
Abstract
Zenia insignis Chun, from the subfamily Dialioideae of the family Fabaceae, is a tree species of significant economic and ecological values. It is a near threatened species as per the IUCN Red List. In this study, we generated a reference genome for Z. insignis, with a contig N50 of 6.02 Mb, a total length of 352.84 Mb, and 97.71% of the sequences assembled into 14 pseudo-chromosomes. The BUSCO assessment score for completeness is 97.30%, and the LAI index assessment score for continuity is 14.57. The present study contributes to provide a reference genome for the subfamily Dialioideae. The reference genome will provide a valuable resource for the phylogenomic studies of the family Fabaceae and facilitate further research on Z. insignis.
Similar content being viewed by others


Background & Summary
Zenia insignis Chun, a member of the subfamily Dialioideae (Fabaceae), is a tree species recognized as one of the Grade-II Wild Plants of National Priority Protection in China (https://www.gov.cn/gongbao/content/2000/content_60072.htm) and is listed as near threatened species on the International Union for Conservation of Nature (IUCN) Red List (http://www.iucnredlist.org/). This species was named by the renowned botanist Woon-Young Chun in honor of Hongjun Ren (H.C. Zen)1. The species is primarily distributed in southern and southwestern China at an altitude range of 200–1000 meters and northern Vietnam2. Zenia insignis boasts a robust root system and strongly resists drought and infertile conditions3. In the karst regions of southern and southwestern China, it has been utilized as an outstanding tree species for afforestation on rocky terrain and as a pioneering species for the control of rocky desertification3,4,5. Additionally, it has significant economic value. The wood of Z. insignis is versatile and suitable for producing doors, boards, agricultural implements, and furniture6,7. The plant’s tender leaves serve as livestock feed and green manure in rice fields5,6,7.
Research on Z. insignis has predominantly concentrated on seedling8,9, reforestation technologies3,4, and phylogenetic studies10,11,12,13,14,15,16,17. Though partial and complete plastid sequences10,11,12,14 and nuclear sequences13,15,16,17 have been utilized in constructing the phylogenetic tree of the family Fabaceae, some deep phylogenetic relationships within Fabaceae remain to be fully elucidated. A chromosome-level genome sequence of Z. insignis would be instrumental in clarifying these phylogenetic relationships. In this study, we have successfully obtained the reference genome of Z. insignis.
In this study, we integrated multiple sequencing data types to assemble and annotate the genome of Z. insignis. Specifically, we used PacBio continuous long reads (CLR) (174.14 Gb, 363.43 × coverage), next-generation sequencing (NGS) reads (68.98 Gb, 143.71 × coverage), Hi-C data (59.63 Gb, 124.45 × coverage), and full-length and NGS transcriptome reads. The resulting reference genome consists of 14 pseudo-chromosomes and spans a total length of 352.84 Mb with a contig N50 of 6.02 Mb. The genome’s LAI index score is 14.57. The GC content of the genome is 34.38%, and the proportion of repeat sequences is 35.47%, with long terminal repeat (LTR) elements being the most prevalent, accounting for 9.03%. We also obtained the plastid genome sequence of Z. insignis with a length of 159,390 bp.
Methods
Genome size estimation and sequencing
Fresh young leaves of Z. insignis were collected from the Kunming Botanical Garden (KBG), Kunming Institute of Botany, Chinese Academy of Sciences (KIB), Yunnan Province, China. The voucher specimen (Collection Number: Yi20005) had been deposited at the Herbaria (KUN) of Kunming Institute of Botany, Chinese Academy of Sciences, Yunnan Province, China. The leaves were immediately flash-frozen in liquid nitrogen to preserve their cellular structure and molecular content. Subsequently, the samples were transferred to an ultra-low temperature freezer maintained at −80 °C for storage.
DNA/RNA extraction, library construction, and sequencing processes were provided by Wuhan Frasergen Bioinformatics Co., Ltd. The major steps are summarized below:
-
1.
