
Abstract
Obesity is a heterogeneous condition not adequately captured by a single adiposity trait. We conducted a multi-trait genome-wide association analysis using individual-level data from 452,768 UK Biobank participants to study obesity in relation to cardiometabolic health. We defined continuous ‘uncoupling phenotypes’, ranging from high adiposity with healthy cardiometabolic profiles to low adiposity with unhealthy ones. We identified 266 variants across 205 genomic loci where adiposity-increasing alleles were simultaneously associated with lower cardiometabolic risk. A genetic risk score (GRSuncoupling) aggregating these variants was associated with a lower risk of cardiometabolic disorders, including dyslipidemia and ischemic heart disease, despite higher obesity risk; unlike an adiposity score based on body fat percentage-associated variants (GRSBFP). The 266 variants formed eight genetic subtypes of obesity, each with distinct risk profiles and pathway signatures. Proteomic analyses revealed signatures separating adiposity- and health-driven effects. Our findings reveal new mechanisms that uncouple obesity from cardiometabolic comorbidities and lay a foundation for genetically informed subtyping of obesity to support precision medicine.
Similar content being viewed by others
Main
Obesity is a major risk factor for a variety of cardiometabolic disease outcomes and is the consequence of intricate interactions between genes and environment1,2,3,4. Genome-wide association studies (GWAS) identified more than 1,000 genetic loci associated with obesity risk5 and pointed to the central nervous system (CNS) as a key player in body weight regulation5,6. Despite these insights into the overarching biology, our understanding of the mechanisms that control body weight is still limited. This could be, in part, because GWASs have so far analyzed one adiposity trait at a time, most often body mass index (BMI). Such single-trait GWASs ignore the vast heterogeneity among individuals with obesity in, for example, etiology, life course trajectory and cardiometabolic comorbidities.
Therefore, while single-trait GWASs have identified numerous loci5, they likely represent only a subset of the mechanisms underlying obesity. Multi-trait GWASs, on the other hand, hold the potential to reveal more layers of the underlying biology. For example, we and others have performed genome-wide searches for loci that uncouple excess adiposity from cardiometabolic risk and identified 87 loci for which the adiposity-increasing allele associates with lower cardiometabolic risk7,8,9,10,11,12,13,14. These uncoupling loci have implicated peripheral mechanisms, such as fat distribution, adipocyte function and differentiation, and inflammation, but not the CNS15.
Here, we build upon our previous work11 and leverage individual-level data from 452,768 European participants in the UK Biobank to perform a comprehensive multi-trait genome-wide screen. We aimed to identify new genetic loci that uncouple adiposity from cardiometabolic comorbidities by analyzing three adiposity and eight cardiometabolic traits, including lipid, glycemic and blood pressure traits. We identified 205 genetic loci, harboring 266 lead variants, where the adiposity-increasing allele is associated with a lower risk of at least one cardiometabolic trait. Follow-up analyses identified genetic subtypes of obesity with distinct pathways, cardiometabolic risk and serum protein profiles.
Results
Genome-wide screen identifies 266 adiposity-increasing alleles with protective effects on cardiometabolic health
We performed a genome-wide screen in up to 452,768 individuals of European ancestry from the UK Biobank to identify adiposity-increasing loci that have protective effects on cardiometabolic health. We analyzed three adiposity traits (BMI, body fat percentage (BFP) and waist-to-hip ratio (WHR)) and eight cardiometabolic traits (total cholesterol (TC), LDL cholesterol (LDL-C), HDL cholesterol (HDL-C), triglycerides (TGs), random glucose and HbA1c levels, systolic blood pressure (SBP) and diastolic blood pressure (DBP)) (Methods, Fig. 1 and Supplementary Table 1). We first created 24 ‘bi-traits’, which combine an adiposity and a cardiometabolic trait into a new phenotype, obtained by subtracting standardized values of one of the eight cardiometabolic traits from standardized values of one of the three adiposity traits. High values for a bi-trait represent high adiposity but low levels of the cardiometabolic trait and vice versa (Methods and Extended Data Fig. 1). We next performed GWAS for each of the 24 bi-traits and each of the 11 single traits from which the bi-traits were derived. Variants associated with bi-traits at genome-wide significance (P < 5 × 10−10), and for which the associations with both corresponding single traits reached nominal significance (P < 10−4) and that also colocalized (when the same underlying signal drives the associations with both single traits as well as with the bi-trait (Methods)) were considered uncoupling variants. As such, we identified 266 unique lead variants located in 205 loci (more than 1 Mb apart) across the 24 bi-traits (Methods and Supplementary Tables 2–4). Of the 205 uncoupling loci we identified, 139 have not previously been reported in the context of cardiometabolic uncoupling, whereas the remaining 66 replicate the majority of the 87 previously reported loci7,8,9,10,11,12,13,14,15. The remaining 21 did not reach significance in our study due to a more stringent significance threshold and differences in study design, traits and disease outcomes studied. Altogether, our analyses identified more than double the number of previously reported loci, in part driven by a larger sample size, but also by the use of individual-level data that allowed us to design new, continuous uncoupling phenotypes, as opposed to summary statistics used in previous studies7,8,9,10,11,14 (Supplementary Tables 2 and 5).

Overall steps and traits analyzed in the study. Bi-traits are obtained by subtracting standardized values of a cardiometabolic trait from an adiposity trait.
A genetic risk score (GRSuncoupling) associated with higher adiposity, but lower cardiometabolic traits
To assess the aggregate effect of the 266 uncoupling lead variants (variants that were associated with increased adiposity and lower cardiometabolic effects or vice versa), we created a genetic risk score (GRS) consisting of the 266 lead variants, GRSuncoupling (Methods and Supplementary Table 6). To compare GRSuncoupling with a proxy for adiposity that captures overall body fat without factoring in the cardiometabolic component, we created another GRS consisting of 647 variants that were significantly (P < 5 × 10−10) associated with BFP, GRSBFP (Methods). We tested the association of both GRSs with 16 adiposity and eight cardiometabolic traits (Methods, Fig. 2a and Supplementary Table 7). Both GRSs were associated with higher values of most adiposity traits; with effect sizes for GRSBFP tending to be generally larger than those for GRSuncoupling; however, compared to the GRSBFP, the GRSuncoupling was associated with a more favorable fat distribution. Specifically, a higher GRSuncoupling was associated with a lower (β < 0) MRI-derived visceral adipose tissue (VAT) to abdominal subcutaneous adipose tissue volume (ASAT) ratio, trunk fat:gluteofemoral fat (GFAT) ratio and liver fat fraction, whereas a higher GRSBFP was associated with a higher WHR, trunk fat:GFAT ratio and liver fat fraction, but not with VAT:ASAT ratio. Consistently, the GRSuncoupling had a larger effect on gynoid than on android fat percentage (android FP), whereas the opposite was seen for the GRSBFP.

a, Estimated per ten-allele change effect sizes of GRS–trait associations in UK Biobank European ancestry population for GRSuncoupling (in magenta) and GRSBFP (in blue). b,c, Estimated per ten-allele change effect sizes of GRS–trait associations in UK Biobank European ancestry population for each cluster-specific GRS (GRS 1–8, in red) and GRSBFP (in gray). Dashed circles indicate β = 0, indicating no association between each GRS and the trait. Points outside the circle represent positive GRS–trait associations, whereas those inside represent negative associations. The effect and reference alleles of GRS2, a cluster associated with lower WHR and higher blood pressure, were flipped to reflect a profile of higher adiposity and facilitate comparison with the other clusters.
For cardiometabolic traits, a higher GRSuncoupling was associated with a healthier profile; that is lower levels of LDL-C, TC, TG, HbA1c, glucose, SBP and DBP and higher HDL-C. A higher GRSBFP, on the other hand, was associated with higher TG, HbA1c, glucose, SBP and DBP and lower HDL-C, but no effect on LDL-C or TC (Fig. 2a and Supplementary Table 7). This association signature of the GRSuncoupling was significantly different from that of the GRSBFP (P < 0.0001) (Fig. 2a and Supplementary Table 7).
In men and women separately, the association of GRSuncoupling with adiposity traits tended to differ for the fat distribution traits. Specifically, the association with a more favorable fat distribution (WHR, VAT:ASAT, trunk fat:GFAT ratio) was more pronounced in women than in men (Extended Data Fig. 2). This was mostly due to a smaller effect on abdominal fat accumulation (waist circumference and android FP) in women compared to men, whereas the effect on peripheral fat accumulation (hip circumference and gynoid FP) was not substantially different between sexes. Despite these differences in fat distribution, associations with cardiometabolic traits were similar in men and women. No sex-specific effects for either anthropometric or cardiometabolic traits were observed for the GRSBFP (Extended Data Fig. 2).
GRSuncoupling associates with protective effect on cardiometabolic outcomes but with increased risk of weight-bearing diseases
To better understand the clinical impact of genetic predisposition to adiposity and cardiometabolic comorbidities, we performed a phoneme-wide association analysis (PheWAS) between each of the two GRSs (GRSuncoupling, GRSBFP) and 10,965 disease outcomes in the UK Biobank (n = 373,747) (Methods, Supplementary Table 8 and Fig. 3). Both scores were associated with increased risk of obesity. While GRSuncoupling is associated with a healthier cardiometabolic profile, it is also associated with an increased risk for other diseases, often weight-bearing diseases, to the same extent as GRSBFP. Specifically, a higher GRSuncoupling was associated with a lower risk of conditions related to lipoprotein metabolism (OR = 0.92, P = 1.4 × 10−89), essential primary hypertension (OR = 0.96, P = 1.7 × 10−27), noninsulin-dependent diabetes (OR = 0.94, P = 5.6 × 10−21), ischemic heart disease (OR = 0.96, P = 7.4 × 10−11), angina (OR = 0.96, P = 1.6 × 10−8) and acute myocardial infarction (OR = 0.96, P = 3.6 × 10−6) (Fig. 3), whereas a higher GRSBFP was associated with increased risk of these conditions; however, a higher GRSuncoupling was associated with an increased risk of noncardiometabolic, weight-related conditions, including cellulitis (OR = 1.05, P = 1.14 × 10−11), gonarthrosis (OR = 1.06, P = 9.9 × 10−25) and varicose veins (OR = 1.08, P = 7.9 × 10−26), similar to GRSBFP (Fig. 3 and Supplementary Table 8).

Phenome-wide association results of disease outcomes and GRSBFP (in blue) and GRSuncoupling (in magenta) performed using the PHEnome Scan ANalysis Tool (PHESANT) in 373,747 European participants. Data are presented as OR ± CI. ORs represent effect size estimates per ten risk-allele increments. Case–control sample sizes for each outcome are presented in Supplementary Table 8.
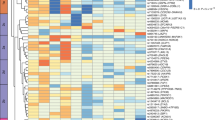
The 266 uncoupling lead variants group into 8 clusters with distinct cardiometabolic signatures
We sought to identify genetically defined subgroups among the 266 uncoupling variants based on similarity of association with adiposity and cardiometabolic traits using NAvMix clustering analyses (Methods). As such, we identified eight clusters with distinct association signatures (Fig. 4). Specifically, variants in three clusters (4,7 and 8) were associated with increased adiposity and protective effects across multiple trait groups, whereas lead variants in the other five clusters were associated with increased adiposity with protective effects on only one of the cardiometabolic trait groups.

Clustering of the 266 uncoupling lead variants using the NAvMix algorithm identified eight clusters. The color coding represents β values ranging from negative (blue) to positive (red). Associations are expressed by the BFP-increasing allele as the effect allele to enable comparison across traits.
