
Abstract
Hsp70 and Hsp90 chaperones and their regulatory cochaperones are critical for maintaining protein homeostasis. Glucose-regulated protein 94 (GRP94), the sole Hsp90 chaperone in the secretory pathway of mammalian cells, is essential for the maturation of important secretory and transmembrane proteins. Without the requirement of cochaperones, the Hsp70 protein BiP controls regulatory conformational changes of GRP94, the structural basis of which has remained elusive. Here we biochemically and structurally characterize the formation of a BiP–GRP94 chaperone complex and its transition to a conformation expected to support the loading of substrate proteins from BiP onto GRP94. BiP initially binds to the open GRP94 dimer through an interaction interface that is conserved among Hsp70 and Hsp90 paralogs. Subsequently, binding of a second BiP protein stabilizes a semiclosed GRP94 dimer, thereby advancing the chaperone cycle. Our findings highlight a fundamental mechanism of direct Hsp70–Hsp90 cooperation, independent of cochaperones.
Similar content being viewed by others


Main
Cellular protein homeostasis depends on the coordinated activity of molecular chaperones1. To this end, the Hsp70 and Hsp90 chaperone families are critical to maintain the integrity of the cellular proteome2,3,4. They are highly conserved across all domains of life, with eukaryotic cells having evolved compartment-specific Hsp70 and Hsp90 paralogs. About 30% of the proteome in eukaryotic cells is processed in the secretory pathway, where the highly abundant BiP and glucose-regulated protein 94 (GRP94) chaperones serve as the exclusive Hsp70–Hsp90 chaperone system5,6,7,8. BiP and GRP94 safeguard protein homeostasis in the endoplasmic reticulum (ER)9,10,11, in post-ER compartments12,13 and in the extracellular space14,15,16,17. BiP engages with exposed hydrophobic residues of newly synthesized or misfolded proteins and accordingly serves a broad substrate spectrum18. GRP94 is a more specialized chaperone, critical for the quality control of a subset of secretory and transmembrane proteins, including key growth factors and immune signaling proteins19. Previous work on Hsp70 and Hsp90 proteins from different organisms and different organelles established a role for Hsp70 BiP proteins in early substrate folding steps and assigned Hsp90 GRP94 proteins a downstream role in substrate maturation and activation20,21,22,23. A direct interaction between Hsp70 and Hsp90 proteins is critical for substrate protein handover24,25,26,27. Mutations impacting conserved Hsp70–Hsp90 interface residues lead to impaired substrate protein folding and growth defects in yeast24,26,28,29. To understand protein homeostasis in the secretory pathway, a detailed molecular understanding of the collaboration between GRP94 and BiP is critical.
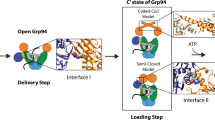
GRP94, like other Hsp90 proteins, consists of an N-terminal ATPase domain (NTD), a middle domain (MD) and a C-terminal dimerization domain (CTD), all connected by flexible linkers30 (Fig. 1a). Hsp90 proteins work as dimers, constitutively dimerized by their CTDs2. Additional dimerization of the NTDs is subject to regulation, where dynamic opening and closing through intermediate states drive substrate protein processing2. Crystal structures of GRP94 show the chaperone in two extreme conformations—an open twisted V31 and a fully closed conformation32 (Fig. 1a). Unique features of GRP94 within the Hsp90 family include its N-terminal ER-targeting sequence (1–21) and the pre-N domain (22–72), required for substrate protein maturation32 and important for the regulation of dimer closure and ATPase activity31,32, as well as an insertion into its ATP lid33. A truncated GRP94 protein, lacking the signal sequence and pre-N domain (GRP94 Δ72), shows increased ATPase activity compared to full-length (FL) GRP94, because of accelerated ATP-driven dimer closure32. Cytosolic Hsp90 proteins rely on cochaperones to promote the conformational cycle of opening and closing of the NTDs2. For GRP94, only two cochaperones have been described, with a role in conferring substrate selectivity34,35. Notably, previous work has demonstrated that BiP acts as a closure-accelerating partner chaperone of GRP94 (refs. 36,37). BiP is an Hsp70-type chaperone with an N-terminal nucleotide-binding domain (NBD) and a C-terminal substrate-binding domain (SBD), which is further subdivided into a β-sheet rich base (SBDβ) and an α-helical lid (SBDα)4,5 (Fig. 1a). An interdomain linker between BiPNBD and BiPSBD provides the basis for allosteric regulation. ATP binding and hydrolysis of the BiPNBD regulate the conformation of the BiPSBD, with the ATP-bound open and the ADP-bound closed conformations representing the most extreme states (Fig. 1a)38,39,40,41. The SBD-open state is characterized by a high on–off rate for substrate proteins and an interaction interface between BiPNBD and BiPSBD38,39,40,41. It is, therefore, also referred to as the domain-docked conformation. Conversely, the domain-undocked conformation is stabilized by substrate protein bound to the closed BiPSBD, which is entirely detached from the BiPNBD39. In its collaboration with GRP94, BiP serves two major roles that are closely connected; BiP delivers substrates to GRP94 and accelerates GRP94 dimer closure36,37,42. Together, these activities should facilitate substrate loading onto GRP94. A recent high-resolution cryo-electron microscopy (cryo-EM) structure provided important insights into substrate transfer between the cytosolic Hsp70–Hsp90 chaperone systems. In the so-called loading complex, resulting from interactions involving Hsp70, Hsp90, the cochaperone Hop and a substrate protein, Hsp90 adopts a semiclosed conformation, where the NTDs have dimerized but are not yet in an ATPase-competent state24. Importantly, Hop organizes the loading complex by bridging Hsp70 and Hsp90, stabilizing the semiclosed Hsp90 conformation and extending the substrate interaction interface of the complex24.

a, Crystal structures of the open GRP94 dimer (PDB 2O1V), the closed GRP94 dimer (PDB 5ULS) and domain-docked BiP (PDB 5E84) and a model of BiP based on the domain-undocked conformation of its homolog DnaK (PDB 2KHO). CL, charged linker; L, linker; H, His6 tag. b, Analytical SEC and SDS–PAGE analysis of complex formation between GRP94 FL and BiP. c, Same analysis as in b for GRP94 Δ72 and BiP. The fraction containing a BiP–GRP94 Δ72 complex (green box) was reapplied to the SEC column. d, Left: chemical crosslinking of GRP94 Δ72noPT and BiPNBD in the presence of ADP or AMP-PNP. Right: quantification of the three different species indicated on the gel (n = 3 independent replicates; mean + s.d.). e, Left: chemical crosslinking of GRP94 Δ72noPT and BiPNBD in the presence of PU-WS13. Right: quantification of the three different species indicated on the gel (n = 3 independent replicates; mean + s.d.). Protein bands on SDS–PAGE gels were visualized by Coomassie staining.
Despite the high conservation and functional similarities between Hsp70–Hsp90 systems across species and organelles, their regulation clearly differs2,19,43. Most prominently, bacteria and organellar compartments lack a bridging cochaperone such as Hop. How BiP is structurally integrated into the GRP94 conformational cycle, in the absence of bridging cochaperones, has remained elusive. Here, we describe two distinct states of the BiP–GRP94 complex. In the ‘preloading complex’, a direct interaction between BiP and the open GRP94 dimer is established through a highly conserved docking site. GRP94 and BiP subsequently move on to a loading complex conformation previously reported for the cytosolic Hsp70–Hsp90–Hop system24 even in the absence of any cochaperone organizing their interaction. Our biochemical and structural data provide insights into a conserved mechanism of chaperone complex formation and priming for substrate transfer from Hsp70 BiP to Hsp90 GRP94 machineries.
Results
A stable complex between BiP and GRP94
To biochemically and structurally characterize the formation of a BiP–GRP94 complex, we reconstituted the interaction between recombinant Strep-tagged GRP94 (residues 23–802) and His6-tagged BiP (residues 27–655) in vitro. Analytical size-exclusion chromatography (SEC) studies showed that BiP elutes in higher-molecular-weight fractions together with the FL GRP94 wild-type protein (Fig. 1b). We then addressed the interaction of BiP and the GRP94-D149N nucleotide-binding mutant, expected to preferentially populate an open dimer conformation, and the GRP94-E103A ATP-hydrolysis mutant, expected to reside mainly in a closed state32,44. Compared to GRP94 wild-type protein, we did not observe major differences in the coelution of BiP with GRP94-D149N or GRP94-E103A (Extended Data Fig. 1a). We also tested the interaction of BiP with GRP94 Δ72, which lacks the pre-N domain (Fig. 1a), and we observed a stable complex, illustrated by the fact that, upon reapplying the complex to the SEC column, BiP and GRP94 Δ72 still coeluted in a single peak (Fig. 1c). By chemical crosslinking, we could further stabilize this complex (Extended Data Fig. 1b) and test the effect of nucleotides on complex formation. These experiments were performed with GRP94 Δ72noPT, where the purification tag (PT) was removed by protease cleavage. In the presence of the nonhydrolyzable ATP analog, AMP-PNP, we saw a trend of reduced BiP–GRP94 complex formation (Extended Data Fig. 2a). In this setup, AMP-PNP affects the conformation of both chaperones, BiP and GRP94. BiP shifts to the SBD-open, domain-docked conformation and GRP94 shifts to the closed conformation (Fig. 1a). To specifically assess the effect of the GRP94 closed state on complex formation, we used a BiP truncation construct comprising only its NBD (BiPNBD). The BiPNBD, irrespective of its nucleotide state, retains GRP94 binding36,37. In analytical SEC experiments, BiPNBD did not coelute with GRP94 Δ72 (Extended Data Fig. 2b). However, in chemical crosslinking experiments, we could visualize a complex between the BiPNBD and GRP94 Δ72noPT. We presume that the bands for this complex are more distinct than with FL BiP because the latter oligomerizes in solution (Fig. 1d and Extended Data Fig. 2a). The addition of ADP did not affect BiPNBD–GRP94 Δ72noPT complex formation, whereas an excess of AMP-PNP, which shifts the GRP94 dimer to the closed conformation32, strongly reduced the interaction (Fig. 1d). The nucleotide dependence of the interaction was recapitulated for GRP94 FL and the GRP94 Δ72 construct (Extended Data Fig. 2c). We then also tested the effect of PU-WS13, a GRP94 ATPase inhibitor that is expected to prevent full closure of the GRP94NTD17,45. Using GRP94 FL or GRP94 Δ72noPT and FL BiP protein, we observed a potential positive effect of the GRP94 inhibitor on chaperone complex formation (Extended Data Fig. 2d). Because crosslinking with BiPNBD yields more distinct bands, we used this construct to assess the effect of PU-WS13, showing that PU-WS13 increases the fraction of GRP94 in complex with BiPNBD (Fig. 1e). Taken together, we characterized the interaction between BiP and GRP94, which is reduced by AMP-PNP and favored by PU-WS13. These data suggest that the complete closure of the GRP94 dimer counteracts binding to BiP.