Genomic DNA was extracted from the young leaves of Z. insignis using the modified cetyltrimethylammonium bromide (CTAB) method18,19. Total RNA was isolated using a Trizol reagent (Invitrogen, CA, USA). The quantity and quality of the DNA/RNA were determined using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
-
2.
The quality-compliant DNA was further tested for purity and integrity using 0.8% agarose gel electrophoresis and a Qubit dsDNA HS Assay Kit on a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). RNA contamination was identified using a 1.5% agarose gel electrophoresis, and purity and integrity were evaluated using a NanoDrop 2000 and a Bioanalyzer 2100 system (Agilent Technologies, CA, USA).
-
3.
According to the manufacturer’s instructions, a short-read library was constructed using the VAHTS Universal DNA Library Prep Kit (Vazyme, Nanjing, China). The library’s quantification and sizing were measured using a Qubit 3.0 Fluorometer and a Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Paired-end sequencing of the short-read library was conducted on an MGI-SEQ 2000 platform (BGI, China).
-
4.
To perform long-read sequencing, the library was prepared in accordance with the PacBio single-molecule real-time (SMRT) protocol, and then sequenced on a Pacific Biosciences Sequel II System in CLR mode.
-
5.
The construction of the Hi-C library commenced with the cross-linking of fresh young leaves of Z. insignis under vacuum infiltration with 3% formaldehyde. Following lysis, digestion, labelling, ligation, purification, adaptor addition, and PCR amplification, the Hi-C libraries were quantified and sequenced on the MGI-SEQ 2000 platform.
-
6.
For short-read transcriptome sequencing, RNA was isolated from Z. insignis young leaves using the QIAGEN kit according to the manufacturer’s instructions. The short-read library was subsequently constructed and sequenced on the MGI-SEQ 2000 platform using paired-end sequencing.
-
7.
The full-length transcriptome sequencing was conducted using the Pacific Biosciences DNA Template Prep Kit 2.0 for library construction (Pacific Biosciences, CA, USA). Subsequently, the SMRT sequencing was performed on a Pacific Biosciences Sequel II platform.
Our study generated 44.14 Gb of raw data for the whole genome, 174.14 Gb of PacBio CLR data, 59.63 Gb of Hi-C data, and 81.92 Mb of full-length transcriptome data (Table 1).
The genome size of Z. insignis was assessed using flow cytometry analysis conducted on a BD FACScalibur flow cytometer (BD Biosciences). The test sample preparation was conducted in accordance with the manufacturer’s guidelines and aligned with established methodologies from previous literature20,21. As a standard reference, we utilized Zea mays L. (maize)22, a species with a well-documented genome size of 2.3 Gb. The analysis yielded a result indicating that the genome size of Z. insignis is approximately 480 ± 4.8 Mb (Fig. 1a).

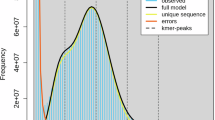
The result of genome size estimation for Zenia insignis. (A) Flow cytometry-based estimation result of Zenia insignis (left peak, the sample) and Zea mays (right peak, the reference). (B) The 21-kmer distribution estimation result was obtained using GenomeScope2. The distribution of 21-kmer exhibited two major peaks, indicating certain heterozygosity within the genome, and high-frequency k-mers with coverage near 1 that was possibly generated by sequencing errors.
A K-mer analysis was conducted on Z. insignis to estimate its genome size. Next-generation sequencing (NGS) data was obtained from Frasergen and processed Fastp v0.19.323. This involved the removal of adapter sequences, the filtration of reads deemed to be of insufficient length, and the elimination of those with low quality. Subsequently, Jellyfish v2.2.1024 was employed to calculate the frequency distribution of the cleaned data using 21-mers. Subsequently, GenomeScope v2.025 was employed to estimate the fundamental characteristics of the genome. The genome size of Z. insignis was estimated to be approximately 364.94 Mb, with a heterozygosity rate of 0.842% (Fig. 1b).
