
Abstract
The sea toad genus Chaunax is a group of small benthic fishes that predominantly inhabiting the deep seas of the Atlantic, Indian, and Pacific Oceans. Although they have the potential to make excellent systems for studies of evolutionary adaptation to deep-sea environments, genomic research on Chaunax has been hindered by a scarcity of high-quality genomic resources. We present a chromosome-scale genome assembly of a Chaunax specimen generated using PacBio long-read sequencing and high-throughput chromosome conformation capture technology. The size of the assembled genome was 706.94 Mb, with a contig N50 of 15.24 Mb and scaffold N50 of 29.42 Mb. Approximately 96.11% of assembled sequences were anchored and oriented onto 24 pseudo-chromosomes. The genome contained 213.47 Mb repetitive sequences, 25,280 protein-coding genes, and 5,090 non-coding RNAs. The high ratio of complete BUSCO genes (97.20%) indicates high quality of genome assembly. The chromosomal-level reference genome of Chaunax sp. provides a preliminary molecular basis for understanding deep-sea adaptation and phenotypic evolution as well as an important reference for whole-genome sequencing of related species.
Similar content being viewed by others
Background &Summary
Members of the genus Chaunax (Lophiiformes: Chaunacidae) are commonly referred to as sea toads, and they are extensively distributed across deep-seas areas at mid- to low latitudes1,2,3. These sea toads are particularly prevalent on the outer continental shelf and the upper continental slope, inhabiting depths ranging from 200 to 2,500 m3,4. Their morphologically distinctive traits include a head that is rounded to slightly cuboidal; a skin texture interspersed with minute, spine-like scales; and the presence of lateral-line neuromasts on both the head and body3. These features are thought to enhance their sensory capabilities, which are crucial for their survival in challenging deep-sea habitats5,6.
Similar to other members of the order Lophiiformes, sea toads are benthic and use their pectoral and pelvic fins for support and locomotion across the seafloor, a movement often likened to ‘walking’7. They use a sit-and-wait strategy to capture their prey and can employ their esca (a specialized lure at the end of the first dorsal fin spine) to entice prey within striking distance8,9. These distinctive morphological and behavioral traits make Chaunax species excellent models for studies of evolutionary adaptations to deep-sea environments.
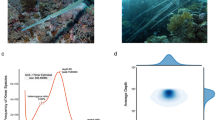
Genetic and evolutionary insights into Chaunax have been limited by a lack of deep-sea sampling and thus genomic data for Chaunacidae. Here, we collected a sea toad specimen in the genus Chaunax from the Zhenbei seamount at a depth of 555.3 m using a submersible vehicle (Faxian) (Fig. 1). The Chaunax specimen had a pinkish-red body with bright-white patches on the dorsal surface, and its skin was covered with a mix of numerous bifurcated and simple spinules. Specialized pectoral fins resembling ‘little feet’ support the body and move quickly across the sea floor. We employed PacBio long-read sequencing, Illumina sequencing, and high-throughput chromosome conformation capture (Hi-C) technology to generate an annotated chromosome-level genome assembly of this Chaunax specimen. This high-quality genome assembly will aid future studies of the deep-sea adaptation and phenotypic evolution of this species, as well as genomic analyses of related species. Our assembly will also advance our understanding of the genomic evolution and phylogenetic relationships within the order Lophiiformes.

In situ observations of the Chaunax sp.
Methods
Sample collection and DNA extraction
The Chaunax specimen was collected using a submersible vehicle (Faxian) from the Zhenbei seamount (15°04′45.888″N, 116°33′55.210″E, 555.3 m deep) in the South China Sea (Fig. 1). The sea toad sample was kept in a closed sample chamber placed inside the sample basket of the submersible. The sample was then immediately stored in liquid nitrogen. All experimental protocols were approved by relevant guidelines and regulations established by the Institutional Animal Care and Use Committee of the Institute of Oceanology, Chinese Academy of Science. Sea toad muscle was used to extract genomic DNA using the sodium dodecyl sulfate method10. The quantity and quality of DNA were determined using agarose gel electrophoresis and a Qubit Fluorometer, respectively.