Assessing the topology of a BiP–GRP94 complex by crosslinking mass spectrometry
To gain initial structural insight into a BiP–GRP94 complex, we performed crosslinking mass spectrometry (XL-MS) experiments. We used disuccinimidyldibutyric urea (DSBU), a homobifunctional crosslinker reactive to primary amines with a spacer length of 12.5 Å. DSBU primarily attaches to lysines but can also react with serines, threonines or tyrosines. We reconstituted complexes between BiP and GRP94 FL or GRP94 Δ72. Upon crosslinking with DSBU, SDS–PAGE gel bands containing high-molecular-weight protein complexes were analyzed by MS (Fig. 2a and Supplementary Tables 1 and 2). In both datasets, the crosslinks between BiPNBD and GRP94 suggest that the two chaperones adopt a similar conformation to their cytosolic paralogs, Hsp70 and Hsp90, in the previously described loading complex24. Figure 2b shows a homology model of BiP and GRP94 based on the cytosolic loading complex (Protein Data Bank (PDB) 7KW7), with BiP in the domain-undocked conformation and GRP94 in a semiclosed state. The majority of BiPNBD–GRP94 crosslinks map to two interaction interfaces (I and II) (Fig. 2bc, Extended Data Fig. 3a and Supplementary Tables 1 and 2), matching interactions described for the cytosolic loading complex. The crosslinks outlining interface I link the GRP94MD helix 455–476 to BiPNBD lobe IIA. The crosslinks that indicate the formation of interface II link the central GRP94NTD helix (residues K114 and K119) and the ATP lid (residues K161 and K168) to the BiPNBD lobe I. Additional crosslinks between BiP residue K213 and GRP94 residues K161 and K168 suggest that the GRP94 ATP lid may be slightly closer to the BiPNBD than shown in the homology model (Fig. 3c and Extended Data Fig. 3a, coral crosslinks). Crosslinks outlining interfaces I and II were also prevalent in XL-MS datasets of BiP and BiPNBD in complex with GRP94 Δ72noPT in the presence of PU-WS13 (Extended Data Fig. 3b and Supplementary Tables 3 and 4).

a, Map of crosslinks identified in the BiP–GRP94 FL (left) and BiP–GRP94 Δ72 (right) samples. b, Homology model of BiP–GRP94 on the basis of the cytosolic loading complex (PDB 7KW7). The ATP lid is indicated in red. Residues indicated as the predicted GRP94 substrate-binding region (dark green) include the amphipathic helix N657–Q668 as identified in the cytosolic Hsp90 paralog and additional residues F398–Y401, K428, I497, E498 and Y575 as identified in Huck et al.32. c, Crosslinks outlining interface I, interface II and the ATP lid–BiPNBD interactions from the BiP–GRP94 FL XL-MS dataset mapped onto the homology model shown in b. BiPSBD is omitted for clarity. d, Crosslinks from the BiPSBD to GRP94 mapped onto the homology model shown in b. BiPNBD is omitted for clarity. e, Summary of XL-MS results. S, Strep tag. Crosslinks to GRP94 protomer A are shown, which represent the closest distances for all residues shown in c,d except for K733.

a, Cartoon model of HT2 misfolding upon covalent conjugation to HyT36. b, Limited proteolysis with trypsin to assess the folding state of HT2 conjugated with HyT36, incubated with the HyT36(-Cl) control or DMSO. c, CD spectroscopy of HT2 conjugated to HyT36 or 6-chlorohexanol. Kinetic measurements at the indicated temperatures are shown. d, Pulldown experiments to study the interaction of His6–BiP with HT2misfolded–HyT36 compared to DMSO control. For quantification, data were normalized to the wild-type BiP elution sample (n = 3 independent replicates; mean ± s.d.). e, Same analysis as in d for GRP94 FL and GRP94 Δ72. f, Sequential pulldown of Strep–GRP94 Δ72 and His6–BiP to isolate a GRP94 Δ72–BiP–HT2misfolded complex. The asterisks in d–f indicate BSA in the interaction buffer. Protein bands on SDS–PAGE gels were visualized by Coomassie staining.
BiP binding to GRP94 is compatible with the open and semiclosed GRP94 conformations, allowing for interactions within interface I or interfaces I and II, respectively (Extended Data Fig. 3c). In the GRP94 closed conformation, the ATP lid would clash with residues of BiPNBD lobe IA, preventing an efficient interaction (Extended Data Fig. 3c). This effect is consistent with our biochemical data showing that the AMP-PNP-bound and, thus, closed GRP94 dimer shows a reduced interaction with the BiPNBD (Fig. 1d). To characterize the conformation of GRP94, we analyzed intra-GRP94 crosslinks. We measured distances between crosslinked residues within one GRP94 protomer and across the GRP94 dimer in the open and the semiclosed GRP94 conformations (Supplementary Tables 1–4). The GRP94MD and GRP94CTD residues exhibit minimal conformational changes between the open and semiclosed states (root-mean-square deviation (r.m.s.d.) of 2.7 Å). Crosslinks within these regions agree with published structures, validating the quality of the crosslinking data (Supplementary Tables 1–4). As these crosslinks cannot distinguish between different GRP94 conformations, we focused on crosslinks between the GRP94NTD and GRP94MD. In the open GRP94 conformation, the central GRP94NTD helix (residue K114) and the GRP94NTD ATP lid (residues K161 and K168) are in close proximity to the GRP94MD helix 455–476, compatible with crosslinks within a single protomer and across the GRP94 dimer between these GRP94NTD and GRP94MD regions (Extended Data Fig. 4a, purple crosslinks). Upon rotation of the GRP94NTD to the semiclosed conformation, the central GRP94NTD helix and the ATP lid move away from the GRP94MD. At the same time, helix H1 is repositioned to participate in the GRP94NTD dimer interface. This repositioning now supports a crosslink between K95 residues across the GRP94 dimer. Additionally, K95 and K97 link to residues of the GRP94MD helix 455–476, within a single protomer and across protomers (Extended Data Fig. 4a, orange crosslinks). Crosslinks supporting either of the two GRP94 conformations were found in all XL-MS datasets (Supplementary Tables 1–4), suggesting that our samples contain open (twisted V) and semiclosed GRP94 dimers.
Crosslinks within the BiPSBD confirm that, when in complex with GRP94, it resides in the closed conformation (Extended Data Fig. 4b and Supplementary Tables 1, 2 and 4), which is typically the substrate-bound state. The closed BiPSBD is fully dissociated from the BiPNBD and free to move around the interdomain linker39,46. Notably, the position of the BiPSBD with respect to GRP94 differs between GRP94 FL and GRP94 Δ72. The majority of BiPSBD–GRP94 Δ72 crosslinks place the BiPSBD in the proximity of the GRP94NTD and suggest an interaction with the N-terminal GRP94 Δ72 PT (Fig. 2a and Supplementary Table 2). The BiP–GRP94 FL XL-MS dataset positions the BiPSBD in close proximity of the GRP94MD and GRP94CTD (Fig. 2d and Supplementary Table 1). Notably, on the basis of previous work, the lumen of the GRP94MD and GRP94CTD dimer contains the predicted substrate-binding region (Fig. 2b)24,32,47. Moreover, this BiPSBD position matches the conformation of the Hsp70–SBDβ resolved in the cytosolic loading complex24, which contained a substrate protein (Supplementary Table 1). Taken together, our data suggest that the BiPSBD extends toward the predicted substrate-binding region of GRP94, even in the absence of substrate (Fig. 2e).
Hydrophobic tagging to form chaperone–substrate complexes
To better understand how GRP94 and a BiP–GRP94 complex interact with substrate proteins, we established interaction studies with a model substrate. The HaloTag 2 (HT2) domain, a self-labeling protein tag48, can covalently conjugate to a hydrophobic tag, which induces HT2 misfolding in cells—a process called hydrophobic tagging (Fig. 3a)49,50,51,52. In mammalian cells, BiP and GRP94 interact with misfolded HT2 (HT2misfolded)-based model substrates targeted to the Golgi apparatus and the ER53,54 (Extended Data Fig. 5).
We characterized the HyT36-induced misfolding process of HT2 in vitro by limited proteolysis using trypsin. Upon incubation of HT2 with HyT36, the protein is more susceptible to tryptic digest, indicating a misfolded state (Fig. 3b). HT2 misfolding is dependent on the covalent conjugation to HyT36 because, in the presence of a control compound, HyT36(-Cl), the protein remains stable (Fig. 3b). In circular dichroism (CD) spectroscopy experiments, we analyzed the kinetics of misfolding (Fig. 3c). The α-helical content of HT2 (measured by an increase in signal at 222 nm) reached its minimum at ~30-min incubation at 37 °C (as opposed to 25 °C), revealing experimental conditions that can be applied in chaperone–substrate interaction studies. Conjugation of HT2 to 6-chlorohexanol, an HT ligand without any functional group, did not affect the stability of the HT2 protein at 25 °C or 37 °C (Fig. 3c).
The interaction of Hsp70-type chaperones with misfolded substrate proteins is well characterized55,56,57. For in vitro experiments, substrate proteins are often destabilized with nonspecific chemical denaturing agents. Upon dilution of the denaturing agent, chaperones can then be added to study chaperone–substrate interactions58,59. With the in vitro hydrophobic tagging system, HT2 misfolding can be induced directly in the presence of the Hsp70 BiP, providing access to HT2misfolded states as they arise. We conjugated HyT36 to HT2 by simple mixing of the two components, followed by the addition of BiP and ATP and finally incubation at 37 °C to induce HT2 misfolding. Under these conditions, HT2misfolded is directly bound by BiP, as observed in pulldown assays (DMSO versus HyT36; Fig. 3d). Deletion of the BiP SBD entirely abolished the BiP–HT2misfolded interaction (Fig. 3d, BiPNBD). A previously described substrate-binding mutant, BiP-V461F (refs. 60,61,62), showed reduced interaction with HT2misfolded. We also tested BiP-I437V, a substitution described to stabilize the closed (domain-undocked) conformation39, and BiP-E243K, a substitution at the BiPNBD–BiPSBD interface, described to have reduced interaction with GRP94 (ref. 37). While BiP-I437V did not consistently affect the binding of the substrate, BiP-E243K showed a trend toward slightly reduced substrate binding (Fig. 3d). We then performed similar experiments with GRP94 FL and GRP94 Δ72, which also bound to HT2 upon misfolding (Fig. 3e). Lastly, we performed a sequential pulldown experiment. By first capturing Strep-tagged GRP94 Δ72 and then His6-tagged BiP, we could isolate a GRP94 Δ72–BiP–HT2misfolded complex (Fig. 3f). In XL-MS experiments including the HT2-based substrate, we unfortunately did not detect crosslinks to HT2, preventing a detailed positioning of the substrate protein within the chaperone complex.