De novo genome assembly
Figure 2 illustrates the methodology employed for the chromosome-level genome assembly of Z. insignis. The objective was to utilize three distinct assemblers, compare their resulting assemblies, and achieve a high-quality assembly. The assemblers employed were Falcon v0.3.026, Canu v2.1.127, and Flye v2.928. The following section provides a summary of the assembly process.
-
1.
The Falcon software was employed to assemble the CLR long reads. The resulting contigs were then polished using long reads with GCpp v2.0.0 (https://github.com/PacificBiosciences/gcpp). Subsequently, the processes above yielded the assembly contig, designated as Assembly_Contig1. Subsequently, Assembly_Contig1 was subjected to further refinement with short reads, employed NextPolish v1.3129. The output from the process was designated as the ‘Falcon_contig’. To eliminate redundancy, the Falcon_contig was processed with Purge_Dups v1.2.530, and the resulting non-redundant contigs were designated as the ‘Falcon_purge_contigs’.
-
2.
The Canu software was employed to assemble the CLR long reads, and the result was subsequently polished with long reads by GCpp. Through these processes, we obtained Assembly_Contig2. Assembly_Contig2 was then polished with short reads using NextPolish, with the output from this step designated as the ‘Canu_contig’. The Canu_contig was subsequently subjected to further processing to eliminate redundancy, with the result that the non-redundant contigs were designated as the ‘Canu_purge_contigs’. Purge_Dups also employed the process of eliminating redundancy.
-
3.
The third assembler employed was Flye, which accepted the error-corrected reads generated by Canu as input. The output from Flye was polished with long reads by GCpp, resulting in the generation of Assembly_Contig3. Subsequently, the contig was polished with short reads using NextPolish, resulting in the ‘Flye_contig’. The Flye_contig was subjected to a process of redundancy elimination using Purge_Dups, resulting in the generation of a set of non-redundant contigs designated as the ‘Flye_purge_contigs’.
-
4.
Six distinct results were obtained after completing the three steps above. The following contigs were identified: Falcon_contig, Canu_contig, Flye_contig, Falcon_purge_contig, Canu_purge_contig, and Flye_purge_contig.

Flowchart of Zenia insignis genome assembly. (A) The pipeline of genome size survey for Zenia insignis. Two methods were used to predict the genome of Zenia insignis: flow cytometry and genome survey. (B) The pipeline of assembly process for three assembly software and two datasets. In this study, three assembly software, Falcon, Canu, and Flye, were utilised to assemble the redundant and non-redundant data, respectively. “Falcon_contig” denotes the result of assembling the redundant dataset using Falcon software, “Falcon_purge_contig” denotes the result of assembling the non-redundant dataset using Falcon software, and so forth. The results obtained from the assembly were evaluated using N50, BUSCO and LAI. (C) The pipeline of Hi-C assembly. Hi-C assembly was first performed on the redundant dataset, and the Falcon_purge_contig dataset was considered the best assembly result through interaction heatmap analysis combined with N50, BUSCO and LAI scoring. Further manual adjustments were made to obtain a high quality genome of Zenia insignis.
