
Abstract
Dysregulation of communication between cells mediates complex diseases such as cancer and diabetes; however, detecting cell–cell communication at scale remains one of the greatest challenges in transcriptomics. Most current single-cell RNA sequencing and spatial transcriptomics computational approaches exhibit high false-positive rates, do not detect signals between individual cells and only identify single ligand–receptor communication. To overcome these challenges, we developed Cell Neural Networks on Spatial Transcriptomics (CellNEST) to decipher patterns of communication. Our model introduces a new type of relay-network communication detection that identifies putative ligand–receptor–ligand–receptor communication. CellNEST detects T cell homing signals in human lymph nodes, identifies aggressive cancer communication in lung adenocarcinoma and colorectal cancer, and predicts new patterns of communication that may act as relay networks in pancreatic cancer. Along with CellNEST, we provide a web-based, interactive visualization method to explore in situ communication. CellNEST is available at https://github.com/schwartzlab-methods/CellNEST.
Similar content being viewed by others
Main
Cell–cell communication (CCC) enables the complex coordination of cells, forming tissues and organs in multicellular organisms and accomplishing critical biological functions; however, aberrant communication among cells or atypical decoding of molecular messages can lead to and promote diseases such as cancer. CCC is involved in several hallmarks of cancer, such as tumor-promoting inflammation, inducing or accessing vasculature and activating invasion and metastasis1,2. It is crucial to pinpoint communication responsible for normal and aberrant cell and tissue function to inform the next generation of therapeutics.
CCC is mediated by ligand–receptor pairs, where a ‘sender’ cell produces ligand proteins that bind to matching receptor molecules on a ‘receiver’ cell2. Common techniques to identify CCC use single-cell RNA sequencing (scRNA-seq) data by matching highly expressed ligand genes from a sender cell type with highly expressed receptor genes from a receiver cell type, prioritizing ligand–receptor pairs with high ‘ligand–receptor coexpression scores’. These scores represent the overall expression of the ligand–receptor pair. After identifying ligand–receptor pairs, these methods diverge by determining confidence in each pair using statistical tests3,4,5, substituting receptor genes with pathways6 or using graph-based approaches7. Others, like CellChat8, use network analysis and pattern recognition approaches. NicheNet7 uses signaling pathway networks and the PageRank algorithm. Despite advances proposed by these methods, detecting CCC remains a major challenge. One major limitation of existing approaches derives from the limited scope of the CCC definition. Rather than being limited to a single ligand–receptor pair, communication may act as a relay network mediated by multiple pairs of cells. A relay network is formed when a ligand from one cell binds to a cognate receptor on another cell and induces the secretion of another ligand that binds to a third cell’s receptor. This signal passing can extend across multiple cells. The frequency of these patterns may indicate higher confidence in CCC detection9,10,11.
Even with single ligand–receptor pair CCC detection, past efforts demonstrated high false-positive and negative rates2, which is in part due to using a single data modality (the transcriptome) from cells. Only 6% of genes exhibit significant expression changes in response to ligands, which may contribute to low accuracy without additional context such as neighboring cells12. This spatial context is lost in scRNA-seq as the method requires tissue dissociation. As CCC is spatially dependent, with juxtacrine and paracrine requiring cells to be in close proximity, scRNA-seq introduces challenges for true single-cell CCC detection instead of cell-type communication13.
Recently, methods such as Scriabin14 and GraphComm15 have been introduced to detect CCC from scRNA-seq data alone and map final results to spatial regions within tissue using corresponding spatial transcriptomic data; however, these approaches incorporate spatial position not to detect CCC, but to validate CCC that has already been identified from dissociated samples. These methods also do not report distant ligand–receptor interactions such as paracrine interactions, which constitute the majority of most ligand–receptor databases. To overcome these limitations, new CCC models that directly integrate the spatial context of gene expression are necessary.
Spatial transcriptomic technologies, such as Visium16 and multiplexed error-robust fluorescence in situ hybridization (MERFISH)17, measure the physical location of cells paired with their transcripts, providing new opportunities to detect CCC. Visium measures transcriptomes of barcoded spots, each 55 μm in diameter and containing approximately 1–10 cells, while the recent launch of Visium HD (high definition) achieves single-cell spatial resolution at 2 μm. Alternatively, MERFISH achieves single-cell resolution, albeit with a smaller subset of genes. Critically, although this data modality promises to better inform CCC detection, there is an urgent need for new analytical approaches beyond single ligand–receptor pair inference.
Although methods have been developed to detect CCC directly from spatial transcriptomic data, most existing methods are unable to detect CCC relay networks at single-cell resolution in situ (Extended Data Table 1). NICHES18 uses k-nearest neighbors to identify proximal cells and calculates their ligand–receptor coexpression scores. NICHES then collapses cells to neighborhoods using principal component analysis to discover niches of communication. COMMOT19 screens CCC in spatial transcriptomics via collective optimal transport. However, COMMOT requires a network pathway list as additional input, which increases its reliance on a priori information. Most of these methods use differentially expressed and variable ligand and receptor genes, only incorporating spatial information to limit potential communication to a neighborhood of cells. Recent methods, including NicheCompass20, Clarify21 and TENET22, model binary CCC between cells or spots and do not differentiate between types of ligand–receptor pairs. Therefore, these methods are unable to identify specific CCC signals and their associated strength across spatial regions of the tissue. HoloNet23 represents a separate class of methods that are constrained for a given target gene and unable to generate an unbiased, global list of active CCC for a given tissue sample. CytoSignal24 multiplies ligand and receptor concentration between pairs of cells to calculate communication scores and uses a permutation test with cell rearrangements; however, CytoSignal combines all ligand–receptor pairs between a pair of cells into a single score, which prevents the method from ranking different ligand–receptor pairs according to their occurrence probabilities. SpaCCC25, Giotto26, TWCOM27 and CellChat’s spatial method28 focus on CCC at the level of cell types or clusters instead of single cells or spots, missing complex communication network components. Moreover, none of these existing methods attempt to identify CCC relay networks, which limits the discovery of large patterns of communication. To address the need for an accurate, high-resolution method capable of predicting complex CCC relay networks, we require a sophisticated pattern-finding algorithm bolstered by deep learning.
To facilitate CCC detection, we can represent communication from spatial transcriptomic data as a knowledge graph, where cells or spots are vertices and edges represent different types of neighborhood relations. As our goal is to predict which relations are probable communication, a deep-learning option to unravel the communication network is a graph neural network (GNN)29. A GNN serves as an effective model for encoding topological structures in graph representations by generating a graph embedding. Variants of GNNs are already being applied to transcriptomic data, including a graph convolutional network for clustering30 and a GNN-based encoder for deconvolution and integration31. A newer addition to the transformer32 family is the graph attention network (GAT), a powerful tool that has already revolutionized other knowledge-graph-based problems, including social networks and molecular structures. As this model requires ground-truth data for supervised model training, we propose using a contrastive learning approach, Deep Graph Infomax (DGI)33, which excels in unsupervised learning problems.
Built with these state-of-the-art advances in artificial intelligence, we present CellNEST, a method that measures cell–cell communication and patterns between individual cells or spots by leveraging a GAT encoder model with DGI contrastive learning. We applied our model to five biological contexts across multiple tissues, species and technologies to map spatially resolved CCC34,35,36. Using new benchmarks for single-cell ligand–receptor pair detection and CCC relay networks, we found that CellNEST outperforms existing methods on both biological samples and synthetic data. We show that CellNEST can not only accurately reconstruct traditional single ligand–receptor signals between cells using both MERFISH and new Visium HD technologies, but also reports potential relay networks of communication based on repeated patterns observed throughout both two-dimensional (2D) and three-dimensional (3D) spatial transcriptomic samples. Of note, applying CellNEST to our cohort of patients with pancreatic ductal adenocarcinoma (PDAC) revealed critical CCC associated with PDAC progression and spatially associated with known PDAC subtypes linked with treatment response and overall survival. As demonstrated, CellNEST is not limited to a single technology or species. Rather, it is a transferable model applicable to data across domains. We believe that CellNEST is a major step forward in accelerating the application of deep learning to spatial transcriptomics and other related knowledge-graph-based contexts. CellNEST is open source and publicly available at https://github.com/schwartzlab-methods/CellNEST with a Singularity image at https://cloud.sylabs.io/library/fatema/collection/cellnest_image.sif.
Results
CellNEST infers communication in spatial transcriptomic data
Ligand–receptor pair-based communication depends on spatial distance; however, the majority of existing tools do not leverage positional information to detect CCC and collapse communicating units to cell types and clusters rather than spots and cells. To overcome these limitations, we developed CellNEST for high-resolution, spatially resolved CCC detection (Fig. 1a).

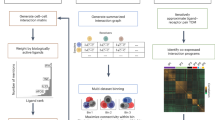
a, A high-level flowchart of the main steps of the CellNEST method. b, Input tissue sample at either spot (for example, Visium; top) or cell resolution (for example, MERFISH, Visium HD; bottom). UMI, unique molecular identifier. c, Input ligand–receptor database containing known ligand and cognate receptor pairings. d, Preprocessing step, where genes with expression above a threshold percentile are considered active (left). Pairwise Euclidean distances between vertices are stored in a physical distance matrix (right). e, Input graph G = (V, E) generation step with V spots or cells as vertices and E edges as neighborhood relations, some of which represent communication (bottom). An input threshold distance is used for the neighborhood formation (blue arrow). From the graph, vertex features are represented as a one-hot vector matrix (top left). The edge feature matrix holds edge feature vectors containing three attributes: pairwise distance, ligand–receptor coexpression score and the ligand–receptor pair identity from the database in c. f, Communication prediction step using a GAT encoder through unsupervised contrastive learning with DGI. g, Output graph step visualizing edges with the highest attention scores. Attention scores range from 0 (white) to 1 (black), where 1 represents the strongest connections. Lower-scoring edges are removed (dashed lines), resulting in subgraphs of communicating vertices. h, Example output showing the flow of communication between tumor-annotated spots (filled squares) with stroma spots (open circles), colored by connected component. i, An example CellNEST-generated histogram showing the frequency of communication through ligand–receptor pairs in the top 20% highest-scoring attention edges. Colors in the histogram correspond to connected components in h. For instance, the most abundant communication, labeled as FN1–RPSA, is found primarily in the blue region. Altogether, CellNEST offers a high-resolution approach for detecting the strength and location of cell–cell communication in tissues.
Given a 2D or 3D spatial transcriptomic dataset at either spot or single-cell resolution and an existing ligand–receptor database, CellNEST scores each intercellular signal based on the coexpression of highly expressed ligand and cognate receptor genes (Fig. 1b–d). CellNEST may optionally incorporate signaling pathways downstream of the receiver’s receptor with ligand–receptor coexpression. To achieve single-cell- and single-spot-level communication identification, CellNEST relies on a GNN, a class of deep-learning-based models, to identify which ligand–receptor pairs are highly probable to exist based on reoccurring patterns of communication in a particular tissue region. For example, transforming growth factor (TGF)β1 signaling is upregulated in tumor cells across various cancers35. This signal occurs multiple times in cancer tissue along the boundary of tumor and nontumor cells, forming a distinct pattern that is not observed in other regions of the same tissue. Deep-learning models excel in detecting such hidden patterns; CellNEST leverages this strength by using a GAT37, an encoder model that records such patterns in the form of a vertex embedding. While some communication may involve a single ligand–receptor pair, more intricate patterns can exist, where CCC acts as a relay network with multiple ‘hops’ between cells9,10,11. CellNEST extends its pattern-finding capabilities to predict frequent arrangements of coexpressed signaling, which may represent relay networks, and supports these predictions with evidence from protein–protein and transcription factor–target gene interactions38,39,40.
