
Abstract
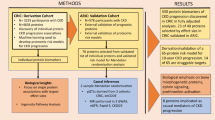
Nearly one-third of the global population is affected by cardio-kidney-metabolic (CKM) diseases; however, the molecular mechanisms underlying CKM diseases are poorly understood. Here we show that tissue proteomics provide critical insights not captured by tissue gene expression or blood proteomics information by performing whole-genome and RNA sequencing and proteomics analysis of human kidney samples (n = 337), and we generated a publicly available database. Via Bayesian co-localization and Mendelian randomization analyses of kidney protein quantitative trait loci and 36 CKM genome-wide association studies, we prioritized 89 proteins for CKM traits. We prioritized relationships that could underlie the interconnectedness of CKM traits and discovered multiple and targetable mechanisms for CKM diseases, including the potential role of kidney angiopoietin-like protein 3 (ANGPTL3) in serum lipid levels and kidney function as well as the role of charged multivesicular body protein 1A in kidney function and hypertension. Notably, we identify pathways with confluence of evidence from genetic loci, tissue gene expression and protein levels for CKM traits. In summary, our large-scale kidney proteomics study uncovers proteins and targetable mechanisms prioritized for CKM diseases.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
Summary statistics for kidney tissue pQTL and eQTL analyses are publicly available at https://susztaklab.com/pl/pQTL. Individual-level clinical metadata and genomics, transcriptomic and proteomics data generated in this study cannot be made publicly available, as participant consent for data sharing was not obtained. Researchers who would like to inspect the raw data may apply for controlled access, subject to approval by our institutional data access committee. Requests should be directed to the corresponding author. Raw data, processed data and metadata from the single-nuclei RNA-seq and spatial RNA-seq have been deposited in the Gene Expression Omnibus (GEO) with accession code GSE211785. The single-nuclei expression and spatial data are also available at https://susztaklab.com/hk_genemap/. No consent was obtained to share individual-level genotype data. Drug–gene interactions were identified using the June 2024 release of the Drug–Gene Interaction Database (DGIdb version 4.2.0; interactions.tsv, genes.tsv and drugs.tsv; https://www.dgidb.org/downloads)38. Public reference resources used include the GRCh38 reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.38), Protein Atlas (https://www.proteinatlas.org/about/download), GTEx (https://gtexportal.org/home/), PDC proteomics dataset (PDC000464; https://pdc.cancer.gov/pdc/study/PDC000464) and Hallmark gene sets from MSigDB (https://www.gsea-msigdb.org). Source data are provided with this paper.
Code availability
All codes used for the analysis are provided via a GitHub repository (https://github.com/daigorohirohama/Human_Kidney_pQTL).
References
Ndumele, C. E. et al. Cardiovascular-kidney-metabolic health: a presidential advisory from the American Heart Association. Circulation 148, 1606–1635 (2023).
Sullivan, K. M. & Susztak, K. Unravelling the complex genetics of common kidney diseases: from variants to mechanisms. Nat. Rev. Nephrol. 16, 628–640 (2020).
Liu, H. et al. Epigenomic and transcriptomic analyses define core cell types, genes and targetable mechanisms for kidney disease. Nat. Genet. 54, 950–962 (2022).
Sheng, X. et al. Mapping the genetic architecture of human traits to cell types in the kidney identifies mechanisms of disease and potential treatments. Nat. Genet. 53, 1322–1333 (2021).
Qiu, C. et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med. 24, 1721–1731 (2018).
Ko, Y. A. et al. Genetic-variation-driven gene-expression changes highlight genes with important functions for kidney disease. Am. J. Hum. Genet. 100, 940–953 (2017).
Eales, J. M. et al. Uncovering genetic mechanisms of hypertension through multi-omic analysis of the kidney. Nat. Genet. 53, 630–637 (2021).
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338 (2023).
Zhang, J. et al. Plasma proteome analyses in individuals of European and African ancestry identify cis-pQTLs and models for proteome-wide association studies. Nat. Genet. 54, 593–602 (2022).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53, 1712–1721 (2021).
