
Abstract
Rosa laevigata is an excellent rose germplasm, highly resistant to aphid, and immune to both rose black spot and powdery mildew disease. It is also a well-known edible plant with a long history of medicinal use in China, having the effects of improving kidney function, inhibiting arteriosclerosis, and reducing inflammation. In this study, we assembled a high-quality chromosome-scale genome for R. laevigata by combining Illumina, PacBio, and Hi-C data, which has a length of approximately 494.2 Mb with a scaffold N50 of 68.6 Mb. A total of 493.2 Mb (99.8%) of the draft genome sequences were anchored on seven pseudochromosomes and two gapless pseudochromosomes were included in the final genome assembly. A total of 37,117 protein-coding genes were predicted, 34,047 of which were functionally annotated. Repeat annotation revealed 659,558 (285.6 Mb) repeat elements, accounting for 57.8% of the genome. The chromosome-scale genome provides valuable information to facilitate comparative genomic analysis of rose family and will accelerate genome-guided breeding and germplasm improvement of both R. laevigata itself and modern roses.
Similar content being viewed by others

Background & Summary
Rosa laevigata Michx. (2n = 2x = 14)1 is an evergreen climbing shrub widely distributed in eastern and southern China, with altitude of 200–1,600 meters. Commonly known as Cherokee rose, R. laevigata is the only species in Rosa sect. Laevigatae of Rosaceae, with glabrous leathery leaves and large fragrant white flowers2 (Fig. 1). On one hand, R. laevigata is an excellent germplasm for the germplasm innovation and improvement of modern rose cultivars, highly resistant to aphid3, and immune to both black spot disease and powdery mildew disease of rose4. On the other hand, it is an edible and traditional herbal medicine in China5. The fruits of R. laevigata, known as “Jin-Ying-Zi” in Chinese, are a main Traditional Chinese Medicines ingredient that is prescribed as a kidney tonic for the treatment of urinary diseases, including urinary incontinence and urinary frequency, as well as menstrual irregularities, leucorrhea, and uterine prolapse6. The roots of R. laevigata possess functions such as clearing heat, detoxifying, cooling blood, promoting blood circulation, dispelling stasis, and relieving pain7. In addition, the roots of R. laevigata are used to treat gynecological infections and diseases of the urinary system6. The leaves of R. laevigata could cure burns, skin tumors and ulcers, while the flowers show efficacy in alleviating cold and heat, as well as contain insecticidal properties8.

Rosa laevigata Michx. (a) flowers and leaves, (b) fruit (photographed by H.Y. Jian).
Recent studies on R. laevigata revealed the presence of phenolic acids, steroids, triterpenoids, phenylpropanoids, and other chemical components in the plant that exhibits diverse pharmacological effects, such as antioxidant, anti-inflammatory, antibacterial, kidney function improvement, immunity enhancement, blood sugar reduction, and anti-tumor properties9,10. R. laevigata is also used in treatments for spermatorrhea, premature ejaculation, urinary incontinence, diarrhea, chronic bronchitis, and chronic kidney disease11. Among over 123 ingredients that have been isolated from different parts of R. laevigata, triterpenoids were regarded as the most significant bioactive substances12, and were useful to combat Alzheimer’s disease (AD)13,14,15.
There are more than 200 species in Rosa16 with varying ploidy levels, ranging from 2n = 2x to 10x17,18. Despite the fact that at least eight published nuclear genome sequences are publicly available at present (https://www.plabipd.de/), some of them are at the draft genome level with relatively low quality, e.g., R. multiflora19. Although draft genomes could provide useful genomic information, the construction of a high-quality genome assembly is a fundamental step for dissecting genomic variations that contribute to exploring the genetic and molecular basis of desirable traits in plants. Thus, in this study, we assembled a high-quality chromosome-scale genome of R. laevigata using Illumina, PacBio, and Hi-C data. The final genome assembly spans a total length of 494.2 Mb, featuring a scaffold N50 size of 68.6 Mb. Additionally, 99.8% (493.2 Mb) of the genome sequence has been successfully anchored on seven pseudochromosomes. The assembled genome sequence consists of repeat elements, which make up 57.8% of the total. The most abundant class of repeat elements is the long terminal repeats (LTRs), which account for 42.1% of the genome. A total of 37,117 protein-coding genes were found using ab initio gene prediction, RNA-seq, and homologous protein evidence. Out of these, 34,074 genes were annotated with their respective functions. We have also detected 151 miRNAs, 1,115 tRNAs, 1,289 rRNAs and 627 snRNAs in the genome of R. laevigata. The high-quality chromosome-scale genome provides valuable resource for exploring key genes and molecular regulatory mechanisms involved in the high resistance to pests and diseases of modern roses on one hand, and in the biosynthesis of important compounds such as triterpenoids on the other, which will facilitate genome-guided breeding and improvement of R. laevigata itself and modern roses.