Illumina sequencing and genome size estimation
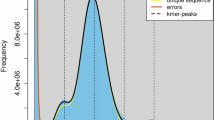
Illumina paired-end libraries with insert sizes of 350 bp were constructed and sequenced on an Illumina NovaSeq6000 platform (Illumina, CA, USA). Low-quality reads and sequencing adaptor-contaminated reads were trimmed using fastp11 with the following parameters: -q 10 -u 50 -y -g -Y 10 -e 20 -l 100 -b 150 -B 150. A total of 32.41 Gb Illumina short reads (clean data) were generated and retained for the genome survey (Table 1). The genome survey of Chaunax sp. was performed using the K-mer method. K-mer analysis was conducted using jellyfish v2.2.712 with an optimal K-value of 17 (Table 2). The K-mer frequency distribution map was generated to estimate the genome size, heterozygosity, and proportion of repetitive sequences using GenomeScope software13. A total of 24,428,214,893 K-mers were obtained using a K-mers peak at a depth of 35 (Table 2). The genome size estimated using the formula Knum/Kdepth was approximately 683.46 Mb. The heterozygosity rate and proportion of repetitive sequences were 0.38% and 36.16%, respectively (Table 2).
PacBio long-read sequencing and Hi-C sequencing
The genomic DNA was fragmented to 20 kb and sequenced using the PacBio Sequel II platform (Pacific Biosciences, USA) following the manufacturer’s protocols. Briefly, the gDNA was sheared using the g-TUBE device (Covaris) to the target fragment size for the construction of 20-kb libraries. Damage repair and end repair were performed using the SMRTbell Damage Repair Kit on the interrupted DNA fragments. After attaching dumbbell adapters, the fragments were digested by exonuclease. BluePippin electrophoresis (Sage Science, MA, USA) was used for size selection of the sequencing library, and the cutoff threshold size was set to 20 kb. A total of 23.67 Gb high-quality HiFi reads were produced using the circular consensus sequencing mode on the PacBio Sequel II platform (Table 1).
A Hi-C library was constructed by chromatin cross-linking, restriction endonuclease (DpnII) cleavage, end repairing and biotin labeling, ligation, and DNA purification and shearing; interacting DNA fragments were captured by streptavidin magnetic beads for library construction. A Qubit 2.0 Fluorometer and Agilent 2100 Bioanalyzer were used to detect the concentration and insert size of the library, and Q-PCR was used to accurately quantify the effective concentration of the library to ensure its quality. The Hi-C sequencing library was sequenced on an Illumina NovaSeq (PE150) platform. A total of 70.63 Gb Hi-C data were generated after quality control (Table 1).
Chromosome-level genome assembly
The high-accuracy PacBio HiFi reads were assembled into the initial set of contigs using Hifiasm v0.1914 with the following parameters: -l 2 -n 4. Sequence contamination in the genome assembly and mitochondrial sequences were removed via comparison of the genome assembly with the nucleotide sequence database (nt) and mitochondrial database (https://ftp.ncbi.nlm.nih.gov/refseq/release/mitochondrion/) from the National Center for Biotechnology Information (NCBI). The initial genome assembly length was 709.63 Mb with a contig number of 354 and N50 of 15.24 Mb (Table 2). The size of the assembled genome nearly matched that of the results of the genome survey, which reflected the high accuracy and integrity of our assembled genomes.
To generate the chromosome-level assembly, the unmapped pair-end reads, singleton reads, and other invalid reads were filtered using HiC-Pro v2.10.015. Only uniquely valid interaction pairs (79.12%) were retained for further assembly. A total of 70.63 Gb clean reads pairs were obtained from the Hi-C library and mapped to the assembled genome using BWA v0.7.1716. The uniquely mapped data were retained, and the clustered contigs were sorted and oriented using LACHESIS software17. After Hi-C assembly and manual heat map adjustment, a total of 679.46 Mb of genome sequences were anchored and oriented onto 24 pseudo-chromosomes (Fig. 2a and Table 3). These pseudo-chromosomes, ranging in size from 13.00 to 35.46 Mb (Fig. 2b and Table 3), comprised approximately 96.11% of the total genome. Finally, the chromosome-level genome assembly with a total length of 706.94 Mb was obtained; this assembly showed a high level of continuity, with a scaffold N50 of 29.42 Mb (Table 2).

Chromosome-level genome assembly of Chaunax sp. (a) Contact map of chromosomal interactions in the Chaunax sp. genome using Hi-C data. (b) Circos plot of 24 chromosomes in Chaunax sp. genome. The tracks from outside to inside are 24 chromosome ideograms, transposable element (TE) density, simple sequence repeat (SSR) density, gene density, GC content, and co-linearity relationship.