We then calculated cluster-specific GRSs, GRS1–GRS8, which aggregate the effects of lead variants in each cluster and tested their association with anthropometric and cardiometabolic traits (Methods and Supplementary Table 6). These cluster-specific GRSs displayed a distinct association signature (Methods, Fig. 2b,c and Supplementary Table 7). For example, GRS4, GRS7 and GRS8 are associated with two or more cardiometabolic trait groups. Specifically, GRS4 is associated with higher overall adiposity (higher BFP, BMI, fat-free mass index (FFMI)) and favorable lipid and glycemic profiles (lower LDL-C, TC, TG, higher HDL-C and lower HbA1c), possibly mediated through a more favorable fat distribution (lower WHR, VAT:ASAT and android FP). GRS7 and GRS8 were mainly associated with lower TG and higher HDL-C, lower HbA1c and blood pressure (Fig. 2b,c and Supplementary Table 7). In addition to the stronger effects on HDL-C and TG, GRS7 differs from GRS8 in its association with adiposity. GRS7 has a strong effect on body fat distribution (for example lower WHR, VAT:ASAT and higher gynoid and lower android FP). On the other hand, GRS8 is associated with higher overall adiposity (BFP, BMI, gynoid and android FP, ASAT and SAT), lower FFMI, without an obvious effect on fat distribution (Fig. 2b and Supplementary Table 7). GRS3 and GRS5 are similarly associated with higher overall adiposity, with GRS3 being associated with lower blood pressure (SBP and DBP) and GRS5 with lower glycemic traits (glucose and HbA1c). GRS1 is associated with greater overall body size (BMI, FFMI and BFP), and with lower TG and higher HDL-C levels, but also with higher levels of LDL-C, TC and HbA1c. Of all cluster-specific GRSs, GRS6 has the strongest effect on overall body size. On the cardiometabolic side, GRS6’s profile is opposite to that of GRS1; as GRS6 is associated with lower LDL-C and TC levels, but also with higher TG and lower HDL-C levels, and high blood pressure and glycemic traits (Figs. 2b,c and 4). Finally, GRS2 is the only cluster where the adiposity effect is driven mainly by an association with WHR. A higher GRS2 is associated with higher gynoid fat accumulation (WHR, VAT:ASAT ratio and trunk fat:GFAT ratio), but lower blood pressure. In terms of disease outcomes, we observed cluster-specific associations consistent with the characteristics of each cluster (Supplementary Table 8).
These findings highlight the potential for GRSs to identify subgroups among individuals with obesity. The GRSuncoupling quantifies people’s risk of obesity without cardiometabolic comorbidities, whereas the GRSBFP quantifies people’s risk of obesity with cardiometabolic comorbidities. The cluster-specific GRSs provide further granularity to this subgroup identification.
GRSuncoupling association signature validated in an independent cohort
We validated the association profiles of GRSuncoupling, the eight cluster-specific GRSs (GRS1–8) and GRSBFP with adiposity and cardiometabolic traits in the Atherosclerosis Risk in Communities (ARIC) study, a population-based cohort (n = 15,792 individuals) (Methods and Supplementary Table 9). Similar to the UK Biobank, in ARIC, GRSuncoupling was associated with a healthier cardiometabolic profile with lower levels (β < 0) for glucose, TC, LDL-C, TG, SBP and DBP and higher HDL compared to GRSBFP. Both GRSs were associated with high adiposity including BMI, waist and hip circumference, and WHR, albeit GRSBFP generally had higher effect sizes (Extended Data Fig. 3a and Supplementary Table 10). Similarly, the association signature of the cluster-specific GRSs with adiposity and cardiometabolic traits corresponded to that of the UK Biobank (Extended Data Fig. 3b,c and Supplementary Table 10).
GRSuncoupling is associated with lower T2D and CHD incidence
The implementation of GRSs in other cohorts is straightforward and allows for the assessment of individuals’ predisposition to comorbidities. We tested associations of GRSuncoupling and GRSBFP with incident coronary heart disease (CHD) and T2D in ARIC and the Mount Sinai BioMe Biobank, an electronic medical record-linked biobank (n = 50,000). A higher GRSuncoupling was associated with a significantly lower risk of both incident CHD (hazard ratio (HR) 0.95; 95% CI 0.92–0.98, per ten-allele change in GRS) and T2D (HR 0.96, 95% CI 0.92–0.99) (Supplementary Table 11), whereas a higher GRSBFP was associated with higher risk of developing T2D (HR 1.04, 95% CI 1.01–1.07), but not CHD (HR 1.01, 95% CI 0.99–1.04).
To evaluate whether lifestyle factors influence the effects of GRSuncoupling and GRSBFP on cardiometabolic disease risk, we stratified ARIC participants by physical activity level (Methods). Within the physically active group, the protective effect of GRSuncoupling on T2D risk was enhanced (HR 0.88, 95% CI 0.80–0.97) (Supplementary Table 12), whereas the adverse association of GRSBFP was attenuated (HR 0.99, 95% CI 0.92–1.05). These findings suggest that physical activity may modulate the effects of GRSuncoupling and GRSBFP on T2D susceptibility. We did not observe the same effect for CHD.
GRSuncoupling already predisposes to higher adiposity but a more favorable cardiometabolic profile early in life
Among 3,457 Danish children and adolescents from the HOLBAEK study (Methods and Supplementary Table 13), the GRSBFP and GRSuncoupling were associated with higher overall adiposity (Extended Data Fig. 4a). Within the population-based cohort subset of 1,811 participants, both GRSs showed associations with BMI, to the same extent as those observed in the UK Biobank (for example, GRSuncoupling: 0.09 in UK Biobank and 0.08 in HOLBAEK) (Supplementary Table 14). Participants with a higher GRSBFP were more likely to present with a dysglycemic profile (higher HOMA-IR, insulin, C-peptide), whereas the GRSuncoupling associated with a neutral glycemic profile and lower alkaline phosphatase (ALP) levels (Extended Data Fig. 4b). Moreover, having a higher GRSuncoupling associated with a lower risk of dyslipidemia (OR 0.89, 95% CI 0.82–0.97) (Extended Data Fig. 4c).
Uncoupling loci and overall adiposity loci have distinct tissue and pathway enrichment profiles
We next performed enrichment analyses for the 266 uncoupling lead variants, using data-driven expression prioritized integration for complex traits (DEPICT) (Methods), to identify the tissues and gene sets in which potential candidate genes may be acting and compared these with the results for the 647 BFP variants. Genes located in BFP-associated loci were mainly enriched in the CNS (P = 0.002), consistent with previous observations for BMI-associated loci6. In contrast, genes located in the uncoupling loci were not enriched in the CNS (P = 0.74) and were mostly enriched in adipose tissue (P = 7 × 10−7), and in cardiovascular (P = 1.4 × 10−5), digestive (P = 7.8 × 10−4), endocrine (P = 2.5 × 10−4) and musculoskeletal systems (P = 2.5 × 10−5) (Fig. 5 and Supplementary Table 15).

DEPICT results were based on summary statistics generated from the association analyses of the 266 lead single-nucleotide polymorphisms (SNPs) with BFP. Tissues enriched after correction for multiple testing at significance threshold (FDR < 0.05) are highlighted in bright blue. The dashed line represents the nominal significance level (P < 0.05). For full results, see Supplementary Table 15.
In gene set enrichment analyses, we replicate previous findings for uncoupling loci, implicating insulin signaling, glucose homeostasis, lipid metabolism, immune and inflammatory response, and pathways related to adipose tissue biology (Supplementary Table 16). In addition, we identify gene sets not previously implicated, including those related to vascular development, skeletal muscle development, liver development, circadian rhythm, sex differentiation, among others, thereby implicating new biological processes.
On the other hand, genes in BFP loci were enriched for neurodevelopment and neuron differentiation including pathways related to brain development and regulatory mechanisms of the nervous system, consistent with previous literature6 (Extended Data Fig. 5 and Supplementary Table 16).
Cluster-specific gene set enrichment analyses implicated biological pathways that are consistent with their cardiometabolic health profile. For example, triglyceride lipase activity was highlighted for cluster 1, cardiovascular-related gene sets for cluster 2, muscle-related gene sets for cluster 4, transcriptional regulation of white adipose tissue regulation for cluster 7 (Extended Data Fig. 6 and Supplementary Tables 16 and 17).
The uncoupling GRSs are defined by distinct plasma proteomic profiles
To further characterize the biological signature of the GRSs, we assessed their association with 2,920 Olink-derived plasma proteins in the UK Biobank (Methods and Supplementary Tables 18 and 19). In line with the finding that 80% of these proteins are associated with measured BMI in this population16, both the GRSBFP and GRSuncoupling were associated (false discovery rate (FDR) < 0.005) with a substantial number of protein levels: 915 (31.3%) and 337 (11.5%), respectively, of which 208 proteins were associated with both GRSs. Of these 208 overlapping proteins, 176 (85%) showed directional consistency in effect estimates, likely reflecting primarily adiposity-driven effects (Extended Data Fig. 7 and Supplementary Table 18). Notable examples include proteins shown to associate most strongly with higher BMI16, such as leptin (LEP), fatty acid binding protein 4 (FABP4) and pro-adrenomedullin (ADM). On the other hand, 32 proteins showed directionally opposing effects, potentially highlighting health-driven effects (Fig. 6). These include proteins involved in lipid transport (for example LDLR, APOA1 and APOF), hormonal status (for example IGFBP1, IGFBP2, SHBG and FGF21) or thermogenesis (for example LDLR, GHR, SHBG, CKB and LAMP2). A total of 129 proteins were associated with GRSuncoupling, but not with GRSBFP (Extended Data Fig. 8 and Supplementary Table 18), including neuropeptides (AGRP, NPY and BDNF), hormones (GCG and ADIPOQ), lipoprotein lipase (LPL) and myostatin (MSTN).

Estimated per ten-allele change effect sizes of GRS–protein associations in UK Biobank European ancestry population for GRSuncoupling (in magenta) and GRSBFP (in blue), for rank-based inverse-normal transformed Olink-derived plasma protein concentrations (n = 32).
Gene prioritization identifies genes implicated in various pathways
To identify putative causal genes within the 205 uncoupling loci, we used 14 bioinformatic and functional genomics tools. We prioritized the gene(s) most likely to be causal in each locus and ranked them based on the number of genomic tools that provided support for the given gene. Of the 1,623 candidate genes, 82 were considered high-scoring genes (Methods and Supplementary Table 20). These include genes previously described to be associated with opposite effects on obesity and cardiometabolic traits, such as PPARG7,17,18,19, FAM13A9,11, PEPD11,19,20,21 and IRS1 (refs. 11,19,22,23). The two highest scoring genes, prioritized by 11 tools, were PCSK1 and SMG6 (Supplementary Table 20). While PSCK1’s role in obesity is well established24,25, there is no obvious functional role for either PCSK1 or SMG6 in uncoupling of obesity from cardiometabolic health.
Other high-scoring genes have been implicated in adipose tissue expandability (PPARG26,27, IRS1 (refs. 28,29,30), RSPO3 (ref. 31), FAM13A32, CTSS33,34, TIMP4 (refs. 35,36), PEPD19,20,21, JMJD1C37, CSK38,39, HLX40, MED19 (ref. 41), SENP2 (ref. 42), MLXIPL43,44, ARNT45,46, PIK3R1 (ref. 47) and PNPLA2 (ref. 48)), insulin secretion and beta-cell function (PIK3R149, GPRC5B50, MEF2D51, FBN1 (ref. 52), LDB1 (ref. 53), SENP2 (ref. 54), MAPT55, PBX1 (ref. 56), beiging of white adipose tissue and brown adipose tissue function (CSK39, SLC22A3 (ref. 57), SENP2 (ref. 58), MED19 (ref. 41), LDB1 (ref. 59), HLX40 and CRHR1 (ref. 60)) and inflammation and fibrosis (PEPD20,21, BCN2 (ref. 61), MST1 (refs. 62,63,64), GPRC5B65, MAFF66, CTSS34,67, NPEPPS68, CSK38 and FBN1 (ref. 52,69)) (Supplementary Table 21). Many of these genes and pathways have been described before in the context of uncoupling11, but we also identify genes involved in pathways that have not previously been implicated, such as hepatic control of glucose homeostasis (ARNT70, CTSS34,71, YWHAB72, FBN1 (ref. 73), LDB1 (ref. 74) and SENP2 (ref. 75)), hepatic lipid accumulation (JMJD1C76, NPEPPS77 and MLXIPL44,78) and skeletal muscle growth and function (PPP3R1 (ref. 79), CTSS80,81, CXXC5 (ref. 82), NPEPPS83, SENP2 (ref. 84) and FBN1 (ref. 69)) (Supplementary Table 21).