Methods
Sample collection, DNA extraction and sequencing
Fresh young leaves of R. laevigata, which had been propagated by cuttings collected from Changshou District of Chongqing Municipality (107°12′53.598″E, 30°10′42.169″N, 350 m), were sampled at the Flower Research Institute of the Yunnan Academy of Agricultural Sciences, Yunnan Province, China, for DNA extraction. At the same time, the tender roots, stems, leaves, and fruits of the same individual were used as source for RNA extraction. All samples were frozen using liquid nitrogen and transported to the laboratory to be stored in an ultra-low-temperature freezer at −80 °C, prior to the DNA and RNA extraction process.
Total high-molecular-weight genomic DNA was extracted from the leaves of R. laevigata using the Tiangen Extraction Kit (Tiangen Biotech, China) that was based on the cetyltrimethylammonium bromide (CTAB) method. The concentration of the DNA extract was ascertained by the Quant-iT PicoGreen assay (Invitrogen, Waltham, MA, USA). The quality and quantity of the DNA samples were assessed using an ultraviolet spectrophotometer at 260 nm and 280 nm wave lengths. The DNA was fragmented with a Covaris M220 Focused-ultrasonicator instrument. Genomic DNA sequencing was conducted at Novogene Co., Ltd., Beijing, China.
Three different genomic DNA sequence libraries were constructed in this study. For the first approach, the DNA PCR-free libraries with insert sizes of 350 bp were constructed by using the NEBNext Ultra DNA library Pre-Kit for Illumina short-reads sequencing. The resulting barcoded libraries were sequenced on an Illumina NovaSeq6000 platform to generate 150-bp paired-end reads. Quality control was carried out on all the obtained reads by trimming adaptors and low-quality reads using fastp v0.23.220. A total of 53 Gb filtered short reads were obtained and used in subsequent data processing steps.
The second approach used the single-molecule real-time (SMRT) PacBio libraries that were constructed by using the PacBio 15-kb protocol and sequenced by using a PacBio Sequel IIe platform. The raw data generated with the PacBio Sequel IIe system were processed through the SMRT Analysis software suite v5.1.0 (https://www.pacb.com/products-and-services/analytical-software/smrt-analysis/), whereas the consensus HiFi reads were produced by the CCS subprogram (https://github.com/PacificBiosciences/ccs) with default parameters, which then generated approximately 36 Gb filtered data.
The last approach would be via the Hi-C library that was generated by using the restriction endonuclease MboI. The MboI-digested chromatin was labeled with biotin-14-dATP, and in situ DNA ligation was performed. The DNA underwent extraction, purification, and shearing. After A-tailing, pull-down, and adapter ligation steps, the DNA library was subjected to sequencing on an Illumina NovaSeq6000 platform. The total filtered data generated from Hi-C library was approximately 99 Gb (Table 1).
RNA extraction and sequencing
Fresh roots, stems, leaves, and fruits of the same R. laevigata individual were used for transcriptome sequencing. Total RNA was extracted using the TRIzol reagent (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s protocol. A 150-bp paired-end RNA-seq library was constructed and sequenced on an Illumina Novaseq6000 platform. After trimming the adapters and low-quality raw sequence reads using fastp, approximately 25 Gb clean data were obtained (Table 1).