Transcriptome sequencing and assembly
RNA was extracted from multiple tissue samples, including muscle, kidney, liver, gonad, and cholecyst, using the TRIzol reagent (Thermo Fisher Scientific). RNA integrity and quality were assessed using agarose gel electrophoresis, and the quantity of RNA was determined using a Qubit Fluorometer. Sequencing libraries were constructed using the NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) per the manufacturer’s instructions. Illumina RNA-seq libraries were prepared and sequenced on an Illumina NovaSeq6000 platform, and 150 bp paired-end reads were generated. After trimming based on the quality scores using Trimmomatic-0.3917, a total of 6.77 Gb of clean transcriptome data were obtained (Table 1). The clean reads were aligned to the sea toad genomes using HISAT2 v2.2.118.
Annotation of repetitive sequences
Transposable elements (TEs) and tandem repeats were annotated via the following workflows. TEs were identified via a combination of homology-based searches and de novo prediction. A de novo repeat library was generated using RepeatModeler2 v2.0.119, along with the two embedded programs RECON v1.0.820 and RepeatScout v1.0.621, all of which were operated using default parameters. Long terminal repeats (LTRs) were identified using LTR_retriever v2.9.022, and they were mainly identified using the predicted results of LTRharvest v1.5.1023 and LTR_FINDER v1.0724. A non-redundant species-specific TE library was constructed by combining the de novo TE sequence library above with the known Dfam v3.5 database, and RepeatClassifier19 was used to classify the prediction results. Finally, RepeatMasker v4.1.225 was used to predict the TEs of the genome based on the constructed repeat sequence database. Tandem repeats were annotated by TRF v4.0926 and MISA v2.127. The results showed that 30.20% of repetitive sequences were annotated in the Chaunax sp. genome, and the total length of the TEs was 143.93 Mb, which comprised 20.36% of the genome; the total length of tandem repeat sequences was 69.54 Mb, which comprised 9.84% of the genome (Table 4).
Gene prediction and functional annotation
Protein-coding genes were annotated using a combination of ab initio prediction, homology-based searches, and RNA sequencing (RNA-seq) (Table 5). Augustus v.3.1.028 and SNAP v2006-07-2829 were used for ab initio gene prediction with default parameters. GeMoMa v.1.730 software was performed for homology-based prediction. The protein sequences of Lophius litulon31, Lophiodes sp.31, Solocisquama erythrina31, Takifugu rubripes32, and Thamnaconus septentrionalis33 were downloaded from the NCBI and figshare database. RNA-seq-based gene prediction was performed by mapping clean RNA-seq reads to the reference genome using HISAT2 v2.2.118, and the transcripts were assembled by StringTie v.1.2.334. GeneMarkS-T v5.135 was used to predict genes based on the assembled transcripts. PASA v2.4.136 software was used to predict genes based on the unigenes (and full-length transcripts from the PacBio HiFi sequencing) assembled by Trinity v.2.1137. Gene models from these different approaches were combined using EVM v1.1.138 and updated by PASA. The final gene models were annotated by blasting sequences against the GenBank NR, eggNOG, GO, KEGG, TrEMBL, KOG, Pfam, and SwissProt databases using an E-value cut-off of 1 × 10−5. We predicted a total of 25,280 protein-coding genes in the Chaunax sp. genome via multiple methods (Table 5), with a gene length of 398,546,511 bp, exon length of 55,947,487 bp, coding sequence length of 42,599,371 bp, and intron length of 342,599,024 bp (Table 6). A total of 23,457 genes (92.79% of the total) were functionally annotated using public databases (Table 7).
Non-coding RNA annotation and pseudogene prediction
For non-coding RNA annotation, tRNAs and rRNAs were predicted using tRNAScan-SE v1.3.139 and Barrnap v0.9 (the parameters deployed in barrnap–kingdom euk)40, respectively; miRNAs, snRNAs, and snoRNAs were identified using Infernal v1.141 software against the Rfam v14.542 database. A total of 2,462 tRNAs, 1,811 rRNAs, 202 miRNAs, 440 snRNAs, and 175 snoRNAs were predicted (Table 8).
GenBlastA v1.0.443 software was used to scan the whole genomes after masking predicted functional genes. Putative candidates were then analyzed by searching for premature stop codons and frame-shift mutations using GeneWise v2.4.144. Finally, a total of 67 pseudogenes were identified, encompassing a combined length of 496,033 bp, with an average length of 7,403 bp (Table 8).
Data Records
The Chaunax sp. genome has been deposited in GenBank under the accession number JBAGJB00000000045 and the BioProject number PRJNA1068823. The Illumina, PacBio HiFi, Hi-C, and RNAseq data have been deposited in the NCBI Sequence Read Archive under the accession number SRR27768100-SRR2776810346. The genome-related annotation files can be accessed through Figshare at https://doi.org/10.6084/m9.figshare.2510047447. The specific accessions are provided in the Material and Methods sections describing the data and analyses.
