA, 300 ng, was used for library preparation with TruSeq Stranded mRNA (Illumina) and sequenced at Genewiz (Azenta Life Sciences). The samples were sequenced using a 2× 150 paired-end configuration. Approximately 30 million 150-nt-long reads per sample were generated. After quality checking with FastQC, a preprocessing step, including trimming and adapter removal, was performed using TrimGalore (v.0.5.0). The trimmed reads were mapped to the GRCh38 reference genome assembly provided by the 10× reference data repository (refdata-cellranger-GRCh38-3.0.0) using STAR (v.2.7.0d), with the parameter outFilterMultimapNmax set to 1 to consider only uniquely mapped reads. Reads were assigned to genes using featureCounts (v.1.6.3), with the parameter minOverlap set to 10 and discarding chimeric reads.
Differential gene expression on bulk RNA-seq data
Data preprocessing, exploration and differential gene expression analyses were conducted using the DESeq2 (v.1.26.0) R package, normalizing for library size using the DESeq2 median of ratios. Exploratory data analysis involved PCA on regularized log(transformed) data. Genes with a corrected P value (Benjamini–Hochberg correction) <0.05 were considered differentially expressed. Differential gene expression results were examined through over-representation analysis (ORA) and GSEA, querying multiple databases including gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome and the Molecular Signatures Database (MSigDB). Both analyses were carried out using the ClusterProfiler (v.4.2.2) package94. GSEA was performed on pre-ranked gene lists (ordered by log2(fold-change (FC)) values) using the MSigDB Hallmark database, with results filtered by a false recovery rate (FDR) <0.05.
Sample and library preparation for single-cell transcriptomics and sequencing
Cell suspensions were diluted in PBS with 0.04% bovine serum albumin to a final concentration of 1 × 106 cells ml−1. Vitality was assessed for each individual sample. Approximately 20,000 cells were loaded on to a single-cell chip for gel bead-in-emulsion generation using the 10x Genomics Chromium Controller (10x Genomics). The 3′-mRNA-seq gene expression libraries were constructed using the Chromium Single Cell 3′ Library & Gel Bead Kit v.2 (10x Genomics) according to the manufacturer’s guidelines. All reactions were performed in a C1000 Touch Thermal Cycler (BioRad Laboratories). Twelve cycles were used for complementary DNA amplification, whereas the number of total cycles for the sample index PCR was calculated based on the cDNA concentration. Amplified cDNA and final libraries were evaluated using a TapeStation 4200 with a high sensitivity chip. ScRNA-seq libraries were sequenced on an Illumina NovaSeq6000 with the standard sequencing protocol of R1 28-, I1 10-, I2 10- and R2 90-nt read length.
scRNA-seq analysis
Sequenced scRNA-seq libraries were demultiplexed and processed using the CellRanger Single-Cell Software Suite (v.3.1.0, 10x Genomics), using either the GRCh38 (hg38) human reference genome or the GRCm38 (mm10) mouse reference genome depending on the experiment. Unique molecular identifiers (UMIs) were quantified to generate a cell-by-gene count matrix for each sample. These matrices were then imported into the R environment (v.4.1.3) and analyzed using Seurat v.4.3.0 (ref. 95). Specifically, cells expressing <200 genes or >8,000 genes were excluded, because a low gene count suggests false positives or low viability, whereas a high count indicates potential doublets. In addition, cells with >20% of transcripts originating from mitochondrial (mt) genes were removed, because they probably represent stressed or dying cells. UMI counts were log(normalized) and scaled by a factor of 10,000. The top 20% most variable genes were selected for further analysis. Cell-cycle scores were assigned using the Cell-CycleScoring function with reference gene lists. Data were scaled and sources of variability were regressed out by including the UMI count, mitochondrial gene percentage and cell-cycle difference as variables in the vars.to.regress argument. Cell-cycle difference was defined as the difference between the S phase and G2/M phase module scores. Downstream analysis was conducted on the top 20 PCs from a total of 50 components computed using the PCA. To mitigate sample-related variability and 10× chemistry version bias, data integration was performed using the Harmony package (v.1.0)96. Uniform manifold approximation and projection (UMAP) dimensionality reduction and the cells neighbor graph were computed with default parameters using the first 30 of 50 Harmony PCs97. Clusters were identified with the original Louvain algorithm through the FindClusters function at various resolutions and cluster markers were determined using the FindAllMarkers function in Seurat. Each cluster was compared with all other cells using Wilcoxon’s rank-sum test for significance. Genes expressed in >0% of cells in either group, with an adjusted P < 10−6 and a log2(FC) > 0.25, were considered to be markers. The Seurat function AddModuleScore was used to calculate the average expression of gene signatures in different cell sets.