Genome survey and assembly
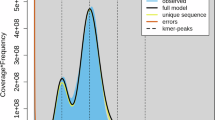
Previous studies have shown that R. laevigata is a diploid plant21. Flow cytometry analysis determined the nuclear DNA content of R. laevigata to be 0.51 pg22, indicating that its genome size is approximately 498.78 Mb (1 pg = 978 Mb). In this study, by employing Jellyfish v2.3.023 and GenomeScope224 with 19-kmer to estimate the genome size, heterozygosity, and repeat content based on Illumina short reads, the genome of R. laevigata was estimated to be 510.2 Mb in size, with a heterozygosity of 0.6% and a repeat content of 53.9% (Fig. 2), which is consistent with the results of previous studies. For the genome de novo assembly of R. laevigata, the bam2fastq v3.0.0 (https://github.com/PacificBiosciences/pbtk#bam2fastx) pipeline was first used to convert the raw data into the fastq format. A primary assembly was then constructed by using hifiasm v0.16.125 with default parameters, while the redundant sequences were removed using purge_dups v1.2.526 with default parameters. The draft genome assembly comprised of 56 contigs, which had a combined length of 494.2 Mb, an N50 size of 63.1 Mb, and a GC content of 38.8% (Table 2).

K-mer analysis of the R. laevigata genome based on the 19-mer frequency distribution.
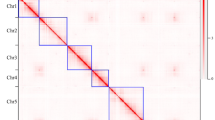
After removing adapter and low-quality sequences, we employed Juicer v1.627 to align the filtered Hi-C data to the primary assembly and generate a deduplicated list of Hi-C reads with default parameters. By using 3D-DNA v20100828, the primary contigs were anchored on the pseudochromosomes, while the heatmap for Hi-C interaction was generated using the 3D-DNA visualize module. The heatmap was further visualized, and manual curation was conducted with Juicebox v2.17.0029. The seven pseudochromosomes with lengths ranging from 49.9 Mb to 88.5 Mb were identified via distinct interaction signals in the Hi-C interaction heatmap (Fig. 3). Among them, two pseudochromosomes were gapless (Supplementary Table 1). The final genome assembly of R. laevigata was 494.2 Mb in size, with a scaffold N50 of 68.6 Mb (Table 2, Fig. 4).

Hi-C contact map at chromosome-scale for the genome assembly of R. laevigata.

Overview of genome features of R. laevigata. Syntenic blocks among inter-chromosome were analysed with MCScanX74. Each layer of the genome map represents the level of (a) gene density, (b) GC content, (c) transposon element density, (d) LTR/Copia density, (e) LTR/Gypsy density, (f) tandem repeat density.
Repeat annotation
Tandem repeats in the final genome assembly were identified by using TRF v4.0930 based on default parameters. By selecting the masked genome sequence as the target genome, transposon element (TE) identification was conducted using a combination approach via de novo and homology. The miniature inverted repeat transposable elements (MITEs) and long terminal repeat (LTR) elements were identified by de novo methods with MITE-Hunter (11–2011)31 and LTR_retriever v2.8.732. LTR_retriever incorporated LTRs predicted from LTRHarvest33 and LTR_FINDER v1.0.734. A species-specific repeat sequence library was also constructed using by RepeatModeler v2.0.335, while homology-based predictions were performed using by RepeatMasker v4.1.4 (http://repeatmasker.org/) and referring to the Repbase v2018 (http://www.girinst.org/server/RepBase/index.php) database. Annotation was carried out for all the repeat sequences with RepeatMasker based on a combined reference library that combined the MITEs, LTRs, species-specific library and homology library. The LTR Assembly Index (LAI) that is embedded in LTR_retriever was used to assess the assembly quality of the genome. A total of 285.6 Mb of repetitive elements were identified, which constituted 57.8% of the genome of R. laevigata. Among these repeats, the most abundant repeating element was the LTRs (42.14%). Within LTRs, Copia and Gypsy were the two most dominant classes in the genome, accounting for 16.9% and 24.7%, respectively (Fig. 5, Table 3).

Repeat landscape plots illustrating the transposon element (TE) accumulation history of R. laevigata genome, based on Kimura distance-based copy divergence analysis. The y-axis represents the percentage of the genome represented by each TE type, while the X-axis represents the sequence divergence at CpG adjusted Kimura substitution level. The legend on the top right corner indicates the colour code used for each type of transposon.