Technical Validation
Genome assembly and annotation completeness evaluation
The completeness of the final genome assembly was evaluated using the Benchmarking Universal Single-Copy Orthologs (BUSCO v5.2.1)48 program, along with the actinopterygii_odb10 database and the Core Eukaryotic Genes Mapping Approach (CEGMA v2.5)49. This yielded 97.06% of the complete BUSCO genes and 99.56% of the core eukaryotic genes (Table 2). The trimmed Illumina short reads and PacBio long reads were mapped against the assembled genome using BWA to evaluate the accuracy of the assembly, and the mapping rates were 99.63% and 99.75%, respectively (Table 3). The BUSCO completeness of the predicted gene models was determined against the actinopterygii_odb10 database under the protein mode, and the orthologous genes of Chaunax sp. contributed 97.20% of the complete genes, indicating the high completeness of the gene annotation (Table 2).
The interaction strength among chromosomes was evaluated and a Hi-C interaction heat map was constructed using HiCPlotter software50. The assembled sequences were anchored and oriented onto 24 pseudo-chromosomes (Fig. 2); within each pseudo-chromosome group, the interaction strength was higher at diagonal positions than at non-diagonal positions, suggesting that the quality of our genome assembly was high.
Code availability
Genome assembly:
(1) Hifiasm: parameter: -l 2 -n 4.
(2) HiC-Pro: all parameters were set as default.
(3) Bwa: parameter: bwa aln && bwa sampe.
(4) LACHESIS: parameter: CLUSTER_MIN_RE_SITES = 328; CLUSTER_MAX_LINK_DENSITY = 2; ORDER_MIN_N_RES_IN_TRUNK = 15; ORDER_MIN_N_RES_IN_SHREDS = 15.
Genome annotation:
(1) RepeatModeler: parameter: BuildDatabase -name && RepeatModeler -pa 12.
(2) LTR_retriever: all parameters were set as default.
(3) LTR_FINDER: parameter: ltr_finder -w 2 -C -D.
(4) RepeatMasker: parameter: repeatmasker -nolow -no_is -norna -engine wublast -parallel 8 -qq.
(5) TRF: parameter: trf 2 7 7 80 10 50 500 -d -h.
(6) MISA: all parameters were set as default.
(7) Augustus: all parameters were set as default.
(8) SNAP: all parameters were set as default.
(9) GeMoMa: parameter: run.sh mmseqs.
(10) Hisat: parameter: hisat2 -- dta -p 10.
(11) Stringtie: parameter: stringtie -p 2.
(12) GeneMarkS-T: all parameters were set as default.
(13) PASA: all parameters were set as default.
(14) Trinity: parameter: Trinity -- genome_guided_bam.
(15) EVidenceModeler: all parameters were set as default.
References
Caruso, J. H. Systematics and distribution of the atlantic chaunacid anglerfishes (Pisces: Lophiiformes). Copeia 1, 153–165, https://doi.org/10.2307/1445616 (1989).
Ho, H. & Shao, K. A new species of Chaunax (Lophiiformes: Chaunacidae) from the western South Pacific, with comment on C. latipunctatus. Zootaxa 2445, 53–61, https://doi.org/10.11646/zootaxa.2445.1.3 (2010).
Ho, H. C., Roberts, C. D. & Stewart, A. L. A review of the anglerfish genus Chaunax (Lophiiformes: Chaunacidae) from New Zealand and adjacent waters, with descriptions of four new species. Zootaxa 3620, 89–111, https://doi.org/10.11646/zootaxa.3620.1.4 (2013).
Caruso, J. H., Ross, S. W., Sulak, K. J. & Sedberry, G. R. Deep-water chaunacid and lophiid anglerfishes (Pisces: Lophiiformes) off the South-Eastern United States. J Fish Biol. 70, 1015–1026, https://doi.org/10.1111/j.1095-8649.2007.01360.x (2007).
Ho, H. C., Kawai, T. & Satria, F. Species of the anglerfish genus Chaunax from Indonesia, with descriptions of two new species (Lophiiformes: Chaunacidae). Raffles B Zool. 63, 301–308, http://zoobank.org/urn:lsid:zoobank.org:pub:8DBC268B-F75B-4EDF-95A5-2441AAD61600 (2015).