Prioritized genes in loci that belong to the same cluster tend to share related pathways. For example, prioritized genes in cluster 7 are implicated in regional adipose expandability, including FAM13A32 and RSPO3 (ref. 31), consistent with lower WHR and improved lipid profile, being the defining characteristics for cluster 7 (Fig. 3 and Supplementary Table 21). Another example is for cluster 3, characterized by lower SBP and DBP, which contains several genes implicated in beiging of white adipose tissue and brown adipose tissue function, including CSK39, HLX40, LDB1 (ref. 59), MED19 (ref. 41) and SENP2 (ref. 58) (Fig. 4 and Supplementary Table 21). Brown adipose tissue has been previously linked to lower odds of cardiometabolic diseases, including hypertension85. The overall protective cluster 8 has a more diverse biological basis with multiple genes implicated in adipose expandability, including PPARG26,86, IRS1 (refs. 23,28,29,30,87), TIMP4 (ref. 36), CTSS34, ARNT45,46 and PIK3R1 (ref. 47). TIMP4 is also implicated in nutrient uptake35 and ARNT and CTSS in hepatic glucose control70,71. Therefore, our prioritized genes are implicated in known and/or novel pathways that plausibly contribute to the uncoupling of adiposity from cardiometabolic risk.
Discussion
Obesity is a highly heterogeneous disease that cannot be captured by one single adiposity trait. Here, we performed a multi-trait gene-discovery analysis to account for heterogeneity in cardiometabolic comorbidities. We designed continuous uncoupling phenotypes that range from high adiposity with a healthy cardiometabolic profile to low adiposity with an unhealthy cardiometabolic profile. GWASs of these new phenotypes identified 266 independent variants across 205 genomic loci where the adiposity-increasing allele is also associated with a lower cardiometabolic trait. Furthermore, the 266 variants cluster into eight groups, each representing a genetic subtype with a distinct cardiometabolic risk profile, pointing to specific underlying pathways.
The genetic uncoupling score that aggregates the uncoupling effects of the 266 variants (GRSuncoupling) was associated with a healthier cardiometabolic profile, distinctly different from that of the genetic adiposity score (GRSBFP). The protective effects of GRSuncoupling may be partially mediated through an association with a more favorable fat distribution characterized by a lower WHR and lower MRI-derived VAT/ASAT and trunk fat/GFAT, in particular among women, compared to GRSBFP. These findings corroborate previous observations with greater power, including the sex-specific observations9,10,12,88,89. With such distinct cardiometabolic risk profiles, these genetic scores may facilitate early risk stratification of individuals with obesity allowing for a timely and personalized prevention. This genetic risk stratification was already apparent in childhood and adolescence. Moreover, the uncoupling score was also associated with lower risk of prevalent and incident T2D and CHD in adulthood; however, while the cardiometabolic risk is reduced among individuals with a high uncoupling score, risk for diseases, such as cellulitis, arthrosis, sleep disorders and phlebitis among others, is comparable to those with a high adiposity score, consistent with a previous report90. These observations underscore that the weight-bearing impact of a high body weight on overall health remains, even when cardiometabolic risk is lower.
In previous studies, we and others have identified three clusters of uncoupling loci11,14. Here, with many new uncoupling loci, we replicate the previously reported clusters with greater delineation and identify several new clusters. For example, in previous studies, one of the uncoupling clusters was linked to favorable fat distribution. We identified two such clusters (4 and 7) that, however, differ in the strength of associations with each of the phenotypes. Most new clusters are characterized by the fact that the adiposity-increasing loci are associated with specific cardiometabolic traits, for example, healthier glycemic profile (cluster 5), lower blood pressure (cluster 3) and more granularity (clusters 1, 6, 7 and 8 with four distinct lipid profiles). Our findings extend current knowledge by underscoring that there is substantial heterogeneity among uncoupling loci. The clusters of loci represent distinct genetic subtypes that suggest a range of mechanisms underlying the uncoupling of obesity from its cardiometabolic comorbidities.
We identified shared and distinct proteomic association signatures for GRSuncoupling versus GRSBFP. For the majority (85%) of the 208 proteins associated with both genetic scores, the direction of the association was consistent across both scores. This suggests that, for these proteins, levels are driven by adiposity. For example, levels of leptin and adipsin/complement factor D, two adipokines known to be elevated in individuals with obesity irrespective of their cardiometabolic health status91, increased with the increase in both uncoupling and adiposity scores. A subset of proteins (15%) showed directionally opposite associations between the two scores, capturing health-driven effects. For example, higher plasma levels of IGFBP1 and IGFBP2 were associated with a higher uncoupling score (higher adiposity and improved cardiometabolic health), but with a lower adiposity score, representing lower adiposity and improved cardiometabolic health, corroborating previous reports demonstrating that lower levels of IGFBP1 and IGFBP2 are associated with hypertriglyceridemia and insulin resistance92,93,94. Also, lower LDLR levels and higher SHBG levels were associated with a higher uncoupling score, consistent with a metabolically healthy state as observed by others95,96,97,98,99,100.
Higher levels of circulating LDLR may indicate increased hepatic LDLR shedding and reduced lipoprotein clearance101. GRSBFP was associated with higher levels of circulating LDLR, which has been linked to elevated plasma triglycerides and LDL-C95,101—factors contributing to cardiometabolic complications such as inflammation, atherosclerosis and myocardial infarction96,97. In contrast, GRSuncoupling and lipid-protective clusters 1, 7 and 8 were associated with lower plasma LDLR, consistent with a healthier metabolic profile. Reduced circulating LDLR in these subtypes may reflect greater hepatic LDLR availability and more efficient lipid clearance, suggesting enhanced hepatic lipid clearance as a potential mechanism underlying the cardiometabolic benefits observed in these subtypes.
Several proteins were exclusively associated with a higher uncoupling score, such as ADIPOQ and LPL. ADIPOQ has been shown to be higher in metabolically healthy individuals, promoting insulin sensitivity and having cardioprotective and anti-inflammatory effects91,102,103, whereas LPL plays a role in triglyceride clearance and lipid distribution, potentially contributing to a metabolically healthy state104,105. Myostatin levels decreased with an increasing uncoupling score. Myostatin is considered a drug target for sarcopenia and muscle mass preservation in combination with weight-loss drugs106,107, implicating skeletal muscle mass and function in metabolic health91.
In contrast to a role of the CNS in overall obesity, tissue enrichment analysis for genes in uncoupling loci pointed to adipose tissue, cardiovascular, digestive, endocrine and musculoskeletal systems, implicating pathways previously reported for uncoupling (insulin signaling, glucose homeostasis, lipid metabolism, immune and inflammatory response and adipose tissue biology)7,11,15, but also revealing new ones (for example vascular development, skeletal muscle development, liver development, kidney development, cartilage development, circadian rhythm, sex differentiation and response to hypoxia).
To pinpoint candidate causal genes within each uncoupling locus, we established a bioinformatics and functional genomics gene prioritization pipeline. The highest scoring genes provide further support for biological processes such as adipose tissue expandability, fat distribution and brown adipose tissue function. Other newly identified genes highlight emerging mechanisms, such as inflammation and fibrosis, hepatic glucose control and lipid accumulation, and muscle function. For example, liver specific ablation of Jmjd1c, a gene prioritized for rs10761785, which is associated with higher BMI, WHR and lower LDL-C and TC levels, decreases lipogenesis and protects from insulin resistance despite obesity in mice76. Knockout of Arnt, a gene prioritized for rs10888393 and associated with increased BFP, high HDL-C and low HbA1c, may have a tissue-specific role affecting adiposity and cardiometabolic health. Fat-specific Arnt knockout mice are leaner and protected against diet-induced glucose intolerance and obesity, whereas hepatocyte-specific Arnt knockout mice have increased fasting glucose and impaired glucose tolerance45,46,70.
Our study has several limitations. First, it was conducted exclusively in individuals of European ancestry, so the generalizability of our findings to other populations remains to be determined. Second, our proteomic profiling analysis was constrained by the set of proteins assayed and may not capture all relevant proteins driving adiposity and/or health effects. Third, gene prioritization relied on existing annotations and previous knowledge, which may bias against novel genes with currently unknown functions. Nonetheless, we employed a comprehensive strategy that integrates predictions from multiple bioinformatic and functional genomic tools to help mitigate these limitations.
Taken together, by designing continuous uncoupling traits, we substantially increased statistical power for discovery, resulting in a more than twofold increase in the number of uncoupling loci identified. Gene prioritization and pathway and protein analyses underscore the importance of a range of peripheral pathways in uncoupling. We provide further support for adipose tissue expandability, insulin secretion and beta-cell function, beiging of white adipose tissue, inflammation and fibrosis, and also highlight mechanisms not previously implicated in uncoupling, such as hepatic lipid accumulation, hepatic control of glucose homeostasis and skeletal muscle growth and function. Individuals with a high genetic uncoupling score display a protective cardiometabolic risk profile despite having a higher risk of obesity. Their profile is distinct from that of individuals with a high overall adiposity score who have an increased cardiometabolic risk. Notably, we show that this risk stratification is already evident in childhood and adolescence. The overall genetic uncoupling score and its eight derived sub-scores advance the genetic subtyping of obesity by delineating distinct cardiometabolic risk signatures. The GRSs corresponding to the eight genetic subtypes can be readily implemented in other populations. These genetic subtypes may form the basis of subtype-stratified treatment, prevention and prognosis and may ultimately contribute to precision medicine in obesity.
Methods
Ethical approval
UK Biobank data access was approved by the UK Biobank through project application number 1251. UK Biobank has obtained approval from a committee and researchers do not need separate approval. The HOLBAEK study was approved by the ethics committee of region Zealand, Denmark (SJ-104) and by the Danish Data Protection Agency (REG-043-2013). For ARIC, all relevant ethical guidelines have been followed and any necessary Institutional Review Board (IRB) and/or ethics committee approvals have been obtained.
Study populations
UK Biobank
The UK Biobank is a prospective cohort study with extensive genetic and phenotypic data, collected in approximately 500,000 individuals, aged between 40–69 years. Participants were enrolled from April 2007 to July 2010 at one of 21 assessment centers across the UK. Baseline information, physical measures and biological samples were collected according to standardized procedures108,109,110. Questionnaires were used to collect health and lifestyle data. Study design, protocols, sample handling and quality control have been described in detail elsewhere108,109,110.
Ancestry
We restricted analyses to individuals of European ancestry, defined by using k-means clustering111. In brief, we calculated principal components and their loadings for 488,377 genotyped participants based on the intersection of ~121,000 quality-controlled variants with the 1000 Genomes Project reference panel (phase 3 v.5). We projected the 1000 Genomes reference panel dataset on the principal-component analysisA (PCA) loadings from the UK Biobank. We then applied k-means clustering to the UK Biobank PCA and the projected 1000 Genomes reference panel dataset, prespecifying four clusters. Individuals that clustered with the EUR 1000 Genomes cluster were assigned as European ancestry.
Phenotypes
We analyzed 11 single traits; three adiposity traits: BMI, BFP and WHR; and eight cardiometabolic traits: TC, LDL-C, HDL-C, TGs, glucose, HbA1c levels, SBP and DBP. All phenotypic data used for analyses were collected at the baseline visit. BMI was calculated as weight (kg) divided by height squared (m2). WHR was created by dividing the waist circumference by the hip circumference. Individuals with waist and hip measurements <50 cm and >150 cm were removed. For individuals on lipid-lowering medication, LDL-C was adjusted by dividing the LDL-C value by 0.7 and TC by dividing by 0.8 (refs. 112,113). TC and TG were log transformed. For the GWAS analysis of glucose and HbA1c, individuals receiving insulin therapy, (n = 4,697) and those with glucose > 15 mmol l−1 or HbA1c > 100 were excluded (n = 804). SBP and DBP were created by calculating the average of two measurements at the baseline visit and adjusted for medication use by adding 15 mm Hg on the SBP value and 10 mm Hg on the DBP value114,115. Upon exploring the data, we identified a subgroup of women recruited in one center, Sheffield, who deviated from the rest of the data. As a result, we excluded 121 women recruited at Sheffield and that had BFP > 55% and BMI > 40 kg m−2. Additionally, women who were pregnant at the time of recruitment were excluded. Finally, 452,768 participants (207,204 men and 245,564 women) were included in the analyses of lipid traits and 448,071 (204,377 men and 243,694 women) for glycemic traits.