BiP binds to the open GRP94 dimer in the preloading complex
To study the topology and conformations of a BiP–GRP94 Δ72 complex, we performed single-particle negative-stain EM. Even though we could not detect any crosslinks to HT2, we hypothesized that the misfolded substrate may stabilize or support the formation of the BiP–GRP94 chaperone complex; therefore, we included HT2misfolded in our sample preparation. To establish a BiP–GRP94 Δ72–HT2misfolded complex, we first incubated BiP with HT2 in the presence of HyT36 and ATP and then added GRP94 Δ72. To enrich the multichaperone–substrate complex, we performed gradient fixation (GraFix)63, which resulted in the formation of a GRP94 dimer (complex 0) and two complexes composed of a GRP94 dimer, HT2 and one or two BiP molecules, respectively (complexes I and II) (Extended Data Fig. 6a). GraFix fraction 12 contained the largest amounts of complex I and II with little amount of free GRP94 dimer and was, therefore, used for negative-stain EM (Extended Data Fig. 6a,b). As expected, reference-free two-dimensional (2D) classifications and ab initio three-dimensional (3D) reconstructions indicated sample heterogeneity (Extended Data Figs. 6c and 7). Nevertheless, the ab initio 3D reconstructions in comparison to known GRP94 structures showed two predominant complexes corresponding to a GRP94 dimer with one (complex I) and two (complex II) BiP proteins bound (Extended Data Fig. 7).
In the complex I reconstruction, we identified a density corresponding to a GRP94 dimer with a gap between the NTDs (Fig. 4a). The additional density attached to GRP94 protomer A resembles a BiPNBD domain with a characteristic globular shape outlining lobes I and II and an indentation corresponding to the nucleotide-binding cleft (Fig. 4a). Accordingly, we chose an open twisted V GRP94 Δ72 structure (PDB 2O1V)31 and the isolated BiPNBD (PDB 6DWS) as the starting structures for molecular modeling. After an initial rigid-body fit, we performed a map-fitting refinement of the complex with Rosetta64,65. Both the total_score and the elect_dens_fast values of the obtained models negatively correlated with the real-space correlation coefficient (RSCC) values (Extended Data Fig. 8a), probably because the models could fit most of the density (Fig. 4b). We presume that the BiPSBD is rather flexible and, therefore, largely not visible in the EM map. A density close to the GRP94NTD protomer B, not accounted for in our model, may correspond to the BiP interdomain linker and/or parts of the BiPSBD (Fig. 4ab). We refer to this state of the BiP–GRP94 complex as the preloading complex, a conformation that exists before dimerization of the GRP94NTD (that is, before the full engagement of GRP94 with substrate protein). As expected, for a BiP–GRP94 complex with GRP94 in the open twisted V conformation, the preloading complex model is supported by our crosslinking data outlining interface I, whereas interface II has not formed yet (Fig. 2b and Extended Data Fig. 8b). The preloading complex depicts the initial engagement of BiP with the open GRP94 dimer, the substrate-loading competent form.

a,b, Negative-stain EM reconstruction (a) and molecular model (b) of the BiP–GRP94 preloading complex. c,d, Negative-stain EM reconstruction (c) and molecular model (d) of the BiP–GRP94 loading complex. The dotted blue circles indicate the potential position of the BiPSBD. e, Crosslinks identified between BiPSBD and GRP94 Δ72 mapped onto the loading complex structure. Green lines indicate crosslinks < 35 Å; black lines indicate crosslinks > 35 Å. The arrow indicates the N terminus of GRP94 that is the attachment point of the PT. f, Left: Ni-NTA pulldown analysis of His6–BiP with GRP94 Δ72, GRP94 FL and GRP94 Δ72noPT (exact sequences of the N termini in Extended Data Fig. 9a). The asterisk indicates BSA in the interaction buffer. Right: quantification of the pulldown experiment (n = 3 independent replicates; mean ± s.d.). Protein bands on SDS–PAGE gels were visualized by Coomassie staining.
BiP bound to a semiclosed GRP94 conformation
In addition to the preloading complex, we obtained a reconstruction that indicated the binding of two BiP molecules to a GRP94 dimer (complex II) and largely resembled the cytosolic loading complex reported by Wang et al.24. Our ab initio reconstruction shows that the GRP94NTD established a contact at the center of the dimer, indicating a semiclosed conformation (Fig. 4c). Furthermore, the NBDs of the two outlined BiP proteins established interactions with the GRP94MD (interface I) and GRP94NTD (interface II). To initiate molecular modeling, we obtained a homology model of GRP94 based on the loading complex (PDB 7KW7)24. For BiP1, we used the isolated NBD (PDB 6DWS) and, for BiP2, we included the BiPSBD β-subdomain using a homology model of a domain-undocked conformation based on the structure of its Escherichia coli homolog DnaK (PDB 2KHO)66. The individual models were placed into the EM reconstruction and structural refinement allowed to fit the extended conformation of BiP2NBD–SBD into the density (Fig. 4d). We refer to this state of the BiP–GRP94 complex as the loading complex, given the similarity to the previously reported Hsp70–Hsp90 structure (Extended Data Fig. 8c)24. The fitting scores and the RSCC for the loading complex as compared to the preloading complex lacked a clear correlation (Extended Data Fig. 8a). This could be explained by the larger size and complexity of the loading complex model and by the fact that, at a lower contour level, the EM reconstruction showed a density connected to BiP1NBD that is not accommodated by the final model (Extended Data Fig. 8a). We did not observe any additional densities that could be assigned to the HT2misfolded, which may be because of its flexibility and size or resolution limits of negative-stain EM.
In both EM reconstructions, the BiPSBD appears to extend toward the NTD of the opposing GRP94 protomer (Fig. 4a–d). In the preloading complex, residual density in combination with the BiPNBD C terminus indicates the position of the BiPSBD (Fig. 4b). In the loading complex, the defined BiP2SBD extends toward the GRP94NTD protomer A and an additional interaction between the BiP interdomain linker and the GRP94NTD of protomer B is formed (Fig. 4d,e). This position of the BiP2SBD with respect to GRP94 and its PT is in agreement with our crosslinking data (Fig. 4e). Upon close inspection of the PT sequence, we identified a hydrophobic stretch, which may serve as a BiP substrate mimetic (Extended Data Fig. 9a). To address the contribution of the GRP94 PT to the interaction, we removed it by protease cleavage and performed comparative pulldown analyses (Fig. 4f). Removal of the tag reduced GRP94 binding to BiP by ~65%, suggesting that the tag contributes to the stability of the chaperone complex. In line with this, we also observed ~75% less GRP94 FL protein pulled down with BiP compared to GRP94 Δ72 (Fig. 4f). The FL protein, while carrying a Strep tag, does not contain the predicted BiP substrate sequence (Extended Data Fig. 9a).
The binding of the BiPSBD to the GRP94 Δ72 PT contributes an additional interaction interface and we presume that it prohibits transition to the fully closed GRP94 conformation, which is expected to induce complex disassembly (Fig. 1d). Taken together, stabilization of the BiP–GRP94 Δ72 complex by this inadvertently engineered interaction allowed us to capture two chaperone complex transition states.
BiPNBD–GRP94 interactions in preloading and loading complex
In the two obtained EM reconstructions, the preferred position of the BiPSBD is determined by the GRP94 Δ72 PT. Given that, in the domain-undocked state, the BiPNBD acts independently of the BiPSBD, we further characterized BiPNBD–GRP94 interactions (Fig. 5a). In the preloading complex, the BiPNBD and GRP94 showed one major contact point with an interface area (GRP94MD:BiPNBD) of 1,072.5 Å2 (Fig. 5b). According to our model, the long GRP94MD helix (residues 455–476) packs against BiPNBD lobe IIA, thereby forming electrostatic interactions involving BiP residues D238, N239 and E243 and GRP94 residues K462, K463, R466 and K467 (Fig. 5b). Additionally, the tip of a GRP94MD β-sheet inserts between BiPNBD lobes IA and IIA. This interaction matches interface I in the previously described cytosolic loading complex24 (Fig. 5b). Mutations impacting interface I impair the interaction and functional cooperation between Hsp70 and Hsp90 chaperones25,26,28,37. Consistently, BiP-E243K (E218 in cytosolic Hsp70) and GRP94-K463E or GRP94-K467E (K414 and K419 in cytosolic Hsp90) strongly reduced BiP–GRP94 Δ72 complex formation in analytical SEC studies and pulldown experiments (Fig. 5c,d). For all three mutant proteins, an ~85% reduction in the amount of GRP94 Δ72 copurified with BiP was observed in pulldown experiments (Fig. 5c).

a, Overview of interfaces I and II in the preloading and loading complex conformations. b, Zoomed-in view of interface I of the BiP–GRP94 preloading complex. Interface I of the cytosolic (Hsp70–Hsp90) loading complex is shown for comparison. The arrow points to the GRP94MD β-sheet loop inserted between BiPNBD lobes IA and IIA. Residues in bold were used for mutational analysis. c, Analytical SEC and SDS–PAGE analysis of complex formation between BiP and GRP94 Δ72 interface I mutants. d, Ni-NTA pulldown analysis of His6-tagged BiP and Strep-tagged GRP94 Δ72 interface I mutants and quantification of the pulldown experiment (n = 3 independent replicates; mean ± s.d.). The asterisk indicates BSA in the interaction buffer. e, Zoomed-in view of interface II of the BiP–GRP94 loading complex. Interface II of the cytosolic loading complex is shown for comparison. f, Ni-NTA pulldown analysis of His6-tagged BiP and Strep-tagged GRP94 Δ72-R116G interface II mutant and quantification of the pulldown experiment (n = 3 independent replicates; mean ± sd). The asterisk indicates BSA in the interaction buffer. Protein bands on SDS–PAGE gels were visualized by Coomassie staining.