After data preprocessing, CellNEST converts spatial transcriptomic data into a graph G = (V, E) with V cells or spots as vertices and E edges as some neighborhood relation between the pair of vertices (Fig. 1e). CellNEST inserts edges between a pair of cells or spots (herein referred to as vertices) i and j if they are proximal neighbors (by default within four spots in spot-based and 300 μm in cell-based experiments), with elevated ligand gene expression in i and elevated receptor gene expression in j (Supplementary Note 1).
G can be massive, containing thousands of vertices with millions of edges based on the number of expressed genes. Notably, E represents neighborhood relations and not CCC, as proximal cells do not always establish communication. Tissue context12, epigenetic factors41 and other signaling pathways7 may influence high ligand–receptor coexpression. CellNEST sifts through these putative relations to predict which edges are more likely to represent communication. For this purpose, we pass G to the core deep-learning module in CellNEST, the ‘communication prediction step’, where a GAT model generates the vertex embedding (Fig. 1f).
The traditional GAT model requires ground-truth data for training an encoder, but this information is unknown from spatial transcriptomic data. We instead chose to implement unsupervised training through DGI33, a contrastive learning approach (Fig. 1f). DGI compares encoder weights derived from the observed network with encoder weights from a ‘corrupted’ network of randomly shuffled and permuted vertices and edges. DGI maximizes weights from the observed network while penalizing weights from the corrupted network. As the model converges, CellNEST assigns higher attention scores to stronger neighborhood relations (Fig. 1g). We use these attention scores to represent communication strength. To retain the most probable intercellular signals, we filter edges, retaining the top 20% of highest-scoring attention edges by default (Supplementary Note 2).
After predicting high-resolution CCC, CellNEST identifies highly communicating regions of tissue in the ‘output graph step’ by determining connected components (Fig. 1g,h). As CellNEST identifies CCC between each vertex along with associated signal strength, we provide a unique visualization that displays vertices colored by densely communicating regions of tissue, along with ligand–receptor pairs as an arrows whose thicknesses are determined by their attention scores (Fig. 1h). To complement the tissue visualization, CellNEST also generates histograms that display the counts of all ligand–receptor pairs in the top edges ranked by attention score and colored by the community they are found in within the tissue (Fig. 1i). With this extensive tool set, CellNEST is fully equipped as an end-to-end framework for spatially resolved CCC detection.
CellNEST pinpoints T cell homing signals in the lymph node
To determine the accuracy of our algorithm, we applied CellNEST to Visium data from a human lymph node34 (Fig. 2a–e). We hypothesized that CellNEST would identify the T cell homing signal of chemokine (C–C motif) ligand 19 with cognate CC-chemokine receptor 7 (CCL19–CCR7) and place this CCC within the T cell zone42. The T cell zone was previously annotated using cell2location34 (Fig. 2a). We applied CellNEST to the entire tissue and ranked all CCC based on their attention scores, keeping the ligand–receptor pairs with the top 20% highest attention scores and located within the T cell zone (Fig. 2b). Among the 12,605 possible ligand–receptor pairs in the database (Supplementary Note 3), CellNEST identified CCL19–CCR7 as the second most abundant pair in the T cell zone, with strict thresholds above 20%. The topmost detected pair was CCL21–CXCR4, another T cell migratory signal43 (Fig. 2b). Of note, while CellNEST found CCL19–CCR7 as a top signal in the T cell zone based on attention score (Fisher’s exact test, P = 9.16 × 10−224), this pair’s coexpression score was not among the highest (Fig. 2c,d and Supplementary Table 1). Increasing attention score thresholds further confirmed T cell zones as the primary location for CCL19–CCR7 (Fig. 2e). Notably, these T cell zones were enriched with top genes encoding proteins downstream of CCR7 signaling, and incorporation of these genes into the model recapitulated these CCCs, further suggesting activation by CCR7 identified by CellNEST (Mann–Whitney U-test, P = 6.42 × 10−164; Fig. 2f,g and Supplementary Fig. 1). This prioritization and localization suggest that CellNEST does not score edges based solely on input ligand and receptor expression. Instead, CellNEST focuses on hidden communication patterns to predict which edges are essential to represent the context of the tissue sample.

a, Human lymph node tissue assayed with Visium and annotated with cell2location34 (n = 4,035 spots). b, Histogram of ligand–receptor pairs (x axis) with the top 20% highest attention scores in T cell zones assigned by CellNEST in descending order of abundance (y axis). CCL19–CCR7 (red text with triangle) is a canonical T cell homing signal. c, Density plot of CCL19–CCR7 attention scores (red) compared to all other ligand–receptor pairs (gray) in T cell zones. d, Density plot of CCL19–CCR7 ligand–receptor coexpression scores (red) compared to all other ligand–receptor pairs (gray) in T cell zones. e, Selection of the top 5,000, 2,500 and 500 CCL19–CCR7 edges with the strongest attention scores (left to right) across the entire tissue. Stronger CCL19–CCR7 communication is found in T cell zones. f, Mean expression of the top 20% expressed genes encoding proteins downstream of CCR7 signaling mapped onto the human lymph node, which aligns with CellNEST-detected regions in T cell zones in e. g, Box and whisker plots comparing mean gene expression from f within (n = 417 spots) or outside (n = 3,618 spots) of T cell zones. Center line, median; box, interquartile range; whiskers, 1.5 × interquartile range; points, outliers. There is elevated expression of CCR7 downstream signaling genes in T cell zones (two-sided Mann–Whitney U-test, P = 6.42 × 10−164). h, Application of COMMOT to the human lymph node, with red arrows indicating CCL19–CCR7 strength. Regions do not align well with T cell zones. i, Application of NICHES to the human lymph node. Using a cluster-based analysis (left), NICHES identified three signals but missed the CCL19–CCR7 signal (right).
To compare the performance of CellNEST against other emerging methods for CCC detection, we applied NICHES, COMMOT, NicheCompass, CytoSignal, CellChat, Giotto and TWCOM to identify CCL19–CCR7 within the T cell zone (Fig. 2h,i, Supplementary Fig. 2a–i and Supplementary Note 4). CellNEST outperformed all methods in localizing CCL19–CCR7 to the T cell zone. In addition to demonstrating robust performance on biological data, CellNEST also shows comparable computational efficiency to existing methods (Supplementary Fig. 2j,k and Supplementary Note 5).
CellNEST’s unique capability extends single ligand–receptor pairs to patterns of communication, which may indicate a relay network or other complex patterns. Although CellNEST detects any type of pattern, for simplicity, we quantified the frequency of two-hop CCC between cells. Two-hop CCC involves a ligand–receptor pair s from an i sender to a j receiver and a ligand–receptor pair t from a j sender to a k receiver (Fig. 3a). Extending outward from the single ligand–receptor pair containing CCL19 identified earlier, we sought to determine potential relay networks associated with this homing signal. CellNEST reported a high abundance of CCL19–CCR7 to CCL21–CXCR4 in the T cell zones (Fig. 3b,c). CCL19–CCR7 and CCL21–CXCR4 are regulated together in various T cell induced activities44, which is concordant with CellNEST finding mostly T cells participating in this predicted relay network and in other top-ranked networks (Fig. 3b,d, Supplementary Fig. 3a and Supplementary Note 6). To further validate the occurrence of these relay networks, we equipped CellNEST with the capability to report confidence scores using experimentally validated protein–protein and transcription factor–target gene interactions from independent databases (Fig. 3e and Supplementary Notes 7 and 8). CellNEST identified these potential relay networks with confidence scores significantly higher than random, suggesting the effectiveness of CellNEST in detecting such patterns (Dunn’s test with Benjamini–Hochberg correction, P = 4.5 × 10−02; Supplementary Fig. 3b).

a, Diagram showing the conceptual difference between a single communication between two vertices (here cells) versus a relay network involving a group of vertices. b, Histogram of the most abundant two-hop relay networks (ligand–receptor–ligand–receptor) in the T cell zone identified by CellNEST, including CCL19–CCR7 (red triangle). c, CCL19–CCR7 to CCL21–CXCR4 within T cell zones in Fig. 2a (red). d, Pie charts showing the proportion of each cell type involved in the CCL19–CCR7 to CCL21–CXCR4 relay network in the T cell zone, from sender (left) to receiver and sender (middle) to second receiver (right). DC, dendritic cell; Endo, endothelial; FDC, follicular dendritic cell; ILC, innate lymphoid cell; NK, natural killer; NKT, natural killer T; TIM3, T-cell immunoglobulin and mucin domain-containing protein 3; TfR, T follicular regulatory; Treg, regulatory T; VSMC, vascular smooth muscle cell. e, Diagram showing relay network confidence scoring by integrating experimental scores of protein–protein interactions from STRING38 and NicheNet39, as well as transcription factor–target interactions from DoRothEA40. f–h, Example synthetic distributions for equidistant (for example, Visium; f), uniformly (for example, MERFISH, Visium HD; g), and mixture of Gaussian and uniformly (for example, MERFISH, Visium HD; h) distributed cells. i,j, Heat maps displaying balanced accuracy of CCC methods on synthetic (i) and diffusion-based models (j) measured at single-cell resolution.
To further evaluate CellNEST’s detection capabilities on data with an established ground truth, we conducted extensive benchmarking across 21 synthetic data setups, which represent different spatial transcriptomic technologies and cell distributions (Fig. 3f–j and Supplementary Fig. 3c,d). CellNEST outperformed all methods across spatial distributions, levels of injected noise, as well as in both nonrelay- and relay-based benchmarks (Supplementary Note 9). Based on this comparative analysis, CellNEST is uniquely equipped to more accurately localize CCC to tissue regions and may complement existing methods for communication detection.
CellNEST maps single-cell communication in the mouse brain
Our synthetic benchmarks suggest CellNEST is uniquely capable of detecting CCC in various spatial transcriptomic technologies. To evaluate CellNEST’s performance on single-cell resolution spatial transcriptomic technologies, we applied CellNEST to MERFISH slides from the hypothalamus preoptic region of female parent and female virgin mice36 (Fig. 4a–d). CellNEST revealed that female parent and virgin tissues varied in spatial distributions of strong communication (Fig. 4a,c). CellNEST identified galanin receptor-associated communication involving Galr1 and Galr2 in parent and virgin mice (Fig. 4b,d), which is consistent with previous studies noting galanin’s association with behavior in the preoptic region36. As well, in both parent and virgin mice, CellNEST identified brain-derived neurotrophic factor (Bdnf)-associated communication (Fig. 4b,d), whose gene expression is linked with temperature sensitivity45. Moreover, CellNEST identified signals unique to the female parent mouse, including signals mediated by oxytocin (Oxt) and its receptor (Oxtr), which form core parenting signals46 (Fig. 4b and Supplementary Fig. 4a–e).