Genotypes
Participants were genotyped on two arrays. The majority (n = ~450,000) were genotyped using the UK Biobank Axiom Array and the remaining participants (n = ~50,000) were genotyped using the UK BiLEVE Array, which has >95% of the variants in common with the UK Biobank Axiom Array. Quality control, performed by the UK Biobank team, included testing for batch, plate and array effects, Hardy–Weinberg equilibrium and discordance across control replicates. Samples of poor quality, with high missingness rate and heterozygosity, were removed110. Missing single-nucleotide polymorphisms (SNPs) were imputed using the UK10K reference panel by the UK Biobank team.
Atherosclerosis Risk in Communities study
The ARIC study is a prospective cohort study of 15,792 individuals, including 11,478 white individuals and 4,314 African American individuals, from four US communities (Forsyth County, NC; Jackson, MS; suburbs of Minneapolis, MN; and Washington County, MD). Participants aged between 45–64 years at baseline and recruited between 1987–1989, received extensive examinations, including medical, social and demographic data. A detailed description of the ARIC study design is published elsewhere116. Adiposity and cardiometabolic traits considered in this study were measured at the baseline visit.
Incident CHD was ascertained through a combination of death certificate reviews, hospital records and annual participant follow-ups to identify hospitalizations and deaths occurring during the previous year117. Incident CHD cases were defined as definite fatal CHD, definite or probable myocardial infarction (MI), silent MI between examinations as determined by ECG, or coronary revascularization. T2D cases were identified at baseline and during follow-up visits using glucose measurements, self-reported physician diagnosis of T2D or use of diabetes medication. T2D was defined in accordance with World Health Organization guidelines as a fasting serum glucose ≥7.0 mmol l−1, a non-fasting serum glucose ≥11.0 mmol l−1 (when fasting samples were unavailable) or the use of blood glucose-lowering medications. Individuals with T2D at baseline were removed from this analysis.
Blood was drawn for DNA extraction at the baseline exam. Genotyping within ARIC was performed on the Affymetrix 6.0 DNA microarray (Affymetrix) and genotype data that passed quality control filters were imputed into the 1000 Genomes phase 3 reference data using IMPUTE v.2.3.2 (refs. 118,119). The ARIC study was approved by the IRBs at each site and written informed consent was obtained from all study participants.
BioMe Biobank
BioMe is an ongoing electronic medical record-linked biobank with more than 60,000 patients enrolled through the Mount Sinai Health System in New York. BioMe is a multiethnic biobank comprising individuals of African, Hispanic, European, Asian and other ancestries. Genotyping data on the Global Screening Array (GSA-24v1-0_A1) is available for 32,595 individuals. The data were cleaned for duplicate samples, discordant sex, heterozygosity rate that exceeded 6 × s.d. from the population mean, call rate <95% at the site and individual level and deviation from the Hardy–Weinberg equilibrium. After quality control (QC), 31,705 individuals and 604,869 variants were retained. Imputation of the GSA array was performed using impute2 (ref. 120) using the 1000 Genomes reference panel. In the BioMe Biobank, CHD cases were identified using ICD-9 and ICD-10 codes, procedure codes for bypass surgery or percutaneous transluminal coronary angioplasty or documentation of abnormal cardiac catheterization. T2D cases were defined using the eMERGE phenotyping algorithm121. Baseline was defined as first outpatient visit after 1 January 2011, with at least 1 year enrolled in the Mount Sinai Health system. Individuals with CHD or T2D cases occurred before or within 1 year of enrollment were classified as prevalence cases and excluded from analyses. The BioMe Biobank received ethics approval from the IRB of the Mount Sinai School of Medicine.
The HOLBAEK study
The HOLBAEK study consists of two Danish cohorts: a cohort from the Children’s Obesity Clinic of the Holbaek Hospital, comprising children and adolescents with a BMI at or above the 90th percentile (BMI s.d. score (SDS) ≥ 1.28, overweight or obesity) based on Danish reference standards122; and a population-based cohort recruited from schools in 11 municipalities across Region Zealand123. In the obesity clinic cohort, anthropometrics were measured at clinical examinations, whereas the population-based group was assessed in a mobile laboratory by medical professionals. Details on the cohort and phenotypic data are published elsewhere124. We considered 23 traits for our cross-sectional analyses (5 binary and 18 continuous). BMI SDS was derived using the least mean squares method, referenced against Danish reference standard122. Waist-to-height ratio (WtHR) SDS and WHR SDS were calculated based on age- and sex-specific reference values from NHANES125. Obesity was defined as having a BMI SDS ≥ 2.33 (99th percentile and above126). Hyperglycemia, insulin resistance, dyslipidemia and hypertension were defined according to published guidelines127,128,129,130. HOMA-IR was calculated as (insulin mU l−1 × glucose mM)/22.5. Exclusion criteria for the current analyses included individuals younger than 5 years or older than 19 years, those taking medications for obesity or diabetes, participants meeting T2D criteria based on fasting plasma glucose levels ≥7.0 mmol l−1 or HbA1c ≥ 48 mmol mol−1, and individuals without genotyping data. The final number of participants of European ancestry analyzed was 1,646 for the obesity clinic cohort (45% boys, median age 11.7 years (Q1 9.6 years–Q3 14.0 years)) and 1,811 for the population-based cohort (43% boys, median age 11.6 years (Q1 8.9 years–Q3 14.5 years))
Genotype data
Genotyping in the HOLBAEK study was conducted using Illumina Infinium HumanCoreExome-12 v.1.0 or HumanCoreExome-24 v.1.1 BeadChips, analyzed on the Illumina HiScan system. Genotype calling was performed using the Genotyping Module (v.1.9.4) within GenomeStudio software (v.2011.1; Illumina). Phasing was done with EAGLE2 (v.2.0.5), and imputation was carried out using PBWT to the Haplotype Reference Consortium (HRC1.1) via the SANGER imputation server. As HRC1.1 does not include insertions and deletions and does not fully overlap with imputed UK Biobank genotype data, up to 20% of GRSBFP and GRSuncoupling variants were not available. We therefore identified high linkage disequilibrium (LD) (R2 > 0.8) proxies, based on LD information from 20,000 unrelated (KING < 0.0884), randomly sampled UK Biobank participants. These participants self-identified as ‘White British’ and clustered with this group in PCA. Genotype QC for this reference panel included filtering on SNP missingness <5% and INFO > 0.3, and we excluded participants with sex chromosome anomalies, sex discrepancies, heterozygosity outliers and genotype call rate outliers. Proxies were identified for 123 of 142 missing variants for GRSBFP and 56 of 58 missing variants for GRSuncoupling, with a median R2UKB of 0.99. The final GRSs therefore included 628 and 264 variants, respectively, with a median INFOHOLBAEK of 0.98. The GRSs were scaled to per ten-allele change.
Statistical analysis
Using linear and logistic regression, we assessed the association between both GRSs and the 23 outcome traits, adjusting for age, sex, four PCs and genetic batch (n = 3). The continuous traits were log transformed (except for BMI, WHR, WHtR, SBP and DBP SDS) and then standardized to unit variance and s.d. of one. All analyses were stratified by cohort (obesity clinic/population) and estimates pooled using inverse-variance weighting. In addition, all analyses were further stratified by sex (Supplementary Table 11). P values were adjusted using Benjamini–Hochberg correction across the 23 traits.
GWAS analyses
Our GWAS analyses aimed to identify variants that uncouple adiposity from its comorbidities. As such, we used 11 single traits: three adiposity traits: BMI, BFP and WHR; and eight cardiometabolic traits: TC, LDL,-C HDL-C, TG, glucose, HbA1c, SBP and DBP. First, we derived residuals for each of the single traits, for men and women separately, using linear regression analyses adjusting for age, age2, genotyping array and sequencing center. Next, the distributions of residuals of the 11 single traits were inverse normalized. The derived s.d. scores have a mean of 0 and a s.d. of 1. We then created pairwise composite traits with one of the anthropometric traits and one of the cardiometabolic traits by subtracting the s.d. scores of the cardiometabolic trait (TC, LDL-C, HDL-C, TG, glucose, HbA1c, SBP and DBP) from those of the adiposity traits (BMI, BFP and WHR), resulting in 24 bi-traits. For example, a BMI–TC bi-trait is created as follows: BMIsdcores – TCsdcores (Extended Data Fig. 1). For any given individual, a positive score for the BMI–TC bi-trait indicates that the person has a relatively higher BMI compared to their TC levels, whereas a negative score means that they have a relatively lower BMI compared to their TC levels. As HDL-C correlates positively with cardiometabolic health, HDL-C needs to be treated differently when creating the bi-traits. We have addressed this by flipping the sign of HDL z-scores before creating the bi-traits. This adjustment ensures that subtracting HDL-C from an adiposity trait results in a bi-trait where a positive or negative score retains the same meaning as for the other bi-traits. In other words, BMI–HDL, is in fact BMI–(–HDL). Therefore, a positive score indicates high BMI and high HDL, which corresponds to high adiposity and a protective cardiometabolic effect. Conversely, a negative score indicates low adiposity and high cardiometabolic risk (low HDL).
For BMI as the adiposity measure, we have eight bi-traits: BMI–TC, BMI–LDL-C, BMI–HDL-C, BMI–TG, BMI–glucose, BMI–HbA1c, BMI–SBP, BMI-DBP. Similarly, for BFP: BFP–TC, BFP–LDL-C, BFP–HDL-C, BFP–TG, BFP–glucose, BFP–HbA1c, BFP–SBP, BFP–DBP, and for WHR: WHR–TC, WHR–LDL-C, WHR–HDL-C, WHR–TG, WHR–glucose, WHR–HbA1c, WHR–SBP and WHR–DBP. As such, we performed 70 mixed model GWAS tests analyzing 24 bi-traits and 11 single traits within men and women separately (35 traits × 2) using BOLT-LMM v.2.4.1131 adjusting for the first ten principal components.
Variants with MAF < 0.1% were removed from the analyses. For imputed variants, an INFO score threshold = 0.3 was used. For each trait, we used METAL to meta-analyze the GWAS results of men and women using fixed effects132. Using LD score regression v.1.0.1 (LDSC), we observed mild inflation, with LDSC intercepts ranging between 1.09 and 1.18 and LDSC ratios indicating that 10–13% of the inflation observed can be ascribed to causes other than a polygenic signal. Therefore, we applied genomic control by adjusting the s.e. for the LDSC intercept. Specifically, for each trait, we used the corrected s.e.: s.e.corrected = s.e.GWAS × sqrt (LDSC intercept) of the sex-specific GWAS as the s.e. column in the inverse-variance weighted meta-analysis of men and women. The new corrected LDSC intercepts ranged from 1.03 to 1.08. LDSC ratio indicated that no more than 5.7% of the inflation observed can be ascribed to causes other than a polygenic signal.
Conditional analyses
To identify additional independent signals in associated loci for the 24 bi-traits, we used GCTA v.1.94.4 (ref. 133). We performed approximate joint and conditional SNP association analyses in each locus, which takes into account LD between SNPs. For each locus, we defined a 2-Mb region encompassing 1 Mb on both sides of the lead SNP. Lead SNPs (P < 5 × 10−10) identified in known long-range high-LD regions were treated as a single large locus in the GCTA analysis134. We used unrelated European ancestry participants from the UK Biobank as the reference sample to acquire conditional P values for association. Conditional independent variants that reached P < 5 × 10−10 were considered as index SNPs. We additionally restricted to SNPs that were genome-wide significant (P < 5 × 10−10) in the original summary statistics.