BUSCO v5.4.531 with 1614 embryophyta single-copy orthologs was employed to assess the completeness of the assembly, the N5032 metric provided insights into the distributions of contigs, and the LTR Assembly Index (LAI) v2.9.033 was employed to evaluate the contiguity of the assembled genome. The LTR Assembly Index (LAI) is a metric that evaluates the continuity of genome assemblies33. It is calculated in two steps. First, raw LAI is calculated as the length of intact long terminal repeats retrotransposons (LTR-RTs)/length of total LTR-RTs. As the proportion of intact LTR-RTs is dynamic due to their amplification and removal, it is assumed that recently generated LTR-RTs are likely to be intact, and ancient LTR-RTs are likely to be degenerated33. Therefore, raw LAI is then corrected using a correction factor estimated from the age of intact LTR-RTs. A higher LAI indicates a greater number of LTR-RTs are correctly assembled, which also indicates a higher quality of the genome. Genomes with LAIs under 10 are considered as draft quality, while genomes above 10 are considered as reference quality33. The results are presented in Tables 2–4. Notably, the Canu_purge assembly exhibited the highest N50 value, the Flye_purge assembly achieved the optimal BUSCO score, and the Falcon_purge_contig assembly exhibited the highest LAI score. In other words, the result generated by Purge_Dups exhibited a better contiguity. Manual inspection using Hi-C reads revealed a low redundancy in Falcon_purge assembly (Supplementary Figure 1). Consequently, the results generated by Purge_Dups were selected for further comparison. A comparison of the Falcon_purge assembly with other results generated by Purge_Dups revealed that it exhibited high contiguity with N50 of 6.02 Mb, BUSCO completeness of 97.30%, and LAI score of 14.57 that is similar to the quality of Arabidopsis thaliana (L.) Heynh (LAI = 14.9)33, suggesting a high-quality reference genome being assembled.
During the chromosome assembly phase, we integrated Hi-C data with the previously assembled contigs to achieve chromosome-level scaffolding. Specifically, Juicer34 was employed to align the Hi-C reads to the contig genome. Subsequently, the 3D-DNA pipeline35 was used to map the contigs onto chromosome-level scaffolds. For a visual representation of the Hi-C data, Juicebox v1.2236 was employed. The results of the chromosome assembly also demonstrated that the Purge_Dups results exhibited lower redundancy, compared to the rest two assemblies (Supplementary Figure 1). The Purge_Dups result was chosen as the optimal choice, and subsequently, the heatmap of the result was manually adjusted (Fig. 3a). The final outcome of this process was the generation of 14 pseudo-chromosomes with a total length of 352.84 Mb, as illustrated in Fig. 3a,b.

The chromosome and plastid information of Zenia insignis. (A) The interaction heat map of Hi-C among chromosomes for the Zenia insignis genome. The Hi-C interaction matrix shows the pairwise correlations among the 14 pseudomolecules, and the presence of a red colouration is indicative of an interaction between the sequences, with the shade of red denoting the magnitude of this interaction. (B) The high-quality genomic landscape of Zenia insignis. The genome diagram has four circles from the outside: gene density, GC content, chromosomal length, and intraspecies collinearity. (C) The visualization of the plastid genome of Zenia insignis using OGDRAW. The outside circle displays genes color-coded according to their functional groups. The second circle illustrates the large single-copy (LSC), inverted repeat (IRA, IRB), and small single-copy (SSC) regions. The third circle delineates the percentage of GC content and genomic positions in kilobase pairs. (D) Compared analysis of plastid for Zenia insignis. Gene(green), CDS (yellow), RNA (red), and IR regions (grey) have been color-coded according to their functional groups. The identity of the two plastid genomes sequences exhibited high overall similarity, except for some regions in non-coding regions.
The GetOrganelle v1.7.3.337 software was employed to assemble the plastid genome, successfully acquiring the complete plastid genome sequence depicted in Fig. 3c. The newly assembled plastid genome spans 159,390 base pairs (bp). The plastid genome is annotated using the PGA tool38, as shown in Fig. 3c, drawn by OGDRAW v1.3.139. For comparative analysis of the plastid genome, we employed the MAFFT40 to align our sequence with a published plastid genome of Z. insignis14 (NC_045299.1) obtained from the National Center for Biotechnology Information(NCBI). As illustrated in Fig. 3d, the alignment result revealed that apart from some single nucleotide polymorphisms (SNPs) in non-coding regions, there were no significant differences in large-scale sequences.