Xu, X. et al. Genetic imputation of kidney transcriptome, proteome and multi-omics illuminates new blood pressure and hypertension targets. Nat. Commun. 15, 2359 (2024).
Ritchie, S. C. et al. Integrative analysis of the plasma proteome and polygenic risk of cardiometabolic diseases. Nat. Metab. 3, 1476–1483 (2021).
Inker, L. A. et al. New creatinine- and cystatin C–based equations to estimate GFR without race. N. Engl. J. Med. 385, 1737–1749 (2021).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
Enroth, S., Hallmans, G., Grankvist, K. & Gyllensten, U. Effects of long-term storage time and original sampling month on biobank plasma protein concentrations. EBioMedicine 12, 309–314 (2016).
Chesnaye, N. C., Carrero, J. J., Hecking, M. & Jager, K. J. Differences in the epidemiology, management and outcomes of kidney disease in men and women. Nat. Rev. Nephrol. 20, 7–20 (2024).
Dhindsa, R. S. et al. Rare variant associations with plasma protein levels in the UK Biobank. Nature 622, 339–347 (2023).
Shao, X. et al. CellTalkDB: a manually curated database of ligand–receptor interactions in humans and mice. Brief. Bioinform. 22, bbaa269 (2021).
Eldjarn, G. H. et al. Large-scale plasma proteomics comparisons through genetics and disease associations. Nature 622, 348–358 (2023).
Hirohama, D. et al. Unbiased human kidney tissue proteomics identifies matrix metalloproteinase 7 as a kidney disease biomarker. J. Am. Soc. Nephrol. 34, 1279–1291 (2023).
Battle, A. et al. Genomic variation. Impact of regulatory variation from RNA to protein. Science 347, 664–667 (2015).
Assum, I. et al. Tissue-specific multi-omics analysis of atrial fibrillation. Nat. Commun. 13, 441 (2022).
He, B., Shi, J., Wang, X., Jiang, H. & Zhu, H. J. Genome-wide pQTL analysis of protein expression regulatory networks in the human liver. BMC Biol. 18, 97 (2020).
Pietzner, M. et al. Synergistic insights into human health from aptamer- and antibody-based proteomic profiling. Nat. Commun. 12, 6822 (2021).
Visscher, P. M. et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Liu, H. et al. Kidney multiome-based genetic scorecard reveals convergent coding and regulatory variants. Science 387, eadp4753 (2025).
Giambartolomei, C. et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics 34, 2538–2545 (2018).
Guan, Y. et al. A single genetic locus controls both expression of DPEP1/CHMP1A and kidney disease development via ferroptosis. Nat. Commun. 12, 5078 (2021).
Ku, E., Lee, B. J., Wei, J. & Weir, M. R. Hypertension in CKD: Core Curriculum 2019. Am. J. Kidney Dis. 74, 120–131 (2019).
Klag, M. J. et al. Blood pressure and end-stage renal disease in men. N. Engl. J. Med. 334, 13–18 (1996).
Jafar, T. H. et al. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition: a patient-level meta-analysis. Ann. Intern. Med. 139, 244–252 (2003).
Zhu, Z. et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 9, 224 (2018).
Raal, F. J. et al. Evinacumab for homozygous familial hypercholesterolemia. N. Engl. J. Med. 383, 711–720 (2020).
Shimizugawa, T. et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J. Biol. Chem. 277, 33742–33748 (2002).
Freshour, S. L. et al. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49, D1144–D1151 (2021).
Raies, A. et al. DrugnomeAI is an ensemble machine-learning framework for predicting druggability of candidate drug targets. Commun. Biol. 5, 1291 (2022).
Xie, J. et al. The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat. Commun. 11, 1600 (2020).
Pietzner, M. et al. Mapping the proteo-genomic convergence of human diseases. Science 374, eabj1541 (2021).
Hoxha, E., Reinhard, L. & Stahl, R. A. K. Membranous nephropathy: new pathogenic mechanisms and their clinical implications. Nat. Rev. Nephrol. 18, 466–478 (2022).