Cell-type annotation
Cell-type annotation was determined using the SingleR package (v.1.8.1)98, leveraging multiple independent reference annotation datasets from both human and mouse sources. For each dataset, we performed dual annotations using both main labels and refined labels. These cell labels were then incorporated into the Seurat object.
Pseudotime analysis
Pseudotime trajectory inference was carried out using Slingshot (v.2.2.1)68 with default parameters, using clusters as label inputs and UMAP embeddings as the reduced dimension input, to construct cell lineages and determine pseudotime starting from the most primitive cluster (HSCs or MPPs).
Over-representation analysis
Cluster marker genes were examined using ORA with the ClusterProfiler R package (v.4.2.2)94, focusing on the Hallmark gene set v.7.0 (from MSigDB) and gene ontology (GO) gene sets. Briefly, we utilized the ego function, setting the cluster’s markers as the query genes and the genes expressed in the dataset under analysis as the gene universe. GO terms with an adjusted P value (Benjamini–Hochberg correction) <0.05 were considered significantly enriched. Query and universe gene sets were converted from gene symbols to ENTREZID using the bitr function in the DOSE package (v.3.20.1)99. Customized dotplots were generated using the ggplot2 R package (v.3.4.4).
GSEA
Downstream analyses were performed on the complete list of marker genes, ranked by decreasing log(FC). GSEA was performed using the GSEA function from the ClusterProfiler R package94 (v.4.2.2), focusing on the Hallmark gene set v.7.0 (from MSigDB) and GO gene sets. GSEA results from all comparisons were consolidated into a single matrix of normalized enrichment scores (NESs). The GSEA test was executed for each set of markers identified by the findmarkers function to retrieve all genes included in the test.
Customized heatmaps were generated using the ggplot2 R package (v.3.4.4).
Integration with public dataset of BM CD34− cells from humans
BMMCs, enriched in the CD34+ HSPCs fraction belonging to the San Raffaele cohort, were integrated with the published dataset from Wu et al.44 (Genome Expression Omnibus (GEO) accession GSE190652) comprising nine patients with VEXAS syndrome and four age-matched controls. Low-quality cells characterized by <500 expressed genes or >20% mtDNA transcripts were removed from all downstream analyses. Data integration was performed using the Harmony package, accounting for both dataset and donor batch effects. UMAP dimensionality reduction and the cells neighbor graph were computed with default parameters using the first 15 of 50 Harmony PCs. Clusters were identified using the Louvain algorithm through the FindNeighbors and FindClusters functions in Seurat, and then they were annotated, leveraging multiple independent reference annotation datasets. Within clusters, analyses were performed to identify DEGs between controls and patients with VEXAS syndrome, as well as controls and each subgroup of patients annotated with a different VEXAS mutation using the FindMarkers Seurat. Genes expressed in >10% of cells in either group, with an adjusted P < 10−6 and a log2(FC) > 0.25, were considered to be markers. These analyses were complemented with GSEA as described above. For both differential expression and GSEA the nominal P values were corrected by the Benjamini–Hochberg procedure to account for multiple hypotheses testing issue. AUCell scores based on the signature of the top-50 markers in the ‘monocyte’ cluster of the xenograft model were calculated. The resulting scores for control and VEXAS samples across and within each of the predicted cell types were compared through a Wilcoxon’s rank-sum test, correcting the nominal P values for the multiple hypotheses testing issue with Holm’s procedure.
Integration with public datasets of BM CD34+ HSPCs from humans
The scRNA-seq San Raffaele dataset was subset to retrieve CD34+ HSPCs, which were combined with HSPCs from Ainciburu et al.72 (GEO accession GSE180298, restricting the analysis to ‘elderly’ samples) and Wu et al.44 (GEO accession GSE190652, CD34+ sorted samples). Low-quality cells characterized by <200 expressed genes or >25% mtDNA transcripts were removed from all downstream analyses. Data integration was performed using the Harmony package, accounting for both dataset and donor batch effects, and every cell was projected on to UMAP lower dimensional space using the top 33 harmonized PCs. Clusters were identified using the Louvain algorithm through the FindNeighbors and FindClusters functions in Seurat, and then they were annotated leveraging multiple independent reference annotation datasets. Within clusters, analyses were performed to identify DEGs between controls and patients with VEXAS syndrome, as well as controls and each subgroup of patients annotated with a different VEXAS mutation using the FindMarkers Seurat. This was achieved through the FindMarkers Seurat function that performs Wilcoxon’s rank-sum tests for each gene expressed by at least 10% of the cells in either group being compared. These analyses were coupled by GSEA, as described above. For both differential expression and GSEA the nominal P values were corrected by the Benjamini–Hochberg procedure to account for multiple hypotheses testing issue. UCell scores based on the signature of the top-50 markers in the ‘Monocyte’ cluster of the xenograft model were calculated. Scores resulting from control and VEXAS samples across and within each of the predicted cell types were compared through a Wilcoxon’s rank-sum test, correcting the nominal P values for the multiple hypotheses testing issue with Holm’s procedure.