Repeat annotation
The RepeatModeler v2.0.141 software was employed to predict the occurrence of repeat sequences within the genome of Z. insignis. RepeatModeler integrates three de novo repeat-finding programs: RECON v1.0842, RepeatScout v1.0.643, and LTRHarvest v5.944/LTR_retriever v2.9.045. For our analysis, we employed rmblast v2.13.0 (https://www.repeatmasker.org/rmblast/) as the search engine. Subsequently, the output from RepeatModeler was processed using RepeatMasker v4.1.446 to identify and mask the repetitive elements within the Z. insignis genome.
Table 5 presents the results of the repeat sequence analysis for Z. insignis. A total of 128.08 Mb were identified as repetitive sequences, constituting 35.47% of the pseudo-chromosome sequences. The analysis revealed four primary classes of transposable elements within the genome: 9.03% for long terminal repeats (LTRs), 3.08% for DNA elements, 1.81% for long interspersed nuclear elements (LINEs), and 0.08% for short interspersed nuclear elements (SINEs).
Gene prediction and functional annotation
A hybrid approach was implemented to achieve high-quality gene prediction, incorporating de novo prediction, homology-based prediction, and RNA-Seq-assisted prediction. We employed a similar pipeline to the AnnoSmk v1.0 pipeline (https://github.com/yanhui-k/AnnoSmk) for gene prediction. Firstly, the RepeatMasker v4.1.446 software was employed to generate repeat evidence. For homology-based prediction, three sets of protein data for Arabidopsis thaliana47, Medicago truncatula Gaertn.48 and Swiss-Prot(https://www.uniprot.org/) were used to construct homologous gene models by genBlastG v1.3849 as protein evidence. For transcriptome evidence, HISAT2 v2.2.150 was employed to map paired-end short transcriptome reads to the Z. insignis genome. Then, the mapped reads were assembled using Trinity v2.11.051. Concurrently, we processed full-length transcriptome data with the IsoSeq 3 v3.4.052 pipeline to produce high-quality consensus transcript sequences. Minimap2 v2.2253 was employed to align both short and long-read data, from which the transcriptome evidence was derived. The integrated repeat, protein, and transcript evidence were then subjected to gene prediction using MAKER2 v3.01.0354. Three rounds of MAKER2 analysis were executed. In the second and third rounds, the output generated through de novo gene prediction using AUGUSTUS v3.4.055 was also included. BUSCO was employed to evaluate the gene prediction results after each round of MAKER2, yielding scores of 46.60%, 96.60%, and 96.80%, respectively (Table 6). Following the third round of MAKER2, the results were input into the PASA pipeline v2.5.056, resulting in the final output. The final BUSCO score for the gene prediction was 97.20% (Table 6). In conclusion, a total of 33,322 gene models were predicted.
The functional annotation of protein-coding genes was conducted using the eggNOG-mapper website (http://eggnog-mapper.embl.de/), which employs the eggNOG-mapper v2.1.1257 and the eggNOG 5 database. This tool utilizes several comprehensive databases, including Pfam58, InterPro59, Gene Ontology (GO)60, and the Kyoto Encyclopedia of Genes and Genomes (KEGG)61, to annotate protein sequences. This analysis resulted in the successful functional annotation of 30,143 protein-coding genes, representing 90.46% of the total gene models Fig. 4.