Coenen, M. J. et al. Phospholipase A2 receptor (PLA2R1) sequence variants in idiopathic membranous nephropathy. J. Am. Soc. Nephrol. 24, 677–683 (2013).
Hoxha, E. et al. Enhanced expression of the M-type phospholipase A2 receptor in glomeruli correlates with serum receptor antibodies in primary membranous nephropathy. Kidney Int. 82, 797–804 (2012).
Sukocheva, O. et al. Current insights into functions of phospholipase A2 receptor in normal and cancer cells: more questions than answers. Semin. Cancer Biol. 56, 116–127 (2019).
Griveau, A. et al. Targeting the phospholipase A2 receptor ameliorates premature aging phenotypes. Aging Cell 17, e12835 (2018).
Yang, B., Shen, F., Zhu, Y. & Cai, H. Downregulating ANGPTL3 by miR-144-3p promoted TGF-β1-induced renal interstitial fibrosis via activating PI3K/AKT signaling pathway. Heliyon 10, e24204 (2024).
Ma, Y. et al. Podocyte protection by Angptl3 knockout via inhibiting ROS/GRP78 pathway in LPS-induced acute kidney injury. Int. Immunopharmacol. 105, 108549 (2022).
Sarwar, N. et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet 379, 1205–1213 (2012).
Swerdlow, D. I. et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a Mendelian randomisation analysis. Lancet 379, 1214–1224 (2012).
Du, X. Y. et al. The potential mechanism of INHBC and CSF1R in diabetic nephropathy. Eur. Rev. Med. Pharm. Sci. 24, 1970–1978 (2020).
Yang, H., Lian, D., Zhang, X., Li, H. & Xin, G. Key genes and signaling pathways contribute to the pathogensis of diabetic nephropathy. Iran. J. Kidney Dis. 13, 87–97 (2019).
Schlosser, P. et al. Transcriptome- and proteome-wide association studies nominate determinants of kidney function and damage. Genome Biol. 24, 150 (2023).
Graham, S. E. et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 600, 675–679 (2021).
Gold, L. et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 5, e15004 (2010).
Niewczas, M. A. et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 25, 805–813 (2019).
Li, G. X. et al. Comprehensive proteogenomic characterization of rare kidney tumors. Cell Rep. Med. 5, 101547 (2024).
Assarsson, E. et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 9, e95192 (2014).
O’Brien, J. J. et al. A data analysis framework for combining multiple batches increases the power of isobaric proteomics experiments. Nat. Methods 21, 290–300 (2024).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Picard Tools. Broad Institute. http://broadinstitute.github.io/picard/ (2018).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
Abecasis, G. R. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Mills, R. E. et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 16, 1182–1190 (2006).
Price, A. L. et al. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 83, 132–135; author reply 135–139 (2008).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Delaneau, O. et al. A complete tool set for molecular QTL discovery and analysis. Nat. Commun. 8, 15452 (2017).
Stegle, O., Parts, L., Piipari, M., Winn, J. & Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 7, 500–507 (2012).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA 100, 9440–9445 (2003).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Wingo, A. P. et al. Sex differences in brain protein expression and disease. Nat. Med. 29, 2224–2232 (2023).
Lee, S., Wu, M. C. & Lin, X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics 13, 762–775 (2012).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly (Austin) 6, 80–92 (2012).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375, S1–3 (2012).
Xu, F. et al. Genome-wide genotype-serum proteome mapping provides insights into the cross-ancestry differences in cardiometabolic disease susceptibility. Nat. Commun. 14, 896 (2023).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Evangelou, E. et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 50, 1412–1425 (2018).
Wallace, C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 17, e1009440 (2021).
Wang, C. et al. Genetic architecture of cerebrospinal fluid and brain metabolite levels and the genetic colocalization of metabolites with human traits. Nat. Genet. 56, 2685–2695 (2024).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Wu, Y. et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 9, 918 (2018).