Identification of genome-wide significant loci
Our genome-wide significance threshold of P < 5 × 10−10 accounted for the analyses of 24 bi-traits, across women and men. To identify genome-wide associated loci and their respective lead SNPs, we proceeded as follows. We started with the independent variants that resulted from the conditional analyses. To define a locus associated with increased adiposity and protective effects on cardiometabolic traits, we retained only variants for which the single-trait GWASs for both traits reached marginal significance, defined as P < 10−4, and for which the association was opposite to the established phenotypic correlation. For example, for BMI and LDL-C, we selected variants for which the BMI–LDL-C bi-trait reached genome-wide significance, and subsequently extracted the variants for which the single-trait associations with BMI and with LDL-C reached P < 10−4 and their direction of association was opposite of the established phenotypical positive association. As such, we identified 1,103 association signals across the 24 bi-traits. Next, we applied a Bayesian divisive clustering algorithm, HyPrColoc135 to determine, for each of the 1,103 association signals, whether the associations across the bi-traits and both single traits colocalize. In the above example, we would only keep loci of which the associations colocalize across the BMI–LDL-C bi-trait, BMI and LDL-C. HyPrColoc is a deterministic Bayesian algorithm that, for a given genomic region, identifies clusters of traits which colocalize at distinct causal variants135. The algorithm also allows for sample overlap for the tested traits and corrects for it. As such, we provided the following input files to HyPrColoc upon testing the colocalization for each of the 1,103 associations: (1) a file with the β values of all the variants to be tested and the traits in consideration; (2) another file with their s.e. values; (3) a 3 × 3 matrix denoting the phenotypic correlation between the bi-trait and its corresponding pair of single traits estimated from the UK Biobank data; (4) an LD matrix for all variants within 1-Mb region around the lead SNPs; and (5) a 3 × 3 matrix with all values equal to 1. We used a 1-Mb region around the input variant (0.5 Mb on each side). Therefore, of 1,103 association signals, we found that 602 association signals corresponding to 425 unique SNPs colocalized across the bi-traits and their corresponding single traits. We subsequently inspected LocusZoom plots for potential overlap between independent loci across the traits and for missed colocalized association signals because HyPrColoc assumes only one ‘causal’ variant per region. This manual inspection led to either narrowing or widening the genomic region of 32 colocalized association signals and performing HyPrColoc again to identify other colocalized variants that were missed.
If more than one variant in the same region was retained for different traits, we chose the lead variant based on the following criteria. If the variants were in high LD (r2 > 0.9), we randomly picked one of them. Otherwise, if more than one variant was significantly associated with related traits, such as BMI–glucose, BFP–glucose and BMI–HbA1c, then we chose the variant that had lower P values for a larger number of traits. Last, if two variants in the same region were each associated with a bi-trait that represents different categories of cardiometabolic traits, we kept both variants even though they were in the same locus. For example, if one variant was associated with BMI–SBP and another with BMI–HbA1c, then we kept both, as the two may be contributing to the uncoupling of adiposity and comorbidities via different mechanisms. In all cases, each variant is represented only once regardless of how many traits the variant is associated with. In total, we retained 266 unique variants in 205 genomic loci. For 152 (57%) of the 266 variants, the effect allele was the minor allele.
Cluster analysis
We used the Noise-Augmented von Mises-Fisher Mixture model (NAvMix) algorithm to cluster the 266 lead variants based on their association with the single traits136. In brief, noise-augmented directional clustering clusters variants based on their proportional associations with different traits. The algorithm outputs a probability of membership for each datapoint (variant) to belong to each cluster. Each datapoint is then assigned to the cluster for which it has the highest probability. The procedure is repeated for a varying number of clusters and the final number of clusters is chosen based on the Bayesian Information Criterion. NAvMix outputs a noise cluster that includes data points (variants) that do not belong to any cluster and are thus considered outliers. We used the effect size (β) of the association of each of the 266 lead variants with each of the 11 single traits as input. We used BFP as a reference, assuming a positive direction of effect to facilitate comparison across traits. The associations with other traits were expressed using the BFP-increasing allele as the effect allele and the BFP-decreasing allele as the alternate allele.
Genetic risk scores and their association with anthropometric and cardiometabolic traits, and phenome-wide association study
We constructed GRSs for all 266 identified variants combined (GRSuncoupling) and for each of the eight clusters separately (GRS1–GRS8) in 373,747 unrelated individuals of European ancestry from the UK Biobank. We also generated a GRS for BFP (GRSBFP) based on 647 lead variants (where 353 (55%) variants had the effect allele as the minor allele) that reached P < 5 × 10−9 (clumped for r2 < 0.1 in a 1-Mb region, MHC region removed) in our single-trait GWAS for BFP in the UK Biobank. All of the ten GRSs were weighted by the effect size estimated for BFP from the GWAS that we performed in the current study. The GRSs were rescaled to per ten-allele change137 to enable comparison across GRSs that consist of different number of SNPs. Cluster 2 was identified by the clustering analysis to be associated with lower WHR. For all analyses that followed the clustering analyses, the effect and reference alleles of genetic variants that were used to construct GRS2 were flipped to reflect a profile of higher adiposity and facilitate comparison with the other clusters. To support implementation of our approach in other cohorts, we provide the code for constructing the GRSs (Supplementary Code) and the variant-specific effect sizes (β values) used in the GRSs (Supplementary Table 6).
To test the association of GRSs with anthropometric and cardiometabolic traits, we performed linear regression analyses for 24 traits, including SBP, DBP, HDL-C, TG, LDL-C, TC, glucose and HbA1c, and the following anthropometric traits: height, WHR, hip circumference, waist circumference, BFP, FFMI (computed as whole body fat-free mass divided by height squared), BMI, gynoid fat percentage, android fat percentage, MRI-measured VAT, ASAT, VAT:ASAT ratio, liver proton density fat fraction (liver fat), trunk fat (total trunk fat volume), GFAT volume and trunk fat:GFAT ratio. GFAT was calculated using VAT, ASAT and the total adipose tissue between the bottom of the thigh muscles to the top of vertebrae T9 (TAT) volume using the following formula138:
GFAT = TAT (between top of T9 and bottom of thigh muscles) – VAT – ASAT. Each of the traits was adjusted for age, sex, genotype array and study site and the first ten principal components in a linear regression model. The resulting residuals were transformed to approximate normality using inverse-normal rank scores before the association testing.
In addition, we performed phenome-wide association (PheWAS) analyses in unrelated individuals of European ancestry from the UK Biobank using the PHEnome Scan ANalysis Tool (PHESANT)139. Analyses were performed using a linear or logistic regression for continuous and binary outcomes, respectively, using the following covariates: age at enrollment, sex, genotyping array and the first ten genetic principal components.
Statistical analysis in ARIC and BioMe
We generated ten GRSs: GRSuncoupling, GRS1–GRS8 and GRSBFP in 9,240 unrelated European ancestry participants of the ARIC study. For the continuous traits, we created rank-based inverse-normal transformed traits adjusting for age, sex and the first ten PCs. We analyzed adiposity and cardiometabolic traits, including hip circumference, waist circumference, WHR, BMI, SBP, DBP, HDL-C, TG, LDL-C, TC and glucose in ARIC. We performed linear regression analyses to test the association of each GRS with the continuous traits. For analyses of incident T2D and CHD, we generated ten GRSs: GRSuncoupling and GRSBFP in 23,208 unrelated individuals of European (n = 8,985), Hispanic (n = 7,984) and African (n = 6,239) ancestry from the BioMe Biobank. We tested the association of each GRS with incident T2D and CHD in BioMe and ARIC using a Cox proportional hazard model after adjusting for age, sex, ancestry (for BioMe) and ten principal components. BioMe and ARIC association results were meta-analyzed using inverse-variance weighted meta-analysis. To further examine how lifestyle factors influence the association between GRSs and cardiometabolic disease risk in ARIC, we assessed physical activity—quantified as total metabolic equivalent (MET) hours per week of moderate-to-vigorous leisure-time physical activity. In the Cox models, we further stratified participants into physically active (top 30% of MET levels) and inactive (bottom 70%) groups to assess whether physical activity level impacted these associations.
Plasma proteomic characterization of the GRSs
We assessed the association of GRSuncoupling, GRS1–GRS8 and GRSBFP with Olink-derived plasma protein measurements in the UK Biobank. This analysis included 30,271 unrelated individuals of European ancestry from the random baseline sample selected by the UK Biobank Pharma Proteomics Project16, specifically those included in Olink batches 1–6. After excluding three proteins with >25% missing data (PCOLCE (62%), NPM1 (73%) and GLIPR1 (>99%)), we included 2,920 proteins with a median missingness of 2.8%. Measurements below the limit-of-detection were included, in line with Olink’s recommendations (https://olink.com/faq/how-is-the-limit-of-detection-lod-estimated-and-handled/). Using linear regression, we assessed the association between the ten GRSs (scaled to a per ten-allele change) and protein measurements (rank-based inverse-normal transformed). Covariates included age at measurement, age squared, sex, UK Biobank assessment center, ten PCs, genotyping array, Olink batch, fasting time at measurement (hours) and time between measurement and processing of the sample by Olink (years). Benjamini–Hochberg adjustment was applied to control the FDR at 0.005 (0.05/10 × GRS) across the 29,200 protein–GRS associations. To distinguish between adiposity- and health-driven associations, we grouped proteins that showed evidence of association with both GRSBFP and one or more of the adiposity-uncoupled GRSs, based on their directional concordance. Moreover, we assessed which proteins uniquely associated with any of the adiposity-uncoupling GRSs, but not GRSBFP.
Gene prioritization analysis
To identify the likely causal gene(s) within each of the identified loci, we used a combination of up to 14 bioinformatics and functional genomics methods. In addition to the annotated ‘nearest gene’, we used a ‘coding proxy’ approach, five bioinformatics tools, and leveraged in vitro adipogenic differentiation dataset with seven measures. For each gene, we summed the number of methods for which it was prioritized. Genes with a score ≥7 were prioritized. The methods used for the gene prioritization score are described below.
Bioinformatics tools
Nearest gene
We used the nearest gene as predicted by Ensembl Variant Effect Predictor (VEP)140.
Coding proxy
For a given lead SNP, we considered variants in high LD (r2 > 0.8) within a 1-Mb window. If one or more of those variants was a coding variant, then the gene(s) in which those coding variants lie were prioritized. We annotated the variants in VEP. A coding variant was defined as any variant with the following annotations: synonymous_variant, missense_variant, inframe_insertion, inframe_deletion, stop_gained, frameshift_variant, splice_donor_variant and splice_acceptor_variant.
ABC-max
We used FUMA141 to select all SNPs in high LD (r2 > 0.8) using as reference panel UK Biobank release 2b 10K Europeans. We intersected the total of 10,095 SNPs (lead SNPs and proxies in high LD) with enhancers and target genes predicted by the Activity-by-Contact (ABC) model142 in the following tissues: adipose, adrenal gland, astrocytes, pancreas, cardiac muscle cell, coronary artery, smooth muscle cell of coronary artery, heart ventricle, hepatocyte, liver, spleen, skeletal muscle myoblast and thyroid gland. Intersection of SNPs and enhancers was carried out using the function intersect from BEDtools v.2.29.2143.
Polygenic Priority Score
We used Polygenic Priority Score v.0.1 (ref. 144), a similarity-based algorithm that uses a broad range of omics data, including scRNA-seq. We used a reference panel of 10,000 randomly selected subjects from the UK Biobank and retrieved the gene with the highest Polygenic Priority Score in each associated loci.
Data-driven expression prioritized integration for complex traits
We used summary statistics generated from the association analyses of the 266 lead SNPs with BFP as input for DEPICT with default parameters145. DEPICT is an integrative tool that uses transcriptome expression (microarray data), pathways and protein–protein interactions to prioritize the most likely causal genes and highlight enriched tissues and pathways.
fastENLOC
We colocalized associated loci with expression quantitative trait loci (eQTLs) from GTEx v.8 (ref. 146) using fastENLOC147, prioritizing genes affected by eQTLs colocalizing at regional colocalization probability (RCP) > 0.1. We colocalized lead SNPs and variants in high LD (r2 > 0.8) with eQTLs in adipose tissue (subcutaneous and visceral), pituitary, brain cortex, brain hypothalamus, brain hippocampus, brain amygdala, adrenal gland, thyroid gland, liver, kidney, pancreas, skeletal muscle, salivary gland and heart (atrial appendage and left ventricle).