a,b, CellNEST-detected communication (green) in tissue from the female parent mouse (n = 5,533 cells; a) with a corresponding histogram of the top 20% strongest ligand–receptor pairs (b). c,d, As in a and b for female virgin mouse tissue (n = 5,606 cells). CellNEST identified signals involving galanin receptor (Galr1 and Galr2) and brain-derived neurotrophic factor (Bdnf) in both parent and virgin mice (black triangle, bold). In contrast, the parenting signal Oxt–Oxtr is exclusively found in the female parent mouse (red triangle in b). e, Cell-type-specific communication zoomed in from the red rectangle in a. f, The corresponding CellNEST-generated histograms from e showing CCC between microglia and excitatory neurons. As in b, Oxt–Oxtr is detected (black triangle, bold) as one of the strongest communications. g,h, One of the most frequent relay-network patterns, Pnoc–Oprd1 to Pnoc–Lpar1 (red triangle), compared to the abundance of other top patterns in a histogram (g) and overlaid on the tissue (red; h). i, 3D MERFISH sample from a female naive mouse (n = 38,372 cells) with CellNEST-detected communication (top), with a zoom-in of two layers for clearer visualization (bottom). j, Histogram of top communication found in i.
Of note, we found that CellNEST could detect communication between two individual cells: a neuron and a microglial cell (Fig. 4e,f). Using the single-cell MERFISH female parent mouse sample with previously annotated cell types, we filtered ligand–receptor pairs identified by CellNEST such that the sender and receiver cells were classified as neurons or microglia only. Upon inspection, we observed a notably high-resolution image with a predicted Oxt–Oxtr interaction between an excitatory neuron and a receiving microglia (Fig. 4e). This ligand–receptor pair establishes communication that contributes to emotional bonding within the female parent mouse47. This communication was well represented across all neuron-microglia communication (Fig. 4f). Together, this analysis suggests that CellNEST can detect precise cell signaling at single-cell resolution rather than solely between pseudobulk cell types.
CellNEST identified potential relay networks that were dominated by prepronociceptin (Pnoc) and delta-type opioid receptor (Oprd1) signals, including Pnoc–Oprd1 to Pnoc–Lpar1 and Pnoc–Oprd1 to Bdnf–Esr1 (Fig. 4g). Of note, CellNEST detected these relay signals in different locations on the tissue than previously detected CCC (Fig. 4h). Pnoc, Oprd1 and Bdnf are linked to behavioral disorders as well as a number of psychiatric affective disorders, such as anxiety, seizure and schizophrenia, so we expect joint activation of these signals48.
Spatial transcriptomic technologies such as MERFISH also may take consecutive slices to infer 3D cell organization (Fig. 4i). We sought to extend our model to 3D data points by combining cells across six such consecutive slides along the bregma axis. As CellNEST uses a graph structure that is not limited to 2D, we extended our edges to incorporate 3D input where the physical distance matrix records pairwise distances of 3D coordinates. When applying CellNEST to a 3D female naive mouse sample, CellNEST detected general communication in the mouse brain with fewer parental signals, likely because this mouse was not exposed to pups36 (Fig. 4i,j). A comparative analysis between 2D (within sections) and 3D (across sections) revealed mostly overlapping CCC, but there did exist between-section CCC interactions which were undetectable in 2D analysis alone, such as Adcyap1–Mc4r, whose proteins are associated with energy homeostasis and anxiety, as well as Oxt–Avpr1a, whose gene expressions have been linked to sex-specific social and emotional behaviors45,49 (Supplementary Fig. 4f,g). CellNEST’s identification of CCC unique to 2D and 3D MERFISH samples revealed the method’s flexibility across dimensions as well as spot- and single-cell-resolution technologies.
CellNEST detects aggressive CCC in lung adenocarcinoma
The tumor microenvironment is a complex and heterogeneous collection of different cell types and signals, where CCC contributes to disease progression. To identify specific regions of tumor tissue associated with cancer-promoting communication, we applied CellNEST to a Visium sample of lung adenocarcinoma (LUAD)35 (Extended Data Fig. 1a). Within the most probable ligand–receptor pairs, CellNEST detected transforming growth factor β-associated communication involving TGFB1 and TGFB2, important in metastasis50, as concentrated near the top out of over 12,605 pairs in the database based on attention scores (Extended Data Fig. 1b); however, CellNEST found apolipoprotein E (APOE)-based communication including APOE–SDC1 as the most strongly occurring CCC. APOE promotes LUAD proliferation and migration and is associated with poor prognosis in patients with lung cancer51. To support the presence of APOE–SDC1 within the tumor region, we observed alignment between the expression of each gene on the tissue with the location of the CCC (Extended Data Fig. 1c–e). Overall, CellNEST observed enriched LUAD-related pathways in the tumor component, including E2F transcription factor upregulation (normalized enrichment score (NES) of 5.73) and G2M checkpoint activation (NES of 5.98)52 (two-sided permutation test, all q < 2.20 × 10−16; Supplementary Fig. 5a–c).
In addition to tumor-localized APOE–SDC1, CellNEST identified other strong communication in different locations of the tissue. Specifically, CellNEST identified enrichment within the lymph node region for gene programs linked to lymph node metastasis, such as T cell modulation (NES of 6.00) and interleukin-10 signaling (NES of 5.93)53, and specifically assigned FN1–RPSA to this region (two-sided permutation test: all q < 2.20 × 10−16; Supplementary Fig. 5d–i). As FN1 and APOE are associated with lymph node metastasis in patients with LUAD, CellNEST may have identified potential disease progression54,55. Separate from the lymph node region, CellNEST identified TGFβ signaling and pathways associated with LUAD stromal regions within the surrounding tumor microenvironment, including epithelial-to-mesenchymal transition (NES of 5.29) and chaperone-mediated autophagy56 (NES of 4.71) (two-sided permutation test: all q < 2.20 × 10−16; Supplementary Figs. 5j–l and 6). Based on these observations, CellNEST is able to deconvolve complex tumor microenvironments, providing insights into how signals may be organized in tissue regions.
Beyond single ligand–receptor interactions, CellNEST predicted PSAP–LRP1 to APOE–LRP1 (Extended Data Fig. 1f,g) along with additional previously unobserved patterns. The tumor-secreted protein prosaposin (PSAP) and APOE signaling pathways share patterns57 and exhibit high gene coexpression in inflammation58, suggesting reliable relay-network detection. Of note, although PSAP is a marker of many cancer types including pancreatic cancer, prostate cancer and lymph node metastasis59,60, the link between LUAD and PSAP is not yet sufficiently explored. As such, CellNEST’s predicted relay networks enable new approaches to reveal complex CCC patterns.
CellNEST recovers signals in invasive colorectal cancer
Emerging spatial transcriptomic assays, such as Visium HD, enable whole-transcriptome sequencing at subcellular resolution. To demonstrate the flexibility and capability of CellNEST on these technologies, we applied CellNEST to a human colorectal cancer sample at 2 μm bin size (Extended Data Fig. 2a,b and Supplementary Note 10). Focusing on a region of interest containing a mixture of invasive cancer and surrounding noncancer cells, we applied CellNEST to an input graph of 6,857,387 ligand–receptor-pair connections.
CellNEST clearly identified the invasive cancer region as a separate network of localized signals (Extended Data Fig. 2b). The topmost abundant ligand–receptor pairs included amyloid precursor protein as a ligand, which promotes growth and proliferation of colon cancer both in vitro and in vivo61 (Extended Data Fig. 2c). The corresponding receptor integrin α6 gene (ITGA6) expression is a useful biomarker for colorectal cancer early detection, and transforming growth factor β receptor type II gene (TGFBR2) alterations promote the formation of colon cancer62,63. Although CellNEST detected these signals at adenoma locations bordering the tissue, there was increased abundance in the invasive cancer region (chi-squared test of dependency, P < 2.2 × 10−16, hypergeometric test of over-representation, P = 1.08 × 10−53; Extended Data Fig. 2b,c and Supplementary Note 11).
CellNEST also predicted several two-hop relay networks of CCC on the tissue surface (Extended Data Fig. 2d,e). In addition to signals between cancer cells, we observed relay networks specific to the tumor microenvironment that promote cancer progression, such as C3–CXCR4 to C3–LRP1 (Extended Data Fig. 2d). CellNEST pinpointed this CCC pattern specifically in the stromal region surrounding the invasive tumor, in contrast to nonrelay CCC, which appeared throughout the tissue (Extended Data Fig. 2e). Complement C3 gene (C3) expression is associated with the colorectal adenocarcinoma microenvironment and prognosis64. CXCR4 binds with stromal cell-derived ligands, and high CXCR4 expression is associated with an increased risk of death and progression in colorectal cancer65. LRP1 encodes a signature protein of radio-resistant colorectal cancer66. Together, our results suggest that CellNEST is a robust method that is applicable to the latest spatial transcriptomic technologies without any modification to the model architecture.
CellNEST finds consistent communication across patients with pancreatic cancer
To evaluate CellNEST’s ability to generalize to other cancer types with heterogeneous regions, we applied CellNEST to pancreatic ductal adenocarcinoma (PDAC) tissues. PDAC is widely recognized as a highly aggressive disease, yet treatment responses can vary widely among patients. There is immense transcriptional diversity defining classical and basal-like subtypes of PDAC that is crucial in explaining treatment heterogeneity. Basal-like tumors exhibit characteristics reminiscent of basal or squamous epithelium, leading to heightened chemoresistance and poorer patient prognosis. Conversely, classical tumors demonstrate transcription factor expression associated with pancreas development, rendering them more responsive to chemotherapy and yielding improved clinical outcomes36.
The PDAC tumor microenvironment is a heterogeneous and dense collection of tumor, stromal and immune cells. Stromal areas with high (activated) or low (deserted) immune activity contribute to divergent regions within tumor tissue. To date, the relationship between divergent regions, transcriptomic subtypes and cell states of PDAC is unclear. To resolve specific cell–cell interactions in this complex disease, we applied CellNEST, which considers tumor and stromal proximity at a high resolution and does not rely solely on highly expressed genes. We evaluated whether CellNEST could detect CCC associated with spatially distinct PDAC transcriptomic subtypes.
We applied CellNEST to Visium data collected from two cases that showed morphological heterogeneity across tissue regions (Fig. 5). Transcriptomic subtypes are known to correlate with tumor morphology. Classical tumors are well differentiated and have a gland-forming morphology, whereas basal-like tumors are moderately to poorly differentiated with non-gland-forming morphology67. Both cases were resectable, stage IIb PDAC tumor samples (PDAC_64630 and PDAC_140694). Sample PDAC_64630 presented several regions of morphologically and transcriptionally distinct tumor subtypes separated by stroma67 (Fig. 5a,b).