Gene prediction and annotation
Gene annotation was carried out by using the repeat masked genome. The protein-coding genes were annotated by incorporating transcriptional evidence, homology-based, and ab initio methods. For transcriptional evidence, fastp was used to trim the data separately, based on different tissues, and the clean data were aligned to the assembled genome of R. laevigata by using HISAT2 v2.2.136. The transcripts were assembled by using Stringtie v2.2.137 based on default parameters, while the transcripts from all samples were merged and subjected to protein-coding sequence prediction and quality filtering via TransDecoder (https://github.com/TransDecoder/TransDecoder/wiki), which is available in PASA v2.4.138. Only complete transcripts were retained for subsequent analysis. The protein sequences from Arabidopsis thaliana39, Crataegus pinnatifida var. major40, Eriobotrya japonica41, Fragaria vesca42, Malus domestica43, Potentilla anserina44, Prunus armeniaca45, Pyrus communis46, Rosa chinensis47, Rosa rugosa48, and Rubus idaeus49 were mapped to the assembled genome using by GeMoMa v1.950 to identify high-quality protein structures. Ab initio gene prediction was carried out by using Augustus v3.3.351, GeneMark-ESSuite v4.5752, and SNAP v2006-07-2853. They were all trained by high-quality transcripts from the previous step. Ab initio gene identification was performed according to the manuals. All predicted gene structures were integrated into a nonredundant gene set by using EVdenceModeler (EVM)38 v1.1.1. The weight values for high-quality RNA-seq transcripts, homologous proteins, and ab initio prediction were set to 10, 7, and 3 (Table 4). The EVM-predicted genes were further corrected by using PASA v2.4.138 to identify the untranslated and alternative splicing regions. In total, 37,117 protein-coding genes were predicted and annotated, with an average gene length of 3,047.31 bp (Table 5).
Functional annotation of the protein-coding genes was performed based on sequence similarity and the identification of conserved domains. For sequence similarity, the protein-coding genes were matched against the Universal Protein Knowledgebase (UniProt) database54, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database55, and the eggNOG56 database using diamond v2.0.1157 with a specified E-value cut-off of 1e-5. For the identification of conserved domains, InterProScan v5.5258 was employed to detect and classify domains and motifs by referring to the Pfam59, SMART60, PANTHER61, PRINTS62, and ProDom63 databases. With both approaches, a total of 34,074 (91.8%) protein-coding genes were functionally annotated (Table 6).
Noncoding RNA was annotated by using RNAmmer v1.264 for rRNA, tRNAscan-SE65 v2.0.9 for tRNA and the cmscan module in Infernal v1.1.266 for miRNA, snRNA. All predictions were performed with the default parameters, with the exception of tRNAs, which were filtered with a score >40. Finally, a total of 3,182 noncoding RNAs were predicted (Table 7), including 1,115 transfer RNAs (tRNAs), 1,289 ribosomal RNAs (rRNAs), 151 micro-RNAs (miRNAs), and 627 small nuclear RNAs (snRNAs).
Data Records
The Illumina short reads, PacBio long-reads, Hi-C sequencing data, and RNA-seq data have been deposited in the National Center for Biotechnology Information Sequence Read Archive (SRA) database with the accession number SRP51180767. The chromosome-scale genome assembly has been deposited in DDBJ/ENA/GenBank under the accession number JBEFKI00000000068. The genome annotation files were submitted to Figshare69.
Technical Validation
To assess the accuracy of the assembly, BWA70 v0.7.17 and Minimap2 v2.2471 were used to remapped the Illumina short reads and PacBio long reads into the final assembled genome. A 99.8% and 99.9% mapping rates of short and long reads was achieved, respectively. The Benchmarking Universal Single-Copy Orthologs (BUSCO, v5.4.772) was used to further evaluate the genome completeness by performing searches against the embryophyte_odb10 database. The BUSCO analysis showed that the final genome sequence contained 99.0% (1,598) complete BUSCOs (including 95.4% (1,540) single-copy BUSCOs, 3.6% (58) duplicated BUSCOs), 0.6% (9) fragmented BUSCOs and 0.4% (7) missing BUSCOs (Table 8). The LTR Assembly Index (LAI) was used to assess the quality of the R. laevigata genome assembly. An LAI value of 25.21 was obtained, indicating that the quality of R laevigata genome assembly qualified the level as a reference genome (Table 9). Merqury v1.373 was used to assess the consensus quality value (QV) of the R. laevigata genome assembly. The QVs were 64.8 and 55.3 estimated with HiFi and Illumina k-mers, respectively (Supplementary Figure 1). The above evaluation results indicate that the R. laevigata genome assembly has high accuracy and integrity.