Burgess, S., Davies, N. M. & Thompson, S. G. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 40, 597–608 (2016).
Xue, A. et al. Unravelling the complex causal effects of substance use behaviours on common diseases. Commun. Med. (Lond.) 4, 43 (2024).
Karczewski, K. J. et al. Pan-UK Biobank GWAS improves discovery, analysis of genetic architecture, and resolution into ancestry-enriched effects. Preprint at medRxiv https://doi.org/10.1101/2024.03.13.24303864 (2024).
Pulit, S. L. et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 28, 166–174 (2019).
Vujkovic, M. et al. A multiancestry genome-wide association study of unexplained chronic ALT elevation as a proxy for nonalcoholic fatty liver disease with histological and radiological validation. Nat. Genet. 54, 761–771 (2022).
Haas, M. E. et al. Machine learning enables new insights into genetic contributions to liver fat accumulation. Cell Genom. 1, 100066 (2021).
Suzuki, K. et al. Genetic drivers of heterogeneity in type 2 diabetes pathophysiology. Nature 627, 347–357 (2024).
Chen, J. et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet. 53, 840–860 (2021).
Wuttke, M. et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 51, 957–972 (2019).
Aragam, K. G. et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 54, 1803–1815 (2022).
Levin, M. G. et al. Genome-wide association and multi-trait analyses characterize the common genetic architecture of heart failure. Nat. Commun. 13, 6914 (2022).
Mishra, A. et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 611, 115–123 (2022).
Hartiala, J. A. et al. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur. Heart J. 42, 919–933 (2021).
van Zuydam, N. R. et al. Genome-wide association study of peripheral artery disease. Circ. Genom. Precis. Med. 14, e002862 (2021).
Pirruccello, J. P. et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun. 11, 2254 (2020).
Nauffal, V. et al. Genetics of myocardial interstitial fibrosis in the human heart and association with disease. Nat. Genet. 55, 777–786 (2023).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Han, S. K. et al. Mapping genomic regulation of kidney disease and traits through high-resolution and interpretable eQTLs. Nat. Commun. 14, 2229 (2023).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13, e1007081 (2017).
Foley, C. N. et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun. 12, 764 (2021).
Gill, D. et al. Common pitfalls in drug target Mendelian randomization and how to avoid them. BMC Med. 22, 473 (2024).
Zuber, V. et al. Combining evidence from Mendelian randomization and colocalization: review and comparison of approaches. Am. J. Hum. Genet. 109, 767–782 (2022).
Koplev, S. et al. A mechanistic framework for cardiometabolic and coronary artery diseases. Nat. Cardiovasc. Res. 1, 85–100 (2022).
Brotman, S. M. et al. Adipose tissue eQTL meta-analysis highlights the contribution of allelic heterogeneity to gene expression regulation and cardiometabolic traits. Nat. Genet. 57, 180–192 (2025).
de Klein, N. et al. Brain expression quantitative trait locus and network analyses reveal downstream effects and putative drivers for brain-related diseases. Nat. Genet. 55, 377–388 (2023).
Abedini, A. et al. Single-cell multi-omic and spatial profiling of human kidneys implicates the fibrotic microenvironment in kidney disease progression. Nat. Genet. 56, 1712–1724 (2024).
Sriworarat, C. et al. Performant web-based interactive visualization tool for spatially-resolved transcriptomics experiments. Biol. Imaging 3, e15 (2023).
Acknowledgements
The work described in this article was supported by the National Institutes of Health (NIH; grants R01 DK105821, R01 DK087635 and R01 DK076077 to K.S. and R01 DK123459 to M.A.N.). This work was also supported by a research grant from the Manpei Suzuki Diabetes Foundation to D.H. The authors acknowledge the use of BioRender.com in creating schematic Figs. 1a and 4e, Extended Data Figs. 7a and 8a and Supplementary Figs. 4 and 9.