Our model of complex II matches the overall topology of the cytosolic loading complex, with slightly modified interaction interfaces between the BiP and GRP94 proteins (Extended Data Fig. 8c and 9b). At the heart of the complex is the semiclosed GRP94, supported by the rotation of the GRP94NTD compared to the preloading complex and by the binding of two BiP molecules (Supplementary Video 1). The resulting interface II from the GRP94NTD–BiP1 interaction indicates conserved hydrophobic contacts with the C-terminal part of the GRP94NTD helix (residues 117–121) and polar contacts with R116 (Fig. 5e). To evaluate the importance of the conserved interface II, we analyzed the GRP94-R116G mutant (R60G in cytosolic Hsp90 and R46G in yeast Hsp90), previously shown to decrease the interaction between cytosolic Hsp70 and Hsp90 proteins and to reduce the fitness of yeast cells24. Pulldown experiments showed an ~30% decrease in the ability of this mutant to bind BiP (Fig. 5f), corroborating the importance of the conserved interface II for the stability of a BiP–GRP94 Δ72 complex. The comparatively small reduction in the bound GRP94 Δ72 mutant protein can be attributed to the fact that interface I is critical for preloading and loading complex conformations, while the role of interface II is limited to stabilization of the loading complex (Fig. 4b). Overall, our data suggest that, through interactions along interface I, BiP establishes the initial contact with GRP94, which is still in an open conformation. Single substitutions at interface I strongly impair the interaction between BiP and GRP94. The binding of a second BiP molecule through interface I and the subsequent formation of interface II stabilize the GRP94 semiclosed state.
Discussion
GRP94 is a structurally dynamic molecular chaperone critical for the folding, maturation and degradation of important secretory and transmembrane proteins19. In the apo or ADP-bound state, the GRP94 dimer resides in an open conformation31,32,36. To tightly interact with its substrate proteins, the dimer closes32. Cytosolic Hsp90 proteins rely on cochaperones for controlled ATP-driven transitioning from the open to the closed state2,30. In the secretory pathway, the Hsp70 protein BiP regulates GRP94 function36,37,42. Extensive characterization of Hsp90 proteins shows that closure intermediate states have a critical role in the chaperone cycle24,67,68,69 and that BiP is critical for the stabilization of such GRP94 states36.
Building on this previous work, our structural and biochemical data provide insight into the BiP–GRP94 preloading and loading complex conformations (Fig. 6). These transition states likely became visible by an interaction of BiP with the PT of GRP94 Δ72 acting as a BiP substrate mimetic and preventing the full closure of the GRP94NTD. In the context of the stabilized complex, we could characterize interactions between BiPNBD and GRP94. On the basis of our findings, we propose a model according to which the stepwise binding of BiP controls GRP94 dimer closure. Initially, one BiP molecule binds to the open twisted V conformation of GRP94 by establishing interactions at interface I. The twisted V conformation indicates the GRP94 apo or ADP-bound state. Furthermore, BiP needs to reside in the ADP-bound, extended conformation to interact with GRP94 (ref. 37), which is consistent with our XL-MS data. The residues within the BiPNBD and GRP94MD critical to this interaction site are highly conserved from bacteria to the different Hsp70 and Hsp90 chaperone systems in mammalian cells24,25,26,28,37, indicating that the formation of the preloading complex may also be conserved. Following this initial contact, additional interactions between BiP and GRP94 are formed along interface II, favoring the loading complex conformation. Importantly, in the loading complex, a second BiP molecule is bound to GRP94, increasing the interface area of the complex. We propose that the simultaneous engagement of both GRP94 protomers in interface II promotes GRP94NTD rotation (Fig. 6). Upon rotation, the GRP94NTD is poised to dimerize and assume the semiclosed conformation. The interactions along interface II thereby define the semiclosed GRP94 state and may underlie the GRP94 closure-accelerating activity of BiP36,37. We presume that, under physiological conditions, GRP94 is bound to ATP in the loading complex to enable the transition to the maturation complex. Interestingly, our data suggest that binding of the GRP94 ATPase inhibitor PU-WS13 (ref. 17) affects the BiP–GRP94 complex. According to the crystal structure of GRP94NTD in complex with the closely related PU-H36 (PDB 8TF0), inhibitor binding rearranges the GRP94NTD ATP lid (residues 161–197) and the GRP94 N-terminal residues (including helix H1) compared to the apo state but does not support full closure of the GRP94NTD17,45. We hypothesize that PU-WS13 stabilizes a GRP94 semiclosed state, which promotes the interaction with BiP.

In the preloading complex, one BiP molecule is bound to an open GRP94 dimer, which is in the apo or ADP-bound state. The binding of a second BiP molecule supports transitioning into the loading complex, where GRP94 is in a semiclosed conformation. ATP binding to GRP94 may support the partial closure of the ATP lid and GRP94NTD dimerization under physiological conditions. A similar effect may be produced by PU-WS13. Both BiP molecules bound to GRP94 reside in the ADP-bound, domain-undocked conformation. Finally, the GRP94 dimer transitions into the fully closed state and BiP concurrently dissociates. We presume that ATP binding to BiP drives this process. The relative position of the GRP94NTD semiclosed dimer interface is indicated in purple.
The cytosolic loading complex is stabilized by the cochaperone Hop through direct contacts with all complex components—Hsp70, Hsp90 and the substrate protein. The semiclosed Hsp90 conformation is defined by direct Hop–Hsp90 interactions and by the binding of two Hsp70 proteins24. Even in the absence of a bridging cochaperone such as Hop, two BiP proteins are important for stabilizing the semiclosed GRP94 conformation. The additional role of Hop in substrate binding may not be required for secretory and transmembrane protein folding or may be substituted by still uncharacterized cochaperones. Given the similarities between the BiP–GRP94 and cytosolic Hsp70–Hsp90 complexes and the conservation of interface residues, we believe that the stepwise BiP (Hsp70) binding model to drive GRP94 (Hsp90) dimer closure (Fig. 6) may also apply to more sophisticated systems, where cochaperones have evolved to support and fine-tune this principal mechanism of Hsp70–Hsp90 cooperation. Consistent with previous work in the Hsp90 field24, our data show that the ATP-bound, fully closed GRP94 does not interact with BiP (Fig. 1d). Closing of the GRP94NTD ATP lid breaks contacts at interface II and subsequent swapping of the GRP94NTD and ATP binding to BiP leaves substrate-bound GRP94 and BiP in the open, domain-docked conformation primed for another round of substrate recruitment (Fig. 6). Agard and colleagues also proposed a stepwise release of Hsp70 proteins during the transition from the loading complex to the so-called maturation complex, which may equally apply to the simpler BiP–GRP94 system24,70 (Fig. 6).
The coordinated interactions between BiP and GRP94 are aimed at transferring substrates from BiPSBD to GRP94. We applied the chemical biology tool hydrophobic tagging to translate chaperone–substrate interactions previously identified in cells53,54 (Extended Data Fig. 5) into an in vitro reconstituted system. This shows that hydrophobic tagging provides an efficient approach for in vitro reconstitution of chaperone–substrate complexes, as demonstrated by our usage for two different types of chaperones, BiP and GRP94 (Fig. 3). Inducing HT2 misfolding in the presence of chaperones may provide an advantage in establishing chaperone–substrate complexes over an experimental setup where protein misfolding and chaperone-binding reactions are separated. BiP and GRP94 individually interacted with HT2misfolded but could also form a multichaperone–substrate complex. One limitation of the HT2-based system is its limited applicability in crosslinking experiments with DSBU and glutaraldehyde. Unfortunately, HT2 could not be visualized in our XL-MS or GraFix EM experiments, which may be because of its low lysine content (seven lysines, 2.3% of its sequence). For comparison, lysine residues make up 9% (58 residues) and 9.7% (77 residues) of the sequences of BiP and GRP94, respectively. Furthermore, HT2 may be shielded by the substrate-binding sites of BiP and GRP94, preventing the formation of crosslinks flanking the substrate-binding regions. The use of HT2 fusion proteins may be an avenue in overcoming this limitation in future research. While we could not visualize a substrate-bound chaperone complex by XL-MS or EM, we expect, by extrapolating from the cytosolic loading complex, a state where the substrate protein is engaged by BiP and GRP94 simultaneously. We presume that interactions between the substrate protein and GRP94 drive the formation of the loading complex, in addition to interactions between GRP94NTD and BiPNBD, further facilitating substrate transfer. A substrate protein would bridge the two chaperones, providing additional interaction interfaces, and may support the transition to the GRP94 semiclosed state. The two orientations of BiPSBD with respect to BiPNBD captured in our samples underline its flexibility in the domain-undocked state. In-solution studies have previously highlighted that the crystal structures of the domain-docked and domain-undocked BiP conformations (Fig. 1a) represent endpoint states39,40. This flexibility of the BiPSBD can facilitate the delivery of a highly diverse range of substrate proteins to GRP94. In the context of the GRP94 FL protein, BiPSBD is in close proximity to GRP94MD and GRP94CTD, suggesting that closed BiPSBD may have a preference for the orientation toward the predicted GRP94 substrate-binding region.
Our negative-stain EM and XL-MS data highlight striking similarities between the comparatively simple BiP–GRP94 chaperone system and their intricate Hsp70–Hsp90 paralogs and point to a highly conserved mode of substrate transfer among Hsp70 and Hsp90 systems. Accordingly, Hsp70 (BiP) proteins have a conserved role, not only in the delivery of substrate proteins but also in the activation of Hsp90 (GRP94) proteins. On the basis of the identified BiP–GRP94 preloading and loading complexes, it will be interesting to define ER-specific regulatory mechanisms. Post-translational modifications may influence transitioning between the different states of the GRP94 chaperone cycle. Regulatory modifications may affect the stability and lifetime of the different BiP–GRP94 complex states and could also change the role of the chaperones from collaborating in protein folding to supporting protein degradation.
Methods
Reagents and antibodies
HyT36 and HyT36(-Cl) were kind gifts from C. Crews and G. Burslem50,51. 6-Chlorohexanol (Sigma-Aldrich, C45008), trypsin (Sigma-Aldrich, T8003), DSBU (Thermo Fisher, A35459), glutaraldehyde (Thermo Fisher), ATP (Sigma-Aldrich, A6419), AMP-PNP (Sigma-Aldrich, A2647), ADP (ARCOS Organics, 164670010), PU-WS13 (MedChemExpress, HY-18680), HALTS1 (N-(3,5-dichloro-2-ethoxybenzyl)-2H-tetrazol-5-amine)71 (ChemBridge Corporation, 9074451), cycloheximide (Sigma-Aldrich, 01810), doxycycline hyclate (Sigma-Aldrich, DD9891), anti-His5 (Santa Cruz Biotechnologies, SC-8036), anti-Strep (Qiagen, 34850), anti-BiP C50B12 (Cell Signaling, 3177), anti-GRP94 (Proteintech, 14700-1-AP), anti-HA tag C29F4 (Cell Signaling 3724), anti-HT (Promega, G9211), goat anti-mouse HRP-coupled secondary antibody (Invitrogen, 31444) and goat anti-rabbit HRP-coupled secondary antibody (Invitrogen, 31460) were from commercial suppliers.
