
Abstract
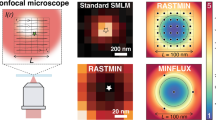
MINFLUX offers a breakthrough in single molecule localization precision, but is limited in field of view. Here we combine centroid estimation and illumination pattern induced photon count variations in a conventional widefield imaging setup to extract position information over a typical micrometer-sized field of view. We show a near two-fold improvement in precision over standard localization with the same photon count on DNA-origami nanostructures and tubulin in cells, using DNA-PAINT and STORM imaging.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
Raw image data and processed conventional SMLM and SIMFLUX localization data is available at https://doi.org/10.4121/uuid:b1078e64-48d5-4f42-a1a8-3386ed14d4c7
Code availability
Software for processing SIMFLUX datasets is available as Supplementary Software. Updates will be made available at https://www.github.com/qnano/simflux
References
Hell, S. W. Microscopy and its focal switch. Nat. Meth. 6, 24–32 (2009).
Huang, B., Babcock, H. & Zhuang, X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell 143, 1047–1058 (2010).
Klein, T., Proppert, S. & Sauer, M. Eight years of single-molecule localization microscopy. Histochem Cell Bio. 141, 561–575 (2014).
Ries, J., Kaplan, C., Platonova, V., Eghlidi, H. & Ewers, H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Meth. 9, 582–587 (2012).
Raulf, A. et al. Click chemistry facilitates direct labeling and super-resolution imaging of nucleic acids and proteins. RSC Adv. 4, 30462–30466 (2014).
Li, H. & Vaughan, J. C. Switchable fluorophores for single-molecule localization microscopy. Chem. Rev. 118, 9412–9454 (2018).
Strauss, S. et al. Modified aptamers enable quantitative sub-10-nm cellular DNA-PAINT imaging. Nat. Meth. 15, 685–688 (2018).
Heydarian, H. et al. Template-free 2D particle fusion in localization microscopy. Nat. Meth. 15, 781–784 (2018).
Grimm, J. B. et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Meth. 12, 244–250 (2015).
Kaufmann, R. et al. Super-resolution microscopy using standard fluorescent proteins in intact cells under cryo-conditions. Nano Lett. 14, 4171–4175 (2014).
Weisenburger, S. et al. Cryogenic optical localization provides 3D protein structure data with Angstrom resolution. Nat. Meth. 14, 141–144 (2017).
Hulleman, C. H., Li, W., Gregor, I., Rieger, B. & Enderlein, J. Photon yield enhancement of red fluorophores at cryogenic temperatures. Chem. Phys. Chem. 19, 1774–1780 (2018).
Rieger, B. & Stallinga, S. The lateral and axial localization uncertainty in super-resolution light microscopy. Chem. Phys. Chem. 15, 664–670 (2014).
Balzarotti, F. et al. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 355, 606–612 (2016).
Heintzmann, R. & Huser, T. Super-resolution structured illumination microscopy. Chem. Rev. 117, 13890–13908 (2017).
Busoni, L., Dornier, A., Viovy, J.-L., Prost, J. & Cappello, G. Fast subnanometer particle localization by traveling-wave tracking. J. Appl. Phys. 98, 064302 (2005).
Wicker, K. Non-iterative determination of pattern phase in structured illumination microscopy using autocorrelations in Fourier space. Opt. Exp. 21, 24692–24701 (2013).
Schnitzbauer, J., Strauss, M. T., Schlichthaerle, T., Schueder, F. & Jungmann, R. Super-resolution microscopy with DNA-PAINT. Nat. Prot. 12, 1198–1228 (2017).
Nieuwenhuizen, R. P. J. et al. Measuring image resolution in optical nanoscopy. Nat. Meth. 10, 557–562 (2013).
Li, Y. et al. Real-time 3D single molecule localization using experimental point spread functions. Nat. Meth. 15, 367–369 (2018).