Furthermore, BUSCO was used to evaluate the completeness of the R. laevigata genome annotation by performing searches against the embryophyte_odb10 database. The BUSCO analysis showed that 95.1% of conserved orthologous genes were complete in the predicted protein coding genes, comprising 90.8% single-copy and 4.3% duplicated genes (Table 10).
Code availability
No in-house code or scripts were used in this study. Commands and pipelines used for data processing were executed using their corresponding default parameters.
References
Yokoya, K. Nuclear DNA Amounts in Roses. Ann. Bot. 85, 557–561 (2000).
Gu, C. Z. & Kenneth R. R. Rosa. In Wu, Z.Y., Raven, P.H. and Hong, D. Y. (Eds.). Flora of China. Science Press, Beijing, China and Missouri Botanical Garden Press, St. Louis. 9, 339–381 (2003).
Fan, Y. L. et al. Screening of Rosa germplasm resources with resistance to aphids. J. Yunnan Univ. Nat. Sci. Ed. 43, 619–628 (2021).
Qiu, X. Q. et al. Powdery mildew resistance identification of wild Rosa germplasms. Acta Hortic. 1064, 329–335 (2015).
Gao, P. Y., Si, X. X., Liu, X. G. & Li, D. Q. Research Progress in Extraction, Purification of Triterpenoids From Rosa laevigata Michx. and Its Anti-Alzheimer’s Disease Activity. Contemp. Chem. Ind. 51, 1196–1200 (2022).
Yuan, J. Q. et al. New Triterpene Glucosides from the Roots of Rosa laevigata Michx. Molecules 13, 2229–2237 (2008).
Dai, H. N. et al. Triterpenoids from roots of Rosa laevigata. Chin. Tradit. Herb. Drugs 47, 374–378 (2016).
Yoshida, T., Tanaka, K., Chen, X. M. & Okuda, T. Tannis of rosaceous medicinal plants. V. Hydrolyzable tannis with dehydrodigalloyl group from Rosa laevigata Michx. Chem. Pharm. Bull. 37, 920–924 (1989).
Fan, X. R., Li, R. R., Lin, M. L., Liao, D. F. & Li, C. Research progress in medicinal parts of Rosa laevigata Michx. Chin. Pharm. J. 53, 1333–1341 (2018).
Huang, Y. L. & Liu, Y. Experimental study on the antineoplastic effect of polysaccharide from fructus Rosae Laevigatae in vitro. Genomics Appl. Biol. 34, 1848–1851 (2015).
An, D. Q., Yan, J. X. & Wang, X. L. UV determination of content of total flavonoids in Rosa laevigata Michx. from different places in different harvest period. J. Anhui Agric. Sci. 43, 79–80+83 (2015).
Li, B. L., Yuan, J. & Wu, J. W. A Review on the Phytochemical and Pharmacological Properties of Rosa laevigata: A Medicinal and Edible Plant. Chem. Pharm. Bull. 69, 421–431 (2021).
Gao, P. et al. Extraction and isolation of polyhydroxy triterpenoids from Rosa laevigata Michx. fruit with anti-acetylcholinesterase and neuroprotection properties. RSC Adv. 8, 38131–38139 (2018).
Choi, S. J. et al. Protective effect of Rosa laevigata against amyloid beta peptide-induced oxidative stress. Amyloid 13, 6–12 (2009).
Jung Choi, S. et al. Ameliorative effect of 1,2-benzenedicarboxylic acid dinonyl ester against amyloidbetapeptide-induced neurotoxicity. Amyloid 16, 15–24 (2009).
Fayaz, F., Singh, K., Gairola, S., Ahmed, Z. & Shah, B. A. A Comprehensive Review on Phytochemistry and Pharmacology of Rosa Species (Rosaceae). Curr. Top Med. Chem. 24, 364–378 (2024).
Jian, H. et al. Decaploidy in Rosa praelucens Byhouwer (Rosaceae) Endemic to Zhongdian Plateau, Yunnan, China. Caryologia 63, 162–167 (2014).
Roberts, A. V., Gladis, T. & Brumme, H. DNA amounts of roses (Rosa L.) and their use in attributing ploidy levels. Plant Cell Rep. 28, 61–71 (2008).
Nakamura, N. et al. Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. 25, 113–121 (2018).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Jian, H. Y. et al. Karyological diversity of wild Rosa in Yunnan, southwestern China. Genet. Resour. Crop Evol. 60, 115–127 (2013).