CS2G depot-specific gene prioritization
We intersected the 266 associated variants and their proxies (r2 > 0.8) with depot-specific chromatin accessible signals in subcutaneous and visceral human primary adipose-derived mesenchymal stem cells (AMSCs). We then performed combined S2G strategy CS2G148 to predict the effector genes. We reported the effector genes with a cutoff of 0.05 on the 1000 Genome and UK Biobank scores. The detailed protocol for AMSC proliferation, induction and differentiation is outlined in Laber et al.149. Overall, AMSCs were obtained from subcutaneous and VAT from patients undergoing a range of abdominal laparoscopic surgeries149 and isolated as previously described150. For a subset of donors, the purity of AMCSs was assessed as previously described151. Each participant gave written informed consent before inclusion and the study was approved by the ethics committee of the Technical University of Munich (study no. 5716/13). Cells were introduced (counted as differentiation day 0) and put into differentiation for 14 days until fully differentiated. Samples are collected at differentiation day 14. Donor genotyping, SNP QC as well as the genotype imputation were performed as previously described152. Nuclei and library preparation for AMSC ATAC-seq were performed as previously described152. ATAC peaks were called by MACS3 (v.3.0.0). After peak calling, narrow peaks from all the samples (n = 15 subcutaneous AMSCs and n = 14 visceral AMSCs) were first combined, then the overlapped intervals were merged into a single interval using BEDtools (v.2.30.0) (function BEDtools merge -I)143.
Overlap with eQTL data from GTeX
For the association and scoring of SNPs with genes we made use of eQTL data from GTEx (v.8) for subcutaneous and VAT as well as enhancer capture HiC data (‘GSE140782_ECHiC.txt.gz’ https://doi.org/10.1038/s41588-020-0709-z), DNase-seq based chromatin accessibility (‘GSE113253_DNase_processed_data.tar.gz’ https://doi.org/10.1038/s41588-019-0359-1) and gene expression data (‘GSE113253_GeneExpr_BM.txt.gz’ https://doi.org/10.1038/s41588-019-0359-1) of hBM-MSC-TERT4 cells subjected to adipocyte differentiation in vitro.
Specifically, the lead SNP or a proxy SNP, which was determined using the R package LDlinkR (https://doi.org/10.3389/fgene.2020.00157) with a threshold of R2 > 0.8, had to fulfill at least one of the four criteria: (1) overlapping with an eQTL in VAT; (2) overlapping with an eQTL in subcutaneous adipose tissue; (3) overlapping with a genomic region that is linked by the enhancer capture data to a promoter region; or (4) overlapping with a DNase1 hypersensitive region. If only the latter case was true, the closest transcription start site was chosen to be the candidate gene. Overlap was determined using the R package GenomicRanges.
For each unique combination of lead SNP and putative candidate gene, we set up a score based on an overlap of the proxy SNPs with an eQTL, overlap of the proxy SNPs with a DNase1 hypersensitive site and its change in accessibility during adipocyte differentiation, overlap of the proxy SNPs with an enhancer region contacting the promoter of the candidate gene and the expression of the candidate gene and its significant change during adipocyte differentiation.
Tissue and gene set enrichment
Tissue and gene set enrichment were performed by DEPICT, using summary statistics generated from the analysis on BFP as input with default parameters145. Both were performed on all uncoupling loci and on cluster-specific loci. Only gene sets with at least ten genes were included. We restricted our analyses to the Gene Ontology, KEGG and REACTOME pathway terms. We used FDR < 0.05 as a threshold for significance when considering all the 266 variants. For the cluster-specific analysis, as the clusters have a small number of variants and therefore less power, we used P < 0.05 as a threshold from the ‘Nominal P value’ output from DEPICT regardless of the FDR value.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Description of the datasets and full results are provided in Supplementary Tables 1–21. UK Biobank data are accessible through the application process outlined at https://www.ukbiobank.ac.uk/enable-your-research. Detailed information on the genetic data provided by UK Biobank is available at http://www.ukbiobank.ac.uk/scientists-3/genetic-data/ and http://biobank.ctsu.ox.ac.uk/crystal/label.cgi?id=100314. Access to the HOLBAEK study data can be granted through appropriate approvals from the Danish Data Protection Agency and the Ethics Committee for Region Zealand, in accordance with patient consent and data processing agreements. The ARIC dataset is accessible through a process outlined here: https://aric.cscc.unc.edu/aric9/researchers/new_to_aric.
Code availability
The R code required to reproduce the GRSs for the genetic obesity subtypes is present as an accompanying supplementary file.
References
Mokdad, A. H. et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 289, 76–79 (2003).
Alberti, K. G. et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120, 1640–1645 (2009).
Hubert, H. B., Feinleib, M., McNamara, P. M. & Castelli, W. P. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation 67, 968–977 (1983).
Must, A. et al. The disease burden associated with overweight and obesity. JAMA 282, 1523–1529 (1999).
Loos, R. J. F. & Yeo, G. S. H. The genetics of obesity: from discovery to biology. Nat. Rev. Genet. 23, 120–133 (2022).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Yaghootkar, H. et al. Genetic evidence for a normal-weight ‘metabolically obese’ phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 63, 4369–4377 (2014).
Yaghootkar, H. et al. Genetic evidence for a link between favorable adiposity and lower risk of type 2 diabetes, hypertension, and heart disease. Diabetes 65, 2448–2460 (2016).
Lotta, L. A. et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet. 49, 17–26 (2017).
Ji, Y. et al. Genome-wide and abdominal MRI data provide evidence that a genetically determined favorable adiposity phenotype is characterized by lower ectopic liver fat and lower risk of type 2 diabetes, heart disease, and hypertension. Diabetes 68, 207–219 (2019).
Huang, L. O. et al. Genome-wide discovery of genetic loci that uncouple excess adiposity from its comorbidities. Nat. Metab. 3, 228–243 (2021).
Martin, S. et al. Genetic evidence for different adiposity phenotypes and their opposing influences on ectopic fat and risk of cardiometabolic disease. Diabetes 70, 1843–1856 (2021).
Coral, D. E. et al. A phenome-wide comparative analysis of genetic discordance between obesity and type 2 diabetes. Nat. Metab. 5, 237–247 (2023).
Abraham, A. et al. Genetic evidence for distinct biological mechanisms that link adiposity to type 2 diabetes: toward precision medicine. Diabetes 73, 1012–1025 (2024).
Loos, R. J. F. & Kilpelainen, T. O. Genes that make you fat, but keep you healthy. J. Intern. Med. 284, 450–463 (2018).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338 (2023).
Sharma, A. M. & Staels, B. Review: peroxisome proliferator-activated receptor γ and adipose tissue–understanding obesity-related changes in regulation of lipid and glucose metabolism. J. Clin. Endocrinol. Metab. 92, 386–395 (2007).
Scott, R. A. et al. Common genetic variants highlight the role of insulin resistance and body fat distribution in type 2 diabetes, independent of obesity. Diabetes 63, 4378–4387 (2014).
Chen, Z. et al. Functional screening of candidate causal genes for insulin resistance in human preadipocytes and adipocytes. Circ. Res. 126, 330–346 (2020).
Pellegrinelli, V. et al. Dysregulation of macrophage PEPD in obesity determines adipose tissue fibro-inflammation and insulin resistance. Nat. Metab. 4, 476–494 (2022).
Pellegrinelli, V. et al. Defective extracellular matrix remodeling in brown adipose tissue is associated with fibro-inflammation and reduced diet-induced thermogenesis. Cell Rep. 42, 112640 (2023).
Kilpelainen, T. O. et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat. Genet. 43, 753–760 (2011).
Araki, E. et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 372, 186–190 (1994).
Nead, K. T. et al. Contribution of common non-synonymous variants in PCSK1 to body mass index variation and risk of obesity: a systematic review and meta-analysis with evidence from up to 331 175 individuals. Hum. Mol. Genet. 24, 3582–3594 (2015).
Jackson, R. S. et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 16, 303–306 (1997).
Rosen, E. D. et al. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 4, 611–617 (1999).
Jones, J. R. et al. Deletion of PPARγ in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl Acad. Sci. USA 102, 6207–6212 (2005).
Laustsen, P. G. et al. Lipoatrophic diabetes in Irs1(-/-)/Irs3(-/-) double knockout mice. Genes Dev. 16, 3213–3222 (2002).
Miki, H. et al. Essential role of insulin receptor substrate 1 (IRS-1) and IRS-2 in adipocyte differentiation. Mol. Cell Biol. 21, 2521–2532 (2001).
Tseng, Y. H., Kriauciunas, K. M., Kokkotou, E. & Kahn, C. R. Differential roles of insulin receptor substrates in brown adipocyte differentiation. Mol. Cell Biol. 24, 1918–1929 (2004).
Loh, N. Y. et al. RSPO3 impacts body fat distribution and regulates adipose cell biology in vitro. Nat. Commun. 11, 2797 (2020).
Fathzadeh, M. et al. FAM13A affects body fat distribution and adipocyte function. Nat. Commun. 11, 1465 (2020).
Taleb, S., Cancello, R., Clement, K. & Lacasa, D. Cathepsin s promotes human preadipocyte differentiation: possible involvement of fibronectin degradation. Endocrinology 147, 4950–4959 (2006).
Zheng, J. et al. Cathepsin S inhibitor reduces high-fat-induced adipogenesis, inflammatory infiltration, and hepatic lipid accumulation in obese mice. Ann. Transl. Med. 10, 1172 (2022).
Sakamuri, S. et al. Absence of tissue inhibitor of metalloproteinase-4 (TIMP4) ameliorates high fat diet-induced obesity in mice due to defective lipid absorption. Sci. Rep. 7, 6210 (2017).
Wu, Y., Lee, M. J., Ido, Y. & Fried, S. K. High-fat diet-induced obesity regulates MMP3 to modulate depot- and sex-dependent adipose expansion in C57BL/6J mice. Am. J. Physiol. Endocrinol. Metab. 312, E58–E71 (2017).
Buerger, F. et al. Depletion of Jmjd1c impairs adipogenesis in murine 3T3-L1 cells. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1709–1717 (2017).
Choi, S. et al. Novel phosphorylation of PPARγ ameliorates obesity-induced adipose tissue inflammation and improves insulin sensitivity. Cell Signal 27, 2488–2495 (2015).
Usui, M., Uno, M. & Nishida, E. Src family kinases suppress differentiation of brown adipocytes and browning of white adipocytes. Genes Cells 21, 302–310 (2016).
Huang, L. et al. Transcription factor Hlx controls a systematic switch from white to brown fat through Prdm16-mediated co-activation. Nat. Commun. 8, 68 (2017).
Dean, J. M. et al. MED19 regulates adipogenesis and maintenance of white adipose tissue mass by mediating PPARγ-dependent gene expression. Cell Rep. 33, 108228 (2020).
Krapf, S. A. et al. SENP2 knockdown in human adipocytes reduces glucose metabolism and lipid accumulation, while increases lipid oxidation. Metab. Open 18, 100234 (2023).
Morigny, P. et al. Interaction between hormone-sensitive lipase and ChREBP in fat cells controls insulin sensitivity. Nat. Metab. 1, 133–146 (2019).
Hurtado del Pozo, C. et al. ChREBP expression in the liver, adipose tissue and differentiated preadipocytes in human obesity. Biochim. Biophys. Acta 1811, 1194–1200 (2011).
Lee, K. Y., Gesta, S., Boucher, J., Wang, X. L. & Kahn, C. R. The differential role of Hif1β/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab. 14, 491–503 (2011).
Jiang, C. et al. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60, 2484–2495 (2011).
Kwok, A. et al. Truncation of Pik3r1 causes severe insulin resistance uncoupled from obesity and dyslipidaemia by increased energy expenditure. Mol. Metab. 40, 101020 (2020).
Haemmerle, G. et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312, 734–737 (2006).
Kaneko, K. et al. Class IA phosphatidylinositol 3-kinase in pancreatic beta cells controls insulin secretion by multiple mechanisms. Cell Metab. 12, 619–632 (2010).
Atanes, P. et al. Identifying signalling pathways regulated by GPRC5B in beta-cells by CRISPR-Cas9-mediated genome editing. Cell. Physiol. Biochem. 45, 656–666 (2018).