a,b, Tissue from patient PDAC_64630 assayed with Visium (n = 1,406 spots) with a hematoxylin and eosin (H&E) stain (a) or colored by PDAC subtype determined with gene signatures (b). c, CellNEST-detected communicating regions from a. CellNEST output graphs show tumor (filled square) and stroma (open circle) regions. Colors correspond to different connected components. d,e, Histogram of the strongest CCC counts (y axis) determined by CellNEST across the whole tissue (d) or tumor-communicating regions (e). Colors correspond to each connected component in c. f,g, Tissue from patient PDAC_140694 assayed with Visium (n = 2,298 spots) with H&E (f) or colored by PDAC subtype determined with gene signatures (g). h,i, CellNEST-detected strongly communicating regions from f (h) with the corresponding histogram of CCC counts (i) colored by each connected component in h. Ligand–receptor pairs highlighted with bold text in d and i represent communications detected by CellNEST in both samples. PLXNB2–MET/MST1R highlighted with red text in e and i represents a communication that is associated with the classical subtype in both samples by CellNEST. j–m, Mean of top 20% downstream signaling gene expression of MET (PLXNB2–MET; j,k) and ITGB4 (LGALS3–ITGB4; l,m) on PDAC_64630 (j,l) and PDAC_140694 (k,m). Gene expression is high in regions where the corresponding CCCs are detected by CellNEST (black boxes).
We first assessed whether CellNEST could identify PDAC-relevant ligand–receptor pairs across the whole tissue. CellNEST reported 411 ligand–receptor pairs out of 12,605 total pairs in the top 20% strongest signals, with the predicted interaction between fibronectin and ribosomal protein SA (FN1–RPSA) as the most abundant with an occurrence of 239 instances. FN1–RPSA was mainly found in the stromal region (Fig. 5c–e and Supplementary Fig. 7a,b). Fibronectin is considered one of the main extracellular matrix constituents of pancreatic tumor stroma, and its high expression associates with more aggressive tumors in patients with resected PDAC68. Ribosomal protein SA is a ribosomal subunit but can also act as a cell surface receptor that regulates pancreatic cancer cell migration69. We observed additional canonical signals, such as TGFβ signaling, which promotes fibrosis and immune evasion in PDAC70, and protein tyrosine phosphatase receptor type F (PTPRF)-associated signaling, whose expression has been implicated in multiple cancers71. We also identified significant enrichment of GAS6–AXL specifically within tumor regions, whose signaling pathway is associated with PDAC tumorigenesis72 (Fisher’s exact test, P = 1.307 × 10−2; Supplementary Table 2).
To determine whether CellNEST could identify consistent tumor-associated CCC across multiple tissues, we applied our model to PDAC_140694 derived from a different patient with similar PDAC subtypes to PDAC_64630 (Fig. 5f–i). PDAC_140694 contained mostly tumor cells with fewer stroma than PDAC_64630. To directly compare communication occurring within each sample, we filtered CellNEST-identified signals in PDAC_64630 to those between tumor spots only or tumor and stromal spots (Fig. 5e,h,i). We found overlapping PDAC-associated CCC between both patients in the top 20 strongest signals along with their downstream signaling genes, including LGALS3–ITGB4, PLXNB2–MET/MST1R, PTPRF–RACK1, TGFB1–ITGB5 and TIMP1–LRP1 (ref. 73) (Fig. 5e,i–m). The high concordance of top signals suggests CellNEST can detect similar communication in similar contexts.
CellNEST reveals subtype-region-specific communication in PDAC
After identifying tumor-wide CCC associated with PDAC, we evaluated whether CellNEST could resolve CCC within specific tissue regions. We annotated tumor regions according to classical and basal-like transcriptomic PDAC subtypes74. Using CellNEST, we detected region-specific communication involving PLXNB2–MET/MST1R primarily in classical regions (Fisher’s exact test, P = 4.02 × 10−25; Fig. 5d,e,i–k, Supplementary Fig. 7c–h and Supplementary Table 3) and ANXA1–EGFR in basal-like regions (Fisher’s exact test, P = 1.79 × 10−3; Supplementary Table 4) across both samples (Supplementary Note 12). PLXNB2 codes for a plexin protein, a member of a family of transmembrane receptors initially recognized for their role in axon guidance. Plexins are known for their key role in tumor CCC, tumor growth, migration and metastasis. Semaphorins are the main ligands of plexin receptors; however, some plexins can also form complexes with other tyrosine-kinase receptors, such as the hepatocyte growth factor receptor encoded by MET75 and RON encoded by MST1R76. To further explore differences in the PLXNB2–MET axis between classical and basal-like tumor cells, we analyzed RNA sequencing data from a library of ten PDAC patient-derived organoid models (Fig. 6a–c). Organoid gene expression confirmed that classical tumors exhibit significantly higher MET expression than basal-like tumors (two-sided Fisher–Pitman permutation test, P = 3.18 × 10−02; Fig. 6a,b and Supplementary Note 13). While the role of plexins is described in other solid tumors, including PDAC, previous studies explored semaphorins as their predominant ligands77. Of note, both NICHES and COMMOT were unable to detect a consistent set of CCC specific to classical or basal-like regions (Supplementary Figs. 8 and 9), underscoring CellNEST’s unique ability to identify subtype-specific CCC.

a, Heat map displaying gene expression of MET, classical-associated genes and basal-like-associated genes74 in PDAC patient-derived organoid models assayed with bulk RNA sequencing (n = 10). b,c, Box and whisker plots comparing gene expression in basal-like (n = 5) versus classical (n = 5) organoids classified with an established subtyping scheme74. Center line, median; box, interquartile range; whiskers, 1.5 × interquartile range; points, outliers. b, MET expression is significantly higher in the classical organoids (two-sided Fisher–Pitman permutation test: P = 3.18 × 10−02). c, Both classical and basal-like organoids express LGALS3 (two-sided Fisher–Pitman permutation test: P = 0.175). d,e, Histogram of the most abundant two-hop relay-network patterns (d) along with the spatial location of FN1–RPSA to FN1–RPSA (e) detected by CellNEST on the PDAC_64630 sample (n = 1,406 spots; filled square, tumor; open circle, stroma), highlighted in red. f, Overview of CellNEST-Interactive. The CellNEST-Interactive display shows a fully interactive network of vertices (cells or spots) connected by ligand–receptor pairs (left). The display features a user interface (right) with options to filter genes and select thresholds for attention scores of communication, as well as a histogram of communication abundance colored by component. Zoomed insets display a sender stroma spot and receiver tumor spot participating in FN1-SDC1 communication in the PDAC_64630 sample.
In contrast to subtype-specific CCC, CellNEST detected LGALS3–ITGB4 in basal-like and classical mixed regions (Fisher’s exact test, P = 0.621; Supplementary Table 5). Galectin-3 (LGALS3) mediates tumor–stroma interactions by activating pancreatic stellate cells78. We observed equally high expression of LGALS3 in both classical and basal-like organoids (two-sided Fisher–Pitman permutation test, P = 0.175; Fig. 6c). To determine the potential impact of these signals, we explored the association between these genes and PDAC using The Cancer Genome Atlas79. All CellNEST-identified genes were classified as ‘unfavorable’ in the context of PDAC, and associated with survival (log-rank test, P = 0.0140 for FN1, P = 8.50 × 10−3 for PLXNB2, P = 1.21 × 10−7 for MET, P = 6.39 × 10−4 for ITGB4 and P = 1.40 × 10−4 for ITGB5). Furthermore, MET, ITGB4 and ITGB5 achieved high antibody staining results for PDAC and their gene expression is considered prognostic by the Human Pathology Atlas80, which highlights them as potential targets for treatment. Of note, CellNEST’s top-identified ligand corroborates previous findings that illustrate the critical role of FN1 as a signaling gene against pancreatic cancer based on survival and gene expression analyses81. Together, these findings suggest that different subtypes of PDAC use distinct tumor-promoting CCC, which may impact patient outcomes.
We next sought to characterize differences between our previously identified ligand–receptor pairs with relay networks within pancreatic tumor tissue. CellNEST predicted FN1–RPSA to FN1–RPSA, COL1A1–SDC1 to FN1–RPSA, and TGFB1–ITGB5 to FN1–RPSA among the most frequently occurring pattern of this type (Fig. 6d,e and Supplementary Fig. 10a,b). These signals promote cell adhesion (FN1 and TGFB1)73, migration (RPSA)69, metastasis (FN1)81, epithelial–mesenchymal transition (COL1A)82 and inflammation (SDC1)83. CellNEST largely localized FN1–RPSA to FN1–RPSA, the most abundant relay network in PDAC_64630, to myofibroblast-like cancer-associated fibroblasts, which are key drivers of fibrosis in the PDAC tumor microenvironment84 (Supplementary Fig. 10c). These results suggest that CellNEST uncovers cascades of adhesion and inflammatory networks that would remain undetected by traditional single ligand–receptor pair analyses.
CellNEST-Interactive is a web-based visualization tool for exploring communication
To help visualize cell–cell communication on tissues, we developed CellNEST-Interactive as a web-based data visualization tool (Fig. 6f and Supplementary Figs. 11 and 12). CellNEST-Interactive features a 3D responsive graph illustrating cells or spots as vertices and ligand–receptor pairs as directed edges. The user is able to specify the number of strongest ligand–receptor pairs which updates connected components and colors on-the-fly. CellNEST-Interactive also displays a corresponding histogram listing each unique ligand–receptor pair stacked by connected components showing their specific region of tissue. The user can visualize a particular gene or ligand–receptor pair on both the 3D graph and the histogram using a fuzzy search feature. CellNEST-Interactive is designed with responsiveness in mind for both mobile and desktop. CellNEST-Interactive is available on GitHub at https://github.com/schwartzlab-methods/CellNEST-interactive.
Discussion
Detecting communication through ligand–receptor interactions is necessary to decipher cellular activity in tissue. Existing scRNA-seq-based computational methods for identifying CCC in tissue samples often produce an extensive number of false positives, as they lack cell–cell proximity information. Recent spatial transcriptomics-based tools either quantify CCC at cell-population resolution, missing critical rare communication events, or do not consider patterns of ligand–receptor usage. We overcome these challenges by introducing CellNEST, which integrates ligand–receptor information with cell location through a graph attention network at single-cell or spot resolution. We quantitatively evaluated CellNEST and found our model to have superior performance against other available methodologies using new benchmarks of 21 different arrangements of synthetic data representing different technologies and species. CellNEST consistently captured known CCC in both healthy and diseased conditions at various resolutions and dimensions. CellNEST predicted subtype-specific CCC across patients with pancreatic ductal adenocarcinoma, with associated genes correlating with survival in independent cohorts from The Cancer Genome Atlas and the Human Pathology Atlas.
Existing spatial transcriptomic methods for detecting CCC, such as COMMOT and NICHES, focus on high coexpression of ligand–receptor pairs and do not attempt to recognize patterns of activity. However, patterns may correlate with tissue regions even when lowly expressed. Using a pattern recognition algorithm may contribute to CellNEST’s advantage over other methods when identifying T cell homing signals in precise locations of T cell zones in human lymph nodes. Moreover, CellNEST uses all genes to identify CCC, orders communication based on learned importance, and spatially pinpoints their location. Notably, CellNEST does not filter out low-variance ligand–receptor pairs, as this would prevent the method from detecting well-characterized genes that belong to informative modules but are stable across the tissue. CellNEST identified expected signals that were not among the most highly expressed, indicating the importance of integrating spatial and molecular information. These unique capabilities of CellNEST help associate CCC with a target disease and its subtypes.