Li, S. Q., Zhang, C. & Gao, X. F. Estimation of nuclear DNA content of 17 Chinese wild rose species by flow cytometry. Plant Sci. J. 35, 558–565 (2017).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Robinson, J. T. et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 6, 256–258.e251 (2018).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199–e199 (2010).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Ou, S. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mob. DNA 10, 48 (2019).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457 (2020).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360, (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210 (2012).
Zhang, T. et al. Cultivated hawthorn (Crataegus pinnatifida var. major) genome sheds light on the evolution of Maleae (apple tribe). J. Integr. Plant Biol. 64, 1487–1501 (2022).
Wang, Y. A draft genome, resequencing, and metabolomes reveal the genetic background and molecular basis of the nutritional and medicinal properties of loquat (Eriobotrya japonica (Thunb.) Lindl). Hortic. Res. 8, 231 (2021).
Edger, P. P. et al. Single-molecule sequencing and optical mapping yields an improved genome of woodland strawberry (Fragaria vesca) with chromosome-scale contiguity. GigaScience 7, 1–7 (2018).
Qin, S. et al. A chromosome-scale genome assembly of Malus domestica, a multi-stress resistant apple variety. Genomics 115, 110627 (2023).
Gan, X. et al. Chromosome-Level Genome Assembly Provides New Insights into Genome Evolution and Tuberous Root Formation of Potentilla anserina. Genes 12, 1993 (2021).
Groppi, A. et al. Population genomics of apricots unravels domestication history and adaptive events. Nat. Commun. 12, 3956 (2021).
Yocca, A. et al. A chromosome-scale assembly for ‘d’Anjou’ pear. G3 14, jkae003 (2024).
Hibrand, L. et al. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants 4, 473–484 (2018).
Chen, F. et al. A chromosome-level genome assembly of rugged rose (Rosa rugosa) provides insights into its evolution, ecology, and floral characteristics. Hortic. Res. 8, 141 (2021).
Sassa, H. et al. Chromosome-scale genome sequence assemblies of the ‘Autumn Bliss’ and ‘Malling Jewel’ cultivars of the highly heterozygous red raspberry (Rubus idaeus L.) derived from long-read Oxford Nanopore sequence data. Plos One 18, e0285756 (2023).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics 19, 1–12 (2018).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467 (2005).
Besemer, J. & Borodovsky, M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33, W451–W454 (2005).
Leskovec, J. & Sosič, R. SNAP: A General-Purpose Network Analysis and Graph-Mining Library. ACM Trans. Intell. Syst. Technol. 8, 1–20 (2016).
The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017).
Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2014).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Schultz, J. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 28, 231–234 (2000).
Mi, H. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 33, D284–D288 (2004).
Attwood, T. K. The PRINTS database: A resource for identification of protein families. Brief. Bioinform. 3, 252–263, (2002).
Bru, C. The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Res. 33, D212–D215 (2004).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 25, 955–964 (1997).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP511807 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBEFKI000000000.1 (2024).
Zhang, Y. H. Genome assembly and annotation files of Rosa laevigata. Figshare https://doi.org/10.6084/m9.figshare.25998949 (2024).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Manni, M. et al. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49 (2012).
Acknowledgements
The authors thank Guodong Li and Chunlin Gao from Yunnan University of Chinese Medicine and Ticao Zhang from Kunming Institute of Botany, Chinese Academy of Sciences for their technical assistance and valuable discussions. This work was financially supported by the Technology Talents and Innovation Team Project of Yunnan Province (No. 202305AS350002 to Jian) and the National Natural Science Foundation of China (No. 31760048 to Zhang).
Author information
Authors and Affiliations
Contributions
Jian H.Y., Yan H.J., Qiu X.Q. and Zhang H. conceived the research and collected materials. Wang Y. assembled the sequences and analyzed the data. Wang Y. and Zhang Y.H. prepare the manuscript. Zhang Y.H. and Jian H.Y. revised the manuscript. All authors read, edited and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Yan, H., Qiu, X. et al. A high-quality chromosome-scale genome assembly of the Cherokee rose (Rosa laevigata). Sci Data 12, 132 (2025). https://doi.org/10.1038/s41597-025-04461-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04461-7