Ho, H. C. & Ma, W. C. Four new species of the frogmouth genus Chaunax (Lophiiformes: Chaunacidae) from Taiwan and the Philippines. Zootaxa 5189, 146–179, https://doi.org/10.11646/zootaxa.5189.1.17 (2022).
Lundsten, L., Johnson, S. B., Cailliet, G. M., Devogelaere, A. P. & Clague, D. A. Morphological, molecular, and in situ behavioral observations of the rare deep-sea anglerfish Chaunacops coloratus (garman, 1899), order Lophiiformes, in the eastern North Pacific. Deep Sea Res. I. 68, 46–53, https://doi.org/10.1016/j.dsr.2012.05.012 (2012).
Miya, M. et al. Evolutionary history of anglerfishes (Teleostei: Lophiiformes): a mitogenomic perspective. BMC Evol. Biol. 10, 58, https://doi.org/10.1186/1471-2148-10-58 (2010).
Moore, T. Y. & Biewener, A. A. Outrun or outmanoeuvre: predator-prey interactions as a model system for integrating biomechanical studies in a broader ecological and evolutionary context. Integr. Comp. Biol. 55, 1188–1197, https://doi.org/10.1093/icb/icv074 (2015).
Natarajan, V. P., Zhang, X. X., Morono, Y., Inagaki, F. & Wang, F. P. A modified SDS-based DNA extraction method for high quality environmental DNA from seafloor environments. Front. Microbiol. 7, 986, https://doi.org/10.3389/fmicb.2016.00986 (2016).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259, https://doi.org/10.1186/s13059-015-0831-x (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125, https://doi.org/10.1038/nbt.2727 (2013).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Bao, Z. R. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276, http://www.genome.org/cgi/doi/10.1101/gr.88502 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, I351–I358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal-repeat retrotransposons. Plant Physiol. 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2017).
Ellinghaus, D., Kurtz, S. & Willhoef, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268, https://doi.org/10.1093/nar/gkm286 (2007).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 25, 4.10.1–4.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Beier, S., Tiel, T., Munch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585, https://doi.org/10.1093/bioinformatics/btx198 (2017).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89, https://doi.org/10.1093/nar/gkw092 (2016).
Liang, Y. S. Protein coding sequences of three Lophiiformes species. figshare. Dataset. https://doi.org/10.6084/m9.figshare.27158106.v1 (2024).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_901000725.2/ (2019).
Bian, L. Thamnaconus septentrionalis genome supporting data. figshare. Dataset. https://doi.org/10.6084/m9.figshare.11874825.v1 (2020).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Tang, S. Y. Y., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43, e78, https://doi.org/10.1093/nar/gkv227 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–U130, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Seemann, T. Barrnap 0.9: Rapid ribosomal RNA prediction. Available at: https://github.com/tseemann/barrnap.
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Grifths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124, https://doi.org/10.1093/nar/gki081 (2005).
She, R., Chu, J. S. C., Wang, K., Pei, J. & Chen, N. S. GenBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res. 19, 143–149, https://doi.org/10.1101/gr.082081.108 (2009).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995, https://doi.org/10.1101/gr.1865504 (2004).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_037577475.1 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP486588 (2024).
Liang, Y. S. Chromosome-level genome assembly and annotation of the sea toad Chaunax sp, figshare. Dataset., https://doi.org/10.6084/m9.figshare.25100474.v2 (2024).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067, https://doi.org/10.1093/bioinformatics/btm071 (2007).
Akdemir, K. C. & Chin, L. HiCPlotter integrates genomic data with interaction matrices. Genome Biol. 16, 198, https://doi.org/10.1186/s13059-015-0767-1 (2015).
Acknowledgements
This research was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB42000000), the National Natural Science Foundation of China (41930533), the National Key R&D Program of China (2022YFC2803800), the Senior User Project of RV KEXUE (KEXUE2020GZ02), and the Science and Technology Program of Nanji Islands National Marine Nature Reserve Administration (JJZBPYCG-2021112901).
Author information
Authors and Affiliations
Contributions
Z.Z. and K.X. conceived the research project. Y.L., Z.Z. and K.X. collected the samples. Y.L., Z.Z. and W.J. performed the bioinformatics analyses. Y.L. and Z.Z. wrote the manuscript. Z.Z., L.J. and K.X. revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhan, Z., Liang, Y., Wei, J. et al. Chromosome-level genome assembly and annotation of a sea toad (Chaunax sp.). Sci Data 11, 1397 (2024). https://doi.org/10.1038/s41597-024-04245-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04245-5