Recent methods alternatively use scRNA-seq data for CCC detection before mapping ligand–receptor pairs to spatial data14,15,35; however, such tools only resolve CCC between adjacent cells or spots and do not discriminate between distant ligand–receptor mechanisms, such as paracrine communication, which constitutes the majority of ligand–receptor databases. In contrast, CellNEST is capable of detecting three major types of communication: autocrine (self-communication), juxtacrine (communication between adjacent cells) and paracrine (communication between nearby, nonadjacent cells). Furthermore, existing machine-learning-based tools such as GraphComm15 use supervised learning, which is difficult to train due to the unavailability of labeled data. To overcome the ground-truth data scarcity problem, CellNEST applies contrastive learning, an unsupervised training approach. This powerful and generalizable architecture enables CellNEST to accommodate data across varying resolutions, two or three dimensions, and healthy or diseased conditions.
To enable this flexibility, CellNEST only requires a ligand–receptor database, and optionally, pathway information with experimental confidence scores for a priori knowledge. CellNEST reports active cell–cell communication and relay networks based on their learned importance and frequency of appearance within the tissue; however, as spatial transcriptomic data are a snapshot of expression, a limitation of CellNEST is that the algorithm cannot determine whether the observed ligand expression is truly caused by receptor activation. CellNEST’s predicted relay-network results are likely events based on learned patterns of frequent coexpressed signals from the data which suggest a strong role of specific CCC in the tissue. CellNEST assigns confidence scores to the proposed relay networks based on a model of intracellular signals downstream of a receptor triggering ligand production. As such, CellNEST generates hypotheses that assist users in identifying candidate ligand–receptor pairs for further validation, which may produce some false-positive results in noisy conditions that impact CCC in a tissue.
CellNEST’s underlying model is flexible with the expectation of integrating additional data types. With the advancement of spatial-omics technologies, future models may incorporate other data modalities to improve CCC detection, such as protein or chromatin accessibility from emerging assays. In addition, an extension of CellNEST may include subcellular information provided by technologies like MERFISH and Xenium. We anticipate methods like CellNEST that take full advantage of the spatial proximity of cells will provide new avenues for determining cellular neighborhoods and their contributions to health and disease.
Methods
CellNEST architecture
CellNEST is an end-to-end solution for processing data directly from a spatial transcriptomic data structure from programs such as Space Ranger, detecting strong signals and patterns of communication within specific regions of tissue, and displaying CCC through an accessible visualization. There are four main steps in the CellNEST workflow: a data preprocessing step, input graph generation step, communication prediction step and output graph generation step (Fig. 1).
Data preprocessing step
CellNEST takes four inputs: a spatial transcriptomic dataset, a ligand–receptor database, a threshold percentile --threshold_gene_exp (for example, 80th or 98th percentile) to select highly expressed genes, and a threshold distance --neighborhood_threshold as a neighborhood cutoff distance (Fig. 1a–c). The default database provided by our model is a combination of the CellChat and NicheNet databases, totaling 12,605 ligand–receptor pairs. For N spots or cells (here called vertices) and M genes in a spatial transcriptomic dataset, CellNEST generates a gene expression matrix \({{\it{A}}}\in {{\mathbb{R}}}^{\it{N}\times \it{M}}\). CellNEST calculates the Euclidean distance between each pair of vertices to generate a physical distance matrix of dimension \({{\it{D}}}\in {{\mathbb{R}}}^{\it{N}\times \it{N}}\). CellNEST uses quantile normalization85,86 on the gene expression matrix to standardize gene distributions across vertices to enable direct comparisons. For each vertex in the gene expression matrix, CellNEST considers genes having expression over --threshold_gene_exp percentile (default 98) as active.
Input graph generation step
After preprocessing, CellNEST generates an input graph G = (V, E), where V (∣V∣ = N) represents the set of vertices and E (where ∣E∣ is typically over 1 × 106) represents the set of neighborhood relations among the vertices in G (Fig. 1d). We add a neighborhood relation between a vertex i and j if the distance between i and j is less than or equal to --neighborhood_threshold. For each ligand l and paired receptor r from the ligand–receptor database, if Ai,l and Aj,r are active, CellNEST will insert a directed edge from i to j. CellNEST allows for multiple edges to represent multiple ligand–receptor pairs between two vertices. Of note, an edge between a pair of vertices does not necessarily mean that a communication is happening along that edge, because CCC is highly context-dependent12 and affected by various epigenetic factors41. An edge is a neighborhood relation representation between a pair of vertices, which CellNEST evaluates as a probable CCC or random coincidence.
We next pass G to the deep-learning module ‘communication prediction step’ through two input feature matrices: a vertex feature matrix \({{{\it{H}}}}_{v}\in {{\mathbb{R}}}^{{F}_{v}\times | V| }\) and an edge feature matrix \({{{\it{H}}}}_{e}\in {{\mathbb{R}}}^{{F}_{e}\times | E| }\). Each column in Hv is a vertex input feature vector (for example, \(\overrightarrow{{\bf{h}}_{i}}\) for vertex i), which represents each cell or spot in the dataset. CellNEST uses a one-hot vector to present each vertex uniquely, so Fv = ∣V∣ (Fig. 1d). Similarly, each column in the edge feature matrix, He, is an edge feature vector representing an edge (neighborhood relation) in G. The edge feature vector has dimension Fe = 3, as it has three attributes (Fig. 1d): physical distance between vertices (for example, di,j from the physical distance matrix \({{\it{D}}}\in {{\mathbb{R}}}^{\it{N}\times \it{N}}\)), ligand–receptor coexpression score for the corresponding edge (for example, Le × Re from Fig. 1c), and the identifier of that ligand–receptor pair from the input database (Fig. 1b). We pass these two input feature matrices to the next step, the ‘communication prediction step’.
Communication prediction step: overview
The CellNEST architecture builds on two main deep-learning concepts: graph attention networks37 (GAT) as encoders and deep graph infomax33 (DGI) to train encoders through contrastive learning (Fig. 1e and Supplementary Fig. 13a). Although GAT-based models are traditionally used with a training set, there is no ground truth for CCC detection, so CellNEST instead uses DGI for training. We provide implementation functions for integrating GAT into the DGI model in our GitHub repository located at https://github.com/schwartzlab-methods/CellNEST/blob/main/CCC_gat.py.
Communication prediction step: graph attention network
The GAT generates a vertex embedding that encodes information about a vertex i in G along with its neighborhood information, here meaning which vertices can i communicate with and through which ligand–receptor pairs. The attention module in the GAT assigns ‘attention scores’ to each edge based on how necessary and sufficient those edges are to capture hidden patterns that together reconstruct the input sample.
Let input vertex feature vectors for vertices i and j be \(\overrightarrow{{\bf{h}}_{i}},\overrightarrow{{\bf{h}}_{j}}\in {{\mathbb{R}}}^{{F}_{v}}\), input edge feature vectors from j to i be \(\overrightarrow{{\bf{e}}_{i,j}}\in {{\mathbb{R}}}^{{F}_{e}}\), and the dimensions of vertex and edge embeddings be \({\it{F}}^{{\prime} }\). The learnable weight matrix for the linear transformation of vertex features is \({{{\it{W}}}}_{v}\in {{\mathbb{R}}}^{{F}_{v}\times {F}^{{\prime} }}\), while the equivalent matrix for edge features is \({{{\it{W}}}}_{e}\in {{\mathbb{R}}}^{{F}_{e}\times {F}^{{\prime} }}\). Then, the attention score for the edge from j to i is
This score indicates the importance of vertex \({\it{j}}^{{\prime} }{{\rm{s}}}\) features to vertex i. Here, the attention \(\overrightarrow{\bf{a}}\) is a learnable parameter, where \(\overrightarrow{\bf{a}}\in {{\mathbb{R}}}^{\it{F}^{\prime} }\). Here we use tanh, as we found increased performance using tanh nonlinearity instead of the parametric rectified linear unit and rectified linear unit activation functions, the latter of which was too unstable (Supplementary Fig. 13b,c). After learning the attention scores, we apply a Softmax normalization over all incoming edges to vertex i from its neighbors Ni using
\({\it{\alpha} }_{i,\;j}^{{\prime} }\) ranges from 0 to 1 in an effort to scale attention scores. We use Softmax normalization for the message propagating principle. Using the normalized attention scores, we obtain a vertex embedding for i with
Here, the GAT generates a vertex embedding matrix \({{{\it{H}}}}_{v}^{{\prime} }\in {{\mathbb{R}}}^{|V| \times {F}^{{\prime} }}\); however, for communication prediction, we use the attention scores rather than the vertex embedding to prioritize edges in set E based on global context. To detect which regions are more active than others in the input sample, we use unnormalized attention scores from equation (1), as these scores are globally comparable across the tissue (Supplementary Fig. 14a). As such, we use the scores obtained by equation (1) directly to represent CCC probability. We can scale these scores between 0 to 1 over all the edges in E such that scores closer to 1 present a higher probability of communication.
CellNEST generally assigns higher attention scores to input edges with high ligand–receptor coexpression scores (Supplementary Fig. 14b–m). Of note, the conventional way of using normalized attention scores cannot achieve this goal (Supplementary Fig. 14a), so CellNEST uses the unnormalized attention scores assigned by the GAT.
Communication prediction step: DGI for encoder training
We apply the contrastive learning model DGI33 to train the GAT in an unsupervised approach. DGI takes the input graph G = (V, E) and applies random permutation, shuffling edges to form a corrupted graph \({G}_{C}=(V,{E}^{{\prime} })\), where \({E}^{{\prime} }\) is the set of corrupted edges (Supplementary Fig. 13a). We store the original input graph as GT. This contrastive learning approach has two branches to handle each version of the input graph: the corrupted branch and the original branch.
Both branches use the same GAT encoder with shared learnable parameters or weight matrices to generate a vertex embedding matrix \({{{\it{H}}}}_{v}^{{\prime} }\in {{\mathbb{R}}}^{| V| \times {F}^{{\prime} }}\). The vertex embedding generated from GT through the original branch is summed to obtain the ‘summary vector’ \(\overrightarrow{\bf{s}}\). This summary vector captures global information content of the entire graph. We use a discriminator function to measure the distance between \(\overrightarrow{\bf{s}}\) from the corrupted graph embedding (negative sample) and the true graph embedding (positive sample). CellNEST maximizes the mutual information between the summary vector and vertex embedding from the true graph by optimizing the Jensen–Shannon divergence between the negative and positive graphs. This divergence distance is related to the generative adversarial network distance33. Through many iterations (approximately 60,000 in our testing), CellNEST eventually converges to a minimal loss, and we save that model state.