Flowchart of Zenia insignis gene prediction. The hybrid approach incorporates de novo prediction, homology-based prediction, and RNA-Seq-assisted prediction.Three evidences, including repeat, protein, and transcript evidence, was generated firstly. The RepeatMasker software was employed to generate repeat evidence. Three sets of protein data for Arabidopsis thaliana, Medicago truncatul and Swiss-Prot were used to construct homologous gene models by genBlastG as protein evidence. The evidence from the transcript was divided into two sections: one comprising NGS transcript data, and the other full-length transcript data. HISAT2 was used to map paired-end short transcriptome reads to the Zenia insignis genome. Thereafter, the mapped reads were retrieved and subsequently assembled using Trinity. The full-length transcriptome data were processed using the IsoSeq 3. Minimap2 was used to align both short and long-read data, from which the evidence of the transcriptome was derived. The integrated repeat, protein, and transcript evidence were then subjected to gene prediction using MAKER2. Three rounds of MAKER2 analysis were executed. In the second and third rounds, the output generated through de novo gene prediction using AUGUSTUS was also included. Following the third round of MAKER2, the results were input into the PASA pipeline. PASA pineline could optimise and refine MAKER2 outputs. The BUSCO was employed to evaluate the gene prediction results after each round of MAKER2 and PASA pipeline.
Data Records
The data associated with this paper have been submitted to the Sequence Read Archive (SRA) at the National Center for Biotechnology Information(NCBI) under the project number PRJNA1124267, including short-read genomic data62,63 (accession number SRR29423585 and SRR29423588), short-read transcriptomic data64,65,66,67,68,69 (accession number SRR29423578-SRR29423581, SRR29423586, and SRR29423587), long-reads data including PacBio long-reads70 (accession number SRR29423589), full-length transcriptomic data71,72 (accession number SRR29423582 and SRR29423583), and Hi-C data73 (accession number SRR29423584).
Genome assembly of Z. insignis have been submitted to Science Data Bank with URL: https://doi.org/10.57760/sciencedb.1093174. Annotation data are available in the Science Data Bank with URL: https://doi.org/10.57760/sciencedb.0782875. Assembly and annotation of the plastid genome have been deposited in the NCBI76 (accession number PP882779).
Technical Validation
As part of our sequencing pipeline, we conducted a rigorous sample quality assessment for both DNA and RNA samples used in the study. The specific procedures and criteria for this assessment are detailed in our paper’s ‘Methods’ section, ensuring that only high-quality samples were selected for subsequent sequencing and analysis.
In our study, we employed two distinct methodologies to estimate the genome size of Z. insignis. The first method involved Flow cytometry analysis, which provided a quick and accurate measurement of the relative DNA content within the cells of Z. insignis. The second method was K-mer analysis, which leverages the frequency distribution of DNA sequences to predict genome size. The details have been mentioned in the ‘Method’ section. Both approaches were crucial in validating the genome size and ensuring the reliability of our genomic data.
In our assessment of the de novo genome assembly for Z. insignis, we utilized a suite of metrics, including N50, BUSCO, and LAI, to evaluate the performance of various assembly software tools. The results of these evaluations are presented in Tables 2–4, which compare the assembly quality, completeness, and continuity across different assemblers. Based on these assessments, Falcon-purged output demonstrated the most favorable balance of completeness and continuity, making it the choice for subsequent downstream analyses. In summary, we have successfully generated a high-quality reference genome for Z. insignis, which may promote further biological and comparative genomic studies of this species.
Code availability
We used all software or pipelines, depending on the tools’ manuals or protocols. The software’s version is described in the methods section, and the parameters are given if not default. The custom code has been submitted to the GitHub website: https://github.com/csykib/genome_assembly_annotation.
References
Chun, W.-Y. A new genus in the Chinese flora. Sunyatsenia 6, 196–198 (1946).
Xu, L.-R. et al. Fabaceae(Leguminosae). Flora of China 10, 27 (Science Press & Missouri Botanical Garden Press, 2010).
Ao, H., He, D., Zhang, Z., Xia, H. & Yang, W. Zenia insignis communities in limestone areas of Guangdong. Trop. Geogr. 17, 275–276+278–282, http://dx.chinadoi.cn/10.13284/j.cnki.rddl.000227 (1997).
Zhang, Z.-P., He, D.-Q., Ao, H.-X., Yang, W.-J. & Wu, H.-M. The characteristic of physiological ecology of the main tree species used for afforestation at limestone regions in North Guangdong. Acta Phytoecol. Sin. 17, 133–142 (1993).