Author information
Authors and Affiliations
Contributions
D.H. performed experiments and analyzed data. E.H., H.L., J.F., A.A., J.L., A.V., Z.L., C.L., S.M., D.S., T.N., N.O. and F.E.M. assisted with data generation and analysis. A.K. and M.A.N. offered experimental and analytical suggestions. K.S. was responsible for overall design and oversight of the experiments. K.S. supervised the experiment. D.H. and K.S. wrote the original draft. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The laboratory of K.S. (including D.H., E.H., H.L., A.A., J.L., A.V., L.Z., C.L., S.M. and S.V.) is supported by Gilead, Novo Nordisk, Novartis, GlaxoSmithKline, BIPI, Regeneron, Genentech and Calico Life Sciences. K.S. is on the scientific advisory board of Pfizer. T.N., N.O. and F.E.M. are employees of Calico Life Sciences. J.F., D.S. and A.K. are employees of Novo Nordisk. All other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Melanie Chan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Ashley Castellanos-Jankiewicz and Sonia Muliyil, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Overview of kidney pQTL analysis: Significance, effect Size, and functional annotation.
(a) Significance level (y-axis) of cis associations according to distance from the TSS (x-axis) for the cognate gene in the cis-window ( ≤ 1 Mb from the gene body) in kidney pQTL analysis. (b) Distance from the TSS (x-axis) versus absolute effect size (y-axis) in kidney pQTL data. (c) MAF versus absolute effect size in kidney pQTL data. (d) Proportion of functional and uncertain SNVs in pQTLs: This pie chart illustrates the breakdown of SNPs identified as lead SNVs for pQTLs, with a majority categorized as having uncertain functional impact and the remainder with annotated functions based on Ensembl VEP analysis. (e) SNV functional annotation categories in pQTLs: Distribution of lead pQTL SNVs across functional categories such as coding variants, regulatory/UTR regions, non-codings (including “non coding transcript exon variant,” “non coding transcript variant,” and “NMD transcript variant”), and intronic/intergenic regions. (f) Effect size distribution by functional annotation in pQTLs: Violin plots showing the distribution of effect sizes of genetic variants on protein levels, segregated by functional annotation. Coding variants demonstrate a significantly higher effect size compared to all other categories. Each dot represents a protein–SNV pair (n = 873). Box plots within violins show the median, 25th and 75th percentiles, and values within 1.5× the interquartile range. Group differences were assessed using one-way ANOVA, followed by Tukey’s HSD test.
Extended Data Fig. 2 Comparison of Kidney Tissue pQTLs between Multi-Ancestry and European Populations.
(a) Venn diagram illustrating the unique and overlapping pQTL proteins identified in kidney tissue pQTL analyses conducted in multi-ancestry and European-only populations. Parentheses indicate the unique SOMAmer counts. (b) Scatter plot showing the correlation of effect sizes for pQTLs between multi-ancestry and European-only analyses. The correlation coefficient was calculated using Spearman’s rho (r), with a two-sided p-value determined by asymptotic t approximation. (c) Scatter plot comparing the -log10(p-values) for pQTLs between multi-ancestry and European-only analyses. The correlation coefficient was calculated using Spearman’s rho (r), with a two-sided p-value determined by asymptotic t approximation. (d) Bar plot showing the frequency of loci associated with the number of proteins in the European-only analysis, indicating the number of proteins per genomic region.
Extended Data Fig. 3 Examples of Pathway Networks Highlighted by pQTLs.