Output graph generation step: overview
CellNEST uses the stochastic optimization algorithm Adam87, which may introduce small variations in the output of multiple runs. As an optional step to increase the accuracy and stability of communication detection, we run each experiment multiple times (default of five) with different seeds and combine the results from each run. Then, we apply postprocessing on the aggregated result to obtain the final output graph (Fig. 1f).
Output graph generation step: ensemble of multiple runs
We obtain the ranks of edges based on the attention scores assigned by each encoder layer for each run. Using the rank product88, we sort by the aggregated rank for each layer. We then merge the results for both attention layers, as existing metapath work on GNNs suggests important characteristics are present in each layer89. This step also accepts a top percentage of communications, --top_percent, as input from the user. By default, we select the top --top_percent = 20% as the most reliable signals for the analyses presented here, as most of the positive CCCs are detected within the top 20% based on synthetic benchmarking (Supplementary Fig. 15). We select this threshold on both layers independently. We must select a cutoff point, as the GAT architecture does not discard any edge by default, only assigning attention scores where a higher score correlates with importance. Optionally, CellNEST provides a cutoff based on median absolute deviations from the median attention (--cutoff_MAD) and skewness of the distribution (--cutoff_z_score) to provide alternative statistical approaches. In addition to filtering the CCC based on cutoff criteria, CellNEST optionally provides confidence intervals using a bootstrapping technique invoked with the confidence_interval command, as well as p values (Supplementary Notes 14 and 15).
Output graph generation step: postprocessing
This step postprocesses the list of strong CCC for better visualization and downstream analysis. We apply a connected component finding algorithm90 on the strongly communicating --top_edge_count (user chosen) edges to generate subgraph components. In this way, we observe subgraphs where all vertices are strongly communicating with at least one other vertex in the community, suggesting a set of vertices localized to specific regions. We provide several visualization outputs to best quantify CellNEST’s predictions using graph, list and tabular formats (Fig. 1g,h). Although we count the number of detected CCCs and sort the ligand–receptor pairs by abundance for histogram generation, we also provide the option (--sort_by_attentionScore) to sort by total attention score, which here resulted in similar rankings (Supplementary Fig. 16). When analyzing relay networks with commands relay_extract, relay_celltype, and relay_confidence, CellNEST outputs relay-network abundance, spatial location, cell-type proportions and confidence scores associated with relay networks using graph, table, pie and bar charts. A detailed list of generated outputs is available on GitHub at https://github.com/schwartzlab-methods/CellNEST/blob/main/vignette/user_guide.md.
Synthetic data preparation for benchmarks
To represent different distributions of cells and spots, we compared methods across three types of benchmarks: equidistant data points (n = 3,000; for example, Visium data), uniformly distributed data points (n = 5,000; for example, MERFISH data) and data points with a mixture of uniform and Gaussian distributions (n = 5,000) representing other complex data types (Fig. 3f–h). To generate the gene expression of each data point, we randomly sampled from Gaussian distributions with varying levels of noise and separate distributions for active and inactive ligand and receptor genes.
We generated 3,000 equidistant data points representing Visium spots, each having 10,000 genes. We assigned 10% of genes as ligand or receptor genes and formed synthetic ligand–receptor pairs with these genes. The synthetic ligand–receptor database generated in this way has ∼1,400 pairs. In this same way, we sampled 5,000 data points from a uniform distribution representing MERFISH cells, each having 350 genes. The synthetic ligand–receptor database generated this way has 100 pairs with 12% of genes acting as ligand or receptor genes to approximate observed proportions17. Last, we sampled 5,000 data points from a mixture of uniform and Gaussian distribution representing single-cell data types, each having 350 genes, with 12% of genes forming ligand–receptor pairs. The synthetic ligand–receptor database generated this way has 100 pairs.
In the mechanistic model, we changed the criteria of neighbor selection. For adding ground-truth connections, we considered a Gaussian distribution around each sender cell such that closer neighbors would have a higher probability of acting as a receiver cell. In this way, we drew ligand–receptor pairs with decreasing probability as a function of distance from a sender cell and set a maximum limit on the number of ligands a receptor can accept.
Notably, while we sought to evaluate standard CCC of a single ligand–receptor pair between spots or cells, we also introduced new benchmarks to test the model’s ability to recognize relay networks by incorporating such patterns in the synthetic data. The relay-based benchmark models a sender cell i sending a type s signal to a receiver cell j, after which j sends a type t signal to a receiver cell k.
Relay-network generation
CellNEST applies contrastive learning for the representation learning of input data. During this process, CellNEST assigns higher attention scores to the CCCs that form repeated relay-network patterns. We record these highly scored CCC through depth-first search. The relay-network assignment algorithm starts at an arbitrary vertex in the CellNEST-derived graph and follows the direction of outgoing edges (CCC) recursively until there are no more outgoing edges or a predefined number of hops is reached. Unless otherwise stated, we here specified two-hop relay networks. CellNEST users may extend the default to n-hops. The flexibility of the relay-network recovery step allows us to apply this process to other method outputs as well, for example, on COMMOT and NICHES (Supplementary Fig. 17a–d).
Intracellular signaling pathway generation
CellNEST builds directed knowledge graphs of signaling pathways from a receptor node down to transcription factors in a manner conceptually similar to SpaTalk91 and FlowSig92. CellNEST searches up to a user-defined maximum hops (default --num_hops = 10 hops for memory considerations). Using breadth-first search from the receptor node, we identify the path to all downstream transcription factor nodes as in SPAGI93, aggregating their gene expression. We provide options to either include the gene expression of the downstream transcription factor only or both the genes encoding proteins in the signaling pathway and the transcription factor genes, weighted or unweighted by the previously calculated positive experimental score values between nodes.
Relay-network confidence scoring
CellNEST assigns a confidence score to each relay network by constructing a putative intracellular network between the receptor and subsequent ligand of the second vertex. CellNEST creates this network using breadth-first search to identify paths that link the receptor protein to a transcriptional activator of the ligand using the aforementioned interaction databases. Due to memory considerations, we prune the protein–protein interaction database by minimum confidence scores at five thresholds from 0.1 to 0.5. CellNEST then computes the first path found from the source to target node for each minimum edge weight. CellNEST nominates the path with the highest cumulative confidence score, which is calculated as the product of the experimental confidence scores reported by the interaction databases along the path.
Spatial transcriptomics of human patients with pancreatic cancer
Two solid tumor biospecimens were collected from the pancreas of two patients with stage IIB PDAC (PDAC_64630, 76-year-old male; PDAC_140694, 83-year-old female). Both biospecimens were collected from the University Health Network Biospecimens Program (Toronto, Canada). Ethical approval was obtained through the University Health Network Research Ethics Board (13-6377). Tumors were collected at the time of resection. Samples were stabilized for approximately 3 h at 4 °C until long-term preservation (embedded in optimal-cutting-temperature compound). Samples were stored at −80 °C until used. The cases were selected according to have >30% tumor cellularity. The regions of interest for capture areas (6.5 × 6.5 mm) were selected, targeting tumor areas with representative subtype morphologies67. The 10-μM cuts were placed into 10x Genomics Visium FFPE spatial gene expression slides from selected trimmed tissue areas. Spatial transcriptomics using the Visium platform was carried out according to manufacturer’s instructions (10x Genomics, part no. 1000200, protocol CG000160 RevB, CG000239 RevD). Sequencing was performed on the Illumina NovaSeq 6000 platform with paired-end reads according to 10x Genomics specifications. Data was processed using Space Ranger (v.2.0.0) and mapped to the GRCH38 v.93 genome assembly.
Annotation of pancreatic cancer samples
Histology categories of tumor and stroma were assigned based on the following features. Tumor: malignant cells arranged in any architecture of glands, cords, strands, solid sheets and single cells67; and stroma: nontumor tissue surrounding tumor cells, composed mainly of fibroblasts, myofibroblasts and collagen fibers94. Transcriptomic subtype annotations were assigned using Loupe Browser v.6.4.1 (10x Genomics) according to the log2 Feature Sum filter using a previously determined subtype gene list74.
Preparation of PDAC patient-derived organoid library
An organoid library with matching whole-transcriptome sequencing from laser microcapture-enriched tumors was established from 44 cases with resectable (stage I/II) and advanced (stage III/IV) PDAC. Tumor transcriptomic subtype classifications were obtained from published data95. Advanced organoids were generated by University Health Network Living Biobank as part of a clinical trial (NCT02750657) and resectable organoids were generated at the Notta Laboratory (CAPCR 13-6377, 21-5648) following established methods96. In brief, organoids were cultured in DMEM/F-12 medium (Fisher, 12634-010) supplemented with B-27 supplement 1× (Life Technologies, 17504-044), GlutaMAX (2 mM; Life Technologies, 35050-061), HEPES (10 mM; Fisher, 15630080), nicotinamide (10 mM; Sigma, N0636-100G), N-acetyl-l cysteine (1.25 mM; Sigma, A9165-5G), gastrin I (10 nM; sigma, G9020-250UG), Noggin (100 ng ml−1; Peprotech, 120-10C-500UG), FGF-10 (100 ng ml−1; Biotechne, NBP2-34927-5UG), A83-01(0.5 μM ml−1; TOCRIS, 2939), Y-27632 (10 μM; Selleckchem, S1049-50MG), EGF (50 ng ml−1; Peprotech, AF-100-15-500UG), CHIR (2.5 μM; Tocris, 4423), Wnt-3a (20% v/v, condition medium by the University Health Network (UHN) Living Biobank), R-spondin1 (30% v/v, condition media by UHN living biobank) and antibiotics, with medium replacement twice a week. Organoids were passaging using TrypLE express enzyme (Thermo Fisher 12605028) at 37 °C until dissociation. After passage 6, RNA was extracted from dissociated organoids. Sequencing libraries were prepared using the Smart-3SEQ protocol97 from 10 ng of RNA. Pools of 20 libraries were sequenced on the Illumina NextSeq 500 using 150 cycles kit v.2 for Single Read 150 on a Mid-Output flow cell.
Development of CellNEST-Interactive
CellNEST-Interactive uses vanilla Javascript and HTML on the front end with Tailwind CSS for styling. We used D3.js for the histogram and Vasco Asturiano’s 3D-force-graph library (which extends off of D3.js and Three.js) for the responsive graph. To obtain the data for display, CellNEST-Interactive uses jQuery to send AJAX requests to the back-end server as well as to deep-copy current graph data. The back end uses the Django framework. After receiving a request from the front end with edge count as a parameter, a Python script reads all CSV records stored locally and returns graphable nodes and edges in JSON format. Necessary files to be read include complete records for cell (or spot), cell coordinates, cell annotations (if available) and the list of top 20% CCC detected by CellNEST. CellNEST-Interactive further processes these data by separating vertices into connected components and assigning colors using NumPy, Pandas, SciPy and Matplotlib libraries. CellNEST-Interactive is available on GitHub at https://github.com/schwartzlab-methods/CellNEST-interactive.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
PDAC spatial transcriptomic data for PDAC_64630 and PDAC_140694 are available on the Gene Expression Omnibus under accession no. GSE262245. The spot annotations for both samples are available on GitHub at https://github.com/schwartzlab-methods/CellNEST_paper_figures/blob/main/NEST_figures_input_PDAC.7z. We obtained spatial transcriptomic data of human lymph nodes from https://www.10xgenomics.com/datasets/human-lymph-node-1-standard-1-0-0, mouse hypothalamic preoptic region from https://doi.org/10.5061/dryad.8t8s248, LUAD from the Gene Expression Omnibus under accession no. GSE189487 and human colorectal cancer (Visium HD) from https://www.10xgenomics.com/datasets/visium-hd-cytassist-gene-expression-libraries-of-human-crc. Processed ligand–receptor and signaling pathway databases are available on GitHub at https://github.com/schwartzlab-methods/CellNEST/tree/main/database.