HSC-enriched analysis on projection of xenograft HSPCs on the BM reference map
HSC-enriched analysis was conducted on a subset of cells annotated as HSCs, MLPs, MPP-MkEry, MPP-MyLy, LMPPs, MEPs or early GMPs. These annotations were obtained by projecting our VEXAS dataset on the BM reference map (https://github.com/andygxzeng/BoneMarrowMap). Cells with <0.5 annotation accuracy were discarded from further analysis. BM projection figures were obtained accordingly (https://github.com/andygxzeng/BoneMarrowMap). AUCell scores for each signature were calculated.
HSPC transduction with lentiviral vectors
The GFP and BFP reporter lentiviral vectors were produced as previously described100. One day after thawing, HSPCs were transduced at a concentration of 1 × 106 cells per ml using a multiplicity of infection of 40 transducing units per ml. Ciclosporin H was added at a concentration of 8 μM immediately before transduction. HSPCs were washed with ten volumes of DPBS without Ca2+ and Mg2+ 48 h later, when base editing was performed.
Quantifications and statistical analyses
The number of biologically independent samples, animals or experiments is indicated by ‘n’. For some experiments, different HSPC donors were pooled to account for donor-related variability and to reach the number of cells needed for the analyses. Data were summarized as the median with the IQR (or range) or the mean ± s.e.m. depending on data distribution. Inferential techniques were applied in the presence of adequate sample sizes (n ≥ 5), otherwise only descriptive statistics were reported. Two-tailed Student’s t-tests were performed throughout the study. The Mann–Whitney U-test was performed to compare two independent groups, whereas, in the presence of more than two independent groups, the Kruskal–Wallis test followed by post-hoc analysis using Dunn’s test was used. Moreover, correction methods were used to address the problem of multiplicity arising from testing simultaneously several hypotheses and to adjust P values. For all analyses the significance threshold was set at 0.05: *P < 0.05; **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact P values are provided in Supplementary Table 9. Analyses were performed using GraphPad Prism v.9.4.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All relevant data are included in the manuscript. The reagents described in this manuscript are available under a material transfer agreement with IRCCS Ospedale San Raffaele and Fondazione Telethon; requests for materials should be addressed to S.F. and R.M. Sequencing data have been deposited in the GEO (accessions GSE272578 and GSE272816). Raw data from the figures have been deposited in the San Raffaele Open Research Data Repository101. Source data are provided with this paper.
Code availability
The scripts for bioinformatic analyses are available at http://www.bioinfotiget.it/gitlab/custom/Molteni_NatMed2024.
References
Martincorena, I. & Campbell, P. J. Somatic mutation in cancer and normal cells. Science 349, 1483–1489 (2015).
Martincorena, I. et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015).
Lee-Six, H. et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478 (2018).
Olafsson, S. et al. Somatic evolution in non-neoplastic IBD-affected colon. Cell 182, 672–684.e11 (2020).
Yizhak, K. et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 364, eaaw0726 (2019).
Colom, B. et al. Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat. Genet. 52, 604–614 (2020).
Martincorena, I. et al. Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041.e21 (2017).
Watson, C. J. et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 367, 1449–1454 (2020).
Williams, N. et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 602, 162–168 (2022).
Avagyan, S. & Zon, L. I. Clonal hematopoiesis and inflammation—the perpetual cycle. Trends Cell Biol. 33, 695 (2023).
Kakiuchi, N. et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 577, 260–265 (2019).
Mitchell, S. R., Gopakumar, J. & Jaiswal, S. Insights into clonal hematopoiesis and its relation to cancer risk. Curr. Opin. Genet. Dev. 66, 63 (2021).
Bernstein, N. et al. Analysis of somatic mutations in whole blood from 200,618 individuals identifies pervasive positive selection and novel drivers of clonal hematopoiesis. Nat. Genet. 56, 1147–1155 (2024).
Mitchell, E. et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 606, 343–350 (2022).
Zink, F. et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 (2017).
Weng, C. et al. Deciphering cell states and genealogies of human haematopoiesis. Nature 627, 389–398 (2024).
Beck, D. B. et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N. Engl. J. Med. 383, 2628–2638 (2020).
Beck, D. B. et al. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA 329, 318 (2023).
Gutierrez-Rodrigues, F. et al. Spectrum of clonal hematopoiesis in VEXAS syndrome. Blood J. 142, 244–259 (2023).
Koster, M. J., Samec, M. J. & Warrington, K. J. VEXAS syndrome—a review of pathophysiology, presentation, and prognosis. J. Clin. Rheumatol. 29, 298–306 (2023).
Zakine, E. et al. UBA1 variations in neutrophilic dermatosis skin lesions of patients with VEXAS syndrome. JAMA Dermatol. 157, 1349 (2021).