Yoshida, M. et al. O-GlcNAcylation of myocyte-specific enhancer factor 2D negatively regulates insulin secretion from pancreatic beta-cells. Biochem. Biophys. Res. Commun. 605, 90–96 (2022).
Lee, T., Yun, S., Jeong, J. H. & Jung, T. W. Asprosin impairs insulin secretion in response to glucose and viability through TLR4/JNK-mediated inflammation. Mol. Cell. Endocrinol. 486, 96–104 (2019).
Toren, E., Liu, Y., Bethea, M., Wade, A. & Hunter, C. S. The Ldb1 transcriptional co-regulator is required for establishment and maintenance of the pancreatic endocrine lineage. FASEB J. 36, e22460 (2022).
Nan, J. et al. SENP2 regulates mitochondrial function and insulin secretion in pancreatic beta cells. Exp. Mol. Med 54, 72–80 (2022).
Wijesekara, N., Goncalves, R. A., Ahrens, R., De Felice, F. G. & Fraser, P. E. Tau ablation in mice leads to pancreatic beta cell dysfunction and glucose intolerance. FASEB J. 32, 3166–3173 (2018).
Kim, S. K. et al. Pbx1 inactivation disrupts pancreas development and in Ipf1-deficient mice promotes diabetes mellitus. Nat. Genet. 30, 430–435 (2002).
Song, W. et al. Organic cation transporter 3 (Oct3) is a distinct catecholamines clearance route in adipocytes mediating the beiging of white adipose tissue. PLoS Biol. 17, e2006571 (2019).
Lee, J. S. et al. SENP2 suppresses browning of white adipose tissues by de-conjugating SUMO from C/EBPβ. Cell Rep. 38, 110408 (2022).
Kepple, J. D. et al. The transcriptional co-regulator LDB1 is required for brown adipose function. Mol. Metab. 53, 101284 (2021).
Lu, B. et al. The corticotrophin-releasing factor/urocortin system regulates white fat browning in mice through paracrine mechanisms. Int J. Obes. (Lond.) 39, 408–417 (2015).
Bobowski-Gerard, M. et al. Functional genomics uncovers the transcription factor BNC2 as required for myofibroblastic activation in fibrosis. Nat. Commun. 13, 5324 (2022).
Wang, T. et al. Mst1 participates in the atherosclerosis progression through macrophage autophagy inhibition and macrophage apoptosis enhancement. J. Mol. Cell. Cardiol. 98, 108–116 (2016).
Maejima, Y. et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 19, 1478–1488 (2013).
Quan, M. et al. MST1 suppresses disturbed flow induced atherosclerosis. Circ. Res. 131, 748–764 (2022).
Kim, Y. J., Sano, T., Nabetani, T., Asano, Y. & Hirabayashi, Y. GPRC5B activates obesity-associated inflammatory signaling in adipocytes. Sci. Signal 5, ra85 (2012).
von Scheidt, M. et al. Transcription factor MAFF (MAF basic leucine zipper transcription factor F) regulates an atherosclerosis relevant network connecting inflammation and cholesterol metabolism. Circulation 143, 1809–1823 (2021).
Xu, S. et al. CTSS modulates stress-related carotid artery thrombosis in a mouse FeCl(3) model. Arterioscler Thromb. Vasc. Biol. 43, e238–e253 (2023).
Dai, C. et al. CD8(+) T and NK cells characterized by upregulation of NPEPPS and ABHD17A are associated with the co-occurrence of type 2 diabetes and coronary artery disease. Front. Immunol. 15, 1267963 (2024).
Jung, T. W. et al. Asprosin attenuates insulin signaling pathway through PKCδ-activated ER stress and inflammation in skeletal muscle. J. Cell. Physiol. 234, 20888–20899 (2019).
Scott, C. H. et al. Hepatic aryl hydrocarbon receptor nuclear translocator (ARNT) regulates metabolism in mice. PLoS ONE 12, e0186543 (2017).
Karimkhanloo, H. et al. Circulating cathepsin S improves glycaemic control in mice. J. Endocrinol. 248, 167–179 (2021).
Ji, L. et al. The 14-3-3 protein YWHAB inhibits glucagon-induced hepatic gluconeogenesis through interacting with the glucagon receptor and FOXO1. FEBS Lett. 595, 1275–1288 (2021).
Romere, C. et al. Asprosin, a fasting-induced glucogenic protein hormone. Cell 165, 566–579 (2016).
Liu, G., Wang, L., Wess, J. & Dean, A. Enhancer looping protein LDB1 regulates hepatocyte gene expression by cooperating with liver transcription factors. Nucleic Acids Res. 50, 9195–9211 (2022).
Dou, X. et al. The protease SENP2 controls hepatic gluconeogenesis by regulating the SUMOylation of the fuel sensor AMPKα. J. Biol. Chem. 298, 101544 (2022).
Viscarra, J. A., Wang, Y., Nguyen, H. P., Choi, Y. G. & Sul, H. S. Histone demethylase JMJD1C is phosphorylated by mTOR to activate de novo lipogenesis. Nat. Commun. 11, 796 (2020).
Huang, B. et al. PSA controls hepatic lipid metabolism by regulating the NRF2 signaling pathway. J. Mol. Cell. Biol. 13, 527–539 (2021).
Herman, M. A. et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484, 333–338 (2012).
Pfluger, P. T. et al. Calcineurin links mitochondrial elongation with energy metabolism. Cell Metab. 22, 838–850 (2015).
Wan, Y. et al. Cathepsin S deficiency improves muscle mass loss and dysfunction via the modulation of protein metabolism in mice under pathological stress conditions. FASEB J. 37, e23086 (2023).
Wan, Y. et al. Cathepsin S activity controls chronic stress-induced muscle atrophy and dysfunction in mice. Cell. Mol. Life Sci. 80, 254 (2023).
Li, G. et al. CXXC5 regulates differentiation of C2C12 myoblasts into myocytes. J. Muscle Res. Cell Motil. 35, 259–265 (2014).
Osana, S. et al. Puromycin-sensitive aminopeptidase is required for C2C12 myoblast proliferation and differentiation. J. Cell. Physiol. 236, 5293–5305 (2021).
Koo, Y. D. et al. SUMO-specific protease 2 (SENP2) is an important regulator of fatty acid metabolism in skeletal muscle. Diabetes 64, 2420–2431 (2015).
Becher, T. et al. Brown adipose tissue is associated with cardiometabolic health. Nat. Med. 27, 58–65 (2021).
Lefterova, M. I., Haakonsson, A. K., Lazar, M. A. & Mandrup, S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 25, 293–302 (2014).
Tamemoto, H. et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 372, 182–186 (1994).
Ahmed, A., Justo, S. & Yaghootkar, H. Genetic scores associated with favourable and unfavourable adiposity have consistent effect on metabolic profile and disease risk across diverse ethnic groups. Diabet. Med. 40, e15213 (2023).
Akbari, P. et al. Multiancestry exome sequencing reveals INHBE mutations associated with favorable fat distribution and protection from diabetes. Nat. Commun. 13, 4844 (2022).
Martin, S. et al. Disease consequences of higher adiposity uncoupled from its adverse metabolic effects using Mendelian randomisation. eLife 11, e72452 (2022).
Petersen, M. C. et al. Cardiometabolic characteristics of people with metabolically healthy and unhealthy obesity. Cell Metab. 36, 745–761 e745 (2024).
Ho, J. E. et al. Protein biomarkers of cardiovascular disease and mortality in the community. J. Am. Heart Assoc. 7, e008108 (2018).
Shah, R. V. et al. Proteins altered by surgical weight loss highlight biomarkers of insulin resistance in the community. Arterioscler. Thromb. Vasc. Biol. 39, 107–115 (2019).
Rajwani, A. et al. Increasing circulating IGFBP1 levels improves insulin sensitivity, promotes nitric oxide production, lowers blood pressure, and protects against atherosclerosis. Diabetes 61, 915–924 (2012).
Mayne, J. et al. Associations between soluble LDLR and lipoproteins in a white cohort and the effect of PCSK9 loss-of-function. J. Clin. Endocrinol. Metab. 103, 3486–3495 (2018).
Alabi, A. et al. Membrane type 1 matrix metalloproteinase promotes LDL receptor shedding and accelerates the development of atherosclerosis. Nat. Commun. 12, 1889 (2021).
Zegeye, M. M. et al. Soluble LDL-receptor is induced by TNF-α and inhibits hepatocytic clearance of LDL-cholesterol. J. Mol. Med. 101, 1615–1626 (2023).
Simo, R., Saez-Lopez, C., Barbosa-Desongles, A., Hernandez, C. & Selva, D. M. Novel insights in SHBG regulation and clinical implications. Trends Endocrinol. Metab. 26, 376–383 (2015).
Laaksonen, D. E. et al. Testosterone and sex hormone-binding globulin predict the metabolic syndrome and diabetes in middle-aged men. Diabetes Care 27, 1036–1041 (2004).
Lapidus, L., Lindstedt, G., Lundberg, P. A., Bengtsson, C. & Gredmark, T. Concentrations of sex-hormone binding globulin and corticosteroid binding globulin in serum in relation to cardiovascular risk factors and to 12-year incidence of cardiovascular disease and overall mortality in postmenopausal women. Clin. Chem. 32, 146–152 (1986).
Krauss, R. M., Fisher, L. M., King, S. M. & Gardner, C. D. Changes in soluble LDL receptor and lipoprotein fractions in response to diet in the DIETFITS weight loss study. J. Lipid Res. 65, 100503 (2024).
Ahl, S. et al. Adiponectin levels differentiate metabolically healthy vs unhealthy among obese and nonobese white individuals. J. Clin. Endocrinol. Metab. 100, 4172–4180 (2015).
Turer, A. T. & Scherer, P. E. Adiponectin: mechanistic insights and clinical implications. Diabetologia 55, 2319–2326 (2012).
Serra, M. C., Ryan, A. S., Sorkin, J. D., Favor, K. H. & Goldberg, A. P. High adipose LPL activity and adipocyte hypertrophy reduce visceral fat and metabolic risk in obese, older women. Obesity 23, 602–607 (2015).
Serra, M. C., Ryan, A. S. & Goldberg, A. P. Reduced LPL and subcutaneous lipid storage capacity are associated with metabolic syndrome in postmenopausal women with obesity. Obes. Sci. Pr. 3, 106–114 (2017).
Rodgers, B. D. & Ward, C. W. Myostatin/activin receptor ligands in muscle and the development status of attenuating drugs. Endocr. Rev. 43, 329–365 (2022).
Nunn, E. et al. Antibody blockade of activin type II receptors preserves skeletal muscle mass and enhances fat loss during GLP-1 receptor agonism. Mol. Metab. 80, 101880 (2024).
Elliott, P., Peakman, T. C. & UK Biobank. The UK Biobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int J. Epidemiol. 37, 234–244 (2008).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Chami, N., Preuss, M., Walker, R. W., Moscati, A. & Loos, R. J. F. The role of polygenic susceptibility to obesity among carriers of pathogenic mutations in MC4R in the UK Biobank population. PLoS Med. 17, e1003196 (2020).
Lu, X. et al. Exome chip meta-analysis identifies novel loci and East Asian-specific coding variants that contribute to lipid levels and coronary artery disease. Nat. Genet. 49, 1722–1730 (2017).
Li, Y. et al. Association of genetic variants related to combined lipid-lowering and antihypertensive therapies with risk of cardiovascular disease: 2 ×2 factorial Mendelian randomization analyses. BMC Med. 22, 201 (2024).
Tobin, M. D., Sheehan, N. A., Scurrah, K. J. & Burton, P. R. Adjusting for treatment effects in studies of quantitative traits: antihypertensive therapy and systolic blood pressure. Stat. Med. 24, 2911–2935 (2005).
Evangelou, E. et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 50, 1412–1425 (2018).
The ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am. J. Epidemiol. 129, 687–702 (1989).
White, A. D. et al. Community surveillance of coronary heart disease in the Atherosclerosis Risk in Communities (ARIC) Study: methods and initial two years’ experience. J. Clin. Epidemiol. 49, 223–233 (1996).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Howie, B., Marchini, J. & Stephens, M. Genotype imputation with thousands of genomes. G3 (Bethesda) 1, 457–470 (2011).