(a) Diagrammatic representation of kidney drug metabolism pathways with pQTL insights: This diagram provides a stylized representation of two critical pathways involved in drug metabolism within the kidney: the glutathione (GSH) pathway and the nucleotide metabolism pathway. It outlines the key steps in the GSH pathway in the left, including synthesis, utilization in the redox cycle, conjugation for detoxification, and the recycling of GSH. Concurrently, it depicts the nucleotide metabolism pathway in the right, focusing on the conversion processes of cytosine and thymine to their respective metabolites and the involvement of ATP in nucleotide synthesis and purine metabolism regulation. KEGG pathway analysis identified significant enrichment for drug metabolism (FDR = 1.6×10^-4) and xenobiotic metabolism (FDR = 7.4×10^-7), with the latter corresponding to drug metabolism in MsigDB. Red boxes signify enzymes that were found to be significant in kidney pQTL analysis. (b) Overview of the complement pathways: This figure provides a streamlined representation of the complement system, including the classical, lectin, and alternative pathways. Initiation of these pathways occurs through the interaction of complement proteins with various molecular structures. KEGG pathway analysis showed significant enrichment for complement activation (FDR = 2.8×10^-5), further supported by MsigDB analysis (FDR = 7.3×10^-3). Proteins associated with cis pQTLs are highlighted in red, accompanied by boxplots illustrating protein expression based on allele type, indicated next to SNV IDs. Each dot represents an individual patient. Box plots show the median, 25th and 75th percentiles, and values within 1.5× the interquartile range (n = 321). Statistical significance was assessed using a two-sided t-test (for two groups) or one-way ANOVA (for three groups). GSSG, Glutathione disulfide; GPX1, GPX7, Glutathione peroxidases 1 and 7; GSR, Glutathione reductase; GSH, Glutathione; GGT5, Gamma-glutamyltransferase 5; GSTA1, GSTM1, GSTM3, GSTM4, GSTM5, GSTT2, GSTT2B, GSTO1, GSTP1, Glutathione S-transferases A1, M1, M3, M4, M5, T2, T2B, O1, P1; CDA, Cytidine deaminase; TYMP, Thymidine phosphorylase; ATP, Adenosine triphosphate; TPMT, Thiopurine S-methyltransferase; NME4, NME7, Non-metastatic cells 4 and 7, related to nucleoside diphosphate kinase activity; MeTG, Methylthioguanine; NDP, Nucleoside diphosphate; C1-INH, C1 inhibitor; CFD, complement factor D; CFH, complement factor H; MASP, mannan-binding lectin-associated serine protease; MBL, mannose-binding lectin.
Extended Data Fig. 4 Integration of Kidney Function GWAS (European ancestry population only) and kidney eQTL and pQTL data using Coloc and Moloc Analysis.
(a) Coloc and moloc analysis overlap for eGFR GWAS, and kidney eQTL, and pQTL: Venn diagram illustrating the intersection between proteins and genes identified as significant in coloc analysis and further refined by moloc analysis. This highlights 8 kidney proteins/genes that were found to be significant in moloc. (b) LocusZoom plots describing the association signals for the ARL6IP5 across eGFR GWAS traits, kidney pQTLs, and eQTLs, with a high posterior probability of association (PPA = 0.99). The lead SNV, rs7039, is also shown. ARL6IP5 serves as a representative example among the 14 proteins/genes found significant in moloc analysis. (c) ARL6IP5 expression with protein and gene level associations: Box plots illustrating the association of lead SNV of ARL6IP5 (rs7039) (x-axis) with ARL6IP5 protein and RNA expression levels (y-axis). Linkage disequilibrium (LD) information is derived from the European population data provided by the 1000 Genomes Project. Each dot represents an individual patient (n = 321 for protein, n = 311 for gene). Box plots show the median, 25th and 75th percentiles, and values within 1.5× the interquartile range. Group differences were assessed using one-way ANOVA.
Extended Data Fig. 5 Comprehensive Analysis of Genetic Influence on Kidney pQTL/eQTL and eGFR GWAS.