Sterling, D. et al. VEXAS syndrome (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) for the dermatologist. J. Am. Acad. Dermatol. https://doi.org/10.1016/j.jaad.2022.01.042 (2022).
Groarke, E. M., Dulau-Florea, A. E. & Kanthi, Y. Thrombotic manifestations of VEXAS syndrome. Semin. Hematol. 58, 230–238 (2021).
Borie, R. et al. Pleuropulmonary manifestations of vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome. Chest 163, 575–585 (2023).
Muratore, F. et al. VEXAS syndrome: a case series from a single‐centecohort of Italian patients with vasculitis. Arthr. Rheumatol. 74, 665–670 (2022).
van der Made, C. I. et al. Adult-onset autoinflammation caused by somatic mutations in UBA1: a Dutch case series of patients with VEXAS. J. Allergy Clin. Immunol. 149, 432–439.e4 (2022).
Georgin‐Lavialle, S. et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large‐scale analysis of a multicentre case series of 116 French patients*. Br. J. Dermatol. 186, 564–574 (2022).
Khitri, M.-Y. et al. Comparison between idiopathic and VEXAS-relapsing polychondritis: analysis of a French case series of 95 patients. RMD Open 8, e002255 (2022).
Diral, E. et al. Case report: cytopenias in VEXAS syndrome—a WHO 2022 based approach in a single-center cohort. Front. Immunol. 15, 1354130 (2024).
Obiorah, I. E. et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. 5, 3203–3215 (2021).
Koster, M. J. et al. Clinical heterogeneity of the VEXAS syndrome. Mayo Clin. Proc. 96, 2653–2659 (2021).
Gurnari, C. et al. Allogeneic hematopoietic cell transplantation for VEXAS syndrome: results of a multicenter study of the EBMT. Blood Adv. 8, 1444–1448 (2024).
Heiblig, M. et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: a retrospective multicenter study. Blood 140, 927–931 (2022).
Mekinian, A. et al. A phase II prospective trial of azacytidine in steroid-dependent or refractory systemic autoimmune/inflammatory disorders and VEXAS syndrome associated with MDS and CMML. Leukemia 36, 2739–2742 (2022).
Loschi, M. et al. Allogeneic stem cell transplantation as a curative therapeutic approach for VEXAS syndrome: a case report. Bone Marrow Transplant. 57, 315–318 (2022).
Diarra, A. et al. Successful allogeneic hematopoietic stem cell transplantation in patients with VEXAS syndrome: a 2-center experience. Blood Adv. 6, 998–1003 (2022).
Campochiaro, C. et al. Successful use of cyclosporin A and interleukin-1 blocker combination therapy in VEXAS syndrome: a single-center case series. Arthr. Rheumatol. 74, 1302–1303 (2022).
Kucharz, E. J. VEXAS syndrome: a newly discovered systemic rheumatic disorder. Reumatologia 61, 123 (2023).
Schulman, B. A. & Wade Harper, J. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10, 319–331 (2009).
Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 19, 59–70 (2018).
Beck, D. B., Werner, A., Kastner, D. L. & Aksentijevich, I. Disorders of ubiquitylation: unchained inflammation. Nat. Rev. Rheumatol. 18, 435–447 (2022).
Groen, E. J. N. & Gillingwater, T. H. UBA1: at the crossroads of ubiquitin homeostasis and neurodegeneration. Trends Mol. Med. 21, 622 (2015).
Ferrada, M. A. et al. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood 140, 1496–1506 (2022).
Wu, Z. et al. Early activation of inflammatory pathways in UBA1-mutated hematopoietic stem and progenitor cells in VEXAS. Cell Rep Med. 4, 101160 (2023).
Ganesan, S. et al. Single-cell genotype-phenotype mapping identifies therapeutic vulnerabilities in VEXAS syndrome. Preprint at bioRxiv https://doi.org/10.1101/2024.05.19.594376 (2024).
Kosmider, O. et al. VEXAS syndrome is characterized by inflammasome activation and monocyte dysregulation. Nat. Commun. 15, 1–17 (2024).
Templé, M. & Kosmider, O. VEXAS syndrome: a novelty in MDS landscape. Diagnostics 12, 1590 (2022).
Vitale, A. et al. VEXAS syndrome: a new paradigm for adult-onset monogenic autoinflammatory diseases. Intern. Emerg. Med. 18, 711–722 (2023).
Nicholson, L. T., Cowen, E. W., Beck, D., Ferrada, M. & Madigan, L. M. VEXAS syndrome—diagnostic clues for the dermatologist and gaps in our current understanding: a narrative review. JID Innov. 4, 100242 (2024).
Chiaramida, A. et al. Sensitivity to targeted UBA1 inhibition in a myeloid cell line model of VEXAS syndrome. Blood Adv. 7, 7445–7456 (2023).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
Doudna, J. A. The promise and challenge of therapeutic genome editing. Nature 578, 229–236 (2020).