Code availability
CellNEST is available at GitHub at https://github.com/schwartzlab-methods/CellNEST, on Zenodo at https://doi.org/10.5281/zenodo.15459529 (ref. 98) or as a Singularity image at https://cloud.sylabs.io/library/fatema/collection/nest_image.sif with a tutorial on GitHub at https://github.com/schwartzlab-methods/CellNEST#vignette. CellNEST-Interactive is available at https://github.com/schwartzlab-methods/CellNEST-interactive and on Zenodo at https://doi.org/10.5281/zenodo.15459868 (ref. 99). Scripts for generating the figures and plots of the manuscript can be found on GitHub at https://github.com/schwartzlab-methods/CellNEST_paper_figures.
References
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov 12, 31–46 (2022).
Armingol, E., Officer, A., Harismendy, O. & Lewis, N. E. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 22, 71–88 (2021).
Vento-Tormo, R. et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353 (2018).
Wang, Y. et al. iTALK: an R package to characterize and illustrate intercellular communication. Preprint at bioRxiv https://doi.org/10.1101/507871 (2019).
Noël, F. et al. Dissection of intercellular communication using the transcriptome-based framework ICELLNET. Nat. Commun. 12, 1089 (2021).
Choi, H. et al. Transcriptome analysis of individual stromal cell populations identifies stroma-tumor crosstalk in mouse lung cancer model. Cell Rep 10, 1187–1201 (2015).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Jin, S. et al. Inference and analysis of cell–cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Thurley, K., Wu, L. F. & Altschuler, S. J. Modeling cell-to-cell communication networks using response-time distributions. Cell Syst. 6, 355–367.e5 (2018).
Lu, H. et al. CommPath: an R package for inference and analysis of pathway-mediated cell-cell communication chain from single-cell transcriptomics. Comput. Struct. Biotechnol. J. 20, 5978–5983 (2022).
Mayer, S. et al. The tumor microenvironment shows a hierarchy of cell-cell interactions dominated by fibroblasts. Nat. Commun. 14, 5810 (2023).
Innes, B. T. & Bader, G. D. Transcriptional signatures of cell–cell interactions are dependent on cellular context. Preprint at bioRxiv https://doi.org/10.1101/2021.09.06.459134 (2021).
Müller, P. & Schier, A. F. Extracellular movement of signaling molecules. Dev. Cell 21, 145–158 (2011).
Wilk, A. J., Shalek, A. K., Holmes, S. & Blish, C. A. Comparative analysis of cell–cell communication at single-cell resolution. Nat. Biotechnol. 42, 470–483 (2024).
So, E., Hayat, S., Kadambat Nair, S., Wang, B. & Haibe-Kains, B. GraphComm: a graph-based deep learning method to predict cell–cell communication in single-cell RNAseq data. Preprint at bioRxiv https://doi.org/10.1101/2023.04.26.538432 (2023).
Marx, V. Method of the year: spatially resolved transcriptomics. Nat. Methods 18, 9–14 (2021).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Raredon, M. S. B. et al. Comprehensive visualization of cell–cell interactions in single-cell and spatial transcriptomics with NICHES. Bioinformatics 39, btac775 (2023).
Cang, Z. et al. Screening cell–cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218–228 (2023).
Birk, S. et al. Quantitative characterization of cell niches in spatially resolved omics data. Nat. Genet. 57, 897–909 (2025).
Bafna, M., Li, H. & Zhang, X. CLARIFY: cell–cell interaction and gene regulatory network refinement from spatially resolved transcriptomics. Bioinformatics 39, i484–i493 (2023).
Lee, Y., Xu, Y., Gao, P. & Chen, J. TENET: triple-enhancement based graph neural network for cell–cell interaction network reconstruction from spatial transcriptomics. J. Mol. Biol. 436, 168543 (2024).
Li, H. et al. Decoding functional cell–cell communication events by multi-view graph learning on spatial transcriptomics. Brief. Bioinform. 24, bbad359 (2023).
Liu, J. et al. CytoSignal detects locations and dynamics of ligand–receptor signaling at cellular resolution from spatial transcriptomic data. Preprint at bioRxiv https://doi.org/10.1101/2024.03.08.584153 (2024).
Ji, B., Wang, X., Qiao, D., Xu, L. & Peng, S. SpaCCC: large language model- based cell-cell communication inference for spatially resolved transcriptomic data. Big Data Min. Anal. 7, 1129–1147 (2024).
Dries, R. et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78 (2021).
Wu, D. & Datta, S. TWCOM: an R package for inference of cell–cell communication on spatially resolved transcriptomics data. Bioinform. Adv. 4, vbae101 (2024).
Jin, S., Plikus, M. V. & Nie, Q. CellChat for systematic analysis of cell–cell communication from single-cell transcriptomics. Nat. Protoc. https://doi.org/10.1038/s41596-024-01045-4 (2024).
Wu, L., Cui, P., Pei, J., Zhao, L. & Guo, X. Graph neural networks: foundation, frontiers and applications. In Proc 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining (eds Zhang, A. & Rangwala, H.) 4840–4841 (ACM, 2022).
Li, J., Chen, S., Pan, X., Yuan, Y. & Shen, H.-B. Cell clustering for spatial transcriptomics data with graph neural networks. Nat. Comput. Sci. 2, 399–408 (2022).
Long, Y. et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat. Commun. 14, 1155 (2023).
Vaswani, A. et al. Attention is all you need. In Advances in Neural Information Processing Systems 30: Annual Conference on Neural Information Processing Systems 2017 (eds von Luxburg, U. et al.) 5998–6008 (NIPS, 2017).
Velickovic, P. et al. Proc. 7th International Conference on Learning Representations (ICLR, 2019).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 40, 661–671 (2022).
Zhu, J. et al. Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics. Exp. Mol. Med. 54, 2060–2076 (2022).
Moffitt, J. R. et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018).
Brody, S., Alon, U. & Yahav, E. How attentive are graph attention networks? Preprint at https://arxiv.org/abs/2105.14491 (2021).
Szklarczyk, D. et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Sang-Aram, C., Browaeys, R., Seurinck, R. & Saeys, Y. Unraveling cell–cell communication with NicheNet by inferring active ligands from transcriptomics data. Nat. Protoc. https://doi.org/10.1038/s41596-024-01121-9 (2025).
Garcia-Alonso, L., Holland, C. H., Ibrahim, M. M., Turei, D. & Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res 29, 1363–1375 (2019).
Smith, R. J. et al. Epigenetic control of cellular crosstalk defines gastrointestinal organ fate and function. Nat. Commun. 14, 497 (2023).
Yan, Y. et al. CCL19 and CCR7 expression, signaling pathways, and adjuvant functions in viral infection and prevention. Front. Cell Dev. Biol. 7, 212 (2019).
Britschgi, M. R., Favre, S. & Luther, S. A. CCL21 is sufficient to mediate DC migration, maturation and function in the absence of CCL19. Eur. J. Immunol. 40, 1266–1271 (2010).
Fu, H., Ward, E. J. & Marelli-Berg, F. M. Mechanisms of T cell organotropism. Cell. Mol. Life Sci. 73, 3009–3033 (2016).
Tan, C. L. et al. Warm-sensitive neurons that control body temperature. Cell 167, 47–59.e15 (2016).
Rich, M. E., deCárdenas, E. J., Lee, H.-J. & Caldwell, H. K. Impairments in the initiation of maternal behavior in oxytocin receptor knockout mice. PloS ONE 9, e98839 (2014).
Loth, M. K. & Donaldson, Z. R. Oxytocin, dopamine, and opioid interactions underlying pair bonding: highlighting a potential role for microglia. Endocrinology 162, bqaa223 (2021).
Lee, Y. S. et al. Repeated exposure to neurotoxic levels of chlorpyrifos alters hippocampal expression of neurotrophins and neuropeptides. Toxicology 340, 53–62 (2016).
Bakoev, S. Y. et al. Genomic signatures of positive selection in human populations of the OXT, OXTR, AVP, AVPR1A and AVR1B gene variants related to the regulation of psychoemotional response. Genes 14, 2053 (2023).
Derynck, R., Turley, S. J. & Akhurst, R. J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 18, 9–34 (2021).
Su, W.-P. et al. Apolipoprotein E expression promotes lung adenocarcinoma proliferation and migration and as a potential survival marker in lung cancer. Lung Cancer 71, 28–33 (2011).
Zheng, Y.-L. et al. Less efficient g2-m checkpoint is associated with an increased risk of lung cancer in African Americans. Cancer Res 65, 9566–9573 (2005).
Peng, J.-M. & Su, Y.-L. Lymph node metastasis and tumor-educated immune tolerance: potential therapeutic targets against distant metastasis. Biochem. Pharmacol. 215, 115731 (2023).
Morita, Y. et al. PP071: Fibronectin up-regulates expression of VEGF-C and increases lymph node metastasis. Oral Oncol 49, S118 (2013).
Lee, Y. S., Yeo, I. J., Kim, K. C., Han, S.-B. & Hong, J. T. Inhibition of lung tumor development in ApoE knockout mice via enhancement of TREM-1 dependent NK cell cytotoxicity. Front. Immunol. 10, 1379 (2019).
Li, F. et al. Identifying the EMT-related signature to stratify prognosis and evaluate the tumor microenvironment in lung adenocarcinoma. Front. Genet. 13, 1008416 (2022).
Liu, A., Fernandes, B. S., Citu, C. & Zhao, Z. Unraveling the intercellular communication disruption and key pathways in Alzheimer’s disease: an integrative study of single-nucleus transcriptomes and genetic association. Alzheimers Res. Ther. 16, 3 (2024).
Van Leent, M. et al. Prosaposin mediates inflammation in atherosclerosis. Sci. Transl. Med. 13, eabe1433 (2021).
Miyahara, Y. et al. Prosaposin, tumor-secreted protein, promotes pancreatic cancer progression by decreasing tumor-infiltrating lymphocytes. Cancer Sci 113, 2548–2559 (2022).
Queisser, A. et al. Comparison of different prostatic markers in lymph node and distant metastases of prostate cancer. Mod. Pathol. 28, 138–145 (2015).
Pandey, P. et al. Amyloid precursor protein and amyloid precursor-like protein 2 in cancer. Oncotarget 7, 19430–19444 (2016).
Beaulieu, J.-F. Integrin α6β4 in colorectal cancer: expression, regulation, functional alterations and use as a biomarker. Cancers 12, 41 (2019).