Plasmids and molecular cloning
The expression plasmids His6–linker–TEV site–GRP94 (73–802) in pET151 and His6–BiP (27–655) in pNIC28-Bsa4 were a kind gift from T. Street37. GRP94 was recloned into pET21d to yield an expression plasmid encoding Strep–linker–TEV site–GRP94 (73–802). Details on the N-terminal PT can be found in Extended Data Fig. 9a. Strep–GRP94 FL was cloned from C2C12 complementary DNA into pET21d by Gibson assembly (New England Biolabs Hifi Builder). His6–BiPNBD (27–411) was cloned from His6–BiP in pNIC28-Bsa4 by Gibson assembly into the same vector. pcDNA5/FRT/TO-EGFP-HT2 (ref. 53) was used as a template to clone HT2–His6 in pET21a. Spot–HT2 was cloned from His6–HT2 into pET22b-SUMO. All point mutations were introduced by site-directed mutagenesis. All primers are listed in Supplementary Table 5.
Protein expression and purification
All proteins were expressed in Escherichia coli BL21(DE3) cells. Protein expression was induced with 250 μM IPTG for 16 h at 18 °C for GRP94 and BiP proteins and for 5 h at 25 °C for His6–SUMO–HT2 and HT2–His6. Cells were harvested by centrifugation, resuspended in 50 mM Na2HPO4 pH 8.0 and 300 mM NaCl for His6–SUMO–HT2 and HT2–His6, 100 mM HEPES pH 8.0, 400 mM NaCl, 10 mM imidazole and 0.5 mM tris(carboxyethyl)phosphine (TCEP) for His6–BiP proteins and 100 mM HEPES pH 8.0, 400 mM NaCl and 0.5 mM TCEP for Strep-tagged GRP94 proteins and lysed by sonication.
For His6-tagged proteins, cleared cell lysate was applied to a HisTrap HP column (Cytiva). The column was washed by applying a stepwise imidazole gradient and the His6-tagged protein was eluted in 150 mM imidazole in lysis buffer. His6–SUMO–Spot–HT2 was digested with His6–Ulp1 and subjected to another round of HisTrap affinity chromatography to isolate Spot–HT2. For Strep-tagged proteins, cleared cell lysate was applied to a StrepTrap HP column (Cytiva) and eluted with 2.5 mM d-desthiobiotin in lysis buffer. In the case where the PT was removed by protease digest with His6–GST-TEV-S219V, GRP94 Δ72 was exchanged into 100 mM HEPES pH 8.0, 150 mM NaCl, and 2 mM DTT. As a final step, all proteins were subjected to SEC using a Superdex 200 16/600 column equilibrated with 50 mM HEPES pH 7.5 and 150 mM NaCl for the HT2 proteins, supplemented with 0.5 mM TCEP for BiP and GRP94 proteins. Fractions containing the protein of interest were concentrated, flash-frozen in liquid N2 and stored at −70 °C.
Hydrophobic tagging
Spot–HT2 or HT2–His6 was incubated with HyT36, HyT36(-Cl) or DMSO for 10 min at room temperature. To induce misfolding, samples were incubated at 37 °C.
Limited proteolysis
First, 10 µM His6–HT2 reacted with 12 µM HyT36 or in the presence of 12 µM HyT36(-Cl) or DMSO was incubated with 0.1 µM trypsin (Sigma-Aldrich) for 15, 30, 60 and 90 min at 37 °C. Reactions were stopped by the addition of SDS–PAGE sample buffer and resolved on SDS–PAGE gels.
CD spectroscopy
CD spectra of 10 µM Spot–HT2 reacted with 10 µM HyT36 or 10 µM 6-chlorohexanol or in the presence of DMSO were recorded in 20 mM Tris pH 7.5 and 100 mM NaCl at 25 °C and 37 °C with a Jasco Spectropolarimeter J-710 connected to the temperature control unit Jasco PFD-350s with a Julabo F-250 compact recirculation cooler at 0.2-nm steps with ten scans. Data were analyzed in GraphPad Prism. Given the presence of DMSO in the samples, spectra were analyzed from 218 to 260 nm.
Sample preparation for EM
To prepare protein complexes for negative-stain EM, 16 µM BiP and 32 µM HT2 reacted with 32 µM HyT36 were incubated for 30 min at 37 °C in 50 mM HEPES pH 7.5, 50 mM NaCl, 2 mM MgCl2 and 50 µM ATP. Then, 16 µM GRP94 was added to the mix and the sample was incubated for another 30 min at 37 °C. Gradients were generated using a range from 5% glycerol in 50 mM HEPES, 50 mM NaCl and 1 mM CaCl2 to 45% glycerol in 50 mM HEPES, 50 mM NaCl, 1 mM CaCl2 and 0.5% glutaraldehyde using the Master Gradient 108 (BioComp Instruments; program: 5–45% sucrose for 45 s). The generated gradient was incubated for 60 min at 4 °C. The protein samples were carefully applied to the gradient and centrifuged for 16 h at 130,000g at 4 °C (Optima XPN-80 Ultracentrifuge, Beckman Coulter). Next, 200 µl fractions were taken from the top of the gradient and the glutaraldehyde was quenched with 20 mM Tris pH 7.5. Gradient fractions were analyzed by SDS–PAGE.
To generate negative-stain grids, 4 µl of the GraFix fraction was applied to copper 400-mesh grids with continuous carbon (EM Sciences) after glow discharge, incubated for 30 s, immediately blotted with filter paper and stained by five successive short incubations of 2% (w/v) uranyl formate (Science Services). The excess stain was removed with filter paper and the grids were dried before imaging.
Collection and processing of negative-stain EM data
Grids were imaged using a Talos L120C microscope (Thermo Fisher) operated at 120 keV with a complementary metal–oxide–semiconductor camera (Ceta 16M). Micrographs were acquired at a nominal pixel size of 1.89 Å and total dose of 25 e− per Å2. Data were processed in cryoSPARC72, using CTFFIND4 for CTF estimation73, blob picker for particle picking and iterative rounds of reference-free 2D classification. Classes representing damaged protein or artifacts were removed in several rounds of 2D classification. Approximately ~434,000 particles corresponding to representative 2D class averages were used for ab initio reconstructions with several classes and heterogeneous refinements (Extended Data Figs. 6b and 7).
Molecular modeling
The preloading state of the GRP94–BiP complex was modeled with the structures of the ‘open’ ADP-bound GRP94 (PDB 2O1V)31 and the structure of BiPNBD (PDB 6DWS) as inputs. The region comprising residues 393–408 of GRP94 was modeled using MODELLER74,75. For the loading complex, a homology model of GRP94 was obtained with the SWISS-MODEL web server76 using the ‘semiclosed’ structure of Hsp90 (PDB 7KW7)24 as a template. This complex encompassed one BiPNBD chain as in the preloading complex and a model of the extended conformation of BiP that also included the SBD (residues 30–635). This structure was built using SWISS-MODEL with the E. coli Hsp70 homolog DnaK (PDB 2KHO)66 as a template. For this model, the region of residues 285–330 was truncated similarly to the open structure.
Before fitting into the EM density, the α-helical C-terminal region of the model of the extended structure of BiP was truncated (residues 530–635 were removed). For the modeling based on the obtained negative-stain EM maps, a first rigid-body fitting was performed with UCSF Chimera77 on the densities obtained for the preloading and loading complexes. Subsequently, a refinement of the fitting was performed with Rosetta’s ‘relax’ application64 with the specific flags for cryo-EM refinement65. We used the crosslinking information corresponding to NBD–NBD, SBD–SBD and NBD–SBD pairs as constraints for the fitting. For all the fittings, the input resolution of the density maps was 20 Å. For both preloading and loading complexes, a Cartesian fitting was used and 5,000 decoys were generated. For the evaluation, Rosetta’s ‘ref2015’ (refs. 78,79) and ‘elec_dens_fast’80,81 score functions were used, the latter with a weight of 10. Additionally, the RSCC was calculated with Rosetta’s ‘density_tools’ application. The interface area was measured with UCSF ChimeraX82. UCSF Chimera82 and PyMol (Molecular Graphics System, version 2.0, Schrödinger) were used for the analysis and visualization of the structures.
Crosslinking of chaperone complexes for SDS–PAGE analysis and XL-MS
All reactions were carried out in 50 mM HEPES pH 7.5, 50 mM NaCl and 2 mM MgCl2. For generating the BiP–GRP94 complex, 4 µM BiP or BiPNBD and 4 µM GRP94 Δ72, GRP94 Δ72noPT or GRP94 FL were incubated for 60 min at 37 °C. For the reactions containing PU-WS13, either 100 µM PU-WS13 or 1% DMSO was incubated with 4 µM GRP94 Δ72noPT for 30 min at 37 °C and then 4.0 µM BiP and 50 µM ATP were added before incubating for an additional 30 min at 37 °C. Samples were brought to room temperature (10 min) and DSBU (Thermo Fisher, A35459) was added to a final concentration of 0.5 mM. Crosslinking reactions were incubated for 20 min at room temperature and quenched with 20 mM Tris pH 7.5; the protein complexes were isolated by SDS–PAGE.
In-gel digestion of DSBU-crosslinked proteins
Gel regions containing crosslinked proteins were excised and washed twice with water and twice with 100 mM ammonium bicarbonate solution. The proteins were reduced with 10 mM TCEP (30 min, 62 °C) while gently shaking and subsequently alkylated by adding 55 mM iodoacetamide (30 min, room temperature, in the dark). The gel slabs were subsequently washed three times with acetonitrile (50 µl each). After the last wash, the gel slabs were completely dried by using a vacuum concentrator (Eppendorf) for 5 min. The dried gel pieces were next incubated with 200 µl of 10 ng µl−1 trypsin (37 °C, 16 h, vigorous shaking). The digestion was stopped by addition of formic acid to a final concentration of 5% (v/v) formic acid. The digestion supernatant was transferred to a fresh Eppendorf tube. The remaining gel pieces were subsequently washed three times with acetonitrile (50 µl each). The supernatants of these washes were combined with the recovered digestion mix and dried in a vacuum concentrator (Eppendorf).
The cleared tryptic digests were then desalted on homemade C18 stag tips as described previously83. Briefly, peptides were immobilized and washed on a two-disc C18 stage tip. After elution from the stage tips, samples were dried using a vacuum concentrator (Eppendorf); the peptides were taken up in 0.1% formic acid solution (10 μl) and directly used for liquid chromatography (LC)–MS/MS experiments, as described below.