Fiumara, M. et al. Genotoxic effects of base and prime editing in human hematopoietic stem cells. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01915-4 (2023).
Lam, D. K. et al. Improved cytosine base editors generated from TadA variants. Nat. Biotechnol. 41, 686–697 (2023).
Neugebauer, M. E. et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nat. Biotechnol. 41, 673–685 (2022).
Gaudelli, N. M. et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 38, 892–900 (2020).
Koblan, L. W. et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 36, 843–846 (2018).
Basso-Ricci, L. et al. Multiparametric whole blood dissection: A one-shot comprehensive picture of the human hematopoietic system. Cytometry A. 91, 952–965 (2017).
Lettera, E. et al. Molecular and phenotypic blueprint of the hematopoietic compartment reveals proliferation stress as a driver of age-associated human stem cell dysfunctions. Preprint at bioRxiv https://doi.org/10.1101/2023.09.15.557553 (2023).
Skirecki, T. et al. Mobilization of stem and progenitor cells in septic shock patients. Sci. Rep. 9, 3289 (2019).
Abdellatif, H. Circulating CD34+ hematopoietic stem/progenitor cells paralleled with level of viremia in patients chronically infected with hepatitis B virus. Regen. Med. Res. 6, 1 (2018).
Santoro, A. et al. Chronic lymphocytic leukemia increases the pool of peripheral blood hematopoietic stem cells and skews differentiation. Blood Adv. 4, 6310 (2020).
Mende, N. & Laurenti, E. Hematopoietic stem and progenitor cells outside the bone marrow: where, when, and why. Exp. Hematol. 104, 9–16 (2021).
Hetz, C., Zhang, K. & Kaufman, R. J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21, 421–438 (2020).
Ferrari, S. et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat. Biotechnol. 38, 1298–1308 (2020).
Scala, S. et al. Hematopoietic reconstitution dynamics of mobilized- and bone marrow-derived human hematopoietic stem cells after gene therapy. Nat. Commun. 14, 1–18 (2023).
Quaranta, P. et al. Circulating hematopoietic stem/progenitor cell subsets contribute to human hematopoietic homeostasis. Blood 143, 1937 (2024).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 19, 1–16 (2018).
Zeng, A. G. X. et al. Precise single-cell transcriptomic mapping of normal and leukemic cell states reveals unconventional lineage priming in acute myeloid leukemia. Preprint at bioRxiv https://doi.org/10.1101/2023.12.26.573390 (2023).
Bogeska, R. et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell. 29, 1273–1284.e8 (2022).
Zeng, A. G. X. et al. Identification of a human hematopoietic stem cell subset that retains memory of inflammatory stress. Preprint at bioRxiv https://doi.org/10.1101/2023.09.11.557271 (2023).
Ainciburu, M. et al. Uncovering perturbations in human hematopoiesis associated with healthy aging and myeloid malignancies at single-cell resolution. eLife 12, e79363 (2023).
Saul, D. et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat. Commun. 13, 4827 (2022).
Purcell, M., Kruger, A. & Tainsky, M. A. Gene expression profiling of replicative and induced senescence. Cell Cycle 13, 3927–3937 (2014).
Pribluda, A. et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 24, 242–256 (2013).
Avagyan, S. et al. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science 374, 768–772 (2021).
MacNamara, K. C. The role of interferon, inflammation and infection in splastic snemia. Blood 134, SCI-34 (2019).
Jakobsen, N. A. et al. Selective advantage of mutant stem cells in human clonal hematopoiesis is associated with attenuated response to inflammation and aging. Cell Stem Cell 31, 1127–1144.e17 (2024).
Schneider, M. et al. Activation of distinct inflammatory pathways in subgroups of LR-MDS. Leukemia 37, 1709–1718 (2023).
Molteni, R. et al. Oncogene-induced maladaptive activation of trained immunity in the pathogenesis and treatment of Erdheim–Chester disease. Blood 138, 1554–1569 (2021).
Biavasco, R. et al. Oncogene-induced senescence in hematopoietic progenitors features myeloid restricted hematopoiesis, chronic inflammation and histiocytosis. Nat. Commun. 12, 4559 (2021).
Kirkland, J. L. & Tchkonia, T. Senolytic drugs: from discovery to translation. J. Intern. Med. 288, 518 (2020).
Pereira, B. I. et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 10, 1–13 (2019).
Pietras, E. M. et al. Chronic interleukin-1 drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 18, 607 (2016).
Matatall, K. A. et al. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep. 17, 2584–2595 (2016).
Song, Y. et al. A highly efficient and faithful MDS patient-derived xenotransplantation model for pre-clinical studies. Nat. Commun. 10, 1–14 (2019).
Nestorowa, S. et al. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31 (2016).