Kho, A. N. et al. Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome-wide association study. J. Am. Med. Inf. Assoc. 19, 212–218 (2012).
Nysom, K., Molgaard, C., Hutchings, B. & Michaelsen, K. F. Body mass index of 0 to 45-y-old Danes: reference values and comparison with published European reference values. Int. J. Obes. Relat. Metab. Disord. 25, 177–184 (2001).
Lausten-Thomsen, U. et al. Reference values for serum total adiponectin in healthy non-obese children and adolescents. Clin. Chim. Acta 450, 11–14 (2015).
Huang, Y. et al. Lipid profiling identifies modifiable signatures of cardiometabolic risk in children and adolescents with obesity. Nat. Med. 31, 294–305 (2025).
Sharma, A. K., Metzger, D. L., Daymont, C., Hadjiyannakis, S. & Rodd, C. J. LMS tables for waist-circumference and waist-height ratio Z-scores in children aged 5-19 y in NHANES III: association with cardio-metabolic risks. Pediatr. Res. 78, 723–729 (2015).
Plesner, J. L. et al. Obesity is associated with vitamin D deficiency in Danish children and adolescents. J. Pediatr. Endocrinol. Metab. 31, 53–61 (2018).
American Diabetes Association. Classification and diagnosis of diabetes: standards of medical care in diabetes-2021. Diabetes Care 44, 2182 (2021).
Frithioff-Bojsoe, C. et al. Glucose metabolism in children and adolescents: population-based reference values and comparisons to children and adolescents enrolled in obesity treatment. Pediatr. Diabetes 20, 538–548 (2019).
De Jesus, M. J. et al. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics 128, S213–S256 (2011).
Flynn, J. T. et al. Clinical practice guideline for screening and management of high blood pressure in children and adolescents. Pediatrics 140, e20171904 (2017).
Loh, P. R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375, S361–363 (2012).
Price, A. L. et al. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 83, 132–135; author reply 135–139 (2008).
Foley, C. N. et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun. 12, 764 (2021).
Grant, A. J., Gill, D., Kirk, P. D. W. & Burgess, S. Noise-augmented directional clustering of genetic association data identifies distinct mechanisms underlying obesity. PLoS Genet. 18, e1009975 (2022).
Cornelis, M. C. et al. Joint effects of common genetic variants on the risk for type 2 diabetes in US men and women of European ancestry. Ann. Intern. Med. 150, 541–550 (2009).
Agrawal, S. et al. Inherited basis of visceral, abdominal subcutaneous and gluteofemoral fat depots. Nat. Commun. 13, 3771 (2022).
Millard, L. A. C., Davies, N. M., Gaunt, T. R., Davey Smith, G. & Tilling, K. Software Application Profile: PHESANT: a tool for performing automated phenome scans in UK Biobank. Int J. Epidemiol. 47, 29–35 (2018).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Fulco, C. P. et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Weeks, E. M. et al. Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases. Nat. Genet. 55, 1267–1276 (2023).
Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2015).
The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Wen, X., Pique-Regi, R. & Luca, F. Integrating molecular QTL data into genome-wide genetic association analysis: probabilistic assessment of enrichment and colocalization. PLoS Genet. 13, e1006646 (2017).
Gazal, S. et al. Combining SNP-to-gene linking strategies to identify disease genes and assess disease omnigenicity. Nat. Genet. 54, 827–836 (2022).
Laber, S. et al. Discovering cellular programs of intrinsic and extrinsic drivers of metabolic traits using LipocyteProfiler. Cell Genom. 3, 100346 (2023).
Skurk, T. & Hauner, H. Primary culture of human adipocyte precursor cells: expansion and differentiation. Methods Mol. Biol. 806, 215–226 (2012).
Raajendiran, A. et al. Identification of metabolically distinct adipocyte progenitor cells in human adipose tissues. Cell Rep. 27, 1528–1540 e1527 (2019).
Glunk, V. et al. A non-coding variant linked to metabolic obesity with normal weight affects actin remodelling in subcutaneous adipocytes. Nat. Metab. 5, 861–879 (2023).
Acknowledgements
This study was conducted using the UK Biobank data under application no. 1251. The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent research center at the University of Copenhagen, partially funded by an unrestricted donation from the Novo Nordisk Foundation (NNF23SA0084103 and NNF18CC0034900). R.J.F.L. is supported by grants from the National Institutes of Health (NIH) (R01DK107786, R01DK110113, R01HG010297, R01HL142302, R01DK075787, R01HL156991, U01HG011723, R01DK123019, R01HL151152 and R01HL158884), the Danish National Research Fund (DNRF161) and the Novo Nordisk Foundation (NNF20OC0059313). Z.W. is partially supported by the NHLBI under grant number R35HL155466. M.C. is supported by the Novo Nordisk Foundation (NNF21SA0072102), the NIH (UM1DK126185 and R01DK102173) and the Weissman Davis and Titlebaum MGH Research Scholar award. K.E.N. was supported by the NIH (R01HL142302, R01HL156991, R01HL163262, R01DK139598, R01DK122503, R01HL151152 and R01DK122503). A.R. is supported by a grant from the Lundbeck Foundation (R335-2019-2195). V.D.O. is supported by a grant from the Danish Diabetes and Endocrine Academy, funded by the Novo Nordisk Foundation (NNF22SA0079901). J.C.H. has received honoraria for expert roles from Novo Nordisk and Rhythm Pharmaceuticals and provides training and treatment of obesity. The ARIC study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, NIH, Department of Health and Human Services, under contract nos. (75N92022D00001, 75N92022D00002, 75N92022D00003, 75N92022D00004 and 75N92022D00005). The authors thank the staff and participants of the ARIC study for their important contributions. The datasets used for the analyses described in this manuscript were obtained from dbGaP under accession phs000223. The Mount Sinai BioMe Biobank is supported by The Andrea and Charles Bronfman Philanthropies and by Federal funds from the NIH (U01HG00638001, U01HG007417 and X01HL134588). Furthermore, analyses were in part supported through the computational and data resources and staff expertise provided by Scientific Computing and Data at the Icahn School of Medicine at Mount Sinai and by the Clinical and Translational Science Awards grant UL1TR004419 from the National Center for Advancing Translational Sciences. Research reported in this publication was also supported by the Office of Research Infrastructure of the NIH under award number S10OD026880 and S10OD030463. Funders played no role in the design, data collection, data analysis or write up of the study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
N.C., Z.W. and R.J.F.L. conceived and designed the analyses; N.C. and Z.W. analyzed the data, R.J.F.L. planned and supervised the study; N.C., Z.W. and R.J.F.L. wrote the manuscript with contributions from V.S. and R.A.J.S.; D.H., V.S. and R.A.J.S. contributed to data analyses; V.D.O., Y.H., H.D., A.R., C.S. and M.C. contributed to gene prioritization analyses; and R.J.F.L., M.H.P., L.A.H., C.E.F., K.E.N., J.C.H. and T.H. contributed to data acquisition. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.C.H. has received honoraria for expert roles from Novo Nordisk and Rhythm Pharmaceuticals and provides training and treatment of obesity. All the other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Francesco Gianfagna, Maria Keller, Marie Piqeyre and Renata Risi for their contribution to the peer review of this work. Primary Handling Editor: Liam Messin, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Schematic Representation of Bi-Trait Phenotype Derivation.
Pairwise difference between BMI and TC z-scores results in a new normally distributed bi-trait BMI–TC.
Extended Data Fig. 2 Sex-specific associations of genetic risk scores with anthropometric and cardiometabolic traits in the UK Biobank.
A. Estimated per 10 allele change effect sizes of GRS–trait associations in UK Biobank European ancestry population for GRSuncoupling (men in light magenta, women in dark magenta) and GRSBFP (men in light blue, women in dark blue). B-C. Estimated per 10 allele change effect sizes of GRS–trait associations in UK Biobank European ancestry population for each cluster-specific GRS (GRS 1–8, men in light magenta, women in dark magenta). The dashed circles are labeled ‘0’, indicating no association between each GRS and the trait. Points outside the circle represent positive GRS–trait associations, while those inside represent negative associations.
Extended Data Fig. 3 Associations of genetic risk scores with anthropometric and cardiometabolic traits in the ARIC study.
A. Estimated per 10 allele change effect sizes of GRS–trait associations in 9,240 unrelated European ancestry participants of the ARIC study for GRSuncoupling (in magenta) and GRSBFP (in blue). B-C. Estimated per 10 allele change effect sizes of GRS–trait associations in UK Biobank European ancestry population for each cluster-specific GRS (GRS 1–8, in red) and GRSBFP (in gray). Dashed circles indicate Beta=0, indicating no association between each GRS and the trait. Points outside the circle represent positive GRS–trait associations, while those inside represent negative associations. The effect and reference alleles of GRS2, a cluster associated with lower WHR and higher blood pressure, were flipped in order to reflect a profile of higher adiposity and facilitate comparison with the other clusters.
Extended Data Fig. 4 Associations of genetic risk scores with anthropometric and cardiometabolic traits in the HOLBAEK study.
A. Estimated per 10 allele change effect size of GRS-anthropometric trait associations for GRSuncoupling (in magenta) and GRSBFP (in blue). B-C. As A, but for associations with continuous and binary cardiometabolic traits, respectively. Continuous traits were standardized to mean 0 and SD 1. Dashed circles in A and B indicate Beta=0, and the dashed line in C indicates odds ratio=1. Points outside the dashed circles in A and B represent positive GRS–trait associations, while those inside represent negative associations. *analysis restricted to the population-based cohort only.
Extended Data Fig. 5 Pathways enriched for GRSuncoupling and GRSBFP loci.
REACTOME, GO, and KEGG results from DEPICT gene set enrichment analyses were used to group pathways into broader categories. DEPICT assigns gene scores reflecting their likelihood of belonging to specific gene sets, then tests enrichment by summing these scores across associated loci and comparing them to sums from matched random loci. Repeated sampling generates a null distribution used to compute adjusted Z-scores, P values, and FDRs. Pathways from REACTOME, GO, and KEGG enriched with nominal non-adjusted P < 0.01 were grouped into broad pathway categories to enable visualization and plotted for each of GRSuncoupling and GRSBFP. The width of each category is proportional to the number of pathways in that category. Redundant pathways were removed. Full results are presented in Supplementary Table 16.
Extended Data Fig. 6 Enriched pathways per cluster.
Cluster-specific DEPICT gene set enrichments specific to REACTOME, GO, and KEGG pathways were grouped into broad pathway categories. DEPICT assigns gene scores reflecting their likelihood of belonging to specific gene sets, then tests enrichment by summing these scores across associated loci and comparing them to sums from matched random loci. Repeated sampling generates a null distribution used to compute adjusted Z-scores, P values, and FDRs. To facilitate visualization of the mostly represented pathways per cluster, we considered pathways with nominal and non-adjusted P values < 1x10−3. FDR values adjusted for multiple testing and full results are presented in Supplementary Table 16.
Extended Data Fig. 7 Plasma proteins (n=176) with directionally consistent associations with the body fat percentage- and uncoupling genetic risk scores in the UK Biobank.
Estimated per 10 allele change effect sizes of GRS-protein associations in UK Biobank European ancestry population for GRSuncoupling (in magenta) and GRSBFP (in blue), for rank-based inverse-normal transformed Olink-derived plasma protein concentrations.
Extended Data Fig. 8 Plasma proteins (n=129) which associate with the uncoupling- but not the body fat percentage genetic risk score in the UK Biobank.
Estimated per 10 allele change effect sizes of GRS-protein associations in UK Biobank European ancestry population for GRSuncoupling (in magenta) and GRSBFP (in blue), for rank-based inverse-normal transformed Olink-derived plasma protein concentrations.
Supplementary information
Supplementary Table
Supplementary Tables 1–21.
Supplementary Code
An R code that generates the uncoupling genetic risk score (GRSuncoupling) and the subtype-specific scores (GRS1–GRS8).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chami, N., Wang, Z., Svenstrup, V. et al. Genetic subtyping of obesity reveals biological insights into the uncoupling of adiposity from its cardiometabolic comorbidities. Nat Med (2025). https://doi.org/10.1038/s41591-025-03931-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-025-03931-0