LC–MS/MS
MS experiments were performed on an Orbitrap LUMOS instrument (Thermo Fisher) coupled to an EASY-nLC 1200 ultraperformance LC (UPLC) system (Thermo Fisher). The UPLC was operated in the one-column mode. The analytical column was a fused silica capillary (75 µm × 28 cm) with an integrated fritted emitter (CoAnn Technologies) packed in house with Kinetex 1.7-µm core–shell beads (Phenomenex). The analytical column was encased by a column oven (Sonation PRSO-V2) and attached to a nanospray flex ion source (Thermo Fisher). The column oven temperature was set to 50 °C during sample loading and data acquisition. The LC was equipped with two mobile phases: solvent A (0.2% formic acid, 2% acetonitrile and 97.8% H2O) and solvent B (0.2% formic acid, 80% acetonitrile and 19.8% H2O). All solvents were of UPLC grade (Honeywell). Peptides were directly loaded onto the analytical column with a maximum flow rate that would not exceed the set pressure limit of 980 bar (usually around 0.4–0.6 µl min−1). Peptides were subsequently separated on the analytical column by running a 67-min or 105-min gradient of solvent A and solvent B. The MS instrument was controlled by the Orbitrap Fusion Lumos Tune application and operated using Xcalibur software. Detailed LC–MS settings can be found in Supplementary Information.
XL-MS data analysis
Thermo RAW files were converted to the mzML or mfg file format using ProteoWizard (version 3.0.23018-60066e9)84 with default settings and submitted to Xisearch (version 1.7.6.7) for further analysis85. For each search, a dedicated database containing only the sequences of the investigated proteins was used. The following settings were applied: MS1 error tolerance of 6 ppm, MS2 error tolerance of 20 ppm, trypsin digestion with two missed cleavages allowed, carbamidomethylation on cysteine as the fixed modification, oxidation on methionine as the variable modification, DSBU as the crosslinker (linked amino acids included lysine, serine, threonine and tyrosine), NH2, OH, Bu and BuUr as linked modifications, a maximum of three modifications per peptide and fragment b and y ions. The resulting candidates from Xisearch were filtered to a 5% false discovery rate (FDR) on the residue pair level using the software xiFDR (version 2.1.5.5)86. Xisearch also calculates a score (reported in Supplementary Tables 1–4), which considers the goodness of the fit of the spectrum to the peptide pair based on several factors such as fragment mass error and number of (crosslinked) fragments85. A penalty is applied for crosslinks to serine, threonine and tyrosine as these are a side reaction of DSBU as compared to lysine and the N terminus of the protein. The mgf files, xiFDR results and FASTA sequences were then imported and visualized by XiView to generate the results (Fig. 2a and Extended Data Fig. 3b)87. Distance measurements were performed in PyMol or ChimeraX. The r.m.s.d. values of GRP94 protomers A and B in PDB 2O1V and the model of GRP94 generated on the basis of PDB 7KW7 were 0.8 Å and 0.7 Å respectively. The r.m.s.d. was calculated in PyMol and the flexible GRP94 charged linker region was omitted for the calculation. We, therefore, report only one value for interprotomer distances in the Supplementary Tables 1–4.
Pulldowns and sequential pulldown
All reactions were carried out in 50 mM HEPES pH 7.5, 50 mM NaCl, 2 mM MgCl2, 50 µM ATP and 3 µM BSA. Then, 8 µM HT2 reacted with 12 µM HyT36 was incubated with 4 µM His6-tagged BiP protein and/or 4 µM Strep-tagged GRP94 protein (as indicated in the individual figures) for 60 min at 37 °C. For pulldowns involving untagged GRP94 Δ72, the PT was removed by incubation with His6–GST–TEV-S219V protease for 2 h at room temperature in 50 mM HEPES pH 7.5, 50 mM NaCl and 1 mM CaCl2. Upon protease cleavage, untagged GRP94 Δ72 protein was isolated from the flowthrough of Streptactin following Ni-NTA purification. Next, 4 µM His6-tagged BiP protein and 4 µM Strep-tagged or untagged GRP94 protein were incubated for 60 min at 37 °C.
For His6-tagged BiP protein pulldown, samples were incubated with Ni-NTA beads (Qiagen) for 60 min at 37 °C, washed three times with 50 mM HEPES pH 7.5, 50 mM NaCl, 10 mM imidazole and 1 mM CaCl2 and finally eluted with 500 mM imidazole in wash buffer. For Strep-tagged GRP94 protein pulldown, samples were applied to StrepTactin 4 flowXT gravity flow columns (IBA), washed three times with 50 mM HEPES pH 7.5, 50 mM NaCl and 1 mM CaCl2 and finally eluted with 50 mM biotin in wash buffer. For the sequential pulldown, the elution of a Strep pulldown described above was used as the input for a Ni-NTA pulldown as described above. All input and elution fractions were applied to SDS–PAGE.
Analytical SEC
BiP and GRP94 proteins were incubated at a final concentration of 8 μM (unless otherwise indicated) for 60 min at 37 °C and applied to a Superose 6 PC 3.2/300 column in 50 mM HEPES pH 7.5, 50 mM NaCl and 1 mM CaCl2 on an Ettan LC system. Indicated fractions were analyzed by SDS–PAGE.
Glutaraldehyde crosslinking
For glutaraldehyde crosslinking experiments, 8 µM GRP94 and 8 µM BiPNBD were incubated separately or together in 50 mM HEPES pH 7.5, 50 mM NaCl and 2 mM MgCl2 in the presence or absence of 1 mM AMP-PNP for 60 min at 37 °C. Samples were brought to room temperature (10 min) and 0.006% glutaraldehyde was added. After 5 min at room temperature, reactions were quenched with 20 mM Tris pH 7.5 and the samples were analyzed by SDS–PAGE and western blot using anti-His5 (Santa Cruz Biotechnologies, SC-8036; 1:1,000 dilution) and anti-Strep (Qiagen, 34850; 1:1,000 dilution) antibodies. Bands were detected by chemiluminescence on a BioRad ChemiDoc imager.
Pulldown from HEK293 cells
Stable HEK293 Flp-in Trex cells (Thermo Fisher, R78007) expressing B4GT–HA–EGFP–HT2 were previously described in Serebrenik et al.51. The cells were grown in 15-cm dishes in DMEM (Invitrogen, Life Technologies, 31966047), 10% FBS (Life Technologies, 10500056) and 1% penicillin–streptomycin (Invitrogen, Life technologies, 15140122) in 5% CO2 at 37 °C. The cells were treated with 0.1 µg ml−1 doxycycline hyclate for 24 h, followed by cycloheximide treatment at 20 µg ml−1 1 h before 10 µM HyT36 or control (DMSO) treatments for an additional 4 h. HALTS1 treatment at 10 µM was performed simultaneously with the doxycycline hyclate treatment. The cells were then lysed in 10 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA and 0.5% Triton X-100 (Fisher Scientific, BP151-100) supplemented with cOmplete protease inhibitor cocktail (Roche, 11836145001). The cell lysates were clarified by centrifugation at 17,000g for 20 min at 4 °C. The supernatant was then incubated with GFP-trap agarose beads (ChromoTek) for 1 h at 4 °C. The beads were washed three times with 10 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA and 0.05% Triton X-100 and bound proteins were eluted by boiling the beads in SDS–PAGE sample buffer for 5 min. Samples were applied to SDS–PAGE gels and transferred to nitrocellulose membranes. After blocking with 5% BSA in Tris-buffered saline with Tween-20, membranes were incubated with primary (anti-BiP 1:1,000, anti-GRP94 1:5,000 and anti-HA 1:5,000 in blocking buffer) and secondary antibody solutions (goat anti-mouse or anti-rabbit HRP-coupled secondary antibodies 1:20,000) and finally developed using a BioRad ChemiDoc Imager.
Statistics and reproducibility
SDS–PAGE gel bands were quantified in ImageLab (BioRad) and the data were analyzed and plotted in Prism (GraphPad). All quantifications were derived from three independent biological replicates with the mean and s.d. shown in the respective figures. Analytical size-exclusion experiments were performed at least twice and yielded consistent results for all runs (examples in Figs. 1b,c and 5c and Extended Data Figs. 1a and 2b). The combined crosslinking–SEC experiment shown in Extended Data Fig. 1b was performed once. Chemical crosslinking and the subsequent SDS–PAGE analysis presented in Extended Data Fig. 2a,c,d was performed at least twice and gave consistent results. The limited proteolysis of HT2 shown in Fig. 3b was repeated more than three times (with varying time points) and yielded consistent results. The representative CD spectroscopy shown in Fig. 3c was repeated twice for samples treated with HyT36 with consistent results and once for each of the control conditions. The sequential pulldown shown in Fig. 3f was repeated twice under the optimized conditions described here with consistent results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The MS proteomics data were deposited to the ProteomeXchange Consortium through the PRIDE88 partner repository with the dataset identifier PXD059917. Negative-stain EM maps were deposited to the EM Data Bank under accession numbers EMD-19600 and EMD-19601. Publicly accessible structures used for molecular modeling and data analysis in this work were obtained from the PDB under accession numbers 2KHO, 2O1V, 5E84, 5E85, 5ULS, 6DWS, 7KW7 and 8TF0. Source data are provided with this paper.
References
Jayaraj, G. G., Hipp, M. S. & Hartl, F. U. Functional modules of the proteostasis network. Cold Spring Harb. Perspect. Biol. 12, a033951 (2020).
Biebl, M. M. & Buchner, J. Structure, function, and regulation of the Hsp90 machinery. Cold Spring Harb. Perspect. Biol. 11, a034017 (2019).
Klaips, C. L., Jayaraj, G. G. & Hartl, F. U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 217, 51–63 (2018).
Rosenzweig, R., Nillegoda, N. B., Mayer, M. P. & Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 20, 665–680 (2019).
Behnke, J., Feige, M. J. & Hendershot, L. M. BiP and its nucleotide exchange factors Grp170 and Sil1: mechanisms of action and biological functions. J. Mol. Biol. 427, 1589–1608 (2015).
Gidalevitz, T., Stevens, F. & Argon, Y. Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta 1833, 2410–2424 (2013).
Eletto, D., Dersh, D. & Argon, Y. GRP94 in ER quality control and stress responses. Semin. Cell Dev. Biol. 21, 479–485 (2010).
Hendershot, L. M., Buck, T. M. & Brodsky, J. L.The essential functions of molecular chaperones and folding enzymes in maintaining endoplasmic reticulum homeostasis. J. Mol. Biol. 436, 168418 (2024).
Vincenz-Donnelly, L. & Hipp, M. S. The endoplasmic reticulum: a hub of protein quality control in health and disease. Free Radic. Biol. Med. 108, 383–393 (2017).
Araki, K. & Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 3, a007526 (2011).
Sun, Z. & Brodsky, J. L. Protein quality control in the secretory pathway. J. Cell Biol. 218, 3171–3187 (2019).
Jin, H., Komita, M. & Aoe, T. The role of BiP retrieval by the KDEL receptor in the early secretory pathway and its effect on protein quality control and neurodegeneration. Front. Mol. Neurosci. 10, 222 (2017).
Yamamoto, K. et al. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 20, 3082–3091 (2001).