Nemkov, T., Hansen, K. C. & D’Alessandro, A. A three-minute method for high-throughput quantitative metabolomics and quantitative tracing experiments of central carbon and nitrogen pathways. Rapid Commun. Mass Spectrom. 31, 663–673 (2017).
Reisz, J. A., Zheng, C., D’Alessandro, A. & Nemkov, T. Untargeted and semi-targeted lipid analysis of biological samples using mass spectrometry-based metabolomics. Methods Mol. Biol. 1978, 121–135 (2019).
Clasquin, M. F., Melamud, E. & Rabinowitz, J. D. LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr. Protoc. Bioinform., https://doi.org/10.1002/0471250953.bi1411s37 (2012).
Pang, Z. et al. MetaboAnalyst 6.0: towards a unified platform for metabolomics data processing, analysis and interpretation. Nucleic Acids Res. 2024, 1–9 (2024).
Kluesner, M. G. et al. EditR: a method to quantify base editing from Sanger sequencing. CRISPR J. 1, 239–250 (2018).
Brinkman, E. K., Chen, T., Amendola, M. & Van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284 (2012).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). 2019 16:12.
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. J. Open Source Softw. 3, 861 (2018).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Yu, G., Wang, L. G., Yan, G. R. & He, Q. Y. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 31, 608–609 (2015).
Soldi, M. et al. Laboratory-scale lentiviral vector production and purification for enhanced ex vivo and in vivo genetic engineering. Mol. Ther. Methods Clin. Dev. 19, 411–425 (2020).
Fiumara, M., Molteni, R., Cavalli, G., Naldini, L. & Ferrari, S. Mechanisms of hematopoietic clonal dominance in VEXAS syndrome. San Raffaele Open Research Data Repository, V1 Preprint at (2025).
Acknowledgements
We thank patients and their families for their consent to donate samples for research, all members of L.N. and S.C.’s laboratory and the clinical units for discussion, the IRCCS San Raffaele Hospital Flow Cytometry facility (FRACTAL), the IRCCS San Raffaele Center for Omics Sciences (COSR) and the Advanced Light and Electron Microscopy BioImaging Center (ALEMBIC) facility (particularly C. Covino and A. Loffreda). We thank C. Asperti, I. Cuccovillo, K. Giannetti, C. Rossi and T. Di Tomaso (SR-Tiget) for supply and purification of mPB HSPCs and F. Midena (SR-Tiget) for help with cell-cycle analyses. This work was supported by grants: to L.N., A. Aiuti and R.D.M. from Fondazione Telethon ETS (SR-Tiget Core Grant); to S.F. and S.S from the Italian Ministry of Health (2021 Ricerca Finalizzata Starting Grant: SG-2021-12374529 to S.F.; 2019 Ricerca Finalizzata Giovani Ricercatori, grant no. GR-2019-12369499 to S.S.); to S.F. from the American Society of Hematology (2024 Global Research Award); to A. Aiuti from the Else Kröner-Fresenius-Stiftung, Germany (prize); and to I.M., G.D.L. and G.C. from the Italian Ministry of University and Research (grant no. 2022 PRIN: 2022APWTE3). A.W. and A.V. conducted the present study as partial fulfillment of their PhD studies. R.D.M. is a New York Stem Cell Foundation Robertson Investigator. Figs. 2a, 3a, and 6a and Extended Data Figs. 2a, 5f,i, 6a, and 7a,b were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
R.M. and M.F. performed research and interpreted data. C.C., A.T., E.D., G.M.B., F.C., G.D.L., L.D. and M.M.C. followed up patients with VEXAS syndrome within their respective clinical units. P.Q. and L.B.R. performed immunophenotypic analyses of patients with VEXAS syndrome and the mouse model, and in vitro differentiation assays under the supervision of S.S. and A. Aiuto. R.A., G.P. and I.M. performed bioinformatic analyses. A.V. performed in vitro experiments and some bioinformatic analyses on the VEXAS mouse model, supervised by S.Z.X. D.S. and A.D. performed metabolomic analyses. E.L., A.W. and D.C. performed some experiments and interpreted data. A. Annoni designed and performed the cytokine profiling study. L. Albano, L. Alessandrini, M.P., E.C. and C.G. provided technical support. S.D. and F.S. performed histopathological analyses. R.D.M. provided data on immunophenotypic analyses of age-matched reference controls. S.C. provided support for the research. R.M. and S.F. coordinated the work. G.C., L.N. and S.F. conceived the study. S.F. supervised the research. M.F., R.M., L.N. and S.F. wrote the manuscript. All authors proofread the manuscript.