Biswas, S. et al. Mutational inactivation of TGFBR2 in microsatellite unstable colon cancer arises from the cooperation of genomic instability and the clonal outgrowth of transforming growth factor β resistant cells. Genes Chromosomes Cancer 47, 95–106 (2008).
Liu, Y. & Wang, X. Tumor microenvironment-associated gene C3 can predict the prognosis of colorectal adenocarcinoma: a study based on TCGA. Clin. Transl. Oncol. 23, 1923–1933 (2021).
Ottaiano, A. et al. Prognostic significance of CXCR4 in colorectal cancer: an updated meta-analysis and critical appraisal. Cancers 13, 3284 (2021).
Lee, K. J. et al. A novel nanoparticle-based theranostic agent targeting LRP-1 enhances the efficacy of neoadjuvant radiotherapy in colorectal cancer. Biomaterials 255, 120151 (2020).
Kalimuthu, S. N. et al. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut 69, 317–328 (2020).
Hu, D. et al. Stromal fibronectin expression in patients with resected pancreatic ductal adenocarcinoma. World J. Surg. Oncol. 17, 29 (2019).
Lefebvre, T. et al. TRPM7/RPSA complex regulates pancreatic cancer cell migration. Front. Cell Dev. Biol. 8, 549 (2020).
Principe, D. R. et al. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res 76, 2525–2539 (2016).
Pehkonen, H., de Curtis, I. & Monni, O. Liprins in oncogenic signaling and cancer cell adhesion. Oncogene 40, 6406–6416 (2021).
Moody, G. et al. Antibody-mediated neutralization of autocrine Gas6 inhibits the growth of pancreatic ductal adenocarcinoma tumors in vivo. Int. J. Cancer 139, 1340–1349 (2016).
Javanshir, H. T. et al. Investigation of key signaling pathways and appropriate diagnostic biomarkers selection between non-invasive to invasive stages in pancreatic cancer: a computational observation. J. Med. Life 15, 1143–1157 (2022).
Oh, K. et al. Coordinated single-cell tumor microenvironment dynamics reinforce pancreatic cancer subtype. Nat. Commun. 14, 5226 (2023).
Toledano, S. & Neufeld, G. Plexins as regulators of cancer cell proliferation, migration, and invasivity. Cancers 15, 4046 (2023).
Cazes, A., Childers, B. G., Esparza, E. & Lowy, A. M. The MST1R/RON tyrosine kinase in cancer: oncogenic functions and therapeutic strategies. Cancers 14, 2037 (2022).
Fard, D., Giraudo, E. & Tamagnone, L. Mind the (guidance) signals! Translational relevance of semaphorins, plexins, and neuropilins in pancreatic cancer. Trends Mol. Med. https://doi.org/10.1016/j.molmed.2023.07.009 (2023).
Zhao, W. et al. Galectin-3 mediates tumor cell-stroma interactions by activating pancreatic stellate cells to produce cytokines via integrin signaling. Gastroenterology 154, 1524–1537 (2018).
Hutter, C. & Zenklusen, J. C. The cancer genome atlas: creating lasting value beyond its data. Cell 173, 283–285 (2018).
Pontén, F., Jirström, K. & Uhlen, M. The Human Protein Atlas—a tool for pathology. J. Pathol. 216, 387–393 (2008).
Ashok, G., Miryala, S. K., Saju, M. T., Anbarasu, A. & Ramaiah, S. FN1 encoding fibronectin as a pivotal signaling gene for therapeutic intervention against pancreatic cancer. Mol. Genet. Genomics 297, 1565–1580 (2022).
Huang, H. et al. Up-regulation of N-cadherin by collagen I-activated discoidin domain receptor 1 in pancreatic cancer requires the adaptor molecule Shc1. J. Biol. Chem. 291, 23208–23223 (2016).
Yablecovitch, D. et al. Serum syndecan-1: a novel biomarker for pancreatic ductal adenocarcinoma. Clin. Transl. Gastroenterol. 13, e00473 (2022).
Shinkawa, T., Ohuchida, K. & Nakamura, M. Heterogeneity of cancer-associated fibroblasts and the tumor immune microenvironment in pancreatic cancer. Cancers 14, 3994 (2022).
Amaratunga, D. & Cabrera, J. Analysis of data from viral DNA microchips. J. Am. Stat. Assoc. 96, 1161–1170 (2001).
Bolstad, B. M., Irizarry, R. A., Åstrand, M. & Speed, T. P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 (2003).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. Preprint at https://arxiv.org/abs/1412.6980 (2014).
Breitling, R., Armengaud, P., Amtmann, A. & Herzyk, P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett 573, 83–92 (2004).
Fu, X., Zhang, J., Meng, Z. & King, I. MAGNN: Metapath aggregated graph neural network for heterogeneous graph embedding. In Proc. The Web Conference 2020 (eds Huang, Y. et al.) 2331–2341 (ACM, 2020).
Pearce, D. J. An Improved Algorithm for Finding the Strongly Connected Components of a Directed Graph (Victoria Univ., 2005).
Shao, X. et al. Knowledge-graph-based cell–cell communication inference for spatially resolved transcriptomic data with SpaTalk. Nat. Commun. 13, 4429 (2022).
Almet, A. A., Tsai, Y.-C., Watanabe, M. & Nie, Q. Inferring pattern-driving intercellular flows from single-cell and spatial transcriptomics. Nat. Methods 21, 1806–1817 (2024).
Kabir, M. H., Patrick, R., Ho, J. W. & O’Connor, M. D. Identification of active signaling pathways by integrating gene expression and protein interaction data. BMC Syst. Biol. 12, 77–87 (2018).
Grünwald, B. T. et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 184, 5577–5592.e18 (2021).
Chan-Seng-Yue, M. et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 52, 231–240 (2020).
Boj, S. F. et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338 (2015).
Foley, J. W. et al. Gene expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. Genome Res 29, 1816–1825 (2019).
Zohora, F. T. & Schwartz, G. schwartzlab-methods/CellNEST: v1.0.0. Zenodo https://doi.org/10.5281/zenodo.15459529 (2025).
Li, J., Zohora, F. T. & Schwartz, G. schwartzlab-methods/CellNEST-interactive: v1.0.0. Zenodo https://doi.org/10.5281/zenodo.15459868 (2025).
Acknowledgements
We thank M. Peralta (Pathology Research Program Laboratory, UHN) for her work on the processing and mounting of pancreatic resections into Visium slides. We thank M. Monajemzadeh (McMaster University) and A. Elqaderi (Notta Laboratory, UHN) for their work annotating Visium slides as well as K. Ng and L. Gong for their work with Visium wet laboratory experiments (Notta Laboratory, UHN). This work was supported by the Canadian Cancer Society Challenge Grant (grant 707484; G.W.S.), the Natural Sciences and Engineering Research Council of Canada (grants RGPIN-2023-04713 and DGECR-2023-00395; G.W.S.), the Social Sciences and Humanities Research Council (grant NFRFE-2022-00681; G.W.S.), the Canada Research Chairs Program (G.W.S.), Canada Foundation for Innovation John R. Evans Leaders Fund (grant 42579; G.W.S.), the Princess Margaret Cancer Foundation (G.W.S.), the Gattuso-Slaight Personalized Cancer Medicine Fund & Research Stimulus Grant 2022 from the Princess Margaret Cancer Foundation (F.N.), Ontario Institute for Cancer Research (F.N.), the Ontario Early Researcher Award (grant ER19-15-205; F.N.), and the University of Toronto’s Eric and Wendy Schmidt AI in Science Postdoctoral Fellowship, a program of Schmidt Sciences.
Author information
Authors and Affiliations
Contributions
G.W.S. conceived and supervised the project. F.T.Z. developed the CellNEST method and software for intercellular communication detection. D.P. contributed to extending the CellNEST method for integrating intracellular signaling pathways and performed relay generating cell-type identification and validation. J.L. developed the CellNEST-Interactive software. F.T.Z. and D.P. ran and analyzed benchmarks. E.F.-F. generated experimental results. F.T.Z., E.F.-F., D.P. and T.G. ran and analyzed data. F.T.Z., D.P., E.F.-F., J.L., T.G. and G.W.S. wrote and edited the paper. All authors reviewed the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Rita Strack, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 CellNEST detects localized signaling in tumor and stromal environments in lung adenocarcinoma tissue assayed with Visium.
CellNEST detects localized signaling in tumor and stromal environments in lung adenocarcinoma tissue assayed with Visium (n = 4,095 spots)35. a, CellNEST-generated communication graph showing regions with strong CCC colored by component. Gray indicates regions with no or weak CCC. The top right red component has thinner arrow widths to accommodate very high communication frequency. The boxes outline three regions: cancer (orange), lymph (green), and stromal (blue) based on prior histological annotations[35]. b, Histogram displaying ligand-pair receptor abundance (y axis) from a, colored by connected component. The APOE–SDC1 and FN1–RPSA signals (black triangles, bold) are exclusively detected by CellNEST. CellNEST also detects many TGFB signals (blue text). c-e, Location of specific tumor and stroma signals found in b. c, Communication from a filtered for APOE–SDC1 signals. This is the most abundant signal and is mainly found in cancer-annotated regions. d,e, Gene expression of APOE (d) and SDC1 (e) on a, found mainly in cancer regions. f,g, Distribution of CellNEST-identified relay-network patterns. f, Histogram showing the abundance of each two-hop relay-network pattern with PSAP–LRP1 to APOE–LRP1 communication highlighted in red. g, The spatial location of the PSAP–LRP1 to APOE–LRP1 pattern from f on the tissue (red).
Extended Data Fig. 2 CellNEST detects localized signaling in tumor and stromal environments in colorectal cancer tissue assayed with Visium HD.
CellNEST detects localized signaling in tumor and stromal environments in colorectal cancer tissue assayed with Visium HD (n = 24,068 cells). a, H& E image of colorectal cancer tissue with adenoma and invasive cancer regions outlined in black. b, CellNEST-detected component graph, where each component is shown with a distinct color and represents a disjoint network of CCC. Component 32 (black boundary) aligns with the invasive cancer region in a. c, Histogram showing the abundance of each CellNEST-detected CCC on the colorectal cancer tissue from a, where each communication is mapped to a particular component in b with a matching color. APP-ITGA6 and APP-TGFBR2 are more frequently found in component 32 (black boxes). d,e, Distribution of relay-network patterns along with their location detected by CellNEST on the tissue in a. d, The most abundant signals detected by CellNEST, with the signal C3-CXCR4 to C3-LRP1 highlighted in red. e, C3-CXCR4 to C3-LRP1 signals on the tissue. This relay pattern is commonly found in the tumor microenvironment region60,61,62, recapitulated by CellNEST. The invasive cancer region from a is outlined in black.
Supplementary information
Supplementary Information
Supplementary Figs. 1–20, Notes 1–15 and Tables 1–6.
Supplementary Tables
Contents of Supplementary Tables 1–5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zohora, F.T., Paliwal, D., Flores-Figueroa, E. et al. CellNEST reveals cell–cell relay networks using attention mechanisms on spatial transcriptomics. Nat Methods 22, 1505–1519 (2025). https://doi.org/10.1038/s41592-025-02721-3
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41592-025-02721-3