(a) Illustration of the use of SMR and HEIDI tests to determine if the same genetic variants influence both kidney pQTL/eQTL and eGFR GWAS, suggesting potential causality. (b, c) Pie charts displaying the number of pQTL proteins and eQTL genes prioritized for their effects on eGFR were supported by SMR and HEIDI analysis, as well as those supported by SMR analysis alone, where HEIDI did not indicate a significant result. (d) Manhattan plot representing chromosomal locations (x-axis) and the strength of the SMR significance for eGFR GWAS, and human kidney pQTL expressed as -log10(SMR Pval) (y-axis). The top 25 most significant protein associations are labeled. The red dashed line represents the significance threshold (P = 6.44×10^-5, Methods). Blue dots denote significant associations in both SMR and HEIDI analyses, while grey dots indicate non-significant associations. (e, f) Venn diagrams illustrating (e) proteins and (f) genes showing significance in colocalization (coloc) and prioritized for their potential effects based on SMR and HEIDI analyses for eGFR GWAS. (g) Combined Venn diagram indicating the overlap between proteins and genes with significant coloc findings and those prioritized based on evidence from both SMR and HEIDI analyses. (h) The UpSet plot shows the overlap of significant proteins across coloc, SMR/HEIDI, and moloc analyses. Bars represent the number of overlapping proteins for each combination of analyses. SMR & HEIDI refers to proteins that are significant in both SMR and HEIDI analyses.
Extended Data Fig. 6 Comparative Analysis of pQTL and eQTL Signatures in Human Kidney.
Miami plot illustrating the comparative landscape of pQTL and eQTL in human kidney tissues. The x-axis represents the chromosomal location of SNVs, while the y-axis indicates the strength of the pQTL and eQTL associations, expressed as -log10(Pvalue). Only protein-coding genes are displayed for eQTLs. The plot is color-coded to distinguish the significance and type of association: red points indicate proteins/genes that are significant in both coloc and SMR/HEIDI analyses, blue points signify proteins/genes significant only in coloc analysis, green points represent proteins/genes significant only in SMR/HEIDI analysis. P values for pQTL and eQTL analyses were obtained using QTLtools (linear regression).
Extended Data Fig. 7 Gene Expression Patterns Related to eGFR Risk Kidney Proteins.
(a) Human kidney single cell analysis experiment using 155,647 nuclei extracted from 18 samples. (b, c) This heatmap details gene expression patterns related to (b) 70 colocalized proteins and (c) 52 proteins that were significant in both SMR and HEIDI tests for eGFR, within kidney nucleus clusters, with distinct groups such as Podo, CNT, and PC maintained. Clusters were merged for Endo (Endo_GC, Endo_Lymphatic, Endo_Peritubular), PT (PT_S1, PT_S2, PT_S3), LOH (segments of the Loop of Henle), DCT (DCT1, DCT2), IC_A (IC_A, IC_B), Stromal (various fibroblasts and stromal cells), and Immune (B cell types, T cell types, NK cells, macrophages and monocytes). Clusters with fewer than 30 nuclei were excluded. Z-scores reflect mean expression levels for each cell type. GPX1 and WARS1 are not represented due to the absence of expression data in this dataset.
Extended Data Fig. 8 Integrated Spatial Transcriptomic Analysis of Human Kidney Sections.
(a) Workflow of integrated spatial analysis, performed on a total of 7 human kidney samples. (b) Hematoxylin and eosin (HE) staining: A representative image of HE-stained human kidney tissue prepared for single-cell spatial RNA-seq. Scale bar, 500 μm. (c, d) Spatial expression in glomeruli: Representative images showing spatial transcriptomics for (c) PLA2R1 (green) and NPHS1 (red), (d) TPPP3 (green) and NPHS1 (red), alongside merged images highlighting co-localization in glomeruli. Scale bars, 250 μm. (e, f) Spatial expression in proximal tubules: Representative images of spatial transcriptomics for (e) DPEP1 (green) and CUBN (red), (f) CHMP1A (green) and CUBN (red), with merged images showing co-expression in proximal tubule cells. Scale bars, 250 μm. Data shown are representative of spatial transcriptomics from n = 7 human kidney samples.
Extended Data Fig. 9 Comprehensive Analysis of Causal Relationships between eGFR, SBP, DBP, LDL-C and HDL-C.