Corresponding authors
Ethics declarations
Competing interests
S.F., L.N., M.F., D.C., A.W. and R.D.M. are inventors of patents on HSPC gene editing. R.M., M.F., G.C., L.N. and S.F. are inventors of patents on VEXAS syndrome. All patents are owned and managed by the San Raffaele Scientific Institute and the Telethon Foundation. L.N. is a founder, quota holder and consultant of GeneSpire and Chroma Medicine. G.C. is currently an employee of Novartis. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Raquel Faria, Alejo Rodriguez-Fraticelli and Iannis Aifantis for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Phenotypic distribution of hematopoietic cells in BM and PB of patients with VEXAS syndrome.
a-d, Percentage of cells within hematopoietic cells (a), myeloid cells (b), lymphoid cells (c) and HSPCs (d) from BM aspirates of patients with VEXAS syndrome and age-matched controls (n = 19, 7). Mean ± s.e.m. Data on age-matched reference controls from59 are shown. e-h, Percentage of cells within total circulating hematopoietic cells (e), myeloid cells (f), lymphoid cells (g) and HSPCs (h) in the blood of patients with VEXAS syndrome and age-matched controls (n = 19, 7). Mean ± s.e.m. i, Number of myeloid, erythroid, megakaryocytic, NK and preT cells obtained upon in vitro differentiation of HSPCs from two patients with VEXAS syndrome (n = 2) or age-matched controls (n = 3). For all panels: Mann-Whitney test.
Extended Data Fig. 2 Transcriptional and functional changes in UBA1mut cells in vitro.
a, Schematic representation of the editing procedure and in vitro experimental design in male human HSPCs. b-c, Western blot analysis of UBA1a, UBA1b, UBA1c protein expression (b), poly-ubiquitylated proteins (Poly-Ub) and BiP expression level (c) in UBA1wt and UBA1mut HSPCs. β-actin was used as protein loading control (n = 2 biological replicates from different HSPC donors). d, Volcano plots showing fold changes of up- (red) and down- (green) regulated genes comparing UBA1mut and UBA1wt HSPCs one (left) or seven (right) days after editing. Non-significant genes are shown in grey. Wilcoxon rank-sum test with Benjamini-Hochberg correction. e, Percentage of male primary T cells carrying the UBA1 edit over time after editing (n = 6 biological replicates). Median with IQR. Wilcoxon matched-pairs signed rank test. f, Percentage of live T cells at the endpoint of the experiment in ‘e’ (n = 6 biological replicates). Median with IQR. Wilcoxon matched-pairs signed rank test. g, Western blot analysis of cleaved caspase-3 in UBA1wt and UBA1mut T cells. β-actin was used as protein loading control. Panel a created with BioRender.com.
Extended Data Fig. 3 VEXAS-causing UBA1 mutation induces pan-lineage perturbation of hematopoietic cell states.
a, Number of human CD45+ cells homed to the BM 3 days after transplantation of UBA1wt or UBA1mut HSPCs (n = 3). Median. b, Percentage of cells within human CD45+ cells in spleen (left) and BM (right) of mice from Fig. 3j (n = 4/group). Mean ± s.e.m. c, Erythroid (left), B-cell (middle), and myeloid (right) pseudotime trajectories starting from the HSC/MPP cluster in UMAP plot (top) and distribution of cells along the trajectory (bottom) from Fig. 4e. d, Significant positively (red) and negatively (blue) enriched ‘Hallmark’ MSigDB signatures comparing GMP-2 versus GMP-1 clusters from Fig. 4e. Wilcoxon rank-sum test with Benjamini-Hochberg correction. e, Projection of CD34+ HSPCs from 100% UBA1wt and 40% UBA1mut scRNA-seq on a BM reference map69. f, Cell abundance in the indicated clusters from ‘e’. g-i, AUcell scores of selected ‘Hallmark’ MSigDB database signatures (g,h) and aged HSC signatures from71,72 (i) within primitive HSCs/MPPs according to projection on the BM reference map in ‘e’. Wilcoxon rank-sum test with Benjamini-Hochberg correction. j, Heatmap showing normalized enrichment scores (NES) for the GSEA performed using the indicated senescence signatures73,74,75 within the HSPC clusters from Fig. 4e. Wilcoxon rank-sum test with Benjamini-Hochberg correction.
Extended Data Fig. 4 Different strengths and natures of VEXAS perturbations of hematopoiesis depending on the type of UBA1 mutation in patients.