Zhang, Y. Growth performance on acid soil and cultivation technique of Zenia insignis. Guangdong Forest. Sci. Tech. 23, 76–79, http://dx.chinadoi.cn/10.3969/j.issn.1006-4427.2007.01.016 (2007).
Hou, L., Li, Y., Li, P., Yang, Y. & Xie, Y. Study of Zenia insignis Chun. comprehensive utilization. Sci. Silvae Sin. 37, 139–143, http://dx.chinadoi.cn/10.11707/j.1001-7488.20010322 (2001).
Yu, G. & Zhang, Y. A preliminary study on the wood fibre form of Zenia insignis introduced in Fujian Province. J. Fujian Forest. Sci. Tech. 25, 31–33, http://dx.chinadoi.cn/10.3969/j.issn.1002-7351.1998.01.006 (1998).
Qin, Y.-R., Jiang, G.-M., Cen, Z.-Y., Chen, Y.-S. & Lan, C.-Y. Research on the seed’s germination characteristic of Zenia insignis as the vanguard tree for afforestation in karst areas. Seed 27, 15–21, http://dx.chinadoi.cn/10.3969/j.issn.1001-4705.2008.12.005 (2008).
Tang, W., Pan, B., Wei, H., Huang, Z. & Lu, Q. Comparative study on the characteristics of seed germination and seedling growth of different Zenia insignis provenances in Guangxi. Seed 38, 93–98, http://dx.chinadoi.cn/10.16590/j.cnki.1001-4705.2019.12.93 (2019).
Bai, H.-R., Oyebanji, O., Zhang, R. & Yi, T.-S. Plastid phylogenomic insights into the evolution of subfamily Dialioideae (Leguminosae). Plant Diver. 43, 27–34, https://doi.org/10.1016/j.pld.2020.06.008 (2021).
Herendeen, P. S. Phylogenetic relationships in Caesalpinioid Legumes: a preliminary analysis based on morphological and molecular data. Advances in legume systematics 10, 37–62 (Royal Botanic Gardens, 2003).
Kajita, T., Ohashi, H., Tateishi, Y., Bailey, C. D. & Doyle, J. J. rbcL and legume phylogeny, with particular reference to Phaseoleae, Millettieae, and allies. Syst. Bot. 26, 515–536, https://doi.org/10.1043/0363-6445-26.3.515 (2001).
Koenen, E. J. M. et al. Large-scale genomic sequence data resolve the deepest divergences in the legume phylogeny and support a near-simultaneous evolutionary origin of all six subfamilies. New Phytol. 225, 1355–1369, https://doi.org/10.1111/nph.16290 (2020).
Lai, Q., Tu, T. & Zhang, D. The complete plastid genome of Zenia insignis Chun (Leguminosae). Mitochondrial DNA B Resour. 4, 2926–2927, https://doi.org/10.1080/23802359.2019.1660266 (2019).
The Legume Phylogeny Working Group. A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny. Taxon 66, 44–77, https://doi.org/10.12705/661.3 (2017).
Zhang, R. et al. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst. Biol. 69, 613–622, https://doi.org/10.1093/sysbio/syaa013 (2020).
Zhao, Y. et al. Nuclear phylotranscriptomics and phylogenomics support numerous polyploidization events and hypotheses for the evolution of rhizobial nitrogen-fixing symbiosis in Fabaceae. Mol. Plant. 14, 748–773, https://doi.org/10.1016/j.molp.2021.02.006 (2021).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Allen, G. C., Flores-Vergara, M. A., Krasynanski, S., Kumar, S. & Thompson, W. F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325, https://doi.org/10.1038/nprot.2006.384 (2006).
Doležel, J. & Bartoš, J. Plant DNA flow cytometry and estimation of nuclear genome size. Ann. Bot. 95, 99–110, https://doi.org/10.1093/aob/mci005 (2005).