Samy, A., Yamano-Adachi, N., Koga, Y. & Omasa, T. Secretion of a low-molecular-weight species of endogenous GRP94 devoid of the KDEL motif during endoplasmic reticulum stress in Chinese hamster ovary cells. Traffic 22, 425–438 (2021).
Tsai, Y. L. et al. Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J. Biol. Chem. 290, 8049–8064 (2015).
Zhang, Y., Liu, R., Ni, M., Gill, P. & Lee, A. S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem. 285, 15065–15075 (2010).
Patel, P. D. et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat. Chem. Biol. 9, 677–684 (2013).
Schneider, M. et al. BiPPred: combined sequence- and structure-based prediction of peptide binding to the Hsp70 chaperone BiP. Proteins 84, 1390–1407 (2016).
Marzec, M., Eletto, D. & Argon, Y. GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 1823, 774–787 (2012).
Moran Luengo, T., Mayer, M. P. & Rudiger, S. G. D. The Hsp70–Hsp90 chaperone cascade in protein folding. Trends Cell Biol. 29, 164–177 (2019).
Alvira, S. et al. Structural characterization of the substrate transfer mechanism in Hsp70/Hsp90 folding machinery mediated by Hop. Nat. Commun. 5, 5484 (2014).
Melnick, J., Dul, J. L. & Argon, Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature 370, 373–375 (1994).
Kirschke, E., Goswami, D., Southworth, D., Griffin, P. R. & Agard, D. A. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157, 1685–1697 (2014).
Wang, R. Y. et al. Structure of Hsp90–Hsp70–Hop–GR reveals the Hsp90 client-loading mechanism. Nature 601, 460–464 (2022).
Doyle, S. M. et al. Intermolecular Interactions between Hsp90 and Hsp70. J. Mol. Biol. 431, 2729–2746 (2019).
Genest, O., Hoskins, J. R., Kravats, A. N., Doyle, S. M. & Wickner, S. Hsp70 and Hsp90 of E. coli directly interact for collaboration in protein remodeling. J. Mol. Biol. 427, 3877–3889 (2015).
Kravats, A. N. et al. Interaction of E. coli Hsp90 with DnaK involves the DnaJ binding region of DnaK. J. Mol. Biol. 429, 858–872 (2017).
Kravats, A. N. et al. Functional and physical interaction between yeast Hsp90 and Hsp70. Proc. Natl Acad. Sci. USA 115, E2210–E2219 (2018).
Nathan, D. F. & Lindquist, S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol. 15, 3917–3925 (1995).
Schopf, F. H., Biebl, M. M. & Buchner, J. The Hsp90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 18, 345–360 (2017).
Dollins, D. E., Warren, J. J., Immormino, R. M. & Gewirth, D. T. Structures of GRP94–nucleotide complexes reveal mechanistic differences between the Hsp90 chaperones. Mol. Cell 28, 41–56 (2007).
Huck, J. D., Que, N. L., Hong, F., Li, Z. & Gewirth, D. T. Structural and functional analysis of GRP94 in the closed state reveals an essential role for the pre-N domain and a potential client-binding site. Cell Rep. 20, 2800–2809 (2017).
Gewirth, D. T. Paralog specific Hsp90 inhibitors—a brief history and a bright future. Curr. Top. Med. Chem. 16, 2779–2791 (2016).
Rosenbaum, M. et al. MZB1 is a GRP94 cochaperone that enables proper immunoglobulin heavy chain biosynthesis upon ER stress. Genes Dev. 28, 1165–1178 (2014).
Liu, B. et al. Folding of Toll-like receptors by the Hsp90 paralogue GP96 requires a substrate-specific cochaperone. Nat. Commun. 1, 79 (2010).
Huang, B. et al. The endoplasmic reticulum chaperone BiP is a closure-accelerating cochaperone of GRP94. Proc. Natl Acad. Sci. USA 119, e2118793119 (2022).
Sun, M., Kotler, J. L. M., Liu, S. & Street, T. O. The endoplasmic reticulum (ER) chaperones BiP and GRP94 selectively associate when BiP is in the ADP conformation. J. Biol. Chem. 294, 6387–6396 (2019).
Yang, J., Nune, M., Zong, Y., Zhou, L. & Liu, Q. Close and allosteric opening of the polypeptide-binding site in a human Hsp70 chaperone BiP. Structure 23, 2191–2203 (2015).
Wieteska, L., Shahidi, S. & Zhuravleva, A. Allosteric fine-tuning of the conformational equilibrium poises the chaperone BiP for post-translational regulation. eLife 6, e29430 (2017).
Marcinowski, M. et al. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat. Struct. Mol. Biol. 18, 150–158 (2011).
Yang, J. et al. Conformation transitions of the polypeptide-binding pocket support an active substrate release from Hsp70s. Nat. Commun. 8, 1201 (2017).
Kotler, J. L. M. & Street, T. O. Mechanisms of protein quality control in the endoplasmic reticulum by a coordinated Hsp40–Hsp70–Hsp90 system. Annu. Rev. Biophys. 52, 509–524 (2023).
Pobre, K. F. R., Poet, G. J. & Hendershot, L. M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: getting by with a little help from ERdj friends. J. Biol. Chem. 294, 2098–2108 (2019).
Huang, B., Friedman, L. J., Sun, M., Gelles, J. & Street, T. O. Conformational cycling within the closed state of GRP94, an Hsp90-family chaperone. J. Mol. Biol. 431, 3312–3323 (2019).
Que, N. L. S., Seidler, P. M., Aw, W. J., Chiosis, G. & Gewirth, D. T.Selective inhibition of Hsp90 paralogs: uncovering the role of helix 1 in GRP94-selective ligand binding. Proteins 93, 654–672 (2025).
Swain, J. F. et al. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell 26, 27–39 (2007).
Verba, K. A. et al. Atomic structure of Hsp90–Cdc37–Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547 (2016).
Encell, L. P. et al. Development of a dehalogenase-based protein fusion tag capable of rapid, selective and covalent attachment to customizable ligands. Curr. Chem. Genomics 6, 55–71 (2012).
Neklesa, T. K. et al. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 7, 538–543 (2011).
Tae, H. S. et al. Identification of hydrophobic tags for the degradation of stabilized proteins. ChemBioChem 13, 538–541 (2012).
Serebrenik, Y. V. et al. Targeted protein unfolding uncovers a Golgi-specific transcriptional stress response. Mol. Biol. Cell 29, 1284–1298 (2018).
Schillinger, J. & Hellerschmied, D. Targeted protein unfolding at the Golgi apparatus. Methods Mol. Biol. 2557, 645–659 (2023).
Hellerschmied, D., Serebrenik, Y. V., Shao, L., Burslem, G. M. & Crews, C. M. Protein folding state-dependent sorting at the Golgi apparatus. Mol. Biol. Cell 30, 2296–2308 (2019).
Raina, K. et al. Targeted protein destabilization reveals an estrogen-mediated ER stress response. Nat. Chem. Biol. 10, 957–962 (2014).
Clerico, E. M., Tilitsky, J. M., Meng, W. & Gierasch, L. M. How Hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J. Mol. Biol. 427, 1575–1588 (2015).
Mayer, M. P. & Gierasch, L. M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 294, 2085–2097 (2019).
Kohler, V. & Andreasson, C. Hsp70-mediated quality control: should I stay or should I go? Biol. Chem. 401, 1233–1248 (2020).
Moran Luengo, T., Kityk, R., Mayer, M. P. & Rudiger, S. G. D. Hsp90 breaks the deadlock of the Hsp70 chaperone system. Mol. Cell 70, 545–552 (2018).
Szabo, A. et al. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc. Natl Acad. Sci. USA 91, 10345–10349 (1994).
Petrova, K., Oyadomari, S., Hendershot, L. M. & Ron, D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 27, 2862–2872 (2008).
Perera, L. A. et al. An oligomeric state-dependent switch in the ER enzyme FICD regulates AMPylation and deAMPylation of BiP. EMBO J. 38, e102177 (2019).
Preissler, S. et al. Physiological modulation of BiP activity by trans-protomer engagement of the interdomain linker. eLife 4, e08961 (2015).
Kastner, B. et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5, 53–55 (2008).
Conway, P., Tyka, M. D., DiMaio, F., Konerding, D. E. & Baker, D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 23, 47–55 (2014).
Wang, R. Y. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 5, e17219 (2016).
Bertelsen, E. B., Chang, L., Gestwicki, J. E. & Zuiderweg, E. R. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl Acad. Sci. USA 106, 8471–8476 (2009).
Hessling, M., Richter, K. & Buchner, J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 16, 287–293 (2009).
Li, J., Richter, K. & Buchner, J. Mixed Hsp90–cochaperone complexes are important for the progression of the reaction cycle. Nat. Struct. Mol. Biol. 18, 61–66 (2011).
Zierer, B. K. et al. Importance of cycle timing for the function of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 23, 1020–1028 (2016).
Noddings, C. M., Wang, R. Y., Johnson, J. L. & Agard, D. A. Structure of Hsp90–p23–GR reveals the Hsp90 client-remodelling mechanism. Nature 601, 465–469 (2022).
Neklesa, T. K. et al. A bidirectional system for the dynamic small molecule control of intracellular fusion proteins. ACS Chem. Biol. 8, 2293–2300 (2013).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Sali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Yang, Z. et al. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J. Struct. Biol. 179, 269–278 (2012).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Park, H. et al. Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J. Chem. Theory Comput. 12, 6201–6212 (2016).
Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031–3048 (2017).
DiMaio, F. et al. Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat. Methods 10, 1102–1104 (2013).
DiMaio, F., Tyka, M. D., Baker, M. L., Chiu, W. & Baker, D. Refinement of protein structures into low-resolution density maps Using Rosetta. J. Mol. Biol. 392, 181–190 (2009).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Mendes, M. L. et al. An integrated workflow for crosslinking mass spectrometry. Mol. Syst. Biol. 15, e8994 (2019).
Fischer, L. & Rappsilber, J. Quirks of error estimation in cross-linking/mass spectrometry. Anal. Chem. 89, 3829–3833 (2017).
Graham, M. L., Combe, C., Kolbowski, L., Fischer, L. & Rappsilber, J.xiVIEW: visualisation of crosslinking mass spectrometry data. J. Mol. Biol. 436, 168656 (2024).
Vizcaino, J. A. et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 (2016).
Acknowledgements
We would like to thank M.-A. Derichs, D. Agranovski and L. Sewald for supporting biochemical experiments. This work was funded by the Sofja Kovaleveskaja Award through the Alexander von Humboldt Foundation endowed by the Federal Ministry of Education and Research. D.H., L.B., L.R., F.K., M.K., S.P. and E.S.-G. were supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; SFB1430, project 424228829). E.S.-G. acknowledges instrumentation funding under the Großgerätinitiative (project 436586093) and the support of Germany’s Excellence Strategy (EXC-2033, project 390677874. Screening and acquisition of EM data were performed at the StruBiTEM Facility of the University of Cologne funded by the DFG (INST 216/949-1 FUGG) with support from M. Gunkel and E. Behrmann.