Gu, L. et al. Molecular resolution imaging by repetitive optical selective exposure. Nat. Meth., https://doi.org/10.1038/s41592-019-0544-2 (2019).
Chmyrov, A. et al. Nanoscopy with more than 100,000 ‘doughnuts’. Nat. Meth. 10, 737–740 (2013).
Stallinga, S. & Rieger, B. Accuracy of the Gaussian point spread function model in 2D localization microscopy. Opt. Exp. 18, 24461–24476 (2010).
Thorsen, R. Ø. et al. Impact of optical aberrations on axial position determination by photometry. Nat. Meth. 15, 989–993 (2018).
Mulliken, J. C. et al. Methods for CCD camera characterization. In Proc. SPIE 2173, Image Acquisition and Scientific Imaging Systems (eds Titus, H. C. and Waks, A.) 73–84 (SPIE, 1994).
Heintzmann, R., Relich, P. K., Nieuwenhuizen, R. P. J., Lidke, K. A. & Rieger, B. Calibrating photon counts from a single image. Preprint at https://arxiv.org/abs/1611.05654 (2019).
Huang, F., Schwartz, S. L., Byars, J. M. & Lidke, K. A. Simultaneous multiple-emitter fitting for single molecule super-resolution imaging. Biomed. Opt. Exp. 2, 1377–1393 (2011).
Smith, C. S. et al. Nuclear accessibility of β-actin mRNA is measured by 3D single-molecule real-time tracking. J. Cell. Biol. 209, 609–619 (2015).
Smith, C. S., Joseph, N., Rieger, B. & Lidke, K. A. Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat. Meth. 7, 373–375 (2010).
Huang, F. et al. Video-rate nanoscopy using sCMOS-camera specific single-molecule localization algorithms. Nat. Meth. 10, 653–658 (2013).
Acknowledgements
J.C. and C.S.S. were supported by the Netherlands Organisation for Scientific Research (NWO), under NWO START-UP project no. 740.018.015 and NWO Veni project no. 16761. T.H., R.Ø.T. and B.R. acknowledge National Institutes of Health (grant no. U01EB021238). F.S. and R.J. acknowledge support by an ERC Starting Grant (MolMap, grant agreement no. 680241). C.S.S. acknowledges a research fellowship through Merton College, Oxford, UK. B.R. acknowledges an ERC Consolidator Grant (Nano@cryo, grant agreement no. 648580). We thank F. Balzarotti for advice on theoretical analysis of localization precision, and D. Jurriens and L. Kapitein for help with imaging cellular samples.
Author information
Authors and Affiliations
Contributions
Imaging experiments were done by T.H., M.S. and F.S. Data analyses were done by J.C. and T.H. Simulations were done by R.Ø.T. and J.C. M.S., F.S. and R.J. provided samples. C.S.S., B.R. and S.S. devised key concepts and supervised the study. T.H., C.S.S., B.R. and S.S. wrote the paper. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Rita Strack was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–21, Supplementary Table and Supplementary Notes 1–7.
Supplementary Software
simflux-publication-release.zip contains the code developed for processing SIMFLUX datasets (source code and binaries and example dataset).
Rights and permissions
About this article

Cite this article
Cnossen, J., Hinsdale, T., Thorsen, R.Ø. et al. Localization microscopy at doubled precision with patterned illumination. Nat Methods 17, 59–63 (2020). https://doi.org/10.1038/s41592-019-0657-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-019-0657-7
This article is cited by
-
Molecular-scale isotropic 3D super-resolution microscopy via interference localization
Nature Methods (2026)
-
4Pi-SIMFLUX: 4Pi single-molecule localization microscopy with structured illumination
Nature Methods (2026)
-
Array detection enables large localization range for simple and robust MINFLUX
Light: Science & Applications (2025)
-
Super-resolution microscopy for structural biology
Nature Methods (2025)
-
MINFLUX achieves molecular resolution with minimal photons
Nature Photonics (2025)