(a) Causality evaluation using GSMR between eGFR and SBP/DBP GWAS. This panel illustrates the evaluation of causality between eGFR and SBP/DBP GWAS using the GSMR method. (b) This panel shows the bidirectional causal relationships between eGFR and SBP/DBP with their respective beta coefficients and p-values. (c) Causality evaluation using GSMR between eGFR and LDL-C/HDL-C/TG GWAS: This panel illustrates the evaluation of causality between eGFR and LDL-C/HDL-C/TG GWAS using the GSMR method. (d) This panel shows the bidirectional causal relationships between eGFR and LDL-C/HDL-C/TG with their respective beta coefficients and p-values. The intensity of the blue color represents the magnitude of the negative beta values. The thickness of the arrows is proportional to -log10(p-value), which reflects the statistical significance. GSMR, generalized summary-data-based Mendelian randomization.
Extended Data Fig. 10 Integrative Genetic Analysis of ANGPTL3: Mendelian Randomization and Colocalization Across Cardio-Kidney-Metabolic Traits.
(a) Colocalization analysis of ANGPTL3 across various cardio-kidney-metabolic GWAS traits. The x-axis represents the colocalization posterior probability, while the y-axis lists the examined traits. Kidney eQTLs, kidney pQTLs, and plasma pQTLs from various studies are represented by different shapes and colors, as indicated in the legend. Dots are colored according to their posterior probability, with grey dots representing loci where posterior probability<0.5. (b) Mendelian Randomization effect sizes and significance for ANGPTL3 across various cardio-kidney-metabolic GWAS traits. The x-axis shows the MR effect size, computed using the Wald ratio method, while the y-axis lists the examined traits. The figure includes eQTLs, kidney pQTLs, and plasma pQTLs from various studies, represented by different shapes and colors as indicated in the legend. The size of the dots corresponds to the -log10(P-value) of the MR effect size. MR P < 2.5×10^-6 was considered statistically significant. Associations not reaching this threshold are represented in grey. ASCVD, atherosclerotic cardiovascular disease; MASH, metabolic dysfunction-associated steatohepatitis; MRI_lvesvi, cardiac magnetic resonance imaging of left ventricular end-systolic volume index; MRI_lvef, cardiac magnetic resonance imaging of left ventricular ejection fraction; DCM, dilated cardiomyopathy; CAD, coronary artery disease; SBP, systolic blood pressure; DBP, diastolic blood pressure; T2D, type 2 diabetes; 2hGlu, 2-hour glucose; GGT, gamma-glutamyl transferase; AST, aspartate aminotransferase; ALT, alanine transaminase; ALP, alkaline phosphatase; whradjbmi, waist-to-hip ratio adjusted for body mass index; TG, triglycerides; ffmi, fat-free mass index; BMI, body mass index; bfpct, body fat percentage; AOR, Aorta; SUF, subcutaneous fat; SKM, skeletal muscle; LIV, Liver.
Supplementary information
Supplementary Information
Supplementary Note and Supplementary Figs. 1–12
Supplementary Tables
Supplementary Tables 1–30. Detailed results including significant pQTLs, co-localization statistics, functional annotations and drug–gene interactions. Each table is provided in a separate tab within the Excel workbook.
Supplementary Data 1
Source Data for Supplementary Figs. 2b and 8a,b.
Source data
Source Data Fig. 1
Statistical Source Data
Source Data Fig. 2
Statistical Source Data
Source Data Fig. 3
Statistical Source Data
Source Data Fig. 4
Statistical Source Data
Source Data Extended Data Fig. 2
Statistical Source Data
Source Data Extended Data Fig. 4
Statistical Source Data
Source Data Extended Data Fig. 7
Statistical Source Data
Source Data Extended Data Fig. 10
Statistical Source Data
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hirohama, D., Fadista, J., Ha, E. et al. The proteogenomic landscape of the human kidney and implications for cardio-kidney-metabolic health. Nat Med 31, 3917–3929 (2025). https://doi.org/10.1038/s41591-025-03872-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-03872-8
This article is cited by
-
Multi-omics data integration from patients with carotid stenosis illuminates key molecular signatures of atherosclerotic instability
Genome Medicine (2026)
-
Advances in risk prediction and treatment strategies for CKM syndrome
Nature Reviews Nephrology (2026)