Funding
Open access funding provided by Universität Duisburg-Essen.
Author information
Authors and Affiliations
Contributions
J.C.B., L.B. and L.R. conducted all the biochemical experiments and prepared the MS samples. F.K. and M.K. processed the MS samples and analyzed the data together with J.C.B., L.B. and D.H. J.D., L.B. and L.R. performed the cell biology experiments. J.C.B and L.C.Z. prepared the samples for EM. L.C.Z. and S.P. performed the EM data collection, processing and analysis. Y.A.-H. carried out the structural modeling. Y.A.-H. and E.S.-G. designed, analyzed and wrote the results and discussion of the computational modeling. D.H. outlined the project and prepared the paper with input from all coauthors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks Lisa Jenkins, Andrew Truman and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Katarzyna Ciazynska, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Additional biochemical data on the interaction of BiP and GRP94.
(a) Analytical SEC and SDS-PAGE analysis of complex formation between GRP94 full-length (FL) ATP-binding mutant D149N, ATP hydrolysis mutant E103A and BiP. (b) Analytical SEC and SDS-PAGE analysis of a BiP-GRP94 Δ72 sample crosslinked with DSBU. Protein bands on SDS-PAGE gels were visualized by Coomassie staining.
Extended Data Fig. 2 Additional chemical crosslinking data on the interaction of BiP and GRP94.
(a) Chemical crosslinking of GRP94 Δ72noPT and BiP in the presence of ADP or AMP-PNP. Protein bands were visualized by Coomassie staining. (b) Analytical SEC and SDS-PAGE analysis of BiPNBD-GRP94 Δ72 complex formation. Protein bands were visualized by Coomassie staining. (c) Top panel: SDS-PAGE of glutaraldehyde crosslinking reactions containing GRP94 Δ72 or FL protein and BiPNBD in the presence and absence of AMP-PNP. Protein bands were visualized by Coomassie staining. Bottom panel: Western blot analysis of glutaraldehyde crosslinking reactions to detect Strep-tagged GRP94 Δ72 or -FL protein and His6-tagged BiPNBD. (d) Chemical crosslinking of GRP94 Δ72noPT (top panel) or GRP94 FL (bottom panel) and BiPNBD in the presence of PU-WS13. Protein bands were visualized by Coomassie staining.
Extended Data Fig. 3 Additional analysis of BiP-GRP94 crosslinks.
(a) Crosslinks outlining interface I, interface II, and the ATP lid – BiPNBD interactions from the BiP-GRP94 Δ72 XL-MS dataset mapped onto the homology model of BiP-GRP94 on the basis of the cytosolic loading complex (PDB ID: 7KW7). (b) Map of crosslinks identified between GRP94 Δ72noPT and BiP (top panel) or the BiPNBD (bottom panel) in the presence of the GRP94 ATPase inhibitor PU-WS13. NTD: N-terminal domain, MD: middle domain, CTD: C-terminal domain, NBD: nucleotide-binding domain, SBD: substrate binding domain, PT: purification tag (which was removed by TEV cleavage), CL: charged linker, L: linker, H: His6-tag. (c) Crosslinks outlining interface I, interface II, and the ATP lid – BiPNBD interactions from the BiP-GRP94 FL XL-MS dataset (comparable to Fig. 2c) mapped onto GRP94 protomer A in the open (2O1V-chain A) and closed (5ULS-chain A) conformation aligned to the semi-closed conformation model shown in Fig. 2b.
Extended Data Fig. 4 Crosslinking mass spectrometry outlines the conformations of BiP and GRP94.
(a) Intra-GRP94 crosslinks mapped onto the open (PDB ID: 2O1V) or semi-closed (homology model based on PDB ID: 7KW7) GRP94 dimer. For simplicity, only MD and NTD are shown. Crosslinks from the MD region in protomer B - K455 (B), K458 (B), K447 (B) to the NTD region within the same protomer and to protomer A are shown - K114 (A/B), K161 (A/B), K95 (A/B), K97 (A/B). Crosslinks shown in purple support the open conformation, crosslinks shown in orange support the semi-closed conformation. (b) Intra-BiPSBD crosslinks between the SBDα and SBDβ (identified in the BiP-GRP94 FL XL-MS dataset, Supplementary Table 1) mapped onto the closed (left panel, PDB ID: 5E85) and open (right panel, PDB ID: 5E84) BiPSBD structure. The BiPSBD domains are aligned based on the SBDβ. Crosslinks between the beta-sheet-rich subdomain, SBDβ (residues K446, K447, K521, K523) and the alpha-helical lid, SBDα (residues K573, K579, K581, K585) outline their close proximity and therefore indicate that the BiPSBD is in the closed conformation.
Extended Data Fig. 5 EGFP-trap pull-down of B4GT-HA-EGFP-HT2 from HEK293 cells.
The Golgi and ER-localized B4GT-HA-EGFP-HT2 fusion protein was pulled down from HEK293 cells. Western blot analysis of input and elution fractions probing for the bait protein, BiP, and GRP94 is shown. HyT36-treatment increased the amounts of BiP and GRP94 pulled down with B4GT-HA-EGFP-HT2, while treatment with the small molecule HALTS1, a HT2-stabilizer, decreased chaperone binding demonstrating that BiP and GRP94 interact with HT2 fusion proteins in cells, depending on the HT2 folding state.
Extended Data Fig. 6 Characterization of negative-stain EM sample.
(a) SDS-PAGE gel analysis of GraFix fractions. Gradient density increases with fraction number. Fraction 12 was analyzed by negative-stain EM. Protein bands were detected by Coomassie staining. (b) Western blot analysis of input and gradient fractions 12, 13, and 14 containing glutaraldehyde. The use of different gels accounts for the different banding pattern compared to (a). The asterisk denotes potentially aggregated HT2 protein. (c) Representative negative-stain EM micrograph and 2D class averages obtained from fraction 12. Scale bar represents 200 nm.
Extended Data Fig. 7 Single particle negative-stain EM image processing pipeline.
Flow chart of classifications and refinements, resulting in reconstructions of the pre-loading and loading complex conformations. Yellow boxes indicate 3D reconstructions that were further used for molecular modeling. The number of particles that went into each reconstruction is indicated, together with their respective percentage share of the initial 434,771 particles. 2D classifications of those particle stacks resulted in the shown respective 2D class averages (scale bar: 160 Å).
Extended Data Fig. 8 Evaluation of pre-loading and loading complex models.
(a) Quality metrics of molecular modeling of the pre-loading and loading complexes. RSCC: Real-space correlation coefficient, RAU: Rosetta arbitrary units. The arrow indicates additional density not filled by the loading complex models. (b) Crosslinks identified between BiPNBD and GRP94 Δ72 mapped onto the pre-loading complex model. GRP94 protomer B is omitted for clarity. Red lines indicate crosslinks > 30 Å, green lines < 30 Å. (c) Side-by-side comparison of the cytosolic (left) and BiP-GRP94 (right) loading complex conformation. For clarity, exclusively the NBDs of Hsp70/BiP and the Hsp90/GRP94 dimer structures are shown.
Extended Data Fig. 9 BiP-GRP94 interaction interfaces in the loading complex conformation.
(a) Sequence alignment of the indicated expression constructs. A sequence stretch that is part of the His6- and Strep-tagged GRP94 Δ72 constructs highlighted in yellow may serve as a BiP substrate mimetic. (b) Zoom-in into interfaces I, I’ and II’ of the BiP-GRP94 loading complex. Compare to Fig. 5b and e.
Supplementary information
Supplementary Information
Supplementary Table 5 and information on LC–MS settings.
Supplementary Table 1
XL-MS dataset of BiP–GRP94 FL. Data tables contain distance measurements (between Cα atoms) of intraprotein and interprotein crosslinks derived from the indicated PDB files. Distances < 30 Å are highlighted in green. Distances < 35 Å are highlighted in yellow. The cutoff applied for Xisearch score was 15. One of two independently obtained datasets is shown. Residues in orange are part of BiPSBD or GRP94CTD, residues in gray are part of GRP94MD and residues in blue are part of the BiPNBD or GRP94NTD.
Supplementary Table 2
XL-MS dataset of BiP–GRP94 Δ72. Data tables contain distance measurements (between Cα atoms) of intraprotein and interprotein crosslinks derived from the indicated PDB files. Distances < 30 Å are highlighted in green. Distances < 35 Å are highlighted in yellow. The cutoff applied for Xisearch score was 15. One of three independently obtained datasets is shown. Residues in orange are part of the BiPSBD or GRP94CTD, residues in gray are part of the GRP94MD and residues in blue are part of the BiPNBD or GRP94NTD.
Supplementary Table 3
XL-MS dataset of BiP–GRP94 Δ72noPT with PU-WS13. Data tables contain distance measurements (between Cα atoms) of intraprotein and interprotein crosslinks derived from the indicated PDB files. Distances < 30 Å are highlighted in green. Distances < 35 Å are highlighted in yellow. The cutoff applied for Xisearch score was 15. Residues in orange are part of the BiPSBD or GRP94CTD, residues in gray are part of the GRP94MD and residues in blue are part of the BiPNBD or GRP94NTD.
Supplementary Table 4
XL-MS dataset of BiPNBD–GRP94 Δ72noPT with PU-WS13. Data tables contain distance measurements (between Cα atoms) of intraprotein and interprotein crosslinks derived from the indicated PDB files. Distances < 30 Å are highlighted in green. Distances < 35 Å are highlighted in yellow. The cutoff applied for Xisearch score was 15. Residues in orange are part of the GRP94CTD, residues in gray are part of the GRP94MD and residues in blue are part of the BiPNBD or GRP94NTD.
Supplementary Video 1
Morphing video between the preloading and loading complex conformation.
Source data
Source Data Figs. 1 and 3–5 and Extended Data Figs. 1, 2 and 8
SEC traces and statistical source data for Figs. 1 and 3–5 and Extended Data Figs. 1, 2 and 8.
Source Data Figs. 1 and 3–5 and Extended Data Figs. 1, 2, 5 and 6
Unprocessed gels for Figs. 1 and 3–5 and Extended Data Figs. 1, 2, 5 and 6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brenner, J.C., Zirden, L.C., Buzuk, L. et al. Conformational plasticity of a BiP–GRP94 chaperone complex. Nat Struct Mol Biol 32, 1947–1958 (2025). https://doi.org/10.1038/s41594-025-01619-0
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41594-025-01619-0