Doležel, J., Greilhuber, J. & Suda, J. Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2, 2233–2244, https://doi.org/10.1038/nprot.2007.310 (2007).
Schnable, P. S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115, https://doi.org/10.1126/science.1178534 (2009).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054, https://doi.org/10.1038/nmeth.4035 (2016).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736, https://doi.org/10.1101/gr.215087.116 (2017).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546, https://doi.org/10.1038/s41587-019-0072-8 (2019).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2019).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921, https://doi.org/10.1038/35057062 (2001).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126, https://doi.org/10.1093/nar/gky730 (2018).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Jin, J.-J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241, https://doi.org/10.1186/s13059-020-02154-5 (2020).
Qu, X.-J., Moore, M. J., Li, D.-Z. & Yi, T.-S. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 50, https://doi.org/10.1186/s13007-019-0435-7 (2019).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64, https://doi.org/10.1093/nar/gkz238 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780, https://doi.org/10.1093/molbev/mst010 (2013).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276, https://doi.org/10.1101/gr.88502 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2017).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. Chapter 4, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s05 (2009).
The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815, https://doi.org/10.1038/35048692 (2000).
Pecrix, Y. et al. Whole-genome landscape of Medicago truncatula symbiotic genes. Nat. Plants 4, 1017–1025, https://doi.org/10.1038/s41477-018-0286-7 (2018).
She, R. et al. genBlastG: using BLAST searches to build homologous gene models. Bioinformatics 27, 2141–2143, https://doi.org/10.1093/bioinformatics/btr342 (2011).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Wang, B. et al. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 7, 11708, https://doi.org/10.1038/ncomms11708 (2016).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491, https://doi.org/10.1186/1471-2105-12-491 (2011).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419, https://doi.org/10.1093/nar/gkaa913 (2020).
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic Acids Res. 51, D418–D427, https://doi.org/10.1093/nar/gkac993 (2022).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nature Genet. 25, 25–29, https://doi.org/10.1038/75556 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462, https://doi.org/10.1093/nar/gkv1070 (2015).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423588 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423585 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423587 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423586 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423581 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423580 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423579 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423578 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423589 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423583 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423582 (2024).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRR29423584 (2024).
Science Data Bank https://doi.org/10.57760/sciencedb.10931 (2024).
Science Data Bank https://doi.org/10.57760/sciencedb.07828 (2024).
NCBI GenBank http://identifiers.org/ncbi/insdc:PP882779 (2024).
Acknowledgements
This research was supported by the Major Program for Basic Research Project of Yunnan Province (202401BC070001), the Youth Talents Special Project of Yunnan Province ‘Xingdian Talents Support Program’ (XDYC-QNRC-2022-0423), the National Natural Science Foundation of China, key international (regional) cooperative research project (No. 31720103903), the Yunnan Provincial Basic Research Program Youth Project (2019FD058), and the National Natural Science Foundation of China (No. 32270247). We are grateful to the Germplasm Bank of Wild Species, particularly the Molecular Biology Experiment Center and the iFlora High Performance Computing Center, the Kunming Botanical Garden, Ms. Ding-Jie Wang, and Mr. Zuo-Ying Xiahou.
Author information
Authors and Affiliations
Contributions
T.-S.Y. and H.L. designed the project. S.-Y.C. and Z.-Y.Y. performed the experiments. S.-Y.C., H.L. and R.Z. analyzed the data. S.-Y.C., T.-S.Y. and H.L. wrote the paper. All Authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, SY., Yang, ZY., Zhang, R. et al. Chromosome-level reference genome assembly for the protected resource plant, Zenia insignis. Sci Data 12, 459 (2025). https://doi.org/10.1038/s41597-025-04780-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04780-9
This article is cited by
-
Chromosome scale assembly unveils genomic structure and gene families of Calotropis procera
Molecular Genetics and Genomics (2025)