a, CD34 expression in cells from patients BM. b, Significant positively enriched ‘Hallmark’ MSigDB signatures comparing monocytes from patients with VEXAS syndrome and age-matched controls from the San Raffaele cohort by bulk RNA-seq. Wilcoxon rank-sum test with Benjamini-Hochberg correction. c, Volcano plot showing up- (red) and down- (blue) regulated metabolites in monocytes from patients with VEXAS syndrome and age-matched controls from the San Raffaele cohort. d, Distribution of cells in G1, S or G2/M phases of the cell cycle within differentiated cell clusters and samples in Fig. 5a according to signatures from ref. 87. e, Significant enriched ‘Hallmark’ MSigDB signatures across annotated clusters from the scRNA-seq dataset comparing patients with VEXAS syndrome from either of the two cohorts and age-matched controls. Wilcoxon rank-sum test with Benjamini-Hochberg correction. f, Heatmaps as in Fig. 4c showing NES for the GSEA performed within the differentiated clusters in patients with VEXAS syndrome compared to controls and segregating patients by mutations. Wilcoxon rank-sum test with Benjamini-Hochberg correction. g, Expression of NR4A1 and SOCS3 genes in patients with VEXAS syndrome and controls across HSPC clusters. Wilcoxon rank-sum test with Holm-Bonferroni correction. h, Heatmap showing NES for the GSEA performed using the indicated senescence signatures73,74,75 within the HSPC clusters comparing patients with VEXAS syndrome and controls. Wilcoxon rank-sum test with Benjamini-Hochberg correction.
Extended Data Fig. 5 Transcriptomic changes at the experimental endpoint in UBA1mut and UBAwt cells from VEXAS and WT mice.
a, Percentage of human cells in the BM of hematochimeric mice transplanted with UBA1mut or UBA1wt HSPCs and euthanized 4 weeks after transplant (n = 3, 4). Median. b-e, Heatmap as in Fig. 4c showing NES for the GSEA performed within clusters in Fig. 6q comparing: VEXAS and WT mice (b); UBA1mut male cells from VEXAS mice and UBA1wt male cells from WT mice (c); UBA1mut male cells and UBA1wt female cells from VEXAS mice (d); UBA1wt male cells and UBA1wt female cells from WT mice (e). f, Schematic representation of the serial transplantation experiment in the competitive setting. g, Percentage of human hematopoietic cells within total live cells in the BM of mice from secondary recipients in f (n = 2). Median. h, Percentage of UBA1mut cells across hematopoietic populations in the pooled BM from mice in g (n = 1). i, Schematic representation of the competitive transplantation experiment mixing UBA1wt HSPCs (labelled with a GFP reporter lentiviral vector) with either UBA1mut or UBA1wt HSPCs (labelled with a BFP reporter lentiviral vector). j, Number of colonies derived from sorted GFP+ HSPCs retrieved from VEXAS and WT mice. k, UMAP plot of murine Lin- HSPCs from the BM of mice transplanted with UBA1mut or UBA1wt HSPCs. Clusters and associated cell types are indicated by name and colors. l, Heatmap as in Fig. 4c showing NES for the GSEA performed within clusters from j. For all panels: Wilcoxon rank-sum test with Benjamini-Hochberg correction. Panels f,i created with BioRender.com.
Extended Data Fig. 6 Allogenic HSCT eradicates UBA1mut cells.
a, Schematic representation of the allo-HSCT experiment in the VEXAS model. b, Percentage of human cells in the BM of hematochimeric VEXAS mice treated or not with busulfan chemotherapy and donor-derived HSPC transplantation (n = 4/group). Median with IQR. c, Percentage of GFP+ (that is, donor-derived) cells within human cells in the BM of mice from b (n = 4/group). The red line indicates the percentage of GFP+ HSPCs in the donor input. Median with IQR. d, Percentage of UBA1mut cells within hematopoietic lineages in mice from b (n = 4/group). Median with IQR. Panel a created with BioRender.com.
Extended Data Fig. 7 Mechanisms of VEXAS pathophysiology and clonal dominance.
a-b, Graphical representations of the hallmarks of VEXAS pathogenesis (a) and of the mechanisms driving clonal dominance (b). Both panels created with BioRender.com.
Supplementary information
Supplementary Information
Supplementary Figs. 1–3 and Discussion.
Supplementary Tables
Supplementary Tables 1–9.
Source data
Source Data Fig. 1
Unprocessed western blots for Fig. 2f–h.
Source Data Fig. 2
Unprocessed western blots for Extended Data Fig. 2b,c,g.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Molteni, R., Fiumara, M., Campochiaro, C. et al. Mechanisms of hematopoietic clonal dominance in VEXAS syndrome. Nat Med 31, 1911–1924 (2025). https://doi.org/10.1038/s41591-025-03623-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-03623-9
This article is cited by
-
Independent mechanisms of inflammation and myeloid bias in VEXAS syndrome
Nature (2026)
-
VEXAS syndrome presenting with features of Sjögren’s disease: a case-based review of VEXAS and autoimmune disease overlap
Clinical Rheumatology (2026)
-
Distinct characteristics of VEXAS-causative UBA1 M41 and recurrent functional non-M41 mutations
Leukemia (2025)
-
Characterization of E1 enzyme dependencies in mutant-UBA1 human cells reveals UBA6 as a novel therapeutic target in VEXAS syndrome
Leukemia (2025)
-
Clonal dominance: mutations in VEXAS syndrome take advantage of inflammation
Nature Reviews Rheumatology (2025)
